


Abstract
Adult stem cell quiescence is critical to ensure regeneration while minimizing tumorigenesis. Epigenetic regulation contributes to cell cycle control and differentiation, but few regulators of the chromatin state in quiescent cells are known. Here we report that the tumor suppressor PRDM2/RIZ, an H3K9 methyltransferase, is enriched in quiescent muscle stem cells in vivo and controls reversible quiescence in cultured myoblasts. We find that PRDM2 associates with >4400 promoters in G0 myoblasts, 55% of which are also marked with H3K9me2 and enriched for myogenic, cell cycle and developmental regulators. Knockdown of PRDM2 alters histone methylation at key promoters such as Myogenin and CyclinA2 (CCNA2), and subverts the quiescence program via global de-repression of myogenesis, and hyper-repression of the cell cycle. Further, PRDM2 acts upstream of the repressive PRC2 complex in G0. We identify a novel G0-specific bivalent chromatin domain in the CCNA2 locus. PRDM2 protein interacts with the PRC2 protein EZH2 and regulates its association with the bivalent domain in the CCNA2 gene. Our results suggest that induction of PRDM2 in G0 ensures that two antagonistic programs—myogenesis and the cell cycle—while stalled, are poised for reactivation. Together, these results indicate that epigenetic regulation by PRDM2 preserves key functions of the quiescent state, with implications for stem cell self-renewal.
INTRODUCTION
Epigenetic regulatory mechanisms play a crucial role in cell fate decisions, whereby global and local controls are imposed on chromatin and result in distinct transcriptional programs. The epi-genome of pluripotent embryonic stem cells (ESC) is highly permissive, accommodating both self-renewal and broad differentiation potential. During development, chromatin configuration becomes progressively restrictive as cells commit and differentiate into specific lineages. Regulation at the level of chromatin is emerging as a primary determinant in the establishment and maintenance of heritable gene expression patterns (1–4).
The global chromatin landscape is controlled by a hierarchy of mechanisms, of which regulation at the level of the basic unit, the nucleosome, is best understood. Interactions of the core nucleosomal histones (H2A, H2B, H3 and H4) leave their N terminal tails accessible to a range of post-translational modifications that are deposited, read or erased by a wide variety of chromatin modifying enzymes, altering the packaging of DNA. Dynamic changes in histone modifications can therefore also alter DNA-transcription factor interactions, and may either accompany or precede transcriptional activation or repression. Thus, the ‘histone code’ embodies gene regulatory information that is embedded in complex cell type- and cell state-specific combinations of histone modifications (5). Typically, in addition to the requisite RNA polymerase II (pol II) binding, transcription activation correlates with tri-methylation of lysine 4 of H3 (H3K4me3), together with histone acetylation (H3K9Ac). By contrast, transcription repression often involves tri-methylation of lysine 27 of H3 (H3K27me3) and di-or tri-methylation of lysine 9 of H3 (H3K9me2/3), through the recruitment of repressive protein complexes.
Heritability of epigenetic information has to meet the challenge of chromatin disassembly and reassembly during DNA synthesis, necessitating cellular memory mechanisms, particularly in adult stem cells (ASC). Adult tissues are comprised of cells in distinct non-proliferating states with distinct functions. In skeletal muscle, differentiated myofibers are permanently arrested (post-mitotic), but a rare population of satellite stem cells enters an alternate cell cycle exit (quiescence or G0), retaining the option to reactivate and repair damage (reviewed in (6)). Recent evidence suggests that rather than a state of passive hibernation entered when nutrients or mitogens are limiting, the quiescence program is actively regulated at transcriptional (7–10) and epigenetic (11–13) levels. Deregulation of quiescence may underlie both tumorigenesis (failure to enter G0 leading to uncontrolled proliferation), as well as degenerative disease (failure to exit G0 leading to loss of progenitor function), necessitating an understanding of mechanisms that control this arrested state.
The mechanisms by which stem cells achieve cellular memory to keep specific regions of their genome repressed but ready to respond to regenerative signals have been emerging over the past decade (14,15). Although ASC exhibit restricted proliferative capacity and potency in comparison to ESC, they also face the opposing demands of stemness versus differentiation. When ASC are quiescent, tissue-specific genes are repressed, yet these cells must activate the appropriate lineage network when called upon to regenerate damaged tissue, restoring not only functional tissue but also a new reserve stem cell pool.
In muscle progenitors or myoblasts, quiescence is associated with repression of lineage determinants both in culture (16,17) and in vivo (18). Myogenic commitment and differentiation are controlled by the MyoD family of muscle regulatory factors (MRFs-MyoD1, Myf5, MyoG, MRF4), in conjunction with Mef2 (19). MyoD couples differentiation to permanent arrest by inducing cell cycle inhibitors p21 and Rb, with coordinate activation of muscle genes by Myogenin (MyoG) (20). Quiescence, however is marked by repression of MyoD, absence of MyoG, p21 and Rb, (16), and induction of Rb2/p130 (21), which together block both myogenesis and S phase entry. Thus, in G0, two antagonistic global programs are reined in, but can be re-activated by extrinsic signals.
Epigenetic changes precede and accompany myogenic gene activation, as MRFs recruit distinct histone modifiers to induce/maintain the muscle program (22,23). During irreversible arrest, tissue-specific and cell cycle genes experience differential epigenetic regulation at the level of histone modification. For example, the MyoG promoter is activated when MyoD recruits p300 HAT, displacing repressive HMTs EZH2 and Suv39h1 (24). However, on cell cycle promoters, Rb and E2F4 associate with Sin3b, HDACs and HMTs to create repressive H3K9me3 marks (25). Rb can also recruit Polycomb complexes, which deposit H3K27me3 marks and permanently silence cell cycle genes during differentiation (26). In human cells, p130 and E2F factors associate with MuvB-like proteins in G0, forming the DREAM complex that is involved in repression of cell cycle genes (27) and in Drosophila, dREAM is implicated in repression of a wide variety of developmental genes (28).
In ESC, cell fate loci are located in ‘bivalent domains’ (marked by both H3K27me3 and H3K4me3), in a transcriptionally repressed state poised for activation (29–31). Lineage commitment correlates with a shift to unique marking by tri-methylation of either H3K27 (silencing) or H3K4 (activation), which underlies and reinforces cell fate choices. ASC are also thought to maintain a chromatin configuration permissive to a (restricted) set of alternate fates (4). Recent reports describe bivalent domains in ASC including muscle SCs (32), but mechanisms of poising are unexplored.
Using genome-wide location analysis, transcriptome analysis and RNAi, we now report that a single regulator, an H3K9 methyl transferase PRDM2, is induced in quiescence and binds to thousands of promoters in quiescent myoblasts, over half of which are also marked by the repressive H3K9me2 mark. PRDM2 acts in two modes on differentiation versus cell cycle target genes—while it represses the activity of the Myogenin promoter in quiescence, this epigenetic regulator also controls association of the PRC2 complex at a novel G0-specific bivalent domain in the CCNA2 gene, preventing silencing. We also present evidence that PRDM2 is found in protein complexes containing EZH2, a key H3K27 methyl transferase involved in silencing. Our results implicate PRDM2 in epigenetic mechanisms promoting establishment and/or maintenance of the reversibly quiescent state, an important feature of ASCs.
MATERIALS AND METHODS
Ethics statement
All animal experiments were performed with the approval of the Institutional Animal Ethics Committee at inStem. Human myoblasts were obtained with informed consent from healthy volunteers in Denmark (see below). Human MSC were purchased from Texas A and M, human dermal fibroblasts were obtained from Lonza and used with the approval of the inStem Institutional Committee for Stem Cell Research.
Cell culture
C2C12 myoblasts (obtained originally from H. Blau, Stanford) were sub-cloned and maintained in growth medium (GM; DMEM + 20% Fetal Bovine Serum (FBS)); differentiation was induced in low mitogen medium (DM: DMEM + 2% horse serum), for 5 days to form mature myotubes (MT); synchronization in G0 was by suspension culture in 1.3% methylcelluose prepared in GM, as described (11,17).
Human primary myoblasts were isolated from muscle biopsies taken from vastus lateralis of three males (18–20 years), with informed consent and approval of the local ethics committee of Region of Southern Denmark. Cultures were established, arrested and differentiated as previously described (33), except that 10% FBS was replaced with 2% Ultroser G (Pall), 2% FBS.
Primary human bone marrow-derived mesenchymal stem cells (MSC) were purchased from Institute for Regenerative Medicine at Scott and White, Texas A and M Health Science Center College of Medicine at passage 1 and expanded in α-MEM containing 20% serum. Quiescence was induced in hMSCs at passage 3–4 by suspension in α-MEM containing 1.3% methylcellulose with 20% serum for 48 h. Under these conditions, MSC undergo synchronous arrest in G0 as for human MBs (Rumman, M., Majumder, A., Harkness, L., Balgopal, V. Pillai, MS, Kassem, M. and J. Dhawan, manuscript in preparation).
Primary human dermal fibroblasts: were expanded in 10% FBS (passage 3–4), grown to 80% confluence and subjected to serum starvation in 0.5% FBS for 48 h to induce quiescence and re-activated with 10% FBS for 24 h.
Mouse muscle satellite cells: primary SC were purified from adult mice as reported by Fukada et al. (34). About 4–6 week old female C57BL/6 mice were used to isolate muscle satellite cells. Briefly, hind limb muscle groups were dissected out, minced and digested in collagenase Type II (Cat# LS4196 Worthington Biochemical, 400U/ml final concentration) for 90 min at 37°C with gentle vortexing after every 15 min. The digested muscle slurry was filtered through 40-µm nylon mesh. The single cell suspension was treated with 0.8% ammonium chloride to lyse RBCs. Muscle mononuclear cells were washed twice with phosphate buffered saline (PBS) and stained with biotinylated anti-VCAM-1 (BD Biosciences, Cat#553331) primary antibody for 30 min, washed with PBS and stained with Streptavidin, Alexa Fluor-488 conjugate (Invitrogen, Cat#S-11223) and CD45-PE (BD Biosciences, Cat#553081) conjugated antibody. Cell sorting was performed on Moflo XPD cytometer using gates for the VCAM-1 positive and CD45 negative population. The gated cell population was sorted directly into Trizol for RNA isolation or into GM for subsequent culturing on Matrigel (BD Biosciences, Cat#354230) coated dishes.
Single muscle fiber analysis
Single skeletal muscle fibers were isolated from the hind limb muscle groups-Soleus, Plantaris, Extensor Digitorum Longus of 6-week old male C57 BL/7 mice. Briefly, the muscle groups were dissected out and treated with Collagenase Type 1 (Cat# LS4196 Worthington 400U/ml final concentration) for 1 h at 37°C. Post collagenase treatment individual muscle fibers were picked and fixed with 4% paraformaldehyde for 15 min at room temperature (RT). For immunostaining, single fibers were washed with 1× PBS twice and mounted on charged slides (Cat#12-550-15, Fisher Scientific). The fibers were permeabilized with 0.5% Triton-X-100 in 1× PBS for 1h at RT followed by blocking with 1× PBS, 0.5% Triton-X-100,10% normal goat serum for 1 h at RT. Primary antibody incubations were performed overnight at 4°C with anti-RIZ1and2 (Cat # ab3790 Abcam) and anti-Pax7 antibody (DSHB). Secondary antibody incubations were for 1 h at RT using fluorescently tagged antibodies from Invitrogen. Antibody incubations were followed by three washes with PBS + 0.05% Tween20. 4',6-Diamidino-2-Phenylindole (DAPI) (Cat# 32670 Sigma) was used to stain the DNA. Confocal images were acquired using Zeiss LSM 510 Meta confocal microscope.
Stable knockdown by RNAi
shRNAs complementary to the target PRDM2 or control GFP transcripts were cloned into mU6 vector. Stable C2C12 myoblast pools expressing shRNAs were generated by transfection and selection in G418. Aliquots of stable transfectants were frozen back after minimal expansion and freshly revived cultures used for only one passage post thawing. Sequences are listed in Supplementary Information. Several hairpins were tested but only one gave reproducible knockdown of PRDM2 transcripts and protein.
RNA interference using siRNA
siRNA (Eurogentec) targeting a different sequence in PRDM2 from the shRNA described above or scrambled control siRNA (Ambion, proprietary sequence) were transfected into C2C12 myoblasts. A total of 300 000 C2C12 cells were seeded in a P100 dish. Twenty-four hours later 400 pmol of siRNA was transfected using Lipofectamine RNAiMax (using manufacturer's protocol). The cells were harvested in RIPA buffer 24 h later and analyzed by western blotting for PRDM2 knockdown and various histone modification marks. Sequences are listed in Supplementary Information.
Over-expression of flag-RIZ constructs
cDNAs for full-length mRIZ1 (1–5127 bp) or mRIZ2 lacking the PR domain (600–5127 bp) were amplified from a mouse RIZ cDNA expression construct (Origene MR227154) and cloned into pEF1α-Fbek3. C2C12 myoblasts in GM were transiently transfected with the Flag-tagged mRIZ1 or mRIZ2 using Lipofectamine and 12 h later were either pulsed with BrdU for 30 min (to detect proliferation) or were switched to FM for 36 h and then processed for detection of Flag-RIZ proteins and BrdU or myogenic markers.
Luciferase activity of promoter-reporter constructs was assayed 24 h after transfection using a dual reporter kit (Promega). Amounts of lysate representing equal protein were used and normalized to transfection efficiency.
Immuno-fluorescence
Myoblasts stably expressing GFPsh or PRDMsh were plated on coverslips, fixed with 3.5% formaldehyde and permeabilized in PBS, 0.2% Triton-X-100. Primary antibodies were diluted in PBS, 0.2% Triton-X-100, 10% horse serum. Secondary antibodies were goat anti-mouse-Alexa fluor 488 or goat anti-rabbit-Alexa fluor 594. Antibody information is detailed in Supplementary Information. Staining was recorded on a Zeiss 510 Meta laser scanning confocal microscope (63×, Plan Apochromat Zeiss objective, 1.4 N.A.; LSM5 software). Images were minimally adjusted for brightness and contrast using Adobe Photoshop 6.0.
Flow cytometric analysis of DNA content
GFPsh and PRDM2sh myoblasts were arrested in G0, harvested, washed in PBS, fixed in ice-cold 80% ethanol, washed and resuspended in PBS + 1%Triton-X-100, 50 μg/ml propidium iodide (PI) and 100 μg/ml DNAse free RNase for 30 min at 37°C. Analysis was performed on a FACS Calibur (BD Bioscience) using doublet discrimination. Data were acquired using CelQuest® and analyzed using FLOWJO®.
Histone Methyl Transferase (H3K9) HMT assay
Flag-tagged mouse RIZ1, RIZ2 or G9a were overexpressed in C2C12 mouse myoblasts. Forty-eight hours post transfection cells were lysed in RIPA buffer. RIZ1/2 and G9a were immunoprecipitated from cleared lysates using Flag antibody (Sigma Cat#F1804) and Protein A/G Plus agarose beads. The immunoprecipitated beads were tested for Histone Methyltransferase (H3K9) activity using an EpiQuik Histone Methyltransferase Activity Assay Kit (Epigentek, Cat#P-3003) as per manufacturer's protocol. Protein in IP samples was quantitated by Amido black staining and HMT Activity was calculated after normalizing to equal protein. G9a and the control enzyme from the kit (0.3 μg) were used as positive controls and empty vector transfected into C2C12 cells was used as a negative control.
Interaction analysis of PRDM2 isoforms with EZH2
HEK293T cells were transfected with MSCVhygro-F-EZH2 (Addgene), pEF1α-BirA-V5 and pEF1α-Fbek plasmids containing either no insert (empty vector control) or mRIZ1 or mRIZ2. Forty-eight hours post transfection, cells were lysed in modified RIPA buffer (50 mM Tris–HCl pH 7.4, 150 mM NaCl, 1% NP40, 0.25% sodium deoxycholate, 1 mM EDTA) containing protease inhibitor cocktail (Roche), 1mM Phenyl methyl sulfonyl fluoride (PMSF), 0.5 mM Dithiothreitol (DTT) for 30 min. Cleared lysates (equal protein) from control and biotin-tagged RIZ transfectants were subjected to streptavidin pull downs using prewashed Dynabeads (M-280 Streptavidin, Cat#11205D, Invitrogen), for 16 h at 4°C. Post incubation, the beads were washed thrice with PBS + 0.5% TX100, bound proteins eluted in 2× Laemmli sample buffer, loaded onto Sodium dodecyl sulphate-polyacrylamide gel electrophoresis gel and subjected to western blotting. The primary antibodies used were anti-biotin HRP (Cat#7075S, Cell Signaling Technology) and anti-EZH2 (Cat#CS203195, Millipore). HRP-conjugated secondary antibodies used were from Jackson ImmunoResearch. Blots were developed using ECL detection reagent (Amersham) and imaged using ImageQuant (GE Amersham).
cDNA microarray analysis (growing myoblasts)
Total RNA was isolated from GFPsh and PRDM2sh myoblasts in growth conditions, fluorescently labeled cDNA synthesized with Cy3 and Cy5 and competitively hybridized to NIA15K mouse cDNA arrays as described (5). Two arrays were used for each sample pair including dye reversal and biological replicates. Slides were scanned using a Molecular Dynamics scanner (Image Quant software). Data was normalized using a Lowess normalization method (TIGR software) and significant genes (false discovery rate <5%) and 1.6-fold cut off, were designated by Significance Analysis of Microarrays (SAM, Stanford).
Affymetrix microarray analysis (Quiescent and Differentiated cells)
Total RNA isolated from G0 arrested and 28 h differentiated muscle cells (GFPsh or PRDM2sh) was converted to cDNA using One-cycle labeling kit and amplified using IVT labeling kit following manufacturer's instructions (Affymetrix). The normalized cRNA was fragmented, hybridized to mouse Affymtrix Gene-chips (430 2.0), washed, stained and scanned as per Affymetrix protocols. The experiment was repeated with three different biological replicates and data analyzed using Affymetrix Gene Chip operating software (GCOS). The data were normalized using PLIER (Affymetrix 2005) algorithms from Avadis and subjected to standard differential expression analysis (MIAME-compliant data is available at GEO accession GSE58676). Genes showing >1.5-fold differential expression with P ≤ 0.05 were selected and a subset validated by real time Q-RT-PCR.
QRT-PCR
Quantitative real time PCR analysis was performed on an ABI 7900HT thermal cycler (Applied Biosystems). cDNA was prepared from 1 μg total RNA using superscript II (Invitrogen) and used in SYBR-Green assay (Applied Biosystems)-each sample was isolated from three independent biological samples and analyzed in triplicate. Amplicons were verified by dissociation curves and sequencing. Primer sequences are listed in the Supplementary Information. Relative abundance of different mRNAs in PRDM2sh G0 myoblasts was calculated with reference to GFPsh G0 myoblasts and normalized to GAPDH levels. Fold change was calculated using normalized cycle threshold value differences 2−ΔΔct.
RNA isolated from human myoblasts was analyzed on QuantStudio 12D Flex (Applied Biosystems), using qbasePlus (Biogazelle) with PGK1 and TBP as reference genes.
Chromatin Immunoprecipitation (ChIP)
Chromatin was isolated and Chromatin Immunoprecipitation (ChIP) performed using antibodies against various histone modifications and H3 as described previously (11). The polyclonal antibody against PRDM2 recognizes both isoforms (detailed antibody information is provided in Supplementary Information). Sonication conditions were optimized for each cellular state separately as the nuclear configuration differs (details are available on request). For ChIP-qPCR analysis, chromatin derived from 106 wild-type C2C12 myoblasts, GFPsh or PRDM2sh myoblasts was processed using antibodies against PRDM2, H3K4me3, H3K9me2/3, H3K9Ac3 and H3K27me3. Fold enrichments were calculated as% enrichment of input chromatin or as fold change over the IgG control. Each sample was analyzed as three technical replicates and data reported are derived from three independent biological experiments.
Re-ChIP analysis
Sequential ChIP was carried out as per manufacturer's instructions using the Re-ChIP-IT kit from Active Motif (Cat#533016). ChIP-1 used anti-H3K4me3 and the eluted chromatin was first checked against known controls and then subjected to ChIP-2 with anti-H3K27me3.
Chromatin-Immunoprecipitation coupled with DNA microarray (ChIP–Chip)
ChIP–Chip analysis was performed using Agilent 244K promoter arrays (Genotypic Technologies, Bangalore) representing 22 170 murine promoters. The array design includes 60-mer probes (Mouse NCBI36/mm8 Assembly) tiled at 200 bp resolution encompassing a 4 kb region surrounding each TSS (−2.5 to +1.5 kb).
Chromatin immuno-precipitation was performed with chromatin isolated from 3 × 106 quiescent myoblasts using ChIP assay kit (Upstate, #17–295) according to the manufacturer's protocol. Briefly, cells were cross-linked using 1% formaldehyde (Fisher Scientific) in GM for 10 min and quenched with 0.125 M glycine (Sigma). Fixed cells were washed well with 1× PBS containing protease inhibitors at 4°C and resuspended in 2 ml lysis buffer supplemented with PMSF, DTT and protease inhibitor cocktail (Roche). Following 15 min incubation on ice, the sample was sonicated using Bioruptor (Diagnode) to obtain fragments of average size 200–600 bp. ChIP assays were performed using 5 μg of polyclonal antibody against PRDM2 (recognizes both isoforms) or H3K9me2 (Abcam, #ab1220). Following crosslink reversal and amplification by ligation-mediated PCR, the enriched and input fractions were labeled with Cy5 and Cy3 dyes respectively (Agilent DNA labeling kit, #5190–0449) and subjected to dual color hybridization at 65°C for 40 h to the promoter arrays. The arrays were washed and scanned and data was extracted using Agilent's feature extraction software.
ChIP–chip data analysis
Normalized enrichment values were determined using DNA Analytics 4.0 (Agilent). Peaks were determined by analyzing the distribution of all the probes in the array to assign p-values to each event and make binding calls using the Whitehead Per-Array Neighborhood Model, which considers the P-value of each probe in conjunction with both its immediate neighbors. A binding event was called if the composite P-value of groups of three probes was less than a set cutoff of 0.05; only peaks called in both the duplicates were retained for analysis. In the experiments reported here, the Pearson correlation coefficient of duplicate ChIP–chip arrays was 0.617.
For calculation of total bound promoters, probe IDs were used. For all comparisons across platforms and Gene Ontology (GO) analysis, unique Unigene IDs were used.
Validation of ChIP–chip data
A subset of 10 promoter regions were validated by real time ChIP-qPCR using sequences called as positively enriched (13 probes) or negatively enriched (10 probes) from the ChIP–chip array. Primer sequences for validation of ChIP–chip data with details of amplicon size are listed in Supplementary Information. Fold enrichments were calculated with respect to input normalized to the IgG control.
Statistical analysis
Other than ChIP–chip, (which represents duplicate arrays), all data represents values derived from at least three biological replicates and is represented as mean ± S.E.M, analysed using Student's two-tailed t-test, where P < 0.05 was taken as significant.
RESULTS
Quiescent cells must execute a balancing act: both proliferation and differentiation are held in abeyance, but must be available for activation during regeneration. The mechanism by which this balance is maintained in G0 is not known, but likely involves chromatin regulation. To test this hypothesis, we analyzed the function of epigenetic modulators that are specifically induced in G0.
PRDM2/RIZ is expressed by quiescent cells in vitro and in vivo
We identified PR domain containing-2/Rb-interacting zinc finger protein (PRDM2/RIZ) as a gene whose expression was upregulated in G0 (10). Two major mRNAs are known-RIZ1 (7.3 kb) has an N terminal PR domain (modified SET/HMT motif) and RIZ2 (7 kb) lacks the PR domain (35) (Supplementary Figure S1). RIZ1-specific as well as total PRDM2/RIZ mRNA was induced in G0 synchronized C2C12 myoblasts (MB), and waned as cells re-enter S phase (Figure 1A left panel). During differentiation to MT, PRDM2 expression was repressed (Figure 1A, right panel). Expression of PRDM2 marked Pax7+ satellite cells (SC) on isolated mouse myofibers (Figure 1B) and purified quiescent mouse SC (Figure 1C), declining during activation or differentiation in vitro (Figure 1D). PRDM2 was detected in presumptive SC in human fetal and adult muscle (Figure 1E and F) and was induced as human primary SC-derived myoblasts, bone marrow-derived MSC and primary fibroblasts entered G0 in culture (Figure 1G–I). Thus, PRDM2 is enriched in reversibly rather than irreversibly arrested cells, suggesting G0-specific functions.
Figure 1.
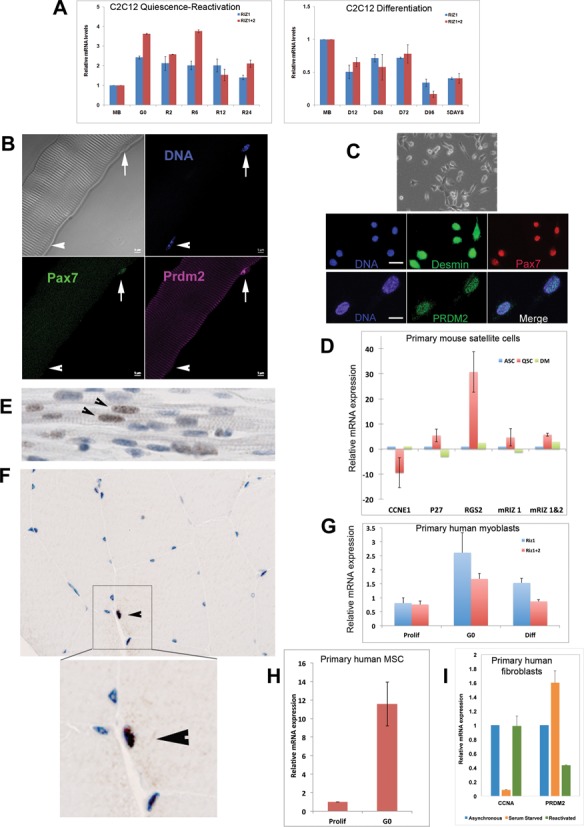
PRDM2 is highly expressed in quiescent cells. (A) PRDM2/RIZ mRNAs are upregulated in C2C12 MBs in G0 and wane during S phase entry (left panel); both transcripts are downregulated during differentiation (right panel). Q-RT-PCR analysis with primers specific to RIZ1 (blue bars); primers to detect both RIZ1 + RIZ2 mRNAs (pink bars); MB: asynchronous cycling MB; G0: quiescent, R2-R24: 2–24 h after reactivation from G0; D12-D96: 12–96 h of differentiation. (B) PRDM2 protein is expressed by Pax7+ mouse SCs (arrow) on isolated myofibers; myonuclei (arrowhead) do not express Pax7 or PRDM2. Note: antibody detects both PRDM2 isoforms (RIZ1/2). (C) PRDM2 expressed by freshly sorted quiescent mouse SC. Top- Phase contrast; Middle- SC markers Desmin and Pax7 (Bar 20 μ); Bottom-PRDM2 is also expressed (Bar 10 μ). (D) Q-RT-PCR analysis of primary SCs: freshly isolated quiescent (QSC, red), activated for 24 h in GM (ASC, blue) or differentiated for 24 h in DM (DM, green). QSC show low Cyc E expression but induction of G0 markers p27, Rgs2, and up-regulation of PRDM2 mRNAs. (E) PRDM2 expression in presumptive SCs (arrowheads) in fetal human muscle sections. (F) PRDM2 in adult human muscle section (arrowhead)—inset shows magnified PRDM2+ SC (brown). (G) Upregulation of PRDM2 mRNAs in cultured G0-synchronized primary human SC. (H) PRDM2 mRNA up-regulation in cultured G0-synchronized primary hMSC. (I) PRDM2 mRNA is induced in serum-starved quiescent human primary fibroblasts, and suppressed by cell cycle reactivation. Values in all graphs represent the mean ± S.E.M., n = 3.
Knockdown of PRDM2 does not hasten the cell cycle, over-expression causes arrest
To analyze the role of PRDM2 in G0 myoblasts, we used shRNAs that target both isoforms and confirmed reduced expression at both mRNA and protein level (Figure 2 A and B). As a known tumor suppressor, loss of PRDM2 might enhance proliferation, as in MCF7 cells (36). However, in MB, knockdown of PRDM2 reduced proliferation as evidenced by reduced cyclin expression (Figure 2C) and fewer S phase cells (Figure 2D, E), and also reduced self-renewal as seen by lower colony formation (Figure 2F).
Figure 2.
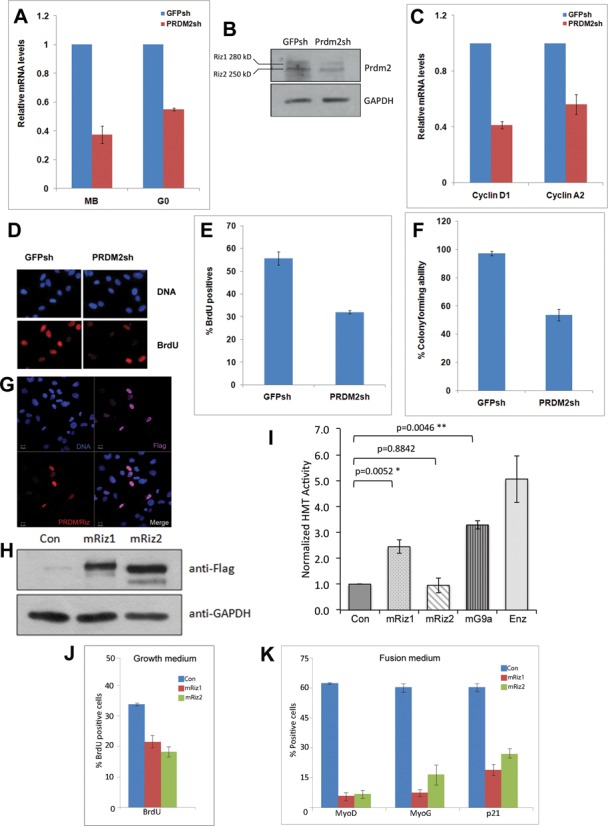
PRDM2 is not a typical quiescence factor: knockdown further slows the cell cycle. Stable knockdown of PRDM2 mRNA and protein in C2C12 myoblasts using shRNA targeting both RIZ1 and RIZ2 mRNAs (control is GFP shRNA). (A) Q-RT-PCR analysis of RNAi in growing and G0 MBs-reduced mRNA. (B) Reduced levels of RIZ1 and RIZ2 proteins (antibody recognizes both isoforms). PRDM2 knockdown cells show reduced proliferation: (C) Reduced CycD1, CycA2 mRNA levels, (D and E) reduced BrdU incorporation and (F) reduced colony formation. (G) Over-expression of Flag-tagged mRIZ1 detected with anti-Flag (purple) or anti-PRDM2 (red). (H) Flag-tagged mRIZ1 and mRIZ2 are expressed at similar levels. (I) H3K9 methyl transferase assay of Flag-tagged mouse RIZ1, RIZ2 and G9a in myoblasts. mRIZ1 shows HMT activity equivalent to mG9a, whereas mRIZ2 does not (Con, empty Flag vector; Enz, purified HMT enzyme (0.3 μg) as positive control; values represent mean + SEM, n = 3 for RIZ1/2, n = 2 for G9a). (J and K) Both RIZ1 and RIZ2 when ectopically expressed suppress BrdU incorporation (values represent mean ± SEM; P < 0.01 for mRIZ1, P < 0.001 for mRIZ2, n = 3) and myogenic markers (P < 0.001, n = 3) compared to control (Con, empty Flag vector).
The PR/SET domain defines the PRDM family of histone modifiers (37). Of the PRDM2 isoforms, RIZ1 (an H3K9 methyl transferase, (38,39) is deleted in cancers, but RIZ2, which lacks the PR domain, is not; targeted deletion of just RIZ1 increased tumor incidence (40). When exogenously expressed in myoblasts both isoforms were nuclear localized (Figure 2G) and expressed to similar levels (Figure 2H). To assess the enzymatic activity of the two isoforms, we performed histone methyl transferase assays on flag-tagged mRIZ1 and mRIZ2 proteins after immuno-precipitation. While mRIZ1 was capable of methylating H3K9 to a similar extent as a proven H3K9 transferase G9a, mRIZ2 did not show detectable activity (Figure 2I). When over-expressed in cycling MB (Figure 2J and K), both mRIZ1-flag and mRIZ2-flag inhibited proliferation (Figure 2J), consistent with the reports that PRDM2/RIZ is a negative regulator of the cell cycle (41). Interestingly, both isoforms also repressed markers of commitment, differentiation and irreversible arrest (MyoD, MyoG, p21) (Figure 2K). Taken together with the down-regulation of this tumor suppressor during differentiation (Figure 1) these findings suggest that induction of PRDM2/RIZ in G0 may prevent inappropriate differentiation and promote reversible arrest.
PRDM2/RIZ knockdown accelerates differentiation
To assess the effects of PRDM2 RNAi on differentiation, we used siRNA and shRNA to target different sequences and examined expression of myogenic markers both in GM and in differentiation medium (DM). Knockdown cells differentiated precociously in GM (Figure 3A and B) and showed MT hypertrophy in DM (Figure 3C), confirming PRDM2 as a repressor of myogenesis. In G0 conditions, control MB remained mono-nucleated, but PRDM2sh cells fused to form syncytia (Figure 3D). Thus in muscle cells, PRDM2 maintains an undifferentiated state.
Figure 3.
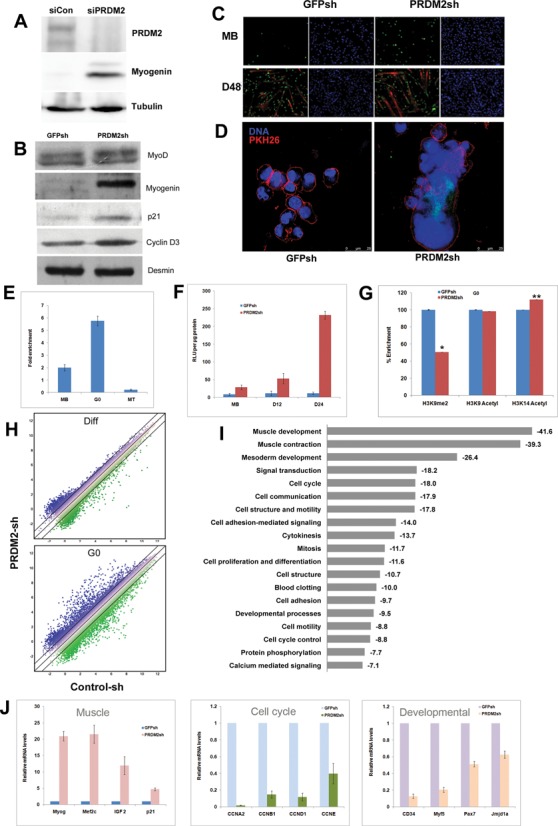
PRDM2 is a master repressor of muscle differentiation in G0: knockdown diverts quiescence program to differentiation. PRDM2 knockdown by siRNA (A) or shRNA (B) leads to precocious induction of early muscle markers, MyoG, p21 and CycD3. (C) Increased MyoG+ nuclei in GM (MB), precocious fusion and larger myosin+ MT after 48 h in DM (D48). (D) Fusion in knockdown cells even in G0-inducing conditions: membrane marker PKH26 (red) reveals multinucleated syncytia (right panel); control cells remain mononucleated (left panel). (E) ChIP analysis shows PRDM2 association with MyoG promoter in MB; this is enhanced in G0 and lost in MT. (F) PRDM2 knockdown de-represses MyoG promoter activity-transfection of a MyoG promoter-luciferase construct into control (GFPsh) or knockdown (PRDMsh) cells in proliferating (MB), 12 or 24 h differentiating conditions (D12, D24). (G) Altered histone marks on MyoG promoter [−120 ± 10 bp] in knockdown (reduced H3K9me2, increased H3K14acetyl) correlate with increased transcriptional activity in PRDM2sh cells in G0-inducing conditions [normalized mean% enrichment ± S.E.M, n = 3; *P < 0.05; **P < 0.005]. (H) Affymetrix gene-arrays reveal only ∼26 genes differentially expressed (≥1.5-fold) between GFPsh and PRDMsh cells when cultured in DM for 28 h (top panel, Supplementary Table S2), but >1400 genes in G0-inducing conditions (bottom panel, top 100 up- and downregulated genes are listed in Supplementary Table S3). (I) Pathway analysis of deregulated transcripts using DAVID (http://david.abcc.ncifcrf.gov/): values represent enrichment scores. Note the substantial representation of GO terms representing muscle specification, development and proliferation. (J) Targeted Q-RT-PCR validation of 12 key transcripts deregulated by PRDM2 knockdown in G0. Myogenic regulators are upregulated (left panel), but regulators of cell cycle (middle panel) and cell fate (right panel) are downregulated, confirming that PRDM2 normally restrains differentiation genes but maintains proliferation and specification genes.
We directly examined a role for PRDM2 in regulating muscle genes. MyoG is a target of several chromatin modulators (22), including the H3K9 methyltransferase Suv39h (42). Using ChIP, we found that PRDM2 associates with the MyoG promoter in undifferentiated MBs (Figure 3E): this association is enhanced in G0 but lost in MTs, consistent with a differentiation-blocking role. We located a consensus PRDM2 binding site (GTTGGC) overlapping a MEF2c site in the critical −200 bp region of the MyoG promoter, suggesting direct binding (43), (Supplementary Figure S2). In PRDM2sh cells cultured in either GM or DM, an ectopic MyoG promoter-luciferase construct was de-repressed (Figure 3F). Since PRDM2 exhibits H3K9 methyl transferase activity (38) (this report Figure 2), we assessed histone modifications at the endogenous MyoG promoter in G0 cells, using ChIP assays. Knockdown cells not only showed reduced H3K9me2, consistent with reduced PRDM2 association, but also increased H3K14Ac (a modification that promotes gene activation) (Figure 3G), correlating with the de-repression of promoter activity (Figure 3F). Overall levels of H3K9-me1/2/3 were unchanged in the PRDM2 knockdown cells (Supplementary Figure S3), suggesting only locus-specific alternations. Together, these results demonstrate that PRDM2 directly targets an early differentiation control hub via repressive histone marks, specifically in G0.
PRDM2 functions peak in G0, knockdown grossly alters quiescence program
To examine the extent and timing of PRDM2's influence, we used stage-specific microarray analysis (Figure 3H and I). In cycling and differentiating conditions, reduced PRDM2 expression had minor effects: only 36 and 26 genes respectively showed altered expression (Supplementary Tables S1 and S2). However, in G0 conditions, PRDM2 knockdown MB showed altered expression of 1420 genes (Supplementary Table S3; data can be retrieved from GEO accession GSE58676). This 50-fold greater effect on the transcriptome suggests that the critical period of PRDM2 function is G0. GO analysis corroborated the phenotypic analysis of the knockdown (Figures 2C–F and 3A–D) revealing that the most highly enriched terms included muscle developmental genes, signaling and the cell cycle (Figure 3I). Of 543 downregulated genes, >50% participate in proliferation (Supplementary Figure S4A), pointing to a major effect of PRDM2 on the cell cycle. About 10% of 877 upregulated genes are involved in differentiation and 2.5% in muscle contraction, consistent with PRDM2's repressive role. GO analysis using a different algorithm that reduces redundancy in large datasets (http://revigo.irb.hr/revigo.jsp) (44) highlighted the enrichment of the muscle regulatory program in the upregulated genes and cell cycle program in the downregulated genes (Supplementary Figure S4B). Thus, PRDM2 functions peak in G0 and knockdown subverts the quiescence program.
Validation of 12 microarray hits by Q-RT-PCR confirmed that loss of PRDM2 promotes myogenesis: MyoG, p21 and Mef2c were strongly induced (Figure 3J, left), as were upstream regulators (IGF2), and downstream targets (myosins, troponins) that build sarcomeres (Supplementary Table S3). Thus, PRDM2 represses the muscle hierarchy at multiple levels. With both cell cycle and myogenesis repressed in G0, this quiescent, undifferentiated state is poised for activation of either program. The role of PRDM2 in promoting the G0 state was evident: induction of differentiation in knockdown cells was coupled with hallmarks of irreversible arrest: Cyclins D1, E, A2, B that are normally repressed in G0 were hyper-repressed (Figure 3J, middle); developmental/cell fate regulators (Pax7, Myf5, CD34, Jmjd1a) that are normally maintained or induced in G0 were repressed (Figure 3J, right). Together, these results demonstrate that PRDM2 preserves reversibility of quiescence, not only by fail-safe repression of differentiation genes, but also by maintaining expression of stem cell and cell cycle genes.
PRDM2 orchestrates a global program by association with 4480 promoters
To investigate potential direct targets of PRDM2, we determined its genome-wide location in G0 MB using ChIP–Chip analysis of Agilent 244K mouse promoter arrays. Duplicate arrays were interrogated with chromatin derived from independent pull-downs using a polyclonal PRDM2 antibody that recognizes both isoforms RIZ1 and RIZ2, and compared with a third array probed for H3K9me2 marks. Figure 4A depicts the binding intensity traces for the entire chromosome 3, showing the high degree of correlation between replicates; Figure 4B shows the CCNA2 locus indicating PRDM2 enrichment specifically at the TSS and in intron 1. The genome-wide analysis revealed that 4480 gene promoters were occupied by PRDM2 in quiescence (P < 0.05), of which 2462 (55%), were co-associated with H3K9me2 marks (Figure 4C). ChIP–chip data is available at GEO accession number GSE58748. In conjuction with the confirmed enzymatic activity of RIZ1, the observation that over half of PRDM2 binding sites in chromatin also show H3K9me2 enrichment, may suggest a role for this tumor suppressor protein in wide-spread chromatin modification. Since the antibody does not distinguish isoforms but only RIZ1 is capable of H3K9 methylation, concievably, the RIZ1 isoform may co-localize with H3K9me2 at common chromatin sites, and sites not enriched for H3K9me2 may represent sites of RIZ2 binding.
Figure 4.
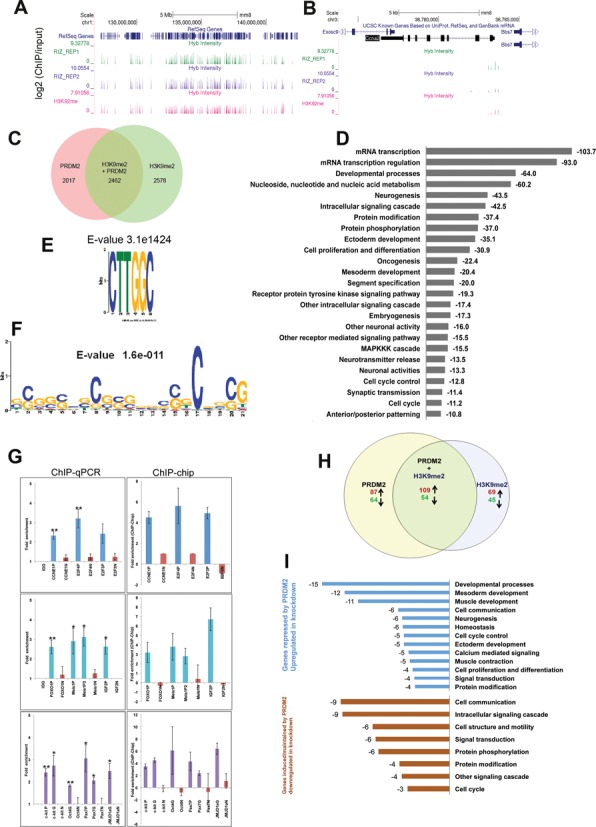
Genome-wide promoter analysis in G0 myoblasts reveals PRDM2 association with a broad program including regulators of the cell cycle, differentiation and stem cell functions. ChIP–Chip analysis (Agilent 244K mouse promoter arrays) using total PRDM2 antibody reveals that PRDM2 proteins associate specifically with thousands of sites in the mouse genome. (A) The three tracks in the schematic depict the entire mouse chr.1 showing the location of binding enrichment (vertical bars) of PRDM2 (RIZ_REP1 and 2) and H3K9me2 from replicate ChIP–chip analysis. Y-axis represents enrichment score (over input). (B) A zoomed view of the CCNA2 locus on chr.3; arrows depict the direction of transcription. Enrichment near TSS and in intron 1 is evident. (C) A total of 4480 promoters (complete list available from GEO [GSE58676]) were bound by PRDM2 in quiescent MB (P < 0.05); 2462 promoters (55%) are also enriched for H3K9me2. (D) Annotation of PRDM2 bound genes using DAVID reveals a broad set of development- and proliferation-related pathways (values represent enrichment scores). (E) Enrichment of known consensus PRDM2 binding site CTTGGC in 860 probes suggests direct binding at these sites. (F) CpG motif is specifically enriched in probes that are most strongly associated with PRDM2 (seven to nine-fold enriched). (G) Validation of global ChIP–chip analysis using targeted ChIP-qPCR of loci encoding positive regulators of myogenic differentiation (middle), cell cycle regulators (top) and stem cell regulators (lower) in G0 MB. Primer coordinates were chosen based on enrichment in genome-wide analysis (see Supplementary Information for details). For each locus, N = negatively enriched region, P = positively enriched promoter region, G = positively enriched gene body region. Left panel: ChIP-qPCR validation [mean ± S.E.M (n = 3), ** P < 0.005, *P < 0.05 compared to respective IgG controls]; Right panel: values derived from ChIP–chip analysis (P < 0.05). (H) Comparison of ChIP–chip analysis with transcriptome analysis reveals potential direct targets. Overlap of genomic locations of PRDM2 association with genes deregulated by PRDM2 knockdown: 314 PRDM2 bound genes show altered regulation suggesting these are direct targets of PRDM2. (I) Pathway analysis of PRDM2-associated and deregulated genes using DAVID enriches developmental and cell cycle genes (values represent enrichment scores). Upper panel (blue bars) represents upregulated and lower panel (brown bars) represents down regulated gene classes.
Pathway analysis of PRDM2-bound promoters using DAVID underscored the broad regulatory function of this epigenetic regulator, showing enrichment of GO terms for control of transcriptional, developmental, differentiation, signaling, oncogenic and cell cycle programs (Figure 4D). Interestingly, the list of PRDM2-associated promoters contained a strong signature of neurogenic and neuro-modulatory genes, suggesting functions in developmental/differentiation programs beyond muscle. GO analysis to evaluate non-redundant terms highlighted the enrichment of myogenic, cell cycle and stem cell programs (Supplementary Figure S5).
Sequence analysis of the bound promoters revealed that 860 probe sets showed a consensus PRDM2 binding site (C/GTTGGC (Figure 4E), suggesting direct binding: 560 of these were co-associated with H3K9me2 in G0. Bio-informatic analysis also suggested indirect binding of PRDM2 at some locations: 208 promoters that were seven to nine-fold enriched by PRDM2 ChIP, [E-value 1.6e−11] (Figure 4F), 70% of which were also enriched for H3K9me2, harbored CpG islands but not canonical PRDM2 binding sites. This finding suggests potential recruitment of PRDM2 by mechanisms other than direct DNA binding and possible cooperation between DNA and histone methylation at these loci.
Targeted ChIP-QPCR analysis (Figure 4G, left panel) of 10 selected promoters showing PRDM occupancy was used to validate the ChIP–Chip analysis (Figure 4G right panel). In these 10 promoters, all 13 regions called as positively enriched and all 10 negatively enriched regions reproducibly yielded ChIP-qPCR enrichment values in concordance with the ChIP–chip analysis. We confirmed that PRDM2 was located not only at MyoG promoter in G0 (Figure 3E), but also at promoters of upstream myogenic regulators (IGF2, Meis1), and at promoters of cyclin and stem cell regulatory genes (Figure 4G).
To determine potential direct transcriptional targets of PRDM2, we compared the transcriptome analysis with the chromatin occupancy analysis of both PRDM2 and H3K9me (Figure 4H and I). Of 1420 genes whose expression was altered in PRDM2 knockdown cells, the promoters of 22.1% were enriched for PRDM2, 19.5% showed H3K9me2 enrichment and 11.5% were co-occupied, in concordance with reports that no single epigenetic/transcriptional regulator is critically required at all its binding sites (45,46). Of the 314 PRDM2-occupied loci that were de-regulated in the PRDM2 knockdown myoblasts, 163 loci were also associated with H3K9me2 (109 were upregulated ≥1.5-fold in PRDM2sh and 54 were downregulated) (Figure 4H, Supplementary Table S4). The observation that most dually enriched promoters are not deregulated is likely a consequence of redundant regulatory mechanisms (45). However, pathway analysis confirmed that developmental, muscle, cell cycle and signaling pathway genes were enriched in the gene sets that were both bound by PRDM2 and deregulated in the knockdown (Figure 4I). Taken together, genome-wide location and expression analysis in G0 suggest that by interacting with promoters that control antagonistic functions (differentiation versus stemness, proliferation versus quiescence), PRDM2 may orchestrate a complex program that maintains cells poised for alternate fates.
PRDM2 maintains expression of stem cell genes in G0
PRDM2 participates in both repressive and activating complexes in MCF7 cells (47). In G0 MBs, PRDM2 not only associates with muscle promoters (MyoG, IGF2) whose expression is enhanced in knockdown cells (Figures 3 and 4), but also with those of pluripotency genes (Oct4, Jmjd1a) and tissue-restricted fate regulators (Pax7, CD34) (Figure 4), whose expression declines. As an H3K9 methyl transferase, PRDM2's regulation of Jmjd1a (an H3K9 demethylase) suggests a feed-back mechanism that may potentiate differentiation in PRDM2sh cells. Loss of SC markers in RNAi cells also suggests that PRDM2 may normally associate with an activating/maintenance complex at these loci; by contrast, at the activated muscle marker genes, PRDM2 may normally participate in a repressive complex.
PRDM2/RIZ prevents silencing of cell cycle genes in reversible arrest
During differentiation, silencing of cell cycle activators by the H3K9 transferase Suv39h reinforces permanent exit (25). However, transient quiescence requires reversible repression of cell cycle genes, necessitating mechanisms to keep them off but poised for re-activation by mitogenic cues. Knockdown of PRDM2 led to hyper-repression of cyclins (Figure 3), suggesting that this chromatin factor may maintain basal activity of these promoters in G0 and/or preserve their competence for future induction. To test this hypothesis, we analyzed the Cyclin A2 (CCNA2) promoter (Cell Cycle Regulatory Element) for repressive (H3K9me2) versus silencing (H3K27me3) marks. We also assessed H3K4me3, since this normally activating mark correlates with CCNA2 repression in G0 (11) and CCNA2 was 50-fold downregulated in PRDM2 knockdown cells (Figure 3J). In control cells (GFPsh) in G0, CCNA2 CCRE was more enriched for H3K4me3 and H3K9me2 than for H3K27me3 (Figure 5A). However, PRDM2sh cells showed a switch in modifications: H3K27me3 was strongly enriched, coinciding with reduced H3K9me2 and H3K4me3. Thus, PRDM2 may block the deposition of silencing marks on cell cycle genes in G0, preserving their potential for future reactivation.
Figure 5.
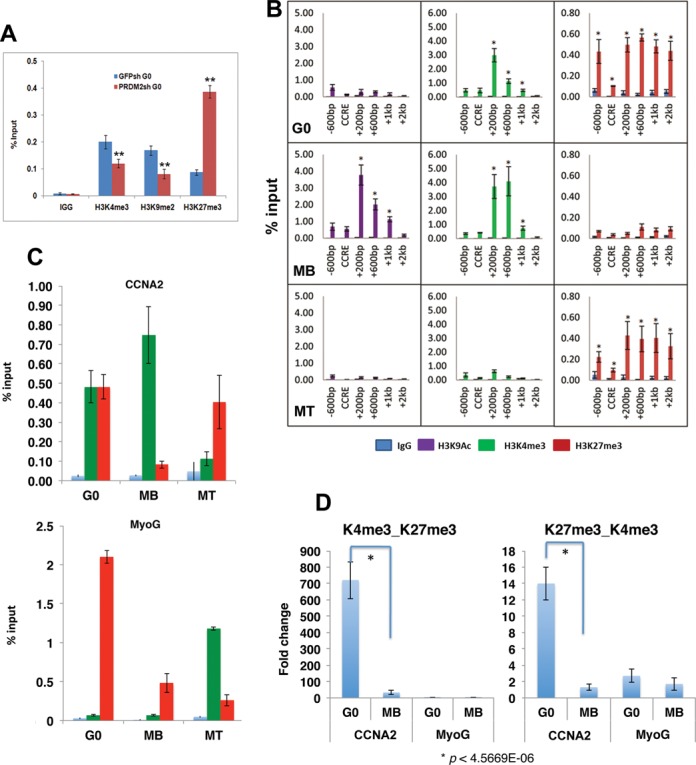
PRDM2 regulates histone modifications at the CCNA2 locus: Identification of a quiescent-specific bivalent domain in intron 1 of the mouse CyclinA2 gene. (A) PRDM2 prevents deposition of silencing marks on CCNA2 promoter. ChIP-qPCR analysis of histone modifications at the cell cycle regulated element of the CCNA2 promoter (CCRE, +9 to +175) in control and PRDM2 knockdown MBs. In G0 conditions, reduced PRDM2 leads to increased levels of silencing marks (H3K27me3) and loss of activation marks (H3K4me3), coincident with reduced H3K9me2, **P < 0.005. (B) Histone modification profile of the −600 to +2000 region of CCNA2 gene in different cellular states by ChIP-qPCR. Activating marks (H3K9Ac [purple], H3K4me3 [green]) dominate in MB (middle panel) and silencing marks (H3K27me3) dominate in MT (bottom panel). Uniquely, in G0 (top panel) CCNA2 exhibits little H3K9Ac, but both H3K4me3 and H3K27me3, suggesting a balanced repressed state. The region between +200 and +600 is subject to greater alterations in histone marks than at the known CCRE (mean ±SEM, n = 3; *P < 0.0001). (C) The CCNA2 intron 1 (+200 to +600) is bivalently marked (H3K4me3 + H3K27me3) specifically in G0 (top panel), but singly marked in MB (H3K4) and MT (H3K27) in keeping with the active and silenced transcriptional state of CCNA2 locus respectively. The MyoG promoter is not dually marked in any state (bottom panel)-silencing marks increase in G0. (D) Validation of the G0-specific bivalent domain by sequential or re-ChIP analysis. Chromatin pulled down with anti-H3K4me3 was re-precipitated with anti-H3K27me3 (left) or anti-K27 followed by anti-K4 (right) to assess whether the two marks are present on the same set of chromatin fragments or two distinct sets. The Y-axis represents fold enrichment of H3K27me3 from an initial H3K4me3 pulldown. CCNA2 (+200 to +600 region) but not MyoG promoter shows dual marking, and only in G0 cells not MB.
A quiescence-dependent bivalent domain in the CCNA2 gene
To assess whether PRDM2 regulates silencing-associated marks specifically in G0, we first profiled histone modifications on the CCNA2 gene (region from −600 bp to +2 kb) in different states (Figure 5B). Of the activating marks, H3K9Ac is enriched around the TSS (−100 bp to +1 kb region) only in MB where the gene is expressed, but not in either G0 or MT; H3K4me3 is enriched in both G0 and MB but not in MT, as reported (11). The repressive H3K27me3 mark is enriched in both states where CCNA2 is not expressed (G0, MT), but not in MB. Integrating H3K4me3 and H3K27me3 marks across the CCNA2 gene in three states, a ‘bivalent domain’ enriched for both activating and repressive marks was found between +200 bp to +1 kb, specifically in G0 (Figure 5C). In MB and MT, this domain resolves to singly marked status in accordance with CCNA2 expression status: in proliferating MB, the activating H3K4me3 mark is retained, while differentiated MT retain the repressive K27me3 mark (Figure 5C, top). Bivalent marking was not observed at MyoG promoter/TSS in any state (Figure 5C, bottom), indicating specificity for CCNA2 promoter in G0. To eliminate the possibility that dual marking reflects population heterogeneity, bivalency was further confirmed by sequential ChIP (‘re-ChIP’) analysis (Figure 5D).
To map the kinetics of histone modifications at the bona fide CCNA2 bivalent domain, we used ChIP analysis of synchronized cell cycle re-entry from G0 revealing that bivalency resolves at G1/S (Supplementary Figure S6A) when CCNA2 expression rises (Supplementary Figure S6B), after which only H3K4me3 is retained. This dynamic chromatin regulation of CCNA2 is distinct from transcriptional control of the S-phase specific CCRE (48) revealing epigenetic control of a key regulator of DNA replication, specifically in G0.
To determine whether other cell cycle promoters are also bivalently marked, we examined the CCND1 promoter and TSS during proliferation, quiescence and differentiation. We observed that while H3K4 marks were strongly retained in G0 although the locus is transcriptionally repressed, neither promoter nor TSS showed equivalently strong H3K27 marking to the extent seen for CCNA2 (Supplementary Figure S7). Thus, the individual cyclin loci may experience distinct epigenetic regulation in G0 and the bivalent domain in CCNA2 may represent a specific cell cycle regulatory node.
A novel Polycomb response element (PRE) in CCNA2 intron 1 binds PRC2 members and represses reporter gene activity
Cyc A is implicated as a direct target for Polycomb proteins in flies (49,50), but such regulation has not been documented in mammals. Given the intronic enrichment of H3K27me3 (Figure 5), we analyzed the entire mouse CCNA2 locus for PRE-like sequences. In Drosophila, PREs associate with factors PHO, GAF and DSP1 (51), whose vertebrate homologs, HMGB2, YY1, ThPOK respectively (52–54) are known. We found two regions containing YY1/GAF consensus sites along with a Drosophila PHO consensus motif (55), in CCNA2 introns 1 and 2 (Regions 1 and 2 respectively) (Figure 6A). When cloned upstream of luciferase reporter, both Regions 1 and 2 showed significant repressive activity, equivalent to a known repressor element in the EVX2–HoxD13 region (56) (Figure 6B), suggesting a potential role as PREs. ChIP analysis determined that core PRC2 member Suz12 was enriched at Region 1 and not Region 2 in G0 (Figure 6C). Thus, an intronic element in the mouse CCNA2 gene contains a PRE-like sequence that can recruit PRC2 members, and repress gene activity in reporter assays.
Figure 6.
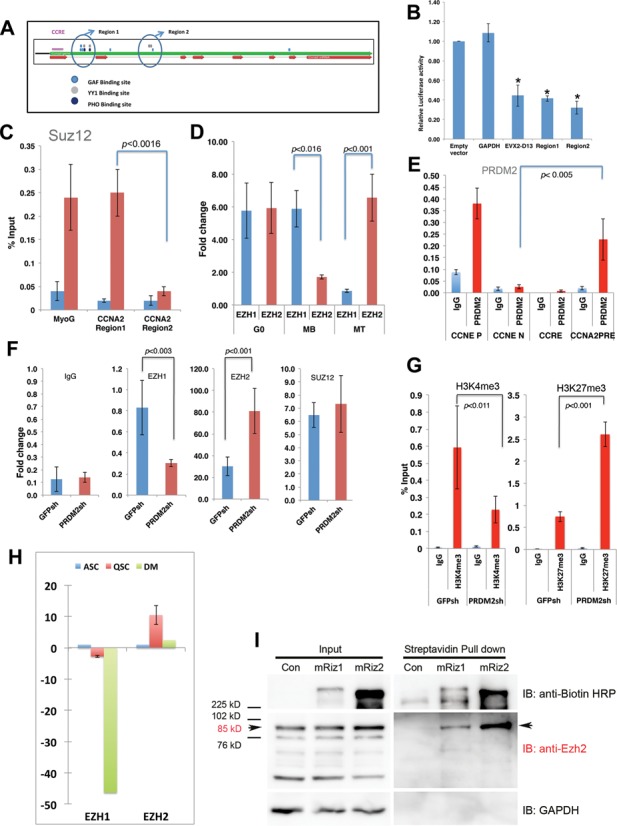
A PRE-like element in the CCNA2 bivalent domain shows cell state-specific EZH1 and EZH2 binding that is regulated by PRDM2. (A) Schematic of CCNA2 locus: exons-red arrows; introns-thin red lines. YY1 binding sites (GCCATHWY), PHO binding sites (CNGCCATNDNND) and GAF binding sites (GGGAAGG, GAGGGG, GAGAG) are clustered in two distinct regions in introns 1 and 2 (Region 1 and 2) respectively). (B) Cloned Regions 1 and 2 repress luciferase reporter activity in transient transfection assays, compared to empty vector control. Positive control for repressor activity is a known repressive element from EVX2–HoxD13 region; negative control is GAPDH gene (500 bp). Values represent normalized reporter activity (mean ± S.D., n = 3; *P < 0.002). (C) SUZ12 (core member of PRC2 complex) is associated only with CCNA2 Region 1 and not Region 2 (SUZ12 occupancy at MyoG TSS serves as a positive control) (mean ± S.E.M, n = 3). (D) The putative PRE in Region 1 shows balanced occupancy of EZH1 and EZH2 in G0, but EZH1 occupancy dominates in MB, EZH2 dominates in MT (mean ± S.E.M, n = 3). (E) PRDM2 binds the CCNA2 gene only at the putative PRE (+200 to +600), not at the CCRE. CCNE P and CCNE N are positive and negative controls respectively for PRDM2 binding (derived from ChIP–chip and validated by ChIP-qPCR) from CCNE locus. (F) Knockdown of PRDM2 alters PRC2 complexes at the CCNA2 PRE in G0: reduced EZH1 and increased EZH2 enrichment; Suz12 enrichment is unaltered. (G) Knockdown of PRDM2 leads to reduced H3K4me3 and increased H3K27me3 marks at the CCNA2 PRE in G0, correlating with increased EZH2 occupancy (mean ± S.E.M., n = 3). (H) Expression of EZH2, like PRDM2 (see Figure 1) is enriched in quiescent mouse SCs. Q-RT-PCR analysis of EZH1 and EZH2 in freshly sorted cells held in quiescent (QSC), activated (ASC) or differentiated (DM) conditions. (I) PRDM2 interacts with EZH2. Biotin-tagged PRDM2 isoforms (mRIZ1 or mRIZ2) or the empty vector (Con) were transfected into HEK cells, pulled down with streptavidin beads and probed for the presence of RIZ, EZH2 or GAPDH (negative control) using immuno-blotting. Input samples contained several isoforms of EZH2, but only the 85 kD full-length EZH2 protein was specifically detected in RIZ1/2 pull downs. Data depicted is one of three biological replicates.
Balanced occupancy of the CCNA2 PRE by EZH1 and EZH2 is specific to G0
Since Suz12 was associated with the CCNA2 PRE in G0, we determined the expression of PRC members in different states. PRC2 members EZH1, EZH2, SUZ12 and EED were all induced in G0 compared to MB and MT (Supplementary Figure S8) and interestingly, PRDM2 was also associated with their promoters. PRC1 members (CBX family) were also induced in G0. Thus, quiescence is uniquely associated with increased expression of PRC1/2 members.
Correlating with dynamic changes in epigenetic marks at CCNA2 locus, occupancy of the putative PRE by PRC2 members also varied. EZH1 and EZH2 are H3K27 methyl transferases that co-regulate ASC function (57). While EZH1 was enriched in MB and EZH2 was enriched in MT, G0 MBs showed balanced enrichment of both EZH1 and EZH2 (Figure 6D). EZH1 is known to associate with active loci (58,59). Hence, association of EZH1 on a silent CCNA2 gene in G0 suggests a mechanism for poising the locus for activation.
PRDM2 maintains the EZH1-EZH2 balance at the CCNA2 PRE in G0
The original ChIP–chip analysis indicated that PRDM2 associates with the CCNA2 intron (Figure 4B). Using a gene-specific ChIP assay, we found that PRDM2 is specifically associated with the CCNA2 PRE-like element, but not with the well-studied CCRE (Figure 6E). Importantly, PRDM2 knockdown affected association of EZH1 and EZH2 at the intronic PRE, altering the balance by decreasing EZH1 and increasing EZH2 (Figure 6F). In addition, PRDM2 knockdown also altered the poised or bivalent marking, with reduced H3K4 and increased H3K27. Finally, EZH2 is more highly expressed in quiescent satellite cells (Figure 6H). As EZH2 is the major H3K27 methyl transferase of PRC2 complex, increased chromatin association combined with increased H3K27me3 levels at the PRE and increased repression of CCNA2 in PRDM2sh indicates a functional interplay between EZH2 and PRDM2 in G0. Specifically, PRDM2 appears to blunt the accumulation of EZH2 at the PRE-like element in CCNA2.
PRDM2 knockdown disrupts the bivalent status of CCNA2 PRE
PRDM2 clearly helps to preserve CCNA2 expression in quiescence since CycA2 mRNA levels were repressed ∼50-fold more when PRDM2 expression was depleted (Figure 3). To determine if PRDM2 contributes to G0-dependent histone marks, we examined H3K4me3 and H3K27me3 at CCNA2 PRE-like element: PRDM2sh cells showed reduced H3K4me3 and increased H3K27me3 levels (Figure 6G) suggesting that PRDM2 normally contributes to maintaining the bivalent status of the PRE in G0. Enhanced H3K27me3 levels in PRDM2sh correlate with, and likely result in, hyper-repression of CCNA2 expression. Taken together, increased H3K27 marking and increased EZH2 association at the PRE in PRDM2 knockdown cells, strongly implicate PRDM2 in controlling the bivalent domain in G0.
PRDM2 proteins interact with EZH2
The control of EZH2 association at the PRE by PRDM2 suggests a possible interaction of these two chromatin proteins. To determine if PRDM2 directly interacts with EZH2, we employed pull down assays. Biotin-tagged mRIZ1 or mRIZ2 were expressed in HEK cells, pulled down using streptavidin-conjugated beads, followed by immuno-blotting for EZH2 or a control protein (GAPDH). Interestingly, both isoforms were specifically found in complexes containing the 85 kD EZH2 protein (Figure 6I). Since the shRNA targets both isoforms, either RIZ1 or RIZ2 or both, might be involved in limiting the deposition of H3K27 marks on the CCNA2 PRE-like element, perhaps by sequestering EZH2. Taken together, our observations provide evidence for a PRDM2-dependent mechanism that maintains CCNA2 in a poised state in G0, by maintaining a balance of EZH1 and EZH2 at a newly identified PRE-like element.
Our findings establish PRDM2 as a global regulator of the quiescent state (Figure 7A), where it controls the choice between alternate forms of arrest. As exemplified by analysis of CCNA2, PRDM2 may maintain regulatory nodes in an epigenetic state that is repressed but poised for activation through control of histone methylation, via regulation of the PRC2 complex (Figure 7B).
Figure 7.
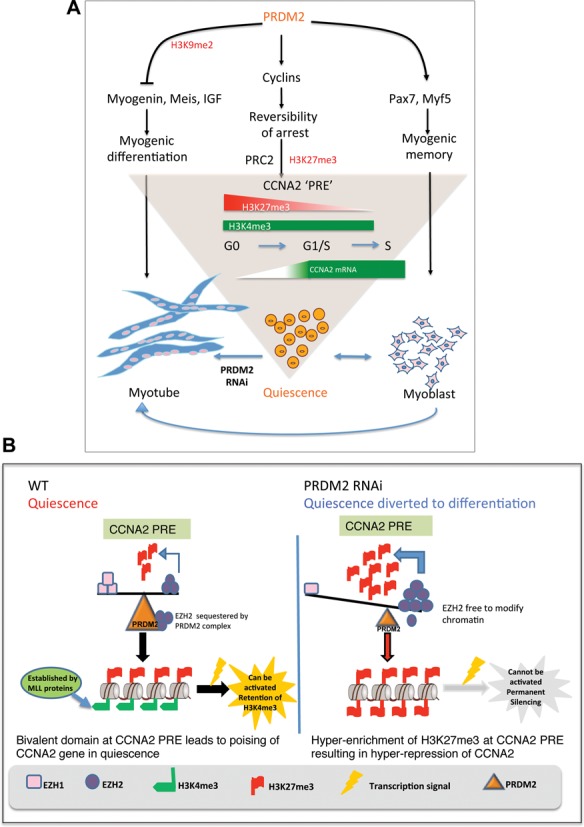
Model showing PRDM2 as a master regulator of quiescence—regulation of CCNA2 poising in reversible arrest via PRC2 dependent bivalent domain. (A) PRDM2 choreographs a genome-wide program to keep quiescent cells in a poised state by repressing myogenic networks to prevent differentiation (left), inducing/maintaining myogenic specification factors (right) and preserving reversibility of the cell cycle program via control of balanced methylation at a newly defined element in CCNA2 intron (center). Knockdown of PRDM2 subverts the quiescence program towards differentiation-coupled irreversible arrest. (B) PRDM2 prevents PRC2-mediated silencing of CCNA2 by sequestering EZH2 and preventing H3K27me3 accumulation at the CCNA2 PRE-like element. Thereby, G0 cells are held in suspended animation, poised to return to active proliferation, with a subsequent option to differentiate if conditions are conducive. PRDM2 knockdown permits EZH2 to accumulate at the PRE, leading to increased H3K27me3, silencing CCNA2 and diverting quiescence toward differentiation.
DISCUSSION
In this study, we reveal the role of an epigenetic regulator PRDM2/RIZ in cellular quiescence. We show expression of this tumor suppressor gene in presumptive muscle SCs in vivo and ex vivo on isolated myofibers. Induction during reversible G0 in mouse and human MBs, multipotent human MSC and fibroblasts in culture suggests that PRDM2 may play a role in a common quiescence-induced program, but our studies have focused on its role in muscle cell quiescence. We demonstrate that in C2C12 MB, PRDM2 plays a G0-specific repressive role targeting MyoG, a control hub for muscle differentiation. We provide evidence that PRDM2 coordinates a genome-wide program controlling >1400 transcripts and associating with >4000 promoters, 55% of which are also marked with repressive H3K9me2 marks. We establish that PRDM2 is a global repressor of myogenesis in G0 and targets the key differentiation regulator Myogenin, but also networks both upstream and downstream of MyoG. We also show that in G0, PRDM2 preserves expression of stem cell factors and prevents silencing of cell cycle genes. Finally, we uncover a new mechanism by which PRDM2 orchestrates control of CCNA2 in G0, by targeting a quiescence-specific bivalent domain via regulation of the PRC2 complex. Our findings support a model for active epigenetic regulation of the quiescent state in muscle cells, where a single regulator PRDM2 coordinates repression of differentiation with protection of cell cycle and stem cell regulators, to preserve reversibility of arrest.
PRDM2 is not required for arrest per se but controls the type of arrest program
As a known tumor suppressor, reduced PRDM2 may be expected to enhance proliferation. However, knockdown of PRDM2 did not activate cell division and even led to hyper-suppression of cell cycle gene expression, while over-expression of either RIZ1 or two isoform suppressed both proliferation and differentiation (Figure 1). Together, these results indicate that in myoblasts, PRDM2 is not required for arrest per se, but for entry into undifferentiated reversible G0, and in its absence, cells enter differentiation-associated terminal arrest. The bifurcation of arrest programs appears to be regulated by the Notch pathway, since G0 fibroblasts enter senescence-associated terminal arrest when depleted of the Notch target repressor Hes1, (60), while muscle SCs null for Notch effector RBPJ are diverted to differentiation-associated terminal arrest (9,61). Our findings implicate PRDM2 in control of this regulatory node. Interestingly, promoters of two Notch pathway genes (Notch1, Hey1) are both bound by PRDM2 and deregulated in the PRDM2 knockdown cells.
Permanent arrest of MTs is preserved by an Rb-controlled tumor suppressive network (26,62,63). By recruiting PRC2, Rb silences cell cycle genes and maintains terminal arrest (64). Intriguingly, PRDM2 was identified as an Rb-interacting protein (65), and we find it also associates with the Rb promoter. Thus, in muscle, while Rb orchestrates irreversible arrest (66), PRDM2 plays a key role in reversible arrest, perhaps by negating Rb. Interestingly, PRDM2 also associated with promoters of E2Fs (E2F6 and E2F7, Supplementary Table S5) that induce/maintain quiescence along with p130 (27), suggesting a complex balancing mechanism. For example, E2F6 participates in distinct activating and repressive chromatin complexes in G0 versus cycling NIH3T3 cells. In G0 MB, PRDM2 associates with the E2F6 promoter and E2F6 expression is de-repressed 1.8-fold in PRDM2sh MB. Regulation of the quiescence-regulating E2F factors supports a role for PRDM2 in G0. Of 196 E2F target genes (http://bioinfo-out.curie.fr/projects/rbpathway), 44 genes (22%) were also bound by PRDM2 in G0, including core components of PRC2 silencing complex—EED, EZH2, YY1, SUZ12. It is tempting to speculate that the overlap of PRDM2 targets with those of quiescence-associated E2F factors may contribute to interplay with the Rb-E2F network.
PRDM2 targets a key differentiation control hub
MyoG is activated early in myogenesis by a chromatin network recruited by MyoD and Meis/Pbx (23); other epigenetic factors (Suv39h1, EZH2, YY1) repress MyoG in MB (24). A consensus PRDM2 binding motif in the MyoG promoter (Supplementary Figure S2) shows increased PRDM2 association in G0 (Figure 3), correlates with increased MyoG promoter activity, and reduced H3K9me2 in PRDMsh MBs. In cycling MBs, Suv39h represses MyoG expression (25). Conceivably, in G0 MBs, PRDM2 may augment/replace Suv39h to repress MyoG and also differentiation.
PRDM2 is a fail-safe repressor of myogenesis in quiescence
Myogenin is a key control point in skeletal muscle differentiation, as evidenced by the presence of determined myoblasts but absence of differentiated myofibers in MyoG−/− mice (67). Thus, PRDM2's repression of MyoG alone may be sufficient to repress differentiation in G0. However, the finding that PRDM2 is associated with a number of muscle-specific promoters, many of which are deregulated in the knockdown cells, is suggestive of a wider role. Of the few reported targets of PRDM2, the IGF1 promoter is directly repressed via H3K9me2 in leukemic cells (39). IGF1 and IGF2 signaling promote myogenesis, and in G0 MBs, PRDM2 knockdown induces IGF2, suggesting that control of this pathway contributes to repression of differentiation. PRDM2 associates with promoters of 27% of genes in the IGF signaling pathway, >50% of which are marked by H3K9me2.
PRDM2 also associates with a spectrum of muscle-specific promoters, from transcription factors (MyoG, MEF2c, MyoD, Foxo1), to cytoskeletal elements (myosins, troponin, tropomyosin). Many sarcomeric components were co-associated with PRDM2 and repressive H3K9me2 in G0. PRDM2 may not only target networks at the level of MyoG, but also upstream and downstream, indicating a fail-safe repression of differentiation in G0. Thus, the myoblast quiescence program may rely on dominant epigenetic repression of muscle genes, coordinated by factors such as PRDM2.
Interestingly, several neurogenic genes involved in neural fate and signaling were also co-marked by PRDM2 and H3K9me2 and de-regulated by PRDM2 knockdown, suggesting broad suppression of differentiation programs.
PRDM2 protects cell cycle and stem cell regulatory genes in G0
In G0, both cell cycle genes and muscle genes are normally repressed. However, in PRDM2sh MB, cell cycle genes are hyper-repressed while myogenic genes are hyper-activated (Figure 3J), indicating a switch from undifferentiated arrest to differentiated arrest. In MT, cell cycle genes are permanently silenced (transcriptionally repressed, H3K27me3 marked) via PRC2 (64), which is not seen in G0 MB. Knockdown of PRDM2 altered CCNA2 promoter marking to that typical of irreversible exit: increased H3K27me3, reduced H3K4me3 (Figure 6). In addition to cyclin A2, both PRDM2 and H3K9me2 were enriched at promoters of cyclins A1, B, D, E, G, I, J and L in G0 (GEO GSE58676). Although our analysis of the CCND1 promoter did not reveal strong bivalent marking (Supplementary Figure S7), expression of cyclins D, E and B was also suppressed in the PRDM2 knockdown (Figure 3). Thus, it is conceivable that while repressive, sustained H3K9 methylation by PRDM2 (or other HMTs) may also maintain a basal activity or poising of other cell cycle genes in G0, protecting these key targets from permanent silencing, possibly by modulating PRC2 activity (Figures 5 and 6).
Intriguingly, PRDM2 also associates with promoters of stem cell and pluripotency markers (Figure 4G) such as HSC marker c-kit, pluripotency factor Oct4, and self-renewal factor Jmjd1a (68). PRDM2 also regulates muscle determinant Myf5, and Pax7 (Figure 3J), a SC survival/specification factor that recruits remodeling complexes to muscle promoters (69). PRDM2 knockdown inhibits expression of these SC genes, consistent with a role in maintaining their expression in G0. Association of PRDM2 with promoters of PRC2 members in G0 suggests coordinated control of cell cycle and stem cell genes. PRC2 mediates repression of developmental genes, is required for maintenance of potency and self-renewal (70) and conditional removal of EZH2 in Pax7+ SC leads to loss of self-renewal (71). Taken together with the reduced colony forming ability of PRDM2sh cells, these observations underscore a link between quiescence and self-renewal.
Although the methyltransferase activity of PRDM2 is well established (38,72) and the crystal structure of its SET domain determined (73), the specificity for histone methylation state has not been unambiguously established. In vitro HMT assays of transfected Hela cells (74) and C2C12 cells (Figure 2I) clearly indicate that RIZ1 can transfer methyl groups to H3K9. While the relationship of PRDM2 occupancy to H3K9me2 marks is correlative, our global analysis revealing their widespread co-association may indicate a role for this chromatin factor in mediating this modification directly or indirectly.
PRDM2 regulates association of PRC2 at a novel CCNA2 regulatory element
Unexpectedly for a tumor suppressor, PRDM2 preserves basal cyclin expression in G0. We therefore explored the mechanism by which this epigenetic regulator may keep genes poised while repressed. Bivalent chromatin domains (dually methylated at H3K43Me and H3K273Me) in fate-specification genes in ESC are associated with poising and these domains resolve to exhibit unique histone marks during lineage commitment (29). Bivalent marking has been recently demonstrated in ASCs (75) including muscle SCs (32), suggesting conserved mechanisms of poising. Intriguingly, our findings suggest that such epigenetic mechanisms are preserved even in the C2C12 cell line which was originally derived from adult SCs (76).
We chose CCNA2 gene to probe the PRDM2-regulated poising control mechanism since PRDM2 was earlier reported to interact with Rb and CCNA2 is a key target of the Rb-E2F-PRC2 axis. Our finding that PRDM2 associates with many E2F targets and key PRC2 genes underscored these links. PRC2-mediated Cyc A regulation in flies suggested the potential for conserved regulatory mechanisms. Our analysis uncovered new mechanisms controlling CCNA2: (i) CCNA2 locus undergoes cell cycle-dependent epigenetic modulation at a newly defined intronic element distinct from the well-known CCRE, (ii) the intronic element has repressive activity and binds a unique combination of PRC2 members specifically in G0, suggesting PRE-like function, (iii) bivalent histone marks at the putative PRE are converted to activation marks in cycling MBs where CCNA2 is expressed and silencing marks in MTs where CCNA2 is repressed, (iv) bivalent marks as well PRC2 association are lost in PRDM2sh cells. Importantly, using re-ChIP analysis, we also demonstrate that the novel element is truly bivalent, and cannot be attributed to cellular heterogeneity.
Interaction of PRDM2 and EZH2 proteins in muscle cells
Rather than mediating de novo gene silencing, evidence suggests that PRC2 plays a major role to maintain the repressive status of its targets (77). We, and others, have shown that repression of CCNA2 during G0 in myoblasts is mediated by Brm/Brg, CCRE-binding repressors, under the control of a Trx protein MLL5 (11,48). Our new findings show that both isoforms of PRDM2 interact with EZH2, whose association at the PRE (downstream of the CCRE) is increased in PRDM2 RNAi cells along with hyper-repression of CCNA2. Of the two H3K27 methyl transferases in the PRC2 complex, EZH2 is reported to be a much more active enzyme than EZH1 (78). The balanced enrichment of EZH1/EZH2 association with the PRE in G0 (Figure 6) and the shift to greater EZH2 enrichment in PRDM2 RNAi cells (reflecting the normal MT pattern), are consistent with hyper-repression of CCNA2. Our findings suggest a model where association of MLL5 and Brm/Brg at the CCRE during entry into quiescence, may initiate repression of CCNA2, while presence of PRDM2 at the PRE may prevent silencing by limiting association of EZH2, maintaining the poised state and competence for reactivation by mitogen stimulation. Establishing the timing of these events will be of importance in understanding the relative contributions of each of these factors. Together with our earlier findings, our observations point to CCNA2 as a regulatory node in quiescence targeted by MLL5, Brm/Brg, PRDM2 and PRC2.
PRDM2 may define a quiescence program
PRDM proteins have been implicated as molecular switches in cell fate choices. PRDM1 mediates selection of slow versus fast skeletal muscle (79), PRDM6 between differentiation and proliferation in smooth muscle (80), PRDM14 between self-renewal and lineage commitment in ESC (81) and PGC (82), and PRDM16 between brown fat and skeletal muscle (83). Our results suggest that PRDM2 regulates a switch between reversible versus irreversible arrest in muscle cells, by regulating association of the PRC complex at a novel element in a key S-phase control gene, CCNA2 (Figure 7). Interestingly, many PRDM2 bound genes are reported to be targets of PRDM14, a regulator of ESC pluripotency (81), suggestive of a potential role in self-renewal (Supplementary Figure S9). Not all PRDM family members have intrinsic histone modification activity (84), and may function via their interacting partners. While the two PRDM2 isoforms differ in their intrinsic HMT activity, they are both able to interact with pRb (35,40) and EZH2 (Figure 6). We speculate that the two isoforms may compete for these partners, which may control the extent of repressive marking of specific loci. Further, the complexes in which PRDM2 is found may differ at myogenic and cell cycle promoters, which may explain the differential regulation of these programs by a single regulator.
CONCLUSIONS
In summary, we provide evidence that PRDM2 is required for maintaining quiescence not only by preventing inappropriate differentiation but also by protecting cell cycle and stem cell genes from silencing. We propose that PRDM2/RIZ choreographs a genome-wide quiescence-associated program that maintains cells in a poised state and by holding two antagonistic programs in abeyance, preserves the option for alternate fates- a return to active proliferation or differentiation.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank Sindhu Subramaniam (identification of PRDM2 in G0), M.B. Vinay Kumar (hMSC), Genotypic Technologies (Agilent promoter arrays), M.B. Madhavi for help with Affymetrix analysis, D. Palakodeti (ChIP–chip bioinformatics), Surabhi Srivastava for helpful discussions, B. Moncharmont (hRIZ1 cDNA), M. Walsh and R. Taneja (Flag-G9a cDNA), C-M. Chiang, S. Galande and I. Siddiqi for comments on the manuscript.
Author contributions: S.C., D.P., R.K.M. and J.D. designed research; S.C. performed genome-wide analysis and phenotypic analysis of PRDM2 RNAi; D.P. investigated the CCNA2 bivalent domain and PRC2 role; S.C., D.P., A.S., H.G., M.R., M.S.P., R.A., P.S., J.S. and H.D.S. performed research and with J.D. analyzed data; S.C., D.P. and J.D. wrote the paper. The authors declare no conflict of interest.
FUNDING
Council of Scientific and Industrial Research (CSIR) doctoral Fellowships (to S.C., D.P., H.G., A.S., P.S. and M.R.); Department of Biotechnology (DBT) postdoctoral Fellowship (to M.S.P.); Institute for Stem Cell Biology and Regenerative Medicine Career Development Fellowship (to R.A.); Wellcome Trust International Senior Research Fellowship [073629] (to J.D.); CSIR Network Program on Cell and Tissue Engineering (to J.D.); Indo-Australia Biotechnology Fund [BT/Indo-Aus/05/36/2010] (DBT) (to R.K.M. and J.D.); Indo-Denmark Collaborative Grant [BT/IN/Denmark/02/PDN/2011] (DBT) (to H.D.S. and J.D.); and core support from CSIR to CCMB and the DBT to InStem. Funding for open access charge: Government of India, Department of Biotechnology.
Conflict of interest statement. None declared.
REFERENCES
- 1.Li M., Liu G.H., Izpisua Belmonte J.C. Navigating the epigenetic landscape of pluripotent stem cells. Nat. Rev. Mol. Cell Biol. 2012;13:524–535. doi: 10.1038/nrm3393. [DOI] [PubMed] [Google Scholar]
- 2.Young R.A. Control of the embryonic stem cell state. Cell. 2011;144:940–954. doi: 10.1016/j.cell.2011.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goldberg A.D., Allis C.D., Bernstein E. Epigenetics: a landscape takes shape. Cell. 2007;128:635–638. doi: 10.1016/j.cell.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 4.Mikkers H., Frisen J. Deconstructing stemness. EMBO J. 2005;24:2715–2719. doi: 10.1038/sj.emboj.7600749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jenuwein T., Allis C.D. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 6.Relaix F., Zammit P.S. Satellite cells are essential for skeletal muscle regeneration: the cell on the edge returns centre stage. Development. 2012;139:2845–2856. doi: 10.1242/dev.069088. [DOI] [PubMed] [Google Scholar]
- 7.Yusuf I., Fruman D.A. Regulation of quiescence in lymphocytes. Trends Immunol. 2003;24:380–386. doi: 10.1016/s1471-4906(03)00141-8. [DOI] [PubMed] [Google Scholar]
- 8.Coller H.A., Sang L., Roberts J.M. A new description of cellular quiescence. PLoS Biol. 2006;4:e83. doi: 10.1371/journal.pbio.0040083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bjornson C.R., Cheung T.H., Liu L., Tripathi P.V., Steeper K.M., Rando T.A. Notch signaling is necessary to maintain quiescence in adult muscle stem cells. Stem Cells. 2012;30:232–242. doi: 10.1002/stem.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Subramaniam S., Sreenivas P., Cheedipudi S., Reddy V.R., Shashidhara L.S., Chilukoti R.K., Mylavarapu M., Dhawan J. Distinct transcriptional networks in quiescent myoblasts: a role for Wnt signaling in reversible vs. irreversible arrest. PLoS One. 2013;8:e65097. doi: 10.1371/journal.pone.0065097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sebastian S., Sreenivas P., Sambasivan R., Cheedipudi S., Kandalla P., Pavlath G.K., Dhawan J. MLL5, a trithorax homolog, indirectly regulates H3K4 methylation, represses cyclin A2 expression, and promotes myogenic differentiation. Proc. Natl. Acad. Sci. U.S.A. 2009;106:4719–4724. doi: 10.1073/pnas.0807136106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Evertts A.G., Manning A.L., Wang X., Dyson N.J., Garcia B.A., Coller H.A. H4K20 methylation regulates quiescence and chromatin compaction. Mol. Biol. Cell. 2013;24:3025–3037. doi: 10.1091/mbc.E12-07-0529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu S., Tao Y. Interplay between chromatin modifications and paused RNA polymerase II in dynamic transition between stalled and activated genes. Biol. Rev. Camb. Philos. Soc. 2013;88:40–48. doi: 10.1111/j.1469-185X.2012.00237.x. [DOI] [PubMed] [Google Scholar]
- 14.Wagers A.J., Weissman I.L. Plasticity of adult stem cells. Cell. 2004;116:639–648. doi: 10.1016/s0092-8674(04)00208-9. [DOI] [PubMed] [Google Scholar]
- 15.Jaenisch R., Young R. Stem cells, the molecular circuitry of pluripotency and nuclear reprogramming. Cell. 2008;132:567–582. doi: 10.1016/j.cell.2008.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Milasincic D.J., Dhawan J., Farmer S.R. Anchorage-dependent control of muscle-specific gene expression in C2C12 mouse myoblasts. In Vitro Cell Dev. Biol. Anim. 1996;32:90–99. doi: 10.1007/BF02723040. [DOI] [PubMed] [Google Scholar]
- 17.Sachidanandan C., Sambasivan R., Dhawan J. Tristetraprolin and LPS-inducible CXC chemokine are rapidly induced in presumptive satellite cells in response to skeletal muscle injury. J. Cell Sci. 2002;115:2701–2712. doi: 10.1242/jcs.115.13.2701. [DOI] [PubMed] [Google Scholar]
- 18.Le Grand F., Grifone R., Mourikis P., Houbron C., Gigaud C., Pujol J., Maillet M., Pages G., Rudnicki M., Tajbakhsh S., et al. Six1 regulates stem cell repair potential and self-renewal during skeletal muscle regeneration. J. Cell Biol. 2012;198:815–832. doi: 10.1083/jcb.201201050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weintraub H. The MyoD family and myogenesis: redundancy, networks, and thresholds. Cell. 1993;75:1241–1244. doi: 10.1016/0092-8674(93)90610-3. [DOI] [PubMed] [Google Scholar]
- 20.Halevy O., Novitch B.G., Spicer D.B., Skapek S.X., Rhee J., Hannon G.J., Beach D., Lassar A.B. Correlation of terminal cell cycle arrest of skeletal muscle with induction of p21 by MyoD. Science. 1995;267:1018–1021. doi: 10.1126/science.7863327. [DOI] [PubMed] [Google Scholar]
- 21.Carnac G., Fajas L., L'Honore A., Sardet C., Lamb N.J., Fernandez A. The retinoblastoma-like protein p130 is involved in the determination of reserve cells in differentiating myoblasts. Curr. Biol. 2000;10:543–546. doi: 10.1016/s0960-9822(00)00471-1. [DOI] [PubMed] [Google Scholar]
- 22.Zhang C.L., McKinsey T.A., Olson E.N. Association of class II histone deacetylases with heterochromatin protein 1: potential role for histone methylation in control of muscle differentiation. Mol. Cell. Biol. 2002;22:7302–7312. doi: 10.1128/MCB.22.20.7302-7312.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sartorelli V., Caretti G. Mechanisms underlying the transcriptional regulation of skeletal myogenesis. Curr. Opin. Genet. Dev. 2005;15:528–535. doi: 10.1016/j.gde.2005.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guasconi V., Puri P.L. Chromatin: the interface between extrinsic cues and the epigenetic regulation of muscle regeneration. Trends Cell Biol. 2009;19:286–294. doi: 10.1016/j.tcb.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ait-Si-Ali S., Guasconi V., Fritsch L., Yahi H., Sekhri R., Naguibneva I., Robin P., Cabon F., Polesskaya A., Harel-Bellan A. A Suv39h-dependent mechanism for silencing S-phase genes in differentiating but not in cycling cells. EMBO J. 2004;23:605–615. doi: 10.1038/sj.emboj.7600074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blais A., van Oevelen C.J., Margueron R., Acosta-Alvear D., Dynlacht B.D. Retinoblastoma tumor suppressor protein-dependent methylation of histone H3 lysine 27 is associated with irreversible cell cycle exit. J. Cell Biol. 2007;179:1399–1412. doi: 10.1083/jcb.200705051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Litovchick L., Sadasivam S., Florens L., Zhu X., Swanson S.K., Velmurugan S., Chen R., Washburn M.P., Liu X.S., DeCaprio J.A. Evolutionarily conserved multisubunit RBL2/p130 and E2F4 protein complex represses human cell cycle-dependent genes in quiescence. Mol. Cell. 2007;26:539–551. doi: 10.1016/j.molcel.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 28.Korenjak M., Kwon E., Morris R.T., Anderssen E., Amzallag A., Ramaswamy S., Dyson N.J. dREAM co-operates with insulator-binding proteins and regulates expression at divergently paired genes. Nucleic Acids Res. 2014;42:8939–8953. doi: 10.1093/nar/gku609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bernstein B.E., Mikkelsen T.S., Xie X., Kamal M., Huebert D.J., Cuff J., Fry B., Meissner A., Wernig M., Plath K., et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 30.Alder O., Lavial F., Helness A., Brookes E., Pinho S., Chandrashekran A., Arnaud P., Pombo A., O'Neill L., Azuara V. Ring1B and Suv39h1 delineate distinct chromatin states at bivalent genes during early mouse lineage commitment. Development. 2010;137:2483–2492. doi: 10.1242/dev.048363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Azuara V. Profiling of DNA replication timing in unsynchronized cell populations. Nat. Protoc. 2006;1:2171–2177. doi: 10.1038/nprot.2006.353. [DOI] [PubMed] [Google Scholar]
- 32.Liu L., Cheung T.H., Charville G.W., Hurgo B.M., Leavitt T., Shih J., Brunet A., Rando T.A. Chromatin modifications as determinants of muscle stem cell quiescence and chronological aging. Cell Rep. 2013;4:189–204. doi: 10.1016/j.celrep.2013.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sellathurai J., Cheedipudi S., Dhawan J., Schroder H.D. A novel in vitro model for studying quiescence and activation of primary isolated human myoblasts. PLoS One. 2013;8:e64067. doi: 10.1371/journal.pone.0064067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fukada S., Higuchi S., Segawa M., Koda K., Yamamoto Y., Tsujikawa K., Kohama Y., Uezumi A., Imamura M., Miyagoe-Suzuki Y., et al. Purification and cell-surface marker characterization of quiescent satellite cells from murine skeletal muscle by a novel monoclonal antibody. Exp. Cell Res. 2004;296:245–255. doi: 10.1016/j.yexcr.2004.02.018. [DOI] [PubMed] [Google Scholar]
- 35.Liu L., Shao G., Steele-Perkins G., Huang S. The retinoblastoma interacting zinc finger gene RIZ produces a PR domain-lacking product through an internal promoter. J. Biol. Chem. 1997;272:2984–2991. doi: 10.1074/jbc.272.5.2984. [DOI] [PubMed] [Google Scholar]
- 36.Gazzerro P., Abbondanza C., D'Arcangelo A., Rossi M., Medici N., Moncharmont B., Puca G.A. Modulation of RIZ gene expression is associated to estradiol control of MCF-7 breast cancer cell proliferation. Exp. Cell Res. 2006;312:340–349. doi: 10.1016/j.yexcr.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 37.Dillon S.C., Zhang X., Trievel R.C., Cheng X. The SET-domain protein superfamily: protein lysine methyltransferases. Genome Biol. 2005;6:227. doi: 10.1186/gb-2005-6-8-227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim K.C., Geng L., Huang S. Inactivation of a histone methyltransferase by mutations in human cancers. Cancer Res. 2003;63:7619–7623. [PubMed] [Google Scholar]
- 39.Pastural E., Takahashi N., Dong W.F., Bainbridge M., Hull A., Pearson D., Huang S., Lowsky R., DeCoteau J.F., Geyer C.R. RIZ1 repression is associated with insulin-like growth factor-1 signaling activation in chronic myeloid leukemia cell lines. Oncogene. 2007;26:1586–1594. doi: 10.1038/sj.onc.1209959. [DOI] [PubMed] [Google Scholar]
- 40.Steele-Perkins G., Fang W., Yang X.H., Van Gele M., Carling T., Gu J., Buyse I.M., Fletcher J.A., Liu J., Bronson R., et al. Tumor formation and inactivation of RIZ1, an Rb-binding member of a nuclear protein-methyltransferase superfamily. Genes Dev. 2001;15:2250–2262. doi: 10.1101/gad.870101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.He L., Yu J.X., Liu L., Buyse I.M., Wang M.S., Yang Q.C., Nakagawara A., Brodeur G.M., Shi Y.E., Huang S. RIZ1, but not the alternative RIZ2 product of the same gene, is underexpressed in breast cancer, and forced RIZ1 expression causes G2-M cell cycle arrest and/or apoptosis. Cancer Res. 1998;58:4238–4244. [PubMed] [Google Scholar]
- 42.Mal A.K. Histone methyltransferase Suv39h1 represses MyoD-stimulated myogenic differentiation. EMBO J. 2006;25:3323–3334. doi: 10.1038/sj.emboj.7601229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Medici N., Abbondanza C., Nigro V., Rossi V., Piluso G., Belsito A., Gallo L., Roscigno A., Bontempo P., Puca A.A., et al. Identification of a DNA binding protein cooperating with estrogen receptor as RIZ (retinoblastoma interacting zinc finger protein) Biochem. Biophys. Res. Commun. 1999;264:983–989. doi: 10.1006/bbrc.1999.1604. [DOI] [PubMed] [Google Scholar]
- 44.Supek F., Bosnjak M., Skunca N., Smuc T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One. 2011;6:e21800. doi: 10.1371/journal.pone.0021800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Laresgoiti U., Apraiz A., Olea M., Mitxelena J., Osinalde N., Rodriguez J.A., Fullaondo A., Zubiaga A.M. E2F2 and CREB cooperatively regulate transcriptional activity of cell cycle genes. Nucleic Acids Res. 2013;41:10185–10198. doi: 10.1093/nar/gkt821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee H.M., Zhang H., Schulz V., Tuck D.P., Forget B.G. Downstream targets of HOXB4 in a cell line model of primitive hematopoietic progenitor cells. Blood. 2010;116:720–730. doi: 10.1182/blood-2009-11-253872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Garcia-Bassets I., Kwon Y.S., Telese F., Prefontaine G.G., Hutt K.R., Cheng C.S., Ju B.G., Ohgi K.A., Wang J., Escoubet-Lozach L., et al. Histone methylation-dependent mechanisms impose ligand dependency for gene activation by nuclear receptors. Cell. 2007;128:505–518. doi: 10.1016/j.cell.2006.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Coisy M., Roure V., Ribot M., Philips A., Muchardt C., Blanchard J.M., Dantonel J.C. Cyclin A repression in quiescent cells is associated with chromatin remodeling of its promoter and requires Brahma/SNF2alpha. Mol. Cell. 2004;15:43–56. doi: 10.1016/j.molcel.2004.06.022. [DOI] [PubMed] [Google Scholar]
- 49.Martinez A.M., Colomb S., Dejardin J., Bantignies F., Cavalli G. Polycomb group-dependent Cyclin A repression in Drosophila. Genes Dev. 2006;20:501–513. doi: 10.1101/gad.357106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Martinez A.M., Cavalli G. The role of polycomb group proteins in cell cycle regulation during development. Cell Cycle. 2006;5:1189–1197. doi: 10.4161/cc.5.11.2781. [DOI] [PubMed] [Google Scholar]
- 51.Ringrose L., Rehmsmeier M., Dura J.M., Paro R. Genome-wide prediction of Polycomb/Trithorax response elements in Drosophila melanogaster. Dev. Cell. 2003;5:759–771. doi: 10.1016/s1534-5807(03)00337-x. [DOI] [PubMed] [Google Scholar]
- 52.Brown J.L., Fritsch C., Mueller J., Kassis J.A. The Drosophila pho-like gene encodes a YY1-related DNA binding protein that is redundant with pleiohomeotic in homeotic gene silencing. Development. 2003;130:285–294. doi: 10.1242/dev.00204. [DOI] [PubMed] [Google Scholar]
- 53.Bocan T.M., Mueller S.B., Brown E.Q., Lee P., Bocan M.J., Rea T., Pape M.E. HMG-CoA reductase and ACAT inhibitors act synergistically to lower plasma cholesterol and limit atherosclerotic lesion development in the cholesterol-fed rabbit. Atherosclerosis. 1998;139:21–30. doi: 10.1016/s0021-9150(98)00046-x. [DOI] [PubMed] [Google Scholar]
- 54.Matharu N.K., Hussain T., Sankaranarayanan R., Mishra R.K. Vertebrate homologue of Drosophila GAGA factor. J. Mol. Biol. 2010;400:434–447. doi: 10.1016/j.jmb.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 55.Mihaly J., Mishra R.K., Karch F. A conserved sequence motif in Polycomb-response elements. Mol. Cell. 1998;1:1065–1066. doi: 10.1016/s1097-2765(00)80107-0. [DOI] [PubMed] [Google Scholar]
- 56.Vasanthi D., Nagabhushan A., Matharu N.K., Mishra R.K. A functionally conserved Polycomb response element from mouse HoxD complex responds to heterochromatin factors. Sci. Rep. 2013;3:3011. doi: 10.1038/srep03011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ezhkova E., Lien W.H., Stokes N., Pasolli H.A., Silva J.M., Fuchs E. EZH1 and EZH2 cogovern histone H3K27 trimethylation and are essential for hair follicle homeostasis and wound repair. Genes Dev. 2011;25:485–498. doi: 10.1101/gad.2019811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mousavi K., Zare H., Wang A.H., Sartorelli V. Polycomb protein Ezh1 promotes RNA polymerase II elongation. Mol. Cell. 2012;45:255–262. doi: 10.1016/j.molcel.2011.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stojic L., Jasencakova Z., Prezioso C., Stutzer A., Bodega B., Pasini D., Klingberg R., Mozzetta C., Margueron R., Puri P.L., et al. Chromatin regulated interchange between polycomb repressive complex 2 (PRC2)-Ezh2 and PRC2-Ezh1 complexes controls myogenin activation in skeletal muscle cells. Epigenetics Chromatin. 2011;4:16. doi: 10.1186/1756-8935-4-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sang L., Coller H.A., Roberts J.M. Control of the reversibility of cellular quiescence by the transcriptional repressor HES1. Science. 2008;321:1095–1100. doi: 10.1126/science.1155998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mourikis P., Sambasivan R., Castel D., Rocheteau P., Bizzarro V., Tajbakhsh S. A critical requirement for notch signaling in maintenance of the quiescent skeletal muscle stem cell state. Stem Cells. 2012;30:243–252. doi: 10.1002/stem.775. [DOI] [PubMed] [Google Scholar]
- 62.Asp P., Blum R., Vethantham V., Parisi F., Micsinai M., Cheng J., Bowman C., Kluger Y., Dynlacht B.D. Genome-wide remodeling of the epigenetic landscape during myogenic differentiation. Proc. Natl. Acad. Sci. U.S.A. 2011;108:E149–E158. doi: 10.1073/pnas.1102223108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Endo T., Nadal-Ginard B. Reversal of myogenic terminal differentiation by SV40 large T antigen results in mitosis and apoptosis. J. Cell Sci. 1998;111:1081–1093. doi: 10.1242/jcs.111.8.1081. [DOI] [PubMed] [Google Scholar]
- 64.Blais A., Dynlacht B.D. E2F-associated chromatin modifiers and cell cycle control. Curr. Opin. Cell Biol. 2007;19:658–662. doi: 10.1016/j.ceb.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Buyse I.M., Shao G., Huang S. The retinoblastoma protein binds to RIZ, a zinc-finger protein that shares an epitope with the adenovirus E1A protein. Proc. Natl. Acad. Sci. U.S.A. 1995;92:4467–4471. doi: 10.1073/pnas.92.10.4467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Huh M.S., Parker M.H., Scime A., Parks R., Rudnicki M.A. Rb is required for progression through myogenic differentiation but not maintenance of terminal differentiation. J. Cell Biol. 2004;166:865–876. doi: 10.1083/jcb.200403004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hasty P., Bradley A., Morris J.H., Edmondson D.G., Venuti J.M., Olson E.N., Klein W.H. Muscle deficiency and neonatal death in mice with a targeted mutation in the myogenin gene. Nature. 1993;364:501–506. doi: 10.1038/364501a0. [DOI] [PubMed] [Google Scholar]
- 68.Loh Y.H., Zhang W., Chen X., George J., Ng H.H. Jmjd1a and Jmjd2c histone H3 Lys 9 demethylases regulate self-renewal in embryonic stem cells. Genes Dev. 2007;21:2545–2557. doi: 10.1101/gad.1588207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.McKinnell I.W., Ishibashi J., Le Grand F., Punch V.G., Addicks G.C., Greenblatt J.F., Dilworth F.J., Rudnicki M.A. Pax7 activates myogenic genes by recruitment of a histone methyltransferase complex. Nat. Cell Biol. 2008;10:77–84. doi: 10.1038/ncb1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lee T.I., Jenner R.G., Boyer L.A., Guenther M.G., Levine S.S., Kumar R.M., Chevalier B., Johnstone S.E., Cole M.F., Isono K., et al. Control of developmental regulators by Polycomb in human embryonic stem cells. Cell. 2006;125:301–313. doi: 10.1016/j.cell.2006.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Juan A.H., Derfoul A., Feng X., Ryall J.G., Dell'Orso S., Pasut A., Zare H., Simone J.M., Rudnicki M.A., Sartorelli V. Polycomb EZH2 controls self-renewal and safeguards the transcriptional identity of skeletal muscle stem cells. Genes Dev. 2011;25:789–794. doi: 10.1101/gad.2027911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Peters A.H., Kubicek S., Mechtler K., O'Sullivan R.J., Derijck A.A., Perez-Burgos L., Kohlmaier A., Opravil S., Tachibana M., Shinkai Y., et al. Partitioning and plasticity of repressive histone methylation states in mammalian chromatin. Mol. Cell. 2003;12:1577–1589. doi: 10.1016/s1097-2765(03)00477-5. [DOI] [PubMed] [Google Scholar]
- 73.Wu H., Min J., Lunin V.V., Antoshenko T., Dombrovski L., Zeng H., Allali-Hassani A., Campagna-Slater V., Vedadi M., Arrowsmith C.H., et al. Structural biology of human H3K9 methyltransferases. PLoS One. 2010;5:e8570. doi: 10.1371/journal.pone.0008570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Congdon L.M., Sims J.K., Tuzon C.T., Rice J.C. The PR-Set7 binding domain of Riz1 is required for the H4K20me1-H3K9me1 trans-tail ‘histone code’ and Riz1 tumor suppressor function. Nucleic Acids Res. 2014;42:3580–3589. doi: 10.1093/nar/gkt1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Voigt P., Tee W.W., Reinberg D. A double take on bivalent promoters. Genes Dev. 2013;27:1318–1338. doi: 10.1101/gad.219626.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yaffe D., Saxel O. A myogenic cell line with altered serum requirements for differentiation. Differentiation. 1977;7:159–166. doi: 10.1111/j.1432-0436.1977.tb01507.x. [DOI] [PubMed] [Google Scholar]
- 77.Riising E.M., Comet I., Leblanc B., Wu X., Johansen J.V., Helin K. Gene silencing triggers polycomb repressive complex 2 recruitment to CpG islands genome wide. Mol. Cell. 2014;55:347–360. doi: 10.1016/j.molcel.2014.06.005. [DOI] [PubMed] [Google Scholar]
- 78.Margueron R., Li G., Sarma K., Blais A., Zavadil J., Woodcock C.L., Dynlacht B.D., Reinberg D. Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol. Cell. 2008;32:503–518. doi: 10.1016/j.molcel.2008.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.von Hofsten J., Elworthy S., Gilchrist M.J., Smith J.C., Wardle F.C., Ingham P.W. Prdm1- and Sox6-mediated transcriptional repression specifies muscle fibre type in the zebrafish embryo. EMBO Rep. 2008;9:683–689. doi: 10.1038/embor.2008.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Davis C.A., Haberland M., Arnold M.A., Sutherland L.B., McDonald O.G., Richardson J.A., Childs G., Harris S., Owens G.K., Olson E.N. PRISM/PRDM6, a transcriptional repressor that promotes the proliferative gene program in smooth muscle cells. Mol. Cell. Biol. 2006;26:2626–2636. doi: 10.1128/MCB.26.7.2626-2636.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chia N.Y., Chan Y.S., Feng B., Lu X., Orlov Y.L., Moreau D., Kumar P., Yang L., Jiang J., Lau M.S., et al. A genome-wide RNAi screen reveals determinants of human embryonic stem cell identity. Nature. 2010;468:316–320. doi: 10.1038/nature09531. [DOI] [PubMed] [Google Scholar]
- 82.Grabole N., Tischler J., Hackett J.A., Kim S., Tang F., Leitch H.G., Magnusdottir E., Surani M.A. Prdm14 promotes germline fate and naive pluripotency by repressing FGF signalling and DNA methylation. EMBO Rep. 2013;14:629–637. doi: 10.1038/embor.2013.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kajimura S., Seale P., Kubota K., Lunsford E., Frangioni J.V., Gygi S.P., Spiegelman B.M. Initiation of myoblast to brown fat switch by a PRDM16-C/EBP-beta transcriptional complex. Nature. 2009;460:1154–1158. doi: 10.1038/nature08262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hohenauer T., Moore A.W. The Prdm family: expanding roles in stem cells and development. Development. 2012;139:2267–2282. doi: 10.1242/dev.070110. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.