

Abstract
Multimeric interactions that occur in biology provide impetus for chemists to explore new types of synthetic multivalent ligands that alter cellular functions by mechanisms inaccessible to natural substances. While many different molecules such as peptides, antibody fragments, carbohydrates and organic moieties have been used in developing multimeric ligands, it is worth exploring other important molecular types that have hardly been tested in developing multimeric compounds. Peptoids are one such class of compounds with highly facile synthesis as well as much better biologically amenable qualities. Recently, we identified two HCC4017 lung cancer cell targeting peptoids. Here we explore the possibility of synthesizing multimers of these compounds completely through a solid phase synthesis approach. We have synthesized mini-libraries of homodimers, homotrimers and most importantly, heterodimers of our lung cancer specific compounds. The idea is to develop series of compounds that only differs by the linker portion, which is readily adjustable within the library. The purpose of this is to find the optimal distance between each monomeric unit of the multimer that allows them to perfectly interact with their individual biological targets displayed on the cell surface. Future screens of these minilibraries will identify the multimers with improved binding affinities.
Keywords: peptoids, receptor targeting, homodimers, homotrimers, heterodimers
INTRODUCTION
Multivalent interactions are well known phenomena that occur in both the natural and synthetic worlds and possess elements of structural complexity not present in their monovalent counterparts. They can be described as the simultaneous interaction of multiple components of a ligand on an entity that has multiple “hot spots” to bind with. The ability of multimeric cell-surface receptors to recognize natural or non-natural multimeric ligands has been a major crossroads in biological and chemical research, providing the basis for mechanisms of both agonizing and antagonizing biological processes. Following nature’s design in producing multivalent forms of ligands to enhance the binding affinity through the avidity effect, there have been many attempts to develop multimeric synthetic compounds to be used as potential drugs. As the structural features of these synthetic multimers can be manipulated, these compounds are suggested as better approaches to successfully compete with natural ligand–receptor interactions.
In recent years, the conventional approaches—such as elucidating the function of a single receptor—are now shifting toward understanding how systems of interacting proteins control cellular responses. Carefully designed multimeric compounds can be very useful in probing higher order biological mechanisms, helping to answer questions such as how the extent of receptor clustering influences their function, and how important these systems are for cell survival and growth.1,2 Multivalent ligands, with their ability to organize, activate, or inhibit multiple receptors, provide an excellent platform to address these questions in systems biology.3 However, there are many parameters that influence the mechanism of interaction for the ligand such as the identity of the binding elements, the structure of the scaffold, the number of binding groups, and the density of binding elements.3 When considering the diversity of these variables, multimeric ligand systems that can be: (1) readily and economically synthesized, (2) flexible for modification and optimization, and (3) highly compatible to applications in biological systems such as mice and humans, are of extreme importance.
Many types of synthetic multivalent ligands have been developed over the years, using structural moieties such as peptides,4–8 truncated versions of antibodies,9,10 carbohydrate analogues,11 and small organic moieties12 as inhibitors and effectors of high complexity.3,13,14 In particular, the literature in the synthetic multivalency arena is highly enriched with a wide variety of interesting chemical strategies to develop peptide dimers and high molecular weight multimers4,15 and dendrimers.5 Simple lysine residues to complex polymeric materials have been introduced as central linker portions to hold each monomeric unit of these multimers together. However, there are downsides to be considered with each of these molecular classes. Peptides can encounter problems with immunogenicity and serum instability. For antibody-based systems, the production costs and handling difficulties are major concerns in addition to drawbacks such as low tissue penetration, longer circulation, and immunogenicity associated with their high molecular weight.16 Modifications on small organic molecules and carbohydrate analogs to form ideal multimers often pose huge synthetic challenges that are time and resource consuming. Given this situation, it is important to explore alternative molecular classes with facile and straightforward syntheses that can facilitate rapid and economical development of multimeric compounds that are not only applicable in “test-tube levels,” but highly amenable to biological systems. Here we report an initial step of such an attempt with the rapid and complete on-bead development of homodimers, homotrimers, and more importantly heterodimers of HCC4017 human lung cancer-specific peptoids that we recently identified.
Peptoids are an emerging class of peptidomimetics with unique characteristics, which make them an excellent choice for developing adaptable synthetic multivalent systems as probes and drugs. This is mainly because of their vast chemical space available for solid phase synthesis, ease of modifications and optimizations, and unique biologically amenable properties that may allow them to rapidly and conveniently translate to clinical use. Peptoids (oligo-N-substituted glycines) are closely related to peptides except that the side chains extend from the main chain nitrogen rather than the α-carbon (Supporting Information Figure 1).17–20 These oligomers are protease resistant, more cell permeable, achiral, and adopt different conformations than peptides.17,19,21–24 Peptoid synthesis is straightforward and can be conveniently conducted on solid phase (Supporting Information Figure 1).20 To add one residue (equivalent to an amino acid of a peptide) all that is needed are two chemical steps that can be completed within 2 × 15 sec microwave pulses.25,26 Bromoacetic acid coupling brings the two carbon unit where Br can then be replaced by any amine group (Supporting Information Figure 1).20 The availability of a wide-range of low-cost and commercially available organic amines dramatically expands the repertoire of chemical space that can be used in the optimization of lead compounds in contrast to peptides, antibodies, carbohydrates, and small organic molecules. Large combinatorial libraries of peptoids (in millions) can be synthesized easily, inexpensively, and rapidly.27–31 Peptoids are also a rich source of protein-binding, functionally active ligands19,28–37 and are nonimmunogenic in mice.38 Furthermore, peptoid literature has already been enriched enough to prove that vast varieties of structural features such as, macrocycles,39–53 nanostructures,54,55 glycopeptoids,56–58 metallopep-toids,59,60 extended peptoids,61 cyclic betapeptoids,62 as well as peptide–peptoid63 mixed units can be conveniently extracted from peptoid scaffolds. These peptoid scaffolds display unique and very interesting folding patterns23,24,64–69 along with various types of structure–function relationships.22 Of note, a few peptoid dimers have already been reported for important biological targets.29,70,71 Based on these findings, we have decided to explore the possibility of complete on-bead development of adaptable “mini-libraries” of peptoid multimers that can be used to identify the best candidates as future drugs and research probes.
EXPERIMENTAL DESIGN
Recently, we have identified two HCC4017 lung cancer-specific peptidomimetics named JM79 and JM81 using a combinatorial cell surface binding assay (Figure 1; Matharage et al., in preparation). The sequences of both compounds contain three common amino acids at the C-terminus (Met, D-Lys, and Lys), followed by a 5-mer peptoid portion which we believe is the important region of interest for target recognition. JM79 was selected to develop a series of homodimers (Figure 2A) and homotrimers (Figure 2B). JM81, which was isolated as another compound targeting the same HCC4017 cell surface, gives us a unique opportunity to develop heterodimers (Figure 2C). Heterodimers could target two different hot spots on the same receptor or, more importantly, two receptor types.7 A heterodimer that binds to two different receptor types can bring unique advantages, such as affecting two signaling pathways at once with greater specificity and efficacy. Therefore, we took the advantage of having two compounds selected against one cell surface, using both in our multimeric compound minilibrary design to develop homomultimers and heteromultimers.
FIGURE 1.
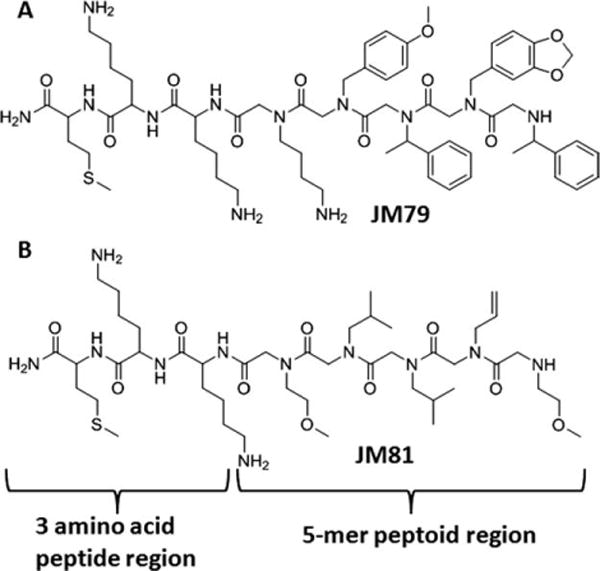
Chemical structures of two high specific HCC4017 lung cancer targeted peptoids JM79 and JM81.
FIGURE 2.
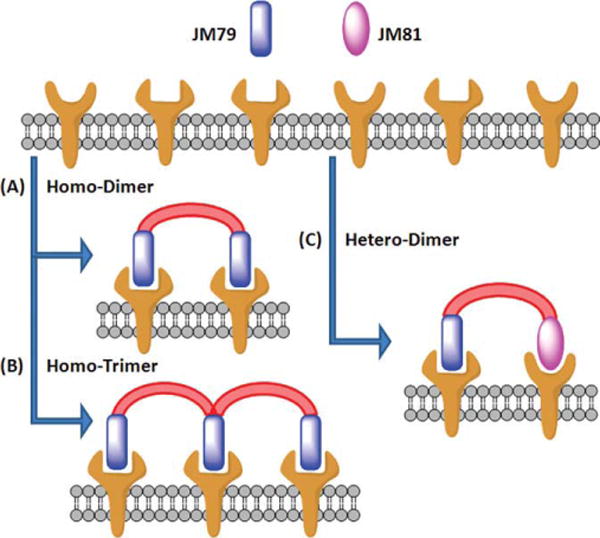
Schematic representation of various multimer forms of ligands that can target multiple cell surface receptors (A) homodimer, (B) homotrimer, and (C) heterodimer.
In both homomultimer and heteromultimer development approaches, the biggest challenge is uncovering the distance between two of the same and/or different receptors on the cell surface as it depends on the expression levels of targeted receptors. In other words, the golden question is; how can one develop a proper linker to bring either multiple copies of the same compound, or multiple different compounds, together to achieve the proper distance while allowing each monomeric unit of the multimer to interact accurately with their targeted receptors? In fact, the question is the same for developing multimers targeting multiple hot spots on the same receptor as well, unless the structures of the targeted receptors are available. Previous attempts on developing multimers have reported various opinions about the actual distances between two receptors on the cell surface and are highly ambiguous.3,7,14,72–74 These different opinions and observations are understandable because the density of a particular receptor on a cell surface is highly dependent on the receptor type, cell type, and physiological condition. Therefore, finding a globally optimized linker to bring together two or more receptor targeted compounds is an extremely difficult problem that is almost impractical to handle. This is especially true for highly complex cancer cells that behave differently than normal cells.
Considering all the factors previously described, we pursued a strategy to develop homodimers and heterodimers with variable linker regions that can be manipulated easily so that each scaffold (homodimer, homotrimer, and heterodimer) can be synthesized as a minilibrary of compounds. These can then be screened for binding and functional activity, identifying the best compounds for each series in future studies. Conducting these types of “screens” with multimeric compounds differing only at the linker region is the most practical way to pick the best compounds when considering receptor expression variability mentioned above. Based on this rationale, we decided to attempt the whole synthesis to completion on solid phase (resin beads), as it provides an avenue for rapid and economical development of series of compounds at once. We initially decided to develop multimers with relatively shorter linkers and after evaluating their binding potencies, the other ranges of (higher order) linkers would be incorporated.
MATERIALS AND METHODS
Materials
The following materials were obtained from the listed commercial sources: Novasyn TGR Resin, all Fmoc- and Boc-protected amino acids from EMD4Biosciences (Gibbstown, NJ); Fmoc-AEEAc-OH from Bachem (Torrance, CA); 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU), and N-Hydroxybenzotriazole (HOBt) from Anaspec (San Jose, CA). All primary amines, bromoacetic acid, diisopropylcarbodiimide (DIC), N,N-diisopropylethylamine (DIPEA), piperidine, trifluoroacetic acid (TFA), and dimethyl formamide (DMF) from Sigma-Aldrich (Milwaukee, WI); dichloromethane (DCM) and acetonitrile from Honeywell Burdick & Jackson (Morristown, NJ).
Methods
General Synthesis Procedures
Peptide Portions
Manual Fmoc-based solid phase peptide synthesis method was used for all peptide portions with each synthesis using a 100 mg of Novasyn TGR resin. Initially, resin beads were swelled in DMF (10× resin volume) for 45–60 min. Unless otherwise noted, all peptide coupling steps were performed by mixing a Fmoc-protected amino acid (5n) with coupling reagents [HBTU (4.9n), HOBt (5n), DIPEA (10n) where n = the loading level (molar amount) of the resin used] in 1 mL of DMF and treated in a disposable reaction vessel (Intavis) gently shaken for 2 h. After washing with DMF (10 times with 10× resin volume), Fmoc deprotection was carried out by treatment with 20% (v/v) piperidine in DMF, 2.5 mL (for 10 min, twice with shaking) followed by another DMF wash as before. For the first amino acid (cysteine), each Boc-Gly-OH, and Fmoc-AEEAc-OH additions, the reactions were allowed to proceed for 8 h to overnight. Each compound was synthesized with a C-terminal cysteine, regardless of the sequence, to use them in future attachments of fluorescein, imaging agents or drugs using maleimide chemistry.
Peptoid Portions
Each peptoid unit was coupled using the two successive reactions, shown in Supporting Information Figure 1, by performing microwave-assisted synthesis protocol. First, beads were treated with 2 M bromoacetic acid and 3.2 M DIC, shaken gently for 30 sec, and the coupling was performed for 15 sec in a microwave oven set to deliver 10% power. The reaction mixture was then gently shaken again for 15 sec, and the microwave step was repeated. Following a subsequent DMF wash (10 times with 10× resin volume), the primary amine (2 M) was treated and mixed gently for 15 sec. The same microwave procedure was used to assist the reaction that was used for the bromoacetylation.
Cleavage and Purification
On completion of each compound, the TFA/triisopropylsilane (TIS)/ddH2O [95:2.5:2.5 (v/v/v); 2 mL] cleavage cocktail was treated for 2 h to remove all TFA labile protecting groups and cleave the compound from the resin. The cleavage solution was retrieved in a 15 mL conical vial along with 2 mL of DCM washes (3×) off the resin to recover any residual product. The majority of TFA and DCM were then evaporated by blowing air over the solution for approximately 15–45 min. The resulting residue was resuspended in ddH2O/acetonitrile (50/50, v/v) and MALDI mass analysis was conducted to confirm the product. The compound was purified by HPLC and lyophilized to obtain pure product, which was stored either in dry powder form or dissolved in dimethyl sulfoxide (DMSO) as a highly concentrated stock solution. Applied Biosystems Voyager—6115 mass spectrometer was used in positive reflector mode to acquire matrix-assisted laser desorption/ionization (MALDI-TOF) mass spectra. Alpha-Cyano-4-hydroxycinnamic acid was used as matrix. High performance liquid chromatography (HPLC) purification was performed in a Waters 1525 Binary HPLC system connected to Waters 2487 Dual λ Absorbance Detector.
Synthesis of JM79.D1-4
First, Fmoc-Cys(Trt)-OH was coupled to the beads, followed by coupling of Fmoc-Lys(Fmoc)-OH as the central linker [Scheme 1 (1–2)], using the procedure described in Peptide Portions section. Then, both Fmoc groups were removed simultaneously, and the linker portions were added. Linker portions were built by coupling different numbers of Fmoc-β-Ala-OH units producing four different compounds {[Scheme 1(2–3)]: n = 0, 1, 2, and 3 units for JM79.D1, JM79.D2, JM79.D3, and JM79.D4, respectively}. Then, Fmoc-Met-OH, Fmoc-D-Lys(Boc)-OH, and Fmoc-Lys(Boc)-OH were coupled again as described in Peptide Portions section, adding all residues to both arms of the central lysine [Scheme 1(3–4)]. After final Fmoc removal, the peptoid units Nlys, N4mob, Nmba, Npip, and Nmba were coupled [Scheme 1(4–5)] by following the microwave-assisted peptoid synthesis protocol described in Peptoid Portions section, completing two JM79 moieties on two arms of the central lysine. Final products were cleaved off the resin, analyzed and purified as described in Cleavage and Purification section.
SCHEME 1.
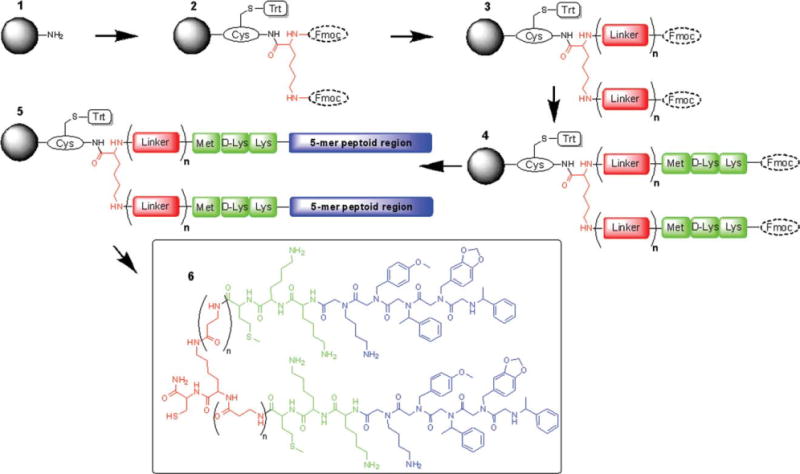
Solid phase synthesis strategy to build JM79 homodimers. The reagents and reaction conditions used are as follows: (1–2) a. Fmoc-Cys(Trt)-OH, HOBt, HBTU, DIPEA, b. 20% piperidine in DMF, c. Fmoc-Lys(Fmoc)-OH, HOBt, HBTU, DIPEA. (2–3) a. 20% piperidine in DMF, b. Fmoc-β-Ala-OH, HOBt, HBTU, DIPEA, (n = 0, 1, 2, or 3 additions). (3–4) a. 20% piperidine in DMF, b. addition of peptide portion (standard solid phase peptide synthesis (SPPS) protocol to add Met, D-Lys, Lys). (4–5) a. 20% piperidine in DMF, b. addition of 5-mer peptoid portion (standard microwave-assisted peptoid synthesis). (5–6) 95% TFA, 2.5% TIS, 2.5% H2O.
Synthesis of JM79.T1 and JM79.T2
After the initial cysteine coupling, Fmoc-Lys(ivDde)-OH and Fmoc-Lys(Fmoc)-OH were coupled [Scheme 2(1–3)] following the procedure described in Peptide Portions section. Then both Fmoc groups (20% piperidine in DMF) as well as the ivDde [hydrazine/DMF 5/95 v/v; 2.5 mL (for 10 min three times)] were removed. The linker portions of the two compounds were built by coupling either Fmoc-β-Ala-OH (JM79.T1) alone, or coupling Fmoc-AEEAc-OH in addition to Fmoc-β-Ala-OH (JM79.T2), again following the procedure described in Peptide Portions section [Scheme 2(3–4)]. Then, Fmoc-Met-OH, Fmoc-D-Lys(Boc)-OH, and Fmoc-Lys(Boc)-OH were coupled as described in Peptide Portions section [Scheme 2(4–5)]. The peptoid units Nlys, N4mob, Nmba, Npip, and Nmba were coupled [Scheme 2(5–6)] following the microwave-assisted peptoid synthesis protocol described in Peptoid Portions section, completing three JM79 moieties on three arms of the central two-lysine scaffold. Final products were cleaved off the resin, analyzed and purified as described in Cleavage and Purification section.
SCHEME 2.
Solid phase synthesis strategy to build JM79 homotrimers. The reagents and reaction conditions used are as follows: (1–2) a. Fmoc-Cys(Trt)-OH, HOBt, HBTU, DIPEA, b. 20% piperidine in DMF, c. Fmoc-Lys(ivDde)-OH, HOBt, HBTU, DIPEA. (2–3) a. 20% piperidine in DMF, b. Fmoc-Lys(Fmoc)-OH, HOBt, HBTU, DIPEA. (3–4) a. 5% hydrazine in DMF, v/v, b. 20% piperidine in DMF, c. Fmoc-β-Ala-OH (n = 6 Fmoc-AEEAc-OH addition), HOBt, HBTU, DIPEA. (4–5) a. 20% piperidine in DMF, b. addition of peptide portion (standard SPPS protocol to add Met, D-Lys, Lys). (5–6) a. 20% piperidine in DMF, b. addition of 5-mer peptoid portion (standard microwave-assisted peptoid synthesis). (6–7) 95% TFA, 2.5% TIS, 2.5% H2O (final product shown with AEEAc-OH).
Synthesis of JM79-81.HD1-3
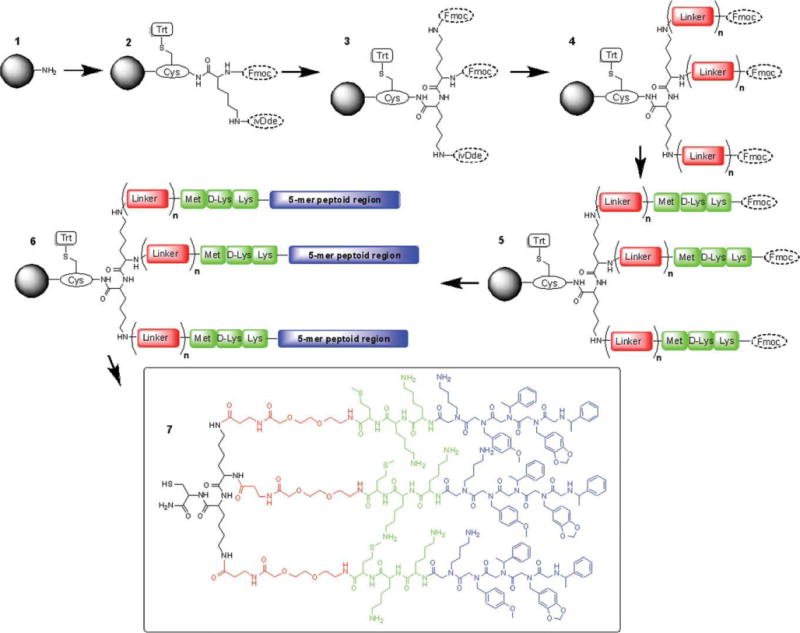
After the initial cysteine coupling, Fmoc-Lys(ivDde)-OH was coupled [Scheme 3(1–2)], followed by Fmoc removal, using the procedure described in Peptide Portions section. Then, as the peptide portion of JM79, Fmoc-Met-OH, Fmoc-D-Lys(Boc)-OH, and Fmoc-Lys(Boc)-OH were also coupled as described in Peptide Portions section [Scheme 3(2–3)]. The peptoid portion of JM79 was coupled [Scheme 3(3–4)] using Cho and Kwon et al. protocol. Briefly, for each of the bromoacetylation steps, 1.0 M bromoacetic acid and 1.1 M DIC in DMF were treated at 35°C with shaking for 6 min. The primary amines were coupled using the microwave-assisted procedure described in Peptide Portions section. After the JM79 was fully completed, Boc-Gly-OH was coupled [Scheme 3(3–4)] as described in Peptide Portions section. Then, ivDde was removed [hydrazine/DMF 5/95, v/v; 2.5 mL (for 10 min three times)] and the linker portion was added [Scheme 3(4–5)] by coupling 1, 2, or 3 Fmoc-AEEAc-OH moieties (JM79.HD1, JM79.HD2, and JM79.HD3) as described in Peptide Portions section. Once again, Fmoc-Met-OH, Fmoc-D-Lys(Boc)-OH, and Fmoc-Lys(Boc)-OH were coupled [Scheme 3(5–6)] as described in Peptide Portions section as the peptide portion of the JM81, The peptoid units Nmea, Nleu, Nleu, Nall, and Nmea were coupled [Scheme 3(6–7)] following the microwave-assisted peptoid synthesis protocol described in Peptoid Synthesis section. Finally, Boc-Gly-OH was coupled [Scheme 3(6–7), as described in General Synthesis Procedure section completing the JM81. Final products were cleaved off the resin, analyzed and purified as described in Cleavage and Purification section.
SCHEME 3.
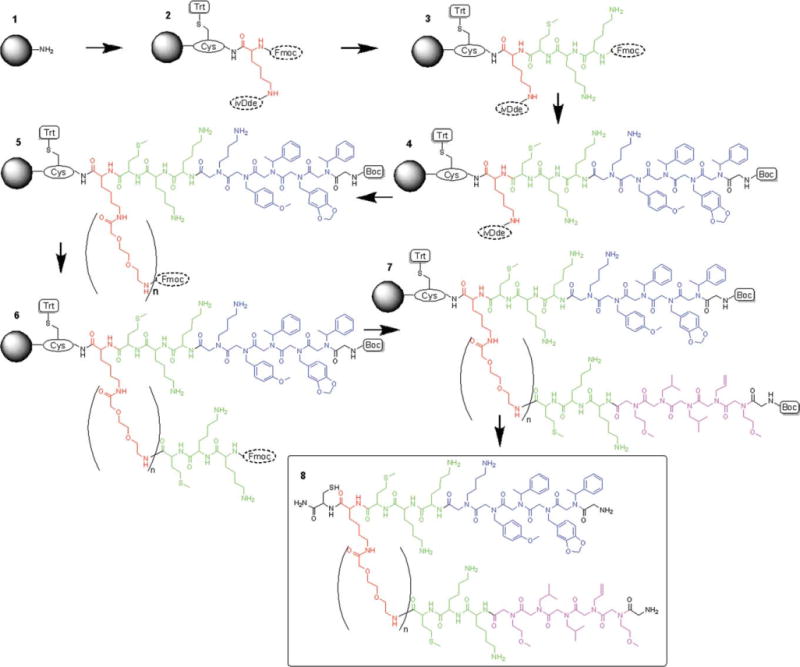
Solid phase synthesis strategy to build JM79-JM81 heterodimers. (1–2) a. Fmoc-Cys(Trt)-OH, HOBt, HBTU, DIPEA, b. 20% piperidine in DMF, c. Fmoc-Lys(ivDde)-OH, HOBt, HBTU, DIPEA, (2–3) a. 20% piperidine in DMF, b. addition of peptide portion of JM79 (Met, D-Lys, Lys). (3–4) a. 20% piperidine in DMF, b. addition of 5-mer peptoid portion of JM79 (follow Cho et al. protocol), c. Boc-Gly-OH, HOBt, HBTU, DIPEA. (4–5) a. 5% hydrazine in DMF v/v, b. Fmoc-AEEAc-OH, HOBt, HBTU, DIPEA, (n = 1, 2, or 3 additions). (5–6) a. 20% piperidine in DMF, b. addition of peptide portion of JM81 (Met, D-Lys, and Lys). (6–7) a. 20% piperidine in DMF, b. addition of 5-mer peptoid portion of JM81 (standard microwave-assisted protocol), c. Boc-Gly-OH, HOBt, HBTU, DIPEA. (7–8) 95% TFA, 2.5% TIS, 2.5% H2O.
RESULTS
Development of JM79 Homodimers
The previous strategy developed by the Kodadek lab for peptoid homodimers in synthesizing a VEGFR2 binding compound was to have a central Lys linker attached to the resin first and then build the two monomeric units on two arms simultaneously.29 Here, we further advance this strategy of developing various multimers on the resin beads. For the synthesis of complex multimers, such as what we are planning to perform here, the selection of a proper solid support is critical. An ideal resin should not only have a lower loading capacity to accommodate the growing of a larger molecule on each active center but also higher swelling properties to efficiently mix and bring together all ingredients needed for each reaction. Therefore, we selected the NovaSyn TGR resin, which has a lower loading level of 0.2–0.3 mmol/g with excellent swelling properties in DMF. All the amino acid couplings were carried out using general Fmoc SPPS (see General Synthesis Procedures section) and peptoid units were built using microwave-assisted peptoid synthesis procedure (see Synthesis of JM79.D1-4 section). Before we started building our desired molecule, we preferred to have Cys coupled at the C-terminus as the sulfhydryl group can be readily modified with well established maleimide chemistry [Scheme 1(1–2)]. This can be used to attach fluorophores, drugs, or imaging agents to the improved peptoids, lending them flexibility in many applications in both therapeutic and diagnostic sides. We avoided having this modifiable Cys at the N-terminus of the sequence (coupling at the end of the synthesis) because both JM79 and JM81 compounds were displayed with free N-termini during our initial screens and modifications closer to this end could affect binding and activity of the final molecule.
We have selected Lys as the central linker that holds two JM79 units together for several reasons. Lys coupling using Fmoc-based SPPS is very well established and more importantly several combinations of amine-protected Lys types are commercially available. In addition, we have already demonstrated the use of Lys as a facile central linker for peptoid homodimers29 and our initial focus is to build multimers with relatively short linkers. Therefore, Fmoc-Lys(Fmoc)-OH was coupled as the central linker [Scheme 1(1–2)]. After removal of both Fmoc groups, a variable linker portion was created by adding β-alanine as the spacer [Scheme 1(2–3)] on both the α-amine and ɛ-amine of the central Lys. Fmoc-β-Ala-OH was chosen as it can be coupled as any other amino acid using general SPPS and adds two carbons to the back bone without bringing any complexity. This is very important for our design where we are planning to build “mini-libraries” of multimers, which only differ at this linker region and should be easily manipulated. Four compounds were synthesized by coupling 0, 1, 2, or 3 Fmoc-β-Ala-OH moieties. It is important to note that in each compound, two β-alanine moieties will be added to both arms of the central Lys and the actual total loading of β-Alanine moieties are 0, 2, 4, or 6, respectively. Next was the addition of three amino acids that are common in both compounds (Met, D-Lys, and Lys) [Scheme 1(3–4)]. Then the 5-mer peptoid region was constructed following the standard microwave-assisted protocol [Scheme 1(4–5)] to obtain final homodimeric compounds [Scheme 1(6)] fully synthesized on-bead. All four compounds synthesized were confirmed by mass spectra (MS) and purified by HPLC (Supporting Information Sections 2.1 and 4.1).
Development of JM79 Homotrimers
The basic approach is similar to the homodimer in that all three monomers are built simultaneously on a central linker attached to the bead. Again, considering the even larger size of the molecules growing on the resin, NovaSyn TGR resin was chosen. The initial Cys was coupled as described earlier on the NovaSyn TGR resin [Scheme 2(1–2)] at the C-terminus. To build three monomeric units simultaneously, three free amine active centers need to be displayed on the central linker. This was achieved by the consecutive coupling of two Lys residues, in which the second Lys was coupled onto the α-amine of the first Lys [Scheme 2(1–3)]. To appropriately build this central scaffold, the α-amine and ɛ-amine of the first Lys have to be orthogonally protected, so we decided to use Fmoc-Lys(ivDde)-OH [Scheme 2(1–2)]. Fmoc and ivDde are an excellent pair of orthogonal protecting groups as Fmoc can be readily removed without affecting the ivDde.75 After coupling Fmoc-Lys(ivDde)-OH, the Fmoc group on the α-amine was removed and Fmoc-Lys(Fmoc)-OH was coupled [Scheme 2(2–3)]. Next, both Fmoc and ivDde were removed, revealing three free amine active centers on the central linker that the three monomeric units of JM79 can be built on.
When introducing the linker portions in the initial homotrimer development plan, we wanted to synthesize two compounds with two different linkers [Scheme 2(3–4)]. On the first compound (JM79.T1), we decided to use Fmoc-β-Ala-OH as the linker, which is similar to the homodimer synthesis previously [Scheme 2(3–4)]. Even though this will add three β-alanine moieties in between the central linker and JM79 monomeric units, it is a relatively short spacer. Therefore, for the second homotrimer (JM79.T2), we introduced Fmoc-AEEAc-OH [{2-[2-(Fmoc-amino)-ethoxy]-ethoxy}acetic acid] which, as with any other amino acid, can be coupled under general SPPS conditions [Scheme 2(3–4)]. AEEAc has better water soluble properties and will introduce an eight-carbon, PEG-type moiety. In JM79.T2 synthesis, AEEAc was coupled in addition to having β-alanine so that all three JM79 monomers will be properly displayed away from each other. After addition of the three amino acid portion of the JM79 [Scheme 2(4–5)], the 5-mer peptoid region was constructed following the standard microwave-assisted protocol [Scheme 2(5–6)] to obtain final homotrimeric compounds [Scheme 2(7)]. The whole synthesis was performed on-bead and both compounds were confirmed by MS and purified by HPLC (Supporting Information Sections 2.2 and 4.2).
Development of JM79-JM81 Heterodimers
The critical difference in the heterodimer synthesis, as compared to previous on-bead homomultimer synthesis where both monomeric units were built simultaneously on the two arms of the central Lys, is that we use two different sequences. The idea is to first build JM79 on the α-amine of the central Lys and then build JM81 on the ɛ-amine, completing the whole process on-bead (Figure 3 and Scheme 3). Here, the lower loading level of the resin is much more critical than that of homomultimer synthesis as we are going to build the 8-mer JM79 to the completion and then start growing the second arm (JM81) on the ɛ-amine of the base of the central linker (Scheme 3). This implies that the steric hindrance is different here than when synthesizing two sequences simultaneously as in the homomultimer synthesis. Here also, due to the need for a resin that has low loading and good swelling properties, we continued to use NovaSyn TGR resin. In addition, there are two other strategic factors that should be considered: (1) the central lysine has to be orthogonally protected and (2) the second protecting group should be resistant to conditions used for synthesizing JM79.
FIGURE 3.
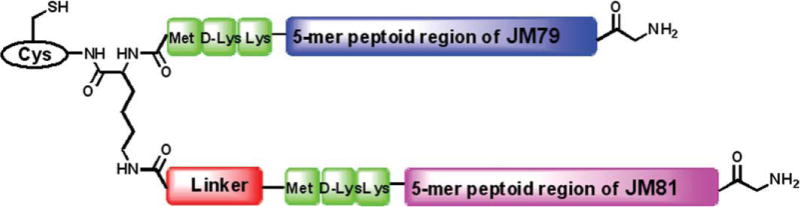
Schematic representation of the structure of the heterodimer that consist of both JM79 and JM81 sequences.
Using the same basic idea as the homomultimers, the synthesis started with an initial Cys at the C-terminal and a central Lys to build each arm on [Scheme 3(1–2)]. Once again, we coupled Fmoc-Lys(ivDde)-OH as the central linker with two orthogonal protection groups. After Fmoc removal from the α-amine, JM79 was synthesized using standard SPPS protocol [Scheme 3(2–3)] and a 5-mer peptoid addition using the microwave-assisted protocol for both bromoacetylation and primary amine addition (see Synthesis of JM79.D1-4 Section). At the end of the JM79 synthesis, the N-terminus was capped using Boc-Gly-OH, again under normal SPPS conditions. This not only makes the capping process much easier, as it is a simple amino acid coupling but also displays a primary amine at the N-terminus increasing water solubility. The next step is removal of the ivDde group at which point mass spectra revealed an anomaly after cleavage in the form of +40 mass units. We originally mistook this peak for the potassiated version of the compound before ivDde removal. Cho and Kwon et al. reported the same exact problem with the synthesis of cyclic peptoids using Fmoc-Dpr(ivDde)-OH under the same microwave-assisted conditions, in which ivDde was not removed with hydrazine treatment, giving an additional +40 mass unit peak.40 They tested several conditions and optimized the synthesis for bromoacetylation at reduced concentrations of reagents (1 M bromoacetic acid and 1.1 M DIC), performed at 35°C for 5 min to avoid this side reaction. Hence, we revised the protocol and introduced the reported conditions by Cho et al. to synthesize the 5-mer peptoid portion of the JM79 [Scheme 3(3–4): See Synthesis of JM79-81.HD1-3 Section]. At the end of the JM79 synthesis, the N-terminus was again capped with Boc-Gly-OH as described earlier [Scheme 3(3–4)]. The ivDde group was removed by treatment with 5% hydrazine in DMF [Scheme 3(4–5)] and the mass spectra showed no side reaction product at +40 mass units.
We then introduced the variable linker region to the ɛ-amine of the central lysine by coupling Fmoc-AEEAc-OH [Scheme 3(4–5)]. At this point we have synthesized three compounds which only differ by one, two, or three AEEAc groups. This will help to screen for an optimized linker for the heterodimers that allows effective interaction of JM79 and JM81 as a single molecule with two different targets on the cell surface. If none of the above AEEAc group combinations are enough to produce the optimized linker, we can simply follow the same procedure to synthesize different combinations of AEEAc groups in the future. After the completion of this linker portion, again the same standard methods of SPPS [Scheme 3(5–6)] and peptoid microwave-assisted protocols [Scheme 3(6–7)] were followed to build the full JM81 sequence [Scheme 3(8)]. Finally, all three heterodimers were fully validated by mass spectroscopy and purified by HPLC (Supporting Information: Sections 2.3, 3.1, and 4.3).
DISCUSSION
In this study, we mainly focused on optimizing a full solid phase heterodimer synthesis protocol, as homodimerization approaches have been reported for many systems in peptides and a few in peptoids. Yields of 55–65% were observed for heterodimer synthesis as determined by HPLC. We believe that these yields are substantial, especially when considering the fact that they were fully synthesized on solid phase without a single solution phase reaction. If solution phase reactions are involved with time consuming purification steps, rapid development of minilibraries of these compounds becomes more challenging and nonpractical. Solid phase synthesis is highly convenient and better suited for this type of minilibrary synthesis of multimeric compounds. This is because the main focus of developing these minilibraries of multimers is to identify the optimized linker lengths. Therefore, the synthesis procedure should be mainly optimized for facile development of series of compounds at reasonable yields and once the best candidates are identified, large-scale synthesis of those individual compounds can be optimized for higher yields.
Throughout this multimer synthesis approach, our strategy was to keep the simplicity in designing the synthesis of these complex molecules. For example, in every addition of a building block—whether it was for the peptoid region, peptide region, linker region, or even capping at N-terminus—the bond formed was always an amide bond (except the nucleophilic substitution reaction used in amine substitution to bromine in the peptoid region, which is also a well established reaction). Our main focus was the building of the linker region, which should be readily adaptable to synthesize more compounds in the future, facilitating further “fine tuning” of these multimers. We were very careful to restrict the type of spacer to Fmoc-based moieties that can be conveniently used within the general SPPS described in Peptide Portions Section. In this regard, Fmoc-β-Ala-OH was chosen as the shorter spacer and Fmoc-AEEAc-OH as the longer spacer. Six-carbon long Fmoc-ɛ-Ahx-OH will be another choice, even though it might introduce more hydrophobic character to the overall molecule. More interestingly, peptoid units can also be used as spacers in building this linker region, and the synthesis can be continued using simple peptoid synthesis protocols described in Peptoid Synthesis Section. We did not use this strategy for this initial attempt as it is more suitable for fine tuning, which comes after finding the rough distances using relatively longer spacers as we did here. There are huge varieties of amines with various properties such as being smaller, hydrophilic, hydrophobic or bulkier, readily available to build the linker region.18 These could introduce simple to highly constrained structural features, including stable helices, depending on the type of linker that need to be produced.
When considering the overall multimer synthesis process, once the linker portion is optimized, we need to pay attention to the feasibility of bringing together the actual biologically active ligand to complete the whole process. The use of peptoids as drug leads or probes targeting biological systems has been discussed extensively in the Introduction Section, and has many implications in the multimer development arena also. In particular, the straightforward and flexible peptoid syntheses protocols are invaluable in this type of application. For example, in the heterodimer synthesis attempt, we experienced that the well established microwave condition was not the best approach for bromoacetylation step due to unwanted product formation with ivDde. But, a very appropriate reaction condition was already established in the peptoid literature to solve this problem, indicating the availability of large chemical space for peptoid synthesis.40 In addition, there are several other reaction conditions reported to perform both of these two basic reaction steps in peptoid synthesis.18,76 This again suggests how the versatility and ease of peptoid chemistry readily supports these types of complex applications as compared to many other molecular types available.
Studies are planned to evaluate the effects of these multimers on HCC4017 lung cancer cells as compared to their monomeric units. As the targets for both JM79 and JM81 are yet to be identified, use of these multimers in probing receptor mechanisms is not available at this point. However, these can be applied to see how many different downstream signaling pathways are affected as compared to monomeric units. This is an interesting and unique application and it would even support parallel studies that are directed towards finding out their targets on the HCC4017 lung cancer cell surface.
In conclusion, we have synthesized “minilibraries” of homodimers, homotrimers, and heterodimers of previously identified HCC4017 lung cancer-specific peptoid compounds JM79 and JM81. This demonstrates the ability of fully synthesizing complex molecules on solid phase using peptoids with the combination of standard SPPS approaches.
Supplementary Material
Acknowledgments
This work was supported by the funding from UT Southwestern Medical Center. We thank Funmilayo Adebesin for supplying Bocdiaminobutane for heterodimer synthesis.
Footnotes
Additional Supporting Information may be found in the online version of this article.
References
- 1.Spencer DM, Wandless TJ, Schreiber SL, Crabtree GR. Science. 1993;262:1019–1024. doi: 10.1126/science.7694365. [DOI] [PubMed] [Google Scholar]
- 2.Pruschy MN, Spencer DM, Kapoor TM, Miyake H, Crabtree GR, Schreiber SL. Chem Biol. 1994;1:163–172. doi: 10.1016/1074-5521(94)90006-x. [DOI] [PubMed] [Google Scholar]
- 3.Kiessling LL, Gestwicki JE, Strong LE. Curr Opin Chem Biol. 2000;4:696–703. doi: 10.1016/s1367-5931(00)00153-8. [DOI] [PubMed] [Google Scholar]
- 4.Mourez M, Kane RS, Mogridge J, Metallo S, Deschatelets P, Sellman BR, Whitesides GM, Collier RJ. Nat Biotechnol. 2001;19:958–961. doi: 10.1038/nbt1001-958. [DOI] [PubMed] [Google Scholar]
- 5.Sadler K, Tam JP. J Biotechnol. 2002;90:195–229. doi: 10.1016/s1389-0352(01)00061-7. [DOI] [PubMed] [Google Scholar]
- 6.Vagner J, Handl HL, Gillies RJ, Hruby VJ. Bioorg Med Chem Lett. 2004;14:211–215. doi: 10.1016/j.bmcl.2003.09.079. [DOI] [PubMed] [Google Scholar]
- 7.Vagner J, Xu L, Handl HL, Josan JS, Morse DL, Mash EA, Gillies RJ, Hruby VJ. Angew Chem Int Ed Engl. 2008;47:1685–1688. doi: 10.1002/anie.200702770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vidal M, Liu WQ, Gril B, Assayag F, Poupon MF, Garbay C. J Soc Biol. 2004;198:133–137. [PubMed] [Google Scholar]
- 9.Pluckthun A, Pack P. Immunotechnology. 1997;3:83–105. doi: 10.1016/s1380-2933(97)00067-5. [DOI] [PubMed] [Google Scholar]
- 10.Todorovska A, Roovers RC, Dolezal O, Kortt AA, Hoogenboom HR, Hudson PJ. J Immunol Methods. 2001;248:47–66. doi: 10.1016/s0022-1759(00)00342-2. [DOI] [PubMed] [Google Scholar]
- 11.Kiessling LL, Pohl NL. Chem Biol. 1996;3:71–77. doi: 10.1016/s1074-5521(96)90280-x. [DOI] [PubMed] [Google Scholar]
- 12.Kaae BH, Harpsoe K, Kastrup JS, Sanz AC, Pickering DS, Metzler B, Clausen RP, Gajhede M, Sauerberg P, Liljefors T, Madsen U. Chem Biol. 2007;14:1294–1303. doi: 10.1016/j.chembiol.2007.10.012. [DOI] [PubMed] [Google Scholar]
- 13.Mammen M, Choi S, Whitesides GM. Angew Chem Int Ed. 1998;37:2754–2794. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 14.Kiessling LL, Gestwicki JE, Strong LE. Angew Chem Int Ed Engl. 2006;45:2348–2368. doi: 10.1002/anie.200502794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davis NE, Karfeld-Sulzer LS, Ding S, Barron AE. Biomacromolecules. 2009;10:1125–1134. doi: 10.1021/bm801348g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aina OH, Sroka TC, Chen ML, Lam KS. Biopolymers. 2002;66:184–199. doi: 10.1002/bip.10257. [DOI] [PubMed] [Google Scholar]
- 17.Simon RJ, Kania RS, Zuckermann RN, Huebner VD, Jewell DA, Banville S, Ng S, Wang L, Rosenberg S, Marlowe CK, et al. Proc Natl Acad Sci USA. 1992;89:9367–9371. doi: 10.1073/pnas.89.20.9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Culf AS, Ouellette RJ. Molecules. 2010;15:5282–5335. doi: 10.3390/molecules15085282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zuckermann RN, Kodadek T. Curr Opin Mol Ther. 2009;11:299–307. [PubMed] [Google Scholar]
- 20.Zuckermann RN, Kerr JM, Kent SBH, Moos WH. J Am Chem Soc. 1992;114:10646–10647. [Google Scholar]
- 21.Kwon YU, Kodadek T. J Am Chem Soc. 2007;129:1508–1509. doi: 10.1021/ja0668623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fowler SA, Blackwell HE. Org Biomol Chem. 2009;7:1508–1524. doi: 10.1039/b817980h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shah NH, Butterfoss GL, Nguyen K, Yoo B, Bonneau R, Rabenstein DL, Kirshenbaum K. J Am Chem Soc. 2008;130:16622–16632. doi: 10.1021/ja804580n. [DOI] [PubMed] [Google Scholar]
- 24.Yoo B, Kirshenbaum K. Curr Opin Chem Biol. 2008;12:714–721. doi: 10.1016/j.cbpa.2008.08.015. [DOI] [PubMed] [Google Scholar]
- 25.Olivos HJ, Alluri PG, Reddy MM, Salony D, Kodadek T. Org Lett. 2002;4:4057–4059. doi: 10.1021/ol0267578. [DOI] [PubMed] [Google Scholar]
- 26.Gorske BC, Jewell SA, Guerard EJ, Blackwell HE. Org Lett. 2005;7:1521–1524. doi: 10.1021/ol0502984. [DOI] [PubMed] [Google Scholar]
- 27.Figliozzi GM, Goldsmith R, Ng SC, Banville SC, Zuckermann RN. Methods Enzymol. 1996;267:437–447. doi: 10.1016/s0076-6879(96)67027-x. [DOI] [PubMed] [Google Scholar]
- 28.Alluri PG, Reddy MM, Bachhawat-Sikder K, Olivos HJ, Kodadek T. J Am Chem Soc. 2003;125:13995–14004. doi: 10.1021/ja036417x. [DOI] [PubMed] [Google Scholar]
- 29.Udugamasooriya DG, Dineen SP, Brekken RA, Kodadek T. J Am Chem Soc. 2008;130:5744–5752. doi: 10.1021/ja711193x. [DOI] [PubMed] [Google Scholar]
- 30.Simpson LS, Burdine L, Dutta AK, Feranchak AP, Kodadek T. J Am Chem Soc. 2009;131:5760–5762. doi: 10.1021/ja900852k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lim HS, Archer CT, Kodadek T. J Am Chem Soc. 2007;129:7750–7751. doi: 10.1021/ja072027p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alluri P, Liu B, Yu P, Xiao X, Kodadek T. Mol Biosyst. 2006;2:568–579. doi: 10.1039/b608924k. [DOI] [PubMed] [Google Scholar]
- 33.Lim HS, Cai D, Archer CT, Kodadek T. J Am Chem Soc. 2007;129:12936–12937. doi: 10.1021/ja075469+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reddy Mm, Kodadek T. Proc Natl Acad Sci USA. 2005;102:12672–12677. doi: 10.1073/pnas.0501208102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shores KS, Udugamasooriya DG, Kodadek T, Knapp DR. J Proteome Res. 2008;7:1922–1931. doi: 10.1021/pr7006889. [DOI] [PubMed] [Google Scholar]
- 36.Zuckermann RN, Martin EJ, Spellmeyer DC, Stauber GB, Shoemaker KR, Kerr JM, Figliozzi GM, Goff DA, Siani MA, Simon RJ, et al. J Med Chem. 1994;37:2678–2685. doi: 10.1021/jm00043a007. [DOI] [PubMed] [Google Scholar]
- 37.Lim HS, Reddy MM, Xiao X, Wilson J, Wilson R, Connell S, Kodadek T. Bioorg Med Chem Lett. 2009;19:3866–3869. doi: 10.1016/j.bmcl.2009.03.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Astle JM, Udugamasooriya DG, Smallshaw JE, Kodadek T. Int J Pept Res Ther. 2008;14:223–227. [Google Scholar]
- 39.Yoo B, Shin SB, Huang ML, Kirshenbaum K. Chemistry. 2010;16:5528–5537. doi: 10.1002/chem.200903549. [DOI] [PubMed] [Google Scholar]
- 40.Cho S, Choi J, Kim A, Lee Y, Kwon YU. J Comb Chem. 2010;12:321–326. doi: 10.1021/cc9001857. [DOI] [PubMed] [Google Scholar]
- 41.Comegna D, Benincasa M, Gennaro R, Izzo I, De Riccardis F. Bioorg Med Chem. 2010;18:2010–2018. doi: 10.1016/j.bmc.2010.01.026. [DOI] [PubMed] [Google Scholar]
- 42.Guo L, Zhang D. J Am Chem Soc. 2009;131:18072–18074. doi: 10.1021/ja907380d. [DOI] [PubMed] [Google Scholar]
- 43.Hjelmgaard T, Faure S, Caumes C, De Santis E, Edwards AA, Taillefumier C. Org Lett. 2009;11:4100–4103. doi: 10.1021/ol9015767. [DOI] [PubMed] [Google Scholar]
- 44.De Cola C, Licen S, Comegna D, Cafaro E, Bifulco G, Izzo I, Tecilla P, De Riccardis F. Org Biomol Chem. 2009;7:2851–2854. doi: 10.1039/b905665c. [DOI] [PubMed] [Google Scholar]
- 45.Kwon YU, Kodadek T. Chem Biol. 2007;14:671–677. doi: 10.1016/j.chembiol.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 46.Kwon YU, Kodadek T. Chem Commun (Camb) 2008:5704–5706. doi: 10.1039/b812735b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vercillo OE, Andrade CK, Wessjohann LA. Org Lett. 2008;10:205–208. doi: 10.1021/ol702521g. [DOI] [PubMed] [Google Scholar]
- 48.Rivera DG, Wessjohann LA. Molecules. 2007;12:1890–1899. doi: 10.3390/12081890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Holub JM, Jang H, Kirshenbaum K. Org Lett. 2007;9:3275–3278. doi: 10.1021/ol071169l. [DOI] [PubMed] [Google Scholar]
- 50.Shin SB, Yoo B, Todaro LJ, Kirshenbaum K. J Am Chem Soc. 2007;129:3218–3225. doi: 10.1021/ja066960o. [DOI] [PubMed] [Google Scholar]
- 51.Rivera DG, Wessjohann LA. J Am Chem Soc. 2006;128:7122–7123. doi: 10.1021/ja060720r. [DOI] [PubMed] [Google Scholar]
- 52.Nnanabu E, Burgess K. Org Lett. 2006;8:1259–1262. doi: 10.1021/ol052867d. [DOI] [PubMed] [Google Scholar]
- 53.Mattern RH, Tran TA, Goodman M. J Pept Sci. 1999;5:161–175. doi: 10.1002/(SICI)1099-1387(199904)5:4<161::AID-PSC177>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 54.Nam KT, Shelby SA, Choi PH, Marciel AB, Chen R, Tan L, Chu TK, Mesch RA, Lee BC, Connolly MD, Kisielowski C, Zuckermann RN. Nat Mater. 2010;9:454–460. doi: 10.1038/nmat2742. [DOI] [PubMed] [Google Scholar]
- 55.Lee BC, Chu TK, Dill KA, Zuckermann RN. J Am Chem Soc. 2008;130:8847–8855. doi: 10.1021/ja802125x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Seo J, Michaelian N, Owens SC, Dashner ST, Wong AJ, Barron AE, Carrasco MR. Org Lett. 2009;11:5210–5213. doi: 10.1021/ol9021468. [DOI] [PubMed] [Google Scholar]
- 57.Comegna D, De Riccardis F. Org Lett. 2009;11:3898–3901. doi: 10.1021/ol901524k. [DOI] [PubMed] [Google Scholar]
- 58.Yuasa H, Honma H, Hashimoto H, Tsunooka M, Kojima-Aikawa K. Bioorg Med Chem Lett. 2007;17:5274–5278. doi: 10.1016/j.bmcl.2006.12.075. [DOI] [PubMed] [Google Scholar]
- 59.Maayan G, Ward MD, Kirshenbaum K. Chem Commun (Camb) 2009:56–58. doi: 10.1039/b810875g. [DOI] [PubMed] [Google Scholar]
- 60.Fisher AE, Naughton DP. J Inorg Biochem. 2004;98:343–346. doi: 10.1016/j.jinorgbio.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 61.Combs DJ, Lokey RS. Tetrahedron Lett. 2007;48:2679–2682. doi: 10.1016/j.tetlet.2007.02.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Roy O, Faure S, Thery V, Didierjean C, Taillefumier C. Org Lett. 2008;10:921–924. doi: 10.1021/ol7030763. [DOI] [PubMed] [Google Scholar]
- 63.Kawakami T, Murakami H, Suga H. J Am Chem Soc. 2008;130:16861–16863. doi: 10.1021/ja806998v. [DOI] [PubMed] [Google Scholar]
- 64.Butterfoss GL, Renfrew PD, Kuhlman B, Kirshenbaum K, Bonneau R. J Am Chem Soc. 2009;131:16798–16807. doi: 10.1021/ja905267k. [DOI] [PubMed] [Google Scholar]
- 65.Gorske BC, Stringer JR, Bastian BL, Fowler SA, Blackwell HE. J Am Chem Soc. 2009;131:16555–16567. doi: 10.1021/ja907184g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chongsiriwatana NP, Patch JA, Czyzewski AM, Dohm MT, Ivankin A, Gidalevitz D, Zuckermann RN, Barron AE. Proc Natl Acad Sci USA. 2008;105:2794–2799. doi: 10.1073/pnas.0708254105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhu WL, Song YM, Park Y, Park KH, Yang ST, Kim JI, Park IS, Hahm KS, Shin SY. Biochim Biophys Acta. 2007;1768:1506–1517. doi: 10.1016/j.bbamem.2007.03.010. [DOI] [PubMed] [Google Scholar]
- 68.Gorske BC, Blackwell HE. J Am Chem Soc. 2006;128:14378–14387. doi: 10.1021/ja065248o. [DOI] [PubMed] [Google Scholar]
- 69.Baldauf C, Gunther R, Hofmann HJ. Phys Biol. 2006;3:S1–9. doi: 10.1088/1478-3975/3/1/S01. [DOI] [PubMed] [Google Scholar]
- 70.Dohm MT, Seurynck-Servoss SL, Seo J, Zuckermann RN, Barron AE. Biopolymers. 2009;92:538–553. doi: 10.1002/bip.21309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lee SG, Chmielewski J. Chembiochem. 2010;11:1513–1516. doi: 10.1002/cbic.201000248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li S, McGuire MJ, Lin M, Liu YH, Oyama T, Sun X, Brown KC. Mol Cancer Ther. 2009;8:1239–1249. doi: 10.1158/1535-7163.MCT-08-1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Oyama T, Sykes KF, Samli KN, Minna JD, Johnston SA, Brown KC. Cancer Lett. 2003;202:219–230. doi: 10.1016/j.canlet.2003.08.011. [DOI] [PubMed] [Google Scholar]
- 74.Zhou X, Chang YC, Oyama T, McGuire MJ, Brown KC. J Am Chem Soc. 2004;126:15656–15657. doi: 10.1021/ja0446496. [DOI] [PubMed] [Google Scholar]
- 75.Chhabra SR, Hothi B, Evans DJ, White PD, Bycroft BW, Chan WC. Tetrahedron Lett. 1998;39:1603–1606. [Google Scholar]
- 76.Burkoth TS, Fafarman AT, Charych DH, Connolly MD, Zuckermann RN. J Am Chem Soc. 2003;125:8841–8845. doi: 10.1021/ja0352101. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.