

Abstract
The MAPKs ERK1/2 respond to nutrients and other insulin secretagogues in pancreatic β-cells and mediate nutrient-dependent insulin gene transcription. Nutrients also stimulate the mechanistic target of rapamycin complex 1 (mTORC1) to regulate protein synthesis. We showed previously that activation of both ERK1/2 and mTORC1 in the MIN6 pancreatic β-cell-derived line by extracellular amino acids (AAs) is at least in part mediated by the heterodimeric T1R1/T1R3, a G protein-coupled receptor. We show here that AAs differentially activate these two signaling pathways in MIN6 cells. Pretreatment with pertussis toxin did not prevent the activation of either ERK1/2 or mTORC1 by AAs, indicating that Gi is not central to either pathway. Although glucagon-like peptide 1, an agonist for a Gs-coupled receptor, activated ERK1/2 well and mTORC1 to a small extent, AAs had no effect on cytosolic cAMP accumulation. Ca2+ entry is required for ERK1/2 activation by AAs but is dispensable for AA activation of mTORC1. Pretreatment with UBO-QIC, a selective Gq inhibitor, reduced the activation of ERK1/2 but had little effect on the activation of mTORC1 by AAs, suggesting a differential requirement for Gq. Inhibition of G12/13 by the overexpression of the regulator of G protein signaling domain of p115 ρ-guanine nucleotide exchange factor had no effect on mTORC1 activation by AAs, suggesting that these G proteins are also not involved. We conclude that AAs regulate ERK1/2 and mTORC1 through distinct signaling pathways.
In pancreatic β-cells, the activities of the MAPKs ERK1/2 mirror the demand on the cells to produce insulin. ERK1/2 integrate short- and long-term nutrient-sensing information and secretagogue stimulation primarily to regulate insulin gene transcription (1–6). The mechanistic target of rapamycin complex 1 (mTORC1) coordinates energy and growth signals with the availability of amino acids (AAs) and glucose to ensure synthesis of preproinsulin and other proteins (7–12). AAs regulate insulin secretion by incompletely defined mechanisms thought to require their uptake and metabolism (13, 14). AA uptake is also important for mTORC1 activation, and cells apparently use multiple mechanisms to modulate mTORC1 in response to extracellular and intracellular AAs (15). In searching for the link between AAs and these β-cell signaling pathways, we recently found that AAs stimulate ERK1/2 and mTORC1 activities through the T1R1/T1R3 taste receptor, a G protein-coupled receptor (GPCR) (16).
T1R1/T1R3 is a heterodimer, one of several class C GPCRs, including the metabotropic glutamate, Ca2+-sensing, and G protein receptor C 6A receptors, that are sensitive to AAs (17). T1R1/T1R3 was identified as mediating umami taste in gustatory neurons and is an AA sensor in the intestine (18–20). In gustatory neurons, T1R1/T1R3 signals to the G protein gustducin, a Gi family member. The taste receptor signaling pathway delineated in taste neurons involves G protein gustducin-βγ subunits activating phospholipase C-β and increasing inositol trisphosphate. Inositol trisphosphate activates receptors on the Ca2+ storage compartment, elevating cytoplasmic free Ca2+ and activating a transient receptor potential cation channel. Monovalent cations enter by this or other mechanisms and depolarize cells, which opens voltage-sensitive Ca2+ channels to promote further Ca2+ entry (21). A similar pathway has been suggested to occur in the gut (22). However, T1R1/T1R3 is widely expressed.
To explore mechanisms of ERK1/2 and mTORC1 regulation by T1R1/T1R3 in β-cells, we examined the effects of AAs, ligands for receptors that regulate several different G protein family members, and inhibitors of signaling by G proteins on the activities of these kinases in MIN6 cells. We report that T1R1/T1R3 regulates both ERK1/2 and mTORC1 in MIN6 cells but does so using different signaling pathways that are both distinct from that reported to sense taste in gustatory neurons.
Materials and Methods
Materials
Chemicals were obtained from the following sources: glucagon-like peptide 1 amide fragment 7–36, human (GLP-1), exendin-4 (EXD-4), individual AAs, UK14304, inosine monophosphate (IMP) from Sigma-Aldrich; human epidermal growth factor (EGF) from Gemini Bio-Products; pertussis toxin from Invitrogen; Fura-2AM from Molecular Probes; essential AAs (EAAs) (50× stock) and nonessential AAs (NEAAs) (100× stock) from either Invitrogen or MP Biomedicals; and the Gq inhibitor UBO-QIC was the gift of Dr V. Slepak (University of Miami, Miami, Florida) (23). The p115 ρ-guanine nucleotide exchange factor (GEF) regulator of G protein signaling (RGS) domain plasmid and SRE.L luciferase plasmid were obtained from Dr Paul Sternweis and colleagues (24) and Dr Silvio Gutkind (25). Antibodies were as follows: mouse monoclonal phosphorylated (p) ERK1/2 (human ERK1, Thr202/Tyr204) antibody (number M8159) from Sigma; rabbit anti-ERK1/2 polyclonal antibody (Y691), as described (26); p-S6 kinase (S6K) (T389) (number 9206L), S6 (number 2317S), pS6 (S235/236) (number 2211S), pS6 (S240/244) (number 5364S), and rabbit anti-p4E-binding protein 1 (4EBP1; T37/46) (number 2855P) antibodies from Cell Signaling. The AA mixture used for most studies included 0.1 mM Gly and the following L-AAs: 0.1 mM Ala, 0.6 mM Arg, 0.1 mM Asn, 0.1 mM Asp, 0.1 mM Cys, 0.5 mM Gln, 0.1 mM Glu, 0.2 mM His, 0.4 mM Ile, 0.4 mM Leu, 0.4 mM Lys, 0.1 mM Met, 0.2 mM Phe, 0.1 mM Pro, 0.1 mM Ser, 0.4 mM Thr, 0.1 mM Trp, 0.2 mM Tyr, and 0.4 mM Val (totaling 4.7 mM). EAAs included the following: Arg, Cys, His, Ile, Leu, Lys, Met, Phe, Thr, Trp, Tyr, and Val at the same concentrations. The NEAA mixture included Gly, Ala, Asn, Asp, Glu, Pro, and Ser.
Cell culture and treatment
DMEM was used to culture MIN6 and HeLa cells (27) at 37°C in 10% CO2. DMEM was supplemented with 10% fetal bovine serum, 10 mM HEPES (pH 7.4), 50 mM sodium pyruvate, 10.2 mM L-glutamine, 25 mM glucose, 2.5 mM β-mercaptoethanol, penicillin (100 U/mL), and streptomycin (0.1 mg/mL). MIN6 cells stably expressing either nontarget short hairpin RNA (shRNA) or T1R3 shRNA were made as described in (16). Human islets were provided by the Islet Resource Facility supported by the University of Alabama, Birmingham, Comprehensive Diabetes Center (Figure 1B, body mass index-36). Prior to stimulation, MIN6 cells were incubated for 2 hours in Krebs-Ringer's bicarbonate solution (KRBH) supplemented with 4.5 mM glucose and 0.1% BSA. HeLa cells were in KRBH for 30–35 minutes prior to stimulation. Cells were then stimulated with mixtures of AAs specified above or as indicated in figure legends.
Figure 1.

AAs activate ERK1/2 and mTORC1. MIN6 cells (A, C, and D) were placed in KRBH without AAs (see Materials and Methods) and 4.5 mM glucose for 2–3 hours prior to stimulation with AAs for the times indicated on each panel. Human islets (B) were treated similarly except that 2 mM glucose was used. After treatment, cell lysates were resolved on gels and transferred to nitrocellulose for immunoblotting of pERK1/2, pS6K, pS6 (240/244), or p4EBP1 as indicated. ERK1/2, S6K, and S6 were loading controls used to normalize the p protein signals to total protein amount (eg, pS6K/S6K). The value for the untreated control signal is set to 1. A, Stimulation with AAs for the indicated times and is representative of 10 experiments. Quantification of 4 is shown in Supplemental Figure 1A. B, Stimulation of islets with AAs for 2 minutes. One of two experiments with islets from different donors is shown. C, Stimulation with individual AAs at 0.25, 0.5, or 5 mM for 2 minutes. One of six experiments is shown. D, Stimulation with individual AAs at 1 mM for 30 minutes. Immunoblots show one of three experiments is shown. Bar graphs are means of quantification ± SEM (n = 3). *, P < .05; **, P < .01, using one-way ANOVA.
Immunoblotting
Cells were lysed by vortexing in 137 mM NaCl, 20 mM Tris (pH7.5), 10% glycerol, 1% Triton X-100, 0.2 mM sodium vanadate, 1 mM sodium fluoride, 1 mM phenylmethylsulfonylfluoride, 0.4 mg/mL pepstatin A, 0.4 mg/mL leupeptin, 4 mg/mL Nα-p-tosyl-L-arginine methyl ester, 4 mg/mL Nα-p-tosyl-L-lysine chloromethyl ketone, 4 mg/mL Nα-benzoyl-L-arginine methyl ester, and 4 mg/mL soybean trypsin inhibitor. Lysates were stored at −80°C and, upon thawing, were sedimented at 16 000 × g for 10 minutes at 4°C to remove insoluble material. Lysate proteins were separated in 10% polyacrylamide gels in sodium dodecyl sulfate and transferred to nitrocellulose for immunoblotting. Either enhanced chemiluminescence and film or LI-COR Odyssey infrared imaging was used to detect immunoreactivity. Quantitation of the immunoblots was performed using Odyssey version 3 software.
Intracellular Ca2+ assays
MIN6 cells were incubated in KRBH containing 5 μM fura 2-AM for 1 hour as described (16) and then washed and equilibrated in fresh KRBH for 30 minutes. dimethylsulfoxide (DMSO) or UK14304 in DMSO was added to cells at the beginning of the equilibration period. Changes in intracellular Ca2+ were measured every 0.74 seconds for 2 minutes by dual excitation of fura 2 at 340/11 and 380/20 nm (center/bandpass) and emission at 508/20 nm using a Synergy2 microplate reader (BioTek) and Gen5 software. Treatments were in triplicate for each experiment. To determine changes in free intracellular Ca2+, we normalized the basal 340:380 ratio by averaging the value of the basal conditions before stimulation. The average value was subtracted from the pre- and poststimulation values.
Detection of cAMP using an intracellular cAMP sensor
The exchange proteins activated by cAMP-based bioluminescence resonance energy transfer (BRET) sensor for cAMP (CAMYEL) was as described (28). MIN6 cells were infected with a retrovirus expressing CAMYEL and selected for expression using G418 as described (28). BRET assays were performed on the microplate reader as above. Emission signals at 485/20 and 528/20 nm (center/bandpass) were measured every 0.8 seconds for 1 minute (basal) and for an additional 4 minutes after stimulation.
Luciferase assay
A mutated serum-response element (SRE), SRE.L, was used to monitor the activation of G12/13 (25). The pGL4 vector containing the firefly luciferase gene driven by the SRE.L promoter (0.22 mg) was transfected into HeLa cells. The pCMV5-myc vector either empty or containing the RGS domain of p115 ρGEF (24) were also transfected into these cells using LipofectAMINE (Life Technologies, Inc). After transfection, cells were incubated for 16 hours in serum-free medium and then stimulated with 10 μM sphingosine 1-phosphate (S1P), an agonist for the G12/13-linked S1P receptor for 5 hours as a control. Assays measuring luciferase activity were performed with the dual luciferase kit (Promega).
Results
Most of the 20 AAs activate ERK1/2 and mTORC1 in MIN6 cells
To explore the relationship between ERK1/2 and mTORC1 activities and AA sensing, we characterized the responses of these kinases to AA mixtures and individual AAs. For these experiments MIN6 cells or human islets were placed in AA-free medium (KRBH) with 4.5 mM glucose (2 mM for islets) for 2 hours prior to treatment. This basal condition was chosen to approximate physiological glucose concentrations.
A mixture of AAs (4.7 mM total; see Materials and Methods) rapidly but transiently activated ERK1/2 in MIN6 cells, as assessed with antibodies to the activating phosphorylation sites. The response peaked between 2 and 5 minutes and was usually not detectable after 15 minutes (Figure 1A, quantified in Supplemental Figure 1A) (16). The rapid response to AAs contrasted with the slower activation of ERK1/2 by glucose, which occurs primarily between 10 and 30 minutes (1). AAs also activated ERK1/2 in intact human islets (Figure 1B). Of individual AAs, most activated ERK1/2 and several displayed concentration optima below 5 mM (Figure 1C). Differences in ERK1/2 activity in basal glucose resulted in a variable relative (fold) activation by ligands, accounting for the observed range of relative maximal responses among experiments. Detailed comparisons of mouse and human T1R1/T1R3 identified differences in the responses to alanine and serine, among other AAs, that were shown to be due to sequence differences in the venus flytrap extracellular domains of the receptor in mouse and human (29). Thus, specific features reflected in the response of MIN6 mouse cells may not be uniformly found in other species.
AAs also activated mTORC1 over the same range of concentrations but displayed a different pattern of selectivity (Figure 1D). Leucine was one of the poorer ERK1/2-activating AAs, yet it stimulated mTORC1, measured as the phosphorylation of its substrate p70 ribosomal protein S6K, the S6K substrate S6, or the mTORC1 substrate 4EBP1 (Figure 1, C and D) (30). Other AAs tested individually, alanine, serine, asparagine, arginine, and glutamine (Figures 1D and 2B and Supplemental Figure 1B), also activated mTORC1, although less well than the AA mixture (31). EAAs at concentrations found in some growth media stimulated ERK1/2 less well than NEAAs, whereas the opposite was the case for mTORC1-EAA activated better than NEAAs (Figure 2, C and D). Glutamine enhanced mTORC1 activation by EAAs but less so ERK1/2. The effect of glutamine was less pronounced in combination with NEAAs.
Figure 2.

Comparison of AA activation ERK1/2 and mTORC1 in MIN6 cells. A, Stimulation with Ala (lanes 1–5) or Ser (lanes 7–11) at 0.05, 0.16, 0.5, 1, or 5 mM for 2 minutes or unstimulated (lane 6). One of four (for Ser) or three (for Ala) experiments are shown. B, Stimulation with 0.25, 0.5, 1, or 5 mM Ala or Ser for 30 minutes. Top panel, Immunoblots for one of three experiments. Bottom panel, Quantification (n = 3). *, P < .05, **, P < .01, using one-way ANOVA with Fisher's posttest. Stimulation of cells with 0.5 mM Gln, EAA, or NEAA, with or without Gln for 2 minutes (C) and 30 minutes (D), which is representative of four (C) and six (D) experiments. Cells were preincubated for 1 hour without or with 1 mM Gln (Gln loaded) with the indicated dilutions of the AA mixture for 2 minutes (E) or 30 minutes (F), which is representative of three (E) and six (F) independent experiments. Numbers under blots are normalized to total ERK1/2 and fold change relative to the control as in Figure 1 (C and E) or S6 (D and F). Gln (1- to 7-fold), EAA (4- to 25-fold), NEAA (13- to 147-fold), Gln/EAA (6- to 31-fold), and Gln/NEAA (3- to 188-fold).
Numerous reports suggest that the influx of AAs into cells is important for the activation of mTORC1 (11, 15). In particular, expression of a bidirectional AA transporter regulates mTORC1 activation by importing EAAs into cells in exchange for the export of glutamine. To test this, cells were exposed to 1 mM glutamine prior to stimulation with an AA mixture. Preloading MIN6 cells with glutamine enhanced the activation of mTORC1 by the AA mixture (Figure 2F). In contrast, glutamine pretreatment reduced ERK1/2 activation by extracellular AAs, probably reflecting glutamine-induced negative feedback (32) or perhaps stimulation of antagonistic pathways (Figure 2E).
Differential activation of ERK1/2 and mTORC1
We showed previously that the response of ERK1/2 and mTORC1 to AAs in MIN6 cells was largely, but not completely, mediated by the GPCR T1R1/T1R3 (16). Further confirming those findings, here we show that stable knockdown of T1R3 in MIN6 cells (see triplicate samples in Figure 3C) reduced ERK1/2 activation by alanine (Figure 3A) and also reduced mTORC1 activation by a mixture of AAs (Figure 3B). A distinguishing characteristic of T1R1/T1R3 is that its activation by AAs can be enhanced by IMP (33). We examined the effects of IMP on ERK1/2 and mTORC1 stimulation by dilutions of the AA mixture and individual AAs (Figure 3, D and E, and Supplemental Figure 2, A and B). ERK1/2 were stimulated by one-eighth to one-fourth the concentration of AAs in growth medium, but a response could be detected with one-16th the concentration of AAs by including IMP (Figure 3D). IMP increased mTORC1 activation by AAs at 1/4 and 1/2 the concentration in growth medium (Figure 3E). IMP alone had little effect on the phosphorylation of either S6K or ERK1/2. In addition, activation of ERK1/2 by AAs was sensitive to glucose concentration, with ERK1/2 stimulation by AAs much stronger at 4.5 mM glucose and above (Supplemental Figure 3, A–C). In contrast, mTORC1 was activated almost equally well in the presence of 0 or 4.5 mM glucose.
Figure 3.
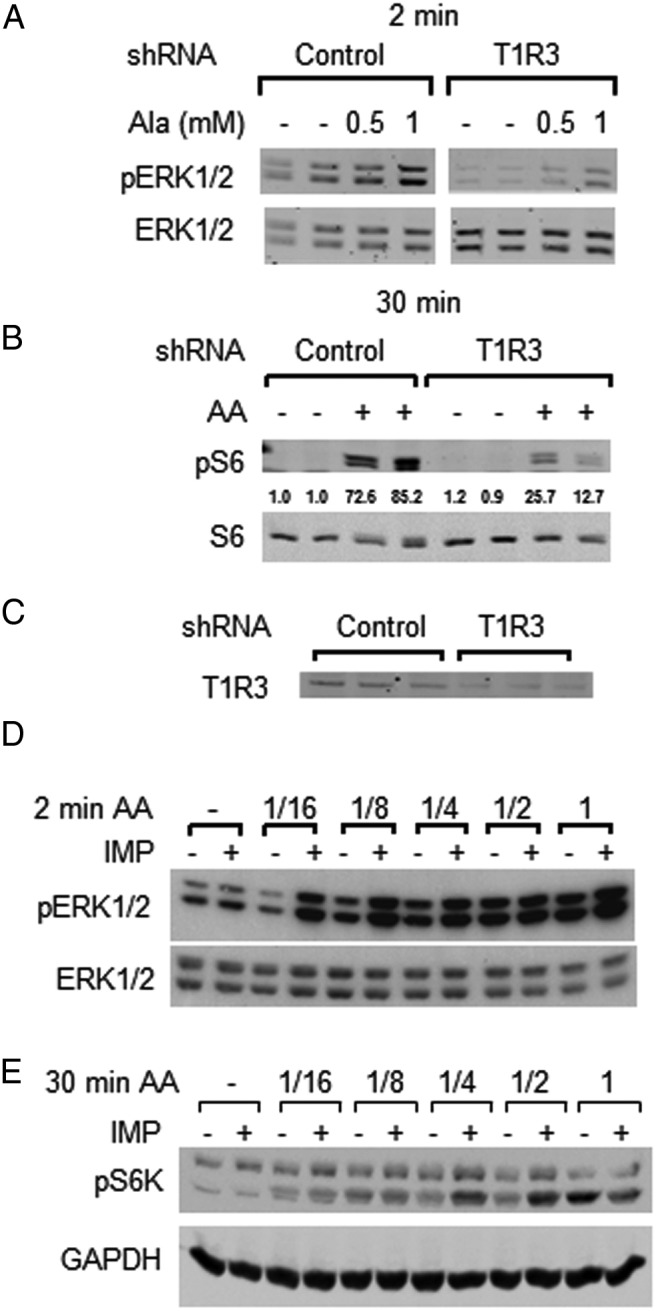
AAs activate ERK1/2 and mTORC1 through T1R1/T1R3. MIN6 cells were preincubated in KRBH and ERK1/2, pERK1/2, and S6 and pS6 (S235–236) and were immunoblotted in lysate proteins. MIN6 cells stably expressing either control nontarget shRNA or T1R3 shRNA were stimulated with the indicated concentrations of Ala for 2 minutes (A) and the AA mixture for 30 minutes (B). Numbers under the blot are the fold change in pS6/S6 as in Figure 1. One of two (A) or three (B) independent experiments is shown. C, Immunoblots of T1R3 in triplicate lysates of MIN6 cells expressing control nontarget shRNA or T1R3 shRNA as in panels A and B. Stimulation with the indicated dilutions of AAs with or without 2.5 mM IMP for 2 minutes (D) or 30 minutes (E), which is representative of three (D) or two (E) independent experiments (also see Supplemental Figure 2, A and B). GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Increased intracellular Ca2+ is required for activation of ERK1/2 but not mTORC1
Intracellular Ca2+ was manipulated with diazoxide to prevent membrane depolarization, nifedipine, an L-type Ca2+ channel blocker, or by removing extracellular Ca2+ to compare the impact on AA stimulation of ERK1/2 and mTORC1. Diazoxide reduced basal ERK1/2 activity and its activation by AAs (Figure 4A). It also inhibited ERK1/2 activation by carbachol, which acts primarily on the Gq/11-coupled M3 muscarinic receptor, and had a smaller effect on EGF-induced ERK1/2 activation (Figure 4B). Diazoxide also inhibited ERK1/2 activation by GLP-1, an agonist for the Gs-coupled GLP-1 receptor and, to a lesser extent, by EXD-4, a GLP-1 analog (Figure 4, A and B). Effects on the K(ATP) channel by these ligands probably occur, at least in part, through modulation by lipid-derived second messengers, as suggested by earlier analyses showing that insulin secretion was blocked by diazoxide, even when there was little change in the ATP to ADP ratio (14, 34). Removal of Ca2+ from the medium prevented ERK1/2 activation by carbachol and AAs (16).
Figure 4.

Effects of Ca2+ on activation of ERK1/2 and mTORC1. A, Cells were preincubated in KRBH with 4.5 mM Glc for 2 hours, treated with 100 μM diazoxide (DZ) or vehicle (DMSO) for 15 minutes, and then stimulated with 100 μM carbachol (Carb), AAs, or 100 nM EXD-4 for 2 minutes prior to harvest. One of four experiments is shown. B, Cells treated as in panel A were stimulated with AAs, 100 nM GLP-1, or 100 ng/mL EGF as indicated for either 2 or 30 minutes. One of two experiments is shown. Numbers under blots are fold change as in Figure 1. C, Cells were placed in fresh KRBH with or without Ca2+ plus 2 mM EGTA for 5 minutes before stimulation with AAs for 30 minutes. One of five experiments is shown. D, Cells preincubated in KRBH were stimulated with AAs or 100 μM Carb for the indicated times. pS6K and pS6 (S235/236) were immunoblotted. One of two experiments is shown.
We previously observed that nifedipine slightly reduced AA-stimulated mTORC1 activity (16). Because Ca2+ entry is reported to be required for mTORC1 activation by AAs in HeLa cells (35), we further investigated the importance of Ca2+ entry on this pathway in MIN6 cells. As seen in Figure 4B, diazoxide had little effect on mTORC1 activity in response to AAs. This contrasts with the effects of diazoxide, which interferes with ERK1/2 activation (Figure 4, A and B). Reducing extracellular Ca2+ had little or no effect on the ability of AAs to induce the phosphorylation of S6K or S6 (Figure 4C), suggesting the absence of a Ca2+ requirement for mTORC1 activation in MIN6 cells. mTORC1 activity appears greater using pS6 as the readout because cAMP-dependent protein kinase and other kinase pathways have some ability to phosphorylate S6 (36). Carbachol, which activates Gq/11 and increases intracellular free Ca2+, rapidly activated ERK1/2 (16, 37) (Figure 4A) but did not detectably activate mTORC1 between 30 seconds and 30 minutes of stimulation (Figure 4D). We also reevaluated our earlier finding that the MEK1/2 inhibitor U0126 partially inhibited mTORC1 in light of the identification of some off-target effects of the drug (38, 39). A comparison of U0126 with a different inhibitor PD0325901 indicated that both strongly inhibited ERK1/2 phosphorylation (Supplemental Figure 2C). Under the same conditions, both compounds inhibitied mTORC1 activity weakly, although we have observed stronger inhibition in other cell types.
AAs do not detectably stimulate accumulation of cAMP in MIN6 cells
It has been suggested that T1R1/T1R3 may couple to Gs and elevate cAMP in some settings (40). To determine whether that is the case, we compared changes in cAMP elicited by AAs with those induced by GLP-1, which signals through cAMP. GLP-1 caused a clear increase in cAMP within 1 minute of hormone exposure (Figure 5A). No change in cAMP was detected in response to AAs over 4 minutes, a time when ERK1/2 activation has peaked.
Figure 5.

G proteins and activation of ERK1/2 and mTORC1 by AAs. A, Intracellular cAMP was measured in MIN6 cells stably expressing the CAMYEL BRET sensor. Cells in KRBH were stimulated with AAs (n = 4) or GLP-1 (n = 5). Data were analyzed as in Materials and Methods. B and C, MIN6 cells were treated with or without 50 ng/mL of pertussis toxin (PTX) for 18 hours in normal growth medium before incubation in KRBH with 4.5 mM Glc ± 50 ng/mL pertussis toxin (PTX) for 2 hours. After a 15-minute treatment with 10 μM UK14304 or vehicle (DMSO), cells were stimulated with AAs for 2 minutes (B) or 15 minutes (C). pERK1/2, pS6K, or pS6 (240/244) was immunoblotted. D and E, Cells treated as in panels B and C were loaded with fura 2, pretreated with 10 μM UK14304, and stimulated with AAs, as indicated to measure effects on intracellular Ca2+. One of three (panels B, D, and E) or four (panel C) experiments is shown. F and G, MIN6 cells in KRBH with 4.5 mM Glc for 2 hours were treated with or without 300 nM of the Gq inhibitor UBO-GIC for 15 minutes prior to the addition of AAs for 2 minutes (F) or 30 minutes (G). Lysate proteins were immunoblotted for pERK1/2 and ERK1/2 (F) or pS6K and S6K (G) (n = 3 for panels F and G). G, Bar graph, means ± inhibitor; AA stimulation relative to control + or − inhibitor. Numbers obtained as described in Figure 1. **, P < .01. H, HeLa cells were cotransfected with pGL4 vectors encoding SRE.L-dependent firefly luciferase and pCMV5 vector, either empty or encoding the p115 ρGEF RGS domain. Amounts of p115 ρGEF RGS domain plasmid DNA were as indicated. Cells were placed in serum-free medium for 16 hours prior to preincubation in KRBH for 30 minutes and then stimulated with AAs for 30 minutes. **, P < .01, student's t test. Top panel, Means of pS6K/S6K were expressed as fold increase compared with starved cells without p115 ρGEF RGS DNA. Middle panel, Immunoblots showing pS6K, S6K, and actin. Bottom panel, As indicated, cells were stimulated with 10 μM S1P for 5 hours and lysed, and firefly luciferase activities were measured. The means of activities are presented as fold stimulation relative to firefly luciferase activity from cells transfected with control (empty) pCMV5 vector in the absence of drug. This panel demonstrates the inhibitory effect of the RGS domain in a system that is dependent on G12/13.
Inactivation of Gi with pertussis toxin does not interfere with activation of ERK1/2 or mTORC1 by AAs
As noted earlier, T1R1/T1R3 is thought to work through a member of the Gi family in several systems (21). To test the possibility that Gi mediated AA activation of ERK1/2 or mTORC1, we pretreated cells with pertussis toxin to block signaling downstream of Gi and examined the activation of the two kinase pathways. AAs activated ERK1/2 equally well in the absence or presence of pertussis toxin (Figure 5B). We showed previously that the α2-adrenergic receptor agonist UK14304, which activates Gi, inhibits glucose-stimulated ERK1/2 activity, and inhibition is reversed by pretreatment with pertussis toxin (41). Similarly, UK14304, an α2-adrenergic agonist, inhibited AA-stimulated ERK1/2 activity, and inhibition was blocked by the pertussis toxin. The pertussis toxin had no discernable effect on AA-stimulated mTORC1 activity in MIN6 cells (Figure 5C) or in HeLa cells (Supplemental Figure 4A). These results suggest that Gi is not mediating the actions of AAs through T1R1/T1R3. Also in agreement with this assessment, UK14304 neither stimulated nor inhibited the mTORC1 activity in the MIN6 cells. Because we found that AAs increased intracellular Ca2+, we examined the effects of the pertussis toxin and UK14304 on this response. The AA-induced increase in Ca2+ was attenuated by UK14304 (Figure 5D). The α2-adrenergic agonist alone did not elevate intracellular Ca2+. As expected, the pertussis toxin reduced the inhibitory effect of UK14304 but did not block an increase in Ca2+ caused by AAs (Figure 5E).
Inactivation of Gq with UBO-QIC differentially inhibits AA stimulation of ERK1/2 and mTORC1
The Ca2+ requirement for AA stimulation of ERK1/2 suggested that T1R1/T1R3 might be working through a Gq family member. To test this possibility, we pretreated MIN6 cells with 300 nM UBO-QIC to interfere with signaling downstream of Gq (23, 42) and examined the activation of the two kinase pathways. AAs activated ERK1/2 poorly in the presence of UBO-QIC (Figure 5F). At 5 minutes, ERK1/2 activation in the presence of the inhibitor was reduced by nearly 75%, suggesting that Gq is required for the maximal stimulation of ERK1/2. On the other hand, UBO-QIC had no discernable effect on AA-stimulated mTORC1 activity in MIN6 cells (Figure 5G) or over a range of concentrations in HeLa cells (Supplemental Figure 4B). Stimulation in the presence and absence of the inhibitor was not different, suggesting that Gq is not the primary mediator of the actions of AAs on mTORC1. We also inhibited G12/13 family members in HeLa cells by expressing the RGS domain of p115 ρGEF (24) and found no effect on mTORC1 activation by AAs (Figure 5H).
Discussion
The regulation of ERK1/2 and mTORC1 by nutrients, growth factors, and other ligands is essential to maintain key functions of animal cells. Using knockdown experiments and antagonists, we showed that the GPCR T1R1/T1R3 contributes to activation of both of these pathways by AAs in MIN6 cells and human islets (16, 17). The signaling events we have characterized here for activation of ERK1/2 and mTORC1 by AAs are dissimilar from mechanisms described for T1R1/T1R3 in other systems, which have identified Gi as the essential heterotrimeric G protein in the pathway (21). Instead, activation of Gi antagonizes ERK1/2 (41) and has no effect on mTORC1 in MIN6 cells, nor does pertussis toxin interfere with the activation of either pathway by AAs, leading us to conclude that Gi is not important for the regulation of these kinases by T1R1/T1R3. A second key event reported to be initiated by T1R1/T1R3 is the elevation of cytoplasmic free Ca2+ (21). Intracellular free Ca2+ is increased by AAs in MIN6 cells. Ca2+ is important for ERK1/2 activation by AAs and glucose, but is not essential for activation of mTORC1. Thus, the Ca2+ requirement distinguishes the mechanism of activation of ERK1/2 from that activating mTORC1. Inhibition of Gq reduces ERK1/2 but not mTORC1 activation by AAs. These findings demonstrate a second mechanistic distinction in pathways regulating ERK1/2 and mTORC1 in MIN6 cells. T1R1/T1R3 signals to ERK1/2 through effects on intracellular Ca2+ mediated at least in part through Gq. On the other hand, we found no evidence that Gq or Ca2+ mediates the actions of AAs on mTORC1. We also found no evidence that the activation of mTORC1 by AAs is dependent on Gi, Gq, or G12/13 proteins in HeLa cells. Another possibility was that autocrine insulin signaling might have stimulated mTORC1; however, neither diazoxide nor the α2-adrenergic agonist UK14304 reduced the AA-induced phosphorylation of p70 S6K to an appreciable extent, although both inhibit insulin secretion and the phosphorylation of ERK1/2.
The signaling pathways that lead to mTORC1 activation may branch through the actions of arrestins, other RGS proteins, or a variety of scaffolds for other signaling proteins, either acting through or independently of G proteins (43–48). Of relevance to the β-cell system, β-arrestin 1 is reported to mediate GLP-1-induced insulin secretion (49), and arrestins have been implicated in insulin action, metabolic flux, and obesity (45, 50). Ligand-specific effects that induce distinct receptor conformations and consequent signals are possible and have been demonstrated for other GPCRs; in some cases, molecular mechanisms are suggested by structural studies (eg, reference 51). Signaling kinetics may also contribute to the different responses of the two pathways (52). Rapid AA uptake, compared with the several minutes needed for maximal mTORC1 stimulation, may allow intracellular sensors to contribute substantially. Regardless, depleting T1R1/T1R3 eliminates most of the signal to both ERK1/2 and mTORC1, and it is therefore likely that the divergence between the activation of ERK1/2 and mTORC1 represents branched signaling pathways downstream of the receptor.
Organization of mTORC1 components on endosomal and lysosomal membranes is thought to be a key step in control of its activity by AAs (53, 54). Our earlier studies indicated that depletion of T1R1/T1R3 alters the organization of lysosomes in cells and interferes with optimal mTORC1 activation (16). Activation of mTORC1 by AAs may depend on coincidence monitoring of two inputs, one of which is AA ligation of T1R1/T1R3. Studies of mTORC1 activation in other systems suggest that AA uptake, effects of AAs at lysosomes, and glutaminolysis may also influence mTORC1 activity (11, 15, 53, 55, 56). Unraveling these pathways and determining the mechanisms of pathway choice will be central to our understanding of how AAs control cell growth and the cell's specialized activities.
Acknowledgments
We thank Jonathan Senkler (Des Moines University) for technical assistance; Ildiko Lingvay (Department of Internal Medicine, University of Texas Southwestern Medical Center) for helpful discussions; Elma Zaganjor, Andrés Lorente Rodríguez, and Michael Kalwat (former and current members of Cobb Laboratory) for comments on the manuscript; and Dionne Ware for administrative assistance.
Present address for M.L.G.: StemSynergy Therapeutics, Nashville, Tennessee.
This work was supported by Grant DK55310 from the National Institutes of Health and Grant I1243 from the Robert A. Welch Foundation (to M.H.C.) and a grant from the Cancer Prevention and Research Institute of Texas (jointly to M.H.C. and E.M.R.). Some of the later work was also supported by funds from the Iowa Osteopathic Education and Research (to E.M.W.). E.M.W. and M.L.G. were supported in part by a mentor-based postdoctoral fellowship from the American Diabetes Association during early stages of this work.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- AA
- amino acid
- BRET
- bioluminescence resonance energy transfer
- DMSO
- dimethylsulfoxide
- EAA
- essential AA
- 4EBP1
- 4E-binding protein 1
- EGF
- epidermal growth factor
- EXD-4
- exendin-4
- GEF
- guanine nucleotide exchange factor
- GLP-1
- glucagon-like peptide 1 amide fragment 7–36, human
- GPCR
- G protein-coupled receptor
- IMP
- inosine monophosphate
- KRBH
- Krebs-Ringer's bicarbonate solution
- mTORC1
- mechanistic target of rapamycin complex 1
- NEAA
- nonessential AA
- p
- phosphorylated
- RGS
- regulator of G protein signaling
- shRNA
- short hairpin RNA
- S6K
- S6 kinase
- S1P
- sphingosine 1-phosphate
- SRE
- serum-response element.
References
- 1. Khoo S, Cobb MH. Activation of MAP kinase by glucose is not required for insulin secretion. Proc Natl Acad Sci USA. 1997;94:5599–5604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Benes C, Poitout V, Marie JC, Martin-Perez J, Roisin MP, Fagard R. Mode of regulation of the extracellular signal-regulated kinases in the pancreatic β-cell line MIN6 and their implication in the regulation of insulin gene transcription. Biochem J. 1999;340:219–225. [PMC free article] [PubMed] [Google Scholar]
- 3. Petersen HV, Jensen JN, Stein R, Serup P. Glucose induced MAPK signalling influences NeuroD1-mediated activation and nuclear localization. FEBS Lett. 2002;528:241–245. [DOI] [PubMed] [Google Scholar]
- 4. Khoo S, Griffen SC, Xia Y, Baer R, German MS, Cobb MH. Regulation of insulin gene transcription by extracellular-signal regulated protein kinases (ERK) 1 and 2 in pancreatic β cells. J Biol Chem. 2003;278:32969–32977. [DOI] [PubMed] [Google Scholar]
- 5. Lawrence MC, McGlynn K, Park BH, Cobb MH. ERK1/2-dependent activation of transcription factors required for acute and chronic effects of glucose on the insulin gene promoter. J Biol Chem. 2005;280:26751–26759. [DOI] [PubMed] [Google Scholar]
- 6. Lawrence MC, McGlynn K, Shao C, et al. Chromatin-bound mitogen-activated protein kinases transmit dynamic signals in transcription complexes in β-cells. Proc Natl Acad Sci USA. 2008;105:13315–13320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gleason CE, Lu D, Witters LA, Newgard CB, Birnbaum MJ. The role of AMPK and mTOR in nutrient sensing in pancreatic β-cells. J Biol Chem. 2007;282:10341–10351. [DOI] [PubMed] [Google Scholar]
- 8. Mori H, Inoki K, Opland D, et al. Critical roles for the TSC-mTOR pathway in β-cell function. Am J Physiol Endocrinol Metab. 2009;297:E1013–E1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rachdi L, Balcazar N, Osorio-Duque F, et al. Disruption of Tsc2 in pancreatic β cells induces β cell mass expansion and improved glucose tolerance in a TORC1-dependent manner. Proc Natl Acad Sci USA. 2008;105:9250–9255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yang J, Chi Y, Burkhardt BR, Guan Y, Wolf BA. Leucine metabolism in regulation of insulin secretion from pancreatic β cells. Nutr Rev. 2010;68:270–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Avruch J, Long X, Ortiz-Vega S, Rapley J, Papageorgiou A, Dai N. Amino acid regulation of TOR complex 1. Am J Physiol Endocrinol Metab. 2009;296:E592–E602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dazert E, Hall MN. mTOR signaling in disease. Curr Opin Cell Biol. 2011;23:744–755. [DOI] [PubMed] [Google Scholar]
- 13. Wiederkehr A, Wollheim CB. Mitochondrial signals drive insulin secretion in the pancreatic β-cell. Mol Cell Endocrinol. 2012;353:128–137. [DOI] [PubMed] [Google Scholar]
- 14. Doliba NM, Wehrli SL, Vatamaniuk MZ, et al. Metabolic and ionic coupling factors in amino acid-stimulated insulin release in pancreatic β-HC9 cells. Am J Physiol Endocrinol Metab. 2007;292:E1507–E1519. [DOI] [PubMed] [Google Scholar]
- 15. Nicklin P, Bergman P, Zhang B, et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell. 2009;136:521–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wauson EM, Zaganjor E, Lee A, et al. The G protein-coupled receptor T1R1/T1R3 regulates mTORC1 and autophagy. Mol Cell. 2012;47:851–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wauson EM, Lorente-Rodriguez A, Cobb MH. Minireview: nutrient sensing by G protein-coupled receptors. Mol Endocrinol. 2013;27:1188–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Adler E, Hoon MA, Mueller KL, Chandrashekar J, Ryba NJ, Zuker CS. A novel family of mammalian taste receptors. Cell. 2000;100:693–702. [DOI] [PubMed] [Google Scholar]
- 19. Matsunami H, Montmayeur JP, Buck LB. A family of candidate taste receptors in human and mouse. Nature. 2000;404:601–604. [DOI] [PubMed] [Google Scholar]
- 20. Mace OJ, Affleck J, Patel N, Kellett GL. Sweet taste receptors in rat small intestine stimulate glucose absorption through apical GLUT2. J Physiol. 2007;582:379–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang Y, Hoon MA, Chandrashekar J, et al. Coding of sweet, bitter, and umami tastes: different receptor cells sharing similar signaling pathways. Cell. 2003;112:293–301. [DOI] [PubMed] [Google Scholar]
- 22. Rozengurt E, Sternini C. Taste receptor signaling in the mammalian gut. Curr Opin Pharmacol. 2007;7:557–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Karpinsky-Semper D, Volmar CH, Brothers SP, Slepak VZ. Differential effects of the Gβ5-RGS7 complex on muscarinic M3 receptor-induced Ca2+ influx and release. Mol Pharmacol. 2014;85:758–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wells CD, Gutowski S, Bollag G, Sternweis PC. Identification of potential mechanisms for regulation of p115 ρGEF through analysis of endogenous and mutant forms of the exchange factor. J Biol Chem. 2001;276:28897–28905. [DOI] [PubMed] [Google Scholar]
- 25. Sahai E, Alberts AS, Treisman R. ρA effector mutants reveal distinct effector pathways for cytoskeletal reorganization, SRF activation and transformation. EMBO J. 1998;17:1350–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Boulton TG, Cobb MH. Identification of multiple extracellular signal-regulated kinases (ERKs) with antipeptide antibodies. Cell Regulation. 1991;2:357–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Oh E, Heise CJ, English JM, Cobb MH, Thurmond DC. WNK1 is a novel regulator of Munc18c-syntaxin 4 complex formation in soluble NSF attachment protein receptor (SNARE)-mediated vesicle exocytosis. J Biol Chem. 2007;282:32613–32622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jiang LI, Collins J, Davis R, et al. Use of a cAMP BRET sensor to characterize a novel regulation of cAMP by the sphingosine 1-phosphate/G13 pathway. J Biol Chem. 2007;282:10576–10584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Toda Y, Nakagita T, Hayakawa T, et al. Two distinct determinants of ligand specificity in T1R1/T1R3 (the umami taste receptor). J Biol Chem. 2013;288:36863–36877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xu G, Marshall CA, Lin TA, et al. Insulin mediates glucose-stimulated phosphorylation of PHAS-I by pancreatic β cells. An insulin-receptor mechanism for autoregulation of protein synthesis by translation. J Biol Chem. 1998;273:4485–4491. [DOI] [PubMed] [Google Scholar]
- 31. Xu G, Kwon G, Cruz WS, Marshall CA, McDaniel ML. Metabolic regulation by leucine of translation initiation through the mTOR-signaling pathway by pancreatic β-cells. Diabetes. 2001;50:353–360. [DOI] [PubMed] [Google Scholar]
- 32. Duan L, Cobb MH. Calcineurin increases glucose activation of ERK1/2 by reversing negative feedback. Proc Natl Acad Sci USA. 2010;107:22314–22319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nelson G, Chandrashekar J, Hoon MA, et al. An amino-acid taste receptor. Nature. 2002;416:199–202. [DOI] [PubMed] [Google Scholar]
- 34. Kakei M. Receptor-operated regulation of ATP-sensitive K+ channels via membrane phospholipid metabolism. Curr Med Chem. 2003;10:235–243. [DOI] [PubMed] [Google Scholar]
- 35. Gulati P, Gaspers LD, Dann SG, et al. Amino acids activate mTOR complex 1 via Ca2+/CaM signaling to hVps34. Cell Metab. 2008;7:456–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gregory JS, Boulton TG, Sang B-C, Cobb MH. An insulin-stimulated ribosomal protein S6 kinase from rabbit liver. J Biol Chem. 1989;264:18397–18401. [PubMed] [Google Scholar]
- 37. Jain S, Ruiz de Azua I, Lu H, White MF, Guettier JM, Wess J. Chronic activation of a designer G (q)-coupled receptor improves β cell function. J Clin Invest. 2013;123:1750–1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Freeman MR, Kim J, Lisanti MP, Di VD. A metabolic perturbation by U0126 identifies a role for glutamine in resveratrol-induced cell death. Cancer Biol Ther. 2011;12:966–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wauson EM, Guerra ML, Barylko B, Albanesi JP, Cobb MH. Off-target effects of MEK inhibitors. Biochemistry. 2013;52:5164–5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. He W, Yasumatsu K, Varadarajan V, et al. Umami taste responses are mediated by α-transducin and α-gustducin. J Neurosci. 2004;24:7674–7680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gibson TB, Lawrence MC, Gibson C, et al. Inhibition of glucose-stimulated activation of extracellular signal-regulated protein kinases (ERK) 1 and 2 by epinephrine in pancreatic β cells. Diabetes. 2006;55:1066–1073. [DOI] [PubMed] [Google Scholar]
- 42. Inamdar V, Patel A, Manne BK, Dangelmaier C, Kunapuli SP. Characterization of UBO-QIC as a Gα inhibitor in platelets. Platelets. 2015;1–8. doi: 10.3109/09537104.2014.998993. [DOI] [PubMed] [Google Scholar]
- 43. Reiter E, Ahn S, Shukla AK, Lefkowitz RJ. Molecular mechanism of β-arrestin-biased agonism at seven-transmembrane receptors. Annu Rev Pharmacol Toxicol. 2012;52:179–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nakajima K, Wess J. Design and functional characterization of a novel, arrestin-biased designer G protein-coupled receptor. Mol Pharmacol. 2012;82:575–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Patwari P, Lee RT. An expanded family of arrestins regulate metabolism. Trends Endocrinol Metab. 2012;23:216–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ross EM, Wilkie TM. GTPase-activating proteins for heterotrimeric G proteins: regulators of G protein signaling (RGS) and RGS-like proteins. Annu Rev Biochem. 2000;69:795–827. [DOI] [PubMed] [Google Scholar]
- 47. Wang X, Zeng W, Soyombo AA, et al. Spinophilin regulates Ca2+ signalling by binding the N-terminal domain of RGS2 and the third intracellular loop of G-protein-coupled receptors. Nat Cell Biol. 2005;7:405–411. [DOI] [PubMed] [Google Scholar]
- 48. Ghamari-Langroudi M, Digby GJ, Sebag JA, et al. G-protein-independent coupling of MC4R to Kir7.1 in hypothalamic neurons. Nature. 2015;520:94–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sonoda N, Imamura T, Yoshizaki T, Babendure JL, Lu JC, Olefsky JM. β-Arrestin-1 mediates glucagon-like peptide-1 signaling to insulin secretion in cultured pancreatic β cells. Proc Natl Acad Sci USA. 2008;105:6614–6619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Stockli J, James DE. Insulin action under arrestin. Cell Metab. 2009;9:213–214. [DOI] [PubMed] [Google Scholar]
- 51. Nygaard R, Zou Y, Dror RO, et al. The dynamic process of β(2)-adrenergic receptor activation. Cell. 2013;152:532–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ross EM. Coordinating speed and amplitude in G-protein signaling. Curr Biol. 2008;18:R777–R783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell. 2010;141:290–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Korolchuk VI, Saiki S, Lichtenberg M, et al. Lysosomal positioning coordinates cellular nutrient responses. Nat Cell Biol. 2011;13:453–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y, Sabatini DM. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H-ATPase. Science. 2011;334:678–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Duran RV, Oppliger W, Robitaille AM, et al. Glutaminolysis activates Rag-mTORC1 signaling. Mol Cell. 2012;47:349–358. [DOI] [PubMed] [Google Scholar]