

Abstract
Despite our extensive understanding of water-soluble protein folding kinetics, much less is known about the folding dynamics and mechanisms of membrane proteins. However, recent studies have shown that for relatively simple systems, such as peptides that form a transmembrane α-helix, helical dimer, or helix-turn-helix, it is possible to assess the kinetics of several important steps, including peptide binding to the membrane from aqueous solution, peptide folding on the membrane surface, helix insertion into the membrane, and helix-helix association inside the membrane. Herein, we provide a brief review of these studies and also suggest new initiation and probing methods that could lead to improved temporal and structural resolution in future experiments.
Keywords: membrane, membrane peptide, protein folding, stopped-flow kinetics, fluorescence spectroscopy
INTRODUCTION
The study of how water-soluble proteins fold has reached a mature stage wherein advanced theoretical and experimental approaches are routinely applied to examine the folding dynamics of various protein systems.1–9 As a result, we now know a great deal about the mechanisms by which many proteins fold in aqueous solution. In contrast, our current understanding of how membrane proteins fold, specifically the kinetic and mechanistic aspects of the process, is limited. This is partially due to the fact that many of the standard biophysical techniques used to study the structure and folding dynamics of water-soluble proteins are not readily applicable to membrane proteins because this class of proteins requires a hydrophobic rather than a polar environment to fold. For this reason, recent efforts have been devoted to developing new strategies and techniques that can be used to initiate and probe the protein folding processes in a membrane environment. Herein, we provide a concise review of recent studies in this area, with a focus on the folding kinetics of membrane peptides.
Membrane peptides perform a wide range of biological activities and functions, such as viral fusion,10 molecular translocation,11 cell signaling,12 and drug delivery.13 In addition, the two-stage model14 of membrane protein folding suggests that secondary structure formation precedes the formation of tertiary structure. Thus, short peptides that fold into well-defined secondary structures in a membrane environment constitute invaluable model systems for studying the folding mechanisms of membrane proteins. In the following sections, we will first provide an overview of the techniques available to initiate and probe the folding process of membrane peptides. We will then discuss recent studies on the folding kinetics and mechanism of monomeric α-helices, helical dimers, helix-turn-helix motifs, and proteins inside model membranes.
INITIATION AND PROBING METHODS
For the study of protein folding/unfolding kinetics in aqueous solution, many initiation methods are available, such as rapid mixing,15 laser-induced temperature-jump (T-jump)16,17 or pH-jump,18 and photo-triggering.19,20 However, the choice of methods to initiate membrane protein folding kinetics is limited. In practice, the most commonly used initiation method is based on solution mixing. If a membrane peptide is soluble and unstructured in aqueous solution, but folds into a well-defined secondary or tertiary structure in the presence of lipid membranes, it is possible to induce or initiate folding by simply mixing two aqueous solutions (via stopped-flow technique), one containing the peptide of interest and the other containing a membrane mimetic (e.g., vesicles). Similarly, if the membrane binding affinity of a peptide exhibits a strong dependence on a particular solution property, such as pH, the peptide-membrane interaction kinetics can also be initiated by rapidly mixing two solutions that will lead to a change in the desired property of the solution. Most, if not all previous studies on the folding kinetics of membrane peptides have employed such mixing techniques. One major challenge with this initiation method is that if the peptide or protein in question is insoluble in water, detergent micelles and denaturants are required.21–30
Due to its high sensitivity and ease of application, fluorescence spectroscopy is most commonly used to probe the folding and conformational kinetics of membrane peptides and proteins. In particular, the intrinsic fluorophore, tryptophan (Trp), has been widely used to probe the structure and dynamics of membrane protein folding because its fluorescence spectrum and quantum yield are sensitive to the local environment.31 In addition, various extrinsic fluorophores, such as p-cyanophenylalanine (PheCN),32 carboxytetramethyl rhodamine (TAMRA),33 carboxylfluorescein (FAM),34 6-dodecanoyl-2-dimethylaminonaphthalene (Laurdan),35 and fluorescein (FI),36 have also been used to investigate the kinetics of folding/unfolding and association of membrane peptides. Moreover, fluorescence measurements based on fluorescence resonance energy transfer (FRET) have been used to report on the folding kinetics of membrane peptides and proteins as well. Since FRET can provide distance information, it is more advantageous than conventional fluorescence spectroscopy in terms of correlating the observed kinetics to specific conformational changes. Specifically, the following FRET pairs have been used to investigate the folding kinetics and mechanisms of peptides or proteins in a membrane environment: naphthalene/dansyl, pyrene/dansyl,37 PheCN/Trp,38 Trp/dansyl,39 Trp/DBO,40 and Trp/Laurdan.41 Besides fluorescence spectroscopy, other kinetics methods, including stopped-flow circular dichroism (CD),42 hydrogen/deuterium (H/D) exchange,43 SDS-PAGE,44 and time-resolved electron paramagnetic resonance (EPR) spectroscopy,45 are powerful tools for investigating the mechanism of membrane peptide and protein folding.
1. FOLDING KINETICS OF MONOMERIC α-HELICES
Many naturally occurring and designed peptides can spontaneously associate with, insert into, or extend across lipid membranes. In many cases, when bound to a membrane, these peptides adopt a well-defined structure. For example, due to their amphipathic nature, many antimicrobial peptides (AMPs)46 are disordered in aqueous solution but fold into an α-helical conformation in the presence of a membrane. Thus, such peptides constitute great model systems for studying and elucidating the folding dynamics and mechanisms of secondary structure elements of membrane proteins. However, many membrane-binding peptides, including AMPs, can oligomerize and/or form higher-order structures when the peptide to lipid (P/L) ratio reaches a certain threshold.47 Therefore, great care must be taken in the design and interpretation of folding experiments involving membranes. In this section, we review recent studies on the kinetics of membrane-mediated α-helix folding with a focus only on experimental investigations beginning from an aqueous solution where the peptide of interest is unfolded.
For instance, using stopped-flow fluorescence spectroscopy, Tucker et al.48 have studied the membrane-mediated folding kinetics of magainin 2 (sequence: GIGKFLHSAKKFGKAFVGQIMNS), an AMP that is isolated from the skin of the African clawed frog, Xenopus laevis,49 in vesicles consisting of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoglycerol (POPG) and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC). By adding a PheCN residue to the N-terminus and replacing Phe12 with Trp, they created a mutant of magainin 2 (referred to as magainin-2-P1) that contains the PheCN-Trp FRET pair.38 As shown (Figure 1), their stopped-flow Trp fluorescence kinetics obtained via either direct excitation (i.e., with λex = 290 nm) or indirect excitation (i.e., through the mechanism of FRET by selectively exciting PheCN with λex = 240 nm) of the Trp fluorophore revealed that the fluorescence signals change as a function of time in a double-exponential manner, wherein the fast and slow phase reports on the rates of peptide binding and insertion, respectively. However, the relative amplitude of the fast phase in the stopped-flow kinetics obtained with indirect excitation is larger than that obtained with direct excitation. Since helix formation will lead to a change in the separation distance between the donor and acceptor, they concluded that the underlying coil-to-helix transition occurs rapidly upon peptide binding to the membrane surface. However, they were unable to resolve the rate of α-helix folding as this faster process occurs immediately after a slower kinetic event (i.e., the bimolecular peptide-membrane binding process). This is consistent with the fact that monomeric α-helices typically fold on microsecond or faster timescales.50–52 Using stopped-flow fluorescence spectroscopy and the same FRET pair, Tang et al.53 showed that the membrane-induced helix folding of another AMP, mastopran X, follows the same mechanism. Moreover, by inducing a-helix nucleation in the solution phase through the use of a double histidine mutant of mastopran X and metal ions, they further demonstrated that helix formation is not a prerequisite for binding of the peptide to the membrane surface; instead, its role is to stabilize the membrane-bound state. Similarly, the study of Contantinescu et al.,54 which used a brominated acyl chain to quench the Trp fluorescence, also indicated that the helix folding step of another AMP, melittin, is tightly coupled to the binding of membranes composed of either POPC, POPG/POPC, or POPC/cholesterol. Thus, taken together, these studies support the following kinetic scheme for AMP-membrane interactions:
Figure 1.

Stopped-flow fluorescence traces of magainin-2-P1 obtained with different excitation wavelengths, as indicated. Smooth lines are global fits to the following equation, S(t) = A – B1•exp(−t/t1) – B2•exp(−t/t2), with t1 = 11 ms and t2 = 99 ms, respectively. For λex = 240 nm, A = 0.929, B1 = 0.109, and B2 = 0.014; for λex = 290 nm, A = 0.875, B1 = 0.067, and B2 = 0.060. The larger B1 value in the case of 240 nm excitation indicates that the folding transition occurs in this kinetic phase. Reprinted with permission from Ref [48]. Copyright 2006 American Chemical Society.
This model indicates that the unfolded peptide (PU) first binds to the membrane (M), yielding a surface-bound state (PUM)S, which rapidly converts to another surface-bound state (PHM)S, where the peptide is in an α-helical conformation. Even as kS approaches the diffusion-limited value, the corresponding peptide-membrane binding rate is expected to be significantly slower than kH (i.e., the actual α-helical folding rate of the peptide). Thus, kH cannot be independently determined from the stopped-flow measurements. Conversely, the formation of the final membrane-bound state (PHM)I, where the α-helix becomes more deeply inserted into the membrane, occurs on a slower timescale than initial binding and helix formation. Depending on experimental conditions, the membrane-bound α-helices can further associate to form higher-order structures. Similarly, the values of the membrane binding and insertion rate constants depend on peptide sequence and membrane composition.37,54–56
Another very useful model system for studying membrane peptide folding kinetics is the pH low insertion peptide (pHLIP) introduced by Engelman and co-workers.26 At neutral or basic pH, pHLIP is unfolded and either remains in the solution phase or binds to the membrane surface as an extended chain, but acidic pH induces the folding and insertion of pHLIP into the membrane bilayer. Taking advantage of this property of pHLIP, several groups have studied its membrane-mediated folding kinetics using a rapid pH change as a conformational trigger.57–61 For example, using this method, Tang et al.62 studied the membrane-binding and folding kinetics of pHLIP in POPC vesicles at different initial pH conditions by monitoring the fluorescence of the Trp residue. They found that the stopped-flow fluorescence kinetics in all cases are complex and require at least three exponentials to fit (Figure 2). Based on these results, they proposed a kinetic model describing the underlying pHLIP-membrane interaction in which the initial bimolecular binding process produces at least two distinguishable surface-adsorbed conformational states. Depending on pH, these states can become further stabilized on the membrane surface or evolve into transmembrane (TM) α-helices. Interestingly, they found that when starting from a membrane surface-bound state, the membrane insertion kinetics of pHLIP become slower. Subsequently, using stopped-flow Trp fluorescence and CD techniques, Andreev et al.63 examined the membrane insertion kinetics of pHLIP from the surface-bound states in more detail. They found that in response to a pH drop from 8 to 4, the surface-bound pHLIP molecules undergo a rapid (about 0.1 s) helix folding event, which is followed by a much slower (about 100 s) insertion process that involves several distinct intermediate steps. In addition, by systemically varying the pHLIP sequence, Karabadzhak et al.64 showed that the membrane insertion rate of pHLIP depends on the number of protonatable residues at the inserting end of the peptide.
Figure 2.

Stopped-flow Trp fluorescence kinetics of pHLIP obtained under different mixing conditions. Magenta: the process was initiated by mixing equal volumes of pHLIP in 10 mM phosphate buffer (pH 8.0) with POPC vesicles (pH 8.0). Red: the process was initiated by mixing equal volumes of pHLIP in 10 mM phosphate buffer (pH 8.0), POPC vesicles (pH 8.0), and a HCl solution (pH 1.6). Blue: the process was initiated via a pH-jump from 8.0 to 4.0 by mixing equal volumes of a pre-equilibrated peptide-vesicle solution (pH 8.0) with a HCl solution (pH 1.9). Reprinted with permission from Ref [62]. Copyright 2008 American Chemical Society.
Finally, many designed TM α-helical peptides have been introduced,65–69 serving as model systems to study peptide-membrane interactions, particularly, factors that control the stability, structure and orientation of the TM α-helical state. While an in-depth discussion of studies in this area is beyond the scope of this Review, we believe that these deigned peptides are also great models for investigating the kinetics and mechanism of membrane-mediated a-helix folding.
2. FOLDING KINETICS OF HELIX DIMERS
Specific helix-helix interactions inside lipid bilayers, such as lateral association70–73 and head-to-head dimerization,74,75 guide the folding and assembly of many integral membrane proteins and their complexes and, as a result, have been the focus of a large number of experimental and computational studies.72,76 However, there are only a few studies that specifically focus on the kinetic aspects of the helix-helix interaction inside a membrane environment.
In one such study, Tang et al.77 investigated the dimerization kinetics of a water-soluble TM homodimer, anti-aIIb,78 in POPC/POPG vesicles using stopped-flow fluorescence spectroscopy. To dissect various kinetic steps involved in anti-aIIb-membrane interactions, which include membrane binding, insertion, and dimerization, they employed a two-fluorophore strategy using either the native Trp fluorescence in anti-αIIb or the fluorescence arising from PheCN in anti-αIIb-PheCN, an anti-αIIb mutant wherein the Trp residue was mutated to PheCN. As shown (Figure 3), rapid mixing of an aqueous solution containing either anti-aIIb or anti-αIIb-PheCN with a POPC/POPG vesicle solution produced stopped-flow fluorescence kinetic traces consisting of distinct phases. In a self-consistent manner, they assigned these kinetic phases to membrane binding, insertion, and dimerization of anti-αIIb. By globally fitting the stopped-flow kinetics obtained at different P/L ratios to an expanded two-stage model, they were able to further extract the respective rate constants of these kinetic steps. Specifically, they determined the membrane-insertion rate constant of the anti-αIIb monomer to be 2.4 ± 0.7 s−1, which is in good agreement with those measured for other membrane helical peptides,38,48,53,58,61–64,79 and the insertion rate constant of the anti-αIIb dimer to be 0.5 ± 0.1 s−1, which is comparable to (~0.24 s−1) a helical membrane protein, diacylglycerol kinase.80 More importantly, they found that the helix-helix association kinetics inside a membrane environment occur on a time scale of a few seconds, which is several orders of magnitude slower than expected for a diffusion-limited dimerization process.
Figure 3.

Stopped-flow kinetics of anti-αIIb (black) and anti-αIIb-PheCN (blue) upon association with POPC/POPG vesicles (0.86 mg/mL). In both cases, the final peptide concentration was 2.5 μM. The smooth lines are fits of this data to a sequential kinetic model consisting of binding, insertion and dimerization steps. Reprinted with permission from Ref [77]. Copyright 2009 American Chemical Society.
In another study, Anbazhagan et al.21 investigated the dissociation kinetics of the glycophorin A (GpA) dimer in detergent/SDS micelles using a stopped-flow FRET strategy that employs FI as a donor and TAMRA as an acceptor. By rapidly diluting a detergent/dodecyl maltoside (DDM) solution containing a 1:1 mixture of FI-labaled GpA and TAMRA-labeled GpA with SDS micelles at varying concentrations, they found that the time-depended FRET signals, which were attributed to GpA dimer dissociation, follow a double-exponential function. In addition, they found that while the rate constants of these two phases show a strong dependence on SDS concentration (e.g., at χSDS = 0.95, k1 = 0.126 s−1 and k2 = 0.026 s−1), their relative amplitudes do not. While double-exponential kinetics can arise from either a parallel or sequential dissociation mechanism, they suggested that it is likely that the fast phase represents an initial destabilization step of the GpA dimer prior to helix–helix separation.
Besides helix-helix association in a parallel fashion, many small peptides can assemble into linear dimers that span the length of a bilayer.81–88 For example, the functional state of gramicidin A corresponds to a helical dimer arranged in a head-to-head manner.89,90 However, a comprehensive examination of its folding kinetics and mechanisms are still lacking.76,86,91 Moreover, many membrane peptides and proteins need to dimerize or oligomerize to perform their functions, thus studying and understanding the kinetics and mechanism of peptide-peptide association inside membranes is an important undertaking; we expect that more studies in this area will emerge.
3 FOLDING KINETICS OF HELIX-TURN-HELIX MOTIFS
The helical-hairpin or helix-turn-helix (HTH) motif plays important structural and functional roles in both globular and integral membrane proteins.92,93 While the folding dynamics and mechanism of water-soluble helical hairpins have been extensively studied and well-understood,94 it is only recently that the membrane-mediated folding kinetics of a few HTH models have emerged,95–99 including the hemagglutinin fusion peptide (HAfp).
HAfp corresponds to the N-terminal domain of the trimeric HA2 hemagglutinin glycoprotein of the influenza virus.100 Several NMR studies have shown that HAfp can adopt different conformations in model membranes, including a HTH conformation, depending on pH, and can even induce membrane fusion. For example, the NMR structures of Han et al.100 indicate that HAfp forms an inverted V-shape consisting of an N-terminal α-helix and an extended conformation in the C-terminal region at pH 7.4, which, upon lowering the pH to 5, the C-terminal segment converts to a short 310-helix. On the other hand, the NMR studies of Bax and coworkers101,102 reveal that HAfp folds predominantly into a helical hairpin structure (or HTH) in DPC micelles at pH 7, while in acidic conditions HAfp samples three conformations (i.e., helical hairpin, L-shaped, and extended) with the helical hairpin structure being dominant (~80%).
Recently, we have carried out a series of stopped-flow Trp fluorescence measurements on the wild type (sequence: GLFGAIAGFIENGWEGMIDGWYG-NH2) and a mutant (i.e., Phe3/PheCN) of HAfp in POPC and POPC/POPG vesicles,103 aiming to provide a better understanding of its membrane-mediated folding mechanism. We found that the stopped-flow kinetic traces obtained by rapidly mixing HAfp with 100 nm POPC vesicles at varying P/L ratios can be fit by a three-exponential function. Interestingly, except the slowest phase, which accounts for only about less than 10% of the total signal, the other two kinetic phases showed a clear dependence on the P/L ratio, indicating that they arise from second order kinetics. While a sequential mechanism, which involves peptide binding to the membrane followed by peptide dimerization and subsequent insertion deeper into the membrane, would produce kinetics with two of the three rates being dependent on peptide concentration, we quickly dismissed this model as (1) it cannot explain the observed dependence of the rates on the P/L ratio and (2) HAfp is not known to dimerize or oligomerize under the current experimental conditions. Therefore, we sought to use a parallel kinetic scheme, as indicated below, to interpret the stopped-flow results.
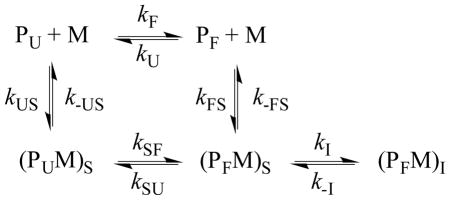
The key assumption underlying this model was that HAfp exists in a dynamic equilibrium between a folded (PF) and an unfolded (PU) conformation in aqueous solution, with the equilibrium highly favoring the unfolded state. Upon interaction with a membrane (M), the peptide first becomes surface-bound where the unfolded state (PUM)S changes to the folded state (PFM)S, which further converts to the final membrane-inserted state, (PFM)I. Because PF has a much smaller population than PU, and (kF + kU) −1 is expected to be on the microsecond timescale,94 its membrane-binding kinetics can only be detected when kFS > kUS. While this is an attractive model, it is impractical to use it to directly fit the stopped-flow kinetics as it contains too many variables. To simplify the analysis, we further assumed that the folding/unfolding rate (i.e., kSF + kSU) of HAfp on the membrane is too fast to be observed. Under these conditions, the above scheme is effectively reduced to a simpler kinetic model, which contains two second-order binding events (manifested by kUS and kFS), followed by a first-order insertion process (manifested by kI). To determine these kinetic parameters, we globally fit the stopped-flow kinetic signals obtained under pseudo first-order conditions, F(t, CL), obtained at different lipid concentrations (CL) to the following equation:
| (1) |
where k3 = kI + k-I ≈ kI. As indicated (Figure 4), this equation fits the stopped-flow kinetics traces of HAfp well and yields membrane binding rate constants (Table 1) that are consistent with reported values for peptides of similar size.52,53,61–63,77 In addition, the membrane insertion rate constant, 3.1 s−1, is in agreement with other helical membrane peptides.53,77
Figure 4.

Stopped-flow kinetics obtained upon mixing HAfp with 100 nm POPC vesicles at different final lipid concentrations, as indicated. The Trp fluorescence was directly excited using an excitation wavelength of 290 nm and the final peptide concentration was 2.5 μM. Smooth lines are fits of this data to Eq. 1 and the fitting parameters are listed in Table 1. For clarity, these kinetic traces have been offset.
Table 1.
Parameters obtained from globally fitting the stopped-flow kinetics of HAfp and HAfp-F3CN to Eq. 1. The bimolecular rate constants were calculated using the lipid concentration and the relative amplitudes are average values.
| HAfp (λex = 290 nm) | HAfp-F3CN (λex = 290 nm) | HAfp-F3CN (λex = 290 nm) | ||
|---|---|---|---|---|
| kFS (M−1 s−1) | 4.8×105 | 3.7×105 | 3.7×105 | |
| k-FS (s−1) | ~0 | ~0 | ~0 | |
| kUS (M−1 s−1) | 1.4×105 | 1.4×105 | 1.4×105 | |
| k-US (s−1) | 0.14 | 0.14 | 0.14 | |
| k3 (s−1) | 3.1 | 5.5 | 5.5 | |
|
|
36.0 | 34.4 | 50.5 | |
|
|
57.7 | 60.9 | 42.6 | |
|
|
6.3 | 4.7 | 6.9 |
To further test the validity of this parallel kinetic model, we also carried out stopped-flow measurements on the Phe3/PheCN mutant (referred to hereafter as HAfp-F3CN). As shown (Figure 5), the stopped-flow kinetics of HAfp-F3CN obtained by directly exciting (i.e., λex = 290 nm) the Trp fluorophores, can also be satisfactorily described by Eq. 1. In addition, the rate parameters obtained from the fits are similar to those obtained with the wild type (Table 1), indicating that the Phe to PheCN mutation has not significantly changed the membrane-interaction property of the peptide. Furthermore, by using the determined rate constants and only varying the amplitudes we were able to satisfactorily fit the stopped-flow kinetics of HAfp-F3CN obtained by indirectly exciting the Trp fluorophores via FRET (i.e., λex = 240 nm) (Figure 6). More importantly, the relative amplitudes of the three kinetic phases obtained in this case (Table 1) are consistent with the above proposed HAfp-membrane interaction mechanism. For example, if the slowest kinetic component (i.e., k3) in the stpped-flow kinetics corresponds to the folding process of the surface-bond state (i.e., from (PUM)S to (PFM)S), instead of the proposed membrane insertion of (PFM)S, one would expect to be significantly increased when an excitation of 240 nm is used. However, this is not observed.
Figure 5.

Stopped-flow kinetics obtained upon mixing HAfp-F3CN with 100 nm POPC vesicles at different final lipid concentrations, as indicated. The Trp fluorescence was directly excited using an excitation wavelength of 290 nm and the final peptide was 2.5 μM. Smooth lines are fits of this data to Eq. 1 and the fitting parameters are listed in Table 1. For clarity, these kinetic traces have been offset.
Figure 6.

Stopped-flow kinetics obtained upon mixing HAfp-F3CN with 100 nm POPC vesicles at different final lipid concentrations, as indicated. The Trp fluorescence was indirectly excited via FRET using an excitation wavelength of 240 nm and the final peptide was 2.5 μM. Smooth lines are fits of this data to Eq.1 and the fitting parameters are listed in Table 1. For clarity, these kinetic traces have been offset.
It is interesting to note that the membrane-mediated folding mechanism of HAfp uncovered here is similar to that of intrinsically disordered proteins (IDPs).104,105 It has been shown that the accumulation of the folded state of an IDP can be realized through either a binding-induced folding pathway from the unfolded state or an equilibrium shift mechanism wherein binding decreases the folded population.106 Additionally, when the HTH of interest is a part of a larger protein, for example in the case the diphtheria toxin T-domain,107 the underlying folding and membrane insertion mechanism can become more complicated.98
4 FOLDING KINETICS OF PROTEINS
The above examples provide clear indications that membrane-mediated folding processes are complex, even at the secondary structure level. Thus, it is more difficult to study and understand the tertiary folding mechanisms of integral membrane proteins. Despite many technological challenges, recent studies have yielded many insights into the folding dynamics of this class of proteins.108,109 While a comprehensive discussion of this topic is beyond the scope of this Review, we briefly describe a few examples to highlight the rapid growth of this important research field. In this regard, one of the most studied α-helical membrane proteins is bacteriorhodopsin (bR), which consists of seven TM α-helical segments (A–G). For example, Booth and coworkers110 have examined the refolding kinetics of bR in DOPC membranes from a partially denatured state in SDS micelles. By examining kinetics obtained from intrinsic and extrinsic fluorophores, which were introduced at different sites of the protein, they proposed that bR folds in a two-stage manner, where one phase (6.7 s−1) represents partitioning of helix-D with the lipid head groups and the second (0.33 s−1) represents TM insertion. Additionally, they concluded that the insertion of a preformed core is essential for the protein to fold correctly inside the membrane. More recently, Curnow et al. have carried out F-value analysis111 to determine the folding transition state structure ensemble of bR. Their results suggested that helix-B is native-like in the transition state and acts as a folding nucleus,112 whereas helix-G is formed on the native side of the major folding free energy barrier.113 Among β-sheet membrane proteins, outer membrane proteins (Omps), in particular the 8-stranded OmpA β-barrel of Escherichia coli, have emerged as great model systems to study the membrane-mediated protein folding mechanism as they fold spontaneously from a chemical-denatured state upon dilution with lipid vesicles and/or detergent micelles.114,115 For example, in a series of studies, Tamm and coworkers116,117 have proposed a folding mechanism of OmpA wherein four β-hairpins insert and translocate across the membrane concurrently. Damachi et al.118 reported refolding times (0–5 s) of individual β-hairpins in OmpG, and suggested that these β-hairpins fold and insert into the membrane in a hierarchical manner. Furthermore, when OmpA folds in vivo it requires periplasmic chaperones as well as the barrel assembly machinery (BAM) complex. Indeed, recent studies by Patel et al.119 and Gessmann et al.120 have shown that the kinetics of OmpA folding and insertion were accelerated when the preformed bilayers also contained the protein, BamA. Since phosphoethanolamine head groups of the bilayer are thought to impose a kinetic barrier on OmpA folding, their results suggest that BamA can decrease this barrier.120 Other than studying the folding properties of β-barrel motifs, great effort has also gone into investigating the kinetics and mechanism of membrane-mediated interactions between β-sheets.121 For example, Fu et al.122 and Ling et al.123 have employed non-linear infrared spectroscopy to investigate how membranes affect the amyloid formation kinetics of the human islet amyloid polypeptide (hIAPP).
SUMMARY AND PERSPECTIVES
Lipid bilayers offer a unique environment for a wide variety of peptides and proteins to fold, interact, and function. Numerous studies have been carried out to investigate and understand how and why folding occurs in this environment. While we now know a great deal about the thermodynamic principles and factors that govern the structure and stability of membrane peptides and proteins,124,125 we know much less about the underlying folding kinetics and mechanisms. This is because the membrane environment is dynamic, heterogeneous, crowded, and consists of both polar and hydrophobic regions, which all present experimental challenges for initiating and probing protein folding events with desired temporal and structural resolution. In this Review, we briefly describe the recent progress in the study of membrane-mediated folding kinetics of peptides that form well-defined simple structural elements, such as the α-helix, helix-helix dimer, and helix-turn-helix. It is clear that even for such simple systems, arriving at a molecular-level understanding of each and every kinetic step, including binding, folding, insertion, and association (for dimer and oligomer formation), is not a trivial task. It is also clear that in order to move the field forward, new experimental methods that can offer better time and structural resolution are needed. For example, several techniques, including laser-induced temperature-jump,126 laser-induced pH-jump,127 and phototriggering based on cleavage of a tetrazine128 or a photocage,129 can potentially be used to induce or initiate a membrane protein folding process on the nanosecond or microsecond timescale. In addition, by using a photoresponsive structural constraint, it is possible to initiate a folding or unfolding process from a well-selected conformational state. To improve the structural resolution of folding kinetic studies, we believe that the following approaches, many of which have already been used to study the equilibrium structures of membrane peptides and proteins, are promising: using (1) a local vibrational mode, such as that arising from an isotopically-labeled amide unit130–132 or an unnatural amino acid side chain,126,133–135 and resonance Raman scattering of a Trp probe,136 to gain site-specific structural and environmental information; (2) the coupling between two site-specific vibrators to probe tertiary interactions;137 (3) multiple fluorophores to simultaneously probe conformational changes of the protein of interest in different regions;138 (4) multi-fluorophore FRET to reveal correlated conformational motions;138 and (5) various nonlinear techniques, such as two-dimensional (2D) infrared (IR),139 second harmonic generation,140 and sum frequency generation141–145 spectroscopy.
Acknowledgments
We gratefully acknowledge financial support from the National Institutes of Health (GM-065978). B.N.M is supported by the NIH Ruth Kirschtein National Research Service Award Predoctoral Fellowship (F31AG046010).
References
- 1.Gelman H, Gruebele M. Q Rev Biophys. 2014;47:95–142. doi: 10.1017/S003358351400002X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dill KA, MacCallum JL. Science. 2012;338:1042–1046. doi: 10.1126/science.1219021. [DOI] [PubMed] [Google Scholar]
- 3.Serrano AL, Waegele MM, Gai F. Protein Sci. 2012;21:157–170. doi: 10.1002/pro.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lindorff-Larsen K, Piana S, Dror RO, Shaw DE. Science. 2011;334:517–520. doi: 10.1126/science.1208351. [DOI] [PubMed] [Google Scholar]
- 5.Baldwin RL. Annu Rev Biophys. 2008;37:1–21. doi: 10.1146/annurev.biophys.37.032807.125948. [DOI] [PubMed] [Google Scholar]
- 6.Englander SW, Mayne L, Krishna MMG. Q Rev Biophys. 2008;40:287–326. doi: 10.1017/S0033583508004654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rose GD, Fleming PJ, Banavar JR, Maritan A. Proc Natl Acad Sci USA. 2006;103:16623–16633. doi: 10.1073/pnas.0606843103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dobson CM. Nature. 2003;426:884–890. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- 9.Kubelka J, Hofrichter J, Eaton WA. Curr Opin Struct Biol. 2004;14:76–88. doi: 10.1016/j.sbi.2004.01.013. [DOI] [PubMed] [Google Scholar]
- 10.Donald JE, Zhang Y, Fiorin G, Carnevale V, Slochower DR, Gai F, Klein ML, DeGrado WF. Proc Natl Acad Sci USA. 2011;108:3958–3963. doi: 10.1073/pnas.1019668108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Henriques ST, Castanho MARB, Pattenden LK, Aguilar MI. Biopolymers. 2010;94:314–322. doi: 10.1002/bip.21367. [DOI] [PubMed] [Google Scholar]
- 12.Beevers AJ, Dixon AM. Chem Soc Rev. 2010;39:2146–2157. doi: 10.1039/b912944h. [DOI] [PubMed] [Google Scholar]
- 13.Rajendran L, Knölker HJ, Simons K. Nat Rev Drug Discov. 2010;9:29–42. doi: 10.1038/nrd2897. [DOI] [PubMed] [Google Scholar]
- 14.Popot JL, Engelman DM. Biochemistry. 1990;29:4031–4037. doi: 10.1021/bi00469a001. [DOI] [PubMed] [Google Scholar]
- 15.Roder H, Maki K, Cheng H. Chem Rev. 2006;106:1836–1861. doi: 10.1021/cr040430y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dyer RB, Gai F, Woodruff WH, Gilmanshin R, Callender RH. Acc Chem Res. 1998;31:709–716. [Google Scholar]
- 17.Markiewicz BN, Jo H, Culik RM, DeGrado WF, Gai F. The Journal of Physical Chemistry B. 2013;117:14688–14696. doi: 10.1021/jp409334h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Callender R, Dyer RB. Chem Rev. 2006;106:3031–3042. doi: 10.1021/cr050284b. [DOI] [PubMed] [Google Scholar]
- 19.Ihalainen JA, Paoli B, Muff S, Backus EHG, Bredenbeck J, Woolley GA, Caflisch A, Hamm P. Proc Natl Acad Sci U S A. 2008;105:9588–9593. doi: 10.1073/pnas.0712099105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tucker MJ, Abdo M, Courter JR, Chen J, Brown SP, Smith AB, Hochstrasser RM. Proc Natl Acad Sci USA. 2013;110:17314–17319. doi: 10.1073/pnas.1311876110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Anbazhagan V, Cymer F, Schneider D. Arch Biochem Biophys. 2010;495:159–164. doi: 10.1016/j.abb.2010.01.006. [DOI] [PubMed] [Google Scholar]
- 22.Booth PJ, Farooq A. Eur J Biochem. 1997;246:674–680. doi: 10.1111/j.1432-1033.1997.00674.x. [DOI] [PubMed] [Google Scholar]
- 23.Fisher LE, Engelman DM, Sturgis JN. J Mol Biol. 1999;293:639–651. doi: 10.1006/jmbi.1999.3126. [DOI] [PubMed] [Google Scholar]
- 24.Fleming KG, Ackerman AL, Engelman DM. J Mol Biol. 1997;272:266–275. doi: 10.1006/jmbi.1997.1236. [DOI] [PubMed] [Google Scholar]
- 25.Huang KS, Bayley H, Liao MJ, London E, Khorana HG. J Biol Chem. 1981;256:3802–3809. [PubMed] [Google Scholar]
- 26.Hunt JF, Rath P, Rothschild KJ, Engelman DM. Biochemistry. 1997;36:15177–15192. doi: 10.1021/bi970147b. [DOI] [PubMed] [Google Scholar]
- 27.Riley ML, Wallace BA, Flitsch SL, Booth PJ. Biochemistry. 1997;36:192–196. doi: 10.1021/bi962199r. [DOI] [PubMed] [Google Scholar]
- 28.Schlebach JP, Woodall NB, Bowie JU, Park C. J Am Chem Soc. 2014;136:16574–16581. doi: 10.1021/ja508359n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tulumello DV, Deber CM. Biochemistry. 2009;48:12096–12103. doi: 10.1021/bi9013819. [DOI] [PubMed] [Google Scholar]
- 30.Turner GJ, Chittiboyina S, Pohren L, Hines KG, Correia JJ, Mitchell DC. Biochemistry. 2009;48:1112–1122. doi: 10.1021/bi801799j. [DOI] [PubMed] [Google Scholar]
- 31.Clayton AH, Sawyer WH. Eur Biophys J. 2002;31:9–13. doi: 10.1007/s002490100182. [DOI] [PubMed] [Google Scholar]
- 32.Tucker MJ, Oyola R, Gai F. Biopolymers. 2006;83:571–576. doi: 10.1002/bip.20587. [DOI] [PubMed] [Google Scholar]
- 33.Karabadzhak AG, An M, Yao L, Langenbacher R, Moshnikova A, Adochite RC, Andreev OA, Reshetnyak YK, Engelman DM. ACS Chem Biol. 2014;9:2545–2553. doi: 10.1021/cb500388m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jungbauer LM, Yu C, Laxton KJ, Ladu MJ. J Mol Recognit. 2009;22:403–413. doi: 10.1002/jmr.948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Macháň R, Jurkiewicz P, Olżyńska A, Olšinová M, Cebecauer M, Marquette A, Bechinger B, Hof M. Langmuir. 2014;30:6171–6179. doi: 10.1021/la5006314. [DOI] [PubMed] [Google Scholar]
- 36.Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C. J Immunol Methods. 1995;184:39–51. doi: 10.1016/0022-1759(95)00072-i. [DOI] [PubMed] [Google Scholar]
- 37.Möglich A, Joder K, Kiefhaber T. Proc Natl Acad Sci USA. 2006;103:12394–12399. doi: 10.1073/pnas.0604748103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tucker MJ, Oyola R, Gai F. J Phys Chem B. 2005;109:4788–4795. doi: 10.1021/jp044347q. [DOI] [PubMed] [Google Scholar]
- 39.Schuler B, Lipman EA, Steinbach PJ, Kumke M, Eaton WA. Proc Natl Acad Sci USA. 2005;102:2754–2759. doi: 10.1073/pnas.0408164102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Khan YR, Dykstra TE, Scholes GD. Chem Phys Lett. 2008;461:305–309. [Google Scholar]
- 41.Loura LMS, Prieto M, Fernandes F. Eur Biophys J. 2009;39:565–578. doi: 10.1007/s00249-009-0532-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ge N, Zhang X, Keiderling TA. Biochemistry. 2010;49:8831–8838. doi: 10.1021/bi1008936. [DOI] [PubMed] [Google Scholar]
- 43.Demmers JA, Haverkamp J, Heck AJ, Koeppe RE, Killian JA. Proc Natl Acad Sci USA. 2000;97:3189–3194. doi: 10.1073/pnas.050444797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rath A, Glibowicka M, Nadeau VG, Chen G, Deber CM. Proc Natl Acad Sci USA. 2009;106:1760–1765. doi: 10.1073/pnas.0813167106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nusair NA, Mayo DJ, Dorozenski TD, Cardon TB, Inbaraj JJ, Karp ES, Newstadt JP, Grosser SM, Lorigan GA. Biochim Biophys Acta. 2012;1818:821–828. doi: 10.1016/j.bbamem.2011.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fjell CD, Hiss JA, Hancock REW, Schneider G. Nat Rev Drug Discov. 2012;11:37–51. doi: 10.1038/nrd3591. [DOI] [PubMed] [Google Scholar]
- 47.Melo MN, Ferre R, Castanho MARB. Nature Reviews Microbiology. 2009;7:245–250. doi: 10.1038/nrmicro2095. [DOI] [PubMed] [Google Scholar]
- 48.Tucker MJ, Tang J, Gai F. J Phys Chem B. 2006;110:8105–8109. doi: 10.1021/jp060900n. [DOI] [PubMed] [Google Scholar]
- 49.Zasloff M. Proc Natl Acad Sci USA. 1987;84:5449–5453. doi: 10.1073/pnas.84.15.5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mukherjee S, Waegele MM, Chowdhury P, Guo L, Gai F. J Mol Biol. 2009;393:227–236. doi: 10.1016/j.jmb.2009.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mukherjee S, Chowdhury P, DeGrado WF, Gai F. Langmuir. 2007;23:11174–11179. doi: 10.1021/la701686g. [DOI] [PubMed] [Google Scholar]
- 52.Huang CY, Klemke JW, Getahun Z, DeGrado WF, Gai F. J Am Chem Soc. 2001;123:9235–9238. doi: 10.1021/ja0158814. [DOI] [PubMed] [Google Scholar]
- 53.Tang J, Signarvic RS, DeGrado WF, Gai F. Biochemistry. 2007;46:13856–13863. doi: 10.1021/bi7018404. [DOI] [PubMed] [Google Scholar]
- 54.Constantinescu I, Lafleur M. Biochim Biophys Acta. 2004;1667:26–37. doi: 10.1016/j.bbamem.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 55.Zhao H, Mattila JP, Holopainen JM, Kinnunen PKJ. Biophys J. 2001;81:2979–2991. doi: 10.1016/S0006-3495(01)75938-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bradrick TD, Philippetis A, Georghiou S. Biophys J. 1995;69:1999–2010. doi: 10.1016/S0006-3495(95)80070-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.An M, Wijesinghe D, Andreev OA, Reshetnyak YK, Engelman DM. Proc Natl Acad Sci U S A. 2010;107:20246–20250. doi: 10.1073/pnas.1014403107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Andreev OA, Engelman DM, Reshetnyak YK. Front Physiol. 2014;5:97. doi: 10.3389/fphys.2014.00097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Guo L, Gai F. Biophys J. 2010;98:2914–2922. doi: 10.1016/j.bpj.2010.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Reshetnyak YK, Andreev OA, Segala M, Markin VS, Engelman DM. Proc Natl Acad Sci USA. 2008;105:15340–15345. doi: 10.1073/pnas.0804746105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Weerakkody D, Moshnikova A, Thakur MS, Moshnikova V, Daniels J, Engelman DM, Andreev OA, Reshetnyak YK. Proc Natl Acad Sci U S A. 2013;110:5834–5839. doi: 10.1073/pnas.1303708110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tang J, Gai F. Biochemistry. 2008;47:8250–8252. doi: 10.1021/bi801103x. [DOI] [PubMed] [Google Scholar]
- 63.Andreev OA, Engelman DM, Reshetnyak YK. Mol Membr Biol. 2010;27:341–352. doi: 10.3109/09687688.2010.509285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Karabadzhak AG, Weerakkody D, Wijesinghe D, Thakur MS, Engelman DM, Andreev OA, Markin VS, Reshetnyak YK. Biophys J. 2012;102:1846–1855. doi: 10.1016/j.bpj.2012.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Almeida PF, Ladokhin AS, White SH. Biochim Biophys Acta. 2012;1818:178–182. doi: 10.1016/j.bbamem.2011.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Im W, Brooks CL. Proc Natl Acad Sci USA. 2005;102:6771–6776. doi: 10.1073/pnas.0408135102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ladokhin AS, White SH. Biochemistry. 2004;43:5782–5791. doi: 10.1021/bi0361259. [DOI] [PubMed] [Google Scholar]
- 68.Rankenberg JM, Vostrikov VV, Greathouse DV, Grant CV, Opella SJ, Koeppe RE. Biochemistry. 2012;51:10066–10074. doi: 10.1021/bi301394z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wimley WC, White SH. Biochemistry. 2000;39:4432–4442. doi: 10.1021/bi992746j. [DOI] [PubMed] [Google Scholar]
- 70.Hong H, Blois TM, Cao Z, Bowie JU. Proc Natl Acad Sci USA. 2010;107:19802–19807. doi: 10.1073/pnas.1010348107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li R, Gorelik R, Nanda V, Law PB, Lear JD, DeGrado WF, Bennett JS. J Biol Chem. 2004;279:26666–26673. doi: 10.1074/jbc.M314168200. [DOI] [PubMed] [Google Scholar]
- 72.Psachoulia E, Marshall DP, Sansom MSP. Acc Chem Res. 2010;43:388–396. doi: 10.1021/ar900211k. [DOI] [PubMed] [Google Scholar]
- 73.Ulmschneider JP, Smith JC, Ulmschneider MB, Ulrich AS, Strandberg E. Biophys J. 2012;103:472–482. doi: 10.1016/j.bpj.2012.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rawat SS, Kelkar DA, Chattopadhyay A. Biophys J. 2004;87:831–843. doi: 10.1529/biophysj.104.041715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Xie X, Al-Momani La, Reiß P, Griesinger C, Koert U. FEBS Journal. 2005;272:975–986. doi: 10.1111/j.1742-4658.2004.04531.x. [DOI] [PubMed] [Google Scholar]
- 76.Wanasundara SN, Krishnamurthy V, Chung SH. The Journal of Physical Chemistry B. 2011;115:13765–13770. doi: 10.1021/jp2084583. [DOI] [PubMed] [Google Scholar]
- 77.Tang J, Yin H, Qiu J, Tucker MJ, DeGrado WF, Gai F. J Am Chem Soc. 2009;131:3816–3817. doi: 10.1021/ja809007f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Caputo GA, Litvinov RI, Li W, Bennett JS, DeGrado WF, Yin H. Biochemistry. 2008;47:8600–8606. doi: 10.1021/bi800687h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Aprilakis KN, Taskent H, Raleigh DP. Biochemistry. 2007;46:12308–12313. doi: 10.1021/bi7010674. [DOI] [PubMed] [Google Scholar]
- 80.Lorch M, Booth PJ. J Mol Biol. 2004;344:1109–1121. doi: 10.1016/j.jmb.2004.09.090. [DOI] [PubMed] [Google Scholar]
- 81.Blake S, Capone R, Mayer M, Yang J. Bioconjugate Chem. 2008;19:1614–1624. doi: 10.1021/bc800180z. [DOI] [PubMed] [Google Scholar]
- 82.Kelkar DA, Chattopadhyay A. Biochim Biophys Acta. 2007;1768:2011–2025. doi: 10.1016/j.bbamem.2007.05.011. [DOI] [PubMed] [Google Scholar]
- 83.Sobko AA, Vigasina MA, Rokitskaya TI, Kotova EA, Zakharov SD, Cramer WA, Antonenko YN. J Membrane Biol. 2004;199:51–62. doi: 10.1007/s00232-004-0674-y. [DOI] [PubMed] [Google Scholar]
- 84.Mukherjee S, Chattopadhyay A. Biochemistry. 1994;33:5089–5097. doi: 10.1021/bi00183a012. [DOI] [PubMed] [Google Scholar]
- 85.Schönknecht G, Althoff G, Junge W. J Membrane Biol. 1992;126:265–275. doi: 10.1007/BF00232323. [DOI] [PubMed] [Google Scholar]
- 86.Oconnell AM, Koeppe RE, Andersen OS. Science. 1990;250:1256–1259. doi: 10.1126/science.1700867. [DOI] [PubMed] [Google Scholar]
- 87.Brunner J, Richards FM. J Biol Chem. 1980;255:3319–3329. [PubMed] [Google Scholar]
- 88.Bamberg E, Lauger P. J Membr Biol. 1973;11:177–194. doi: 10.1007/BF01869820. [DOI] [PubMed] [Google Scholar]
- 89.Gu H, Lum K, Kim JH, Greathouse DV, Andersen OS, Koeppe IIRE. Biochemistry. 2011;50:4855–4866. doi: 10.1021/bi200136e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Clement NR, Gould JM. Biochemistry. 1981;20:1544–1548. doi: 10.1021/bi00509a021. [DOI] [PubMed] [Google Scholar]
- 91.Chitta RK, Rempel DL, Gross ML. J Am Soc Mass Spectrom. 2009;20:1813–1820. doi: 10.1016/j.jasms.2009.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nagy A, Turner RJ. Biochem Bioph Res Co. 2007;356:392–397. doi: 10.1016/j.bbrc.2007.02.149. [DOI] [PubMed] [Google Scholar]
- 93.Dong G, Chakshusmathi G, Wolin SL, Reinisch KM. Embo J. 2004;23:1000–1007. doi: 10.1038/sj.emboj.7600115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Du D, Gai F. Biochemistry. 2006;45:13131–13139. doi: 10.1021/bi0615745. [DOI] [PubMed] [Google Scholar]
- 95.Chakraborty H, Tarafdar PK, Klapper DG, Lentz BR. Biophys J. 2013;105:2495–2506. doi: 10.1016/j.bpj.2013.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ladokhin AS. Toxins. 2013;5:1362–1380. doi: 10.3390/toxins5081362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Haque ME, Chakraborty H, Koklic T, Komatsu H, Axelsen PH, Lentz BR. Biophys J. 2011;101:1095–1104. doi: 10.1016/j.bpj.2011.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kyrychenko A, Posokhov YO, Rodnin MV, Ladokhin AS. Biochemistry. 2009;48:7584–7594. doi: 10.1021/bi9009264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ladokhin AS. Meth Enzymol. 2009;466:19–42. doi: 10.1016/S0076-6879(09)66002-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Han X, Bushweller JH, Cafiso DS, Tamm LK. Nat Struct Biol. 2001;8:715–720. doi: 10.1038/90434. [DOI] [PubMed] [Google Scholar]
- 101.Lorieau JL, Louis JM, Bax A. Biopolymers. 2013;99:189–195. doi: 10.1002/bip.22102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lorieau JL, Louis JM, Schwieters CD, Bax A. Proc Natl Acad Sci USA. 2012;109:19994–19999. doi: 10.1073/pnas.1213801109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Smith-Dupont KB. PhD Dissertation. 2013. [Google Scholar]
- 104.Dogan J, Gianni S, Jemth P. Phys Chem Chem Phys. 2014;16:6323–6331. doi: 10.1039/c3cp54226b. [DOI] [PubMed] [Google Scholar]
- 105.Sugase K, Dyson HJ, Wright PE. Nature. 2007;447:1021–1025. doi: 10.1038/nature05858. [DOI] [PubMed] [Google Scholar]
- 106.Wright PE, Dyson HJ. Curr Opin Struct Biol. 2009;19:31–38. doi: 10.1016/j.sbi.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Vargas-Uribe M, Rodnin MV, Ladokhin AS. Biochemistry. 2013;52:7901–7909. doi: 10.1021/bi400926k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lanyi JK. Annu Rev Physiol. 2004;66:665–688. doi: 10.1146/annurev.physiol.66.032102.150049. [DOI] [PubMed] [Google Scholar]
- 109.Tamm LK, Hong H, Liang B. Biochim Biophys Acta. 2004;1666:250–263. doi: 10.1016/j.bbamem.2004.06.011. [DOI] [PubMed] [Google Scholar]
- 110.Compton ELR, Farmer NA, Lorch M, Mason JM, Moreton KM, Booth PJ. J Mol Biol. 2006;357:325–338. doi: 10.1016/j.jmb.2005.12.042. [DOI] [PubMed] [Google Scholar]
- 111.Fersht AR, Sato S. Proc Natl Acad Sci USA. 2004;101:7976–7981. doi: 10.1073/pnas.0402684101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Curnow P, Booth PJ. Proc Natl Acad Sci U S A. 2009;106:773–778. doi: 10.1073/pnas.0806953106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Curnow P, Di Bartolo ND, Moreton KM, Ajoje OO, Saggese NP, Booth PJ. Proc Natl Acad Sci U S A. 2011;108:14133–14138. doi: 10.1073/pnas.1012594108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Tamm LK, Arora A, Kleinschmidt JH. J Biol Chem. 2001;276:32399–32402. doi: 10.1074/jbc.R100021200. [DOI] [PubMed] [Google Scholar]
- 115.Thoma J, Bosshart P, Pfreundschuh M, Müller DJ. Structure. 2012;20:2185–2190. doi: 10.1016/j.str.2012.10.006. [DOI] [PubMed] [Google Scholar]
- 116.Kleinschmidt JH, Tamm LK. J Mol Biol. 2002;324:319–330. doi: 10.1016/s0022-2836(02)01071-9. [DOI] [PubMed] [Google Scholar]
- 117.Kleinschmidt JH, Tamm LK. Biochemistry. 1999;38:4996–5005. doi: 10.1021/bi9824644. [DOI] [PubMed] [Google Scholar]
- 118.Damaghi M, Köster S, Bippes CA, Yildiz Ö, Müller DJ. Angew Chem, Int Ed. 2011;50:7422–7424. doi: 10.1002/anie.201101450. [DOI] [PubMed] [Google Scholar]
- 119.Patel GJ, Kleinschmidt JH. Biochemistry. 2013;52:3974–3986. doi: 10.1021/bi400103t. [DOI] [PubMed] [Google Scholar]
- 120.Gessmann D, Chung YH, Danoff EJ, Plummer AM, Sandlin CW, Zaccai NR, Fleming KG. Proc Natl Acad Sci USA. 2014;111:5878–5883. doi: 10.1073/pnas.1322473111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wimley WC, Hristova K, Ladokhin AS, Silvestro L, Axelsen PH, White SH. J Mol Biol. 1998;277:1091–1110. doi: 10.1006/jmbi.1998.1640. [DOI] [PubMed] [Google Scholar]
- 122.Fu L, Ma G, Yan ECY. J Am Chem Soc. 2010;132:5405–5412. doi: 10.1021/ja909546b. [DOI] [PubMed] [Google Scholar]
- 123.Ling YL, Strasfeld DB, Shim SH, Raleigh DP, Zanni MT. J Phys Chem B. 2009;113:2498–2505. doi: 10.1021/jp810261x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Rath A, Deber CM. Annual Review of Biophysics. 2012;41:135–155. doi: 10.1146/annurev-biophys-050511-102310. [DOI] [PubMed] [Google Scholar]
- 125.Stanley AM, Fleming KG. Arch Biochem Biophys. 2008;469:46–66. doi: 10.1016/j.abb.2007.09.024. [DOI] [PubMed] [Google Scholar]
- 126.Nagarajan S, Taskent-Sezgin H, Parul D, Carrico I, Raleigh DP, Dyer RB. J Am Chem Soc. 2011;133:20335–20340. doi: 10.1021/ja2071362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hamm P. The Journal of Physical Chemistry B. 2014 [Google Scholar]
- 128.Tucker MJ, Courter JR, Chen J, Atasoylu O, Smith AB, Hochstrasser RM. Angewandte Chemie (International ed in English) 2010;49:3612–3616. doi: 10.1002/anie.201000500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Markiewicz BN, Culik RM, Gai F. Sci China Chem. 2014 doi: 10.1007/s11426-014-5225-5. [DOI] [PMC free article] [PubMed]
- 130.Ghosh A, Qiu J, DeGrado WF, Hochstrasser RM. Proc Natl Acad Sci USA. 2011;108:6115–6120. doi: 10.1073/pnas.1103027108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Woys AM, Lin YS, Reddy AS, Xiong W, de Pablo JJ, Skinner JL, Zanni MT. J Am Chem Soc. 2010;132:2832–2838. doi: 10.1021/ja9101776. [DOI] [PubMed] [Google Scholar]
- 132.Manor J, Mukherjee P, Lin YS, Leonov H, Skinner JL, Zanni MT, Arkin IT. Structure. 2009;17:247–254. doi: 10.1016/j.str.2008.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Pazos IM, Ghosh A, Tucker MJ, Gai F. Angew Chem Int Ed Engl. 2014;53:6080–6084. doi: 10.1002/anie.201402011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Waegele MM, Culik RM, Gai F. J Phys Chem Lett. 2011;2:2598–2609. doi: 10.1021/jz201161b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Urbanek DC, Vorobyev DY, Serrano AL, Gai F, Hochstrasser RM. The Journal of Physical Chemistry Letters. 2010;1:3311–3315. doi: 10.1021/jz101367d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Sanchez KM, Kang G, Wu B, Kim JE. Biophys J. 2011;100:2121–2130. doi: 10.1016/j.bpj.2011.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Remorino A, Korendovych IV, Wu Y, DeGrado WF, Hochstrasser RM. Science. 2011;332:1206–1209. doi: 10.1126/science.1202997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Rogers JMG, Lippert LG, Gai F. Anal Biochem. 2010;399:182–189. doi: 10.1016/j.ab.2009.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Mukherjee P, Kass I, Arkin IT, Arkin I, Zanni MT. Proc Natl Acad Sci USA. 2006;103:3528–3533. doi: 10.1073/pnas.0508833103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Lu R, Gan W, Wu B-h, Zhang Z, Guo Y, Wang H-f. The Journal of Physical Chemistry B. 2005;109:14118–14129. doi: 10.1021/jp051565q. [DOI] [PubMed] [Google Scholar]
- 141.Wilhelm MJ, Sheffield JB, Gonella G, Wu Y, Spahr C, Zeng J, Xu B, Dai HL. Chem Phys Lett. 2014;605:158–163. [Google Scholar]
- 142.Ding B, Laaser JE, Liu Y, Wang P, Zanni MT, Chen Z. The Journal of Physical Chemistry B. 2013;117:14625–14634. doi: 10.1021/jp408064b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Liu W, Wang Z, Fu L, Leblanc RM, Yan ECY. Langmuir. 2013;29:15022–15031. doi: 10.1021/la4036453. [DOI] [PubMed] [Google Scholar]
- 144.Engel MFM, vandenAkker CC, Schleeger M, Velikov KP, Koenderink GH, Bonn M. J Am Chem Soc. 2012;134:14781–14788. doi: 10.1021/ja3031664. [DOI] [PubMed] [Google Scholar]
- 145.Chen X, Hua W, Huang Z, Allen HC. J Am Chem Soc. 2010;132:11336–11342. doi: 10.1021/ja1048237. [DOI] [PubMed] [Google Scholar]