

Abstract
Immunoglobulin G4-related disease (IgG4-RD) is a fibroinflammatory condition that derives its name from the characteristic finding of abundant IgG4+ plasma cells in affected tissues, as well as the presence of elevated serum IgG4 concentrations in many patients. In contrast to fibrotic disorders, such as systemic sclerosis or idiopathic pulmonary fibrosis in which the tissues fibrosis has remained largely intractable to treatment, many IgG4-RD patients appear to have a condition in which the collagen deposition is reversible. The mechanisms underlying this peculiar feature remain unknown, but the remarkable efficacy of B cell depletion in these patients supports an important pathogenic role of B cell/T cell collaboration. In particular, aberrant T helper type 2 (Th2)/regulatory T cells sustained by putative autoreactive B cells have been proposed to drive collagen deposition through the production of profibrotic cytokines, but definitive demonstrations of this hypothesis are lacking. Indeed, a number of unsolved questions need to be addressed in order to fully understand the pathogenesis of IgG4-RD. These include the identification of an antigenic trigger(s), the implications (if any) of IgG4 antibodies for pathophysiology and the precise immunological mechanisms leading to fibrosis. Recent investigations have also raised the possibility that innate immunity might precede adaptive immunity, thus further complicating the pathological scenario. Here, we aim to review the most recent insights on the immunology of IgG4-RD, focusing on the relative contribution of innate and adaptive immune responses to the full pathological phenotype of this fibrotic condition. Clinical, histological and therapeutic features are also addressed.
Keywords: IgG4, IgG4-related disease, immunology, pathogenesis, review
Introduction
IgG4-related disease (IgG4-RD) is a relapsing–remitting fibroinflammatory condition characterized clinically by tumefactive lesions, serum IgG4 elevation in most, but not all cases, and a prompt response to glucocorticoids (GCs). IgG4-RD was described originally in the pancreas in 2001 as ‘sclerosing pancreatitis’, now referred to as type I IgG4-related autoimmune pancreatitis (AIP) 1. Shortly thereafter, however, the identification of a variety of extra-pancreatic organ involvement linked by unique histopathological features led to the recognition that AIP was part of a systemic condition 2–4. Awareness of this new fibroinflammatory condition has expanded substantially since the first characterization of pancreatic involvement in 2001, but IgG4-RD still represents an underdiagnosed entity. Understanding of the global epidemiology of IgG4-RD remains largely incomplete 1,2. A Japanese survey calculated that IgG4-related AIP affects 2·2 cases per 100 000 individuals and has a predilection for middle-aged to elderly men. However, the multi-organ nature of the disease, the fact that AIP represents only a minority of cases and the likelihood that even many diagnoses of AIP are missed, this figure is almost certainly an under-estimate of the true prevalence of IgG4-RD 5.
Similarly, few immunological studies have been reported to date, and the pathogenesis of IgG4-RD remains unknown. In general theory, an antigen-driven immune response is supposed to drive both B cell commitment to IgG4 production and secretion of profibrotic cytokines by activated T lymphocytes. Indeed, rituximab (RTX), an anti-CD20 monoclonal antibody, has been proved to induce rapid clinical responses in patients with IgG4-RD, further suggesting an important pathogenic contribution of the B cell compartment 6–8. Here, we aim to review the clinical and pathophysiological features of IgG4-RD, focusing on the most recent immunological acquisitions in the field.
Overview of clinical manifestations
IgG4-RD typically affects middle-aged to elderly men, with sporadic reports of paediatric cases 9. The clinical presentation is usually indolent, with signs and symptoms becoming evident over months or even years. High, spiking fevers and other manifestations of systemic inflammation that mimic infections are classically absent, but weight loss can occur during the subclinical period. A longstanding history of allergies is present in 30–40% of patients at diagnosis, but symptoms that overlap with allergic conditions are also reported in some IgG4-RD patients without histories of atopy. These include bronchial asthma, chronic rhinitis and eczema 10.
IgG4-RD is characterized by pseudotumour-like lesions involving single or multiple organs. Different organs might be affected at the same time or one after the other. Clinical manifestations are largely non-specific and vary according to the spectrum of organs involved. Indeed, IgG4-RD might be asymptomatic or present with signs and symptoms related to the mechanical compression exerted by the fibrotic masses on local structures. IgG4-RD has been described in virtually every anatomical region (Table1) 11–19, but the most common manifestations include type I AIP, chronic periaortitis, retroperitoneal fibrosis (Ormond’s disease) and salivary or lacrymal gland swelling (Mikulicz’s disease), conditions regarded as single independent entities for decades.
Table 1.
The spectrum of IgG4-related disorders (IgG4-RD): synopsis of clinical manifestations, differential diagnoses, pathology features and therapeutic approaches. A consensus on hystopathological features diagnostic for IgG4-RD has not been reached for all possible affected organs
| Organ involvment (Ref.) | Nomenclature (previous name) | Clinical presentation |
|---|---|---|
| Head and Neck | ||
| Orbits and periorbital tissue 31–33 | IgG4-related orbital disease (orbital inflammatory pseudotumour) | Exophthalmos, haemianopsia, ocular movement restriction, ptosis, headache, scleritis, xerophthalmia |
| Salivary glands and lacrymal glands 34,35 | IgG4-related sialoadenitis (Mikulicz’s disease, Kuttner’s tumour) | Parotid or submandibular gland swelling, xerostomia |
| Thyroid gland 19 | IgG4-related thyroiditis (Riedel’s rhyroiditis) | Neck pressure, neck mass, malaise, recurrent laryngeal nerve palsy, hypothyroidism |
| Ear, nose and throat 17,36 | IgG4-related sinusitis/midline destructive lesion/pharyngitis | Nasal crusting, nasal obstruction, rhinorrhoea, polyposis, sinusitis, nasal septum perforation, laryngeal obstruction, middle ear effusion |
| Thorax | ||
| Lungs 11,12 | IgG4-related lung disease | Cough, sputum, dyspnoea, chest pain |
| Pleura 12 | IgG4-related pleural disease | Chest pain, dyspnoea, pleural effusion |
| Mediastinum 46 | IgG4-related mediastinitis (fibrosing mediastinitis) | Mediastinal mass, compression of mediastinal structures, dyspnoea, chest pain |
| Breast 50 | IgG4-related mastitis | Painless breast mass |
| Abdomen and pelvis | ||
| Retroperitoneum 27–30 | IgG4-related retroperitoneal fibrosis (Ormond’s disease) | Back/flank pain, leg oedema, hydronephrosis, deep venous thrombosis, varicocele |
| Pancreas 1,21,22 | IgG4-related autoimmune pancreatitis (type1 autoimmune pancreatitis) | Obstructive jaundice, diabetes mellitus, abdominal pain, pale stool, malabsorption |
| Biliary tree/gallbladder 23,24 | IgG4-related sclerosing cholangitis | Jaundice, weight loss, abdominal pain |
| Liver 24 | IgG4-related hepatitis (hepatic inflammatory pseudotumour) | Mainly asymptomatic, transaminitis, hepatic mass |
| Kidney 37–39 | IgG4-related tubule-interstitial nephritis/glomerulonephritis | Elevated serum creatinine, proteinuria, haematuria, nephritic/nephrotic syndrome |
| Gastrointestinal tract 49 | IgG4-related gastrointestinal disease | Epigastric pain, chronic/acute abdominal pain, intestinal obstruction or dysmotility, nausea |
| Mesentere 47 | IgG4-related sclerosing mesenteritis | Abdominal pain, abdominal mass, vomiting, nausea |
| Prostate 44 | IgG4-related prostatitis | Mass, lower urinary tract symptoms, dysuria |
| Testis 45 | IgG4-related epididymo-orchitis (testicular inflammatory pseudotumour) | Scrotal mass, scrotal pain |
| Nervous system | ||
| Central nervous system 43 | Dementia, hemiparesis, multi-focal neurological defects | |
| Pituitary gland 14 | IgG4-related hypophysitis | Hypopituitarism, diabetes insipidus, headache |
| Peripheral nerves 42 | IgG4-related neuropathy | Sensory-motor polyneuropathy, multiplex mononeuritis, perineural mass |
| Meninges 15,16 | IgG4-related pachymeningitis (hypertrophic pachymeniningitis) | Headache, cranial nerve palsies, vision disturbance, motor weakness, limb numbness, sensorineural hearing loss, seizures |
| Cardiovascular system | ||
| Heart and pericardium 41 | IgG4-related cardiac disease | Acute chest pain (coronary syndrome), dyspnoea, pericardial rub |
| Aorta 30,40 | IgG4-related periaortitis (chronic periaortitis/inflammatory aortic aneurism) | Back pain, leg oedema, bruits, acute aneurysm rupture |
| Lymph nodes 13 | IgG4-related lymphadenopathy | Usually asymptomatic, lymph node enlargement |
| Skin 18 | IgG4-related skin disease | Skin plaques, subcutaneous nodules, brown papules, dermatitis |
| Bone 48 | IgG4-related disease of the bone | Headache, tinnitus, skull base destructive lesion, tumefactive sinus lesion |
| Differential diagnoses | Pathology features: IgG4/HPF diagnostic for IgG4-RD*† | Therapeutic strategies |
|---|---|---|
| Lymphoma–granulomatosis with polyangiitis–sarcoidosis–Graves’ orbitopathy, Sjögren’s syndrome | >10/HPF† | GC–MTX–CTX–RTX–BTZ–surgery |
| Lymphoma–Sjögren’s syndrome–sialodocholithiasis–sarcoidosis | >100/HPF* | GC–AZA–MTX–RTX |
| Thyroid lymphoma–differentiated thyroid carcinoma–other malignancies | >10/HPF† | GC–MTX–RTX |
| Allergic disease–Churg–Strauss syndrome–granulomatosis with polyangiitis–chronic infections–sarcoidosis | >10/HPF† | GC |
| Malignancy–sarcoidosis–granulomatosis with polyangiitis–infections–interstitial lung disease–inflammatory miofiroblastic tumour–Churg–Strauss syndrome | >50/HPF (surgical specimen) >20/HPF (biopsy)* | GC–AZA–CTX–RTX–BTZ |
| Mesothelioma–infections | >50/HPF* | GC |
| Lymphoma–sarcoidosis–histoplasmosis–malignancies–mycobacterial infection | >10/HPF† | GC |
| Malignancies–mastitis | >10/HPF† | GC–surgery |
| Lymphoma–sarcoma- Erdheim–Chester disease–periaortitis–idiopathic retroperitoneal fibrosis | >30/HPF* | GC–AZA–MMF–RTX |
| Pancreatic cancer–Type II autoimmune pancreatitis | >50/HPF (surgical specimen) >10/HPF (biopsy)* | GC–MTX–AZA–RTX |
| Pancreatic cancer–cholangiocarcinoma–primary sclerosing cholangitis | >50/HPF (surgical specimen) >10/HPF (biopsy)* | GC–AZA–MMF–RTX |
| Cholangiocarcinoma–hepatocellular carcinoma–autoimmune hepatitis | >50/HPF (surgical specimen) >10/HPF (biopsy)* | GC–RTX |
| Lymphoma–renal-cell carcinoma–drug-induced tubulointerstitial nephritis–vasculitis–systemic lupus erythematosus | >30/HPF (surgical specimen) >10/HPF (biopsy)* | GC–surgery |
| Malignancies–GERD | >10/HPF† | GC |
| Lymphoma–fibromatosis–peritoneal carcinosis | >10/HPF† | GC |
| Benign prostatic hypertrophy–malignancies–infections | >10/HPF† | GC |
| Seminoma–lymphoma–inflammatory miofibroblastic tumour | >10/HPF† | Surgery |
| Central nervous system vasculitis–infections–malignancies | >10/HPF† | GC |
| Neoplasms–histiocytosis–primary hypophisytis–sarcoidosis | >10/HPF† | GC |
| POEMS syndrome–metabolic neuropathy–vasculitis–other demyelinating neuropathies | >10/HPF† | GC |
| Chronic infections–lymphoma–Langherhans-cell histiocytosis–giant-cell arteritis–sarcoidosis–idiopathic hypertrophic pachymeniningitis | >10/HPF* | GC–CTX–MTX–RTX |
| Infections–inflammatory pericarditis–cardiac mixoma–acute coronary syndrome | >10/HPF† | GC |
| Takayasu arteritis–giant cell arteritis–lymphoma–infectious aortitis–sarcoidosis–histiocytosis | >50/HPF* | GC–MTX–RTX |
| Multi-centric Castleman disease–lymphoma–systemic lupus erythematosus–sarcoidosis–infections | >100/HPF* | GC–RTX |
| Cutaneous lymphoma–drug eruption–psoriasis vulgaris–multi-centric Castleman disease | >200/HPF* | GC–RTX |
| Malignancy–osteomielitis | >10/HPF† | GC |
For the organs included in the ‘Consensus Statement on the pathology of IgG4-RD’3* a ‘highly suggestive’ diagnosis of IgG4-RD requires (i) specific cut-offs of IgG4+ plasma cells/high-power field (HPF)*, (ii) an IgG4+/IgG+ plasma cell ratio > 40% and (iii) two or more of the following histological features: dense lymphoplasmacytic infiltrate, storiform fibrosis and obliterative phlebitis. For the organs not included in the ‘Consensus Statement’, ‘Comprehensive diagnostic criteria’4† are still adopted. In this case, a ‘definite’ diagnosis of IgG4-RD requires (i) an infiltration of > 10 IgG4+ plasma cells/HPF† and an IgG4+/IgG+ plasma cells ratio > 40%, together with (ii) diffuse/localized swelling or masses in single or multiple organs and (iii) serum IgG4 concentrations > 135 mg/dl. Abbreviations: glucocorticoids (GC: 0·5–1 mg/kg/day); methotrexate (MTX: 10–20 mg/week); cyclophosphamide (CTX: 500–1200 mg/m2/month); rituximab (RTX: 1000 mg in two doses 15 days apart); azathioprine (2–2·5 mg/kg/day); mycophenolate mofetil (MMF: 750–1000 mg twice daily); bortezomib (BTZ: 1·5 mg/m2/week). IgG4-RD = immunoglobulin G4-related disorders; HPF = high-power field; GERD = gastroesophageal reflux disease.
IgG4-related AIP, the most frequently recognized manifestation of IgG4-RD, presents typically with obstructive jaundice, weight loss or abdominal pain (Fig. 1a) 20–22. Many patients are misdiagnosed initially as having adenocarcinoma of the pancreas – sometimes only after the performance of a modified pancreatectomy. Several cases of pancreatic cancer have been reported in patients with previous IgG4-related AIP, but a clear relationship between the two conditions still needs to be fully verified 23,24. Secondary diabetes mellitus and malabsorption might complicate longstanding pancreatic disease. In 25% of cases, AIP is associated with gallbladder and bile duct involvement 25. Gallbladder disease, known as ‘IgG4-related lymphoplasmacytic cholecystitis’, is generally asymptomatic and not associated with gallstones. Conversely, IgG4-related sclerosing cholangitis is likely to present with jaundice and is often difficult to differentiate from cholangiocarcinoma 22,25,26.
Figure 1.
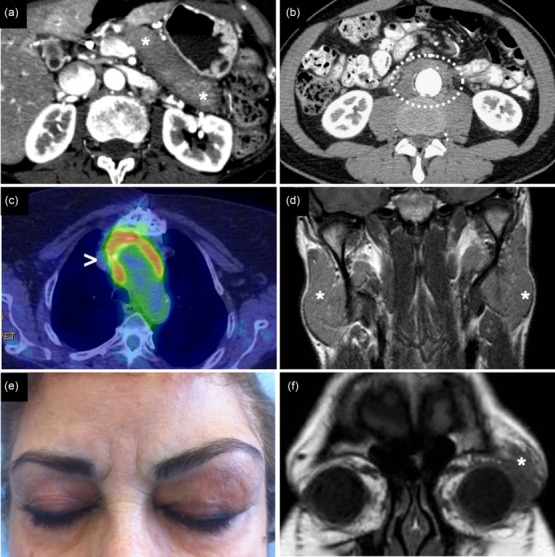
Clinical and radiological presentation of IgG4-related disease (IgG4-RD). (a) IgG4-related autoimmune pancreatitis: computed tomography scan showing a ‘sausage-like’-shaped pancreas with a surrounding rim of hypodense tissue (asterisks). (b) Retroperitoneal fibrosis with periaortic involvement (circle). (c) Inflammatory aneurism of the thoracic aorta showing 18fluoro-deoxyglucose uptake on positron emission tomography (arrowhead). (d) Magnetic resonance showing bilateral parotid enlargement due to IgG4-RD (asterisks). (e,f) Clinical and radiological appearance of IgG4-related orbital pseudotumour (asterisk).
IgG4-related retroperitoneal fibrosis classically affects the connective tissue around the abdominal aorta (‘chronic periaortitis’) and the periureteral areas (Fig. 1b) 27–30. Depending on disease localization, retroperitoneal fibrosis might be asymptomatic or present with back pain, flank pain, dysuria, haematuria, leg or scrotal oedema. Aneurysmal dilatation and hydronephrosis represent the most feared complications of retroperitoneal involvement, and might require surgical approaches to prevent aortic dissection or renal failure, respectively. Chronic periaortitis may also involve the thoracic aorta, occasionally leading to aortic dissection (Fig. 1c). Salivary and lacrymal gland involvement generally causes facial and orbital swelling (Fig. 1d). Sicca symptoms are often less severe in IgG4-RD than in Sjögren’s syndrome and, when present, typically respond well to immunosuppressive therapy 31–35. Orbital pseudotumours affect the lacrymal gland most often but can also occur in other orbital regions. Extra-ocular muscle involvement (frequently termed ‘orbital myositis’ in the days before the recognition of IgG4-RD) can present with exophthalmos and the restriction of ocular movements 32,33 (Fig. 1e,f).
Atypical presentations of IgG4-RD that lack pseudotumour-like lesions should be borne in mind, including tubulointerstitial nephritis, glomerulonephritis, midline destructive lesions, interstitial lung disease, pleural and pericardial effusion 11,36. In particular, renal involvement might present with variable degrees of proteinuria, haematuria, renal failure or hypocomplementaemia 37–39. Finally, life-threatening presentations such as rupture of inflammatory aneurysms, coronary syndromes, pachymeningitis and acute neurological events have also been described occasionally 30,40–42. A comprehensive review of the organs involved by IgG4-RD, common clinical presentation and differential diagnoses is detailed in Table1 43–50.
Diagnosis
Definitive diagnosis of IgG4-RD requires both histopathological confirmation and clinicopathological correlation. Serological and radiological features lack adequate sensitivity and specificity for diagnostic purposes. Histological examination is also mandatory to exclude neoplastic or inflammatory conditions that can mimic IgG4-RD.
Serological findings
Serological findings in patients with IgG4-RD are largely non-specific. Acute phase reactants such as the erythrocyte sedimentation rate and C-reactive protein are usually (but not always) elevated to a moderate degree. Peripheral blood eosinophilia and increased serum IgE occur in almost 30% of patients 10. Some patients have positive low-titre anti-nuclear antibodies, but the presence of anti- Sjögren’s syndrome-related antigen A (Ro/SSA), anti-La/Sjögren’s syndrome-related antigen B (SSB) and anti-neutrophil cytoplasmic antibodies (ANCA) antibodies strongly implicate other immune-mediated conditions.
High serum IgG4 concentrations occur in 60–70% of patients, typically in those with multi-organ involvement 4. Unfortunately, elevation in serum IgG4 can be associated with a host of conditions with the potential to mimic IgG4-RD, e.g. systemic vasculitides (particularly granulomatosis with polyangiitis), connective tissue disorders (e.g. the lupus spectrum of conditions), infections and malignancies 51. Therefore, measurement of serum IgG4 should be regarded only as a useful screening tool, not as a ‘stand-alone’ diagnostic marker.
An analogous principle pertains to the use of serum IgG4 concentrations as a longitudinal biomarker. Although serial measurement of serum IgG4 concentrations are often useful in the assessment of disease activity, they should never be used as the sole determinant of treatment decisions. IgG4 production in the cerebrospinal fluid has also been suggested as a diagnostic biomarker for IgG4-related pachymeningitis, but histopathological evaluation, whenever possible, remains mandatory 52. Other immunoglobulins, including IgM, IgA and other IgG subclasses, are frequently elevated in IgG4-RD, although generally not to the extent of IgG4 2. High concentrations of IgG subclasses other than IgG4, e.g. IgG1 and IgG3, may account for the hypocomplementaemia observed in a minority of IgG4-RD patients, because IgG4 itself typically binds complement poorly 2. Hyperviscosity syndromes have been reported in rare cases 53.
Circulating CD19+CD20−CD27+CD38+ plasmablasts, the precursors of tissue resident plasma cells, have been proposed as the best available indicator of IgG4-RD disease activity, but larger studies are required in order to validate this test 54.
Radiological findings
Radiological findings are largely non-specific and are not sufficient to distinguish IgG4-RD from the neoplastic condition presenting with mass-forming lesions. IgG4-related AIP is the sole exception, because computed tomography and magnetic resonance imaging classically show a ‘sausage-shaped’ pancreas with a halo of oedematous tissue (Fig. 1a). These benign features are supported further by the aspect of the pancreatic gland on endoscopic ultrasonography and on histological examination through endoscopic core biopsy. 18Fluoro-deoxyglucose positron emission tomography reliably identifies active inflammation in IgG4-RD and might be useful both for staging purposes and for assessing disease response to treatment 55 (Fig. 1c).
Pathology
Histopathological hallmarks of IgG4-RD are (i) a dense storiform fibrosis, (ii) obliterative pheblitis, (iii) a lymphoplasmacytic infiltrate rich in IgG4+ plasma cells and (iv) a mild to moderate eosinophilic infiltrate (Fig. 2a–f) 3,4. Epithelial or endothelial damage is not typical until end-stage disease, when fibrosis subverts the parenchymal structure and impairs organ function (Fig. 2a). Neutrophils, necrosis and granulomas are classically absent, and their presence should prompt the exclusion of other potential differential diagnoses.
Figure 2.
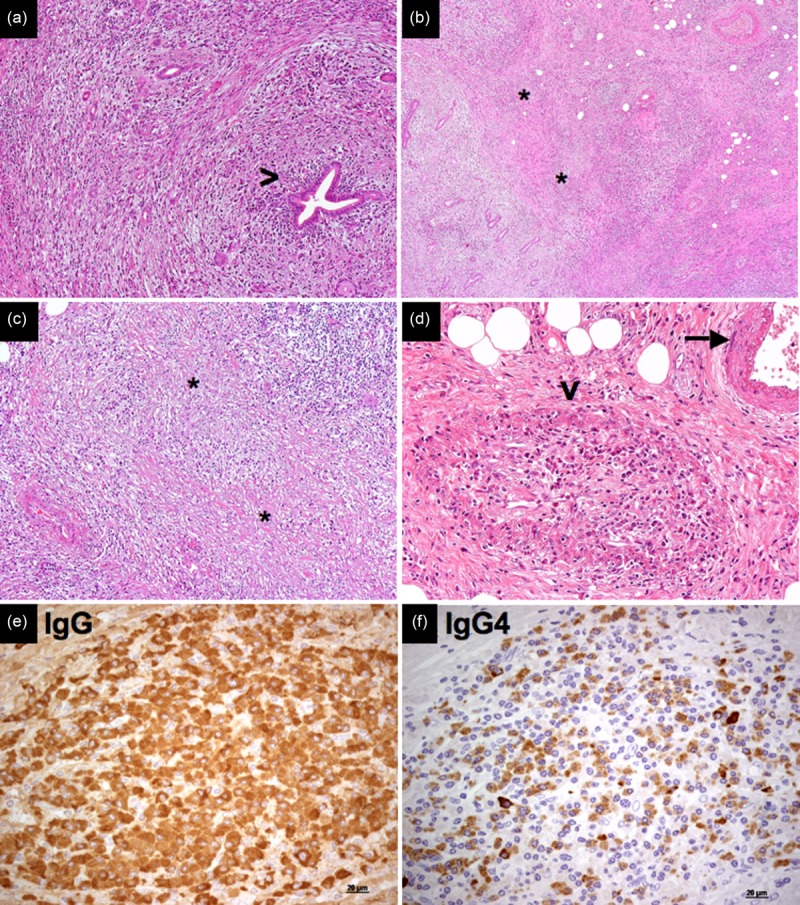
Pathological features of immunoglobulin (Ig)G4-related disease. (a) Pancreatic ducts are not affected by the fibroinflammatory infiltrate in IgG4-related autoimmune pancreatitis (arrowhead; haematoxylin and eosin, magnification ×100). (b,c) Areas of storiform fibrosis in IgG4-related autoimmune pancreatitis [asterisks; haematoxylin and eosin, magnification ×40 (b) and ×100 (c)]. (d) Obliterative phlebitis: an obliterated vein surrounded by an inflammatory nodule (arrowhead), next to an intact artery (arrow) (haematoxylin and eosin, magnification ×200). (e,f) Immunohistochemistry for IgG (e) and IgG4 (e) on sequential sections shows an IgG4/IgG ratio > 40% (magnification ×40).
Storiform fibrosis
‘Storiform fibrosis’ is a hallmark of IgG4-RD. The term ‘storiform’ refers to an irregularly whorled organization of the collagen bundles that can be observed in any organ affected by IgG4-RD (Fig. 2b,c). Storiform fibrosis is not usually prominent in lacrymal glands and lymph node involvement, although this finding has also been reported in those tissues. Storiform fibrosis is likely to be triggered by the activation of myofibroblasts following profibrotic stimuli provided by the inflammatory infiltrate. Some authors have hypothesized that the absence of a lymphoplasmacytic infiltrate in tissue biopsies marks the progression of IgG4-RD from an ‘active’ phase, characterized by abundant numbers of cell of the B and T cell lineages, to an ‘end-stage’ phase, characterized chiefly by fibrosis with few or no IgG4+ plasma cells 8. Even in the earliest stages of disease, however, some degree of fibrosis is typically present. These considerations should be borne in mind, because pathologists might encounter different stages of IgG4-RD due to bioptic procedures that do not always provide adequate sample sizes (e.g. needle biopsies) or areas of active disease.
Obliterative phlebitis
A unique feature of IgG4-RD is obliterative phlebitis. Obliterative phlebitis refers to the partial or complete occlusion of the lumina of small- and medium-sized veins by the lymphoplasmacytic infiltrate and by extrinsic compression (Fig. 2d). The presence of obliterative phlebitis should be sought thoroughly because of its diagnostic value, ideally through the use of elastin stains targeting the internal elastic lamina of the vessel. In contrast to systemic vasculitides such as granulomatosis with polyangiitis, microscopic polyangiitis and polyarteritis nodosa, leucytoklastic vasculitis with vessel wall necrosis and fibrin deposition is not observed in IgG4-RD. Obliterative arteritis has been reported, particularly in the lung 3.
Lymphoplasmacytic infiltrate
Lymphoplasmacytic infiltrate is composed of polyclonal/oligoclonal B and T lymphocytes. B lymphocytes tend to be organized in germinal centres (Fig. 3a), while T lymphocytes are spread throughout the fibrotic tissue (Fig. 3b). Immunohistochemistry is essential for the diagnosis of IgG4-RD because it allows the demonstration of IgG4+ plasma cells and evaluation of the IgG4+/IgG+ ratio (Fig. 2e,f). Of note, as IgG4+ plasma cells might be found in other inflammatory, neoplastic and infectious conditions, organ specific cut-offs have been defined in order to increase the diagnostic accuracy of the immunohistochemical examination (Table1) 3. Indeed, a correct diagnosis should not rely on immunohistochemistry alone, but also consider the other characteristic histological features.
Figure 3.
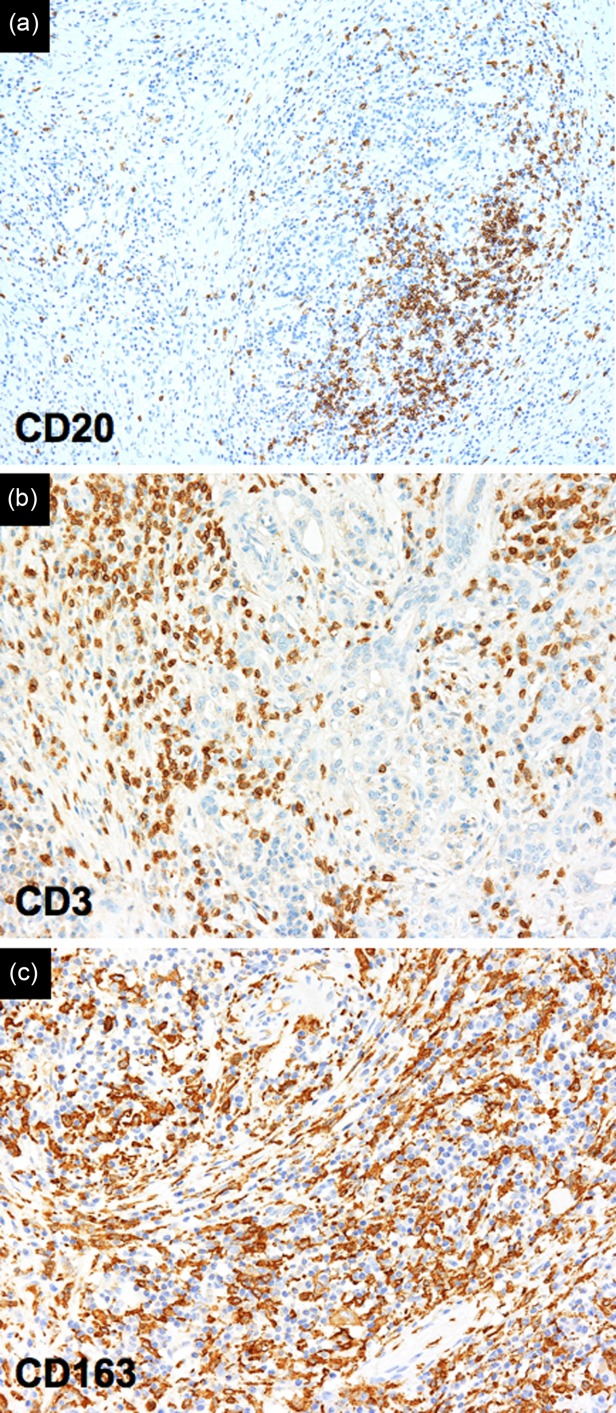
Inflammatory infiltrate in IgG4-related disease. Immunohistochemistry reveals CD20+ B lymphocytes organized in follicular structures (a), CD3+ T lymphocytes (b), and CD163+ M2 macrophages (c) spread throughout the fibrotic tissue (magnification ×100).
Tissue eosinophilia and macrophages
Eosinophils are present in at least 50% of IgG4-RD lesions and might dominate the histological picture in those cases of orbital or upper respiratory tract involvement, sometimes termed ‘eosinophilic angiocentric fibrosis’ 56. Macrophages are usually detectable within the fibroinflammatory infiltrate, but the presence of granulomas argues strongly against the diagnosis of IgG4-RD (Fig. 3c) 3,4.
Treatment
Treatment is not always necessary in patients with IgG4-RD, and watchful waiting is prudent in some asymptomatic cases. Conversely, when vital organs are involved or patients become symptomatic, aggressive treatment is needed because IgG4-RD can lead to serious organ dysfunction and failure. Response to treatment might be assessed by means of the IgG4-RD Responder Index, a validated tool for monitoring clinical, radiological and serological outcomes of IgG4-RD 57.
According to the recently released International Consensus Statement on the Treatment of IgG4-RD 58, GCs represent the first line therapy because they lead to dramatic clinical responses in the majority of cases with both pancreatic and extra-pancreatic disease. One typical approach is to treat with prednisolone (0·6 mg/kg per day for 2–4 weeks), followed by a gradual taper over a period of 3–6 months once remission is achieved 59. IgG4-RD has, however, a marked tendency to relapse during or after GC tapers, especially in cases of elevated serum IgG4 at baseline, multi-organ involvement and history of disease relapse 59. Thus, a variety of GC-sparing agents have been employed in different anatomical districts as remission-maintenance drugs (e.g. azathioprine, mycophenolate mofetil, methotrexate, cyclophosphamide and bortezomib), with alternate results 59,60 (Table1). More recently, RTX has been proved to induce swift clinical responses and a selective decline of serum IgG4 subclass concentrations in patients with recurrent or refractory disease 6,7. Finally, when urgent decompression is needed, as in the case of biliary duct or ureteral strictures, temporary stenting or surgical approaches remain the strategy of choice for preventing serious organ damage.
All in all, a major determinant of the responsiveness to immunosuppressive treatments is probably the degree of inflammatory cells within the affected organs. Indeed, researchers have hypothesized that mass-forming IgG4-RD lesions are more likely to shrink in the presence of a prominent lymphoplasmacytic infiltrate (‘active fibroinflammation’), rather than in the presence of tightly organized collagen bundles in which both inflammatory cells and myofibroblasts are rare (‘acellular end-stage fibrosis’ or ‘fibrotic scar’) 8. Further studies are needed to provide reliable and standardized guidelines for the long-term management of IgG4-RD.
Immunopathology of IgG4-related disease
Little is known with certainty about the pathogenic events that initiate IgG4-RD. The evidence of common histopathological features shared by different unrelated organs suggests that IgG4-RD might be an antigen-driven inflammatory condition leading ultimately to tissue fibrosis. However, a number of unsolved questions still need to be addressed, including: (i) the pathophysiological importance of IgG4 antibodies; (ii) the characterization of the putative microbial or self-antigen; (iii) the imbalance between T helper type 1 (Th1), Th2 and regulatory T cell activation; (iv) the role of innate immunity; and (v) the immunological mechanisms leading to fibrosis.
The role of IgG4 antibodies
IgG4 antibodies represent the least abundant IgG subclass in healthy individuals (1–4% of total IgG), and their role in IgG4-RD is still controversial 2. IgG4 antibodies are supposed to be produced after long-term antigen exposure in response to interleukin (IL)-4 and IL-10 61, but the molecular mechanisms that drive the IgG4 class-switch are not fully understood. Indeed, part of these mechanisms (e.g. Th2 cytokines) might also increase serum IgE levels, as exhibited by many IgG4-RD patients 62.
In contrast to other IgG subclasses, heavy chains in each IgG4 molecule have non-covalent associations and inefficient disulphide bridges due to a single amino acid difference in the hinge region (a serine in lieu of a proline) (Fig. 4) 63,64. As a result, hemi-IgG4 molecules – one heavy chain covalently associated with one light chain – have the propensity to dissociate from each other and reassociate randomly with distinct hemi-IgG4 molecules that have different antigen-binding specificities. This phenomenon is known as ‘Fab arm exchange’ (Fig. 4). Fab arm exchange requires a reducing environment to occur, but the precise location of the exchange in vivo has not been determined 64. This half-antibody exchange, unique to the IgG4 isoclass, generates functionally ‘bi-specific’ antibodies that are capable of binding two different antigens but rarely associate with each other to form large immune complexes 65,66. IgG4 antibodies also have limited ability to form immune responses because of their low affinity for both Fc receptors and the C1 complement molecule 67. For these reasons, IgG4 has been viewed traditionally as a ‘non-inflammatory’ molecule, the primary function of which is to dampen rather than to incite or accelerate chronic immune activation. In short, despite the importance of IgG4 implied by the current name of this disease, the IgG4 molecule is probably not the disease driver. This is consistent with what has long been known about IgG4: that IgG4 antibodies are induced by allergy treatments designed to induce tolerance and protect allergic patients from anaphylactic reactions by competing with allergen specific IgE 68,69.
Figure 4.

Molecular basis of the ‘Fab-arm’ exchange and physiopathological properties of immunoglobulin (Ig)G4 antibodies.
Nevertheless, several pieces of evidence suggest a pathogenic role for IgG4 antibodies, including the correlation between serum IgG4 and disease severity 2, the observation of IgG4 immune complexes in IgG4-related tubulointerstitial nephritis 69 and the possibility of complement activation through the lectin pathway 70. ANCA of the IgG4 subclass have been demonstrated to bind Fc-gamma receptors and to contribute (at least in vitro) to neutrophil activation in ANCA-associated vasculitides 71,72. IgG4 antibodies directed against desmoglein 1 and the metalloproteinase ADAMTS13 have been also implicated in the pathogenesis of pemphigus vulgaris and thrombotic thrombocytopenic purpura, respectively 73,74. Furthermore, IgG4 as well as IgE might, theoretically, bind to Fc receptors on macrophages and eosinophils, foster internalization of extracellular antigens and facilitate their presentation to CD4+ T lymphocytes 75. Finally, recombinant monoclonal IgG4 antibodies cloned from circulating plasmablasts of patients with active IgG4-RD demonstrate self-reactivity in immunofluorescence and enzyme-linked immunosorbent assay (ELISA) experiments on Hep2 and HeLA cells, respectively, even though no definitive antigen specificity has been identified to date 77. The identification of putative target antigens will improve our understanding of the pathogenic role of IgG4 antibodies in IgG4-RD.
Putative microbial and self-antigens
Putative autoantigens have been proposed as targets of an antibody response in a proportion of patients with IgG4-related AIP and sialoadenitis 76–83. These antigens include proteins expressed in duct epithelia (carbonic anhydrase II and IV) and in acinar cells (lactoferrin, amylase-α-2A, pancreatic trypsinogens and pancreatic secretory trypsin inhibitor). However, these autoantigens are expressed only in a minority of organs potentially involved by IgG4-RD, and have been shown to induce autoantibodies in different autoimmune disorders 79, thus lacking adequate specificity for IgG4-RD. Indeed, these antigens might simply represent a preferential target during inflammatory processes in general, and their pathogenic role has yet to be proved. Molecular mimicry between Helicobacter pylori and pancreatic self-proteins has been also proposed as an additional pathogenic mechanism in IgG4-related AIP 84, but these results have not yet been replicated and must be interpreted cautiously.
B lymphocytes
The B cell compartment of patients with IgG4-RD has been studied extensively, because IgG4 elevation in the serum and the abundance of IgG4+ plasma cells in the biopsies initially suggested an underlying lymphoproliferative condition. Moreover, B cell depletion therapy has been shown recently to induce a prompt clinical improvement in patients with IgG4-RD, supporting a central pathogenic role of B lymphocytes in this fibrotic condition 6–8.
However, although IgG4-RD has been associated with an increased risk of malignant lymphoid transformation, immunohistochemistry and in-situ hybridization for kappa-lambda light chains restriction failed to identify monoclonal plasma cells populations in the affected tissues 3,4,13. On the contrary, cerebrospinal fluid analysis of subjects with IgG4-related pachymeningitis revealed the presence of oligoclonal IgG4 bands, suggesting an antigen-driven immune response 85,86. Indeed, next-generation sequencing analysis on biopsy samples and on peripheral blood of IgG4-RD patients demonstrated an oligoclonal expansion of somatically hypermutated IgG4+ B cell clones, further supporting antigen-specific affinity maturation 76,87.
The expanded B cell clones detected on peripheral blood correspond to a population of circulating plasmablasts, identified by flow cytometry as CD19+CD20− CD27+CD38+ cells. Circulating plasmablasts are not distinguishable from small-sized lymphocytes on blood smear, and arise classically in germinal centres after affinity maturation from CD20+ naive precursors. Finally, after circulating into the bloodstream, plasmablasts home to inflammatory niches or to the bone marrow, where they differentiate into antibody-secreting short- or long-lived plasma cells 88. Plasmablasts can circulate for prolonged periods in the setting of chronic antigenic stimulation or autoimmune diseases, but are generally observed in only low concentrations in the peripheral blood of healthy individuals 89–94. Of note, plasmablasts expanded in IgG4-RD patients decrease sharply after RTX-induced remission and re-emerge during relapse, thus correlating tightly with disease activity. Interestingly, re-emerging plasmablasts express a distinct V–J repertoire from that of the clones that dominated at the time of initial presentation (‘clonal divergence’) 76, indicating repeated rounds of mutation and selection driven by a specific antigen. Recent evidence raises the possibility that plasmablasts expanded in IgG4-RD patients might belong to a subset of regulatory B cells, capable of secreting IgG4 antibodies and IL-10 in response to chronic antigen stimulation 95,96. However, although plasmablasts share some surface markers with regulatory B cells, functional assays for IL-10 production failed to confirm this hypothesis, and expansion of regulatory B cells in the peripheral blood of patients with IgG4-RD has not been confirmed 76,95.
Activated IgG4+ B cells and plasmablasts could contribute to the pathogenesis of IgG4-RD either directly through autoantibody production or indirectly through the activation of pathogenic CD4+ T cells, presumably serving as effective antigen-presenting cells. Indeed, the high degree of somatic hypermutation and the ‘clonal divergence’ seen in IgG4+ plasmablasts also suggest extensive T helper cell-dependent processes at different stages of disease. In theory, B lymphocytes might also directly sustain extracellular matrix deposition through myofibroblast activation, the production of profibrotic cytokines [e.g. IL-6 and transforming growth factor (TGF)-β] and the secretion of stimulatory autoantibodies 8,97–99, but these hypotheses have never been assessed in IgG4-RD.
T lymphocytes
IgG4-RD has been considered a Th2/T regulatory-driven condition for quite a long time. Indeed, the presence of a dense fibrotic tissue and of abundant IgG4+ plasma cells are consistent with an underlying ‘modified Th2 immune-response’, which is associated classically with the production of both Th2 (e.g. IL-4 and IL-13) and T regulatory cytokines (e.g. TGF-β and IL-10) 100,101. In this scenario, IL-13 and TGF-β are thought to drive the deposition of extracellular matrix by activated fibroblasts, while IL-4 and IL-10 are considered the major inducer of IgG4 class-switch in naive B lymphocytes 100,101. The presence of allergic symptoms, peripheral blood eosinophilia, serum IgE and IgG4 elevation in many patients at diagnosis further supports this hypothesis 10.
However, accurate analysis of circulating T cells for Th1/Th2/T regulatory polarization has led to conflicting results, and showed an expansion of Th2 memory CD4+ T cells only in IgG4-RD patients with a concomitant history of atopy 102,103. Similarly, molecular and immunohistochemical analyses of IgG4-RD lesions identified variable amounts of Th1, Th2 and T regulatory cytokines, but failed to identify the exact cellular source of these molecules 100. Therefore, direct evidence for a role of Th1/Th2/T regulatory cells in IgG4-RD pathogenesis is still lacking. Indeed, this would require the documentation of Th1/Th2/T regulatory cytokines within T cells that infiltrate affected tissues.
T cells might also contribute to IgG4-RD pathogenesis through a dysregulated follicular T helper cell activity. For instance, altered expression of IL-21 by follicular T helper cells has been associated with autoantibody production in other autoimmune diseases 104, and elevated IL-21 messenger RNA expression has been linked recently to ectopic germinal centres within lacrymal and salivary glands from patients with IgG4-RD 105. It is conceivable that memory and follicular CD4+ T helper cell-dependent processes orchestrate IgG4-RD both by secreting profibrotic cytokines and by inducing IgG4 class-switch, plasmablast expansion and production of autoantibodies. These hypotheses require further investigation.
The lesson from B cell depletion
As reported previously, treatment with anti-CD20 monoclonal antibody induces a prompt clinical response in patients with IgG4-RD and a drastic reduction in the number of circulating oligoclonally expanded plasmablasts 54,76. Plasmablasts do not express CD20, the molecular target of RTX and, thus, they are likely to decrease in the peripheral blood because of failure to replete CD20+ precursors. These data support a potential pathogenic role of IgG4+ B lymphocytes (or B cells in general), and possibly IgG4 immunoglobulins, but the precise mechanisms through which RTX could affect putative T cell-mediated profibrotic processes in IgG4-RD remain elusive. Indeed, the encouraging results obtained recently with RTX in IgG4-RD, as well as in other fibrotic conditions and T cell-driven disorders (such as systemic sclerosis, interstitial lung disease and multiple sclerosis) have expanded the overly simplistic view about B cells as simply precursors of antibody-producing plasma cells 8,106–108. B lymphocytes might act as functional antigen-presenting cells to Th2 effector cells, maintain CD4+ memory T cells by providing antigen-independent factors and promote the proliferation of pathogenic T cells 109–111. Thus, apart from depleting CD20+ B cells, RTX is likely to interfere with more important B cell/T cell cross-talk processes through the elimination of a major B-cell type required for continuous antigen presentation to T cells and for the maintenance of T cell activation. In this sense, it is tempting to speculate that B cell depletion in IgG4-RD ultimately abrogates the secretion of profibrotic cytokines (e.g. IL-4, IL-13, IL-10 and others) by pathogenic T cell populations.
Macrophages
Macrophages are tissue-resident monocytes specialized in phagocytosis and initiation of innate immunity. Two subtypes of macrophages have been identified to date: the classically activated (M1) macrophages, stimulated by Th1 responses, and the alternatively activated (M2) macrophages, induced by Th2 derived IL-4 and IL-13, as well as T regulatory-derived IL-10 112. Alternatively activated macrophages have been implicated in the pathogenesis of IgG4-RD because they are known to contribute to angiogenesis, immunomodulation, wound-healing and fibrosis by secreting a variety of molecules, including profibrotic factors such as TGF-β and platelet-derived growth factor (PDGFP) 113. A direct profibrotic role has been suggested by Furukawa, who showed a correlation between CD163+ M2 macrophages infiltration and the amount of tissue fibrosis in biopsies of patients with Mikulicz’s disease 114. Alternatively, Watanabe demonstrated that macrophages from patients with IgG4-related AIP induce IgG4 production by B cells upon stimulation of Toll-like receptors (TLRs) and nucleotide-binding oligomerization domain receptors (NOD)-like receptors 115. IgG4 production was found to be dependent upon B cell-activating factor (BAFF) secretion by activated monocytes in that study 115. Although more robust evidence is needed in order to drive definitive conclusions, these preliminary results suggest that macrophages might be important in the pathogenesis of IgG4-RD.
Basophils
Basophils, the least common of the circulating granulocytes, are known to be involved in allergic inflammatory reactions as well as in immune responses to parasitic infections. Indeed, basophils express IgE receptors on their surface, function as antigen-presenting cells in Th2 responses and release large amounts of different soluble mediators in response to allergen proteins or helminth antigens, including histamine, heparin and proteolytic enzymes 116. Moreover, basophils are considered the most important source of IL-4 together with T cells 117,118. The role of basophils in IgG4-RD remains largely unexplored. A single study demonstrated that activation of TLRs expressed on basophils from patients with IgG4-RD induced IgG4 production by B cells from healthy controls. As reported for monocytes in a previous study, enhanced IgG4 production was associated with BAFF and IL-13 production by activated basophils 119. These data suggest that TLR-mediated innate immune responses may play a role in the development of IgG4-RD by inducing T cell-independent IgG4 responses.
Eosinophils
Tissue eosinophilia is a histological hallmark of IgG4-RD and peripheral blood eosinophilia has been described in approximately 30% of patients with IgG4-RD 10. This information, together with the common findings of atopy and elevated serum IgE levels in a proportion of patients, led to the premature conclusion that allergic mechanisms drive IgG4-RD 2. However, eosinophilia and IgE elevation have also been observed in non-atopic subjects, and are probably induced by processes inherent to IgG4-RD itself (e.g. Th2 cytokines, such as IL-4 and IL-5) rather than atopy per se 10,102). Either way, activated eosinophils might contribute to IgG4-RD by promoting fibrosis through the production of TGF-β, PDGF and IL-13 120 by sustaining IgG4+ plasma cell survival in inflammatory niches 88 or by up-regulating class II major histocompatibility complex (MHC) and presenting antigens to CD4+ T cells 75. These hypotheses have never been investigated to date, and the pathogenic role of eosinophils in IgG4-RD remains controversial.
Possible pathogenic model
Experimental evidence supports a pathogenic model of IgG4-RD in which B cell/T cell collaboration orchestrates a chronic, self-perpetuating immune response against a specific antigen (whether microbial, environmental or self). In this scenario, T helper cell polarization is probably determined by signals from the innate immune system, and IgG4 class-switch is aimed to dampen chronic inflammation (Fig. 5). A crucial step in the pathogenesis of IgG4-RD might, therefore, be naive T cell activation following antigen presentation by cognate antigen-specific naive or memory B cells, eosinophils or macrophages. Once activated, putative pathogenic T helper and T regulatory cells are thought to produce an inflammatory cytokine milieu that includes IFN-γ, IL-4, IL-10, IL-5, IL-13 and TGF-β. IL-4 and IL-10 may drive preferential class-switch of antigen-specific B cells to IgG4 and IgE, and induce their expansion. IL-5, IL-13 and TGF-β could lead to the activation of eosinophils, fibroblasts, and alternatively activated macrophages. Activated antigen-specific T cells may, in turn, facilitate germinal centre formation and recruitment of increasing numbers of somatically hypermutated B cell clones, thereby setting up a vicious cycle of mutual activation between B and T lymphocytes. In this sense, B cell depletion might eliminate a major cell type required for antigen presentation to T cells, leading to loss of activated T cells, reduced production of profibrotic cytokines and (ultimately) abrogation of collagen secretion by myofibroblasts. RTX might also target the precursors of short-lived plasma cells, thus leading to a rapid decline of serum IgG4 levels. In turn, disease relapse might be due to CD20– memory B cells that survive RTX therapy, re-present antigens to pathogenic T cells and generate re-emerging plasmablasts. Disease relapse ultimately corresponds to a renewed extracellular matrix deposition by activated fibroblasts 8.
Figure 5.
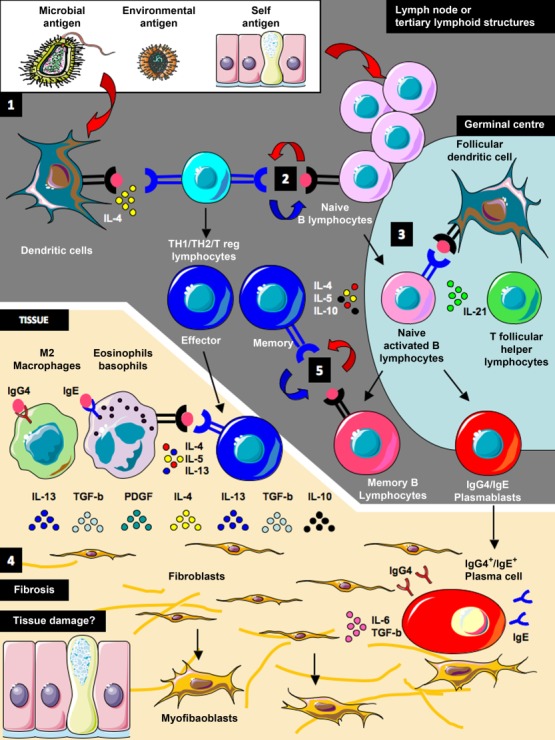
Pathogenic model for immunoglobulin (Ig)G4-related disease. Antigen presentation, T cell and B cell activation presumably occur in lymph nodes or tertiary lymphoid structures originating in inflamed tissues (1). Dendritic and naive/memory B cells might present antigens to CD4+ T lymphocytes triggering their activation (2). Local signals from the innate immune system might determine T helper cell polarization and differentiation into effector or memory T cells. Activated naive B cells migrate to the germinal centre where they undergo somatic hypermutation and affinity maturation, and differentiate into memory B cells or plasmablasts (3). IgG4/IgE class-switch probably occurs under the influence of cytokines produced by activated CD4+ Th2 or T regulatory cells. Effector CD4+ T cells migrate to inflamed tissues, where they are thought to drive the fibroinflammatory process by producing a variety of profibrotic cytokines [such as interleukin (IL)-4, IL-10, IL-13, transforming growth factor (TGF)-β], and by inducing M2 macrophages differentiation and eosinophil activation (4). IgG4 antibodies might have an antinflammatory role but, together with IgE, they might also facilitate antigen capture by innate immune cells through Fc receptor binding and presentation to T cells. In theory, IgG4+ plasma cells in the tissue might also have a profibrotic role through the production of IL-6 (4). All these concomitant events lead ultimately to fibroblast activation, generation of myofibroblasts and extracellular matrix deposition. Whether the fibroinflammatory infiltrate that occurs in IgG4-RD lesions also causes tissue damage and further generation of self-antigens is not known (4). By depleting CD20+ precursors, rituximab might eliminate both short-lived plasma cells and a major B cell type required for antigen presentation to T cells. This, in turn, leads to loss of activated T cells and profibrotic cytokines, and to a reduction in fibroblast activation. Disease relapse corresponds to a new plasmablast expansion, and to a renewed extracellular matrix deposition. Whether re-emerging plasmablasts observed at disease relapse differentiate de novo from naive B cells or from CD20- memory B cells that survive rituximab therapy is not known (5). Indeed memory B cells might de-novo present antigens to pathogenic naive T cells, sustaining disease recurrence (5).
Conclusion
IgG4-RD remains an often-overlooked clinical entity, but awareness of this new fibroinflammatory condition is increasing. In the present work we have reviewed the most recent advances pertaining to the immunopathology of IgG4-RD, and describe how the innate and the adaptive immune system might synergize to ultimately drive the characteristic fibrotic alterations observed in the affected tissues. In the last decade, substantial work has begun to dissect the mechanisms of B cell/T cell collaboration leading to aberrant IgG4 production and to tissue fibrosis. More recently, seminal work has highlighted a possible pathogenic role of macrophages and basophils, thus further complicating the oversimplified view about IgG4-RD as a B cell-/T cell-mediated disorder. Understanding the immune dysregulation that occurs in IgG4-RD will foster our knowledge about the human immune system in general and about potential therapeutic approaches for other fibrotic conditions.
Acknowledgments
The authors thank Prof. John Stone (Rheumatology Unit - Massachusetts General Hospital) for helpful input and review of the manuscript. This work was supported in part by a grant from the “Fondazione Italiana per la Ricerca sull’ Artrite” (FIRA Onlus 2014).
Footnotes
Disclosure
The authors declare no conflicts of interest.
References
- Hamano H, Kawa S, Horiuchi A. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J Med. 2011;344:732–8. doi: 10.1056/NEJM200103083441005. [DOI] [PubMed] [Google Scholar]
- Stone JH, Zen Y, Deshpande V, et al. IgG4-related disease. N Engl J Med. 2012;366:539–51. doi: 10.1056/NEJMra1104650. [DOI] [PubMed] [Google Scholar]
- Deshpande V, Zen Y, Chan JK. Consensus statement on the pathology of IgG4-related disease. Mod Pathol. 2012;25:1181–92. doi: 10.1038/modpathol.2012.72. [DOI] [PubMed] [Google Scholar]
- Umehara H, Okazaki K, Masaki Y, et al. Comprehensive diagnostic criteria for IgG4-related disease (IgG4-RD), 2011. Mod Rheumatol. 2012;22:21–30. doi: 10.1007/s10165-011-0571-z. [DOI] [PubMed] [Google Scholar]
- Kanno A, Nishimori I, Masamune Aetal, et al. Nationwide epidemiological survey of autoimmune pancreatitis in Japan. Pancreas. 2012;41:835–9. doi: 10.1097/MPA.0b013e3182480c99. [DOI] [PubMed] [Google Scholar]
- Carruthers MN, Topazian MD, Khosroshahi A, et al. Rituximab for IgG4-related disease: a prospective, open-label trial. Ann Rheum Dis. 2015 doi: 10.1136/annrheumdis-2014-206605. 74:1171–7. [DOI] [PubMed] [Google Scholar]
- Khosroshahi A, Bloch DB, Deshpande V, Stone JH. Rituximab therapy leads to rapid decline of serum IgG4 levels and prompt clinical improvement in IgG4-related systemic disease. Arthritis Rheum. 2010;62:1755–62. doi: 10.1002/art.27435. [DOI] [PubMed] [Google Scholar]
- Della-Torre E, Feeney E, Deshpande V, et al. B-cell depletion attenuates serological biomarkers of fibrosis and myofibroblast activation in IgG4-related disease. Ann Rheum Dis. 2014 doi: 10.1136/annrheumdis-2014-205799. doi: 10.1136/annrheumdis-2014-205799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griepentrog GJ, Vickers RW, Karesh JW, Azari AA, Albert DM, Bukat CN. A clinicopathologic case study of two patients with pediatric orbital IgG4-related disease. Orbit. 2013;32:389–91. doi: 10.3109/01676830.2013.822899. [DOI] [PubMed] [Google Scholar]
- Della-Torre E, Mattoo H, Mahajan VS, Carruthers M, Pillai S, Stone JH. Prevalence of atopy, eosinophilia, and IgE elevation in IgG4-related disease. Allergy. 2014;69:269–72. doi: 10.1111/all.12320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue D, Zen Y, Abo H. Immunoglobulin G4-related lung disease: CT findings with pathologic correlations. Radiology. 2009;251:260–70. doi: 10.1148/radiol.2511080965. [DOI] [PubMed] [Google Scholar]
- Khan ML, Colby TV, Viggiano RW, Fonseca R, et al. Treatment with bortezomib of a patient having hyper IgG4 disease. Clin Lymphoma Myeloma Leuk. 2010;10:217–9. doi: 10.3816/CLML.2010.n.034. [DOI] [PubMed] [Google Scholar]
- Cheuk W, Chan JK. Lymphadenopathy of IgG4-related disease: an underdiagnosed and overdiagnosed entity. Semin Diagn Pathol. 2012;29:226–34. doi: 10.1053/j.semdp.2012.07.001. [DOI] [PubMed] [Google Scholar]
- Leporati P, Landek-Salgado MA, Lupi I, Chiovato L, Caturegli P. IgG4-related hypophysitis: a new addition to the hypophysitis spectrum. J Clin Endocrinol Metab. 2011;96:1971–80. doi: 10.1210/jc.2010-2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu LX, Della-Torre E, Stone JH, Clark SW. IgG4-related hypertrophic pachymeningitis: clinical features, diagnostic criteria, and treatment. JAMA Neurol. 2014;71:785–93. doi: 10.1001/jamaneurol.2014.243. [DOI] [PubMed] [Google Scholar]
- Carruthers R, Carruthers M, Della-Torre E. IgG4-related disease and other causes of inflammatory meningeal disease. Semin Neurol. 2014;34:395–404. doi: 10.1055/s-0034-1390388. [DOI] [PubMed] [Google Scholar]
- Reder L, Della-Torre E, Stone JH, Mori M, Song P. Clinical manifestations of IgG4-related disease in the pharynx: case series and review of the literature. Ann Otol Rhinol Laryngol. 2014;124:173–8. doi: 10.1177/0003489414549574. [DOI] [PubMed] [Google Scholar]
- Tokura Y, Yagi H, Yanaguchi H. IgG4-related skin disease. Br J Dermatol. 2014;171:959–67. doi: 10.1111/bjd.13296. [DOI] [PubMed] [Google Scholar]
- Dahlgren M, Khosroshahi A, Nielsen GP, Deshpande V, Stone JH, et al. Riedel’s thyroiditis and multifocal fibrosclerosis are part of the IgG4-related systemic disease spectrum. Arthritis Care Res (Hoboken) 2010;62:1312–8. doi: 10.1002/acr.20215. [DOI] [PubMed] [Google Scholar]
- Stone JH, Khosroshahi A, Deshpande V. Recommendations for the nomenclature of IgG4-related disease and its individual organ system manifestations. Arthritis Rheum. 2012;64:3061–7. doi: 10.1002/art.34593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimosegawa T, Chari ST, Frulloni L, et al. International consensus diagnostic criteria for autoimmune pancreatitis: guidelines of the International Association of Pancreatology. Pancreas. 2011;40:352–8. doi: 10.1097/MPA.0b013e3182142fd2. [DOI] [PubMed] [Google Scholar]
- Kamisawa T, Takuma K, Egawa N, Tsuruta K, Sasaki T, et al. Autoimmune pancreatitis and IgG4-related sclerosing disease. Nat Rev Gastroenterol Hepatol. 2010;7:401–9. doi: 10.1038/nrgastro.2010.81. [DOI] [PubMed] [Google Scholar]
- Pezzilli R, Vecchiarelli S, Di Marco MC. Pancreatic ductal adenocarcinoma associated with autoimmune pancreatitis. Case Rep Gastroenterol. 2011;5:378–85. doi: 10.1159/000330291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta R, Khosroshahi A, Shinagare S, et al. Does autoimmune pancreatitis increase the risk of pancreatic carcinoma?: a retrospective analysis of pancreatic resections. Pancreas. 2013;42:506–10. doi: 10.1097/MPA.0b013e31826bef91. [DOI] [PubMed] [Google Scholar]
- Ghazale A, Chari ST, Zhang L, et al. Immunoglobulin G4-associated cholangitis: clinical profile and response to therapy. Gastroenterology. 2008;134:706–15. doi: 10.1053/j.gastro.2007.12.009. [DOI] [PubMed] [Google Scholar]
- Joshi D, Webster GJ, et al. Biliary and hepatic involvement in IgG4-related disease. Aliment Pharmacol Ther. 2014;40:1251–61. doi: 10.1111/apt.12988. [DOI] [PubMed] [Google Scholar]
- Khosroshahi A, Carruthers MN, Stone JH. Rethinking Ormond’s disease: ‘idiopathic’ retroperitoneal fi brosis in the era of IgG4-related disease. Medicine (Balt) 2013;92:82–91. doi: 10.1097/MD.0b013e318289610f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zen Y, Onodera M, Inoue D, et al. Retroperitoneal fibrosis: a clinicopathologic study with respect to immunoglobulin G4. Am J Surg Pathol. 2009;33:1833–9. doi: 10.1097/pas.0b013e3181b72882. [DOI] [PubMed] [Google Scholar]
- Zen Y, Kasashima S, Inoue D, et al. Retroperitoneal and aortic manifestations of immunoglobulin G4-related disease. Semin Diagn Pathol. 2012;29:212–18. doi: 10.1053/j.semdp.2012.07.003. [DOI] [PubMed] [Google Scholar]
- Stone JR. Aortitis, periaortitis, and retroperitoneal fibrosis, as manifestations of IgG4-related systemic disease. Curr Opin Rheumatol. 2011;23:88–94. doi: 10.1097/BOR.0b013e3283412f7c. [DOI] [PubMed] [Google Scholar]
- Wallace ZS, Deshpande V, Stone JH. Ophthalmic manifestations of IgG4-related disease: single-center experience and literature review. Semin Arthritis Rheum. 2013;43:806–17. doi: 10.1016/j.semarthrit.2013.11.008. [DOI] [PubMed] [Google Scholar]
- McNab AA, McKelvie P. IgG4-related ophthalmic disease. Part I: background and pathology. Ophthal Plast Reconstr Surg. 2015;31:83–8. doi: 10.1097/IOP.0000000000000363. [DOI] [PubMed] [Google Scholar]
- McNab AA, McKelvie P. IgG4-related ophthalmic disease. Part II: clinical aspects. Ophthal Plast Reconstr Surg. 2015;31:167–78. doi: 10.1097/IOP.0000000000000364. [DOI] [PubMed] [Google Scholar]
- Geyer JT, Ferry JA, Harris NL. Chronic sclerosing sialadenitis (Kuttner tumor) is an IgG4-associated disease. Am J Surg Pathol. 2010;34:202–10. doi: 10.1097/PAS.0b013e3181c811ad. [DOI] [PubMed] [Google Scholar]
- Himi T, Takano K, Yamamoto M, Naishiro Y, Takahashi H, et al. A novel concept of Mikulicz’s disease as IgG4-related disease. Auris Nasus Larynx. 2012;39:9–17. doi: 10.1016/j.anl.2011.01.023. [DOI] [PubMed] [Google Scholar]
- Della-Torre E, Mattoo H, Mahajan VS. IgG4-related midline destructive lesion. Ann Rheum Dis. 2014;73:1434–6. doi: 10.1136/annrheumdis-2014-205187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornell LD, et al. IgG4-related kidney disease. Semin Diagn Pathol. 2012;29:245–50. doi: 10.1053/j.semdp.2012.07.004. [DOI] [PubMed] [Google Scholar]
- Kawano M, Mizushima I, Yamaguchi Y. Immunohistochemical characteristics of IgG4-related tubulointerstitial nephritis: detailed analysis of 20 Japanese cases. Int J Rheumatol. 2012;2012:609795. doi: 10.1155/2012/609795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawano M, Saeki T, Nakashima H, et al. Proposal for diagnostic criteria for IgG4-related kidney disease. Clin Exp Nephrol. 2011;15:615–26. doi: 10.1007/s10157-011-0521-2. [DOI] [PubMed] [Google Scholar]
- Kajander H, Paavonen T, Valo T, Tarkka M, Mennander AA, et al. Immunoglobulin G4-positive ascending thoracic aortitis may be prone to dissection. J Thorac Cardiovasc Surg. 2012;146::1449–55. doi: 10.1016/j.jtcvs.2012.09.039. [DOI] [PubMed] [Google Scholar]
- Holmes BJ, Delev ND, Pasternack GR, Halushka MK. Novel cause of sudden cardiac death: IgG4-related disease. Circulation. 2012;125:2956–7. doi: 10.1161/CIRCULATIONAHA.111.061002. [DOI] [PubMed] [Google Scholar]
- Inoue D, Zen Y, Sato Y. IgG4-related perineural disease. Int J Rheumatol. 2012;2012:401890. doi: 10.1155/2012/401890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regev K, Nussbaum T, Cagnano E, Giladi N, Karni A, et al. Central nervous system manifestation of IgG4-related disease. JAMA Neurol. 2014;71:767–70. doi: 10.1001/jamaneurol.2014.40. [DOI] [PubMed] [Google Scholar]
- Buijs J, Maillette de Buy Wenniger L, van Leenders G. Immunoglobulin G4-related prostatitis: a case–control study focusing on clinical and pathologic characteristics. Urology. 2014;83:521–6. doi: 10.1016/j.urology.2013.10.052. [DOI] [PubMed] [Google Scholar]
- Bösmüller H, von Weyhern CH, Adam P, Alibegovic V, Mikuz G, Fend F, et al. Paratesticular fibrous pseudotumor–an IgG4-related disorder? Virchows Arch. 2011;458:109–13. doi: 10.1007/s00428-010-0995-4. [DOI] [PubMed] [Google Scholar]
- Peikert T, Shrestha B, Aubry MC. Histopathologic overlap between fibrosing mediastinitis and IgG4-related disease. Int J Rheumatol. 2012;;2012:207056. doi: 10.1155/2012/207056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvarani C, Valli R, Boiardi L, Pipitone N, Nicoli F, Muratore F, et al. IgG4-associated sclerosing mesenteritis. Clin Exp Rheumatol. 2011;29:S79–80. [PubMed] [Google Scholar]
- Bittencourt AG1, Pereira LV, Cabral F, Jr, Halang Fde S, Gonçalves Mde C, Bento RF. IgG4-related sclerosing disease of the temporal bone. Otol Neurotol. 2013;34:e20–1. doi: 10.1097/MAO.0b013e31827f1948. [DOI] [PubMed] [Google Scholar]
- Koizumi S, Kamisawa T, Kuruma S. Immunoglobulin G4-related gastrointestinal diseases, are they immunoglobulin G4-related diseases? World J Gastroenterol. 2013;19:5769–74. doi: 10.3748/wjg.v19.i35.5769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chougule A, Bal A, Das A, Singh G, et al. IgG4 related sclerosing mastitis: expanding the morphological spectrum of IgG4 related diseases. Pathology. 2015;47:27–33. doi: 10.1097/PAT.0000000000000187. [DOI] [PubMed] [Google Scholar]
- Carruthers MN, Khosroshahi A, Augustin T, Deshpande V, Stone JH. The diagnostic utility of serum IgG4 concentrations in IgG4-related disease. Ann Rheum Dis. 2014;74:14–8. doi: 10.1136/annrheumdis-2013-204907. [DOI] [PubMed] [Google Scholar]
- Della-Torre E, Galli L, Franciotta D. Diagnostic value of IgG4 indices in IgG4-related hypertrophic pachymeningitis. J Neuroimmunol. 2013;266:82–6. doi: 10.1016/j.jneuroim.2013.10.008. [DOI] [PubMed] [Google Scholar]
- Wong PC, Fung AT, Gerrie AS, et al. IgG4-related disease with hypergammaglobulinemic hyperviscosity and retinopathy. Eur J Haematol. 2013;90:250–6. doi: 10.1111/ejh.12059. [DOI] [PubMed] [Google Scholar]
- Wallace ZS, Mattoo H, Carruthers M, et al. Plasmablasts as a biomarker for IgG4-related disease, independent of serum IgG4 concentrations. Ann Rheum Dis. 2015;74:190–5. doi: 10.1136/annrheumdis-2014-205233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebbo M, Grados A, Guedj E, et al. Usefulness of 2-[18F]-fluoro-2-deoxy-D-glucose-positron emission tomography/computed tomography for staging and evaluation of treatment response in IgG4-related disease: a retrospective multicenter study. Arthritis Care Res (Hoboken) 2014;66:86–96. doi: 10.1002/acr.22058. [DOI] [PubMed] [Google Scholar]
- Deshpande V, Khosroshahi A, Nielsen GP, Hamilos DL, Stone JH, et al. Eosinophilic angiocentric fibrosis is a form of IgG4-related systemic disease. Am J Surg Pathol. 2011;35:701–6. doi: 10.1097/PAS.0b013e318213889e. [DOI] [PubMed] [Google Scholar]
- Carruthers MN, Stone JH, Deshpande V, Khosroshahi A. Development of an IgG4-RD responder index. Int J Rheumatol. 2012;;2012:259408. doi: 10.1155/2012/259408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khosroshahi A, Wallace ZS, Crowe J, et al. International consensus guidance statement on the treatment of IgG4-related disease. 2015. Arthritis Rheumatol; doi: 10.1002/art.39132. [DOI] [PubMed]
- Khosroshahi A, Stone JH. Treatment approaches to IgG4-related systemic disease. Curr Opin Rheumatol. 2011;23:67–71. doi: 10.1097/BOR.0b013e328341a240. [DOI] [PubMed] [Google Scholar]
- Della-Torre E, Campochiaro C, Bozzolo EP, et al. Methotrexate for maintenance of remission in IgG4-related disease. Rheumatology. 2015 doi: 10.1093/rheumatology/kev244. in press. [DOI] [PubMed] [Google Scholar]
- Tsuboi H, Matsuo N, Iizuka M. Analysis of IgG4 class switch-related molecules in IgG4-related disease. Arthritis Res Ther. 2012;14:R171. doi: 10.1186/ar3924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King C, Tangye SG, Mackay CR, et al. T follicular helper (TFH) cells in normal and dysregulated immune responses. Annu Rev Immunol. 2008;26:741–66. doi: 10.1146/annurev.immunol.26.021607.090344. [DOI] [PubMed] [Google Scholar]
- Aalberse RC, Stapel SO, Schuurman J, Rispens T. Immunoglobulin G4: an odd antibody. Clin Exp Allergy. 2009;39:469–77. doi: 10.1111/j.1365-2222.2009.03207.x. [DOI] [PubMed] [Google Scholar]
- Rispens T, Ooijevaar–de Heer P, Bende O, Aalberse RC. Mechanism of immunoglobulin G4 Fab-arm exchange. J Am Chem Soc. 2011;133:10302–11. doi: 10.1021/ja203638y. [DOI] [PubMed] [Google Scholar]
- van der Neut Kolfschoten M, Schuurman J, Losen M. Anti-inflammatory activity of human IgG4 antibodies by dynamic Fab arm exchange. Science. 2007;317:1554–57. doi: 10.1126/science.1144603. [DOI] [PubMed] [Google Scholar]
- Aalberse RC, Schuurman J, et al. IgG4 breaking the rules. Immunology. 2002;105:9–19. doi: 10.1046/j.0019-2805.2001.01341.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bindon CI, Hale G, Bruggemann M, Waldmann H. Human monoclonal IgG isotypes differ in complement activating function at the level of C4 as well as C1q. J Exp Med. 1988;168:127–42. doi: 10.1084/jem.168.1.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aalberse RC, Van Milligen F, Tan KY, Stapel SO. Allergen-specific IgG4 in atopic disease. Allergy. 1993;48:559–69. doi: 10.1111/j.1398-9995.1993.tb00749.x. [DOI] [PubMed] [Google Scholar]
- Durham SR, Emminger W, Kapp A. Long-term clinical efficacy in grass pollen–induced rhinoconjunctivitis after treatment with SQ-standardized grass allergy immunotherapy tablet. J Allergy Clin Immunol. 2010;125:131–8. doi: 10.1016/j.jaci.2009.10.035. [DOI] [PubMed] [Google Scholar]
- Muraki T, Hamano H, Ochi Y, et al. Autoimmune pancreatitis and complement activation system. Pancreas. 2006;32:16–21. doi: 10.1097/01.mpa.0000188308.75043.e4. [DOI] [PubMed] [Google Scholar]
- Holland M, Hewins P, Goodall M, Adu D, Jefferis R, Savage CO, et al. Anti-neutrophil cytoplasm antibody IgG subclasses inWegener’s granulomatosis: a possible pathogenic role for the IgG4 subclass. Clin Exp Immunol. 2004;138:183–92. doi: 10.1111/j.1365-2249.2004.02566.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain A, Pankhurst T, Goodall M. Chimeric IgG4 PR3-ANCA induces selective inflammatory responses from neutrophils through engagement of Fcγ receptors. Immunology. 2009;128:236–44. doi: 10.1111/j.1365-2567.2009.03108.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari S, Mudde GC, Rieger M, et al. IgG subclass distribution of anti-ADAMTS13 antibodies in patients with acquired thrombotic thrombocytopenic purpura. J Thromb Haemost. 2009;7:1703–10. doi: 10.1111/j.1538-7836.2009.03568.x. [DOI] [PubMed] [Google Scholar]
- Rock B, Martins CR, Theofilopoulos AN, et al. The pathogenic effect of IgG4 autoantibodies in endemic pemphigus foliaceus (fogo selvagem) N Engl J Med. 1989;320:1463–9. doi: 10.1056/NEJM198906013202206. [DOI] [PubMed] [Google Scholar]
- Padigel UM, Hess JA, Lee JJetal, et al. Eosinophils act as antigen-presenting cells to induce immunity to Strongyloides stercoralis in mice. J Infect Dis. 2007;196:1844–51. doi: 10.1086/522968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattoo H, Mahajan VS, Della-Torre E. De novo oligoclonal expansions of circulating plasmablasts in active and relapsing IgG4-related disease. J Allergy Clin Immunol. 2014;134:679–87. doi: 10.1016/j.jaci.2014.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okazaki K, Uchida K, Ohana M, et al. Autoimmune-related pancreatitis is associated with autoantibodies and a Th1/Th2-type cellular immune response. Gastroenterology. 2000;118:573–81. doi: 10.1016/s0016-5085(00)70264-2. [DOI] [PubMed] [Google Scholar]
- Aoki S, Nakazawa T, Ohara H, et al. Immunohistochemical study of autoimmune pancreatitis using anti-IgG4 antibody and patients’ sera. Histopathology. 2005;47:147–58. doi: 10.1111/j.1365-2559.2005.02204.x. [DOI] [PubMed] [Google Scholar]
- Nishimori I, Miyaji E, Morimoto K, Nagao K, Kamada M, Onishi S, et al. Serum antibodies to carbonic anhydrase IV in patients with autoimmune pancreatitis. Gut. 2005;54:274–81. doi: 10.1136/gut.2004.049064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aparisi L, Farre A, Gomez-Cambronero L. Antibodies to carbonic anhydrase and IgG4 levels in idiopathic chronic pancreatitis: relevance for diagnosis of autoimmune pancreatitis. Gut. 2005;54:703–9. doi: 10.1136/gut.2004.047142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohr JM, Faissner R, Koczan D, et al. Autoantibodies against the exocrine pancreas in autoimmune pancreatitis: gene and protein expression profiling and immunoassays identify pancreatic enzymes as a major target of the inflammatory process. Am J Gastroenterol. 2010;105:2060–71. doi: 10.1038/ajg.2010.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo T, Takizawa S, Tanaka S, et al. Amylase α-2A autoantibodies: novel marker of autoimmune pancreatitis and fulminant type 1 diabetes. Diabetes. 2009;58:732–7. doi: 10.2337/db08-0493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asada M, Nishio A, Uchida K, et al. Identification of a novel autoantibody against pancreatic secretory trypsin inhibitor in patients with autoimmune pancreatitis. Pancreas. 2006;33:20–6. doi: 10.1097/01.mpa.0000226881.48204.fd. [DOI] [PubMed] [Google Scholar]
- Frulloni L, Lunardi C, Simone R, et al. et al. Identification of a novel antibody associated with autoimmune pancreatitis. N Engl J Med. 2009;361:2135–42. doi: 10.1056/NEJMoa0903068. ] [DOI] [PubMed] [Google Scholar]
- Della-Torre E, Passerini G, Furlan R. Cerebrospinal fluid analysis in immunoglobulin G4-related hypertrophic pachymeningitis. J Rheumatol. 2013;40:1927–9. doi: 10.3899/jrheum.130678. [DOI] [PubMed] [Google Scholar]
- Della-Torre E, Bozzolo EP, Passerini G, et al. IgG4-related pachymeningitis: evidence of intrathecal IgG4 on cerebrospinal fluid analysis. Ann Intern Med. 2012;156:401–3. doi: 10.7326/0003-4819-156-5-201203060-00025. [DOI] [PubMed] [Google Scholar]
- Maillette de Buy Wenniger LJ, Doorenspleet ME, Klarenbeek PL, et al. Immunoglobulin G4+ clones identified by next-generation sequencing dominate the B cell receptor repertoire in immunoglobulin G4 associated cholangitis. Hepatology. 2013;57:2390–8. doi: 10.1002/hep.26232. [DOI] [PubMed] [Google Scholar]
- Hiepe F, Dörner T, Hauser AE, Hoyer BF, Mei H, Radbruch A, et al. Long-lived autoreactive plasma cells drive persistent autoimmune inflammation. Nat Rev Rheumatol. 2011;7:170–8. doi: 10.1038/nrrheum.2011.1. [DOI] [PubMed] [Google Scholar]
- Harada Y, Kawano MM, Huang Netal. Identification of early plasma cells in peripheral blood and their clinical significance. Br J Haematol. 1996;92:184–91. doi: 10.1046/j.1365-2141.1996.300835.x. [DOI] [PubMed] [Google Scholar]
- Odendahl M, Jacobi A, Hansen A. Disturbed peripheral B lymphocyte homeostasis in systemic lupus erythematosus. J Immunol. 2000;165:5970–9. doi: 10.4049/jimmunol.165.10.5970. [DOI] [PubMed] [Google Scholar]
- Fink K, et al. Origin and function of circulating plasmablasts during acute viral infections. Front Immunol. 2012;3:1. doi: 10.3389/fimmu.2012.00078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobi AM, Odendahl M, Reiter K. Correlation between circulating CD27high plasma cells and disease activity in patients with systemic lupus erythematosus. Arthritis Rheum. 2003;48:1332–42. doi: 10.1002/art.10949. [DOI] [PubMed] [Google Scholar]
- Odendahl M, Keitzer R, Wahn Uetal, et al. Perturbations of peripheral B lymphocyte homoeostasis in children with systemic lupus erythematosus. Ann Rheum Dis. 2003;62:851–8. doi: 10.1136/ard.62.9.851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerkman PF, Rombouts Y, van der Voort EI. Circulating plasmablasts/plasmacells as a source of anticitrullinated protein antibodies in patients with rheumatoid arthritis. Ann Rheum Dis. 2013;72:1259–63. doi: 10.1136/annrheumdis-2012-202893. [DOI] [PubMed] [Google Scholar]
- van de Veen W, Stanic B, Yaman G, et al. IgG4 production is confined to human IL-10-producing regulatory B cells that suppress antigen-specific immune responses. J Allergy Clin Immunol. 2013;131:1204–12. doi: 10.1016/j.jaci.2013.01.014. [DOI] [PubMed] [Google Scholar]
- Sumimoto K, Uchida K, Kusuda T, et al. The role of CD19+ CD24high CD38high and CD19+ CD24high CD27+ regulatory B cells in patients with type 1 autoimmune pancreatitis. Pancreatology. 2014;14:193–200. doi: 10.1016/j.pan.2014.02.004. [DOI] [PubMed] [Google Scholar]
- François A, Chatelus E, Wachsmann D, et al. B lymphocytes and B-cell activating factor promote collagen and profibrotic markers expression by dermal fibroblasts in systemic sclerosis. Arthritis Res Ther. 2013;15:R168. doi: 10.1186/ar4352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lighaam LC, Aalberse RC, Rispens T, et al. IgG4-related fibrotic diseases from an immunological perspective: regulators out of control? Int J Rheumatol. 2012;;2012:789164. doi: 10.1155/2012/789164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato S, Hayakawa I, Hasegawa M, Fujimoto M, Takehara K. Function blocking autoantibodies against matrix metalloproteinase-1 in patients with systemic sclerosis. J Invest Dermatol. 2003;120:542–7. doi: 10.1046/j.1523-1747.2003.12097.x. [DOI] [PubMed] [Google Scholar]
- Mahajan VS, Mattoo H, Deshpande V. IgG4-related disease. Annu Rev Pathol. 2014;9:315–47. doi: 10.1146/annurev-pathol-012513-104708. [DOI] [PubMed] [Google Scholar]
- Wynn TA, et al. Fibrotic disease and the TH1/TH2 paradigm. Nat Rev Immunol. 2004;4:583–94. doi: 10.1038/nri1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattoo H, Della-Torre E, Mahajan VS, Stone JH, Pillai S. Circulating Th2 memory cells in IgG4-related disease are restricted to a defined subset of subjects with atopy. Allergy. 2014;69:399–402. doi: 10.1111/all.12342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi H, Uchida K, Taniguchi Tetal. Circulating naïve and CD4+CD25high regulatory T cells in patients with autoimmune pancreatitis. Pancreas. 2008;36:133–40. doi: 10.1097/MPA.0b013e3181577553. [DOI] [PubMed] [Google Scholar]
- King C, Tangye SG, Mackay CR. T follicular helper (TFH) cells in normal and dysregulated immune responses. Annu Rev Immunol. 2008;26:741–66. doi: 10.1146/annurev.immunol.26.021607.090344. [DOI] [PubMed] [Google Scholar]
- Maehara T, Moriyama M, Nakashima H. Interleukin-21 contributes to germinal centre formation and immunoglobulin G4 production in IgG4-related dacryoadenitis and sialoadenitis, so-called Mikulicz’s disease. Ann Rheum Dis. 2012;71:2011–9. doi: 10.1136/annrheumdis-2012-201477. [DOI] [PubMed] [Google Scholar]
- Jordan S, Distler JH, Maurer B, et al. Effects and safety of rituximab in systemic sclerosis: an analysis from the European Scleroderma Trial and Research (EUSTAR) group. Ann Rheum Dis. 2014;74:1188–94. doi: 10.1136/annrheumdis-2013-204522. [DOI] [PubMed] [Google Scholar]
- Keir GJ, Maher TM, Ming D, et al. Rituximab in severe, treatment-refractory interstitial lung disease. Respirology. 2014;19:353–9. doi: 10.1111/resp.12214. [DOI] [PubMed] [Google Scholar]
- Castillo-Trivino T, Braithwaite D, Bacchetti P, Waubant E, et al. Rituximab in relapsing and progressive forms of multiple sclerosis: a systematic review. PLOS ONE. 2013;8:e66308. doi: 10.1371/journal.pone.0066308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford A, Macleod M, Schumacher T, Corlett L, Gray D. Primary T cell expansion and differentiation in vivo requires antigen presentation by B cells. J Immunol. 2006;176:3498–506. doi: 10.4049/jimmunol.176.6.3498. [DOI] [PubMed] [Google Scholar]
- Pillai S, Mattoo H, Cariappa A. B cells and autoimmunity. Curr Opin Immunol. 2011;23:721–31. doi: 10.1016/j.coi.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr TA, Shen P, Brown S. B cell depletion therapy ameliorates autoimmune disease through ablation of IL-6-producing B cells. J Exp Med. 2012;209:1001–10. doi: 10.1084/jem.20111675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon S, et al. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- Wynn TA, Barron L. Macrophages: master regulators of inflammation and fibrosis. Semin Liver Dis. 2010;30:245–57. doi: 10.1055/s-0030-1255354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa S, Moriyama M, Tanakaa A. Preferential M2 macrophages contribute to fibrosis in IgG4-related dacryoadenitis and sialoadenitis, so-called Mikulicz’s disease. Clin Immunol. 2015;156:9–18. doi: 10.1016/j.clim.2014.10.008. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Yamashita K, Fujikawa S, et al. Activation of toll-like receptors and NOD-like receptors is involved in enhanced IgG4 responses in autoimmune pancreatitis. Arthritis Rheum. 2012;64:914–24. doi: 10.1002/art.33386. [DOI] [PubMed] [Google Scholar]
- Sokol CL, Medzhitov R, et al. Role of basophils in the initiation of Th2 responses. Curr Opin Immunol. 2010;22:73–7. doi: 10.1016/j.coi.2010.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul WE, Zhu J. How are T(H)2-type immune responses initiated and amplified? Nat Rev Immunol. 2010;10:225–35. doi: 10.1038/nri2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Xu W, Wilson M. Immunoglobulin D enhances immune surveillance by activating antimicrobial, proinflammatory and B cell-stimulating programs in basophils. Nat Immunol. 2009;10:889–98. doi: 10.1038/ni.1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T, Yamashita K, Sakurai T, et al. Toll-like receptor activation in basophils contributes to the development of IgG4-related disease. J Gastroenterol. 2013;48:247–53. doi: 10.1007/s00535-012-0626-8. [DOI] [PubMed] [Google Scholar]
- Huaux F, Liu T, McGarry B, Ullenbruch M, Xing Z, Phan SH, et al. Eosinophils and T lymphocytes possess distinct roles in bleomycin-induced lung injury and fibrosis. J Immunol. 2003;171:5470–81. doi: 10.4049/jimmunol.171.10.5470. [DOI] [PubMed] [Google Scholar]