

Abstract
Interleukin-25 (IL-25) and IL-33, which belong to distinct cytokine families, induce and promote T helper type 2 airway inflammation. Both cytokines probably play a role in asthma, but there is a lack of direct evidence to clarify distinctions between their functions and how they might contribute to distinct ‘endotypes’ of disease. To address this, we made a direct comparison of the effects of IL-25 and IL-33 on airway inflammation and physiology in our established murine asthma surrogate, which involves per-nasal, direct airway challenge. Intranasal challenge with IL-33 or IL-25 induced inflammatory cellular infiltration, collagen deposition, airway smooth muscle hypertrophy, angiogenesis and airway hyper-responsiveness, but neither increased systemic production of IgE or IgG1. Compared with that of IL-25, the IL-33-induced response was characterized by more sustained laying down of extracellular matrix protein, neoangiogenesis, T helper type 2 cytokine expression and elevation of tissue damping. Hence, both IL-25 and IL-33 may contribute significantly and independently to asthma ‘endotypes’ when considering molecular targets for the treatment of human disease.
Keywords: asthma, interleukin-25, interleukin-33, remodelling
Introduction
Asthma is one of the most common lifelong chronic diseases. The prevalence of asthma in developed countries is approximately 10% in adults and even higher in children whereas, in developing countries, the prevalence is lower but increasing rapidly.1Asthma is characterized by chronic inflammation of conducting airways, remodelling of airway walls, airway hyper-responsiveness to non-specific stimuli and episodic exacerbations of airway obstruction. Recently, epithelium-derived thymic stromal lymphopoietin (TSLP), interleukin-33 (IL-33) and IL-25 have received attention with regard to their possible roles in initiating and propagating T helper type 2 (Th2) airway inflammation in asthma. These cytokines are perceived to operate upstream of production of Th2 cytokines such as IL-4 and IL-13 and may therefore have greater potential as therapeutic targets.2
Interleukin-33 is a member of the IL-1 family of cytokines, and has been associated with the promotion of systemic Th2-type responses. In mice, IL-33 is abundantly expressed in the lungs in the course of ovalbumin (OVA) -induced airway inflammation,3 whereas exogenous administration of IL-33 exacerbates allergen-induced airway inflammation.4 Patients with asthma have elevated concentrations of IL-33 in induced sputum and serum.5 Interleukin-25 (IL-17E), in contrast, belongs to the IL-17 cytokine family. It has also been implicated in the promotion of Th2 immune responses. Murine airways challenged directly with IL-25 develop epithelial cell hyperplasia, mucous hypertrophy and hyper-reactivity.6,7 Production of IL-25 is also elevated in human asthma.8
Hence, epithelium-derived IL-25 and IL-33 both have the potential capacity to induce and promote Th2-type airway inflammation and induce remodelling and pathophysiological changes, including non-specific hypersensitivity in the airways.9 Consequently, when considering them as molecular targets and potential endotypic markers in human asthma, it is important to distinguish their functions, as well as their origins as fully as possible. Existing literature suggests a more prominent role for IL-33 in epithelially driven airway inflammation and differential effects of these mediators on T cells and dendritic cells.10,11 Recent genome-wide studies suggest a more pronounced influence of IL-33 in generating the asthma phenotypes.12
To clarify the roles of IL-33 and IL-25 in inducing airway inflammation, remodelling and hyper-reactivity, we used an established murine surrogate of asthma to compare, in parallel, the effects of these mediators using direct challenge, with incorporation of a ‘classical’ OVA-induced surrogate necessitating previous IgE sensitization as a positive control. We hypothesized that it is possible to identify distinct effects of these mediators on the immunopathological and immunophysiological changes which characterize asthma.
Materials and methods
Animals
Female BALB/c mice (8–10 weeks old) were purchased from Vital River Laboratories (Beijing, China) and housed in a pathogen-free mouse facility located in the Department of Laboratory Animal Sciences, Capital Medical University, Beijing, China. All animal studies were carried out strictly under protocols approved by the Institutional Animal Care and Use Committee at Chinese Capital Medical University (the approval number: 2010-X-104). Every effort was made to minimize the number of animals used and the animals' suffering.
IL-33, IL-25 and allergen challenge
To compare the differences between IL-33-induced and IL-25-induced airway changes, mice were randomly distributed into four groups including a group serially challenged with 50 μg/dose of OVA as a positive control,7,13 a group administered saline as a negative control, a group serially challenged with IL-257 and a group serially challenged with IL-33.14 In brief, mice challenged with OVA were first sensitized by intraperitoneal injection of 100 μg/dose OVA (Sigma-Aldrich, Beijing, China, emulsified in Al[OH]3) on days 0 and 12.7 These mice were then further challenged daily intranasally on days 18 to 23 with 50 μg/dose of OVA (OVA50) in 50 μl saline. Mice in the IL-33-challenged and IL-25-challenged groups were not sensitized or exposed to OVA, but were subjected to daily nasal challenge from days 18 to 23 with recombinant mouse IL-33 [mIL-33, R&D Systems (Abingdon, UK), Minneapolis, MN, 100 ng in 50 μl saline (=1·1 × 10−7 m)] or IL-25 [mIL-25, R&D Systems, 2 μg in 50 μl saline (=2·2 × 10−6 m)]. Subsequently, the mice in each group were further challenged intranasally with OVA, IL-33 or IL-25 every 2 days for a further 30 days. Some mice in each group were observed for a further 17 days after cessation of the challenges. Control mice were intraperitoneally injected with same amount of Al[OH]3 then nasally challenged with normal saline at corresponding time-points (Fig.1a).
Figure 1.
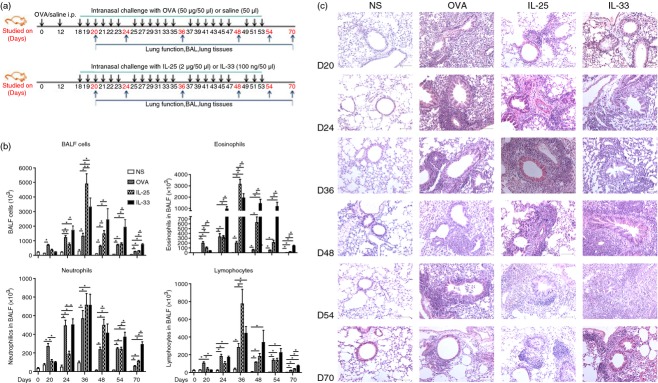
(a) Schedule of murine challenge. (b) Numbers of total cells, eosinophils, neutrophils and lymphocytes in bronchoalveolar lavage (BAL) fluid from interleukin-33 (IL-33), IL-25, ovalbumin (OVA) and saline (NS) -challenged mice at various time-points. Bars show the mean ± SEM (n = 5 in each group at each time-point). *P < 0·05. (c) Representative photomicrographs of haematoxylin & eosin-stained lung sections (original magnification ×20).
Lung function measurements
Lung function was assessed on days 20, 24, 36, 48, 54 and 70 during the challenge period, 24 hr after the most recent intranasal challenge. A baseline was established by measurements at day 0. Airway responsiveness was determined by changes in lung resistance in anaesthetized and tracheostomized mice in response to increasing concentrations of aerosolized methacholine (0–48 mg/ml) using the FlexiVent system (Scireq Inc., Montreal, QC, Canada) as described previously.7,15,16
Bronchoalveolar lavage fluid collection and characterization of cellular infiltrates
After lung function measurements, total cell counts in bronchoalveolar lavage fluid collected from mice were enumerated as previously described.7,15,16
Lung histology
Using standard methods, resected left lung tissue was fixed in 10% neutral-buffered formalin for 24 hr, then dissected into blocks, embedded in paraffin, and 5-μm sections were cut. Sections were stained with haematoxylin & eosin (H&E) for detection of inflammatory cell infiltration and periodic acid-Schiff (PAS) (Beijing Yi Li Fine Chemistry Co Ltd, Beijing, China) to detect mucus deposition. PAS staining was scored to assess the degree of goblet cell hyperplasia as described previously.7,17
A Masson's trichrome kit (Nanjing Mindit Biochemistry Co Ltd, Nanjing, China) was used to observe collagen accumulation.7,13 Congo red (Beijing Solarbio Technology Co Ltd, Beijing, China) staining was used for detection of eosinophil infiltration.7 Digital photographs of four bronchioles per tissue section were taken at ×40 magnification and Image-Pro Plus was used to quantify Congo red-positive eosinophils.
Lung immunohistochemistry
Immunohistochemistry was used to detect smooth muscle and vascular endothelial cells. The primary monoclonal antibodies against mouse α-smooth muscle actin (1 : 200) and CD31 (endothelial cells) (1 : 20) were purchased from Abcam (Hong Kong, China).7 Image-Pro Plus software was used to measure the total numbers of CD31+ vascular endothelial cells per unit area of entire lung sections and the thickness of the smooth muscle layer in large- and middle-sized airways.7
Preparation of lung tissue homogenates and quantification of total collagen
Resected right lung tissue was weighed then homogenized in PBS containing 1% Triton X-100 and a protease inhibitor cocktail tablet (Roche Diagnostics GmbH, Mannheim, Germany). After centrifugation to remove debris, the supernatant was collected for measurement of cytokines and chemokines. The Sircol collagen assay (Biocolor, Belfast, UK) was used to measure total soluble collagen according to the manufacturer's instructions and then normalized for tissue weight.7
Cytokine and chemokine analysis
The concentrations of IL-4, IL-5, IL-13, IL-25, Il-33, TSLP, transforming growth factor-β and eotaxin in supernatants of the right lung homogenates were measured using ELISA kits (eBioscience, San Diego, CA); basic fibroblast growth factor (bFGF) and vascular endothelial growth factor (VEGF) were detected using ELISA kits (RayBiotech, Inc, Atlanta, GA); interferon-γ (IFN-γ), IL-6 and the murine IL-8 homologue (CXCL1/KC) were measured using a commercial BDTM™ cytometric bead array platform (BD Biosciences Pharmingen, San Diego, CA) following the manufacturers' instructions. The concentrations of serum total IgE and IgG1 antibodies were measured by ELISA (eBioscience).7
Statistical analysis
The software package GraphPad Prism 5.01 (GraphPad, San Diego, CA) was used for all data analyses and preparation of graphs. In general, within-group variability was analysed using two-way analysis of variance. Between-group comparisons at specific time-points were performed using the Student's unpaired t-test. Data are expressed as the mean ± SEM. For all tests, P < 0·05 was considered significant.
Results
Inflammatory cellular infiltration of the lung parenchyma tissues and airways
Per-nasal, direct airway challenge with IL-33 and IL-25 in naive mice and OVA sensitized mice caused florid infiltration of inflammatory cells into the airway lumen and peribronchial and perivascular regions of the lung parenchyma (Fig.1b,c). Most of the lumen infiltrating cells were eosinophils, neutrophils and lymphocytes with eosinophils predominating (Fig.1b) and persisting for the entire observation period. Significant IL-33-induced cellular infiltration occurred slightly earlier (day 24) and persisted longer (day 70) compared with that of IL-25 (Fig.1b). H&E staining of lung sections revealed in comparison with saline-challenged control mice, extensive mucous hypertrophy with peribronchiolar and perivascular inflammation in all challenged groups from day 24, which peaked between days 36 and 48, then progressively declined at days 54 and 70. In contrast, in IL-33-challenged mice these changes were still evident at these later time-points, even at day 70, although attenuated compared with the peak (Fig.1c).
Eosinophil infiltration
Congo red staining revealed abundant eosinophil infiltration into the airway peribronchial and perivascular regions of the IL-33-challenged mice, which was evident from day 20 and persisted up to day 70 (Fig.2). OVA-induced lung eosinophil infiltration peaked at day 36, then rapidly declined (Fig2). IL-25-induced lung eosinophilia also peaked at day 36, then declined more slowly, remaining significant up to day 70. In IL-33-challenged mice, eosinophil infiltration peaked by day 24 and persisted until day 54. Even at day 70, 17 days after cessation of challenge, IL-33-induced airway eosinophilia was still statistically greater than that caused by OVA- and IL-25 challenge (Fig.2).
Figure 2.

(a) Representative photomicrographs of Congo red+ eosinophils in lung sections from interleukin-33 (IL-33), IL-25, ovalbumin (OVA) and saline (NS) -challenged mice at various time-points (original magnification ×40). (b) Numbers of Congo red+ cells as percentages of the total nucleated cells. Bars show the mean ± SEM (n = 5 in each group at each time-point). *P < 0·05.
Airway goblet cell hyperplasia
Staining with PAS showed that IL-33 inhalation challenge induced florid mucous hyperplasia in the bronchial epithelium (Fig.3), which was similar in terms of extent and duration to that induced by IL-25. At all time-points after 24 days, the changes in the IL-25 challenge and IL-33 challenge mice were significantly greater than those observed in the OVA-challenged mice (Fig.3).
Figure 3.

(a) Representative photomicrographs of periodic acid-Schiff (PAS) stained lung sections from interleukin-33 (IL-33), IL-25, ovalbumin (OVA) and saline (NS) -challenged mice at various time-points (original magnification ×20). (b) Mucus score based on PAS staining (see Materials and methods). Bars show the mean ± SEM (n = 5 in each group at each time-point). *P < 0·05.
IL-33 increased extracellular lung matrix protein deposition
Masson's trichrome blue staining of lung sections and quantitative detection of total lung collagen by biochemical assay showed significantly elevated collagen deposition around the airways and vasculature in IL-33-, IL-25- and OVA-challenged animals at 48 days and later time-points (Fig.4). While the IL-25- and OVA-challenged animals showed similar profiles of increased lung collagen content with time, only in the IL-33-challenged animals did this change persist following cessation of challenge at day 70, at which time-point it was still significantly elevated compared with the IL-25- and OVA-challenged mice (Fig.4). There was no significant change in lung collagen content in the saline-challenged animals throughout the experiment.
Figure 4.

(a) Representative photomicrographs of Masson's Trichrome Blue-stained lung sections from interleukin-33 (IL-33), IL-25, ovalbumin (OVA) and saline (NS) -challenged mice at various time-points (original magnification ×20). (b) Total lung collagen (μg/100 mg of total protein). Bars show the mean ± SEM (n = 5 in each group at each time-point). *P < 0·05.
IL-33-induced airways smooth muscle hypertrophy
Image analysis of immunoreactivity for α-smooth muscle actin showed that OVA, IL-25 and IL-33 challenge caused transient smooth muscle hypertrophy/hyperplasia, significant at days 36, 48 and 54 and with a similar extent and time–course, (Fig.5). Quantitative analysis demonstrated that the thickening of the airway smooth muscle in the IL-33-challenged animals was slightly but significantly greater than that of the IL-25-challenged animals at day 36.
Figure 5.

(a) Representative photomicrographs of α-smooth muscle actin immunoreactivity in lung sections from interleukin-33 (IL-33), IL-25, ovalbumin (OVA) and saline (NS) -challenged mice at various time-points (original magnification ×20). (b) Quantitative analysis of thickness of α-smooth muscle actin-positive airway smooth muscle. Bars show the mean ± SEM (n = 5 in each group at each time-point). *P < 0·05.
IL-33-induced increased pulmonary vascularity
Analysis of CD31 immunoreactivity showed that all three challenge regimens induced a significant increase in the numbers of CD31+ vascular endothelial cells, evident from day 36 (Fig.6) and persisting at the end of the experiments. Again the effects of each challenge were quantitatively and temporally similar, although the IL-33-challenged animals retained significantly greater airway vascularity at day 70, 17 days after cessation of challenge, compared with the IL-25- and OVA-challenged animals.
Figure 6.
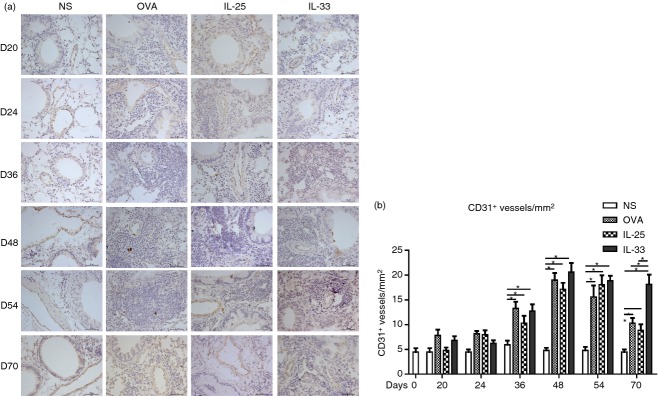
(a) Representative photomicrographs of CD31+ peribronchial blood vessels in lung sections from interleukin-33 (IL-33), IL-25, ovalbumin (OVA) and saline (NS) -challenged mice at various time-points (original magnification ×40). (b) CD31+ vessels per unit area of sections. Bars show the mean ± SEM (n = 5 in each group at each time-point). *P < 0·05.
IL-33 challenge did not increase IgE or IgG1 expression
Intraperitoneal OVA sensitization and subsequent per-nasal OVA challenge induced a marked increase in the concentrations of total serum IgE and IgG1 (the murine equivalent of human IgG4) detectable by day 20, peaking at day 36 and persisting until the end of the study (Fig.7). In contrast, per-nasal challenge with IL-33 or IL-25 did not significantly alter the concentrations of total serum IgE or IgG1 throughout the entire duration of the experiments (Fig.7).
Figure 7.

Concentrations of IgE and IgG1 in serum and cytokines, chemokines and other mediators in lung homogenates from interleukin-33 (IL-33), IL-25, ovalbumin (OVA) and saline (NS) -challenged mice at various time-points. Bars show the mean ± SEM (n = 5 in each group at each time-point). *P < 0·05.
IL-33 challenge increased cytokine and chemokine expression in the lung parenchyma
ELISA and cytometric bead array data from analysis of lung homogenates demonstrated that, whereas the OVA and IL-25 challenge regimens caused modestly elevated expression of the ‘signature’ Th2 cytokines IL-4, IL-5 and IL-13, but not the ‘signature’ Th1 cytokine IFN-γ. Interleukin-33 challenge induced marked local production of IL-4, IL-5 and IL-13, the concentrations of which were typically two to three times higher than those induced by OVA and IL-25 challenge from days 24 until 70 (Fig.7).
Interleukin-33 challenge also induced marked, significantly elevated expression of IL-25 itself and TSLP (two cytokines also thought to a play pivotal role in the initiation of asthma), which peaked at day 48 and persisted without decline until day 70 (Fig.7). Although challenge with IL-25 or OVA also induced elevated expression of either TSLP or TSLP and IL-25, the quantities were much less than those induced by IL-33-challenge. Conversely, as we previously reported,7 both IL-25 and OVA challenge were able to induce increased production of IL-33 (Fig.7).
Although all three stimuli induced increased expression of eotaxin, the peaks of IL-33 and IL-25-induced eotaxin expression were at day 48, whereas the OVA-induced peak was slightly earlier (at day 36, Fig.7). In addition, the zenith of IL-33-induced eotaxin expression persisted up to day 54 before declining, whereas that induced by OVA and IL-25 challenge declined rapidly (Fig.7). Induced production of the chemokine KC (IL-8) was comparable with all three stimuli whereas compared with OVA and IL-33, IL-25 did not significantly increase IL-6 production (Fig.7).
Finally, although all three challenge regimens were able to increase local concentrations of the pro -fibrotic and angiogenetic cytokines transforming growth factor-β, bFGF and VEGF, the lung concentration of bFGF in IL-33-challenged mice was significantly greater than that in the IL-25- or OVA-challenged mice from day 36 onwards until the end of the experiments, whereas the production of VEGF in the IL-33-challenged mice was elevated compared with IL-25-challenged animals at days 54 and 70 (Fig.7).
IL-33 challenge induced airway hyper-responsiveness and increased tissue damping
Ovalbumin challenge induced a significant elevation of methacholine-induced airway resistance relatively early at day 20 although this did not persist at later time-points. The IL-33 challenge induced a similar effect but this was more delayed (from day 36) and more sustained (up to day 54), whereas IL-25 challenge marginally increased airway hyper-responsiveness over the same time period (Fig.8a).
Figure 8.

(a) Airway (Rn) resistance of mice from interleukin33 (IL-33), IL-25, ovalbumin (OVA) and saline (NS) -challenged mice at the indicated time-points. (b) Tissue damping (G) parameter at the same time-points. Data are presented as the mean ± SEM (n = 5 in each group at each time-point). ***P < 0·05 versus IL-25-, OVA- and NS-challenged mice, **P < 0·05 versus OVA- and NS-challenged mice, *P < 0·05 versus NS-challenged mice.
Similarly, tissue damping (G), a parameter that is closely related to tissue resistance and reflects the dissipative properties of the lung tissues was significantly elevated in the OVA-, IL-25- and IL-33-challenged mice. With this parameter the effect of IL-33 was apparent earlier (by day 20) and persisted until the end of the experiments (day 70), whereas methacholine hyper-responsiveness had resolved by this time-point (Fig.8b).
Discussion
In this study we have taken the opportunity to compare the effects of direct airways challenge with IL-33 and IL-25 alongside a conventional animal asthma surrogate involving OVA sensitization and challenge. The concentrations of IL-25 and IL-33 used here were optimized in the preliminary experiments in which 2 μg of IL-25 and 100 ng of IL-33 were found to be the lowest effective dosages causing detectable airway inflammatory changes in the mice. One general caveat when comparing the effects, in terms of magnitude and time–course, of these two mediators on the airways in vivo is that different concentrations of IL-25 and IL-33 were delivered per-nasally to the airways, and it is not possible precisely to determine the fates of these mediators (for example, their clearance half times) following delivery. Notwithstanding this, our regimen offers the opportunity to compare acute and sub-acute changes in airway inflammatory cellular infiltration as well as remodelling and recovery phases. We have made a number of novel observations. First, IL-33 alone is able to increase airway smooth muscle thickness, as can IL-25,7 this might at least partly reflect its enhancement of interstitial fibrosis because previous studies suggest that myofibroblasts contribute to smooth muscle hypertrophy.18 This may also at least partly explain why IL-33 increases airway hyper-responsiveness, as shown here and in a previous study.19 Second, we have shown that IL-33 can increase angiogenesis in the airways (previously this potential has been inferred indirectly from experiments on umbilical vein endothelial cells in vitro20), which may again at least partly reflect its induction of local VEGF and bFGF synthesis potentially by various airway structural and infiltrating inflammatory cells.9
The cytokine IL-33 is essentially an ‘alarmin’,21 constitutively expressed in lymphoid tissues, epithelial barriers and the central nervous system and released in response to injury and infection (in asthma this might include injury from pollutants, viral infections and allergen-derived and other proteases via the uric acid ‘sensor’).22 The cytokine IL-25, although also derived from epithelial cells. is also produced by a wide range of infiltrating inflammatory cells relevant to asthma (macrophages, eosinophils, mast cells, basophils and both CD4+ and CD8+ T cells).23 Precise stimuli for IL-25 production in asthma are yet to be elucidated (local matrix metalloproteinase is one candidate stimulus,24 and helminthic infection is a clear stimulus for the gut mucosal epithelium).25 A further complication, as we have shown here and in a previous study is that either cytokine can induce elevated expression of the other.7 This is also congruent with a study26 showing that blockade of the effects of IL-33 on the murine airways with a blocking antibody or a soluble form of its ST2 receptor abrogated airways IL-25 and TSLP production in an OVA challenge-based surrogate. This tendency for IL-33 and IL-25 to be mutually cross-inducing might cloud the issue of identifying specific endotypes of human asthma that depend primarily on induction of synthesis of one cytokine or the other, which is why a detailed study of their differential downstream effects in asthma is potentially illuminating.
With respect to the downstream similarities and differences in the effects of these two cytokines relevant to asthma pathogenesis, in our experiments both induced a marked cellular and in particular eosinophilic infiltration of the airways with florid associated mucous hypertrophy and airway smooth muscle hypertrophy/hyperplasia. Both also induced the laying down of interstitial collagen and angiogenesis, but these effects were sustained only with IL-33 challenge. A similar, sustained effect of IL-33 in inducing lay down of collagen has previously been reported in an allergen sensitization/challenge surrogate of asthma, and may at least partly reflect a direct effect of IL-33 in enhancing collagen production by fibroblasts.27,28 Sustained angiogenesis may reflect the ability of IL-33 to induce the production of angiogenesis-promoting mediators by a variety of airway structural and inflammatory cells, as mentioned above.9 Interleukin-33 challenge also resulted in elevated and sustained release of the asthma-relevant cytokines IL-4, IL-5 and IL-13, as well the asthma-relevant CCL chemokine eotaxin into the airways. Although for many years labelled as ‘signature’ cytokines of Th2-type T cells, it is now clear that these cytokines may originate, in whole or part, not from T cells but from activation of type 2 innate lymphoid cells within the airway mucosa. Indeed IL-33 has been shown to activate type 2 innate lymphoid cells in murine airways, causing rapid secretion of IL-5 and IL-13,14 and is more potent in this regard than IL-25.19,29 Interleukin-33 has also been shown to activate resident mast cells, which are also potential sources of cytokines such as IL-4, in an IgE-independent fashion.30,31 Similarly, sustained local release of eotaxin may at least partly reflect a direct effect of IL-33 on airway fibroblasts.32–34 Finally it is of interest to note that, although IL-4 and IL-13 are only cytokines known to be capable of switching B cells to IgE synthesis, all of the observed pathophysiological changes effected by both IL-25 and IL-33 in this system appear to be completely mechanistically independent of IgE (which is not to say that both cytokines might not affect IgE-dependent mechanisms in some asthma ‘endotypes’ indeed IL-33 has been reported to play a key role in regulating the production of allergen-specific IgE in humans, with IL-25 and TSLP playing a subsidiary role33).
Finally, underlining the functional consequences of these distinct differences between the activities if IL-33 and IL-25 in regulating airway inflammation, cytokine expression, remodelling and neoangiogenesis, while airway challenge with both cytokines caused a transient increase in bronchial hyper-responsiveness, only IL-33 challenge produced a suggestion of prolonged, increased tissue damping, which may at least partly reflect its longer term effects on airway angiogenesis and the laying down of extracellular matrix proteins.
Although there are important differences between these cytokines, they are also important for the initiation and development of asthma. Data from experiments using mouse models provide evidence for their synergistic interaction. In an OVA challenge-based surrogate, blockade of the effects of IL-33 on the murine airways with a blocking antibody or a soluble form of its ST2 receptor abrogated airway IL-25 and TSLP production26 and in a house dust mite model of airway inflammation, IL-33, TSLP and IL-25 were all elevated but neutralizing IL-25 completely ablated this early innate response to allergen challenge.35 It is known that expression of the two cytokines increased in the airways from the patients with asthma, but the interaction between the two cytokines in patients is less clear than in murine models of asthma, so better understanding of the similarities, differences and relationships of IL-25 and IL-33 may help to improve our understanding of pathogenetic mechanisms and to identify novel targets for therapeutic intervention.
In summary, these data suggest clearly distinct contributions of IL-25 and IL-33 to the pathophysiological changes that accompany asthma and that both may be legitimate molecular targets for addressing symptoms and perhaps even influencing the natural history of the disease.
Acknowledgments
We are grateful for the financial support from the National Natural Science Foundation of China (81102250, 81270153, 81373177, 81471594), the Science and Technology Project of the Beijing Municipal Education Commission (KM201110025005), PhD Programs Foundation of Ministry of Education of China (20101107110003), Capital Health Development of Scientific Research (Shou-Fa: 2011-1004-01), Key Projects in the National Science & Technology Pillar Program during the Twelfth Five-year Plan Period (2012BAI05B01,2012BAI05B02) and the Program of Beijing Municipality Health System for High-Level Talent (2011-2-04).
Glossary
- bFGF
basic fibroblast growth factor
- IFN-γ
interferon-γ
- IL-25
interleukin-25
- IL-25R
interleukin-25 receptor
- OVA
ovalbumin
- PAS
periodic acid-Schiff
- Th2
T helper type 2
- TSLP
thymic stromal lymphopoietin
- VEGF
vascular endothelial growth factor
Disclosures
The authors declare no conflict of interest.
References
- Farahani R, Sherkat R, Hakemi MG, Eskandari N, Yazdani R. Cytokines (interleukin-9, IL-17, IL-22, IL-25 and IL-33) and asthma. Adv Biomed Res. 2014;3:127. doi: 10.4103/2277-9175.133249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams CM, Rahman S, Hubeau C, Ma HL. Cytokine pathways in allergic disease. Toxicol Pathol. 2012;40:205–15. doi: 10.1177/0192623311430694. [DOI] [PubMed] [Google Scholar]
- Hayakawa H, Hayakawa M, Kume A, Tominaga S. Soluble ST2 blocks interleukin-33 signaling in allergic airway inflammation. J Biol Chem. 2007;282:26369–80. doi: 10.1074/jbc.M704916200. [DOI] [PubMed] [Google Scholar]
- Kurowska-Stolarska M, Kewin P, Murphy G, et al. IL-33 induces antigen-specific IL-5+ T cells and promotes allergic-induced airway inflammation independent of IL-4. J Immunol. 2008;181:4780–90. doi: 10.4049/jimmunol.181.7.4780. [DOI] [PubMed] [Google Scholar]
- Hamzaoui A, Berraies A, Kaabachi W, Haifa M, Ammar J, Kamel H. Induced sputum levels of IL-33 and soluble ST2 in young asthmatic children. J Asthma. 2013;50:803–9. doi: 10.3109/02770903.2013.816317. [DOI] [PubMed] [Google Scholar]
- Hurst SD, Muchamuel T, Gorman DM, et al. New IL-17 family members promote Th1 or Th2 responses in the lung: in vivo function of the novel cytokine IL-25. J Immunol. 2002;169:443–53. doi: 10.4049/jimmunol.169.1.443. [DOI] [PubMed] [Google Scholar]
- Yao XJ, Huang KW, Li Y, et al. Direct comparison of the dynamics of IL-25- and ‘allergen’-induced airways inflammation, remodelling and hypersensitivity in a murine asthma model. Clin Exp Allergy. 2014;44:765–77. doi: 10.1111/cea.12298. [DOI] [PubMed] [Google Scholar]
- Tang W, Smith SG, Beaudin S, Dua B, Howie K, Gauvreau G, O'Byrne PM. IL-25 and IL-25 receptor expression on eosinophils from subjects with allergic asthma. Int Arch Allergy Immunol. 2014;163:5–10. doi: 10.1159/000355331. [DOI] [PubMed] [Google Scholar]
- Bartemes KR, Kita H. Dynamic role of epithelium-derived cytokines in asthma. Clin Immunol. 2012;143:222–35. doi: 10.1016/j.clim.2012.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iijima K, Kobayashi T, Hara K, Kephart GM, Ziegler SF, McKenzie AN, Kita H. IL-33 and thymic stromal lymphopoietin mediate immune pathology in response to chronic airborne allergen exposure. J Immunol. 2014;193:1549–59. doi: 10.4049/jimmunol.1302984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oboki K, Nakae S, Matsumoto K, Saito H. IL-33 and airway inflammation. Allergy Asthma Immunol Res. 2011;3:81–8. doi: 10.4168/aair.2011.3.2.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffatt MF, Gut IG, Demenais F, et al. A large-scale, consortium-based genomewide association study of asthma. N Engl J Med. 2010;363:1211–21. doi: 10.1056/NEJMoa0906312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan SJ, Lloyd CM. Prolonged allergen challenge in mice leads to persistent airway remodelling. Clin Exp Allergy. 2004;34:497–507. doi: 10.1111/j.1365-2222.2004.01895.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartemes KR, Iijima K, Kobayashi T, Kephart GM, McKenzie AN, Kita H. IL-33-responsive lineage CD25+ CD44hi lymphoid cells mediate innate type 2 immunity and allergic inflammation in the lungs. J Immunol. 2012;188:1503–13. doi: 10.4049/jimmunol.1102832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonasson S, Hedenstierna G, Hedenstrom H, Hjoberg J. Comparisons of effects of intravenous and inhaled methacholine on airway physiology in a murine asthma model. Respir Physiol Neurobiol. 2009;165:229–36. doi: 10.1016/j.resp.2008.12.005. [DOI] [PubMed] [Google Scholar]
- Riesenfeld E, Allen GB, Bates JH, Poynter ME, Wu M, Aimiand S, Lundblad LK. The temporal evolution of airways hyperresponsiveness and inflammation. J Allergy Ther. 2012;1:1–7. doi: 10.4172/2155-6121.S1-005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickel EA, Siegel LA, Yoon BR, et al. Identification of functional roles for both IL-17RB and IL-17RA in mediating IL-25-induced activities. J Immunol. 2008;181:4299–310. doi: 10.4049/jimmunol.181.6.4299. [DOI] [PubMed] [Google Scholar]
- Davies DE, Wicks J, Powell RM, Puddicombe SM, Holgate ST. Airway remodeling in asthma: new insights. J Allergy Clin Immunol. 2003;111:215–25. doi: 10.1067/mai.2003.128. 226. [DOI] [PubMed] [Google Scholar]
- Barlow JL, Peel S, Fox J, et al. IL-33 is more potent than IL-25 in provoking IL-13-producing nuocytes (type 2 innate lymphoid cells) and airway contraction. J Allergy Clin Immunol. 2013;132:933–41. doi: 10.1016/j.jaci.2013.05.012. [DOI] [PubMed] [Google Scholar]
- Choi YS, Choi HJ, Min JK, et al. Interleukin-33 induces angiogenesis and vascular permeability through ST2/TRAF6-mediated endothelial nitric oxide production. Blood. 2009;114:3117–26. doi: 10.1182/blood-2009-02-203372. [DOI] [PubMed] [Google Scholar]
- Grotenboer NS, Ketelaar ME, Koppelman GH, Nawijn MC. Decoding asthma: translating genetic variation in IL33 and IL1RL1 into disease pathophysiology. J Allergy Clin Immunol. 2013;131:856–65. doi: 10.1016/j.jaci.2012.11.028. [DOI] [PubMed] [Google Scholar]
- Hara K, Iijima K, Elias MK, et al. Airway uric acid is a sensor of inhaled protease allergens and initiates type 2 immune responses in respiratory mucosa. J Immunol. 2014;192:4032–42. doi: 10.4049/jimmunol.1400110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papazian D, Hansen S, Würtzen PA. Airway responses towards allergens – from the airway epithelium to T cells. Clin Exp Allergy. 2014 doi: 10.1111/cea.12451. doi: 10.1111/cea.12451. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Goswami S, Angkasekwinai P, Shan M, et al. Divergent functions for airway epithelial matrix metalloproteinase 7 and retinoic acid in experimental asthma. Nat Immunol. 2009;10:496–503. doi: 10.1038/ni.1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliphant CJ, Barlow JL, McKenzie AN. Insights into the initiation of type immune response. Immunology. 2011;134:378–85. doi: 10.1111/j.1365-2567.2011.03499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HY, Rhee CK, Kang JY, et al. Blockade of IL-33/ST2 ameliorates airway inflammation in a murine model of allergic asthma. Exp Lung Res. 2014;40:66–76. doi: 10.3109/01902148.2013.870261. [DOI] [PubMed] [Google Scholar]
- Saglani S, Lui S, Ullmann N, et al. IL-33 promotes airway remodeling in pediatric patients with severe steroid-resistant asthma. J Allergy Clin Immunol. 2013;132:676–85. doi: 10.1016/j.jaci.2013.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saglani S, Lloyd CM. Eosinophils in the pathogenesis of paediatric severe asthma. Curr Opin Allergy Clin Immunol. 2014;14:143–8. doi: 10.1097/ACI.0000000000000045. [DOI] [PubMed] [Google Scholar]
- Klein WR, Kleinjan A, van Nimwegen M, Bergen I, de Bruijn M, Levani Y, Hendriks RW. Pulmonary innate lymphoid cells are major producers of IL-5 and IL-13 in murine models of allergic asthma. Eur J Immunol. 2012;42:1106–16. doi: 10.1002/eji.201142018. [DOI] [PubMed] [Google Scholar]
- Hsu CL, Neilsen CV, Bryce PJ. IL-33 is produced by mast cells and regulates IgE-dependent inflammation. PLoS ONE. 2010;5:e11944. doi: 10.1371/journal.pone.0011944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd CM. IL-33 family members and asthma – bridging innate and adaptive immune responses. Curr Opin Immunol. 2010;22:800–6. doi: 10.1016/j.coi.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato E, Nelson DK, Koyama S, Hoyt JC, Robbins RA. Inflammatory cytokines modulate eotaxin release by human lung fibroblast cell line. Exp Lung Res. 2001;27:173–83. doi: 10.1080/019021401750069401. [DOI] [PubMed] [Google Scholar]
- Teran LM, Mochizuki M, Bartels J, Valencia EL, Nakajima T, Hirai K, Schroder JM. Th1- and Th2-type cytokines regulate the expression and production of eotaxin and RANTES by human lung fibroblasts. Am J Respir Cell Mol Biol. 1999;20:777–86. doi: 10.1165/ajrcmb.20.4.3508. [DOI] [PubMed] [Google Scholar]
- Kurokawa M, Matsukura S, Kawaguchi M, et al. Expression and effects of IL-33 and ST2 in allergic bronchial asthma: IL-33 induces eotaxin production in lung fibroblasts. Int Arch Allergy Immunol. 2011;155(Suppl. 1):12–20. doi: 10.1159/000327259. [DOI] [PubMed] [Google Scholar]
- Gregory LG, Jones CP, Walker SA, Sawant D, Gowers KH, Jones GA, McKenzie AN, Lloyd CM. IL-25 derives remodelling in allergic airways disease induced by house dust mite. Thorax. 2013;68:82–90. doi: 10.1136/thoraxjnl-2012-202003. [DOI] [PMC free article] [PubMed] [Google Scholar]