

Abstract
Protein-protein interactions (PPIs) are critical in numerous biological processes including signaling transduction, function regulations, and disease development. To regulate PPIs has been thought to be challenging due to their highly dynamic and expansive interfacial areas. Nonetheless, successful examples have been reported of targeting PPI using small molecules, peptides, and proteins. Peptides, especially macrocyclic peptides have proven to be a particularly useful tool to inhibit PPIs for their exquisite potency, stability and selectivity. Herein we review the recent developments of this area of research, focusing on the macrocyclic peptides isolated from natural products, identified from library screening, and rationally designed based on structures, as PPI regulators.
Keywords: macrocyclic peptide, protein-protein interface, rational design, inhibitor
Introduction
Protein-protein interactions (PPIs) regulate numerous essential processes of life, including replication and transcription of nucleic acids, protein biosynthesis, signal transduction, metabolic cycle, and also viral diffusion and virus survival in host cell.1–5 Hence, extensive researches focusing on regulation of PPI have been carried out in the last decade.6–8
Current PPI mediators are mainly divided into three groups: small molecules, linear/cyclic peptides, and proteins.9–12 In this review, we focus on the macrocyclic peptide PPI mediators as they have the following advantages. Firstly, peptides are large enough to render an extensive, continuous binding surface that can competitively inhibit PPIs.13, 14 The polypeptide backbones usually make them more soluble in water than small molecule organic compounds. Secondly, when compared to proteins, peptides can be more readily synthesized, characterized and purified.14, 15 Finally, compared to linear peptides, cyclic peptides are: (A) more rigid, therefore demand less entropy penalty upon association with their targets, which in turn elevates their binding affinities (Figure 1); and (B) more stable in complex biological environments due to enhanced resistance to proteolytic degradation.14–16
Figure 1.

Cyclic peptides are more rigid than their linear counterparts upon binding to proteins, leading to more potent inhibitors of PPIs.
In term of their functions, macrocyclic peptide PPI regulators can be generally described as activators and inhibitors. An activator can facilitate protein-protein associations,17 thereby increasing the PPI-mediated cellular response. For example, peptide A (Figure 2) was found to activate human melanocortin 3 receptor (hMC3R), an inhibitory autoreceptor on the surface of proopiomelanocortin neurons.17–20 Meanwhile, the macrocyclic peptide inhibitors suppress the PPI-induced response by disrupting the protein-protein complex formation. For example, macrocyclic peptide mimetic B (Figure 2) showed significant inhibition of the interaction between mixed lineage leukemia 1 (MLL1) and its mediator menin with a Ki of 4.7 nM.21 In this mini-review, representative macrocyclic peptide PPI regulators identified from natural products and peptide library screening, as well as developed base on known protein structures are discussed.
Figure 2.
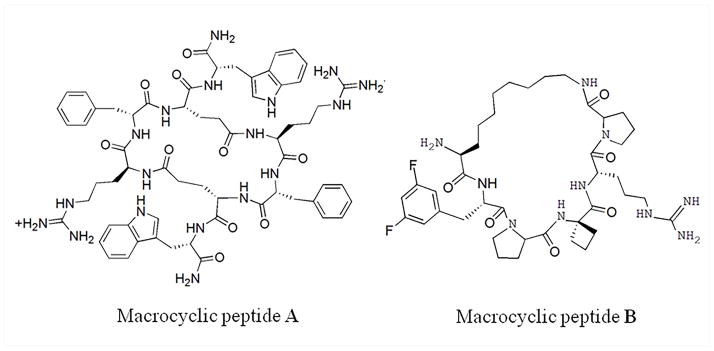
The chemical structures of macrocyclic peptide agonist, A, and antagonist, B, of protein-protein interactions.
Macrocyclic peptides isolated from natural products
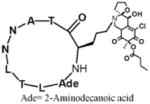
Kallikrein kinin system regulates angiocarpy, urinary and nervous systems, and other physiological functions.22 Sunflower trypsin inhibitor-1 (SFTI-1) isolated from Escherichia coli has been shown to be an inhibitor of Kallikrein-5 (KLK5) by blocking its interaction with protease activated receptor-2 (PAR-2).23 Due to the difficulty of their purification, SFTI-1 analogues 1 (Table 1) was prepared using solid-phase peptide synthesis (SPPS) with an overall 25% isolated yield.23 The SFTI-1 analogue, macrocyclic peptide 1, was stabilized by the introduction of the disulfide linkage. This effective inhibition to KLK5 was monitored by the decreased Ca2+ influx. And this stronger inhibitory activity implies the potential therapeutic application to atopic dermatitis.23 P53, “the guardian of the genome”, is well known for its role as an administrator of cell proliferation, growth and apoptosis.24 Over 50% of human tumors contain p53 mutations.24 Oncogene murine double minute-2 (MDM2) can tightly bind to the DNA-binding domain of p53, consequently regulating transcription of the genes and apoptosis.25 Duncan et al. found a macrocyclic peptide, chlorofusin (2) (Table 1), derived from Microdochium caespitosum can inhibit the p53-MDM2 interaction by competing with p53 binding with MDM2.26 The titration of 2 in the dissociation-enhanced lanthanide fluorescent immunoassay (DELFIA)-modified enzyme linked immunosorbent assay (ELISA) determined the inhibitory potency of 2 with an IC50 value of 4.6 μM.26 For the p53-MDM2 interaction, there have been a number of elegant studies on potent small molecule and peptidomimetic inhibitors which have been reviewed elsewhere.27, 28
Table 1.
The sequences of macrocyclic peptides isolated from natural products.
| Peptides | PPI Targets | Peptide Sequences |
|---|---|---|
| 1 | (PAR-2)-KLK5 |
|
| 2 | p53-MDM2 |
|
Macrocyclic peptides selected from library screening
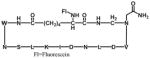
Developing highly efficient anti-human immunodeficiency virus (HIV) drug remains a formidable challenge to scientists around the world. In recent years, inhibiting HIV type 1 (HIV-1) integrase (IN) that inserts viral cDNA into the human genome has gained much interests.13 One of widely known IN-related proteins is its cellular cofactor, LEDGF/p75 that facilitates IN binding to viral cDNA and makes HIV-1 replication.29 By screening a cyclic peptide library derived from the LEDGF/p75 361–370 fragment, peptide 3 (Table 2) was identified to be a potent IN inhibitor. Peptide 3 effectively binds to IN, inhibiting its catalytic activity in vitro with high stability (effective even after eight days).30
Table 2.
The sequences of macrocyclic peptides selected from library screening.
| Peptides | PPI Targets | Peptide Sequences |
|---|---|---|
| 3 | IN-LEDGF/p75 |
|
| 4 | CtBP1-CtBP2 |
|
| 5 | TNFα-TNFR1 |
|
| 6 | p53-HDM2 |

|
As a result to search for transcription factors PPI regulators, macrocyclic peptide 4 was selected from screening of a 64 million-membered combinatory cyclic peptides library. Peptide 4 disrupts the NADH-dependent C-terminal binding protein (CtBP) dimerization.28 It was shown that peptide 4 modulates the maintenance of mitotic fidelity in breast cancer cells (strongly dependent on glycolysis) by breaking the CtBP dimerization without affecting the mitotic fidelity of cells (independent of glycolysis).31 The effect of peptide 4 on disrupting of CtBP dimerization was confirmed using microscopy in COS-7 monkey kidney cells co-transfected YFP–CtBP and CFP–CtBP vectors. 28
Tumor necrosis factor-alpha (TNFα) is a pleiotropic inflammatory cytokine related to many disease developments including tuberculosis, septic shock, and various chronic inflammatory disorders.32, 33 TNFα-induced response starts with the binding of TNFα trimer to the extracellular domain of TNFα receptor 1 (TNFR1) followed by inhibitory protein release.33 Hence blocking the TNFα-TNFR1 interaction is a potential strategy to regulate the TNFα signaling. By building and screening the bicyclic peptide library, Lian et al. found peptide 5 (Table 2) can strongly bind to TNFα and inhibit the TNFα-TNFR1 interaction, significantly extending cell life.34 Biotinylated TNFα was immobilized on the Neutravidin-coated microtiter plate before incubated with horseradish peroxidase (HRP)-conjugated TNFR1 with different concentrations of peptide 5. The amount of HRP–TNFR1 bound to each well was then quantitated by an ELISA assay. Peptide 5 was found to inhibit TNFα-TNFR1 with an IC50 value of 3.1 ± 0.3 μM.34
As mentioned above, since its discovery, p53 has been an important target for anti-cancer drug development. As the negative regulator of p53, human double minute-2 (HDM2) compromises the cancer-preventing functions of p53. Intensive research efforts to develop disruptors of the association of p53 and HDM2 have been successfully carried out, including some macrocyclic peptides as inhibitors to the PPI between p53 and HMD2.9, 35 A β-Hairpin peptidomimetic, peptide 6 (Table 2), with 6-chloro group incorporated in its Aib-Pmp-Trp23-Glu indole side chain, was identified from screening a phage display peptide library.36 Using an ELISA assay, the authors found that peptide 6 competes with p53 by recognizing the same binding pocket on the surface of HDM2, consequently inhibits the p53-HDM2 association with IC50 value of 5 ± 1 nM.36
Macrocyclic peptides developed by structure-based rational design
Toll-like receptors (TLRs) are a protein family and an integral part of innate and adaptive immune systems. All TLR response is activated by their respective signal molecules, pathogen-associated molecular patterns (PAMPs).37 It is known that deficiency or excess of TLR4-mediated immune response, dependent on the heterodimer of TLR4 and myeloid differentiation factor 2 (MD2) formation, both can result in pathological disorders.38, 39 Recently, a number of reports have appeared on a variety of agonists and antagonists for regulation of the TLR4-MD2 interaction, including synthetic molecules, linear peptides and cyclic peptides.40, 41 Most recently, Gao et al. compared linear peptides with cyclic peptides in the regulation of TLR4 signaling and found that macrocyclic peptides 7 and 8 (Table 3) cause synergistic activation with lipopolysaccharide (LPS) to turn on TLR4 signaling monitored by nitric oxide production.42 It is worth noting that these macrocyclic peptides were rationally designed by computer simulation. Using the Rosetta program, the authors designed de novo MD2 mimics that target the TLR4-MD2 protein-protein interface.42 In a separate study, macrocyclic peptides designed based on the natural sequence of MD2 were also found to inhibit the LPS-induced proinflammatory cytokine release.41 Experimental and computational simulation results demonstrated that the MD2 mimic macrocyclic peptide 9 (Table 3) can occupy the MD2-binding region on the TLR4 surface, blocking the LPS-induced inflammation signal.41 Using direct biophysical binding assays, Liu et al. validated that the inhibitor peptide 9 is selective and specific for TLR4 in the presence of other TLRs.43 Peptide 7 and 8 both can synergistically enhance the LPS-induced TLR4 signal, while their corresponding linear peptides showed little effects. This observation suggests that the disulfide bond bridged macrocyclic peptide scaffold is essential for their agonistic activities. As a comparison, macrocyclic peptide 9, a truncated functional region of human MD2, can inhibit the TLR4-MD2 interaction, leading to specific inhibition of the TLR4 signaling. Exact mechanism of the different functions of peptide 7, 8 and 9 remains elusive at present. Nonetheless, structurally homologous ligands caused either agonistic or antagonistic effects to various TLRs have been observed with their native ligands as well,44 implying minimal conformational regulations of these innate immune receptors may result in opposite inflammatory response in cells.
Table 3.
The sequences of macrocyclic peptides developed by structure-based rational design.
| Peptides | PPI Targets | Peptide Sequences |
|---|---|---|
| 7 | TLR4-MD2 |
|
| 8 | TLR4-MD2 |
|
| 9 | TLR4-MD2 |
|
| 10 | CagL-αvβ3 |
|
| 11 | coat protein-integrin |
|
| 12 | SPSB2-iNOS |
|
Helicobacter pylori is a widely studied gastric bacterium and carcinogen. H. pylori targets gastric cells using the RGD sequence (-Arg-Gly-Asp-) of the protein cytotoxin-associated gene L (CagL) that binds to integrins α5β1 and αvβ3 on the host cell surface.45 An approach for H. pylori related gastric diseases like chronic atrophic gastritis and peptic ulcers, is to obstruct the CagL-αvβ3 interaction. For example, a cyclic RGD derivative 10 (Table 3) designed as CagL functional mimics occupies the binding region of CagL-αvβ3 PPI and recognizes αvβ3.45 A cell adhesion assay showed that macrocyclic peptide 10 has higher binding affinity with integrins α5β1 and αvβ3 than CagL.45 Moreover, the NMR measurements and molecular dynamics (MD) calculations demonstrated that the inhibition of the CagL-αvβ3 interaction mainly relies on secondary structure elements, further identified the RGD sequence is favored.45 In addition, the cyclic peptide 11 (Table 3) with RGD sequence was also used to phage delivery in tumor cells recently. Phage VIII, amplified by isopropyl β-D-1-thiogalactopyranoside (IPTG) and decorated with peptides 11 on its major coat protein, can bind with integrins on tumor cells surface and consequently internalized by host cells.46 The amount of 11 decorated can be modulated by IPTG levels and the internalized phage can be quantified by fluorescent microscopy imaging.46
Another example is the macrocyclic peptide inhibitor of the inducible nitric oxide synthase (iNOS). In immune system, nitric oxide controlled by iNOS is an essential molecule to host body for its role of safety alarm when meeting with exogenous threats. It is known that iNOS is regulated by splA kinase and ryanodine receptor (SPRY) domain-containing suppressor of cytokine signaling box protein2 (SPSB2).47 Inhibiting the association of protein SPSB2 and iNOS is a reasonable strategy. Macrocyclic peptide 12 (Table 3) containing the specific binding sequence of iNOS, DINNN, can bind with SPSB2 instead of iNOS in bone marrow-derived macrophages (BMDM) lysates. It is developed to inhibit the interaction of SPSB2-iNOS successfully with potential to be an anti-infective drug.47
Common strategies of ring introduction
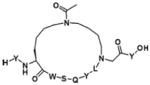
Finally, we will discuss a few commonly used methods employed in the cyclic peptides synthesis. In many naturally occurring cyclic peptides, the rings are formed through disulfide bonds between cysteine residues. However, disulfide bonds may not be maintained under physiological conditions. The “click chemistry”48 has been widely used as a synthetic method to generate macrocyclic peptides.49 A good example is that Kawamoto et al. used Huisgen1,3-dipolar cycloaddition reaction to synthesize triazole-stapled B-cell leukemia/lymphoma 9 (BCL9) α-helical peptides 13 (Table 4), which can inhibit the interaction between BCL9 and β-catenin.50 In addition, utilizing “amine cross-linkages”51 (cyclization through a secondry amine) instead of disulfide bonds becomes an alternative method.51 For instance, the WP9QY (Table 4) derivative peptide 14 (Table 4), with amine cross-linkage replacement of disulfide bond, resulted in higher stability and less deactivaition in vitro. With the improvement in enzymatic stability, the WP9QY derivative 14 can be used to target the interaction of receptor activator of NF-κB (RANK) and receptor activator of NF-κB ligand (RANKL) by binding to RANKL and inhibit RANKL-induced abnormal immunization. Another choice is to replace disulfide bonds by “hydrophobic cross-links”52 (cyclization linked by aliphatic hydrocarbon) to facilitate peptide folding.52 Upon cross-linking with olefin metathesis catalyzed by ruthenium complex, the macrocyclic compound 15 (Table 4) turns into inhibitor of interaction between human adaptor protein 14-3-3 and virulence factor exoenzyme S (ExoS) with strongly enhanced binding affinity with 14-3-3.52 Finally and importantly, two classic examples using olefin metathesis to generate hydrocarbon-stapled peptides are presented here. Walensky et al. successfully synthesized “stapled” peptide 16 (Table 4) with stable α-helix conformation.53 This peptide was able to recognize the BH2-binding pocking on the surface of pro-survival oncogenic protein, B-cell CLL/lymphoma 2 (BCL-2), activating the apoptotic pathway to kill leukemia cells.53 Similarly, peptide 17 (Table 4) was designed by Beak et. al. for inhibiting MDM2-p53 interaction with an all-hydrocarbon staple that conformationally and proteolytically stabilized the short peptide.54 Overall, the cross-link strategy is becoming a useful tool to provide potent peptide PPI inhibitors.
Table 4.
The sequences of macrocyclic peptides synthesized by different cyclization methods.
| Peptides | PPI Targets | Peptide Sequences |
|---|---|---|
| 13 | BCL9-(β-catenin) |
|
| 14 | RANK-RANKL |
|
| 15 | ExoS-(14-3-3) |
|
| 16 | (BCL-2)-BAK |
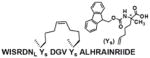
|
| 17 | p53-MDM2 |
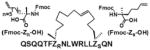
|
| 18(WP9QY) | RANK-RANKL |
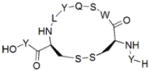
|
Outlook
In this mini review, we briefly discuss macrocyclic peptides as PPI modulators, including representative macrocyclic peptides isolated from natural products, selected from library screening, and obtained through structure-based designs. New technologies such as computer simulation55 provide clear guidance for rational designs and preliminary structure–activity relationship information. We expect to see even more remarkable progress of this research area in the next a few years. With more information available for previously hard-to-get protein complex structures and more computing powers, rationally targeting protein-protein interfaces by macrocyclic peptide may prove to be a generally applicable method to generate new candidates for drug developments. Nonetheless, challenges remains: first, some macrocyclic peptides are hard to synthesize. The synthesis is particularly difficulty with those macrocyclic peptides with more than one ring in a single molecule. Furthermore, the stereochemistry and backbone substitution patterns of unnatural macrocyclic peptides may pose further challenge in their library preparation. Second, the complex crystal structure of macrocyclic peptide and protein can be difficult to get, restricting the further understanding of the mechanism between the targeted receptors and their macrocyclic peptide ligands. This is especially true for some protein groups such as transmembrane proteins, which compose a substantial portion of relevant drug targets. Third, average macrocyclic peptides are still larger than conventional drug-like small molecules. This is helpful when one targets PPIs as they can make more extensive contact with the protein target and can do so without excessive entropic penalty. However, their elevated molecular weights make bioavailability less optimal, which need to be taken into consideration during further therapeutic developments.
Acknowledgments
We thank the National Institutes of Health (R01GM101279 and R01GM103843) and Tsinghua University for financial supports.
References
- 1.McGhee JD, Hippel PHV. J Mol Biol. 1974;86:469–489. doi: 10.1016/0022-2836(74)90031-x. [DOI] [PubMed] [Google Scholar]
- 2.Zhang AX, Wassarman KM, Ortega J, Steven AC, Storz G. Mol Cell. 2002;9:11–22. doi: 10.1016/s1097-2765(01)00437-3. [DOI] [PubMed] [Google Scholar]
- 3.Pawson T, Nash P. Gene Dev. 2000;14:1027–1047. [PubMed] [Google Scholar]
- 4.Lomberk G, Mathison AJ, Grzenda A, Seo S, Demars CJ, Rizvi S, Bonilla-Velez J, Calvo E, Fernandez-Zapico ME, Iovanna J, Buttar NS, Urrutia R. J Biol Chem. 2012;287:13026–13039. doi: 10.1074/jbc.M112.342634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.He YP, Nakao HH, Tan SL, Polyak PJ, Neddermann P, Vijaysri S, Jacobs BL, Katze MG. J Virol. 2002;76:9207–9217. doi: 10.1128/JVI.76.18.9207-9217.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cusick ME, Klitgord N, Vidal M, Hill DE. Hum Mol Genet. 2005;14:R171–R181. doi: 10.1093/hmg/ddi335. [DOI] [PubMed] [Google Scholar]
- 7.Vidal M. Febs Lett. 2009;583:3891–3894. doi: 10.1016/j.febslet.2009.11.024. [DOI] [PubMed] [Google Scholar]
- 8.White TC, Berny MA, Robinson DK, Yin H, DeGrado WF, Hanson SR, McCarty OJT. Febs J. 2007;274:1481–1491. doi: 10.1111/j.1742-4658.2007.05689.x. [DOI] [PubMed] [Google Scholar]
- 9.Fischer PM. Int J Pept Res Ther. 2006;12:3–19. doi: 10.1007/s10989-006-9016-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arkin MR, Wells JA. Nat Rev Drug Discov. 2004;3:301–317. doi: 10.1038/nrd1343. [DOI] [PubMed] [Google Scholar]
- 11.Angeles Bonache M, Balsera B, Lopez-Mendez B, Millet O, Brancaccio D, Gomez-Monterrey I, Carotenuto A, Pavone LM, Reille-Seroussi M, Gagey-Eilstein N, Vidal M, de la Torre-Martinez R, Fernandez-Carvajal A, Ferrer-Montiel A, Teresa Garcia-Lopez M, Martin-Martinez M, Jesus Perez de Vega M, Gonzalez-Muniz R. ACS Comb Sci. 2014;16:250–258. doi: 10.1021/co500005x. [DOI] [PubMed] [Google Scholar]
- 12.Granier S, Terrillon S, Pascal R, Demene H, Bouvier M, Guillon G, Mendre C. J Biol Chem. 2004;279:50904–50914. doi: 10.1074/jbc.M405089200. [DOI] [PubMed] [Google Scholar]
- 13.Maes M, Loyter A, Friedler A. Febs J. 2012;279:2795–2809. doi: 10.1111/j.1742-4658.2012.08680.x. [DOI] [PubMed] [Google Scholar]
- 14.Benyamini H, Friedler A. Future Med Chem. 2010;2:989–1003. doi: 10.4155/fmc.10.196. [DOI] [PubMed] [Google Scholar]
- 15.Katz C, Levy-Beladev L, Rotem-Bamberger S, Rito T, Rudiger SGD, Friedler A. Chem Soc Rev. 2011;40:2131–2145. doi: 10.1039/c0cs00029a. [DOI] [PubMed] [Google Scholar]
- 16.Gavenonis J, Sheneman BA, Siegert TR, Eshelman MR, Kritzer JA. Nat Chem Biol. 2014;10:716–722. doi: 10.1038/nchembio.1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mayorov AV, Cai M, Palmer ES, Liu Z, Cain JP, Vagner J, Trivedi D, Hruby VJ. Peptides. 2010;31:1894–1905. doi: 10.1016/j.peptides.2010.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Butler AA. Peptides. 2006;27:281–290. doi: 10.1016/j.peptides.2005.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cone RD. Endocr Rev. 2006;27:736–749. doi: 10.1210/er.2006-0034. [DOI] [PubMed] [Google Scholar]
- 20.Mayorov AV, Cai MY, Chandler KB, Petrov RR, Van Scoy AR, Yu ZR, Tanaka DK, Trivedi D, Hruby VJ. J Med Chem. 2006;49:1946–1952. doi: 10.1021/jm0510326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou H, Liu L, Huang J, Bernard D, Karatas H, Navarro A, Lei M, Wang S. J Med Chem. 2013;56:1113–1123. doi: 10.1021/jm3015298. [DOI] [PubMed] [Google Scholar]
- 22.Swedberg JE, Nigon LV, Reid JC, de Veer SJ, Walpole CM, Stephens CR, Walsh TP, Takayama TK, Hooper JD, Clements JA, Buckle AM, Harris JM. Chem Biol. 2009;16:633–643. doi: 10.1016/j.chembiol.2009.05.008. [DOI] [PubMed] [Google Scholar]
- 23.Shariff L, Zhu Y, Cowper B, Di WL, Macmillan D. Tetrahedron. 2014;70:7675–7680. [Google Scholar]
- 24.Levine AJ, Momand J, Finlay CA. Nature. 1991;351:453–456. doi: 10.1038/351453a0. [DOI] [PubMed] [Google Scholar]
- 25.Oliner JD, Pietenpol JA, Thiagalingam S, Gvuris J, Kinzler KW, Vogelstein B. Nature. 1993;362:857–860. doi: 10.1038/362857a0. [DOI] [PubMed] [Google Scholar]
- 26.Duncan SJ, Gruschow S, Williams DH, McNicholas C, Purewal R, Hajek M, Gerlitz M, Martin S, Wrigley SK, Moore M. J Am Chem Soc. 2001;123:554–560. doi: 10.1021/ja002940p. [DOI] [PubMed] [Google Scholar]
- 27.Wang W, Hu Y. Med Res Rev. 2012;32:1159–1196. doi: 10.1002/med.20236. [DOI] [PubMed] [Google Scholar]
- 28.Liu M, Pazgier M, Li C, Yuan W, Li C, Lu W. Angew Chem Int Edit. 2010;49:3649–3652. doi: 10.1002/anie.201000329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cherepanov P, Maertens G, Proost P, Devreese B, Van Beeumen J, Engelborghs Y, De Clercq E, Debyser Z. J Biol Chem. 2003;278:372–381. doi: 10.1074/jbc.M209278200. [DOI] [PubMed] [Google Scholar]
- 30.Hayouka Z, Hurevich M, Levin A, Benyamini H, Iosub A, Maes M, Shalev DE, Loyter A, Gilon C, Friedler A. Bioorgan Med Chem. 2010;18:8388–8395. doi: 10.1016/j.bmc.2010.09.046. [DOI] [PubMed] [Google Scholar]
- 31.Birts CN, Nijjar SK, Mardle CA, Hoakwie F, Duriez PJ, Blaydes JP, Tavassoli A. Chem Sci. 2013;4:3046–3057. doi: 10.1039/c3sc50481f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kawakami M, Cerami A. J Exp Med. 1981;154:631–639. doi: 10.1084/jem.154.3.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen GQ, Goeddel DV. Science. 2002;296:1634–1635. doi: 10.1126/science.1071924. [DOI] [PubMed] [Google Scholar]
- 34.Lian W, Upadhyaya P, Rhodes CA, Liu Y, Pei D. J Am Chem Soc. 2013;135:11990–11995. doi: 10.1021/ja405106u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bautista AD, Appelbaum JS, Craig CJ, Michel J, Schepartz A. J Am Chem Soc. 2010;132:2904–2096. doi: 10.1021/ja910715u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Garcia-Echeverria C, Chene P, Blommers MJJ, Furet P. J Med Chem. 2000;43:3205–3208. doi: 10.1021/jm990966p. [DOI] [PubMed] [Google Scholar]
- 37.Beutler B. Nature. 2004;430:257–263. doi: 10.1038/nature02761. [DOI] [PubMed] [Google Scholar]
- 38.Akira S, Uematsu S, Takeuchi O. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 39.Due MR, Piekarz AD, Wilson N, Feldman P, Ripsch MS, Chavez S, Yin H, Khanna R, White FA. J Neuroinflamm. 2012;9:200. doi: 10.1186/1742-2094-9-200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cheng K, Wang X, Zhang S, Yin H. Angew Chem Int Edit. 2012;51:12246–12249. doi: 10.1002/anie.201204910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Slivka PF, Shridhar M, Lee G-i, Sammond DW, Hutchinson MR, Martinko AJ, Buchanan MM, Sholar PW, Kearney JJ, Harrison JA, Watkins LR, Yin H. ChemBioChem. 2009;10:645–649. doi: 10.1002/cbic.200800769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gao M, London N, Cheng K, Tamura R, Jin J, Schueler-Furman O, Yin H. Tetrahedron. 2014;70:7664–7668. doi: 10.1016/j.tet.2014.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu L, Ghosh N, Slivka PF, Fiorini Z, Hutchinson MR, Watkins LR, Yin H. ChemBioChem. 2011;12:1827–1831. doi: 10.1002/cbic.201100211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Park BS, Song DH, Kim HM, Choi BS, Lee H, Lee JO. Nature. 2009;458:1191–1195. doi: 10.1038/nature07830. [DOI] [PubMed] [Google Scholar]
- 45.Conradi J, Huber S, Gaus K, Mertink F, Royo Gracia S, Strijowski U, Backert S, Sewald N. Amino Acids. 2012;43:219–232. doi: 10.1007/s00726-011-1066-0. [DOI] [PubMed] [Google Scholar]
- 46.Choi DS, Jin HE, Yoo SY, Lee SW. Bioconjugate Chem. 2014;25:216–223. doi: 10.1021/bc4003234. [DOI] [PubMed] [Google Scholar]
- 47.Yap BK, Leung EWW, Yagi H, Galea CA, Chhabra S, Chalmers DK, Nicholson SE, Thompson PE, Norton RS. J Med Chem. 2014;57:7006–7015. doi: 10.1021/jm500596j. [DOI] [PubMed] [Google Scholar]
- 48.Kolb HC, Finn MG, Sharpless KB. Angew Chem Int Edit. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 49.Saludes JP, Morton LA, Ghosh N, Beninson LA, Chapman ER, Fleshner M, Yin H. ACS Chem Biol. 2012;7:1629–1635. doi: 10.1021/cb3002705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kawamoto SA, Coleska A, Ran X, Yi H, Yang CY, Wang S. J Med Chem. 2012;55:1137–1146. doi: 10.1021/jm201125d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Aoki K, Maeda M, Nakae T, Okada Y, Ohya K, Chiba K. Tetrahedron. 2014;70:7774–7779. [Google Scholar]
- 52.Glas A, Bier D, Hahne G, Rademacher C, Ottmann C, Grossmann TN. Angew Chem Int Edit. 2014;53:2489–2493. doi: 10.1002/anie.201310082. [DOI] [PubMed] [Google Scholar]
- 53.Walensky LD, Kung AL, Escher I, Malia TJ, Barbuto S, Wright RD, Wagner G, Verdine GL, Korsmeyer SJ. Science. 2004;305:1466–1470. doi: 10.1126/science.1099191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Baek S, Kutchukian PS, Verdine GL, Huber R, Holak TA, Lee KW, Popowicz GM. J Am Chem Soc. 2011;134:103–106. doi: 10.1021/ja2090367. [DOI] [PubMed] [Google Scholar]
- 55.Joce C, Stahl JA, Shridhar M, Hutchinson MR, Watkins LR, Fedichev PO, Yin H. Bioorg Med Chem Lett. 2010;20:5411–5413. doi: 10.1016/j.bmcl.2010.07.103. [DOI] [PMC free article] [PubMed] [Google Scholar]