

Abstract
The pathogenesis of age-related macular degeneration involves chronic oxidative stress, impaired degradation of membranous discs shed from photoreceptor outer segments and accumulation of lysosomal lipofuscin in retinal pigment epithelial (RPE) cells. It has been estimated that a major part of cellular proteolysis occurs in proteasomes, but the importance of proteasomes and the other proteolytic pathways including autophagy in RPE cells is poorly understood. Prior to proteolysis, heat shock proteins (Hsps), agents that function as molecular chaperones, attempt to refold misfolded proteins and thus prevent the accumulation of cytoplasmic protein aggregates. In the present study, the roles of the Hsp70 molecular chaperone and proteasomal and lysosomal proteolytic pathways were evaluated in human RPE cells (ARPE-19). The Hsp70 and ubiquitin protein levels and localization were analysed by Western blotting and immunofluorescense. Confocal and transmission electron microscopy were used to detect cellular organelles and to evaluate the morphological changes. Hsp70 levels were modulated using RNA interference and overexpression techniques. Cell viability was measured by colorimetric assay. The proteasome inhibitor MG-132 evoked the accumulation of perinuclear aggregates positive for Hsp70, ubiquitin-protein conjugates and the lysosomal membrane protein LAMP-2. Interestingly, the hsp70 mRNA depletion significantly increased cell death in conjunction with proteasome inhibition. We found that the accumulation of lysosomes was reversible: a cessation of proteasome inhibition led to clearance of the deposits via a mechanism believed to include autophagy. The molecular chaperone Hsp70, proteasomes and autophagy have an important regulatory role in the protein turnover of human RPE cells and may thus open new avenues for understanding degenerative processes in retinal cells.
Keywords: autophagosome, Hsp70, lysosome, proteasome, proteolysis, retinal pigment epithelium
Introduction
Cells and tissues are regularly exposed to different kinds of acute and chronic environmental stresses. Retinal pigment epithelial (RPE) cells must endure a high level of oxidative stress because of their high oxygen consumption, high levels of polyunsaturated lipids and long periods of exposure to light [1]. Oxidative stress refers to the progressive cellular damage caused by reactive oxygen species, which contributes to protein misfolding and evokes functional abnormalities during cellular senescence of the RPE cells [1, 2]. A disturbance of cellular homeostasis in the post-mitotic RPE cells is involved in the development of age-related macular degeneration (AMD), which is the most common cause of blindness in developed countries [1, 3].
The efficient removal of misfolded proteins from the cytoplasm is critical for cellular survival and adaptation. Cells can avoid the accumulation of potentially toxic misfolded protein aggregates by accessing a battery of mechanisms. One of the most important of these is the suppression of aggregate formation by heat shock proteins (Hsps), which function as molecular chaperones in cells [4]. The transcriptionally and post-transcriptionally regulated expression of Hsps can be promoted by many different kinds of chemical or physical stresses [5–10]. The Hsps are divided into different families (Hsp90, Hsp70, Hsp60, Hsp40 and small Hsps), according to their function, molecular size and cellular localization [11].
Cellular proteins are continuously synthesized and degraded during the lifespan of the cell [12]. The control of protein turnover is particularly important in post-mitotic cells, where the accumulation of malfunctioning proteins may be highly detrimental to cell survival [13]. Four different systems take care of protein degradation in eukaryotic cells: (i) proteasomes, which degrade the majority of long- and short-lived normal and abnormal intracellular proteins, (ii) mitochondrial proteases, which degrade the majority of mitochondrial proteins, (iii) calcium-activated calpains, which degrade membrane and cytoskeletal proteins and several membrane-associated enzymes and (iv) autophagy and lysosomes, which degrade whole cell organelles, membrane proteins and endocytosed proteins [14–17]. During RPE cell senescence, the accumulation of lysosomal lipofuscin correlates well with the path-ogenesis of AMD [1]. Many studies have demonstrated the importance of lysosomes for proteolysis in RPE cells [18–23]. The accumulated lipofuscin probably has multiple secondary detrimental effects on RPE cells. It has been shown that lipofuscin prevents cellular renewal and promotes the misfolding of intracellular proteins by increasing the sensitivity of RPE cells to oxidative stress [23, 24].
Macroautophagy, microautophagy and chaperone-mediated autophagy are the principal pathways to deliver cytoplasmic material for lysosomal degradation in mammalian cells [25, 26]. During macroautophagy, cytoplasmic structures are segregated within autophagosomes, which deliver their contents to lysosomes, while in microautophagy, small portions of cytoplasm enter lysosomes via invagination of the lysosomal membrane. One of the molecular chaperones, Hsc73, delivers specific proteins into lysosomes during chaperone-mediated autophagy [25, 26].
Although the connection between stress and degeneration of RPE cells is well known, the role of proteasomes and autophagy in protein turnover in RPE cells is largely unexplored. Hsps are closely linked to proteasome-mediated proteolysis, and also to lysosomal protein degradation [25, 26]. Should the Hsp-linked protein folding fail, misfolded proteins are first tagged with a polypeptide called ubiquitin (Ub) and subsequently transferred to proteasomes for degradation [13, 27]. In fact, it is believed that up to 80–90% of all intracellular proteins are degraded by the ubiquitin-proteasome proteolysis machinery [15, 16]. However, once the capacity of Hsps to correct proteins is exceeded, or the ubiquitin-proteasome pathway experiences functional limitations, protein aggregates are formed at the cell periphery and delivered along the microtubulus network via dynein-dependent retrograde transport to a perinuclear location. The aggregates form aggresomes, which may be cleaned through autophagy [13, 28, 29].
Our goal was to study the influence of the Hsp70 molecular chaperone and lysosomes on the formation and clearance of protein aggregates in proteasome inhibitor-treated human RPE cells. We show that proteasome inhibition causes an intense perinuclear accumulation of Hsp70, Ub protein conjugates and LAMP-2-positive aggregates in human retinal pigment cells (ARPE-19). Interestingly, we observed that the perinuclear accumulation of aggregates is a reversible process, because the cessation of proteasome inhibition leads to the removal of the deposits. Our results suggest that autophagy also participates in the clearance of the accumulated deposits.
Methods
Cell culture and treatments
ARPE-19 human RPE cells were obtained from the American Type Culture Collection (ATCC). The cells were grown to confluency in a humidified 10% CO2 atmosphere at 37°C in Dulbecco’s MEM/Nut MIX F-12 (1:1) medium (Life Technologies, Paisley, UK) containing 10% inactivated foetal bovine serum (Hyclone, Logan, UT, USA), 100 units/ml penicillin (Cambrex, Charles City, IA, USA), 100 μg/ml streptomycin (Cambrex) and 2 mM L-glutamine (Life Technologies). For proteasome inhibition, the cells were exposed to 10 μM MG-132 proteasome inhibitor (Calbiochem, San Diego, CA, USA) for up to 48 hrs. The lysosomes were alkalized with 10 mM NH4Cl or 100 nM bafilomycin A1 (Sigma-Aldrich, Steinheim, Germany) for up to 48 hrs. In all of the 48-hr ammonium chloride exposures, ammonium chloride-containing medium was replaced with fresh equivalent after 24 hrs. The formation of autophagosomes was inhibited with 10 mM 3-methyladenine (Sigma-Aldrich) for up to 48 hrs.
Western blotting
For Western blotting, whole cell extracts (10 or 20 μg of protein) were run in 10% SDS-PAGE gels and then transferred to nitrocellulose membranes (Amersham Biosciences, Pittsburgh, PA, USA) by using a semi-dry transfer apparatus (Bio-Rad Laboratories, Hercules, CA, USA). Ponceau S staining (Sigma, St. Louis, MO, USA) was used to ensure equal loading of proteins. The membranes were blocked for 1.5 hrs in 3% fat-free dry milk in 0.3% Tween 2/BS at room temperature (RT). Subsequently, the membranes were incubated overnight at 4°C with mouse monoclonal Hsp70 antibody (StressGen, Ann Arbor, MI, USA) or rabbit polyclonal ubiquitin antibody (DakoCytomation, Glostrup, Denmark) diluted (1:5000 or 1:500, respectively) in 0.5% bovine serum albumin in 0.3% Tween 2/BS. After 3 × 10 min. washes with 0.3% Tween 2/BS, the membranes were incubated for 2 hrs at RT with horseradish peroxidase-conjugated antibodies (Amersham Biosciences). Both secondary antibodies were diluted 1:20 000 in 3% fat-free dry milk in 0.3% Tween 2/BS. Before detection, all of the membranes were washed as before. Protein-antibody complexes were detected with an enhanced chemiluminescence method (Millipore, Billerica, MA, USA).
Immunofluorescence
The cells were fixed with fresh formaldehyde prepared from paraformaldeyhyde (1%, in PBS) for 10 min. at RT. To determine the localization of Hsp70, ubiquitin, LAMP-1 and LAMP-2, the permeabilized (0.2% Triton X-100 in PBS for 10 min. at 4°C) samples were blocked with 3% fat-free dry milk in 0.05% Tween/PBS for 30 min. at RT. Thereafter, the samples were incubated with primary antibody diluted in 0.05% Tween/PBS for 1 hr at RT (Hsp70: rat monoclonal, 1:50, ubiquitin: rabbit polyclonal, 1:80, LAMP-1: H4A3, mouse monoclonal, 1:35, LAMP-2: H4B4, mouse monoclonal, 1:20). The Hsp70 antibody was from Cell Signaling Technology (Danvers, MA, USA), ubiquitin antibody from DakoCytomation and LAMP antibodies were from the Developmental Studies Hydrioma Bank (Iowa City, IA, USA). After 3 × 5 min. washes with PBS, the samples were incubated in secondary antibody diluted 1:1 000 in 0.05% Tween/PBS for 1 hr at RT (Hsp70: anti-rat Alexa Fluor 488, ubiquitin: anti-rabbit Alexa Fluor 488, LAMP-1 and 2: anti-mouse Alexa Fluor 488 or anti-mouse Alexa Fluor 594) and washed as before. All secondary antibodies were from Invitrogen (Carlsbad, CA, USA). The nuclei were stained in all samples by incubation with Hoechst 33258 dye (0.5 μM/ml) for 10 min. at RT followed by 4 × 1 min. washes with PBS. Images were captured using a Nikon Eclipse TE300 microscope equipped with a Nikon E995 digital camera or with a confocal microscope (Nikon Eclipse TE 300).
Transmission electron microscopy
Cell culture samples were prefixed with 2.5% glutaraldehyde in 0.1 M phosphate buffer pH 7.4 for 2 hrs at RT. After 15 min. washing in 0.1 M phosphate buffer, the samples were post-fixed in 1% osmium tetraoxide and 0.1 M phosphate buffer for 1 hr, and again washed with phosphate buffer for 15 min. prior to standard ethanol dehydration. Subsequently, the samples were infiltrated and embedded in LX-112 resin. Polymerization was carried out at 37°C for 24 hrs and at 60°C for 48 hrs. The sections were examined with a JEOL-1200EX transmission electron microscope (Jeol, Tokyo, Japan) at 80 kV.
Attenuation of the Hsp70 gene expression by RNA interference
siRNA designed for human Hsp70 gene (HspA1A) and negative control siRNA were purchased from Ambion (Austin, TX, USA). The negative control siRNA was an oligonucleotide with no homology to any known human gene. The siRNAs were transfected into ARPE-19 cells using siPORT ™Amine transfection reagent (Ambion) following the manufacturer’s instructions. Two different concentrations of siRNAs (30 nM and 100 nM) were tested (data not shown) and an optimal concentration was selected (30 nM). The duration of the siRNA treatment was 24 hrs. Maximal expression of Hsp70 was achieved using heat shock treatment: incubation in a 42°C water bath for 1 hr, followed by a 5h recovery in the cell culture incubator prior to sample collection. The decrease in Hsp70 expression was monitored by Western blotting as described above.
Green fluorescent protein (GFP) -Hsp70 fusion plasmid construction
The functional open reading frame (ORF) of the human Hsp70 A1A gene (NCBI GeneBank accession no. NM_005345) was amplified from DNase-treated (DNase I, Roche, Basel, Switzerland) total RNA, extracted (Eurozol reagent, Euroclone, Wetherby, UK) from human ARPE-19 cells. Initially, mRNA was reverse-transcribed (MultiScribe reverse transcriptase, Applied Biosystems, Foster City, CA, USA) and Hsp70 cDNA was amplified with a high-fidelity DNA polymerase (Phusion Hot start DNA polymerase, Finnzymes, Espoo, Finland). The primers were 5′-ATA CTC GAG ATA TGG CCA AAG CCG C (forward) and 5′-AAT AAG CTT GCT AAT CTA CCT CCT CAA TGG TG (reverse), containing the target sites for restriction endonucleases XhoI and HindIII and stuffers for in-frame ligation. The sites for translation initiation and termination are depicted in boldface.
The sticky ends for the amplified Hsp70 ORF as well as the multiple cloning site of the vector pEGFP-C1 (Clontech, Mountain View, CA, USA) were produced with the above-described restriction endonucleases (MBI Fermentas, Vilnius, Lithuania). Ligated (T4 DNA Ligase, Roche) DNA forming a fusion gene of GFP and Hsp70 was transfected into competent DH5αE. coli cells, which were prepared using the protocol of Inoue et al. [30], propagated and purified [31]. The integrity of the construct was determined initially by restriction endonuclease digestion analysis and finally sequencing the junction sites and the entire inserted Hsp70 ORF.
The plasmid was transfected into a subconfluent culture of ARPE-19 cells using FuGENE 6 transfection reagent (Roche) following the manufacturer’s instructions. The function of the fusion protein was initially monitored using RT-PCR amplification of a 365-bp fragment flanking one junction and by Western blotting detecting the 887-aa GFP-Hsp70 fusion protein.
Confocal microcopy
ARPE-19 cells were grown on 8-well glass slides (Nunc ™, Rochester, NY, USA). Before the plating of the cells, the glasses were treated with collagen Type IV (BD) (50 μg/ml in 20 mM acetic acid) for at least 1 hr and rinsed with phosphate buffer. The cells were grown for 24 hrs before transfection to achieve a 50–70% confluency. Briefly, 4 μg/ml of the plasmid, pEGFP-hHsp70, was added to diluted Fugene 6 transfection reagent (Roche) following the manufacturer’s instructions. Drug treatments were performed 48 hrs after transfection. The proteasome inhibitor MG-132 was added at 10 μM and incubated for 24 hrs. To neutralize acidic organelles, the cells were treated with 10 mM ammonium chloride for 24 or 48 hrs after the pEGFP-hHsp70 transfection and MG-132 treatment. When the treatment lasted for 48 hrs, ammonium chloride-containing medium was replaced with fresh equivalent after 24 hrs.
The samples were examined using a Nikon Eclipse TE 300 confocal microscope with a 100 × oil immersion objective. The EGFP stain was visualised at an excitation wavelength of 488 nm while emission wavelength was 525 nm. The lysosomes were stained using LysoTracker® Red DND-99 (Molecular Probes, Eugene, OR, USA), diluted to 50 nM in medium, for 20 min. For LysoTracker, the excitation and emission wavelengths were 577 and 590 nm, respectively.
Lysosome isolation
The lysosome fractions from ARPE-19 cells were isolated as previously described [32]. Briefly, the cells were harvested by trypsination and disrupted by nitrogen cavitation, and the post-nuclear supernatant was obtained by low-speed centrifugation. Subsequent fractionation by a sequence of two discontinuous gradient ultracentrifugations yielded pure lysosomal fractions. The purity of these fractions was also measured. The lysosomal marker fl-hexosaminidase was measured fluorimetrically with the substrate 4-methylumbelliferyl-2-acetamido-2-deoxy-β-D-glucopyranoside. The mitochondrial marker succinate dehydrogenase was measured photometrically with p-iodonitrotetrazolium violet. Lactate dehydrogenase, the cytosomal marker, was determined by measuring the oxidation of NADH with pyruvate as substrate. Alkaline phosphodiesterase, the plasma membrane marker, was assayed photometrically with the substrate 5′monophosphate p-nitrophenylester. UDP-galactosyltransferase, a marker for ER and golgi, was determined radiometrically with [galactose-4,5–3H(N)]-uridine diphosphate as substrate and N-acetylglucosamine as acceptor. DNA, the marker for nuclei, was measured fluorimetrically with bisbenzimide H33258. Protein concentrations were determined by the Lowry procedure.
Viability staining of cells
For the determination of the viability and death rates, cells were stained with fluorescein diacetate (FDA, which permeates the living cell membrane and stains the cells a green colour) and propidium iodide (PI, which stains the nuclei of dead cells a red colour) (both from Sigma) [33]. The cells were monitored and dead cells were counted within a fixed area with a fluorescence microscope (Olympus IX71, light source Olympus Cell MT-10, Olympus, Tokyo, Japan). The statistically significant differences were identified with the Mann–Whitney U-test (n= 9).
Results
Proteasome inhibition increased Hsp70 and ubiquitin levels and evoked a juxtanuclear accumulation of aberrant deposits
To study the effect of proteasome inhibition in human ARPE-19 RPE cells, the cells were either non-stressed or exposed to 10 μM MG-132 for 24 hrs. The levels and localization of Hsp70 and ubiquitinated protein conjugates were then analysed. A robust increase in the Hsp70 protein level and elevated levels of ubiquitinated proteins were seen in Western blots in response to MG-132 exposure (Fig. 1A).
Figure 1.
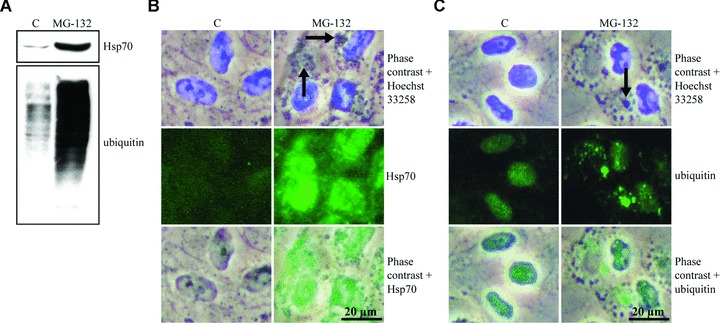
Hsp70 protein expression and ubiquitin-protein conjugate levels. (A) Total proteins (20 μg) of whole cell extracts were examined by Western blot using antibodies against the Hsp70 and ubiquitin of control (C) cells, and cells exposed to 10 μM MG-132 for 24 hrs. Phase contrast microscopy and immunofluorescence analysis of (B) Hsp70 (green) and (C) ubiquitin (green) in control cells, and cells exposed to 10 μM MG-132 for 24 hrs. Nuclei were stained with Hoechst 33258 dye (blue). Arrows point to protein aggregates.
The cellular localization of Hsp70 was studied by phase contrast and immunofluorescence microscopy. A robust accumulation of perinuclear deposits was observed in the phase contrast photographs (Fig. 1B). The Hsp70 staining was observed throughout the nucleus and the cytoplasm, being most intense in the nucleus and in the perinuclear area in MG-132-treated cells (Fig. 1B). In control cells there was faint Hsp70 immunostaining everywhere. Cells treated with MG-132 displayed an increase in mainly perinuclear Ub-immunoreactivity ranging from a cloud-like appearance to a granular morphology (Fig. 1C). The strongest Ub-labelling was detected in the largest cytoplasmic granules, but not in the smaller aggregates. The nuclei of MG-132-treated cells also exhibited an increase in diffuse and granular Ub immunoreactivity. In control cells, moderate labelling for Ub was seen in the nuclei, whereas only faint or no labelling at all was detected in the cytoplasmic areas (Fig. 1C).
Proteasome inhibition-induced juxtanuclear deposits were modified by hsp70 mRNA silencing and were stained with lysosomal membrane proteins
To study the role of Hsp70 in protein aggregation, ARPE-19 cells were treated with non-silencing or silencing hsp70 siRNA constructs, and either non-stressed or exposed to 10 μM MG-132 for 24 and 48 hrs. For evaluating the hsp70 siRNA efficacy, the cells were incubated at 42°C for 1 hr and then allowed to recover in a cell culture incubator for 5 hrs prior to sample collection. As expected, Hsp70 expression decreased in response to hsp70 mRNA silencing. This was monitored by Western blotting in heat-shocked cells (Fig. 2A). The ultrastructure of the cells was examined using transmission electron microscopy. In control cells, no abnormalities were observed, whereas MG-132 evoked a prominent perinuclear accumulation of deposits (Fig. 2B). Both negative and positive siRNA constructs had an effect on the morphology of the perinuxclear deposits but this was much stronger in response to positive hsp70 siRNA treatment. Electron-dense droplets were observed inside the deposits in siRNA-treated cells (Figs 2B and 4C). These dark areas might be lipid-containing droplets that bind osmium during the sample preparation.
Figure 2.
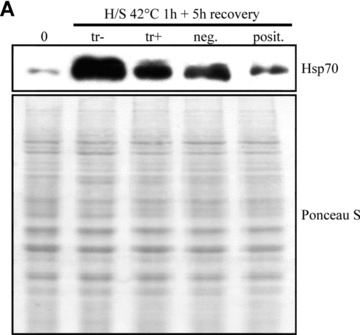
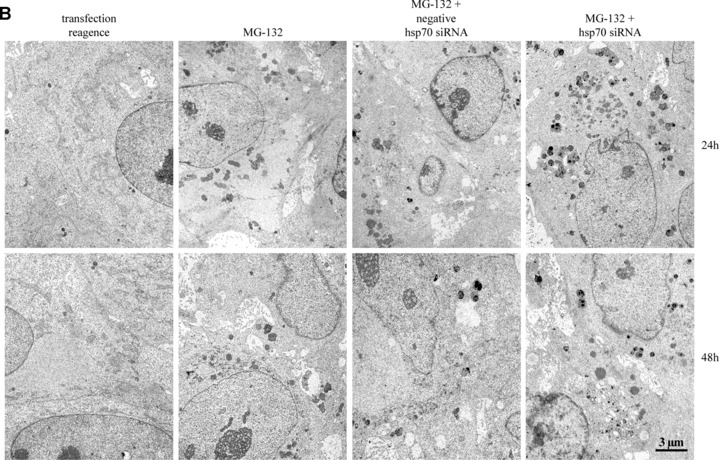
(A) Western blotting analysis for evaluating hsp70 mRNA interference efficacy in ARPE-19 cells. The cells were heat-shocked at 42°C for 1 hr and then allowed to recover for 5 hrs prior to the detection of Hsp70 expression levels. Tr stands for transfection reagent, neg. for negative (non-silencing) siRNA and posit. for Hsp70 (silencing) siRNA. Ponceau S staining was used to ensure the equal loading of proteins. (B) Transmission electron microscopy of transfection reagent positive cells, cells exposed solely to 10 μM MG-132 or with negative or positive hsp70 RNA interference oligonucleotides for 24 and 48 hrs. Note that the hypotonic fixation condition causes swelling artefacts, but does not disturb the interpretation of the results.
Figure 4.
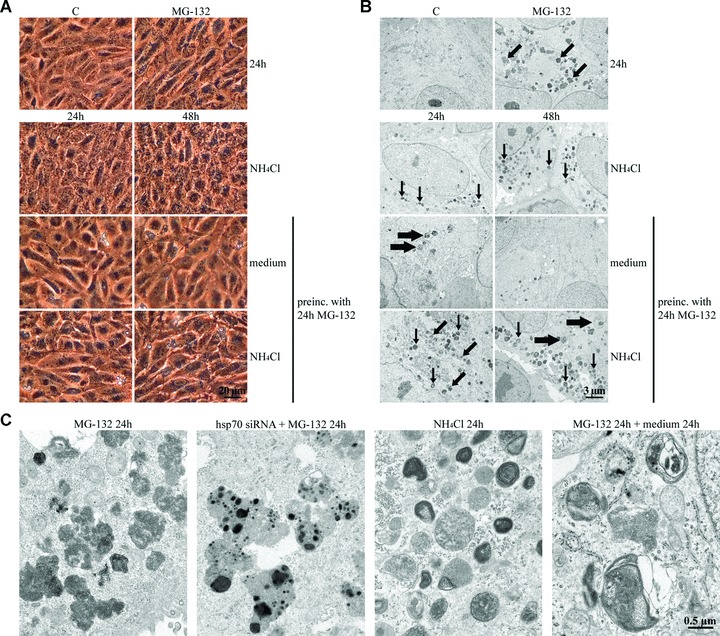
(A) Phase contrast and (B) transmission electron microscopy of ARPE-19 control cells (C), cells exposed to either 10 μM MG-132 for 24 hrs or 10 mM NH4Cl for 24 and 48 hrs, and cells preincubated with 10 μM MG-132 and then allowed to remain in normal cell culture medium with or without 10 mM NH4Cl for 24 and 48 hrs. Small arrows point to enlarged lysosomes, medium-sized arrows point to protein aggregates and large arrows show autophagosome-like structures. (C) Transmission electron microscopy of the cells exposed solely to 10 μM MG-132 or with hsp70 RNA interference oligonucleotides or exposed solely to NH4Cl for 24 hrs or cells that were preincubated with 10 μM MG-132 and then allowed to recover for 24 hrs.
We estimated that these perinuclear deposits might be lysosomal. Therefore, specific lysosomal membrane proteins, LAMP-1 and LAMP-2 [34], were analysed in cells exposed to MG-132 for 12 and 24 hrs. Phase contrast microscopy and immunofluorescence analyses revealed that Hsp70- and ubiquitin-associated perinuclear deposits (Fig. 1) were strongly LAMP-2-positive and partly LAMP-1-positive (Fig. 3).
Figure 3.
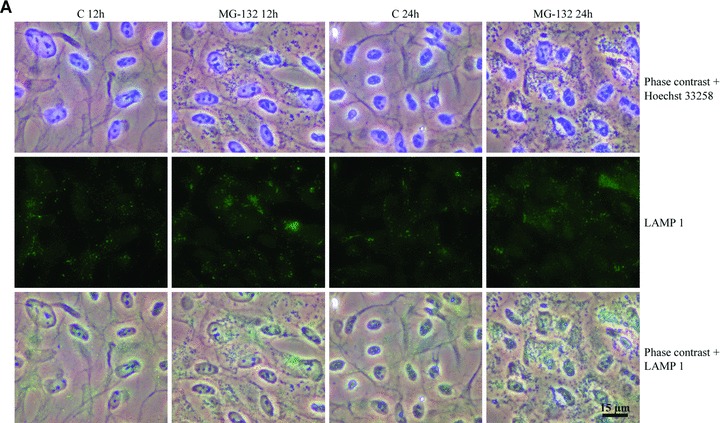

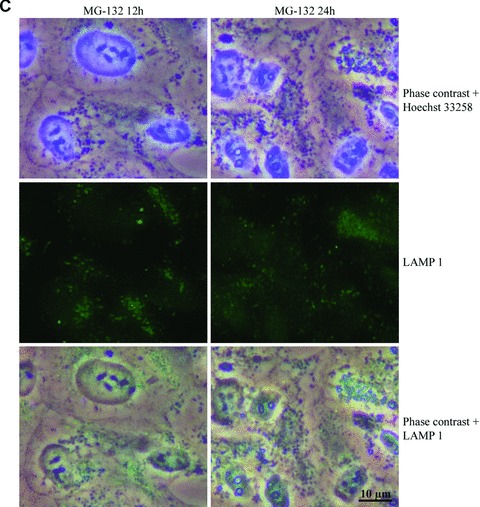
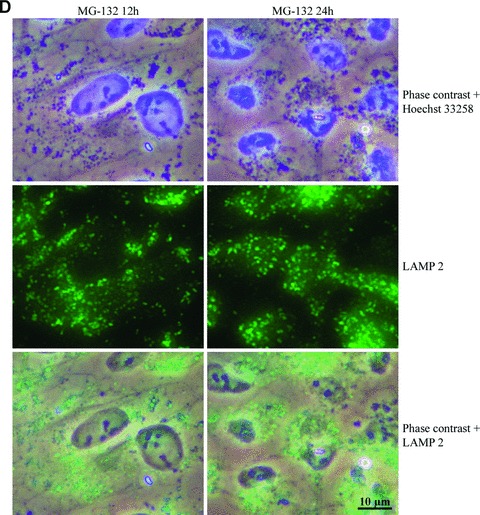
Phase contrast microscopy and immunofluorescence analysis of (A) LAMP-1 and (B) LAMP-2 in control cells (C 12h, C 24h), and cells exposed to 10 μM MG-132 for 24 hrs. Nuclei were stained with Hoechst 33258 dye (blue). (C) and (D) represent higher magnification of the LAMP-1 and LAMP-2 stainings related to (A) and (B) images, respectively. Scale bars 15 μm and 10 μm.
Clearance of perinuclear Hsp70 and ubiquitin-associated lysosomes
Next, we wanted to determine whether the accumulated lysosomes would disappear if the cells were switched to normal medium after proteasome inhibition. The ARPE-19 cells were either non-stressed or preincubated with 10 μM MG-132 for 24 hrs and then allowed to remain in normal culture medium or in a medium containing 10 mM NH4Cl, to neutralize lysosomal pH, for 24 or 48 hrs. The accumulated lysosomes disappeared under normal recovery culture conditions (24 hrs) and membrane-bound organelles resembling autophagic vacuoles were observed in the cytoplasm (Fig. 4). When the cells were allowed to recover for 48 hrs, all of the deposits and autophagic vacuoles disappeared. After the 24-hr post-incubation with ammonium chloride, the cells contained lysosomes of different sizes. The swelling of the lysosomes was due to the ammonium chloride. After the 48-hr post-incubation, the number of MG-132-induced accumulated lysosomes was smaller, and autophagic-vacuole-like structures were also observed. (Fig. 4B) These findings support our earlier observations that the proteasome-inhibition-induced perinuclear Hsp70 and ubiquitin-positive deposits (Fig. 1) are encapsulated in lysosomes (Fig. 3).
We also wanted to evaluate protein aggregation and proteolysis at earlier time points in response to proteasome inhibition. The cells were exposed to either 10 μM MG-132 for 12 hrs or preincubated with 10 μM MG-132 for 12 hrs and then allowed to remain in normal cell culture medium for 12 and 24 hrs. Perinuclear lysomal deposits were observed already after 12 hrs of proteasome inhibition (Fig. 5A).
Figure 5.
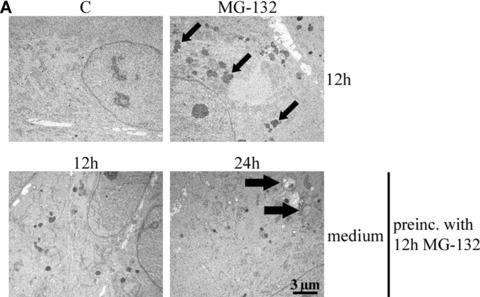
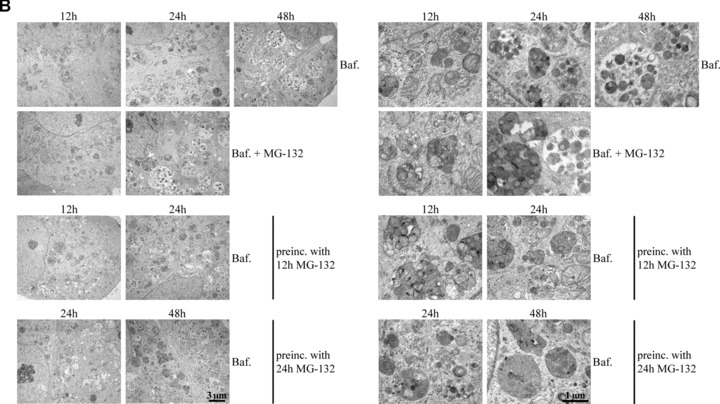
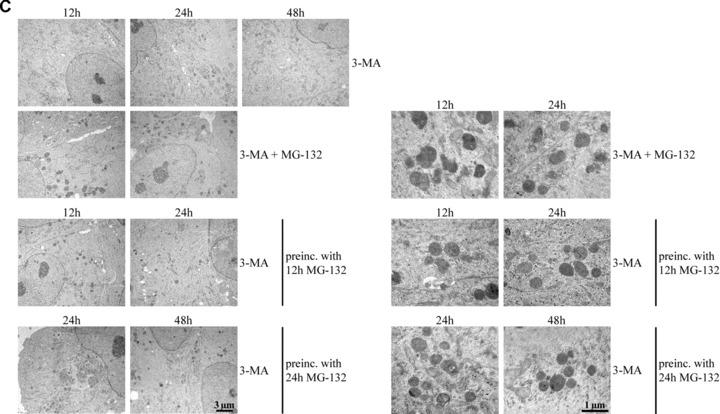
(A) Transmission electron microscopy of ARPE-19 control cells (C), cells exposed to either 10 μM MG-132 for 12 hrs or preincubated with 10 μM MG-132 for 12 hrs and then allowed to remain in normal cell culture medium for 12 and 24 hrs. Small arrows point to enlarged lysosomes and large arrows indicate autophagosome-like structures. (B) Transmission electron microscopy of the cells exposed solely to 100 nM bafilomycin (Baf) for 12, 24 and 48 hrs or 100 nM Baf together with 10 μM MG-132 for 12 and 24 hrs. In addition, the cells were preincubated with 10 μM MG-132 for 12 and 24 hrs and then allowed to recover for up to 48 hrs with 100 nM Baf. (C) Transmission electron microscopy of the cells exposed solely to 10 mM 3-methyladenine (3-MA) for 12, 24 and 48 hrs or 10 mM 3-MA together with 10 μM MG-132 for 12 and 24 hrs. In addition, the cells were preincubated with 10 μM MG-132 for 12 and 24 hrs and then allowed to recover for up to 48 hrs with 10 mM 3-MA. In pictures B and C, the right panels show magnifications of altered structures. Scale bars in all pictures are 3 μm or 1 μm.
Ammonium chloride might be a rather non-specific inhibitor of lysosomes. To confirm the lysosomal nature of the vesicles accumulating during proteasome inhibition, we used a more specific inhibitor of lysosomal proteolysis, namely bafilomycin (Baf). The cells were exposed solely to 100 nM Baf for 12, 24 and 48 hrs, or to 100 nM Baf together with 10 μM MG-132 for 12 and 24 hrs. Moreover, the cells were preincubated with 10 μM MG-132 for 12 and 24 hrs and then allowed to recover for up to 48 hrs with 100 nM Baf. The number of autophagy-like structures increased in response to lysosomal inhibition in the RPE cells (Fig. 5B). Transmission electron micrographs showed that proteasome inhibitor-induced accumulated lysosomes had disappeared, and membrane-bound organelles resembling autophagic vacuoles were observed in the cells post-incubated with Baf (Fig. 5B).
To test whether autophagy plays a role in the clearance of the deposits after proteasome inhibition, we used 3-methyladenide (3-MA) to inhibit autophagy. The cells were exposed solely to autophagosome inhibitor 10 mM 3-MA for 12, 24 and 48 hrs, or 10 mM 3-MA together with 10 μM MG-132 for 12 and 24 hrs. Moreover, the cells were preincubated with 10 μM MG-132 for 12 and 24 hrs and then allowed to recover for up to 48 hrs with 10 mM 3-MA. As expected, no autophagosome-like vacuoles were observed in any of the 3-MA treatments. However, perinuclear lysosomal-like deposits remained in cells post-incubated with 3-MA. (Fig. 5C).
Hsp70 localizes to cytosol in lysosome-rich areas
Because hsp70 mRNA silencing caused changes in perinuclear lysosomes, we wanted to investigate whether Hsp70 would localize with lysosomes. Therefore, a green fluorescent protein (GFP) -Hsp70 overexpression vector was created. The construct efficacy, with two different concentrations (2 μg/ml and 4 μg/ml), was analysed by Western blotting after 2 and 5 days of transfection in ARPE-19 cells. The highest GFP-Hsp70 levels were observed after 2 days of transfection. Therefore, the duration of transfection was 2 days in further experiments (Fig. 6A). The ARPE-19 cells were either non-stressed or preincubated with 10 μM MG-132 for 24 hrs and then transferred to normal culture medium for 24 or 48 hrs. The lysosomes were stained using LysoTracked Red. Confocal microscopy analyses showed the number of acidic LysoTracker-positive vesicles in both control cells and in cells treated with the proteasome inhibitor (Fig. 6B). Intense staining of GFP-Hsp70 was seen in both the cytoplasm and the nucleus in non-stressed cells and in cells exposed to lysosomal or proteasomal inhibition. In addition, our results suggest that GFP-Hsp70 is associated with the lysosomal-rich cytosol (Fig. 6B and C). The ARPE-19 cells treated with MG-132 for 24 hrs and allowed to recover for 24 hrs in normal cell culture medium showed early autophagosome-like structures merging with lysosomes (Fig. 6B and C), whereas the cells allowed to recover for 48 hrs showed acidic late autophagosome-like structures (Fig. 6D). The regulatory role of heat shock cognate protein 73 (Hsc73) is well known in chaperone-mediated autophagy [35]. However, it is not known whether inducible Hsp70 participates in lysosomal proteolysis.
Figure 6.
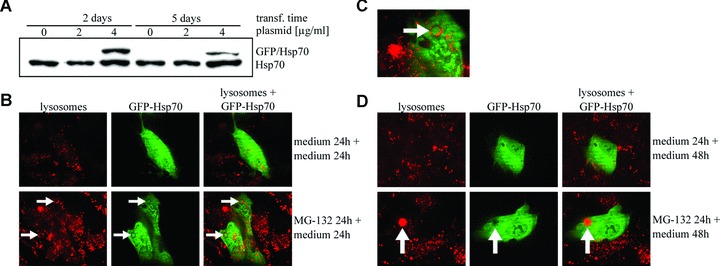
(A) Western blotting analysis for evaluating GFP-Hsp70 transfection efficacy in ARPE-19 cells. The transfection (with two different plasmid concentrations) was allowed to continue for 2 or 5 days prior to detecting the Hsp70 and GFP-Hsp70 expression levels. Ponceau S staining was used to ensure equal loading of proteins. (B and D) Confocal microscopy analyses from the control cells and cells preincubated with 10 μM MG-132 and then allowed to recover with normal medium for 24 or 48 hrs. (C) Magnification of Hsp70 positive stained early autophagosomal structures. Arrows show early autophagosome-like structures, which are merging with lysosomes (B and C). In picture D, arrows show late autophagosome-like structures.
Next, we wanted to confirm an Hsp70 association with lysosomes. Therefore, the cells were exposed to 10 μM MG-132 for 24 hrs and immunostained with Hsp70 and LAMP-2, then analysed by confocal microscopy that showed strong Hsp70 staining with LAMP-2 in the ARPE-19 cells (Fig. 7). In addition, we isolated lysosomes from cells that were either non-stressed or exposed to 10 μM MG-132 for 24 hrs or exposed to 10 μM MG-132 for 24 hrs and then allowed to recover in normal cell culture medium up to 48 hrs. Indeed, we detected Hsp70 in the purified lysosome fractions, especially in lysosomes isolated from proteasome inhibitor-treated cells (Fig. 8). In line with our previous findings of disappeared perinuclear deposits (Figs 4A, B and 5A), the Hsp70 expression levels were lower in lysosomal fractions isolated from cells exposed to 10 μM MG-132 for 24 and then allowed to recover for 48 hrs (Fig. 8). In these lysosome fractions, the lysosomal marker β-hexosaminidase was always more than 100-fold enriched compared with the initial homogenate. Markers of other subcellular fractions, i.e. succinate dehydrogenase for mitochondria, lactate dehydrogenase for cytosol, alkaline phosphodiesterase for plasma membranes, UDP-galactosyltransferase for microsomes and DNA for nuclei, were not detectable in the purified lysosomal fractions (Table 1).
Figure 7.
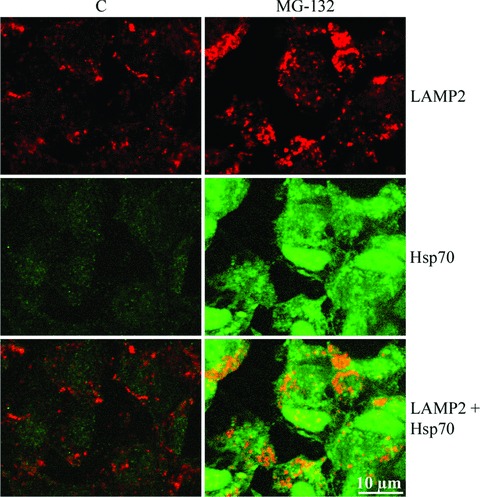
Confocal microscopy analyses of the control cells and cells incubated with 10 μM MG-132 for 24 hrs. LAMP-2 staining is visualized with red fluorescent dye and Hsp70 with green. Scale bar is 10 μm.
Figure 8.
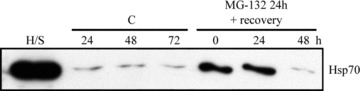
Western blotting analysis (10 μg protein/lane) of the Hsp70 expression levels from isolated lysosome fractions from the control cells (C) or cells exposed to 10 μM MG-132 for 24 or cells exposed to 10 μM MG-132 for 24 and then allowed to recover for up to 48 hrs. H/S stands for heat shock and represents a positive control.
Table 1.
Purification of lysosomes from cultured ARPE-19 cells
| Marker | Marker for | Sample | Homogenate (specific activity) | Lysosomal Fraction (specific activity) |
|---|---|---|---|---|
| β-Hexosaminidase | Lysosomes | Control / 24h | 2.4 mU/mg protein | 261.2 mU/mg protein |
| EC 3.2.1.30 | Control / 48h | 2.5 mU/mg protein | 273.5 mU/mg protein | |
| MG-132 / 24h | 2.4 mU/mg protein | 243.2 mU/mg protein | ||
| MG-132 / 48 | 2.3 mU/mg protein | 241.4 mU/mg protein | ||
| Succinate dehydrogenase | Mitochondria | Control / 24h | 0.75 mU/mg protein | 0.02 mU/mg protein |
| E.C. 1.3.99.1 | Control / 48h | 0.71 mU/mg protein | 0.04 mU/mg protein | |
| MG-132 / 24h | 0.68 mU/mg protein | 0.02 mU/mg protein | ||
| MG-132 / 48 | 0.65 mU/mg protein | 0.03 mU/mg protein | ||
| Lactate dehydrogenase | Cytosol | Control / 24h | 2078 mU/mg protein | n.d. |
| EC 1.1.1.27 | Control / 48h | 1967 mU/mg protein | n.d. | |
| MG-132 / 24h | 1898 mU/mg protein | n.d. | ||
| MG-132 / 48 | 1912 mU/mg protein | n.d. | ||
| Alkaline phosphodiesterase | Plasma membrane | Control / 24h | 628 μU/mg protein | n.d. |
| EC 3.1.4.1 | Control / 48h | 598 μU/mg protein | n.d. | |
| MG-132 / 24h | 605 μU/mg protein | n.d. | ||
| MG-132 / 48 | 611 μU/mg protein | n.d. | ||
| UDP-galactosyltransferase | Microsomes (ER/Golgi) | Control / 24h | 69 nU/mg protein | n.d. |
| E.C. 2.4.1.38 | Control / 48h | 61 nU/mg protein | n.d. | |
| MG-132 / 24h | 65 nU/mg protein | n.d. | ||
| MG-132 / 48 | 64 nU/mg protein | n.d. | ||
| DNA | Nuclei | Control / 24h | 3.48 μg/mg protein | n.d. |
| Control / 48h | 3.46 μg/mg protein | n.d. | ||
| MG-132 / 24h | 3.49 μg/mg protein | n.d. | ||
| MG-132 / 48 | 3.48 μg/mg protein | n.d. |
n.d. not detectable.
Silencing of hsp70 mRNA increases cell death in response to proteasome inhibition
Finally, we wanted to determine how proteasome inhibition and Hsp70 depletion affect cellular viability. The cells were either non-stressed or exposed to 10 μM MG-132 with or without hsp70 mRNA silencing for 24 or 48 hrs. Proteasome inhibition did not markedly affect cellular viability after the 24-hr exposure, but cell death was higher after the 48-hr treatment (Fig. 9). Interestingly, the depletion of Hsp70 levels increased cell death in the proteasome inhibitor-treated ARPE-19 cells (Fig. 9).
Figure 9.
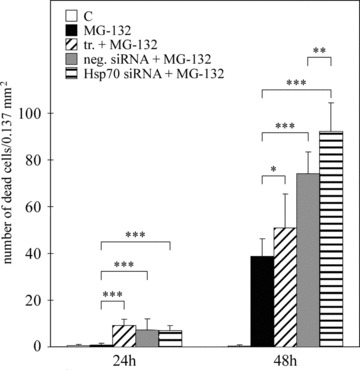
Cell death assay of ARPE-19 cells. The columns represent the number of dead cell/.137 mm2. Cells were either untreated or treated solely with 10 μM MG-132, or with transfection reagent, or with negative or positive hsp70 RNA interference oligonucleotides for 24 and 48 hrs. Variations are represented by standard deviation (n= 9). Mann–Whitney U-test revealed statistically significant differences (*P≤ 0.05, **P≤ 0.01, ***P≤ 0.001).
Discussion
In this study, we demonstrate that Hsp70- and ubiquitin- and LAMP-2-positive deposits induced by proteasome inhibitor MG-132 are autophagocytosed and degraded in human RPE cells. Alkalization of the lysosomes, using 10 mM NH4Cl or 100 nM bafilomycin, slowed down the degradation process. The proteasome inhibitor-induced deposits remained when autophagy was disturbed. The major increase in Hsp70 expression regulated the composition of juxtanuclear protein aggregates in response to protesome inhibition. Hsp70 localized with lysosome-rich cytoplasm. Hsp70 modified the perinuclear aggregation, and the silencing of hsp70 mRNA decreased cellular viability after proteasome inhibition in ARPE-19 cells. Taken together, we believe that the Hsp70 molecular chaperone, lysosomes and proteasomes are closely combined in autophagy-mediated proteolysis in human RPE cells.
One of the main functions of heat-shock proteins is to maintain the correct protein conformation in cells. This is especially important when cells are challenged by stress factors. Hsps also control protein trafficking through the cell organelle membranes and are part of the presentation process of damaged proteins for proteasomal degradation [36]. In line with previous findings [37], abrogation of proteasome-mediated protein degradation caused an intense accumulation of Hsp70 and ubiquitinated protein conjugates, which were colocalized with perinuclear deposits in the ARPE-19 cells. Immunohistochemical analyses have indicated that similar aggregates are enriched amounts of molecular chaperones, including heat shock cognate protein Hsc70 and Hsp40 [13]. Hsp70 was also localized in the cytoplasm and nucleus in the RPE cells. The staining of Hsp70 in nuclei implies that it might have a chaperone function in the nucleus and an autoregulatory role at the transcriptional level via HSF1 transcription factor in response to proteasome inhibition [9].
There is increasing evidence that an efficient clearance of misfolded protein aggregates is essential for cell survival [38–40]. Protein aggregates formed at the cell periphery are delivered along the microtubulus network via dynein-dependent retrograde trafficking to a juxtanuclear location, thus forming aggresomes [13, 28]. The expression of Hsps and the development of aggresomes is part of the cellular defence against misfolded proteins [38, 41]. Here we show that hsp70 mRNA silencing decreased the cellular viability in the proteasome-inhibitor treated cells. At the same time, we observed an increase in the granulation of the perinuclear deposits in response to proteasome inhibition and hsp70 mRNA silencing compared with plain proteasome inhibition. This is evident that Hsp70 regulates the structure of protein aggregates in the cytoplasm probably by the ability to modulate its client protein folding. Previous findings indicate that perinuclear aggregates themselves are nonetheless protective for cells [38, 42]. In this study, we observed that proteasome inhibitor-induced cell death was increased when hsp70 mRNA was silenced. However, the overexpression of Hsp70, via the overexpression vector, did not seem to have any substantial impact on the aggregation process. This is probably attributable to the natural major increase in the Hsp70 expression levels in response to proteasome inhibition.
The reports on ubiquitin localization in aggresomes are inconsistent [13]. In the RPE cells, proteasome inhibition caused juxtanuclear protein aggregates that varied in size and shape. Hsp70 staining often localized strongly with aggregates of different sizes, whereas Ub seemed to consistently localize only with the largest aggregates. The regulatory differences between Hsps and Ub in the aggregation process remain to be studied.
Autophagy is attributable to the turnover of damaged cellular components in response to age-related cellular modifications such as starvation, accumulation of misfolded proteins, oxidative stress and changes in cell volume and in hormonal levels [25, 43, 44]. There are three recognized autophagic pathways: macroautophagy, microautophagy and chaperone-mediated autophagy [45, 46]. Of these, macroautophagy is an inducible form of autophagy that degrades proteins in response to various stress conditions [44]. During macroautophagy, a de novo formed double-membrane seals target proteins or organelles into the autophagosome [47]. Final proteolysis occurs when lysosomes fuse with the autophagosomes and release their enzymes into the vacuole lumen. As far as we are aware, this is the first study to reveal that proteasome inhibitor-induced Hsp70-associated perinuclear protein aggregates are targeted to autophagy in human RPE cells. At the moment, the regulatory mechanisms that mediate the fusion of the autophagosomes with lysosomes are poorly understood. However, recent findings have indicated that Hsp70 is present and modifies the permeabilization of lysosomes [32, 48]. We observed the presence of Hsp70 in lysosome-rich cytoplasm and an increased expression in isolated lysosomal extracts in response to proteasome inhibition. Thus, the inducible Hsp70 might be an important regulator not only for repairing misfolded proteins but also for regulating lysosomal proteolysis, including autophagy, in cells.
The formation of protein aggregates is a hallmark of many neurodegenerative diseases such as Alzheimer’s disease, Parkinson’s disease and many polyglutamine disorders [38]. Several of the proteins identified in the deposits occurring in Alzheimer’s disease are also found in drusens (extracellular protein aggregates) isolated from patients with AMD [49]. One of the main RPE cell functions is to contribute to the outer segment renewal by ingesting and degrading the spent tips of photoreceptor outer segments in lysosomes [50, 51]. Light-generated reactive oxygen species and the lipid-rich tips expose the RPE cells to chronic oxidative stress and protein damage. During aging, lipofuscin accumulates in lysosomes and this disturbs the function of lysosomes and decreases the cellular capacity to degrade damaged proteins and survival against the loading stimuli [51]. In addition, there is one finding indicating that lysosomal lipofuscin may be involved in drusen formation [52]. Interestingly, increased lysosomal LAMP-2A levels in aged tissue restored the macroautophagic and proteasomal pathways to the level observed in corresponding young tissue [53]. Furthermore, mitochondrial functions and cellular ATP levels were restored, and the number of cytosolic oxidized proteins and aggregates of polyubiquitinated proteins were reduced. Preservation of the autophagic activity was associated with a lower intracellular accumulation of damaged proteins, better ability to handle protein damage and improved tissue function. Our findings reveal that Hsp70 plays a central role in improving cellular viability when proteasomal pathways are disturbed in RPE cells. In addition, perinuclear protein aggregates undergo autophagy clearance in proteasome inhibitor-treated RPE cells. These findings open new avenues for understanding the mechanisms of proteolytic processes in retinal cells, and could be useful in the development of novel therapies targeting Hsp70 or the proteins that regulate lysosomal-mediated proteolysis, with the aim of preventing retinal cell deterioration during ageing.
Acknowledgments
This work was supported by the Academy of Finland, the Emil Aaltonen Foundation, the Finnish Cultural Foundation and its North Savo Fund, the Finnish Eye Foundation, the Finnish Eye and Tissue Bank Foundation, the Finnish Eye Science Society, the Finnish Funding Agency for Technology, the Kuopio University Foundation and the National Institutes of Health (EY11515). The authors wish to thank Dr. Ewen MacDonald and Vivian Paganuzzi for checking the language and Päivi Perttula, Virpi Miettinen and Sunna Lappalainen for technical assistance.
References
- 1.Beatty S, Koh H, Phil M, et al. The role of oxidative stress in the pathogenesis of age-related macular degeneration. Surv Ophthalmol. 2000;45:115–34. doi: 10.1016/s0039-6257(00)00140-5. [DOI] [PubMed] [Google Scholar]
- 2.Hjelmeland LM, Cristofolo VJ, Funk W, et al. Senescence of the retinal pigment epithelium. Mol Vis. 1999;5:33. [PubMed] [Google Scholar]
- 3.Young RW. Pathophysiology of age-related macular degeneration. Surv Ophthalmol. 1987;31:291–306. doi: 10.1016/0039-6257(87)90115-9. [DOI] [PubMed] [Google Scholar]
- 4.Nollen EA, Morimoto RI. Chaperoning signaling pathways: molecular chaperones as stress-sensing ‘heat shock’ proteins. J Cell Sci. 2002;115:2809–16. doi: 10.1242/jcs.115.14.2809. [DOI] [PubMed] [Google Scholar]
- 5.Kaarniranta K, Elo M, Sironen R, et al. Hsp70 accumulation in chondrocytic cells exposed to high continuous hydrostatic pressure coincides with mRNA stabilization rather than transcriptional activation. Proc Natl Acad Sci USA. 1998;95:2319–24. doi: 10.1073/pnas.95.5.2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaarniranta K, Oksala N, Karjalainen HM, et al. Neuronal cells show regulatory differences in the hsp70 gene response. Brain Res Mol Brain Res. 2002;101:136–40. doi: 10.1016/s0169-328x(02)00179-1. [DOI] [PubMed] [Google Scholar]
- 7.Kaarniranta K, Ryhanen T, Sironen RK, et al. Geldanamycin activates Hsp70 response and attenuates okadaic acid-induced cytotoxicity in human retinal pigment epithelial cells. Brain Res Mol Brain Res. 2005;137:126–31. doi: 10.1016/j.molbrainres.2005.02.027. [DOI] [PubMed] [Google Scholar]
- 8.Kaarniranta K, Ryhanen T, Karjalainen HM, et al. Geldanamycin increases 4-hydroxynonenal (HNE)-induced cell death in human retinal pigment epithelial cells. Neurosci Lett. 2005;382:185–90. doi: 10.1016/j.neulet.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 9.Abravaya K, Myers MP, Murphy SP, et al. The human heat shock protein hsp70 interacts with HSF, the transcription factor that regulates heat shock gene expression. Genes Dev. 1992;6:1153–64. doi: 10.1101/gad.6.7.1153. [DOI] [PubMed] [Google Scholar]
- 10.Zou J, Guo Y, Guettouche T, et al. Repression of heat shock transcription factor HSF1 activation by HSP90 (HSP90 complex) that forms a stress-sensitive complex with HSF1. Cell. 1998;94:471–80. doi: 10.1016/s0092-8674(00)81588-3. [DOI] [PubMed] [Google Scholar]
- 11.Craig EA, Weissman JS, Horwich AL. Heat shock proteins and molecular chaperones: mediators of protein conformation and turnover in the cell. Cell. 1994;78:365–72. doi: 10.1016/0092-8674(94)90416-2. [DOI] [PubMed] [Google Scholar]
- 12.Goldberg AL, Dice JF. Intracellular protein degradation in mammalian and bacterial cells. Annu Rev Biochem. 1974;43:835–69. doi: 10.1146/annurev.bi.43.070174.004155. [DOI] [PubMed] [Google Scholar]
- 13.Kopito RR. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 2000;10:524–30. doi: 10.1016/s0962-8924(00)01852-3. [DOI] [PubMed] [Google Scholar]
- 14.Mitch WE, Goldberg AL. Mechanisms of muscle wasting. The role of the ubiquitin-proteasome pathway. N Engl J Med. 1996;335:1897–905. doi: 10.1056/NEJM199612193352507. [DOI] [PubMed] [Google Scholar]
- 15.Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–79. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 16.Ciechanover A. Proteolysis: from the lysosome to ubiquitin and the proteasome. Nat Rev Mol Cell Biol. 2005;6:79–87. doi: 10.1038/nrm1552. [DOI] [PubMed] [Google Scholar]
- 17.Cuervo AM, Dice JF. How do intracellular proteolytic systems change with age. Front Biosci. 1998;3:d25–43. doi: 10.2741/a264. [DOI] [PubMed] [Google Scholar]
- 18.Holz FG, Schutt F, Kopitz J, et al. Inhibition of lysosomal degradative functions in RPE cells by a retinoid component of lipofuscin. Invest Ophthalmol Vis Sci. 1999;40:737–43. [PubMed] [Google Scholar]
- 19.Brunk UT, Terman A. Lipofuscin: mechanisms of age-related accumulation and influence on cell function. Free Radic Biol Med. 2002;33:611–9. doi: 10.1016/s0891-5849(02)00959-0. [DOI] [PubMed] [Google Scholar]
- 20.Finnemann SC, Leung LW, Rodriguez-Boulan E. The lipofuscin component A2E selectively inhibits phagolysosomal degradation of photoreceptor phospholipid by the retinal pigment epithelium. Proc Natl Acad Sci USA. 2002;99:3842–7. doi: 10.1073/pnas.052025899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shamsi FA, Boulton M. Inhibition of RPE lysosomal and antioxidant activity by the age pigment lipofuscin. Invest Ophthalmol Vis Sci. 2001;42:3041–6. [PubMed] [Google Scholar]
- 22.Schutt F, Bergmann M, Holz FG, et al. Proteins modified by malondialdehyde, 4-hydroxynonenal, or advanced glycation end products in lipofuscin of human retinal pigment epithelium. Invest Ophthalmol Vis Sci. 2003;44:3663–8. doi: 10.1167/iovs.03-0172. [DOI] [PubMed] [Google Scholar]
- 23.Kaemmerer E, Schutt F, Krohne TU, et al. Effects of lipid peroxidation-related protein modifications on RPE lysosomal functions and POS phagocytosis. Invest Ophthalmol Vis Sci. 2007;48:1342–7. doi: 10.1167/iovs.06-0549. [DOI] [PubMed] [Google Scholar]
- 24.Terman A, Brunk UT. Lipofuscin. Int J Biochem Cell Biol. 2004;36:1400–4. doi: 10.1016/j.biocel.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 25.Martinez-Vicente M, Sovak G, Cuervo AM. Protein degradation and aging. Exp Gerontol. 2005;40:622–33. doi: 10.1016/j.exger.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 26.Terman A, Gustafsson B, Brunk UT. Autophagy, organelles and ageing. J Pathol. 2007;211:134–43. doi: 10.1002/path.2094. [DOI] [PubMed] [Google Scholar]
- 27.Wojcik C. Regulation of apoptosis by the ubiquitin and proteasome pathway. J Cell Mol Med. 2002;6:25–48. doi: 10.1111/j.1582-4934.2002.tb00309.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Iwata A, Riley BE, Johnston JA, et al. HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J Biol Chem. 2005;280:40282–92. doi: 10.1074/jbc.M508786200. [DOI] [PubMed] [Google Scholar]
- 29.Pandey UB, Batlevi Y, Baehrecke EH, et al. HDAC6 at the intersection of autophagy, the ubiquitin-proteasome system and neurodegeneration. Autophagy. 2007;3:643–5. doi: 10.4161/auto.5050. [DOI] [PubMed] [Google Scholar]
- 30.Inoue H, Nojima H, Okayama H. High efficiency transformation of Escherichia coli with plasmids. Gene. 1990;96:23–8. doi: 10.1016/0378-1119(90)90336-p. [DOI] [PubMed] [Google Scholar]
- 31.Sambrook J. Cold Spring Harbor Laboratory Press; 1989. Cold Spring Harbor: [Google Scholar]
- 32.Schutt F, Bergmann M, Holz FG, et al. Isolation of intact lysosomes from human RPE cells and effects of A2-E on the integrity of the lysosomal and other cellular membranes. Graefes Arch Clin Exp Ophthalmol. 2002;240:983–8. doi: 10.1007/s00417-002-0558-8. [DOI] [PubMed] [Google Scholar]
- 33.Chen CT, Bhargava M, Lin PM, et al. Time, stress, and location dependent chondrocyte death and collagen damage in cyclically loaded articular cartilage. J Orthop Res. 2003;21:888–98. doi: 10.1016/S0736-0266(03)00050-0. [DOI] [PubMed] [Google Scholar]
- 34.Eskelinen EL, Tanaka Y, Saftig P. At the acidic edge: emerging functions for lysosomal membrane proteins. Trends Cell Biol. 2003;13:137–45. doi: 10.1016/s0962-8924(03)00005-9. [DOI] [PubMed] [Google Scholar]
- 35.Kaushik S, Cuervo AM. Autophagy as a cell-repair mechanism: activation of chaperone-mediated autophagy during oxidative stress. Mol Aspects Med. 2006;27:444–54. doi: 10.1016/j.mam.2006.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sreedhar AS, Soti C, Csermely P. Inhibition of Hsp90: a new strategy for inhibiting protein kinases. Biochim Biophys Acta. 2004;1697:233–42. doi: 10.1016/j.bbapap.2003.11.027. [DOI] [PubMed] [Google Scholar]
- 37.Pirkkala L, Alastalo TP, Zuo X, et al. Disruption of heat shock factor 1 reveals an essential role in the ubiquitin proteolytic pathway. Mol Cell Biol. 2000;20:2670–5. doi: 10.1128/mcb.20.8.2670-2675.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Taylor JP, Tanaka F, Robitschek J, et al. Aggresomes protect cells by enhancing the degradation of toxic polyglutamine-containing protein. Hum Mol Genet. 2003;12:749–57. doi: 10.1093/hmg/ddg074. [DOI] [PubMed] [Google Scholar]
- 39.Kawaguchi Y, Kovacs JJ, McLaurin A, et al. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell. 2003;115:727–38. doi: 10.1016/s0092-8674(03)00939-5. [DOI] [PubMed] [Google Scholar]
- 40.Holmberg CI, Staniszewski KE, Mensah KN, et al. Inefficient degradation of truncated polyglutamine proteins by the proteasome. EMBO J. 2004;23:4307–18. doi: 10.1038/sj.emboj.7600426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mosser DD, Caron AW, Bourget L, et al. The chaperone function of hsp70 is required for protection against stress-induced apoptosis. Mol Cell Biol. 2000;20:7146–59. doi: 10.1128/mcb.20.19.7146-7159.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bennett EJ, Bence NF, Jayakumar R, et al. Global impairment of the ubiquitin-proteasome system by nuclear or cytoplasmic protein aggregates precedes inclusion body formation. Mol Cell. 2005;17:351–65. doi: 10.1016/j.molcel.2004.12.021. [DOI] [PubMed] [Google Scholar]
- 43.Cuervo AM. Autophagy: in sickness and in health. Trends Cell Biol. 2004;14:70–7. doi: 10.1016/j.tcb.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 44.Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science. 2004;306:990–5. doi: 10.1126/science.1099993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cuervo AM. Autophagy: many paths to the same end. Mol Cell Biochem. 2004;263:55–72. doi: 10.1023/B:MCBI.0000041848.57020.57. [DOI] [PubMed] [Google Scholar]
- 46.Eskelinen EL. Maturation of autophagic vacuoles in Mammalian cells. Autophagy. 2005;1:1–10. doi: 10.4161/auto.1.1.1270. [DOI] [PubMed] [Google Scholar]
- 47.Rubinsztein DC. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature. 2006;443:780–6. doi: 10.1038/nature05291. [DOI] [PubMed] [Google Scholar]
- 48.Daugaard M, Kirkegaard-Sorensen T, Ostenfeld MS, et al. Lens epithelium-derived growth factor is an Hsp70–2 regulated guardian of lysosomal stability in human cancer. Cancer Res. 2007;67:2559–67. doi: 10.1158/0008-5472.CAN-06-4121. [DOI] [PubMed] [Google Scholar]
- 49.Mullins RF, Russell SR, Anderson DH, et al. Drusen associated with aging and age-related macular degeneration contain proteins common to extracellular deposits associated with atherosclerosis, elastosis, amyloidosis, and dense deposit disease. FASEB J. 2000;14:835–46. [PubMed] [Google Scholar]
- 50.Boulton M, Dayhaw-Barker P. The role of the retinal pigment epithelium: topographical variation and ageing changes. Eye. 2001;15:384–9. doi: 10.1038/eye.2001.141. [DOI] [PubMed] [Google Scholar]
- 51.Ambati J, Ambati BK, Yoo SH, et al. Age-related macular degeneration: etiology, pathogenesis, and therapeutic strategies. Surv Ophthalmol. 2003;48:257–93. doi: 10.1016/s0039-6257(03)00030-4. [DOI] [PubMed] [Google Scholar]
- 52.Zhou J, Jang YP, Kim SR, et al. Complement activation by photooxidation products of A2E, a lipofuscin constituent of the retinal pigment epithelium. Proc Natl Acad Sci USA. 2006;103:16182–7. doi: 10.1073/pnas.0604255103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang C, Cuervo AM. Restoration of chaperone-mediated autophagy in aging liver improves cellular maintenance and hepatic function. Nat Med. 2008;14:959–65. doi: 10.1038/nm.1851. [DOI] [PMC free article] [PubMed] [Google Scholar]