

Sphingomyelinase D suppresses Orai current in human T cells and decreases cytokine production, providing a mechanism whereby certain bacteria could inhibit the immune system.
Abstract
Infections caused by certain bacteria including Mycobacterium tuberculosis and Corynebacterium pseudotuberculosis provoke inflammatory responses characterized by the formation of granulomas with necrotic foci—so-called caseous necrosis. The granulomas of infected animals show prominent infiltration by T lymphocytes, and T cell depletion increases host mortality. Notorious zoonotic C. pseudotuberculosis secretes sphingomyelinase (SMase) D, a phospholipase that cleaves off the choline moiety of sphingomyelin, a phospholipid found primarily in the outer leaflet of host cell plasma membranes. Experimental C. pseudotuberculosis strains that lack SMase D are markedly less infectious and unable to spread in hosts, indicating that this enzyme is a crucial virulence factor for sustaining the caseous lymphadenitis infections caused by this microbe. However, the molecular mechanism by which SMase D helps bacteria evade the host’s immune response remains unknown. Here, we find that SMase D inhibits store-operated Ca2+ entry (SOCE) in human T cells and lowers the production of the SOCE-dependent cytokines interleukin-2, which is critical for T cell growth, proliferation, and differentiation, and tumor necrosis factor α, which is crucial for the formation and maintenance of granulomas in microbial infections. SMase D inhibits SOCE through a previously unknown mechanism, namely, suppression of Orai1 current, rather than through altering gating of voltage-gated K+ channels. This finding suggests that, whereas certain genetic mutations abolish Orai1 activity causing severe combined immunodeficiency (SCID), bacteria have the ability to suppress Orai1 activity with SMase D to create an acquired, chronic SCID-like condition that allows persistent infection. Thus, in an example of how virulence factors can disrupt key membrane protein function by targeting phospholipids in host cell membranes, our study has uncovered a novel molecular mechanism that bacteria can use to thwart host immunity.
INTRODUCTION
Infections by zoonotic Corynebacterium pseudotuberculosis and Mycobacterium tuberculosis are characterized by caseous necrosis. C. pseudotuberculosis secretes sphingomyelinase (SMase) D, a virulence factor secreted by some other human bacterial pathogens, and also the active component of certain spider venoms (McNamara et al., 1995; Isbister and Fan, 2011). SMase D cleaves the choline moiety from sphingomyelin (Fig. 1 A) (Souček et al., 1971), a phospholipid found predominantly in the plasma membrane’s outer leaflet, leaving behind ceramide-1-phosphate (C1P). C. pseudotuberculosis is perhaps the most studied model of SMase D in bacterial virulence. Lymph nodes infected with it show prominent infiltration of T lymphocytes (Ellis, 1988; Pépin et al., 1994). These cells play a critical role in the host’s resistance to these microbes, as antibody-mediated depletion of host T cells or of T cell cytokines promotes the spread of infection and increases host mortality (Lan et al., 1999). Intriguingly, experimental C. pseudotuberculosis strains that lack SMase D struggle to establish infections and fail to disseminate throughout infected hosts (McNamara et al., 1994). However, the molecular mechanism by which SMase D evades host immunity has remained unknown.
Figure 1.

SMase D suppresses SOCE in human T lymphocytes. (A) Reaction schemes for SMase D (top) and SMase C (bottom). (B) Fura-2 ratio signals of (26–54) T cells in a single 40× field, where the records show the average signals relative to the resting levels. The upper bar above the panel indicates these treatments: after 2-min rest, cells were treated with either SMase D (SMD, red) or catalytically inactive enzyme (CON, black) for 3 min, and then with 5 µg anti-CD3ε (Ab1) antibody for 2 min, and finally with 5 µg anti-IgG (Ab2) antibody for 18 min; the lower bar shows the Ca2+-containing solution type. (C) Peak and declining phase (at 23 min) values (error bars represent mean ± SEM; n = 5) of Fura-2 ratio signals after antibody stimulation as shown in B. (D) Fura-2 ratio signals of (17–31) T cells as in B, where the upper bar above the panel refers to these treatments: after 2-min rest, cells were treated with SMD (red) or CON (black) for 3 min and 1 µg Tg for 18 min; the lower bar shows the Ca2+-containing solution type. (E) Peak and declining phase (23 min) values (error bars represent mean ± SEM; n = 5) of Fura-2 ratio signals from Tg-stimulated cells for experiments as shown in D. Where present, the asterisks denote statistically significant comparisons as detailed in Materials and methods.
T lymphocyte function depends on Ca2+ signaling (Hogan et al., 2010). Antigen recognition by the T cell receptor (TCR) engenders the production of intracellular inositol 1,4,5 trisphosphate (IP3), which by activating ER IP3 receptor channels causes Ca2+ to leave the ER (Imboden and Stobo, 1985). The resulting ER Ca2+ store depletion mobilizes the Ca2+-sensing molecule Stim1 to activate the store-operated Ca2+ entry (SOCE) channel Orai1 in the plasma membrane (Liou et al., 2005; Roos et al., 2005; Feske et al., 2006; Vig et al., 2006; Zhang et al., 2006). This Orai1-mediated extracellular Ca2+ entry then dramatically amplifies the IP3 receptor–mediated Ca2+ signal and ultimately triggers T cell proliferation, differentiation, cytokine production, and cytotoxic granule release (Hogan et al., 2010). Genetic defects in Orai1 can produce a severe combined immunodeficiency (SCID), underscoring the critical role this channel plays in human immunity (Partiseti et al., 1994; Feske et al., 2006).
T lymphocyte SOCE is supported by endogenously expressed KV1.3 channels (DeCoursey et al., 1984; Matteson and Deutsch, 1984). These channels play a major role in setting the negative resting membrane potential, typically near −50 mV, which drives the entry of Ca2+ ions across cell plasma membranes. Inhibition of KV1.3 channels has been shown to suppress T cell Ca2+ signaling and the critical immune functions it triggers (Cahalan and Chandy, 2009). Our group previously found that, at the −50-mV membrane potential, SMase D treatment can boost the mean fraction of active KV1.3 channels from near 0 to ∼20% (Combs et al., 2013; see also Ramu et al., 2006; Xu et al., 2008; Milescu et al., 2009), which, absent other effects, would be expected to further hyperpolarize the membrane potential and enhance T lymphocyte function. Contrary to this expectation, the effect of SMase D in C. pseudotuberculosis infection is immune suppression. To resolve this apparent conundrum, we investigated the effects of SMase D on human T lymphocytes and found that SMase D actually lowers Ca2+ entry into T lymphocytes rather than boosting it.
MATERIALS AND METHODS
Cell cultures, molecular biology, biochemistry, and reagents
Chinese hamster ovary (CHO) or Jurkat (Clone E6-1) and human T cells were cultured in F12 Kaighn’s or RPMI 1640 media (Invitrogen) supplemented with 10% FBS (Hyclone) at 37°C with 5% CO2. Before recording, CHO cells were trypsinized and resuspended in recording solutions. These cell lines were obtained from ATCC, whereas human peripheral blood T cells were from healthy volunteers through the Penn Immunology Core (IRB protocol 705906). hStim1 and hOrai1 cDNAs (provided by M. Cahalan, University of California, Irvine, Irvine, CA) were cloned into pIRES2-AcGFP vectors. For expression of these constructs, CHO cells were transfected with the Fugene6 (Promega) method 24–48 h before study, and visualization of fluorescence signals was used to identify successful transfection.
All organic chemicals were purchased from Sigma-Aldrich. Thapsigargin (Tg), ionomycin, and valinomycin stock solutions were prepared at 1 mg/ml in DMSO; PMA was prepared at 0.1 mg/ml in DMSO. 5-(4-phenoxybutoxy)psoralen (PAP-1) stock solutions were made at 200 µM in DMSO. Mouse anti–human CD3ε, mouse anti–human CD28, and goat anti–mouse IgG antibodies (all from R&D Systems) were prepared at 1 mg/ml in PBS. N-palmitoyl-ceramide-l-phosphate (Avanti Polar Lipids, Inc.) was prepared as 500-nmol aliquots dried under argon; these aliquots were resuspended in 200 µl of 100% ethanol, which was added drop-wise to an equimolar amount of defatted BSA freshly prepared in calcium-containing solutions (see recipes below) to yield a final lipid concentration of 0.5 mg/ml (Lipsky and Pagano, 1985). Mouse anti–human CD4 antibody conjugated to PerCP-Cy5.5, mouse anti–human CD8 antibody conjugated to APC-Cy7, and CompBeads for compensation controls were purchased from BD. Recombinant bacterial SMase D and an inactive form of this enzyme containing H11A and H47A mutations, as well as recombinant SMase C, were generated as described previously (Ramu et al., 2007) (the SMase D cDNA was provided by S. Billington, University of Arizona, Tucson, AZ). In all experiments, SMase D and SMase C were applied at 0.3 µg/ml, and the inactive form of SMase D was applied at 3 µg/ml.
Ca2+ imaging
For Ca2+-imaging experiments, human T cells suspended at 106 cells/ml in complete RPMI media were exposed for 30 min at 20°C in the dark, with gentle shaking, to 1 µg/ml of the acetoxymethyl ester (AM) form of Fura-2. Fura-2-AM was diluted from 1-mg/ml stock solutions prepared in DMSO, and these stocks were prepared from lyophilized aliquotted powders (Molecular Probes), kept frozen, and used within 1–2 wk. After Fura-2-AM loading, cells were collected by spinning for 4 min at 1,500 g, and then resuspended at 106 cells/ml in complete RPMI media. Cells were incubated for 30 more minutes at 20°C in the dark with gentle shaking to allow for complete de-esterification of Fura-2-AM.
Fura-2 imaging of cells was performed on an inverted microscope (Eclipse-Ti; Nikon) using a 40× oil objective. Fura-2 fluorescence was excited at 340 or 380 nm with a xenon light source housed in a high-speed wavelength switcher (Lamba-DG4; Sutter Instrument) and detected at 510 nm. Images were captured at 0.2 Hz with a Retiga-SRV camera (Q-Imaging) and analyzed on a PC with Elements-AR software (Nikon). The Fura-2 ratio (intensity of the emitted light excited at 340 nm relative to that at 380 nm) was averaged for pixels corresponding to all the cells (19–84 per experiment) in the field of view. Background correction was performed by subtraction of signal from cell-free areas. The 340- and 380-nm signals were corrected for cell autofluorescence by exchanging the bath with solutions containing 10 mM MnCl2 and 1 µg/ml ionomycin to quench the Fura-2 dye at the end of each experiment, and subtracting any remaining fluorescence from the respective signals.
During the above experiments, cells were allowed to settle onto poly-l-lysine–coated glass coverslips mounted on a chamber (RC-24N; Warner Instruments), and bathing solutions were changed by means of a gravity-driven perfusion system. All experiments were performed at room temperature. The 2-mM Ca2+-containing salt solution (2 Ca) contained (mM): 145 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 glucose, and 10 HEPES, with pH adjusted to 7.30 with NaOH. The nominally Ca-free solution (0 Ca; ∼500 nM Ca2+) contained (mM): 145 NaCl, 5 KCl, 0.8 CaCl2, 1 EGTA, 1 MgCl2, 10 glucose, and 10 HEPES, with pH adjusted to 7.30 with NaOH. All perfused reagents were freshly diluted into 500 µl of the desired bathing solution before perfusion.
Data from the Ca2+-imaging experiments were normalized to facilitate comparison between and within the figures. The initial (time = 0) value of the Fura-2 signal for a particular control experiment was used to normalize that tracing and the remaining control tracings, as well as the SMase D–treated tracings. This procedure was followed separately for each stimulus type (anti-CD3ε antibody, Tg, and ionomycin).
Flow cytometry
For flow cytometry experiments, human T cells were suspended at 106 cells/ml in complete RPMI media and exposed for 30 min at 20°C in the dark with gentle shaking to 1 µg/ml of the AM form of Indo-1. Indo-1-AM was diluted from 1-mg/ml stock solutions prepared in DMSO, and these stocks were prepared from lyophilized aliquotted powders (Molecular Probes), kept frozen, and used within 1–2 wk. After Indo-1-AM loading, cells were collected by spinning for 4 min at 300 g, and then resuspended in cold RPMI at 107 cells/ml. Cells were incubated on ice in the presence of CD4 and CD8 antibodies for 20 min, diluted with 9 vol of cold flow salt solution (FSS; recipe follows), collected by spinning for 4 min at 300 g, and then resuspended at 106 cells/ml for analysis. Unstained cell controls and single-stained antibody compensation control beads, the latter prepared according to the manufacturer’s instructions, were prepared in parallel with the multi-stained cells.
Flow cytometry experiments were performed on a flow cytometer running FACSDiva software (LSR II; BD) and housed and maintained by the University of Pennsylvania Flow Cytometry Core Facility. PerCP-Cy5.5 was excited by a 488-nm laser; emission was captured by a 670-nm low-pass filter set behind a 635-nm low-pass dichroic mirror. APC-Cy7 was excited by a 633-nm laser; emission was captured by a bandpass filter centered at 780 nm set behind a 735 low-pass dichroic mirror. Indo-1 was excited by a 355-nm laser; calcium-bound emission was captured by a bandpass filter centered at 405 nm, and calcium-free emission was captured at 530 nm behind a 450-nm low-pass dichroic mirror. Unstained cells were used to set gates for human T cells and define background fluorescence levels for all channels. Indo-1–loaded cells were used to set photomultiplier tube (PMT) voltages for Indo-1 emissions, and single-stained compensation control beads were used to set PMT voltages for antibody-bound fluorophores. Cells were analyzed at speeds of ∼300/s, and post-collection data analysis was performed on FlowJo software (Tree Star). The mean Indo-1 ratio (intensity of the emitted light excited at 405 nm relative to that at 530 nm) was determined for cells binned over 3-s windows. The 405- and 530-nm signals were corrected for cell autofluorescence by the addition of 2 µl of 10 mM MnCl2 and 0.5 µg ionomycin to quench the Indo-1 dye at the end of each experiment, and by subtracting any remaining fluorescence from the respective signals.
All experiments were performed at room temperature in a nominally Ca-free (0 Ca; ∼500 nM Ca2+) FSS containing (mM) 145 NaCl, 5 KCl, 0.8 CaCl2, 1 EGTA, 1 MgCl2, 10 glucose, and 10 HEPES, with pH adjusted to 7.30 with NaOH. Data from the Ca2+-imaging experiments were normalized to facilitate comparison between and within the figures. The initial (time = 0) value of the Indo-1 signal for a particular control experiment was used to normalize that trace and the remaining control traces, as well as the SMase D–treated traces.
Electrophysiology
Channel currents from cells were recorded in the whole-cell configuration with a patch-clamp amplifier (Axopatch 200B; Molecular Devices), filtered at 5 kHz (KV1.3), 2 kHz (CHO Orai1), or 1 kHz (Jurkat Orai1), and sampled at 50 kHz using a Digidata 1322 (Molecular Devices) interfaced to a PC. pClamp 10 software (Molecular Devices) was used for amplifier control and data acquisition. Electrodes were fire polished (2–4 MΩ) and coated with beeswax. Capacitance and series resistance were electronically compensated.
For all KV1.3 studies, membrane potential was held at −100 mV and recordings were started 5 min after the whole-cell configuration was established. KV1.3 steady-state inactivation curves were obtained using a double-pulse protocol where, every 30 s, a 2,550-ms pulse to between −90 and −10 mV at 10-mV intervals was followed by a second 50-ms test pulse to 0 mV. Steady-state inactivation curves were constructed from the peak currents during the second pulse and fit to the following Boltzmann function:
where V1/2 is the midpoint; Z is the slope; c is the fraction of noninactivated channels; and F, R, and T have their usual meaning. G-V relationships (G-V curves) for KV1.3 channels were constructed from isochronic tail currents and fit to the following Boltzmann function:
with symbol meanings as above. For KV channel recordings, the bath solution contained (mM): 145 NaCl, 5 KCl, 0.3 CaCl2, 1 MgCl2, and 10 HEPES, with pH adjusted to 7.30 with NaOH. The electrode solution contained (mM): 140 KCl, 10 EGTA, 1 CaCl2, 1 MgCl2, and 10 HEPES, with pH adjusted to 7.30 with KOH.
For all studies of Orai1 currents expressed in CHO cells, transfected CHO cells in suspension were allowed to settle onto glass coverslips in a bath containing (mM): 150 NaCl, 5 KCl, 0.1 CaCl2, 2 MgCl2, 1 EGTA, and 10 HEPES, with pH adjusted to 7.30 with NaOH, and 1 µM Tg. This solution has a free Ca2+ concentration of ∼20 nM, and recordings in this solution were used to obtain nominally calcium-independent leak traces by stepping from the 0-mV holding potential to test potentials between −120 and 60 mV in 10-mV intervals every 2 s. After collection of these leak traces, Orai1 currents were revealed after replacing the bath solution with a solution containing (mM): 150 NaCl, 5 KCl, 20 CaCl2, 2 MgCl2, and 10 HEPES, with pH adjusted to 7.30 with NaOH, and 1 µM Tg. Calcium-dependent currents were then obtained by stepping from the 0-mV holding potential to test potentials between −120 and 60 mV in 10-mV intervals every 2 s. During tests of the effect of SMase D or the inactive enzyme, cell membrane potential was stepped repeatedly from 0 to −120 mV every 10 s. After enzyme treatment, calcium-dependent and calcium-independent leak traces were recollected as before. Calcium-independent traces were subtracted from calcium-dependent ones to isolate the Orai1 currents. For all Orai1 recordings, the electrode solution contained (mM): 140 CsMeSO3, 8 MgCl2, 1 CaCl2, 10 EGTA, and 10 HEPES, with pH adjusted to 7.30 with MeSO3H.
The approach and solutions used to record expressed Orai1 currents in CHO cells were also used to record native Orai1 currents from Jurkat cells, but with two modifications. First, calcium-dependent and calcium-independent traces were obtained by stepping from the 0-mV holding potential to test potentials between −100 and 60 mV in 20-mV steps every 2 s. Second, Orai1 currents were isolated by subtracting the mean steady-state calcium-independent current at each voltage from the corresponding calcium-dependent current.
ELISA assays of interleukin-2 (IL-2) and tumor necrosis factor α (TNF)
Human T lymphocytes at 2 × 106 cells/ml were exposed to inactive enzyme or SMase D for 5 min in complete RPMI culture medium. Media were then refreshed by centrifuging the cells for 4 min at 1.5 g and resuspending them in fresh media. The cells were then transferred to a 24-well tissue culture plate containing polystyrene beads coated with both anti-CD3 and anti-CD28 antibodies (Life Technologies) to achieve a 1:1 cell/bead ratio and concentrations of 106 cells/ml in 500-µl volumes. Stimulation with the antibody-labeled beads was allowed to proceed for 24 h, whereafter supernatants were collected by centrifugation. The IL-2 or TNF concentrations of supernatant samples diluted at 1:100 in fresh RPMI 10 were determined using an ELISA kit (R&D Systems) with a 96-well plate format. Optical densities of each well at 450 nm were analyzed on an EMax plate reader (Molecular Devices) with wavelength correction set to 570 nm.
Statistics
Statistically significant comparisons are indicated by asterisks in the figure panels. Nearly all figures compare experiments using SMase D with independent control experiments, where a catalytically inactive form of the enzyme was used instead. Generally, these comparisons are made between more than one experimental feature (e.g., the peak height and declining phase of Ca2+-imaging experiments) that are themselves non-independent. In such cases, p-values obtained from t tests were used to evaluate statistical significance, with a P < 0.05 denoted in figure panels with an asterisk. However, some experiments compare three independent conditions: SMase D treatment, C1P treatment, and control. In these cases, one-way ANOVA was first used to evaluate for significance, and asterisks denote post-comparison Bonferroni tests with P < 0.05.
RESULTS
SMase D treatment suppresses the intracellular Ca2+ signal of T lymphocytes
To stimulate human T lymphocyte Ca2+ signaling, we used antibodies against CD3ε (Ab1; a component of the TCR) to mimic antigen recognition by TCRs, and cross-linked these antibodies in situ with an anti-IgG antibody (Ab2) to boost the stimulus strength. This commonly used protocol produced the expected rise of intracellular Ca2+ concentration (Fig. 1 B), as monitored on a fluorescence imaging system with the ratiometric Ca2+ indicator Fura-2 (Grynkiewicz et al., 1985). In the presence of 2 mM of extracellular Ca2+, the Fura-2 Ca2+ signal characteristically peaked and then decayed toward a plateau. Several factors cause the Ca2+ signal to decline and reach a temporary steady state, including deactivation of SOCE channels, refilling of ER, extrusion of Ca2+ by the plasma membrane Ca2+ ATPase, reduction of the driving force for Ca2+ entry after the Ca2+ entry–caused membrane depolarization, and buffering of Ca2+ by other organelles and proteins (Hogan et al., 2010).
To test the effect of SMase D on the Ca2+ signal, we treated T lymphocytes with SMase D before antibody stimulation (Fig. 1 B). Contrary to expectations based on its KV1.3-stimulating effect, SMase D actually lowered the Ca2+ signal. SMase D treatment lowered both the peak and, more modestly, the declining phase of the Fura-2 Ca2+ signal as compared with control cells treated with a catalytically inactive mutant of SMase D (Fig. 1, B and C).
SMase D treatment suppresses SOCE
SMase D could diminish the size of the Fura-2 signal either by directly reducing SOCE across the plasma membrane or by reducing Ca2+ release from the ER stores, which would in turn result in reduced SOCE. To distinguish between these possibilities, we used Tg, an inhibitor of the sarco-/endoplasmic Ca2+ ATPase, to deplete ER Ca2+ stores by blocking their refill, an experimental manipulation known to selectively activate SOCE (Hogan et al., 2010). As expected, when cells treated with inactive SMase D were exposed to 1 µg Tg in the presence of a nominally Ca2+ free solution, the Fura-2 signal rose transiently, reflecting the slow release of Ca2+ from ER stores (Fig. 1 D). Subsequent reperfusion of these cells with the 2-mM Ca2+-containing solution revealed the SOCE component. SMase D treatment markedly suppressed the SOCE component of the Tg-triggered Fura-2 signal (Fig. 1, D and E). This result suggests that SMase D suppresses the Ca2+ signal not primarily by reducing Ca2+ release from ER, as the enzyme still suppresses SOCE when the ER is maximally depleted.
Indeed, SMase D had little effect on releasable Ca2+ of the ER, as cells treated with either active or inactive SMase D showed nearly identical Tg-triggered ER Ca2+ release (Fig. 2, A and B). Similar results were seen with an alternative known method to release intracellular Ca2+, namely, application of a very low concentration of the Ca2+ ionophore ionomycin (Dolmetsch and Lewis, 1994), further supporting the notion that SMase D has little or no direct effect on intracellular Ca2+ store content (Fig. 2, C and D). SMase D evidently suppresses the Fura-2 Ca2+ signal by lowering SOCE across the plasma membrane.
Figure 2.

SMase D does not alter internal store Ca2+ release in human T lymphocytes. (A) Fura-2 ratio signals from experiments shown in Fig. 1 D but with axes rescaled to better show the internal store release component for cells treated with SMase D (SMD, red) or catalytically inactive enzyme (CON, black). (B) Peak values (error bars represent mean ± SEM; n = 5) of Fura-2 ratio signals from Tg-stimulated cells in experiments as shown in A. (C) Fura-2 ratio signals of (38–84) T cells in a single 40× field, where the upper bar above the panel indicates these treatments: after 2-min rest, cells were treated with SMD (red) or CON (black) for 3 min, and then with 1 µg ionomycin (Iono) for 12 min; the lower bar shows the Ca2+-containing solution type. (D) Peak values (error bars represent mean ± SEM; n = 4) of Fura-2 ratio signals from ionomycin-stimulated cells in experiments as shown in C.
SMase D treatment suppresses SOCE in CD4-positive or CD8-positive T lymphocytes
C. pseudotuberculosis infections elicit responses from both CD4-positive and CD8-positive T lymphocytes (Ellis, 1988; Pépin et al., 1994). To evaluate the SMase D sensitivity of these T cell subtypes, T lymphocytes were labeled with fluorophore-conjugated antibodies to CD4 and CD8, and changes in intracellular Ca2+ levels were monitored with the ratiometric indicator Indo-1 during flow cytometry (Grynkiewicz et al., 1985). SMase D strongly suppressed the SOCE component of the Tg-triggered Indo-1 signal, in both the total T lymphocyte population and the CD4-positive and CD8-positive subsets (Fig. 3). Furthermore, these flow cytometry data, covering hundreds of thousands of cells per experiment, document an SMase D effect comparable to that initially observed with Fura-2 Ca2+ imaging, indicating that these previous observations were not made on a rare or peculiar cell subtype.
Figure 3.

SMase D suppresses SOCE in CD4 and CD8 T lymphocyte subsets. (A–C) Indo-1 ratio signals of total T cells (A), CD4-positive T cells (B), and CD8-positive T cells (C), where the upper bar above each panel indicates the following: after 2-min rest, cells were treated with SMase D (SMD, red) or catalytically inactive enzyme (CON, black) for 3 min and 1 µg Tg for 10 min; the lower bar above each panel shows Ca2+-containing solution type before and after the addition of 5 µl of 100 mM CaCl2 at 11 min, which results in a final Ca2+ concentration of ∼2 mM. (D and E) Peak (D) and declining phase (E) values (error bars represent mean ± SEM; n = 5) of indo-2 ratio signals from Tg-stimulated cells for experiments as shown in A–C. Where present, the asterisks denote statistically significant comparisons as detailed in Materials and methods.
Effects of C1P and SMase C on SOCE
In principle, SMase D could suppress SOCE through an intracellular signaling cascade by generating the second messenger C1P (Fig. 1 A). We therefore tested, by means of Ca2+ imaging, whether C1P reproduces the inhibitory effect of SMase D on T lymphocyte SOCE. C1P treatment augmented the Tg-triggered ER store release (Fig. 4, B and D), but it did not suppress SOCE (Fig. 4, A and C). On the other hand, SMase D did not alter the Tg-triggered ER store release but suppressed SOCE (Figs. 1 and 2). Alternatively, SMase D may suppress SOCE by modifying critical interactions between sphingomyelin head groups and membrane proteins. In this case, SMase C may produce a similar effect even though it hydrolyzes sphingomyelin in a different way (generating ceramide instead of C1P; Fig. 1 A; Glenny and Stevens, 1935; Doery et al., 1963). Indeed, we found that SMase C also inhibited Tg-triggered SOCE in T lymphocytes (Fig. 4 E).
Figure 4.

T lymphocyte SOCE is inhibited by SMases but not C1P. (A) Fura-2 ratio signals of (35–45) T cells, where the upper bar above the panel indicates treatments: after 2-min rest, cells were treated with SMase D (SMD, red), C1P (blue), or catalytically inactive enzyme (CON, black) for 3 min and 1 µg Tg for 18 min; the lower bar shows the Ca2+-containing solution type. Cells treated with C1P were also bathed in 0.5 mg/ml of C1P-containing solutions throughout the Tg treatment period. (B) Fura-2 ratio signals from experiments shown in A but with axes rescaled to better show the internal store release component for cells treated with SMD (red), C1P (blue), or CON (black). (C) Peak and declining phase (23 min) values (error bars represent mean ± SEM; n = 5) of Fura-2 ratio signals from Tg-stimulated cells for experiments as shown in A. (D) Peak values (error bars represent mean ± SEM; n = 5) of Fura-2 ratio signals from Tg-stimulated cells in experiments as shown in B. (E) Peak and declining phase values (error bars show mean ± SEM; n = 5) of Fura-2 ratio signals from Tg-stimulated cells treated with SMase C (SMC, orange) or CON (black). Where present, the asterisks denote statistically significant comparisons as detailed in Materials and methods.
Effects of SMase D on Kv1.3 channels
We next turned our attention to certain channels known to support SOCE in T lymphocytes. SMase D could lower Ca2+ influx across the plasma membrane either by inhibiting the SOCE pathway or by reducing the activity of K+ channels such as KV1.3, thereby lowering the electric driving force for Ca2+ entry. KV1.3 undergoes two types of voltage-dependent gating processes: activation and (C-type) inactivation (Cahalan et al., 1985). Although not tested yet, SMase D may lower the fraction of open KV1.3 channels at steady state by affecting their inactivation, thus depolarizing the cell membrane. Orai1 current is sensitive to membrane depolarization because Orai1 channels conduct Ca2+ in an inwardly rectifying manner (Lewis and Cahalan, 1989). We therefore examined how SMase D might affect the steady-state inactivation of KV1.3 channels.
Fig. 5 (A and B) shows KV1.3 currents elicited with double-pulse protocols to examine the extent of steady-state inactivation at various voltages before and after SMase D treatment. Concomitant with its known effect on the G-V curve, SMase D treatment caused a −15-mV shift in the steady-state inactivation curve of KV1.3 in T cells (Fig. 5 C). For a given condition, the channels are expected to exhibit meaningful steady-state activity within the “triangular” window beneath each set of activation and inactivation curves. SMase D caused a hyperpolarizing shift of this window. Judging from this shift alone, SMase D would likely hyperpolarize the membrane potential and thus favor, not impair, Ca2+ influx. If so, agents that hyperpolarize the membrane potential by increasing overall K+ conductance would be expected to have a relatively smaller boosting effect on the Ca2+ signal in cells already treated with SMase D compared with controls.
Figure 5.

Effect of SMase D on C-type inactivation of KV1.3 channels in human T lymphocytes. (A and B) KV1.3 currents elicited by stepping membrane voltage at 30-s intervals from the −100-mV holding potential to a first pulse between −90 and −10 mV in 10-mV increments followed by a second test pulse to 0 mV, before (A) and after (B) the addition of SMase D to the bath solution (0.25 µg/ml). Current traces for −40 mV are colored red; dashed lines indicate zero current levels. (C) G-V curves (squares) and h-infinity curves (circles) of KV1.3 channels in T cells before (black) and after (red) SMase D treatment; error bars represent means ± SEM (n = 5). The curves are fits of Boltzmann functions, yielding: V1/2 = −38.8 ± 0.2 mV and Z = 7.3 ± 0.4 (black circles); V1/2 = −54.4 ± 0.2 mV, Z = 6.3 ± 0.2, and c = 0.04 ± 0.01 (red circles); V1/2 = −26.1 ± 0.4 mV and Z = 3.7 ± 0.2 (black squares); and V1/2 = −42.4 ± 0.6 mV and Z = 4.0 ± 0.3 (red squares).
We tested that prediction with the K+ ionophore valinomycin, insertion of which into the plasma membrane is expected to cause hyperpolarization. In T cells treated with inactive SMase D, as the antibody-triggered Ca2+ signal approached the plateau, the addition of valinomycin induced a sizable Ca2+ transient (Fig. 6 A). Such transient was much smaller after SMase D treatment (Fig. 6, A and B). It was similarly decreased in Tg-stimulated cells (Fig. 6, C and D). These results are consistent with the K+ conductance of SMase D–treated cells being already greater than that of untreated cells. If SMase D’s action on KV1.3 channels indeed produces hyperpolarization, the effect is expected to persist even when KV1.3 channels are blocked, and the cause of the enzyme’s inhibitory effect on Ca2+ influx must be sought elsewhere. To demonstrate this, we blocked KV1.3 channels with PAP-1 (Schmitz et al., 2005). PAP-1 diminished the SOCE component of the Tg-triggered Fura-2 signal in control cells, reflecting the lower driving force for Ca2+ influx when KV1.3 is blocked (Fig. 6, E and F). As predicted, even during this block, SMase D treatment still suppressed the SOCE component of the Tg-triggered Fura-2 signal. Thus, the suppression of SOCE by SMase D cannot be explained by the enzyme’s effects on KV1.3 activity.
Figure 6.

Inhibition of SOCE by SMase D does not depend on decreased K+ conductance. (A) Fura-2 ratio signal of (26–54) T cells in a single 40× field, where the upper bar above the panel indicates these treatments: after 2-min rest, cells were treated with either SMase D (SMD, red) or catalytically inactive enzyme (CON, black) for 3 min, 5 µg anti-CD3ε antibody (Ab1) for 2 min, 5 µg anti-IgG antibody (Ab2) for 18 min, and finally 1 µg valinomycin (Val) for 5 min; the lower bar shows the Ca2+-containing solution type. (B) Peak values (error bars represent mean ± SEM; n = 5) of Fura-2 ratio signals induced by valinomycin as shown in A. (C) Fura-2 ratio signal of (17–31) T cells as in A, where the upper bar above the panel indicates these treatments: after 2-min rest, cells were treated successively with SMD (red) or CON (black) for 3 min, 1 µg Tg for 18 min, and 1 µg valinomycin (Val) for 5 min; the lower bar shows the Ca2+-containing solution type. (D) Peak values (error bars represent mean ± SEM; n = 5) of the valinomycin-induced Fura-2 ratio signals from experiments as shown in C. (E) Fura-2 ratio signals of (19–48) T cells as in A but in the presence of 200 nM PAP-1 throughout, where the upper bar above the panel indicates these treatments: after 2-min rest, cells were treated with either SMD (green) or CON (blue) for 3 min and 1 µg Tg for 18 min; the lower bar shows the Ca2+-containing solution type. (F) Peak and declining phase values (error bars represent mean ± SEM; n = 7) of Fura-2 ratio signals from Tg-stimulated cells in the presence of PAP-1 as shown in E. Where present, the asterisks denote statistically significant comparisons as detailed in Materials and methods.
SMase D treatment suppresses Orai1 current
The above findings suggest that SMase D treatment reduces Ca2+ current through the SOCE pathway, within which Orai1 and Stim1 are essential components (Lewis and Cahalan, 1989; Liou et al., 2005; Roos et al., 2005; Feske et al., 2006; Vig et al., 2006; Zhang et al., 2006). In native T cells, the whole-cell Orai1 current is generally very small, 2–3 pA given a driving force of about −240 mV (Partiseti et al., 1994), too small for us to examine its SMase D sensitivity. We therefore examined Orai1 currents heterologously expressed in CHO cells. Currents were recorded in the presence of 1 µM Tg (to deplete the ER) and 20 mM [Ca2+]ext (to boost current). Fig. 7 A shows Orai1 currents elicited by repeatedly stepping membrane potential from the 0-mV holding potential to −100 mV. Each current trace represents the difference between a matching pair of currents recorded in 20 or 0.1 mM of extracellular Ca2+. The addition of SMase D to the extracellular solution suppressed the current (Fig. 7, A and B). We collected peak and steady-state currents at a range of test potentials before and after SMase D treatment (Fig. 7, C and D) and plotted them against voltage in Fig. 7 E. Judging from the I-V curves, SMase D suppresses current at all voltages. Moreover, we found that SMase D also suppressed native Orai1 currents in Jurkat cells, a leukemic T cell line that expresses larger native Orai1 current than normal T cells (Fig. 7 F). These results show that the SMase D treatment strongly suppresses Orai1 current.
Figure 7.

SMase D suppresses Orai1 currents. (A and B) Orai1 currents (A) and steady-state current time course (B) recorded in the presence of 20 mM [Ca2+]ext, elicited by stepping the voltage from the 0-mV holding potential to −100 mV. After the addition of 0.5 µg/ml SMase D, the current was suppressed. (C and D) Orai1 currents elicited by stepping the membrane voltage from the 0-mV holding potential to between −120 and 60 mV in 10-mV increments before (C) and after (D) the addition of SMase D to the bath solution. Dashed lines in A, C, and D indicate zero current levels, and currents traces are shown after correcting for background currents (i.e., those recorded in a nominally Ca2+-free solution). (E) I-V relations of peak (squares) and steady-state current (circles) before (open) and after (closed) treatment with 0.5 µg/ml SMase D. Peak currents in E were measured as the mean current over a 0.5-ms window 2.0 ms after the start of the voltage step, and steady-state currents in B and E were measured as the mean current of a 40-ms window 100 ms after the start of the voltage step in A and C, or D, respectively. (F) Jurkat whole-cell Orai1 I-V relations of peak (squares) and steady-state current (circles) before (open) and after (closed) treatment with 0.5 µg/ml SMase D. Error bars represent mean ± SEM.
SMase D treatment suppresses production of IL-2 and TNF
Many T cell functions, including the production of key cytokines, depend on Orai1 currents. Two cytokines are important in the present context: IL-2, a signaling molecule important for the growth, proliferation, and differentiation of T lymphocytes (Waldmann, 2006); and TNF, a cytokine that plays a crucial role in the formation and maintenance of granulomas in microbial infections (Aggarwal et al., 2012). Genetic defects in Orai1 impair the production of IL-2 and TNF in stimulated T cells (Feske et al., 2000). In light of these properties and the fact that SMase D inhibits Orai1 current, we tested whether SMase D also decreases production of these key cytokines. Using antibody-coated beads, we stimulated human T lymphocytes in culture to produce IL-2 and TNF. We assayed cell culture supernatants by ELISA for the presence of these cytokines and found that SMase D indeed suppresses IL-2 and TNF production (Fig. 8).
Figure 8.
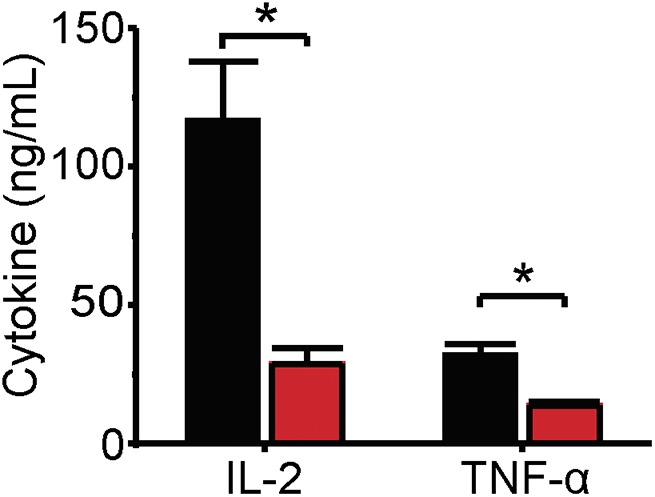
SMase D suppresses IL-2 and TNF production. IL-2 and TNF concentrations (error bars represent means ± SEM; n = 5) in cell culture supernatants of human T lymphocytes stimulated with CD3 and CD28 antibody-coated beads in the presence of SMase D (red) or catalytically inactive enzyme (CON, black). Asterisks denote statistically significant comparisons as detailed in Materials and methods.
DISCUSSION
Host immune systems struggle to clear certain bacterial pathogens. The histology of infected tissues may reveal granulomas—aggregates of multinucleated macrophages, fibroblasts, and lymphocytes—with necrotic foci. These necrotic areas impart a cheese-like texture to gross organ specimens, and this caseous necrosis is characteristic of certain bacterial infections. Although granuloma formation limits dissemination of bacteria, the failure of host immune cells to clear out invading bacteria can ultimately result in latent or chronic infection. Studies of the caseous lymphadenitis of C. pseudotuberculosis show that experimental mutant strains lacking SMase D exhibit dramatically reduced infectiousness and cannot effectively disseminate in host organs. These observations strongly suggest that SMase D helps bacteria to evade host immunity. However, the molecular mechanism by which SMase D produces this effect remains unknown.
In a previous set of biophysical studies, the activation gating of many KV channels was shown to be sensitive to certain SMase enzymes, including bacterial SMase D (Ramu et al., 2006; Xu et al., 2008; Milescu et al., 2009; Combs et al., 2013). SMase D stimulates KV channels by removing the positively charged choline group from sphingomyelin molecules in the outer leaflet of the membrane, making it energetically easier for the positively charged voltage sensor to move to the extracellular side and the channel to become activated (Ramu et al., 2006; Xu et al., 2008; Milescu et al., 2009). In doing so, the enzyme shifts the Q-V and G-V curves in the hyperpolarized direction. SMase D can activate KV1.3 channels, including endogenous KV1.3 channels in human T lymphocytes (Combs et al., 2013). Physiologically, KV1.3 is best characterized in the T lymphocyte system where the channel helps maintain negative resting membrane potentials, ensuring a driving force for Ca2+ entry adequate to trigger various critical immune responses (Cahalan and Chandy, 2009). In fact, KV1.3 channel inhibitors are being developed as effective immunosuppressants to treat autoimmune diseases (Chandy et al., 2004). SMase D markedly increases KV1.3 activity of T lymphocytes near typical resting membrane potentials (−50 mV). Therefore, SMase D would be expected to promote Ca2+ entry and thus lymphocyte activation. However, we found here that SMase D markedly lowers Ca2+ entry in both CD4-positive and CD8-positive lymphocytes by suppressing SOCE. This finding cannot be readily explained by the enzyme’s previously documented stimulating effect on T lymphocyte KV1.3 channels. Given that SMase D treatment shifts the Q-V curve in the hyperpolarized direction, the enzyme could, in principle, lower SOCE by promoting steady-state inactivation, thus causing depolarization and thereby reducing the driving force for Ca2+ entry. However, we find no evidence supporting such a scenario. In fact, even after KV1.3 channels are blocked, SMase D still effectively lowers the Ca2+ signal. These findings imply that SMase D treatment negatively impacts Orai1 activity. Indeed, we find that SMase D strongly suppresses Ca2+ current through both native and heterologously expressed Orai1 channels.
Two types of lipid–channel interaction mechanisms are frequently invoked to explain the effects of lipids on ion channel function: lipid molecules acting indirectly as second messengers, and direct lipid–channel interactions. The best known example of indirect action is PIP2-mediated regulation (Huang et al., 1998). As for direct lipid–channel interactions, native lipid molecules remain bound to KcsA channels, even after the channels are solubilized in detergent-containing solutions and eventually crystallized (Zhou et al., 2001; Valiyaveetil et al., 2002). Furthermore, although common lipids can generally support the folding of KcsA, an appropriate type of lipid head group is critical for KcsA function (Valiyaveetil et al., 2002). In the present case, therefore, one possible scenario is that SMase-generated lipids act as second messengers, triggering an intracellular signaling cascade that leads to Orai1 inhibition. Some prior studies of SMase C have favored this mechanism. For example, SMase C, ceramide (the lipid product of SMase C), and sphingosine have been reported to suppress SOCE in Jurkat T cells (Breittmayer et al., 1994; Lepple-Wienhues et al., 1999; Church et al., 2005). However, other investigations report instead that ceramide and sphingosine boost SOCE in Jurkat T cells by triggering Ca2+ release from intracellular stores (Sakano et al., 1996; Colina et al., 2005b). In this study, we find that exogenous C1P (the lipid product of SMase D) fails to suppress SOCE and instead augments the release of store Ca2+ by Tg. A prior study of Jurkat T cells has documented Ca2+ store release by exogenous C1P (Colina et al., 2005a). Thus, whereas exogenous C1P affects store Ca2+ release, it fails to mimic SMase D’s suppression of SOCE. It is noteworthy that the C1P generated by exogenous SMase D does not appear to break down into other sphingolipids, as SMase D does not increase cell ceramide levels (Feldhaus et al., 2002), and the enzyme generates stoichiometric quantities of C1P from sphingomyelin (Subbaiah et al., 2003). Additionally, sphingomyelin levels recover 5 h after SMase C treatment but show little or no recovery even 20 h after SMase D treatment (Subbaiah et al., 2003).
Hydrolysis of sphingomyelin by SMase D has been shown to suppress the CFTR Cl− channel and to lower the activation voltages of voltage-gated ion channels (Ramu et al., 2006, 2007; Milescu et al., 2009; Combs et al., 2013). The SMase D effect on Kv channels persists even 24 h after treatment (Xu et al., 2008). Exposure to exogenous sphingomyelin or C1P fails to reproduce these functional effects in either channel type (Ramu et al., 2006, 2007). These findings suggest that the channel-impacting lipid molecules remain tightly bound to channel proteins, as they demonstrably do in the KcsA channel. Then implicitly, SMase D affects the channel function by modifying channel–sphingomyelin interactions in situ. This scenario can also explain why SMase D, which generates C1P, and SMase C, which generates a different lipid product (ceramide), both suppress Orai1 activity. Although C1P may, in principle, be broken down to ceramide or other sphingolipids, SMase D does not appear to raise the level of these lipid species, as mentioned above. In any case, further studies are needed to firmly establish the mechanism by which SMases suppress Orai1 activity.
Genetic defects in Orai1 have been identified as a cause of SCID (Partiseti et al., 1994; Feske et al., 2006). The Orai1 mutations in these SCID T cells result in dramatic functional impairments. For example, when stimulated, T cells from these SCID patients show decreased production of key cytokines like IL-2 and TNF (Feske et al., 2000). The former cytokine is crucial for T cell growth, proliferation, and differentiation (Waldmann, 2006), whereas the latter supports the formation and maintenance of granulomas in microbial infections (Aggarwal et al., 2012). In fact, the use of TNF inhibitors in the treatment of human inflammatory diseases is complicated by a risk of reactivation tuberculosis (Wallis, 2008). In the present study, we find that SMase D suppresses Orai1 current and also decreases production of IL-2 and TNF. Thus, C. pseudotuberculosis could, by means of inhibiting Orai1 with SMase D, create an acquired SCID-like condition, allowing the bacteria to avoid clearance by host T lymphocytes. Experimental worsening of this situation, for example, by antibody depletion of host T cells or their cytokines, turns a chronic disease into an acute and highly lethal one (Lan et al., 1998, 1999). A strikingly similar picture is seen in acquired immunodeficiency syndrome, where viral depletion of T cells enables the aggressive spread and high lethality of another organism known for caseous granulomas—M. tuberculosis, which has a C-type SMase activity (Vargas-Villarreal et al., 2003). Although suppression of Orai1 current can account for the mechanism by which SMase D helps to prevent bacterial clearance by the host immune system, SMase D may also affect other signaling processes in lymphocytes. Given that Orai1 channels are present in many other cell types (Hogan et al., 2010), SMase D may impact their functions as well.
The failure of human or animal immune systems to either adequately contain C. pseudotuberculosis in granulomas or to eradicate them from the body has a profoundly negative impact on host health. For example, in some regions of the world the prevalence of caseous lymphadenitis in livestock may be as high as 20%, causing substantial economic losses (Baird and Fontaine, 2007). As a zoonotic organism, C. pseudotuberculosis can be transmitted to humans. On the other hand, M. tuberculosis continues to pose a major threat to human health (Zumla et al., 2013). Worldwide, about two billion people are estimated to be infected with M. tuberculosis, and an estimated 1 in 14 new infections occurs in individuals infected with HIV (Lönnroth and Raviglione, 2008). Treatment of caseous lymphadenitis remains a challenge today, as both antibiotics and vaccines suffer from limited efficacy (Baird and Fontaine, 2007). Antibiotics were originally more successful in treating tuberculosis infections, but their widespread use has led to the emergence of highly resistant strains (Zumla et al., 2013). Thus, our discovery suggests neutralization of bacterial phospholipidases as a new additional strategy to combat these recalcitrant infections, and highlights the importance of appropriate protein–lipid interactions in maintaining normal function of ion channels.
Acknowledgments
We thank S. Billington for SMase D cDNAs; M. Cahalan for Orai1 and Stim1 cDNAs; the Penn Flow Cytometry and Cell Sorting Resource Laboratory and the Penn Human Immunology Core for their services and expertise; and P. De Weer for review of the manuscript.
This study was supported by the National Institutes of Health: a research grant to Z. Lu from the National Institute of General Medical Science (RO1 GM55560) and a fellowship grant to D.J. Combs from the National Institute of Neurological Disorders and Strokes (F31 NS73070). Z. Lu is an investigator of the Howard Hughes Medical Institute.
The authors declare no competing financial interests.
Kenton J. Swartz served as editor.
Footnotes
Abbreviations used in this paper:
- C1P
- ceramide-1-phosphate
- CHO
- Chinese hamster ovary
- IL-2
- interleukin-2
- IP3
- inositol 1,4,5 trisphosphate
- PAP-1
- 5-(4-phenoxybutoxy)psoralen
- SCID
- severe combined immunodeficiency
- SMase
- sphingomyelinase
- SOCE
- store-operated Ca2+ entry
- TCR
- T cell receptor
- Tg
- thapsigargin
- TNF
- tumor necrosis factor α
References
- Aggarwal B.B., Gupta S.C., and Kim J.H.. 2012. Historical perspectives on tumor necrosis factor and its superfamily: 25 years later, a golden journey. Blood. 119:651–665. 10.1182/blood-2011-04-325225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird G.J., and Fontaine M.C.. 2007. Corynebacterium pseudotuberculosis and its role in ovine caseous lymphadenitis. J. Comp. Pathol. 137:179–210. 10.1016/j.jcpa.2007.07.002 [DOI] [PubMed] [Google Scholar]
- Breittmayer J.P., Bernard A., and Aussel C.. 1994. Regulation by sphingomyelinase and sphingosine of Ca2+ signals elicited by CD3 monoclonal antibody, thapsigargin, or ionomycin in the Jurkat T cell line. J. Biol. Chem. 269:5054–5058. [PubMed] [Google Scholar]
- Cahalan M.D., and Chandy K.G.. 2009. The functional network of ion channels in T lymphocytes. Immunol. Rev. 231:59–87. 10.1111/j.1600-065X.2009.00816.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahalan M.D., Chandy K.G., DeCoursey T.E., and Gupta S.. 1985. A voltage-gated potassium channel in human T lymphocytes. J. Physiol. 358:197–237. 10.1113/jphysiol.1985.sp015548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandy K.G., Wulff H., Beeton C., Pennington M., Gutman G.A., and Cahalan M.D.. 2004. K+ channels as targets for specific immunomodulation. Trends Pharmacol. Sci. 25:280–289. 10.1016/j.tips.2004.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Church L.D., Hessler G., Goodall J.E., Rider D.A., Workman C.J., Vignali D.A., Bacon P.A., Gulbins E., and Young S.P.. 2005. TNFR1-induced sphingomyelinase activation modulates TCR signaling by impairing store-operated Ca2+ influx. J. Leukoc. Biol. 78:266–278. 10.1189/jlb.1003456 [DOI] [PubMed] [Google Scholar]
- Colina C., Flores A., Castillo C., Garrido M.R., Israel A., DiPolo R., and Benaim G.. 2005a. Ceramide-1-P induces Ca2+ mobilization in Jurkat T-cells by elevation of Ins(1,4,5)-P3 and activation of a store-operated calcium channel. Biochem. Biophys. Res. Commun. 336:54–60. 10.1016/j.bbrc.2005.08.039 [DOI] [PubMed] [Google Scholar]
- Colina C., Flores A., Rojas H., Acosta A., Castillo C., Garrido M.R., Israel A., DiPolo R., and Benaim G.. 2005b. Ceramide increase cytoplasmic Ca2+ concentration in Jurkat T cells by liberation of calcium from intracellular stores and activation of a store-operated calcium channel. Arch. Biochem. Biophys. 436:333–345. 10.1016/j.abb.2005.02.014 [DOI] [PubMed] [Google Scholar]
- Combs D.J., Shin H.G., Xu Y., Ramu Y., and Lu Z.. 2013. Tuning voltage-gated channel activity and cellular excitability with a sphingomyelinase. J. Gen. Physiol. 142:367–380. 10.1085/jgp.201310986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCoursey T.E., Chandy K.G., Gupta S., and Cahalan M.D.. 1984. Voltage-gated K+ channels in human T lymphocytes: a role in mitogenesis? Nature. 307:465–468. 10.1038/307465a0 [DOI] [PubMed] [Google Scholar]
- Doery H.M., Magnusson B.J., Cheyne I.M., and Sulasekharam J.. 1963. A phospholipase in staphylococcal toxin which hydrolyses sphingomyelin. Nature. 198:1091–1092. 10.1038/1981091a0 [DOI] [PubMed] [Google Scholar]
- Dolmetsch R.E., and Lewis R.S.. 1994. Signaling between intracellular Ca2+ stores and depletion-activated Ca2+ channels generates [Ca2+]i oscillations in T lymphocytes. J. Gen. Physiol. 103:365–388. 10.1085/jgp.103.3.365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis J.A. 1988. Immunophenotype of pulmonary cellular infiltrates in sheep with visceral caseous lymphadenitis. Vet. Pathol. 25:362–368. 10.1177/030098588802500505 [DOI] [PubMed] [Google Scholar]
- Feldhaus M.J., Weyrich A.S., Zimmerman G.A., and McIntyre T.M.. 2002. Ceramide generation in situ alters leukocyte cytoskeletal organization and β 2-integrin function and causes complete degranulation. J. Biol. Chem. 277:4285–4293. 10.1074/jbc.M106653200 [DOI] [PubMed] [Google Scholar]
- Feske S., Draeger R., Peter H.H., Eichmann K., and Rao A.. 2000. The duration of nuclear residence of NFAT determines the pattern of cytokine expression in human SCID T cells. J. Immunol. 165:297–305. 10.4049/jimmunol.165.1.297 [DOI] [PubMed] [Google Scholar]
- Feske S., Gwack Y., Prakriya M., Srikanth S., Puppel S.H., Tanasa B., Hogan P.G., Lewis R.S., Daly M., and Rao A.. 2006. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature. 441:179–185. 10.1038/nature04702 [DOI] [PubMed] [Google Scholar]
- Glenny A.T., and Stevens N.F.. 1935. Staphylococcal toxins and antitoxins. J. Pathol. Bacteriol. 40:201–210. 10.1002/path.1700400202 [DOI] [Google Scholar]
- Grynkiewicz G., Poenie M., and Tsien R.Y.. 1985. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 260:3440–3450. [PubMed] [Google Scholar]
- Hogan P.G., Lewis R.S., and Rao A.. 2010. Molecular basis of calcium signaling in lymphocytes: STIM and ORAI. Annu. Rev. Immunol. 28:491–533. 10.1146/annurev.immunol.021908.132550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C.L., Feng S., and Hilgemann D.W.. 1998. Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by Gβγ. Nature. 391:803–806. 10.1038/35882 [DOI] [PubMed] [Google Scholar]
- Imboden J.B., and Stobo J.D.. 1985. Transmembrane signalling by the T cell antigen receptor. Perturbation of the T3–antigen receptor complex generates inositol phosphates and releases calcium ions from intracellular stores. J. Exp. Med. 161:446–456. 10.1084/jem.161.3.446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isbister G.K., and Fan H.W.. 2011. Spider bite. Lancet. 378:2039–2047. 10.1016/S0140-6736(10)62230-1 [DOI] [PubMed] [Google Scholar]
- Lan D.T., Taniguchi S., Makino S., Shirahata T., and Nakane A.. 1998. Role of endogenous tumor necrosis factor alpha and gamma interferon in resistance to Corynebacterium pseudotuberculosis infection in mice. Microbiol. Immunol. 42:863–870. 10.1111/j.1348-0421.1998.tb02362.x [DOI] [PubMed] [Google Scholar]
- Lan D.T., Makino S., Shirahata T., Yamada M., and Nakane A.. 1999. Tumor necrosis factor alpha and gamma interferon are required for the development of protective immunity to secondary Corynebacterium pseudotuberculosis infection in mice. J. Vet. Med. Sci. 61:1203–1208. 10.1292/jvms.61.1203 [DOI] [PubMed] [Google Scholar]
- Lepple-Wienhues A., Belka C., Laun T., Jekle A., Walter B., Wieland U., Welz M., Heil L., Kun J., Busch G., et al. 1999. Stimulation of CD95 (Fas) blocks T lymphocyte calcium channels through sphingomyelinase and sphingolipids. Proc. Natl. Acad. Sci. USA. 96:13795–13800. 10.1073/pnas.96.24.13795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis R.S., and Cahalan M.D.. 1989. Mitogen-induced oscillations of cytosolic Ca2+ and transmembrane Ca2+ current in human leukemic T cells. Cell Regul. 1:99–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou J., Kim M.L., Heo W.D., Jones J.T., Myers J.W., Ferrell J.E. Jr, and Meyer T.. 2005. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol. 15:1235–1241. 10.1016/j.cub.2005.05.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipsky N.G., and Pagano R.E.. 1985. A vital stain for the Golgi apparatus. Science. 228:745–747. 10.1126/science.2581316 [DOI] [PubMed] [Google Scholar]
- Lönnroth K., and Raviglione M.. 2008. Global epidemiology of tuberculosis: Prospects for control. Semin. Respir. Crit. Care Med. 29:481–491. 10.1055/s-0028-1085700 [DOI] [PubMed] [Google Scholar]
- Matteson D.R., and Deutsch C.. 1984. K channels in T lymphocytes: a patch clamp study using monoclonal antibody adhesion. Nature. 307:468–471. 10.1038/307468a0 [DOI] [PubMed] [Google Scholar]
- McNamara P.J., Bradley G.A., and Songer J.G.. 1994. Targeted mutagenesis of the phospholipase D gene results in decreased virulence of Corynebacterium pseudotuberculosis. Mol. Microbiol. 12:921–930. 10.1111/j.1365-2958.1994.tb01080.x [DOI] [PubMed] [Google Scholar]
- McNamara P.J., Cuevas W.A., and Songer J.G.. 1995. Toxic phospholipases D of Corynebacterium pseudotuberculosis, C. ulcerans and Arcanobacterium haemolyticum: cloning and sequence homology. Gene. 156:113–118. 10.1016/0378-1119(95)00002-N [DOI] [PubMed] [Google Scholar]
- Milescu M., Bosmans F., Lee S., Alabi A.A., Kim J.I., and Swartz K.J.. 2009. Interactions between lipids and voltage sensor paddles detected with tarantula toxins. Nat. Struct. Mol. Biol. 16:1080–1085. 10.1038/nsmb.1679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partiseti M., Le Deist F., Hivroz C., Fischer A., Korn H., and Choquet D.. 1994. The calcium current activated by T cell receptor and store depletion in human lymphocytes is absent in a primary immunodeficiency. J. Biol. Chem. 269:32327–32335. [PubMed] [Google Scholar]
- Pépin M., Pittet J.C., Olivier M., and Gohin I.. 1994. Cellular composition of Corynebacterium pseudotuberculosis pyogranulomas in sheep. J. Leukoc. Biol. 56:666–670. [DOI] [PubMed] [Google Scholar]
- Ramu Y., Xu Y., and Lu Z.. 2006. Enzymatic activation of voltage-gated potassium channels. Nature. 442:696–699. 10.1038/nature04880 [DOI] [PubMed] [Google Scholar]
- Ramu Y., Xu Y., and Lu Z.. 2007. Inhibition of CFTR Cl− channel function caused by enzymatic hydrolysis of sphingomyelin. Proc. Natl. Acad. Sci. USA. 104:6448–6453. 10.1073/pnas.0701354104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roos J., DiGregorio P.J., Yeromin A.V., Ohlsen K., Lioudyno M., Zhang S., Safrina O., Kozak J.A., Wagner S.L., Cahalan M.D., et al. 2005. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J. Cell Biol. 169:435–445. 10.1083/jcb.200502019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakano S., Takemura H., Yamada K., Imoto K., Kaneko M., and Ohshika H.. 1996. Ca2+ mobilizing action of sphingosine in Jurkat human leukemia T cells. Evidence that sphingosine releases Ca2+ from inositol trisphosphate- and phosphatidic acid-sensitive intracellular stores through a mechanism independent of inositol trisphosphate. J. Biol. Chem. 271:11148–11155. [DOI] [PubMed] [Google Scholar]
- Schmitz A., Sankaranarayanan A., Azam P., Schmidt-Lassen K., Homerick D., Hänsel W., and Wulff H.. 2005. Design of PAP-1, a selective small molecule Kv1.3 blocker, for the suppression of effector memory T cells in autoimmune diseases. Mol. Pharmacol. 68:1254–1270. 10.1124/mol.105.015669 [DOI] [PubMed] [Google Scholar]
- Souček A., Michalec C., and Soucková A.. 1971. Identification and characterization of a new enzyme of the group “phospholipase D” isolated from Corynebacterium ovis. Biochim. Biophys. Acta. 227:116–128. 10.1016/0005-2744(71)90173-2 [DOI] [PubMed] [Google Scholar]
- Subbaiah P.V., Billington S.J., Jost B.H., Songer J.G., and Lange Y.. 2003. Sphingomyelinase D, a novel probe for cellular sphingomyelin: effects on cholesterol homeostasis in human skin fibroblasts. J. Lipid Res. 44:1574–1580. 10.1194/jlr.M300103-JLR200 [DOI] [PubMed] [Google Scholar]
- Valiyaveetil F.I., Zhou Y., and MacKinnon R.. 2002. Lipids in the structure, folding, and function of the KcsA K+ channel. Biochemistry. 41:10771–10777. 10.1021/bi026215y [DOI] [PubMed] [Google Scholar]
- Vargas-Villarreal J., Mata-Cárdenas B.D., Deslauriers M., Quinn F.D., Castro-Garza J., Martínez-Rodrĺguez H.G., and Said-Fernández S.. 2003. Identification of acidic, alkaline, and neutral sphingomyelinase activities in Mycobacterium tuberculosis. Med. Sci. Monit. 9:BR225–BR230. [PubMed] [Google Scholar]
- Vig M., Beck A., Billingsley J.M., Lis A., Parvez S., Peinelt C., Koomoa D.L., Soboloff J., Gill D.L., Fleig A., et al. 2006. CRACM1 multimers form the ion-selective pore of the CRAC channel. Curr. Biol. 16:2073–2079. 10.1016/j.cub.2006.08.085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldmann T.A. 2006. The biology of interleukin-2 and interleukin-15: implications for cancer therapy and vaccine design. Nat. Rev. Immunol. 6:595–601. 10.1038/nri1901 [DOI] [PubMed] [Google Scholar]
- Wallis R.S. 2008. Tumour necrosis factor antagonists: structure, function, and tuberculosis risks. Lancet Infect. Dis. 8:601–611. 10.1016/S1473-3099(08)70227-5 [DOI] [PubMed] [Google Scholar]
- Xu Y., Ramu Y., and Lu Z.. 2008. Removal of phospho-head groups of membrane lipids immobilizes voltage sensors of K+ channels. Nature. 451:826–829. 10.1038/nature06618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S.L., Yeromin A.V., Zhang X.H., Yu Y., Safrina O., Penna A., Roos J., Stauderman K.A., and Cahalan M.D.. 2006. Genome-wide RNAi screen of Ca2+ influx identifies genes that regulate Ca2+ release-activated Ca2+ channel activity. Proc. Natl. Acad. Sci. USA. 103:9357–9362. 10.1073/pnas.0603161103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Morais-Cabral J.H., Kaufman A., and MacKinnon R.. 2001. Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 Å resolution. Nature. 414:43–48. 10.1038/35102009 [DOI] [PubMed] [Google Scholar]
- Zumla A., Raviglione M., Hafner R., and von Reyn C.F.. 2013. Tuberculosis. N. Engl. J. Med. 368:745–755. 10.1056/NEJMra1200894 [DOI] [PubMed] [Google Scholar]