

Graphical abstract
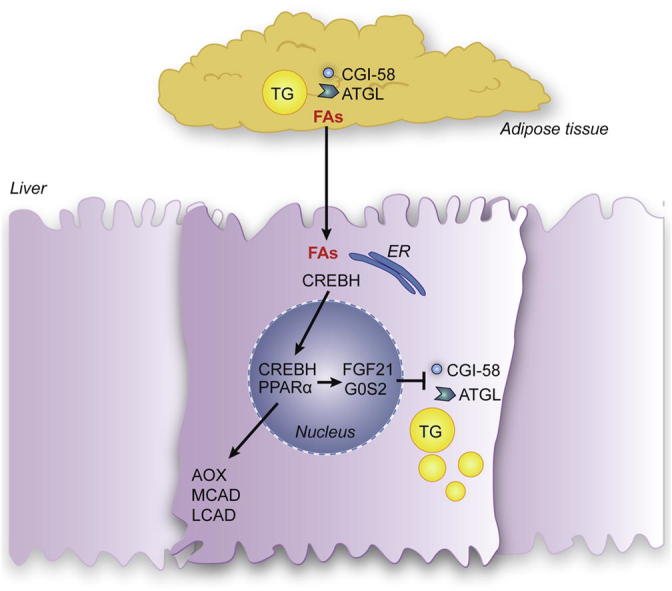
Abbreviations: AT, Adipose tissue; FA(s), fatty acid(s); TG, triacylglycerol; CGI-58, comparative gene identification-58; ATGL, Adipose triglyceride lipase; CGI-58-ATko, AT-selective ablation of CGI-58; ATGL-ATko, AT-specific deficiency of ATGL; LD, lipid droplet; NAFLD, nonalcoholic fatty liver disease; NLSD, Neutral lipid storage disease; PPARα, peroxisome proliferator-activated receptor alpha; CREBH, cAMP-responsive element binding protein, hepatocyte specific; G0S2, G0/G1 Switch Gene 2; FGF21, fibroblast growth factor 21; PGC-1α, PPARgamma co-activator-1alpha; HNF4α, hepatocyte nuclear factor 4alpha; PEPCK, Phosphoenolpyruvate carboxykinase; G6Pase, Glucose-6-phosphatase; ER, endoplasmic reticulum
Keywords: Hepatic steatosis, G0/G1 switch gene 2, Fibroblast growth factor 21, CGI-58, ATGL, Lipolysis, PPARα, CREBH, Obesity
Abstract
Background & Aims
Adipose tissue (AT)-derived fatty acids (FAs) are utilized for hepatic triacylglycerol (TG) generation upon fasting. However, their potential impact as signaling molecules is not established. Herein we examined the role of exogenous AT-derived FAs in the regulation of hepatic gene expression by investigating mice with a defect in AT-derived FA supply to the liver.
Methods
Plasma FA levels, tissue TG hydrolytic activities and lipid content were determined in mice lacking the lipase co-activator comparative gene identification-58 (CGI-58) selectively in AT (CGI-58-ATko) applying standard protocols. Hepatic expression of lipases, FA oxidative genes, transcription factors, ER stress markers, hormones and cytokines were determined by qRT-PCR, Western blotting and ELISA.
Results
Impaired AT-derived FA supply upon fasting of CGI-58-ATko mice causes a marked defect in liver PPARα-signaling and nuclear CREBH translocation. This severely reduced the expression of respective target genes such as the ATGL inhibitor G0/G1 switch gene-2 (G0S2) and the endocrine metabolic regulator FGF21. These changes could be reversed by lipid administration and raising plasma FA levels. Impaired AT-lipolysis failed to induce hepatic G0S2 expression in fasted CGI-58-ATko mice leading to enhanced ATGL-mediated TG-breakdown strongly reducing hepatic TG deposition. On high fat diet, impaired AT-lipolysis counteracts hepatic TG accumulation and liver stress linked to improved systemic insulin sensitivity.
Conclusions
AT-derived FAs are a critical regulator of hepatic fasting gene expression required for the induction of G0S2-expression in the liver to control hepatic TG-breakdown. Interfering with AT-lipolysis or hepatic G0S2 expression represents an effective strategy for the treatment of hepatic steatosis.
Introduction
In mammals, the deposition and mobilization of triacylglycerol (TG) from adipose tissue (AT) plays a central role in energy homeostasis. This steadily recurring metabolic circuit is sensed by the liver and reflected by continuous changes in hepatic TG content and gene expression. Imbalances in this process may lead to pronounced fat accumulation in the liver eventually progressing to non-alcoholic fatty liver disease (NAFLD).
The catabolism of endogenous TG deposited within cellular lipid droplets (LDs) depends on adipose triglyceride lipase (ATGL) [1] and a co-activator designated as comparative gene identification-58 (CGI-58) [2]. Both, mutated ATGL or CGI-58 alleles promote neutral lipid storage disease (NLSD) characterized by TG accumulation in multiple tissues [3] including the liver. In mice, downregulation or absence of ATGL or CGI-58 in the liver provokes marked hepatic fat accumulation [4], [5], [6]. Upon fasting, hepatic TG levels sharply increase due to enhanced TG mobilization from AT and increased fatty acid (FA) flow to the liver. Excess hepatic TG in NAFLD patients substantially originates from AT-derived FAs [7]. However, the functional importance of AT-derived FAs in the regulation of hepatic gene expression is still unclear and challenged by the study of Chakravarthy et al. [8] suggesting that PPARα-regulated gene expression in the liver depends on de novo FA synthesis. The aim of this study was to unravel the in vivo role of exogenously supplied FAs in the regulation of hepatic gene expression and TG homeostasis upon fasting.
Here we show that AT-derived FA supply is critically required to stimulate expression of genes in the liver which are under the dual regulation of PPARα and cAMP-responsive element binding protein H (CREBH) encompassing the lipase inhibitor G0S2 and the endocrine metabolic regulator fibroblast growth factor 21 (FGF21) which has major implications on hepatic TG catabolism and systemic energy homeostasis.
Materials and methods
Mouse models
CGI-58-floxed mice [9] and Fabp4-Cre transgenic mice [10] have previously been described and used for the generation of mice with AT-selective disruption of CGI-58 (CGI-58-ATko). Mice lacking ATGL specifically in AT (ATGL-ATko) were generated by breeding the AdipoQ-Cre transgene onto an ATGL-floxed background [11]. Maintenance of mice is described in detail in the Supplementary Material.
Analysis of body composition
Body composition of non-fasted animals was determined using a calibrated Bruker miniSpec NMR analyzer (Bruker Optics).
Blood parameters
Determination of plasma parameters is described in the Supplementary Material.
PPARα-agonist application and raising plasma FA levels
The established PPARα agonist Wy14643 was injected (50 μg/g body weight) into 12 h fasted mice. After another 2 h fasting the liver was surgically removed for RNA isolation and qRT-PCR. For raising circulating FA levels, an intragastric olive oil gavage (200 μl/mouse) was administered to 12 h fasted mice followed by heparin injection (10 IU/mouse). After another 2 h, blood was collected and liver tissues were harvested.
Analysis of gene expression by qRT-PCR
qRT-PCR and Xbp1-splicing assay was performed as described in the Supplementary Material.
Measurement of tissue TG levels
Tissue lipids were extracted with chloroform and methanol and determined as described in the Supplementary Material.
Western blotting
Western blot analyses were performed according to standard protocols described in the Supplementary Material.
Electron microscopy
Electron microscopy has been performed as previously described [12].
Statistical analysis
Statistical significance was determined by the Student’s unpaired t test (two-tailed). Group differences considered significant for p <0.05 (∗), p <0.01 (∗∗), and p <0.001 (∗∗∗).
Results
AT-derived FAs are critically required to induce G0S2 expression in the liver to regulate hepatic TG catabolism upon fasting
Efficient cellular TG catabolism depends on ATGL and the lipolytic co-activator CGI-58 [1]. To restrict FA release from AT, we generated mice lacking CGI-58 selectively in AT (CGI-58-ATko) by breeding Fabp4-Cre transgenic mice [10] with mice carrying loxP-sites in the Cgi-58 gene [9]. CGI-58 expression was substantially reduced in gonadal white and brown AT of CGI-58-ATko mice (Fig. 1A; Supplementary Fig. 1A) whereas muscle and hepatic CGI-58 mRNA levels were comparable to flox/flox controls. Body weight of CGI-58-ATko mice was similar to flox/flox controls (Supplementary Fig. 1B) whereas body fat mass was 1.4-fold increased paralleled by elevated gonadal and brown AT and reduced liver mass (Supplementary Fig. 1C and D). Fasting provoked a marked decrease in plasma FA (−66%), TG (−58%) and glycerol levels in CGI-58-ATko mice (Table 1). Plasma levels of long-chain saturated and unsaturated FA species were significantly decreased (ranging from −29% up to −73%) (Fig. 1B). The reduction in blood glucose and plasma insulin levels together with lower HOMA-IR indexes in CGI-58-ATko compared to flox/flox mice (Table 1) indicates increased insulin sensitivity. Hepatic glycogen content was markedly reduced (−74%) in 6 h fasted CGI-58-ATko compared to flox/flox mice (3.6 ± 5.6 vs. 13.5 ± 6.1 mg glucose per g tissue, respectively). Together, these data validate CGI-58-ATko mice to study the impact of defective AT-lipolysis on hepatic TG homeostasis and gene expression.
Fig. 1.

AT-derived FAs induce G0S2 expression in the liver to regulate hepatic TG catabolism. (A) Absence of CGI-58 protein expression in white (gonadal) and brown AT (WAT and BAT) of CGI-58-ATko mice. (B) Plasma concentrations of saturated and unsaturated FA species in fasted flox/flox and CGI-58-ATko mice. (C) Hepatic TG levels in non-fasted compared to fasted flox/flox and CGI-58-ATko mice. (D) Transmission electron microscopy of liver sections from fasted mice (Scale bar = 1 μm; LD, lipid droplet; m, mitochondria; n, nucleus). (E) TG hydrolytic activities in liver preparations of non-fasted compared to fasted mice and in the presence of an ATGL-specific inhibitor (ATGLi). (F) Hepatic ATGL and G0S2 mRNA expression in non-fasted compared to fasted mice. (G) Hepatic ATGL and G0S2 protein expression upon fasting. Values are mean ± SD from at least 5 mice per genotype. *p <0.05, **p <0.01, and ***p <0.001 vs. flox/flox; #p <0.05 and ###p <0.001 non-fasted vs. fasted.
Table 1.
Plasma energy metabolites in non-fasted compared to fasted flox/flox and CGI-58-ATko mice on chow. Plasma glucose, insulin and HOMA-IR refer to 6 h fasted mice.
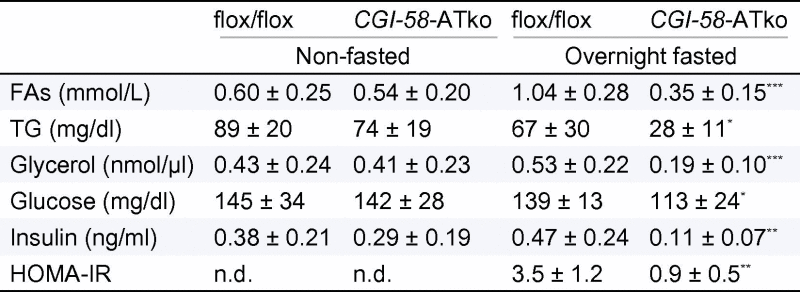 |
Values represent mean ± SD; *p <0.05, **p <0.01, ***p <0.001 (n ⩾6).
Hepatic TG (Fig. 1C) and total cholesterol (Supplementary Fig. 1E) levels were unchanged in non-fasted CGI-58-ATko compared to flox/flox mice whereas TG levels were strongly reduced (−71%) in fasted CGI-58-ATko mice compared to controls. Examination of liver tissue morphology by transmission electron microscopy (Fig. 1D) revealed very low abundance of LDs in liver sections of fasted CGI-58-ATko mice and counting of LDs showed a 9.2-fold reduction in LD numbers of nearly all sizes (Supplementary Fig. 1F). Notably, TG hydrolytic activities (Fig. 1E) were exclusively and markedly increased in liver preparations of fasted CGI-58-ATko compared to control mice, and addition of a specific ATGL inhibitor [13] to tissue preparations reduced activities to flox/flox levels suggesting increased ATGL-mediated TG-breakdown. In accordance with this assumption, hepatic ATGL expression was markedly increased in fasted CGI-58-ATko mice (up to 1.7-fold) whereas expression of the ATGL inhibitor G0S2 [14] was drastically reduced on the mRNA (−3.7-fold) and protein level (−5.5-fold) (Fig. 1F and G). The strong reduction of hepatic G0S2 expression suggests that the increase in hepatic TG breakdown is mainly due to failed induction of hepatic G0S2 expression in the liver. To corroborate this conclusion and to confirm that CGI-58 disruption affected hepatic gene expression via defective AT-lipolysis, we measured G0S2 protein levels in ATGL-ATko mice which significantly impairs AT-lipolysis in the fasted state [15], [16]. ATGL was disrupted in AT by breeding Atgl-floxed mice [11] with AdipoQ-Cre transgenic mice. In line with CGI-58-ATko mice, hepatic G0S2 expression was strongly reduced whereas ATGL expression was increased in fasted ATGL-ATko mice (Supplementary Fig. 1G).
Fasting-induced expression of PPARα-regulated genes in the liver including FGF21 depends on peripheral FA supply
The defect in AT-lipolysis profoundly interfered with hepatic TG catabolism via lack of fasting-induced G0S2 expression in the liver of CGI-58-ATko mice. Hepatic G0S2 mRNA expression is under the regulation of PPARα [17] indicating that AT-derived FAs impact PPARα-regulated gene expression in the liver. As shown in Fig. 2A, hepatic mRNA levels of PPARα and genes regulated or co-regulated by PPARα including FGF21, acyl-CoA oxidase (AOX1), medium-chain acyl-CoA dehydrogenase (MCAD) and long-chain acyl-CoA dehydrogenase (LCAD) were significantly decreased in the liver of overnight-fasted CGI-58-ATko mice (ranging from −35% to −86%) compared to levels of flox/flox mice. In contrast, mRNA expression of PPARα-regulated genes was similar in non-fasted CGI-58-ATko compared to control mice. In line with low mRNA expression, plasma FGF21 concentration was reduced by 48% (Fig. 2B). Next we examined expression of the transcriptional co-activator PGC-1α and hepatocyte nuclear factor 4alpha (HNF4α) which are highly expressed in the liver and bind to FAs [18]. Interestingly, and in contrast to reduced PPARα target gene expression, hepatic PGC-1α and HNF4α expression increased in fasted CGI-58-ATko compared to flox/flox mice on the mRNA (Fig. 2C) and nuclear protein level (Supplementary Fig. 2A and B). Unlike impaired PPARα-target gene expression in the liver, defective AT-lipolysis in CGI-58-ATko mice does not interfere with PPARα-regulated gene expression including FGF21 in cardiac and skeletal muscle (Supplementary Fig. 2C and D), which is in accordance with results of previous studies showing that PPARα-activation in cardiac muscle is not affected by low levels of exogenous FAs [9], [12]. As “proof of principle” we examined PPARα-regulated gene expression in the liver of fasted ATGL-ATko mice which also exhibit a marked defect in AT-lipolysis. AT-specific ATGL disruption showed an essentially identical hepatic mRNA expression profile except that PGC-1α mRNA expression was even more strongly increased (8.1-fold) compared to flox/flox controls (Supplementary Fig. 2E and F) further corroborating the critical role of AT-lipolysis in the regulation of hepatic fasting gene expression. To examine whether these changes in hepatic gene expression are directly mediated by low exogenous FA levels we followed a strategy where we increased circulating FA levels in fasted mice via intragastric olive oil administration followed by heparin injection to release lipoprotein lipase (LPL) thereby increasing TG-breakdown and circulating FA levels. Notably, 2 h after the lipid administration, hepatic mRNA expression of selected PPARα-regulated genes including FGF21 significantly increased in CGI-58-ATko mice (Fig. 2D) and the effect was most pronounced for G0S2. These changes were paralleled by increased FA levels in CGI-58-ATko (and flox/flox) mice implicating that the increase in PPARα target mRNA expression was linked to the raise in circulating FA levels.
Fig. 2.

Hepatic FGF21 and PPARα-regulated gene expression depends on exogenous FA supply. (A) Hepatic mRNA expression of PPARα-regulated genes in non-fasted compared to fasted mice determined by qRT-PCR. (B) Plasma FGF21 concentrations in fasted flox/flox and CGI-58-ATko mice. (C) Hepatic PPARα and HNF4α mRNA expression determined by qRT-PCR. (D) mRNA levels of PPARα-regulated genes in the liver of fasted mice following intragastric olive oil administration and heparin injection which raises plasma FA levels (right panel). (E) Fasting plasma levels of β-hydroxybutyrate (β-HB) and (F) hepatic HMGCS2 mRNA expression. Values are mean ± SD from at least 5 mice per genotype.*p <0.05, **p <0.01, and ***p <0.001 vs. flox/flox; #p <0.05, ##p <0.01, and ###p <0.001 non-fasted vs. fasted; §p <0.05 and §§p <0.01 CGI-58-ATko fasted vs. lipid and heparin administration in CGI-58-ATko mice.
Reduced circulating FGF21 levels and the increase in hepatic PGC-1α and HNF4α expression in CGI-58-ATko mice prompted us to further examine the consequences on energy metabolism. FGF21 induces ketogenic gene expression in the liver and plays a critical role in regulating body temperature [19]. Impaired hepatic FGF21 expression in CGI-58-ATko mice was paralleled by significantly reduced (−66%) plasma ketone body concentrations (β-hydroxybutyrate) (Fig. 2E) and reduced (−69%) mRNA expression of HMG-CoA synthase-2 (Fig. 2F), the rate-limiting enzyme in ketogenesis. Next we tested the impact of fasting on cold adaptation of CGI-58-ATko mice (Supplementary Fig. 3A and B). In contrast to the non-fasted state, fasting provoked a rapid decline in body temperature of CGI-58-ATko mice which disposed us to terminate the experiment. Hepatic gluconeogenesis is under the dual regulation of PGC-1α and HNF4α [20]. mRNA expression of PEPCK and G6P (Supplementary Fig. 3C) was increased in CGI-58-ATko compared to flox/flox controls (1.6- and 1.5-fold, respectively). However, pyruvate tolerance test revealed a moderate decrease in hepatic glucose production of CGI-58-ATko mice (Supplementary Fig. 3D) indicating no gross alterations in the gluconeogenic pathway.
Exogenous FA supply stimulates nuclear CREBH translocation and interferes with the ER stress pathway
The changes in hepatic gene expression of CGI-58-ATko mice prompted us to further explore the underlying mechanisms. Considering the established role of FAs as PPARα ligands it is conceivable that defective AT-lipolysis restricts ligand availability in the liver. Application of the established PPARα ligand Wy14643 further increased hepatic mRNA levels of PPARα-regulated genes (Fig. 3A) in flox/flox control mice (including a 2.9- and 4.5-fold increase in FGF21 and G0S2 expression) whereas mRNA expression of these genes remained low in CGI-58-ATko mice. Failed recovery of hepatic gene expression in CGI-58-ATko mice suggested an additional ligand-independent defect. Global CREBH-deficiency was shown to markedly impair FGF21 and G0S2 mRNA expression in the liver [21], [22] suggesting that these genes are under dual regulation of PPARα and CREBH. Fasting similarly increases CREBH mRNA expression in the liver of flox/flox and CGI-58-ATko mice (Fig. 3B). CREBH-activated gene expression requires translocation of processed microsomal CREBH to the nucleus. Notably, CREBH protein was substantially reduced (−91%) in nuclear preparations derived from liver tissue of fasted CGI-58-ATko compared to flox/flox mice (Fig. 3C) suggesting a defect in CREBH-activated gene expression.
Fig. 3.

Defective AT-lipolysis interferes with nuclear CREBH abundance and ER stress gene expression in the liver. (A) mRNA levels of PPARα-regulated genes in the liver following PPARα agonist (Wy14643) application. (B) Hepatic mRNA expression of CREBH in non-fasted compared to fasted mice. (C) CREBH protein level in nuclear extracts prepared from liver tissue of fasted flox/flox and CGI-58-ATko mice. CREBH signal intensities were normalized to nuclear Lamin A/C. (D) Pdi protein level in liver tissue obtained from fasted flox/flox and CGI-58-ATko mice normalized to GAPDH. (E) Protein levels of the ER stress regulators ATF6, IRE1α, JNK and phosphorylated JNK. Signal intensities were normalized to β-actin and ratio of phosphorylated JNK vs. total JNK was calculated. (F) Ratio of phosphorylated eIF2α and phosphorylated PERK vs. total protein. Values are mean ± SD from at least 5 mice per genotype (for Western blotting n = 3–4). *p <0.05, **p <0.01, and ***p <0.001 flox/flox vs. CGI-58-ATko; #p <0.05, ##p <0.01, and ###p <0.001 non-fasted vs. fasted or vehicle vs. Wy14643.
CREBH cleavage and nuclear translocation is known to be induced by fasting [21] and endoplasmic reticulum (ER) stress [23]. It is conceivable that the fasting-induced enhanced flow of exogenous FAs to the liver can be sensed by hepatocyte ER thereby affecting expression of genes linked to the ER stress pathway. Fasting substantially increased mRNA expression of molecular chaperones and ER stress markers (Supplementary Fig. 4A) encompassing glucose-regulated protein 78 (Grp78/BiP), C/EBP homologous protein (Chop), protein disulfide isomerase (Pdi) and ER DnaJ homologue 4 (Erdj4) in the liver of flox/flox mice (ranging from 1.6- up to 3.3-fold). This induction was absent or less pronounced in the liver of fasted CGI-58-ATko mice. On the protein level, Pdi signal intensity was significantly reduced (−62%) in CGI-58-ATko liver preparations (Fig. 3D) whereas Grp78/BiP protein expression was comparable to flox/flox controls (not shown). To further examine whether fasting interferes with hepatic ER stress we measured protein and/or phosphorylated protein levels of the three ER transmembrane effector proteins of the unfolded protein response (UPR) and their downstream targets including the activating transcription factor-6 (ATF-6), inositol requiring enzyme 1 (IRE1) and PKR-like ER kinase (PERK). ATF-6 protein expression was 1.8-fold increased in liver preparations of fasted CGI-58-ATko compared to flox/flox mice whereas IRE1α levels were comparable to flox/flox samples (Fig. 3E). Hepatic JNK protein levels and ratio of phosphorylated to total JNK were unchanged in CGI-58-ATko mice compared to flox/flox controls which is in accordance with unchanged mRNA levels of the PPARα co-repressors Ncor1 and Nrip1 (Supplementary Fig. 4B) which are under regulation of JNK [24]. Additionally, ratio of phosphorylated to total PERK was moderately increased in liver preparations of CGI-58-ATko mice whereas the ratio of phosphorylated to total eIF2alpha was unchanged (Fig. 3F). Finally, the increase in ATGL expression prompted us to measure protein levels of the forkhead box O1 (FoxO1) transcription factor which has been shown to increase ATGL expression in AT [25]. Insulin stimulates FoxO1-phosphorylation thereby preventing nuclear translocation. Cytosolic FoxO1 protein levels were reduced whereas nuclear levels increased (Supplementary Fig. 4C) indicating that FoxO1 may play a role in the induction of hepatic ATGL expression in fasted CGI-58-ATko mice. Plasma levels of alanine aminotransferase (ALT), as an indicator for liver injury, were similar in CGI-58-ATko compared to flox/flox mice (23.2 ± 6.3 vs. 25.7 ± 9.1).
Impaired AT-lipolysis counteracts hepatic steatosis, reduces hepatic stress and improves systemic insulin sensitivity of CGI-58-ATko mice on high fat diet
Feeding a high fat diet (HFD) comparably increased body weight and fat mass of flox/flox and CGI-58-ATko mice whereas lean mass was moderately reduced in CGI-58-ATko mice (Supplementary Fig. 5A and B). Gonadal AT depots were unchanged whereas brown AT mass was increased and liver mass was reduced in CGI-58-ATko compared to flox/flox mice (Fig. 4A). Plasma FA, glucose and insulin levels and HOMA-IR index were lower in CGI-58-ATko compared to flox/flox mice (Table 2). Hepatic TG levels were reduced (−37%) in CGI-58-ATko mice whereas total cholesterol levels were comparable to flox/flox controls (Fig. 4B). Histomorphological analyses showed the presence of partially large LDs in hepatocytes of both genotypes, whereas LD numbers were 1.6-fold reduced in CGI-58-ATko hepatocytes (Fig. 4C; Supplementary Fig. 5C). Hepatic TG hydrolytic activities and ATGL expression were moderately increased in CGI-58-ATko compared to flox/flox mice (Fig. 4D; Supplementary Fig. 5D and E) whereas mRNA levels of PPARα-regulated genes (Fig. 4E) and plasma FGF21 concentrations (Table 2) remained low in CGI-58-ATko mice on HFD. Similarly, G0S2 protein expression (Fig. 4F) and nuclear abundance of CREBH (Fig. 4G) were reduced in CGI-58-ATko mice compared to controls albeit CREBH levels strongly increased in both genotypes on HFD. Next we investigated the impact of HFD on hepatic stress parameters. Notably, reduced plasma ALT levels (Table 2) together with a significant decrease (−65%) in the ratio of phosphorylated to total JNK (Fig. 4H) indicates that impaired AT-lipolysis counteracts the development of hepatic injury and stress. Furthermore, reduced levels of spliced Xbp-1 mRNA (Fig. 4I) indicate less induction of the UPR. However, levels of other proteins from the UPR (Supplementary Fig. 5F–H) were similar in both genotypes except for a moderate reduction in the ratio of phosphorylated vs. total PERK and decreased Pdi expression in CGI-58-ATko mice. Finally, we investigated the impact of HFD on hepatic insulin signaling. Hepatic ratio of phosphorylated (pSer473) vs. total Akt was mildly increased in CGI-58-ATko mice on chow whereas the ratio was similar in both genotypes on HFD (Supplementary Fig. 5I).
Fig. 4.

Impaired AT-lipolysis counteracts hepatic stress and TG accumulation via promoting hepatic TG catabolism on high fat diet. (A) Organ weights of mice kept on high fat diet (HFD) for 10 weeks starting at the age of 5 weeks. (B) Hepatic TG and TC content upon HFD-feeding in fasted mice. (C) Representative images of hepatic tissue morphology analyzed by transmission electron microscopy (Scale bar = 1 μm; LD, lipid droplet; m, mitochondria; n, nucleus). (D) TG hydrolytic activities in liver tissue of fasted mice. (E) Hepatic mRNA levels of selected PPARα- and CREBH-regulated genes of fasted flox/flox and CGI-58-ATko mice determined by qRT-PCR. (F) Hepatic G0S2 and (G) nuclear CREBH protein expression on chow compared to HFD in fasted mice normalized to β-actin and Lamin A/C, respectively (n = 3). (H) Ratio of phosphorylated JNK vs. total JNK. (I) PCR-amplification of non-spliced and spliced Xbp1 (171 bp and 145 bp, respectively) from hepatic cDNA separated by agarose gel electrophoresis. (J) Simplified scheme depicting the role of AT-derived FAs in the regulation of fasting gene expression in the liver. Values are mean ± SD from at least 5 mice per genotype. *p <0.05, **p <0.01, and ***p <0.001 flox/flox vs. CGI-58-ATko (on chow or HFD). (This figure appears in colour on the web.)
Table 2.
Plasma energy metabolites and hormone levels in fasted flox/flox compared to CGI-58-ATko mice on HFD. Plasma insulin and HOMA-IR refer to 6 h fasted mice.
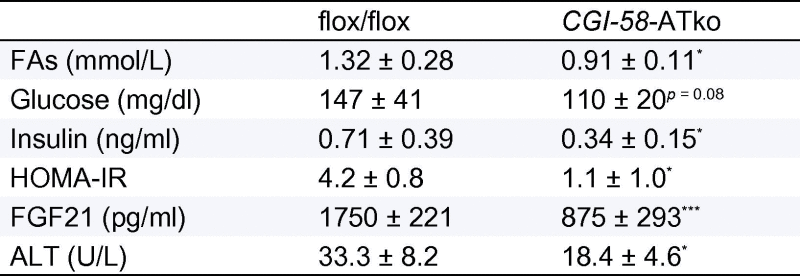 |
Values represent mean ± SD; *p <0.05, **p <0.01, ***p <0.001 (n ⩾5).
Discussion
Fasting is associated with increased activity of transcription factors in the liver which are directly or indirectly activated by FAs [18], including PPARs, CREBH and HNF4α. Yet, the role of AT-derived FAs in the regulation of hepatic gene expression is still unclear. In this study we investigated the functional role of AT-derived FAs in the regulation of hepatic gene expression. We found that defective AT-lipolysis in mice, lacking the lipolytic co-activator CGI-58 (or ATGL) in AT, severely impairs PPARα and CREBH co-regulated gene expression in the liver.
Our study demonstrates that hepatic expression of the lipase inhibitor G0S2 and the metabolic regulator FGF21 essentially depend on FA supply from AT. Recently, G0S2 has emerged as an important metabolic regulator [26], [27] via acting as a lipolytic suppressor. G0S2 is inversely expressed in AT and liver depending on the nutritional state: Fasting lowers G0S2 expression in AT to enhance lipolysis and FA supply. In contrast, G0S2 expression increases in the liver upon fasting to suppress TG catabolism thereby preserving hepatic TG as an energy source or pool for lipoprotein biogenesis or signaling molecules. However, the mechanism and metabolites or hormones regulating G0S2 expression in the liver remain unclear. Our study provides several lines of evidence that G0S2 expression is regulated via exogenous FA flow to the liver: i) Defective AT-lipolysis was associated with the absence of fasting-induced G0S2 mRNA expression in the liver which was reversed by raising fasting plasma FA levels; and ii) Defective AT-lipolysis was linked to low nuclear levels of processed active CREBH. This is in line with the severe reduction in hepatic G0S2 expression of mice globally lacking CREBH [21], [22].
Low hepatic G0S2 expression was paralleled by increased TG hydrolytic activities in liver preparations of CGI-58-ATko mice which was reduced to control levels by incubation with a specific ATGL inhibitor [13] suggesting that the decline in hepatic TG is mainly due to increased ATGL-mediated TG catabolism. In line with this conclusion, liver-specific G0S2 overexpression leads to hepatic steatosis [26], [28]. Liver-specific G0S2 knockdown does not affect fasting plasma FA levels and leads to increased ketogenesis and FAO gene expression by increasing hepatic TG catabolism [26]. Interestingly, and in strong contrast, impaired hepatic G0S2 expression in CGI-58-ATko mice reduced ketone body levels, FAO and ketogenic gene expression indicative for a critical role of AT-derived FAs in the regulation of the hepatic fasting response. Additionally, hepatic glycogen levels increased upon liver-specific G0S2 knockdown whereas glycogen was depleted in fasted CGI-58-ATko mice which may be the consequence of restricted availability of FAs as energy fuel in CGI-58-ATko mice.
Interestingly, it has been reported that the interaction of the nuclear transcription factors PGC-1α and FOXO1 stimulates gluconeogenesis [29] and that PGC-1α induces gluconeogenesis in an HNF4α-dependent manner [20]. Considering the increased nuclear abundance of the aforementioned transcription factors in fasted CGI-58-ATko mice of this study, one would predict increased gluconeogenesis. However, we found that hepatic glucose production was moderately reduced in CGI-58-ATko mice in contrast to mice with hepatic G0S2 knock down, which increases hepatic glucose production in pyruvate tolerance tests. Hepatic glucose formation in the fasted state also involves the utilization of AT-derived glycerol as metabolite and glycogenolysis. It is conceivable that lowered glucose production in response to pyruvate administration in fasted CGI-58-ATko mice involves the deprivation of glycerol and glycogen and possibly restricted FA supply as energy fuel.
Notably, the induction of hepatic TG-breakdown either in mice globally lacking G0S2 or in mice lacking CGI-58 in AT that were fed a HFD increased systemic insulin sensitivity. Furthermore, both mutant mouse models exhibited similarly reduced plasma ALT levels demonstrating that the stimulation of hepatic TG-breakdown per se counteracts the development of metabolic disorders. Similar to impaired G0S2 expression, we show that the induction of hepatic FGF21 expression depends on AT-FA supply. FGF21 expression not only stimulates ketogenesis but also regulates cold adaptation and systemic energy metabolism. The severe defect in cold adaptation observed in fasted CGI-58-ATko mice further demonstrates the critical role of AT-derived FAs in the induction of hepatic FGF21 expression and the regulation of whole body energy homeostasis [19]. Interestingly, hepatic FGF21 expression and serum FGF21 levels have been reported to be significantly increased in obese patients [30] and may involve changes in exogenous FA levels.
In this study we found that PPPAα-ligand treatment of CGI-58-ATko mice did not recover PPARα-regulated gene expression in the liver suggesting a PPARα ligand-independent defect. Recently Kim et al. [31] showed that a mutual transcriptional regulation exists between CREBH and PPARα and that PPARα requires CREBH for trans-activation of FGF21 gene expression. The extremely low protein levels of processed nuclear CREBH in fasted CGI-58-ATko mice suggested that AT-derived FAs are an important energy sensor for the liver, required for CREBH cleavage and nuclear translocation to activate G0S2 and FGF21 gene expression. CREBH was originally discovered as an ER stress regulated gene, albeit its role in ER stress is a matter of discussion [32]. Impaired AT-lipolysis divergently interfered with protein expression and/or activation of the three major regulators of the UPR pathways, including increased ATF6 protein expression and PERK-phosphorylation in CGI-58-ATko mice on chow, implicating that low nuclear abundance of CREBH was not due to classic induction of hepatic ER stress and involves a currently unknown mechanism.
It is conceivable that the increase in LD-formation in the fasted state (which is impaired in CGI-58-ATko mice) is sensed by the ER and triggers CREBH cleavage and nuclear translocation which is required for PPARα- and CREBH-coordinated expression of G0S2 and FGF21 (Fig. 4J) to regulate hepatic and whole body TG homeostasis. The fasting-stimulated induction of G0S2 expression may protect the liver from excessive TG catabolism which preserves TG as energy fuel and reservoir for lipoprotein-TG synthesis under normal diet. Under conditions of increased plasma FA levels, as exemplified in mice fed a HFD, a sustained G0S2-mediated suppression of hepatic TG catabolism may lead to the development of hepatic steatosis and the activation of the UPR. In line with such an assumption, reduced plasma FA levels of CGI-58-ATko mice on HFD were accompanied by increased hepatic TG-breakdown, reduced levels of phosphorylated JNK and PERK together with decreased mRNA levels of spliced Xbp-1 in liver tissue of CGI-58-ATko mice. Additionally, reduced insulin levels and HOMA-IR in CGI-58-ATko mice on HFD suggest that the reduction in circulating FA levels due to impaired AT-lipolysis counteracts the development of whole body and possibly hepatic insulin resistance.
Together, in this study we show that induction of G0S2 and FGF21 expression in the liver depends on FA supply from AT and that plasma FA levels correlate with hepatic G0S2 expression and nuclear abundance of CREBH. Accordingly, a surplus of plasma FAs, linked to obesity and/or dietary fat content, may promote hepatic fat accumulation via sustained suppression of hepatic TG catabolism. Impaired hepatic TG catabolism leads to hepatic steatosis in humans and mice [5], [6], [33]. Inhibition of AT-lipolysis or hepatic G0S2 expression may represent an effective strategy to enhance hepatic TG catabolism thereby counteracting hepatic fat accumulation and NAFLD development.
Financial support
This research was supported by the grants FWF P20602-B05, DK-MCD W1226, P25193, P 25944-B19, J3221-B19 and SFB Lipotox F30-B05 funded by the Austrian Science Fund and the National Institutes of Health grant R01-DK-090166 and a Howard Hughes Medical Institute Physician-Scientist Early Career Award (to E.E.K.).
Conflict of interest
The authors who have taken part in this study declared that they do not have anything to disclose regarding funding or conflict of interest with respect to this manuscript.
Author contributions
D. Jaeger and G. Haemmerle designed the study and were involved in all aspects of the experiments and wrote the manuscript. G. Schoiswohl performed studies with ATGL-ATko mice. M. Schweiger, Renate Schreiber, Nadja Poecher, T. Eichmann, P. Hofer, N. Pollak, R. Schreiber, F. Radner, G. Grabner, and K. Zierler contributed to experiments and acquisition of data. S. Eder guided animal handling. D. Kolb performed electron microscopy. K. Preiss-Landl, A. Lass, R. Zechner, and E. Kershaw discussed the results.
Acknowledgements
We thank Sabrina Huetter for excellent technical assistance and Dr. Ann-Hwee Lee for kindly providing the rabbit polyclonal CREBH-antiserum.
Footnotes
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.jhep.2015.02.035.
Supplementary data
References
- 1.Zechner R., Zimmermann R., Eichmann T.O., Kohlwein S.D., Haemmerle G., Lass A., et al. FAT SIGNALS – Lipases and lipolysis in lipid metabolism and signaling. Cell Metab. 2012;15:279–291. doi: 10.1016/j.cmet.2011.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lass A., Zimmermann R., Haemmerle G., Riederer M., Schoiswohl G., Schweiger M., et al. Adipose triglyceride lipase-mediated lipolysis of cellular fat stores is activated by CGI-58 and defective in Chanarin-Dorfman Syndrome. Cell Metab. 2006;3:309–319. doi: 10.1016/j.cmet.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 3.Schweiger M., Lass A., Zimmermann R., Eichmann T.O., Zechner R. Neutral lipid storage disease: genetic disorders caused by mutations in adipose triglyceride lipase/PNPLA2 or CGI-58/ABHD5. Am J Physiol Endocrinol Metab. 2009;297:E289–E296. doi: 10.1152/ajpendo.00099.2009. [DOI] [PubMed] [Google Scholar]
- 4.Ong K.T., Mashek M.T., Bu S.Y., Greenberg A.S., Mashek D.G. Adipose triglyceride lipase is a major hepatic lipase that regulates triacylglycerol turnover and fatty acid signaling and partitioning. Hepatology. 2011;53:116–126. doi: 10.1002/hep.24006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gauthier N., Abed L., Wu J.W., Wang S.P., Alvarez F., Soni K.G., et al. Deficiency of liver adipose triglyceride lipase in mice. Hepatology. 2011:122–132. doi: 10.1002/hep.24338. [DOI] [PubMed] [Google Scholar]
- 6.Guo F., Ma Y., Kadegowda A.K.G., Xie P., Liu G., Liu X., et al. Deficiency of liver comparative gene identification-58 (CGI-58) causes steatohepatitis and fibrosis in mice. J Lipid Res. 2013;54:2109–2120. doi: 10.1194/jlr.M035519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Donnelly K.L., Smith C.I., Schwarzenberg S.J., Jessurun J., Boldt M.D., Parks E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. 2005;115:1343–1351. doi: 10.1172/JCI23621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chakravarthy M.V., Pan Z., Zhu Y., Tordjman K., Schneider J.G., Coleman T., et al. “New” hepatic fat activates PPARalpha to maintain glucose, lipid, and cholesterol homeostasis. Cell Metab. 2005;1:309–322. doi: 10.1016/j.cmet.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 9.Zierler K.A., Jaeger D., Pollak N.M., Eder S., Rechberger G.N., Radner F.P.W., et al. Functional cardiac lipolysis in mice critically depends on comparative gene identification-58. J Biol Chem. 2013;288:9892–9904. doi: 10.1074/jbc.M112.420620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abel E.D., Peroni O., Kim J.K., Kim Y.B., Boss O., Hadro E., et al. Adipose-selective targeting of the GLUT4 gene impairs insulin action in muscle and liver. Nature. 2001;409:729–733. doi: 10.1038/35055575. [DOI] [PubMed] [Google Scholar]
- 11.Sitnick M.T., Basantani M.K., Cai L., Schoiswohl G., Yazbeck C.F., Distefano G., et al. Skeletal muscle triacylglycerol hydrolysis does not influence metabolic complications of obesity. Diabetes. 2013;62:3350–3361. doi: 10.2337/db13-0500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haemmerle G., Moustafa T., Woelkart G., Büttner S., Schmidt A., van de Weijer T., et al. ATGL-mediated fat catabolism regulates cardiac mitochondrial function via PPAR-α and PGC-1. Nat Med. 2011;17:1076–1085. doi: 10.1038/nm.2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mayer N., Schweiger M., Romauch M., Grabner G.F., Eichmann T.O., Fuchs E., et al. Development of small-molecule inhibitors targeting adipose triglyceride lipase. Nat Chem Biol. 2013;9:785–787. doi: 10.1038/nchembio.1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang X., Lu X., Lombès M., Rha G.B., Chi Y.-I., Guerin T.M., et al. The G(0)/G(1) switch gene 2 regulates adipose lipolysis through association with adipose triglyceride lipase. Cell Metab. 2010;11:194–205. doi: 10.1016/j.cmet.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ahmadian M., Abbott M.J., Tang T., Hudak C.S.S., Kim Y., Bruss M., et al. Desnutrin/ATGL is regulated by AMPK and is required for a brown adipose phenotype. Cell Metab. 2011;13:739–748. doi: 10.1016/j.cmet.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu J.W., Wang S.P., Casavant S., Moreau A., Yang G.S., Mitchell G.A. Fasting energy homeostasis in mice with adipose deficiency of desnutrin/adipose triglyceride lipase. Endocrinology. 2012;153:2198–2207. doi: 10.1210/en.2011-1518. [DOI] [PubMed] [Google Scholar]
- 17.Zandbergen F., Mandard S., Escher P., Tan N.S., Patsouris D., JATkoe T., et al. The G0/G1 switch gene 2 is a novel PPAR target gene. Biochem J. 2005;392:313–324. doi: 10.1042/BJ20050636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jump D.B., Tripathy S., Depner C.M. Fatty acid-regulated transcription factors in the liver. Annu Rev Nutr. 2013;33:249–269. doi: 10.1146/annurev-nutr-071812-161139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Inagaki T., Dutchak P., Zhao G., Ding X., Gautron L., Parameswara V., et al. Endocrine regulation of the fasting response by PPARalpha-mediated induction of fibroblast growth factor 21. Cell Metab. 2007;5:415–425. doi: 10.1016/j.cmet.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 20.Rhee J., Inoue Y., Yoon J.C., Puigserver P., Fan M., Gonzalez F.J., et al. Regulation of hepatic fasting response by PPAR γ coactivator-1 α (PGC-1): Requirement for hepatocyte nuclear factor 4 α in gluconeogenesis. Proc Natl Acad Sci U S A. 2003;100:4012–4017. doi: 10.1073/pnas.0730870100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee J.H., Giannikopoulos P., Duncan S.A., Wang J., Johansen C.T., Brown J.D., et al. The transcription factor cyclic AMP-responsive element-binding protein H regulates triglyceride metabolism. Nat Med. 2011;17:812–815. doi: 10.1038/nm.2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang C., Wang G., Zheng Z., Maddipati K.R., Zhang X., Dyson G., et al. Endoplasmic reticulum-tethered transcription factor cAMP responsive element-binding protein, hepatocyte specific, regulates hepatic lipogenesis, fatty acid oxidation, and lipolysis upon metabolic stress in mice. Hepatology. 2012;55:1070–1082. doi: 10.1002/hep.24783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang K., Shen X., Wu J., Sakaki K., Saunders T., Rutkowski D.T., et al. Endoplasmic reticulum stress activates cleavage of CREBH to induce a systemic inflammatory response. Cell. 2006;124:587–599. doi: 10.1016/j.cell.2005.11.040. [DOI] [PubMed] [Google Scholar]
- 24.Vernia S., Cavanagh-Kyros J., Garcia-Haro L., Sabio G., Barrett T., Jung D.Y., et al. The PPARα-FGF21 hormone axis contributes to metabolic regulation by the hepatic JNK signaling pathway. Cell Metab. 2014;20:512–525. doi: 10.1016/j.cmet.2014.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chakrabarti P., Kandror K.V. FoxO1 controls insulin-dependent adipose triglyceride lipase (ATGL) expression and lipolysis in adipocytes. J Biol Chem. 2009;284:13296–13300. doi: 10.1074/jbc.C800241200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang X., Xie X., Heckmann B.L., Saarinen A.M., Czyzyk T.A., Liu J. Target Disruption of G0/G1 Switch Gene 2 Enhances Adipose Lipolysis, Alters Hepatic Energy Balance, and Alleviates High Fat Diet-Induced Liver Steatosis. Diabetes. 2013;63:934–946. doi: 10.2337/db13-1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heckmann B.L., Zhang X., Xie X., Saarinen A., Lu X., Yang X., et al. Defective adipose lipolysis and altered global energy metabolism in mice with adipose overexpression of the lipolytic inhibitor G0/G1 Switch Gene 2. J Biol Chem. 2013;289:1905–1916. doi: 10.1074/jbc.M113.522011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Y., Zhang Y., Qian H., Lu J., Zhang Z., Min X., et al. The g0/g1 switch gene 2 is an important regulator of hepatic triglyceride metabolism. PLoS One. 2013;8:e72315. doi: 10.1371/journal.pone.0072315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Puigserver P., Rhee J., Donovan J., Walkey C.J., Yoon J.C., Oriente F., et al. Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1alpha interaction. Nature. 2003;423:550–555. doi: 10.1038/nature01667. [DOI] [PubMed] [Google Scholar]
- 30.Gallego-Escuredo J.M., Gómez-Ambrosi J., Catalan V., Domingo P., Giralt M., Frühbeck G., et al. Opposite alterations in FGF21 and FGF19 levels and disturbed expression of the receptor machinery for endocrine FGFs in obese patients. Int J Obes (Lond) 2014;39:121–129. doi: 10.1038/ijo.2014.76. [DOI] [PubMed] [Google Scholar]
- 31.Kim H.B., Mendez R., Zheng Z., Chang L., Cai J., Zhang R., et al. Liver-enriched transcription factor CREBH interacts with peroxisome proliferator-activated receptor α to regulate metabolic hormone FGF21. Endocrinology. 2014;155:en20131490. doi: 10.1210/en.2013-1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee A.-H. The role of CREB-H transcription factor in triglyceride metabolism. Curr Opin Lipidol. 2012;23:141–146. doi: 10.1097/MOL.0b013e3283508fed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang Y., Cohen J.C., Hobbs H.H. Expression and characterization of a PNPLA3 protein isoform (I148M) associated with nonalcoholic fatty liver disease. J Biol Chem. 2011;286:37085–37093. doi: 10.1074/jbc.M111.290114. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.