

Abstract
Background
Ameloblastin (AMBN) is a phosphorylated, proline/glutamine-rich protein secreted during enamel formation. Previous studies have revealed that this enamel matrix protein was present early in vertebrate evolution and certainly plays important roles during enamel formation although its precise functions remain unclear. We performed evolutionary analyses of AMBN in order to (i) identify residues and motifs important for the protein function, (ii) predict mutations responsible for genetic diseases, and (iii) understand its molecular evolution in mammals.
Results
In silico searches retrieved 56 complete sequences in public databases that were aligned and analyzed computationally. We showed that AMBN is globally evolving under moderate purifying selection in mammals and contains a strong phylogenetic signal. In addition, our analyses revealed codons evolving under significant positive selection. Evidence for positive selection acting on AMBN was observed in catarrhine primates and the aye-aye. We also found that (i) an additional translation initiation site was recruited in the ancestral placental AMBN, (ii) a short exon was duplicated several times in various species including catarrhine primates, and (iii) several polyadenylation sites are present.
Conclusions
AMBN possesses many positions, which have been subjected to strong selective pressure for 200 million years. These positions correspond to several cleavage sites and hydroxylated, O-glycosylated, and phosphorylated residues. We predict that these conserved positions would be potentially responsible for enamel disorder if substituted. Some motifs that were previously identified as potentially important functionally were confirmed, and we found two, highly conserved, new motifs, the function of which should be tested in the near future. This study illustrates the power of evolutionary analyses for characterizing the functional constraints acting on proteins with yet uncharacterized structure.
Electronic supplementary material
The online version of this article (doi:10.1186/s12862-015-0431-0) contains supplementary material, which is available to authorized users.
Keywords: Ameloblastin, Evolution, Enamel, Mammals, Purifying selection, Positive selection, Phylomedicine
Background
Ameloblastin (AMBN), amelogenin (AMEL), enamelin (ENAM), amelotin (AMTN), odontogenic, ameloblast associated (ODAM) and SCPP-PQ1, are ameloblast-secreted proteins involved in various steps of the organization and mineralization of the enamel matrix of mammalian teeth. They belong to the large secretory calcium-binding phosphoprotein (SCPP) family (23 members known in humans), which appeared more than 450 million years ago (Ma) then diversified through gene duplication during vertebrate evolution [1–4]. In mammals, non-AMEL proteins account only for 10–20 % of the total enamel proteins, among which AMBN, also known as sheathlin [5, 6] or amelin [7], is the most abundant [8–10]. AMBN mRNA was sequenced in pigs [6], mice [11], humans [12, 13], cattles and guinea-pigs (direct submission to NCBI database). In non-mammals, AMBN cDNA was published only in a crocodile [14] and in the clawed toad [15]. Genomic DNA (gDNA) sequences are known in a lizard [16] and in the coelacanth [17]. These findings indicate that AMBN was present early during vertebrate evolution, and at least in the last common ancestor of sarcopterygians (420 million years ago, Ma [18]). However, the large evolutionary distance among the few AMBN sequences available in non mammalian tetrapods does not allow accurate comparison and further analysis, including new data in non mammalian sarcopterygians are needed to better understand AMBN evolution through such a large geological period.
AMBN is an intrinsically unstructured, enamel matrix protein (EMP), and its functions remain unclear. It has been suggested either to be a structural component of the enamel matrix playing a role in maintaining the prismatic structure of growing enamel crystals at the rod and interrod boundaries [6, 19, 20], or to be involved in ameloblast adhesion [21–23], or to function as a growth factor [24–27] or as a signaling molecule [26, 28]. AMBN possesses two calcium-binding domains that interact with Ca2+ ions after being liberated by proteolysis [29–31]. The transcription start site (TSS) and the promoter region of AMBN were analyzed in the mouse [32]. This region contains cis-acting elements that function both to enhance and suppress transcription, some of them regulating transcription activity in mesenchymal cells in vitro [33]. Cbfa1, the essential transcription factor for osteoblast differentiation, was shown to play an important role in AMBN transcription [34].
It is worth noting that (i) AMBN expression was reported during craniofacial bone development in rats [35], and (ii) in vitro and in vivo experiments have suggested that this protein could also play a role in bone formation and repair [27, 36, 37]. These findings could indicate that AMBN plays a role in early bone formation and modeling, but they have been contradicted by a study showing no implication in bone modeling and repair [38]. Moreover, a possible function in bone development remains elusive as (i) AMBN−/− mice do not exhibit apparent bone phenotype [21], (ii) AMBN is subjected to pseudogenization in birds and in mammalian species, in which the capability to form either enamel or teeth has been lost, e.g., xenarthrans, pangolins and baleen whales, indicating that AMBN is a tooth specific protein [39–41], and (iii) AMBN expression was found to be restricted to teeth in rodents and in a crocodile [9, 14]. Finally, AMBN−/− mice develop severe enamel hypoplasia only, a phenotype that indicates a crucial role of AMBN in enamel formation [21].
Because of its important role in mammalian enamel formation, for long AMBN has been considered a candidate gene for amelogenesis imperfecta hereditary type 2 (AIH2), a human genetic disease [42–46]. In humans, AMBN is located on the long arm of chromosome 4 (4q13-21), containing the gene locus for the autosomal dominant hypoplastic form of AIH2, that affects enamel formation and is the most prevalent amelogenesis imperfecta (AI) type (85 % of all inherited AI) [12, 47]. However, it is only recently that the first case of a disease-associated mutation of AMBN (homozygous exon 6 deletion) was reported in a family having hypoplastic AI [48]. The lack of other cases of AMBN-associated AIH2 strongly contrasts with the important role played by AMBN during amelogenesis. This contradiction could be explained by an autosomal recessive pattern of inheritance as demonstrated in AMBN+/− mice that do not exhibit an apparent dental phenotype [49].
Interestingly, human ameloblastoma, a benign but destructive tumor, expresses AMBN transcripts with tumor-specific mutations [13, 50, 51]. Such expression suggests that AMBN plays a role in epithelial odontogenic tumors. AMBN is also expressed in osteosarcoma cells [27]. In humans and some mammals, amyloidosis has been associated with calcifying epithelial odontogenic tumors (CEOT) and AMBN was found to be highly expressed in these cells [52]. AMBN deficiency was proposed to be the cause of the odontogenic tumors seen in 20 % of the AMBN−/− mice [21]. Also, tumor suppressor genes P21 and P27 are upregulated in in vitro transfected ameloblastoma cells [22].
In recent years, a series of detailed evolutionary analyses of mammalian SCPPs have been conducted (AMEL [53]; ENAM [54]; MEPE [55]; AMTN [56]; DMP1 [57]) with the aim to (i) reveal regions and residues important for the protein function, (ii) predict or validate mutations responsible for genetic diseases, and (iii) understand their mode of evolution, origin and relationships [3, 4]. Here, we perform such an evolutionary analysis of mammalian AMBN sequences in order to predict functionally important sites of the protein and to identify candidate disease-associated mutations responsible for AI in human, an approach called phylomedicine [58].
Material and methods
AMBN sequences and alignment
A total of 56 mammalian full-length AMBN coding sequences, representative of the main lineages, were extracted from the NCBI [59] and Ensembl [60] databases: four published, full-length sequences (human, pig, mouse and rat); two sequences published only in GenBank (cow and guinea-pig), all including the UTRs; 33 computer-predicted sequences, i.e., available from the automatic analysis of sequenced mammalian genomes; and 17 sequences obtained using BLAST from whole genome shotgun data.
The AMBN sequences of armadillo and sloth (Xenarthra), and of aardvark (Afrotheria) are particular because the former is enamel-less in adult and the two others lack enamel, even in juveniles [41, 61]. AMBN being a tooth-specific protein [39, 40] the gene has accumulated mutations after the ability to form enamel was lost, or reduced, in these lineages [41]. Therefore, we performed our evolutionary analyses with two datasets: the complete dataset of 56 sequences and a reduced dataset of 53 functional mammalian AMBN sequences. The published crocodilian sequence (Caiman crocodilus) was also used as outgroup for calculation of the mammalian ancestral sequence. Species names and sequence references are indicated in the Additional file 1.
The coding regions of the AMBN sequences were translated into putative amino acid sequences, aligned to the human sequence using Clustal X 2.0 [62], and checked by hand using Se-Al v.2.0a11 software [63, 64]. Each of the computer-predicted sequences was carefully checked against the published sequences, taking into account the intron/exon boundaries. When necessary, they were completed and/or corrected using (i) Blast search against the Ensembl genome database, (ii) the NCBI trace archives, (iii) the whole genome shotgun (WGS) repository sequences, and (iv) resequencing by performing classical PCR on DNA extracted from alcohol-preserved tissues. In our final alignment only a few residues were missing, representing less than 1 % of the data. The positions with missing data were included in our analyses and treated as “unknown states”.
5' untranslated region (5' UTR) and signal peptides
Taking the published mRNA sequences as templates, we obtained the putative 5' UTR (i.e., exon 1 and beginning of exon 2) of AMBN sequences in 49 genomes using BLAST. Then, using Clustal X 2.0 and Se-Al v.2.0a11, we aligned these sequences against the published mammalian sequences of exons 1 and 2 with particular attention paid on the exon-intron boundaries. The transcription start site (5' end of exon 1) was defined by default, but this region was not useful for our analysis. The 49 sequences were analyzed to identify the ATGs that could be correct translation initiation sites (TIS). We used the DNA functional site miner [65] that predicts functional TIS i.e., possessing the highest Kozak consensus score [66, 67]. All identified putative signal peptides (SPs) were analyzed using the Signal P 3.0 server [68]. This software predicts the location of the three characteristic regions (n-, h- and c- regions) of a SP, the putative cleavage site of the protein, and calculates the probability for each SP to be functional [69].
Phylogenetic reconstruction
The 53 functional mammalian AMBN sequences were aligned based on their amino acid translations using MAFFT [70] within Geneious R6.03 [71]. The alignment was then restricted to the length of the human AMBN sequence by excluding all sites containing gaps in non-human sequences and the final stop codon. This procedure resulted in a dataset containing 1341 nucleotide sites (447 codons/amino acids versus 500 when including gaps). Then the three non-functional sequences (aardvark, armadillo and sloth) were added to this dataset using the program MACSE specifically designed to deal with frameshifts and stop codons [72]. Ambiguously aligned codons were then excluded using Gblocks [73] leading to a dataset with 1239 nucleotide sites (413 codons) for 56 taxa. Maximum likelihood (ML) phylogenetic reconstructions were performed on the two nucleotide alignments under the GTR + GAMMA model using RAxML 7.3.2 [74]. The statistical support for the ML tree was evaluated using 100 bootstrap replications of the initial ML search. The platypus (Ornithorhynchus anatinus) sequence was used as outgroup in all analyses.
Selection analyses
We evaluated the selective constraints acting on AMBN by performing a number of statistical tests based on the ratio of non-synonymous (dN) to synonymous (dS) substitutions (dN/dS or ω) using the codeml program of the PAML 4.7a package [75]. In all analyses, we used the mammalian species tree topology as inferred by Meredith et al. [76], except for the position of the placental root, which follows Romiguier et al. [77]. We first tested for significant among-site variation in dN/dS ratio along the AMBN molecule by using site-specific codon models for the detection of positive selection. This was achieved by comparing the fit of a nearly neutral model (M7) versus a model (M8) that allows a fraction of positively selected sites [78, 79]. These two models were compared using a hierarchical likelihood ratio test (LRT) [80]. A conservation index (CI) was then calculated for each codon from the Bayes Empirical Bayes (BEB) estimated mean values of ω obtained under the M8 model following the procedure proposed by Burk-Herrick et al. [81]. The CI was defined as 1-ω with values of neutral (ω = 1) and positively selected sites (ω > 1) being set to 0 in order to graphically represent site-specific selective constraints along the AMBN molecule [82].
The branch specific variation of dN/dS in AMBN across the mammalian species tree was jointly reconstructed from the complete 56-taxa dataset with divergence times while controlling the effect of life-history traits [83] using the Bayesian framework implemented in the CoEvol program [84]. The dN/dS ratio was estimated under the dsom procedure together with the 24 calibration constraints issued from Benton et al. [85] that were compatible with our taxon sampling to estimate divergence times. The prior on the root node was set to 220 Ma with a standard deviation of 60 Ma following the expert estimate for the split between Monotremata and Placentalia reported on the TimeTree2 website [86]. The values of the three life-history traits incorporated into the analysis (body mass, longevity, and sexual maturity) were extracted from the PanTheria database [87]. Two independent MCMC were run for a total of 2500 cycles sampling points every cycle. The first 500 points of each MCMC were then excluded as the burnin, and inferences were made from the remaining 2000 sampled points.
To further test for significant variations in dN/dS along specific branches, we used the branch model implemented in codeml allowing ω to vary among specific branches and/or clades [88]. Hierarchical LRTs were calculated between the one-ratio model (M0) and alternative models (M2ω, M4ω, M5ω, M8ω), in which branches with a mean dN/dS > 0.6 as estimated in the previous Bayesian reconstruction were allowed to have their own ω (see Table 1).
Table 1.
Results of likelihood ratio tests for positive selection in AMBN
| Models | Hypotheses | LRT | |||
|---|---|---|---|---|---|
| Null hypothesis | Alternative hypothesis | −2ΔLnL (df) | p-value | Sites or branches | |
| Site Models | Sites under M8 (BEB) | ||||
| 53-taxa data set | M7: beta | M8: beta&ω | 42.77 (2) | p < 0.001 | 135 (ω = 1.49 ± 0.06; PP = 0.99) |
| 328 (ω = 1.50 ± 0.04; PP = 1.00) | |||||
| 360 (ω = 1.48 ± 0.12; PP = 0.96) | |||||
| 375 (ω = 1.49 ± 0.06; PP = 0.99) | |||||
| 394 (ω = 1.48 ± 0.11; PP = 0.97) | |||||
| Branch Models | Branches | ||||
| 53-taxa data set | M0: One-ratio model | M2ω: 2-ratio model | 29.89 (1) | p < 0.001 | Background (ω = 0.44), Catarrhines (ω = 1.32) |
| M0: One-ratio model | M5ω: 5-ratio model | 52.16 (4) | p < 0.001 | Background (ω = 0.43), Catarrhines (ω = 1.32), Elephant (ω = 0.73), Elephant shrew (ω = 0.75), Aye aye (ω = 1.13) | |
| 56-taxa data set | M0: One-ratio model | M2ω: 2-ratio model | 18.11 (1) | p < 0.001 | Background (ω = 0.45), Catarrhines (ω = 1.11) |
| M0: One-ratio model | M4ω: 4-ratio model | 19.73 (3) | p < 0.001 | Background (ω = 0.45), Armadillo (ω = 0.67), Sloth (ω = 1.10), Aardvark (ω = 0.76) | |
| M0: One-ratio model | M5ω: 5-ratio model | 39.62 (4) | p < 0.001 | Background (ω = 0.44), Catarrhines (ω = 1.11), Armadillo (ω = 0.67), Sloth (ω = 1.10), Aardvark (ω = 0.76) | |
| M0: One-ratio model | M8ω: 8-ratio model | 58.14 (7) | p < 0.001 | Background (ω = 0.42), Catarrhines (ω = 1.11), Armadillo (ω = 0.68), Sloth (ω = 1.10), Aardvark (ω = 0.76), Elephant (ω = 0.68), Elephant shrew (ω = 0.76), Aye aye (ω = 1.07) | |
BEB Bayes Empirical Bayes, PP Posterior Probability
Ancestral sequence reconstruction
Ancestral sequence reconstruction of mammalian AMBN was computed with FastML [89] on the FastML server [90]. The AMBN sequence of the caiman (Caiman crocodilus) was added to the 53-taxa alignment using MAFFT in order to serve as outgroup. FastML was used for inferring ancestral amino acid sequences at each node of the nucleotide-derived ML tree under the LG + G8 + F model using the marginal reconstruction procedure including indels.
Results
For a better understanding of the results presented below, our current knowledge of AMBN structure (exemplified in humans) and the location of cleavage sites of the protein (identified in pigs) are illustrated in Fig. 1.
Fig. 1.
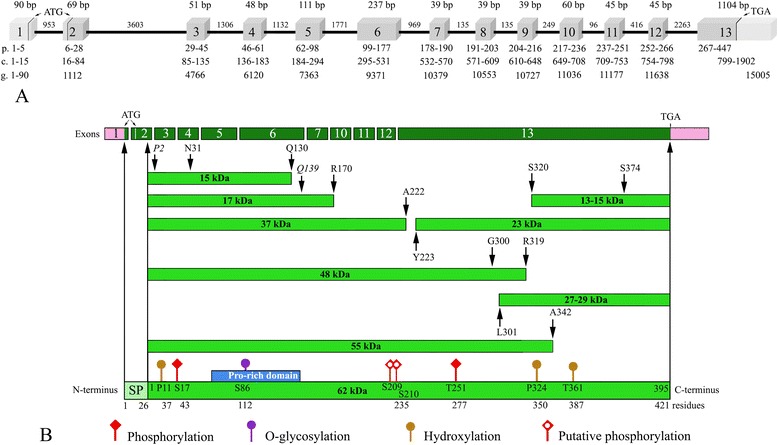
Gene structure and proteolytic cleavage products of ameloblastin. a: Human AMBN. Gene structure based on the reference gene sequence (Genbank accession No NC_000004.11). Exons are numbered from 1 to 13. Two translation initiation sites (ATG) are found: in exon 1 and exon 2. The untranslated regions are in light grey. The number of base pairs (bp) is indicated above each exon. The nomenclatural counts of encoded amino acids (p.), cDNA bp (c.) and gDNA bp (g.) are also indicated below each exon. Introns are symbolized by the black lines (not at scale) with the nucleotide number indicated above. The stop codon (TGA) is located in exon 13. b: Porcine AMBN. Diagram showing the cleavage products and their corresponding location on the protein sequence (after [6, 20, 29, 99, 101, 116]). Top: AMBN exons. Note that exons 8 and 9, which encode 26 residues in humans are lacking in porcine AMBN. Bottom: AMBN protein including either the 26 residues forming the signal peptide-SP (1–421) or not (1–395). Several remarkable residues were identified: two phosphorylated residues (S17, T251) and two putatively phosphorylated residues (S209, S210), two O-glycosylated residues (S86, T361), two hydroxylated prolines (P11, P324), and the proline-rich region. The secreted protein has an apparent molecular weight of 62 kDa; cleavage sites leading to various AMBN fragments found in vivo are indicated in roman characters, two MMP20 cleavage sites found in in vitro studies are in italics
Alignment and sequence comparisons
The 56 AMBN sequences studied here represent 46 families distributed in 19 orders and are representative of the current mammalian phylogenetic diversity (Additional file 1). The alignment of the 53 functional sequences resulted in a 500 amino acid long sequence, when considering methionine in exon 1 (Additional file 2), and including gaps required for accurate alignment. For convenience of presentation, the duplicated exons 9c, 9e, and 9d were not shown: they were nearly identical and only found in a few species (see below).
Sequence comparison revealed a few insertions, the largest being related to the duplication of small exons in 12 species (see below). These additional exons account for a large part of the length variation observed among the sequences (Additional file 2). In the 10 rodent AMBN analyzed, an insertion of 4 or 5 residues (QG/NMAP) was identified in the region encoded by exon 3. This event likely occurred in the rodent ancestral branch after the divergence with lagomorphs (rabbit and pika), which do not possess this insertion. The insertion is located close to a remarkably conserved region and probably results from the duplication of the neighboring PGMAS residues (Additional file 2). In hedgehog, six residues were inserted in the region encoded by exon 13 at positions 365–370, and in marsupials three amino acids (IKQ) were inserted in the region encoded by the 3′ end of exon 2. In addition, a few insertions and deletions (indels) of one to five residues were found distributed in the whole sequences and no sequence repeats were identified, to the exception of the exon duplication reported below.
Additional exons
Human AMBN possesses two exons, exons 8 and 9, which are absent in rodent sequences. Therefore, we screened the DNA region located between exons 7 and 10 in the mammalian genomic sequences to check whether additional exons were present in this region. We show that a gene structure composed of 11 exons (i.e., exons 1 to 7 and 10 to 13) characterizes 41 mammalian AMBN out of the 53 functional sequences investigated. This structure is also conserved in caiman, which indicates that the ancestral mammalian AMBN most probably possessed 11 exons. In 12 species, however, AMBN displays additional exons in this particular region. They are eight simiiform primates (out of nine), three laurasiatherians (horse, microbat and hedgehog), and one afrotherian (elephant shrew) (Additional file 2): one exon in orangutan and microbat; two in humans, chimpanzee, gibbon, squirrel monkey, marmoset and hedgehog; three in elephant shrew; four in baboon and macaque; and six in horse. These additional exons have the same length (39 bp) and are nearly identical in sequence to exon 7 (Additional file 2). These features strongly suggest that these exons originated all from tandem duplications of a short DNA region containing exon 7. This region is the only AMBN location, in which additional exons were identified. The two exons found between exons 7 and 10 in human AMBN were numbered 8 and 9a; therefore, the extra exons found between exon 9a and 10 were numbered 9b, 9c, etc. In the horse, the last duplicated exon was numbered 9e. It is worth noting that closely related species such as shrew vs. hedgehog, megabat vs. microbat, etc. lacked duplicated exons, a finding which strongly suggests that the duplications occurred independently within the different lineages (Additional file 2). However, in primates, duplication of this short DNA region is found only in the Simiiformes (Platyrrhini + Catarrhini), which could indicate that the first duplication likely occurred in the common ancestor of simiiforms, after this lineage diverged from Tarsiiformes, and was subsequently conserved in all simiiform lineages except in gorilla. The presence of these duplicated exons contributes to a substantial increase in length of the encoded protein: from 421 amino acids (aa) on average in the majority of sequences to e.g., 447 aa in humans, 473 in baboon, and 499 aa in horse, which is the largest AMBN mammalian sequence known to date (Additional file 2). These exons also encode two to four prolines, which contribute to enlarge the proline-rich region of the protein.
Analyses of exon/intron boundaries in representative species indicated that the 21 nucleotides on each side of the exons are rather well conserved (Additional file 3). The unchanged nucleotides in these intronic regions could be important for intron splicing.
Translation initiation site (TIS) and signal peptide (SP) analyses
Alignment of the 5' region of the AMBN sequences revealed that the 3' end of exon 1 and the 5' end of exon 2 are well conserved (Fig. 2). Depending on the species, in exon 1, exon 2 and/or in both, DNAFSMiner identified one to five ATGs in the right context i.e., that does not lead to in-frame downstream stop codons. For each sequence analyzed, the first ATG was always predicted as being the functional TIS. In most placentals the putative TIS was located in exon 1, but with a few exceptions (bushbaby, hedgehog, bats and elephant), in which it is found in exon 2 as in marsupials, and monotremes (Fig. 2). In placentals, the TIS was located at the same position in the 3' region of exon 1, with the exception of Bovinae, in which another TIS, was identified upstream. In humans, for instance, DNAFSMiner predicted that the first ATG (starting at position 76, exon 1) is the correct functional TIS (score = 0.844). However, when exon 1 was not included in the analysis, the first ATG in exon 2 (position 106) was also predicted as the functional TIS (score = 0.627). This means that the second TIS could be used if that encoded by exon 1 was lacking, i.e. by using different promoter and transcription initiation sites.
Fig. 2.
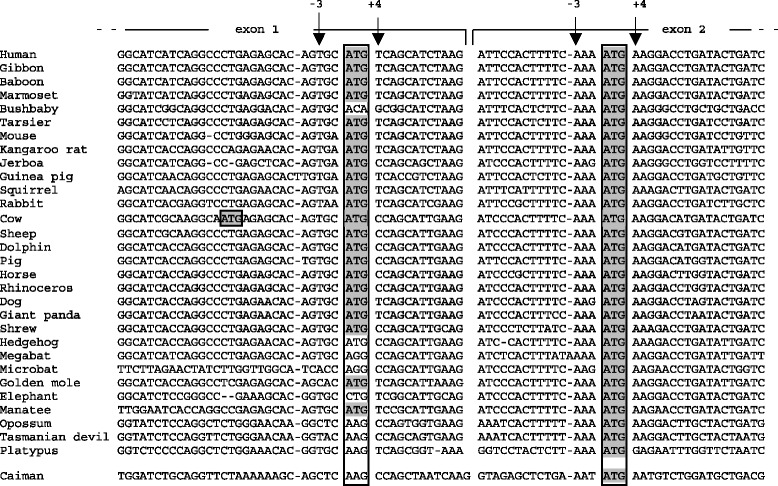
Location of putative translation initiation sites (TIS) in 30 mammalian and one crocodilian AMBN sequences. The arrows (−3, +4) point to nucleotide positions that play an essential role in the initiation of the translation. Two putatively functional TIS (squared ATGs on grey background in exons 1 and 2) are present in placental sequences, but a few exceptions, while only one TIS is identified in marsupials, platypus and crocodile. An additional, putatively functional TIS is present in cow AMBN
Interestingly, the predicted TIS in exon 1 exhibits a weak Kozak consensus as it lacks a purine (A/G) in position −3 while possessing a pyrimidine in position +4. However, such ATG could serve as the initiator codon [66]. In contrast, the ATG located in exon 2 has a strong Kozak consensus (purines in −3 and +4). As a consequence, translation could occur not only at the first (weak) TIS but also at the next (strong) TIS, resulting in the synthesis of two proteins [66]. The additional TIS identified in cow AMBN exon 1 displays also a strong Kozak consensus (a purine at positions −3 and +4), and DNAFSMiner predicted this TIS as the functional one. This additional TIS is also present in the water buffalo Bubalus bubalis and the American bison Bison bison (Bovinae) but is absent in the sheep, goat and Tibetan antelope Pantholops hodgsonii (Antilopinae) AMBN sequences (not shown), which suggests that the CTG to ATG substitution occurred along the Bovinae lineage after its divergence from Antilopinae some 20 Mya [91].
Our alignment indicates that the TIS in exon 2 is conserved in all AMBN analyzed, including that of the caiman, which means an ancient origin for this ATG. In contrast, the ATG in exon 1 was probably recruited in the eutherian ancestor, after an A to T substitution changed the ancestral AAG to ATG (Fig. 2). This ATG appears to have been secondarily lost independently in various lineages or species: ATG to CTG in elephant, ATG to AGG in bats, and ATG to ACA in bushbaby. However, in most primates, glires, most laurasiatherians, and five afrotherians out of six studied, the translated, putative AMBN sequences exhibit two methionines: the methionine encoded by the ATG in exon 1, numbered M1, and the one encoded by the ATG in exon 2, M11. In cow the first methionine is located five residues upstream M1.
The analysis of the putative SPs with SignalP revealed a similar, high probability for the SP starting either at M1 or M11. As a consequence, the SP of most placental AMBN can be either large (26 aa), when starting at M1, or short (16 aa) when beginning at M11. The three typical regions — positively charged n-region, hydrophobic h-region, and polar c-region — were found in both SPs (Additional file 4). The maximal cleavage site probability was always located between Ala26 and Val27. The corresponding predicted cleavage site is strongly supported by the presence of these two residues in all AMBN sequences analyzed, excepted for the platypus, in which Val is replaced with Ile from the same amino acid group (Additional file 2).
Polyadenylation sites (PS)
Several putative alternative polyadenylation signals (PS) were identified in the 3' UTR of the mammalian AMBN sequences (Table 2). This region is large, ranging from 441 to 557 bp in representative species of the main lineages (493 bp in the caiman). The number of PS ranged from two in the platypus to five in several species, including humans, and were numbered PS1 to PS5, from the stop codon onwards. PS were either the highly conserved hexamer AATAAA found at three locations (PS2, PS3, PS5) or the common variant ATTAAA found at two locations (PS1, PS4) (Table 2). In our dataset the latter was less conserved than the canonical signal, and it was not found in the caiman sequence. When both present, PS1 and PS2 are close to one another and can be considered a single PS; PS3 is found in all sequences but the platypus; PS4 is present in rodents and primates, in some laurasiatherians and in marsupials; PS5 is found in all sequences. The caiman AMBN possesses PS2, PS3 and PS5. In marsupials, PS4 and PS5 are close one to each other. Altogether these findings suggest that (i) three PS (PS2, PS3 and PS5) were present in the 3'UTR of the last common ancestral mammalian AMBN and (ii) PS3 was secondarily lost in the platypus lineage.
Table 2.
Location of putative alternative polyadenylation signals (PS) in the 3' UTR of AMBN cDNA sequences
| Species | Length | PS1 | PS2 | PS3 | PS4 | PS5 |
|---|---|---|---|---|---|---|
| 3'UTR | ATTAAA | AATAAA | AATAAA | ATTAAA | AATAAA | |
| Human | 553 | 106-111 | 129-134 | 189-194 | 415-420 | 536-541 |
| Bushbaby | 483 | 77-83 | 100-105 | 157-162 | 344-349 | 466-471 |
| Tree shrew | 553 | 106-111 | 129-134 | 189-194 | 412-417 | 536-541 |
| Mouse | 543 | - | 127-132 | 184-189 | 403-408 | 526-531 |
| Squirrel | 539 | 105-110 | 128-133 | 188-193 | 406-411 | 521-526 |
| Rabbit | 549 | 106-111 | 129-134 | 189-194 | 410-415 | 532-537 |
| Cow | 555 | 106-111 | 129-134 | 189-194 | 419-424 | 538-543 |
| Horse | 556 | 106-111 | 129-134 | 189-194 | - | 539-544 |
| Dog | 557 | 107-112 | 130-135 | 190-195 | - | 540-545 |
| Microbat | 513 | - | 109-114 | 169-174 | 377-382 | 497-502 |
| Elephant | 556 | - | 126-131 | 186-191 | - | 537-542 |
| Opossum | 540 | 121-126 | - | 209-214 | 495-500 | 520-525 |
| Platypus | 441 | 116-121 | - | - | - | 426-431 |
| Caiman | 493 | - | 95-100 | 298-303 | - | 475-480 |
TGA-------------PS1---PS2--------PS3-----------------------PS4--------------PS5--AAAAAAA
The PS are given for 13 representative species of the main mammalian lineages and the crocodile. Bottom: schematical representation of PS locations on the human AMBN sequence from the stop codon (TGA) to the poly(A); an hyphen represents 10 nucleotides
Amino acid composition
EMPs are characterized by their high amount of proline (P) and glutamine (Q), and are included in the P/Q-rich sub-family of SCPPs [92]. In the seven representative species analyzed for AMBN residue composition, including caiman, proline is well represented (14.09 % in average for the full-length sequence and 23.78 % in average for the proline rich region) compared to the 5.1 % encountered in most proteins (Additional file 5). Glutamine (8.7 and 15.7 %, respectively) is also well represented compared to the overall value of 4.0 %. It is worth noting that the percentage of proline in both the full-length sequence and the proline-rich domain is relatively similar from caiman to humans, while the glutamine percentage is decreasing from 11.79 to 6.49 % and from 20.93 to 11.20 %, respectively. These two amino acids represent an average of ca 23 % of the residues in the full-length AMBN sequence and ca 40 % in the proline-rich domain. In the ancestral mammalian AMBN (see below) these amino acids represent ca 24 % (P = 14.1; Q = 9.8 %) and ca 42 % (22.2 + 20.2), respectively (Additional file 5).
Ancestral AMBN and enamel-less species
In addition to its slightly higher P/Q percentage compared to living species, the ancestral mammalian AMBN, calculated from the 53 functional AMBN and using the caiman sequence as outgroup, displays a structure composed of 11 exons (1–7; 10–13), i.e., without duplicated exons between exons 7 and 10. There is a single TIS located in exon 2. Otherwise, the ancestral AMBN sequence is structurally similar to that of most mammalian AMBN and, in particular, possesses all the important positions and domains reported above.
Sloth, armadillo and aardvark sequences were aligned with the ancestral AMBN. This alignment revealed that selective pressures on the AMBN sequence of these enamel-less species were more relaxed in sloth and aardvark than in armadillo (Fig. 3). Sloth and aardvark AMBN show (i) numerous residue substitutions, some having occurred in conserved positions, (ii) premature stop codons resulting from nucleotide substitution, (iii) frameshifts in the region encoded by the large exon 13 resulting from nucleotide insertion or deletion, and (iv) nucleotide substitutions in the splicing sites (not shown). In addition, a large deletion has occurred in the DNA region of aardvark AMBN between exons 5 and 7, and exon 6 has disappeared. Taken together these features indicate that AMBN is no longer functional in these two species and was subjected to pseudogenization. In contrast, armadillo AMBN does not exhibit stop codons and frameshifts, and residue substitutions are less numerous. However, several amino acid substitutions have occurred on conserved positions that were suggested above to play an important role for the protein function (Fig. 3). These data indicate that armadillo AMBN either was recently inactivated or is still active but the encoded protein is probably defective.
Fig. 3.

Alignment of AMBN sequences from three enamel-less mammals with the putative ancestral mammalian AMBN sequence. Exon limits are indicated by vertical lines. The residues conserved during mammalian evolution (see Fig. 5b) are on gray background. In the aardvark genome the region containing exon 6 is deleted. (.): residue identical to the ancestral AMBN residue; (−): indel; (=): unknown amino acid; *: stop codon
Phylogenetic tree
The ML phylogram inferred from the complete 56-taxa nucleotide data set under the GTR + GAMMA model is presented in Fig. 4. This tree illustrates the strong phylogenetic signal contained in AMBN for resolving the phylogeny of mammals. Indeed, the ML topology supports the four major placental clades: Xenarthra, Afrotheria, Laurasiatheria and Euarchontoglires, and the grouping of the latter two in Boreoeutheria. Furthermore, almost all nodes are fully resolved and supported by bootstrap values of more than 90 % with the exception of, for instance, the relationships among laurasiatherian orders or the position of tree shrew within Euarchontoglires (Fig. 4). The ML estimate of the gamma shape parameter (α) was 0.92, which underlines the relative homogeneity of the site-specific substitution rates along the AMBN molecule. Among placentals, strong variations in substitution rates are revealed with contrasted branch lengths observed in the ML tree, with fast evolving taxa such as tenrec and elephant-shrew in Afrotheria, shrew and hedgehog in Laurasiatheria, all Glires except pika and squirrel, and bushbaby in Primates within Euarchontoglires.
Fig. 4.
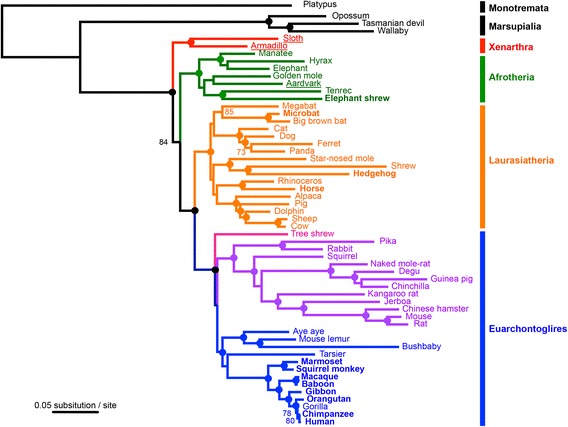
Maximum likelihood tree (GTR + GAMMA model) using the complete dataset of 56 mammalian AMBN sequences. The platypus (Ornithorhynchus anatinus) was used as outgroup. Bootstrap percentages are indicated. Nodes with more than 90 % bootstrap support are indicated with dots and nodes with no value received less than 70 % bootstrap support. Placentals are colored by main clades: Xenarthra (red), Afrotheria (green), Laurasiatheria (orange), Scandentia (pink), Glires (purple) and Primates (blue). Species possessing various copies of exon 7 are indicated in bold (see also Additional file 2). The three enamel-less species are underlined. Branch lengths are expressed as substitutions per site
Site-specific selection constraints and conserved domains
Our amino acid alignment of the 53 functional AMBN sequences revealed that many residues have been unchanged throughout mammalian evolution, some of them being regrouped into short domains (Additional file 2). Other conserved positions include amino acids that can be replaced with residues possessing the same characteristics (conservative positions). Unchanged and conservative positions are subjected to strong selective pressure, which indicates that they are functionally important for the protein. Most of them are located in the N-terminal region of the protein (encoded from exon 2 to beginning of exon 6). Conversely, more variable positions are located in the region encoded by exon 6, exon 7, and for a large part in exon 13 (Additional file 2).
Selective forces acting along the 53 AMBN codon sequences were inferred through non-synonymous vs. synonymous substitution rate (Fig. 5a). The comparison between the nearly neutral model (M7) and the model allowing a fraction of positively selected sites (M8) revealed significant among-site variation in dN/dS with the occurrence of positive selection (p < 0.001; Table 1). Under the M8 model, five codons out of 447 were identified as evolving under significant positive selection (PP > 0.95) during mammalian evolution. These sites are located in the proline rich region encoded by exon 6 at position 135, and in the exon 13 encoded region at positions 328, 360, 375, and 394 (Table 1, Fig. 5b).
Fig. 5.
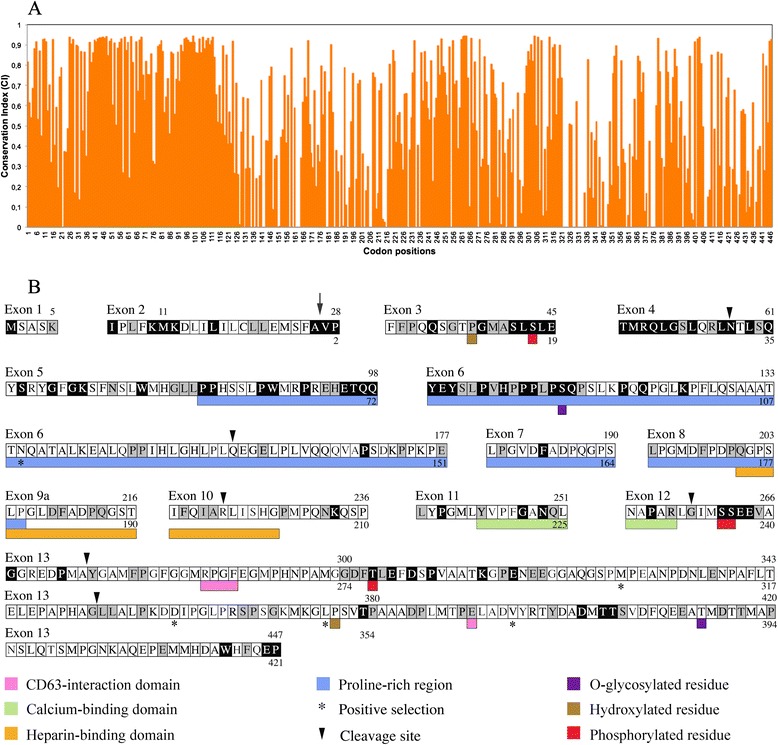
a. Estimation of the codon conservation index along the mammalian AMBN sequences. Positions 1 to 447 refer to the human sequence. Low evolutionary rates i.e., values close to 1, indicate the conserved / important positions, while high evolutionary rates i.e., values close to 0, indicate more variable positions. b Amino acid sequence of human AMBN. Human AMBN is composed of 447 residues when including the 26 residues of the putative leader peptide encoded by exons 1 and 2 (numbers above the residues); otherwise, 421 amino acids constitute the protein (numbers below the residues). Conserved positions are indicated either on black background (unchanged) or on gray background (only replaced with a residue having the same chemical property); variable positions are on white background. Various remarkable domains and remarkable residues are figured in color. The arrow points to the cleavage site of the signal peptide. Arrowheads point to proteolytic sites identified in porcine AMBN. The five sites under significant positive selection are indicated with an asterisk
The site conservation index calculated from the site-specific dN/dS values obtained under model M8 illustrated the variation of the selection constraints across the AMBN (Fig. 5a). Most of the N-ter region (positions 1 to 123, encoded from exon 1 to the 5' region of exon 6), several small regions dispersed in the AMBN sequence (notably positions 233–265, exons 10–12; 300–320, beginning of exon 13; 379–404, mid exon 13) and a short C-ter sequence (442–447, encoded by exon 13) are subjected to high functional constraints. Most of the remaining sequence is characterized by an alternation of high and low selective pressures (Fig. 5a). We identified 161 positions (out of 447 residues in human AMBN) subjected to purifying selection during more than 200 Ma of mammalian evolution pointing to biologically significant residues (Fig. 5b). These constrained positions have certainly important functions, which explains they were either unchanged (80 positions) or they displayed only few substitutions (81 positions). Among these residues are: the cleavage sites N31, G232, Y249 and G326/L327; the hydroxylated P11; the O-glycosylated S86; and the phosphorylated S17, S235, S236 and T277. In contrast, our study identified as variable positions, i.e., having probably no important function, the putative cleavage sites Q130 and R196, hydroxylated P350, and O-glycosylated T387 (Fig. 5b). Out of this dozen of important positions, there are 150 other selectively constrained positions that our evolutionary analysis pointed out as being important for the correct functioning of AMBN. Therefore, we predict that these conserved positions in AMBN, and particularly the unchanged ones, would be potentially responsible for enamel disorder if they were substituted.
Three conserved domains (or motifs) were previously identified as possibly playing an important functional or structural role (Fig. 5b). Between the two CD63-interaction domains the position E364 is conserved, while the RPGF motif is not. Half of the residues of the calcium-binding domain (Y217-R230) and eight residues out of 28 in the heparin-binding domain are conserved, which confirms that these domains are important. In addition to these domains, several conserved residues are located close one to the other in a same region of the protein, representing new motifs of unknown but important function. They are housed principally in the N-terminal region (residues 1–124) of the protein encoded by exons 3, 4, 5 and beginning of exon 6 (Fig. 5b). This large, conserved AMBN region contains important residues: the two first residues that are involved in the cleavage of the signal peptide, the hydroxylated P11, the phosphorylated S17, the O-glycosylated S86 and the cleavage site N31. The residues on both sides of these positions are also conserved, but in the same region other highly conserved amino acids constitute motifs of unknown function. We identified notably two highly conserved peptide sequences: the one is encoded by exons 3 and 4 (35 residues, 29 of them being conserved), and the other encoded by the 3' end of exon 5 and 5' extremity of exon 6 (22 residues conserved), that certainly play an important role remaining to be defined. Interestingly, it is known that the 5' end of exon 6 is alternatively spliced in the few human, rodent and porcine AMBN transcripts available. Our alignment revealed that the residue at the splicing site (Q87 = always encoded by CAG, not shown) is well conserved (Additional file 2). Similarly, several, highly conserved motifs encoded by exons 5 and 13 are highlighted as being probably important for the functionality of AMBN.
In mammals, AMBN does not possess a conserved RGD (integrin-binding) motif; however, we found an RGD sequence in the region encoded by the beginning of exon 13 in the cow and in four primates. This sequence was clearly acquired secondarily during mammalian evolution through the substitution RGG to RGD. The VTKG sequence (heparin-binding motif) is only present in rat, mouse, hamster and kangaroo rat, and was clearly acquired secondarily during rodent evolution through the substitution ATKG to VTKG. However, the TKG sequence is conserved in all eutherians and the lysine, K288, is unchanged (Fig. 5b). The DGEA motif previously described in rats and mice as a possible interacting site with integrins is also present in the hamster AMBN but not in other rodents. This motif was acquired secondarily during murid evolution.
Lineage-specific selection constraints
The estimation of the global dN/dS ratio across the whole tree using the one-ratio branch model (M0) resulted in a ω value of 0.46 showing that AMBN is globally evolving under purifying selection in mammals (Table 1). The joint Bayesian reconstruction of the dN/dS ratio along the phylogeny revealed large variations among mammalian lineages with mean ω values of functional sequences ranging from 0.15 in cat to 1.03 in gibbon (Fig. 6). As expected, elevated ω values were found in the non-functional sequences of aardvark (0.65) and sloth (1.04) as a consequence of relaxed selection constraints, whereas the armadillo has a lower value (0.44). Conversely, some lineages with functional AMBN showed high dN/dS values such as elephant (0.74), elephant shrew (0.73), and aye-aye (0.71). However, AMBN has also evolved under strong purifying selection for instance in rodents with low values of ω observed in this group overall (0.19-0.32).
Fig. 6.
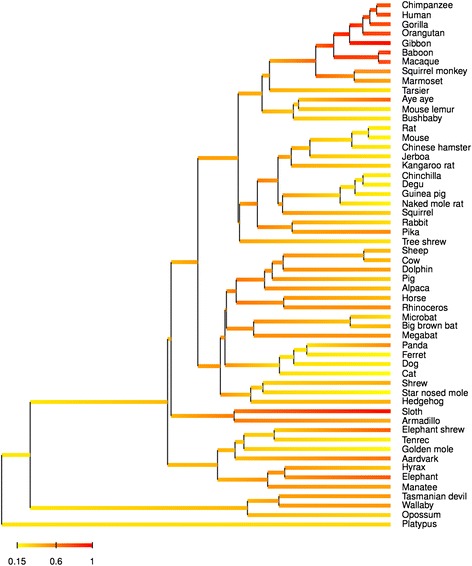
Bayesian reconstruction of dN/dS ratio in AMBN along the mammalian phylogeny. The variation of dN/dS was jointly reconstructed with divergence times while controlling the effect of three life-history traits (body mass, longevity, and maturity). The Bayesian chronogram revealed strong positive selection in catarrihine primates where elevated dN/dS ratios are inferred
The most striking pattern in this reconstruction is nonetheless the high dN/dS ratios inferred along the whole catarrhine subtree, suggesting the occurrence of positive selection on AMBN in this particular group of Primates (Fig. 6). Hierarchical likelihood ratio tests performed between different branch models allowing ω to vary among branches of the phylogeny revealed that strong positive selection has occurred in Catarrhini (Table 1). Indeed, branch model LRTs for positive selection in the catarrhine subtree were strongly significant (p < 0.001) with both the 53-taxa and 56-taxa data sets, in which the dN/dS ratio in Catarrhini was estimated to be respectively 1.32 and 1.11. Finally, among the candidate branches revealed by the previous Bayesian analysis, two were estimated to have ω values higher than 1: in sloth (ω = 1.10), in which AMBN is pseudogenized, and in aye-aye (ω = 1.13), in which it nevertheless appears fully functional.
Discussion
A second translation initiation site (TIS) was recruited in an ancestral eutherian
Two TIS were clearly identified in our analysis as being conserved through mammalian evolution: one in exon 1, the other in exon 2. The latter is the ancestral TIS in tetrapod AMBN; it is the only TIS present in reptiles [14] and frogs [15]. This TIS is homologous to that of other SCPPs [1]. Interestingly, our analysis indicated also that the former TIS was recruited in the last common eutherian ancestor, then conserved in most lineages during more than 100 Ma of evolution [76]. This finding suggests that this additional TIS plays a role in enamel formation in most eutherian species, and contradicts previous studies proposing that the only functional TIS was that in exon 2 [9, 32]. The presence of two (predicted) functional TIS means, however, that they do function through alternative splicing of exon 1 and that this process could play a role in the regulation of the amount of protein synthesized from the mRNA [66, 67]. Such alternative splicing may result in the presence of two AMBN isoforms: one with a short, 16 amino acid long, signal peptide (SP) and the other with a long SP (26 residues), the function of which remains to be tested. The presence of two signal peptides (SP) is quite common in proteins, in which two SP could play a role in the export efficiency of the protein [93]. During amelogenesis, the either use of these SP could regulate AMBN secretion in the forming enamel matrix. For further discussion on this topic we refer the readers to our previous study on ENAM, in which two functional TIS were similarly identified in two exons (exons 3 and 4), resulting in a short and a long SP [54]. The second, short SP of AMBN is homologous to the second SP described in ENAM [1], and sequence similarity in the N-terminal region of these two EMPs allowed to propose that AMBN was derived from an ancestral ENAM after duplication [4]. In fact, AMBN exon 2 and ENAM exon 4 are homologous exons, and both possess 15 nucleotides upstream the TIS. In contrast, AMBN exon 1 and ENAM exon 3, in which is located the first TIS, are not homologous. They were recruited separately during evolution. In ENAM the first TIS was recruited in a mammalian ancestor more than 200 Ma [94], while in AMBN we showed here that it was recruited later, in an eutherian ancestor, more than 100 Ma. In mammals, the convergent recruitment of an additional TIS upstream the ancestral one in two related enamel matrix proteins is, however, intriguing. Indeed, one can wonder whether the second event i.e., the recruitment of a large SP in AMBN, was linked to the presence of two SP in ENAM, and what was the benefit of this feature for enamel microstructure. To our knowledge there are no data in the literature showing to what extend the relative amount of the two EMPs, AMBN and ENAM, is important for enamel microstructure, nor whether the two proteins interact. Here, we speculate that the TIS responsible for encoding a larger SP has appeared at random in AMBN exon 1, then was conserved as providing a better fit with the different regulation of ENAM secretion through the presence of its two SP.
A hotspot for DNA duplication has appeared in placental AMBN
As previously reported in the human AMBN, we identified additional exons of same length and similar sequence in the same region of 12 unrelated eutherian AMBNs. We concluded that these additional exons resulted from tandem duplications of the region containing exon 7 and, therefore, are all homologous. Because the duplication of this short DNA region occurred repeatedly and independently in various lineages, we consider that the AMBN region located downstream exon 7 is a hotspot for the occurrence of such duplication. The process of tandem duplication is known as a major mechanism for gene elongation, but is it advantageous for AMBN function or simply neutral? Repeated duplication of exon 7 was found in terminal taxa, except in simiiform primates, in which the first duplication of this region could have occurred in their common ancestor. The duplicated region was conserved (and some copies were added) in all simiiform gDNA studied excepted in the gorilla genome, in which either the duplicated region was secondarily lost or this AMBN region was badly assembled. Given the presence of these duplicated exons in various taxa and their conservation in simiiform lineages one can wonder whether the region they encode could not have some selective advantage. In addition to enlarge the protein with 13 residues, each exon encodes two to four prolines. Because these duplications do not change much the high percentage of proline in this region, this is the increase in length of this proline-rich region that could be advantageous. Indeed, it has been suggested that the length of the proline-rich region of the EMPs could determine the supramolecular enamel matrix assembly [95].
Alternative polyadenylation signals (PS) were conserved through mammalian evolution
The 3'UTR of a mRNA is important and anomalies in this region of the mRNAs are implicated in a variety of human diseases [96]. Here, we identified two to five PS in the 3' UTR of the sequences analyzed. Three of them were present in the ancestral mammalian AMBN and conserved in all sequences during mammalian evolution. These variant PS could also be selected for regulatory purposes in the concerned sequences. For instance, it is known that mRNAs with multiple PS tend to use noncanonical signals (i.e., ATTAAA) more often than mRNAs with a single PS [97]. The long lasting conservation of the three PS through 200 Ma means that AMBN codes for three mRNAs that differ in their 3' end and play an important role in post-transcriptional regulation. This finding partially confirms previous reports of two AMBN transcripts in rodents and humans: a short sequence ending after PS1 and a large sequence ending after PS5 [9, 11, 13]. In rodents, one of these polyadenylation sites shortens the sequence at the 3' end, which could explain the presence of two transcripts. Here we show that a third transcript sequence ending at PS3 could also exist. The presence of multiple PS is significantly observed in mammalian mRNAs, and alternative polyadenylation may also influence the stability, translation efficiency, or localization of an mRNA [98, 99]. Indeed, mRNAs vary greatly in stability depending on the length of the 3'UTR, which changes a.o., the number of potential binding sites for microRNAs. However, to our knowledge AMBN mRNAs were not found among targets of several miRNAs [100].
Selection analyses highlighted residues and domains important for AMBN function
Our evolutionary analysis revealed that most cleavage sites and most hydroxylated, phosphorylated and O-glycosylated residues either identified or predicted in previous studies are included into the 161 conservative positions that were found here subjected to purifying selection (i.e., functionally constrained) during 200 Ma of mammalian evolution. They are for instance, the hydroxylated P11, phosphorylated S17 (SxE motif) and S86, and the cleavage site N31. These findings, therefore, confirm that these amino acids are important for the AMBN function and, vice versa, given that these positions were previously identified as probably important they support evolutionary analysis as an efficient method for detecting important residues. Similar studies targeting AMEL or ENAM led to the same findings [53, 54]. Interestingly, most of these remarkable residues are adjacent to unchanged amino acids, which indicate that the latter certainly play a role in stabilizing the environment of the crucial residue. Also, our analysis raised some doubts about the potential function of a few positions that were previously predicted as being important in studies of rat and pig AMBN, i.e., some cleavage sites [5, 6, 101, 102], and other hydroxylated and O-glycosylated residues [29, 103–105]. However, conservation of specific sequences could not be necessary for the cleavages to occur.
The proline-rich and calcium-binding domains do not display a large number of unchanged positions but each possesses some conservative positions that probably play a role in the function of the domain. It is known that such domains do not need high sequence conservation but rather a general context appropriate for their function. Proline-rich sequences are known to bind WW and SH3 domains, two small protein modules that mediate protein-protein interactions and are key parts of signaling cascades [106–108]. The calcium-binding site (i.e., an helix-loop-helix structural domain known as EF-hand motif) is supposed either to be involved in mineral nucleation or in calcium-mediated cell binding [23, 29, 30]. Some motifs reported in rodent AMBN, such as VTKG [7, 49, 109] and DGEA [110] are not conserved in mammalian evolution. Given that these motifs are known to interact with cell surface proteins (integrins) they are supposed to serve to link the ameloblasts to the forming enamel matrix. However, their absence in most mammalian lineages suggests that these motifs were only acquired in the rodent lineage ancestor and could have a functional role solely in rodents.
Remarkably, conserved positions and the adjacent conserved residues are however few compared to the 150 positions that were highlighted in our evolutionary analysis, the function of which remain to be characterized. Two important motifs were identified by their high number of conserved residues in the region encoded by exons 3 and 4, on the one hand, and by the 3' end of exon 5 and the 5' extremity of exon 6, on the other hand. Both motifs contain important residues; the first one houses hydroxylated P11, phosphorylated S17 (SxE motif) and cleavage site N31. It remains to be determined whether the other conserved positions are only important in order to keep these residues functional or have other functions. The second motif contains a remarkable residue, a O-glycosylated S86. Here also one can wonder what could be the function of the other conserved positions. In addition, previous studies have revealed that the 15 amino acid sequence encoded by the 5' extremity of exon 6 (including the O-glycosylated S86) is subjected to alternative splicing in rodents, human and pig AMBN through the presence of an intraexonic splicing site [6, 11, 12]. Here we showed that the residue at the splicing site (Q87 = CAG) is conserved in mammalian evolution, which strongly suggests that the two isoforms resulting from the splicing play distinct but crucial roles for the correct functioning of AMBN. In particular, it is worth noting that (i) the short isoform does not contain this O-glycosylated position, suggesting avoiding interaction with the cell membrane is important to be able to fulfill its function, and (ii) deletion of exon 6 is associated with amelogenesis imperfecta [48] with hypoplastic enamel as previously reported in mice lacking AMBN exons 5 and 6 (see below) [111].
Identification of disease-associated mutations potentially leading to AI
A total of 161 positions were identified in mammalian AMBN as conservative positions, i.e., residues that were unchanged or only substituted with a residue having the same properties during more than 200 Ma of mammalian evolution. This finding leads to the prediction that the amino acids located at these positions play an important role. They cannot be changed otherwise the AMBN function linked to this position will be disturbed and will result in a genetic disease with apparent enamel disorder. In fact, the process of natural selection provides us with tests in nature, and our previous evolutionary analyses performed on various SCPPs, including AMELX, ENAM, MEPE, AMTN, and DMP1, have shown that the patterns of long-term evolutionary conservation are crucial for validating human genetic diseases related to residue substitutions [53–57]. Such patterns were recently used for in silico functional diagnoses of non-synonymous single nucleotide variants found in thousands of disease-associated genes (see review in [58]). Given these various findings, we predict that AMBN is an excellent candidate for enamel genetic disease, amelogenesis imperfecta (AI). This prediction confirmed the conclusions previously reached by many authors because (i) AMBN was located on chromosome 4 in a region containing the locus for AIH2, the autosomal hypoplastic form of AI [12, 47], and (ii) the important function of AMBN during amelogenesis, as illustrated by AMBN−/− mice that exhibit severe enamel hypoplasia [49].
During years, mutations of AMBN were never found associated with AI phenotypes until recently when hypoplastic AI was associated with homozygous exon 6 deletion [48]. The reason why other AMBN-associated AI were not identified could be due to an autosomal recessive pattern of inheritance, and this is supported by the fact that AMBN+/− mice do not display any dental phenotype [49]. Another reason could be that AMELX compensate, at least partially, for AMBN deficiency. The two encoding genes are phylogenetically related, they share some similarities, notably in the 5' region, and both proteins possess a proline-rich region [4, 112]. Therefore, in case of homozygous mutation of AMBN compensation by AMELX could contribute to weaken the dental phenotype, which could be hardly detectable.
Deletion of exon 6 leads to the lack of 14 highly conserved and 11 conservative residues, among which the O-glycosylated S86, and a large part of the proline-rich region of the protein. Given this number of sensitive positions predicted by our evolutionary analysis, the mutation is validated as being responsible for the AI and this region as playing an important role during enamel matrix formation.
Lineage-specific positive selection and relaxation of functional constraints
Analyses of lineage-specific selective constraints acting on functional AMBN sequences have revealed a large variation in dN/dS ratio across mammals, even though this gene globally evolved under purifying selection. One of our most prominent results is the strong signal for positive selection detected in catarrhine primates, in which the dN/dS ratio was estimated to be greater than 1. This suggests that episodes of positive selection have occurred throughout the evolutionary history of this sub-clade. Interestingly, this elevated dN/dS ratio correlates with the presence of additional AMBN exons in several of these primate species suggesting that these exon duplications might be adaptive. Apart from Catarrhines, we also found evidence for positive selection in AMBN along the aye-aye (Daubentonia madagascariensis) lineage.
In Primates a correlation between enamel thickness and diet has been reported [113] and enamel thickness is often used to infer the diet of both extant and fossil primates [114, 115]. Adaptive changes related to diet shifts in Primates have been reported in another enamel protein (ENAM) and it has been hypothesized that differences in tooth enamel thickness were correlated with the adaptive evolution of enamelin [116]. Moreover, the aye-aye differs from other lemurs in possessing rodent-like gnawing and ever-growing incisors, and molars with a particularly thick enamel layer [115]. Our results therefore suggest that adaptive changes might have occurred in AMBN of catarrhines and aye-aye in response to selective constraints imposed by dietary adaptation through changes in enamel thickness.
Our analyses also detected traces of relaxed selection in enamel-less species: aardvark, armadillo, and sloth. In adult aardvarks (Afrotheria), armadillos and sloths (Xenarthra), teeth indeed lack enamel. In the nine-banded armadillo (Dasypus novemcinctus), however, at birth the teeth are covered with a thin layer of enamel that is no longer present in adults [61, 117]. In birds and in various tooth- or enamel-less mammals, selective pressures on enamel-specific proteins were relaxed after the ability to form enamel was lost, and the genes were inactivated, becoming pseudogenes [39–41, 118, 119]. As expected from these previous studies, we showed that AMBN has accumulated random deleterious mutations resulting in stop codon and frameshifts in sloth and aardvark, which confirms that AMBN is an enamel-specific protein. In the nine-banded armadillo, the AMBN sequence has not drastically changed, a condition that could be expected when considering the presence of a thin enamel cover in the young, which strongly suggests that AMBN could be expressed during enamel formation prior to birth. However, we identified several substitutions at AMBN positions that were considered important for the function of the protein. Such mutations could lead to the expression of a defective protein resulting in the deposition of a thin (hypoplastic?) enamel layer at the tooth surface, which subsequently disappears after birth through abrasion. Further structural and molecular studies of enamel layer formation in this armadillo species are however needed in order to confirm or infirm this hypothesis.
Conclusions
By adding 50 new sequences to the six previously published sequences, our study improved considerably our knowledge on the gene structure, protein composition and evolution of AMBN in mammals. In particular, we revealed that (i) an additional TIS was recruited in an ancestral eutherian AMBN, allowing the translation of a second, alternative, large SP, (ii) a short DNA sequence including exon 7 was duplicated several times in various eutherian species, increasing the proline-rich region, (iii) several PS were functional, suggesting a regulation process in the 3'UTR, (iv) numerous residues were conserved during more than 200 Ma of mammalian evolution, which strongly suggests that they are structurally and/or functionally important for the correct function of the protein, (v) several conserved residues constituted new domains of predicted high importance, and (vi) AMBN was invalidated in enamel-less species as previously reported [40, 41]. The putative function of some residues identified in previous studies was not confirmed by our analyses, and cannot be generalized to all mammals. Finally, the presence of highly conserved residues indicates that AMBN is a good candidate gene for amelogenesis imperfecta.
Acknowledgements
We are grateful to Géraldine Véron (Muséum national d’Histoire Naturelle, Paris) and François Catzeflis (Institut des Sciences de l’Evolution de Montpellier) who kindly provided ethanol-preserved tissues from some species. We thank sequencing centers for making available assembled genome sequences. The study was financially supported by grants from the Centre National de la Recherche Scientifique (CNRS), the Scientific Council of the Université de Montpellier, and the Université Pierre & Marie Curie, Paris (UMR 7138). Computational resources were provided by the Montpellier Bioinformatics Biodiversity platform of the Labex CeMEB. This is contribution ISEM 2015-148 of the Institut des Sciences de l’Evolution de Montpellier.
Additional files
Names of the 56 mammalian species used in this study. Preferred common names, scientific names, families, orders and references in GenBank are listed in alphabetical order of common names. The AMBN sequence of the crocodilian Caiman crocodilus was used as outgroup. (PDF 95 kb)
Amino acid alignment of the 53 mammalian AMBN used in our evolutionary analysis. The sequences were aligned against the human sequence and are ordered following mammalian relationships. Our alignment led to 500 positions, including gaps. For convenience of presentation our alignment does not include the duplicated exons 9c, 9d and 9e (+9c; +9c-9e) that were found in three species only. The specific length of each AMBN in indicated in brackets at the end of the sequence. The signal peptide (underlined) can start at the methionine located either in the region encoded by exon 1 or exon 2 (M, squared). For a better understanding of SP evolution, the sequences in which the first methionine is lacking are highlighted on grey background. Exon limits are indicated by vertical lines; (.): residue identical to human AMBN residue; (−): indel; (?): unknown amino acid; *: stop codon. (PDF 138 kb)
Comparison of intron/exon boundaries and UTR of twelve mammalian AMBN sequences. For scientific names and references, see Additional file 1. (//): sequence not shown; (?): unknown nucleotide; (.): nucleotide identical to human AMBN nucleotide; (−): indel. (PDF 143 kb)
Prediction of functional signal peptides (SP) in the AMBN sequence using Signal P software. Here the human sequence was used as an example. The high probability for the two sequences (score = 0.998) indicates that the SP encoded either by exon 1 + exon 2 (26 residues) (A) or by exon 2 only (16 residues) (B) are functional. The cleavage site (arrow) does not vary in the two sequences analyzed and exhibits a similar, high probability: 0.971 in A, between positions 26 and 27; 0.965 in B, between positions 16 and 17. The three characteristic regions of SP (n-, h- and c-regions) are indicated. (TIFF 488 kb)
Amino acid composition of AMBN. The amino acid percentages are given for the full length sequence and the proline-rich region for six representative mammalian AMBN, the putative ancestral mammalian sequence, and the crocodile, and are compared to average frequency in most proteins. The percentages of proline and glutamine are in bold characters. (PDF 100 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
FD performed the evolutionary analyses. BG carried out the molecular biology experiments. JYS performed in silico data recovery and sequence alignment. FD and JYS wrote the manuscript.
References
- 1.Kawasaki K, Weiss KM. Mineralized tissue and vertebrate evolution: the secretory calcium-binding phosphoprotein gene cluster. Proc Natl Acad Sci U S A. 2003;100:4060–5. doi: 10.1073/pnas.0638023100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kawasaki K. The SCPP gene repertoire in bony vertebrates and graded differences in mineralized tissues. Dev Genes Evol. 2009;219:147–57. doi: 10.1007/s00427-009-0276-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sire J-Y, Delgado S, Girondot M. The amelogenin story: origin and evolution. Eur J Oral Sci. 2006;114 Suppl 1:64–77. doi: 10.1111/j.1600-0722.2006.00297.x. [DOI] [PubMed] [Google Scholar]
- 4.Sire J-Y, Davit-Béal T, Delgado S, Gu X. The origin and evolution of enamel mineralization genes. Cells Tissues Organs. 2007;186:25–48. doi: 10.1159/000102679. [DOI] [PubMed] [Google Scholar]
- 5.Uchida T, Fukae M, Tanabe T, Yamakoshi Y, Satoda T, Murakami C, et al. Immunochemical and immunocytochemical study of a 15 kDa non-amelogenin and related proteins in the porcine immature enamel: proposal of a new group of enamel proteins “sheath proteins”. Biomed Res. 1995;16:131–40. [Google Scholar]
- 6.Hu CC, Fukae M, Uchida T, Qian Q, Zhang CH, Ryu OH, et al. Sheathlin: cloning, cDNA/polypeptide sequences, and immunolocalization of porcine enamel sheath proteins. J Dent Res. 1997;76:648–57. doi: 10.1177/00220345970760020501. [DOI] [PubMed] [Google Scholar]
- 7.Černý R, Slaby I, Hammarström L, Wurtz T. A novel gene expressed in rat ameloblasts codes for proteins with cell binding domains. J Bone Miner Res. 1996;11:883–91. doi: 10.1002/jbmr.5650110703. [DOI] [PubMed] [Google Scholar]
- 8.Termine JD, Belcourt AB, Christner PJ, Conn KM, Nylen MU. Properties of dissociatively extracted fetal tooth matrix proteins. I. Principal molecular species in developing bovine enamel. J Biol Chem. 1980;255:9760–8. [PubMed] [Google Scholar]
- 9.Krebsbach PH, Lee SK, Matsuki Y, Kozak CA, Yamada KM, Yamada Y. Full-length sequence, localization, and chromosomal mapping of ameloblastin a novel tooth-specific gene. J Biol Chem. 1996;271:4431–5. doi: 10.1074/jbc.271.8.4431. [DOI] [PubMed] [Google Scholar]
- 10.Fong CD, Cerný R, Hammarström L, Slaby I. Sequential expression of an amelin gene in mesenchymal and epithelial cells during odontogenesis in rats. Eur J Oral Sci. 1998;106(Suppl 1):324–30. doi: 10.1111/j.1600-0722.1998.tb02193.x. [DOI] [PubMed] [Google Scholar]
- 11.Simmons D, Gu TT, Krebsbach PH, Yamada Y, MacDougall M. Identification and characterization of a cDNA for mouse ameloblastin. Connect Tissue Res. 1998;39:3–12. doi: 10.3109/03008209809023907. [DOI] [PubMed] [Google Scholar]
- 12.MacDougall M, Simmons D, Gu TT, Forsman-Semb K, Kärrman Mårdh C, Mesbah M, et al. Cloning, characterization and immunolocalization of human ameloblastin. Eur J Oral Sci. 2000;108:303–10. doi: 10.1034/j.1600-0722.2000.108004303.x. [DOI] [PubMed] [Google Scholar]
- 13.Toyosawa S, Fujiwara T, Ooshima T, Shintani S, Sato A, Ogawa Y, et al. Cloning and characterization of the human ameloblastin gene. Gene. 2000;256:1–11. doi: 10.1016/s0378-1119(00)00379-6. [DOI] [PubMed] [Google Scholar]
- 14.Shintani S, Kobata M, Toyosawa S, Fujiwara T, Sato A, Ooshima T. Identification and characterization of ameloblastin gene in a reptile. Gene. 2002;283:245–54. doi: 10.1016/s0378-1119(01)00848-4. [DOI] [PubMed] [Google Scholar]
- 15.Shintani S, Kobata M, Toyosawa S, Ooshima T. Identification and characterization of ameloblastin gene in an amphibian, Xenopus laevis. Gene. 2003;318:125–36. doi: 10.1016/s0378-1119(03)00767-4. [DOI] [PubMed] [Google Scholar]
- 16.Kawasaki K. The SCPP gene family and the complexity of hard tissues in vertebrates. Cells Tissues Organs. 2011;194:108–12. doi: 10.1159/000324225. [DOI] [PubMed] [Google Scholar]
- 17.Kawasaki K, Amemiya CT. SCPP genes in the coelacanth: tissue mineralization genes shared by sarcopterygians. J Exp Zoolog B Mol Dev Evol. 2014;322:390–402. doi: 10.1002/jez.b.22546. [DOI] [PubMed] [Google Scholar]
- 18.Hedges SB. Vertebrates (Vertebrata) In: Hedges SB, Kumar S, editors. The TimeTree of Life. New York: Oxford University Press; 2009. pp. 309–14. [Google Scholar]
- 19.Nanci A, Zalzal S, Lavoie P, Kunikata M, Chen W-Y, Krebsbach PH, et al. Comparative immunochemical analyses of the developmental expression and distribution of ameloblastin and amelogenin in rat incisors. J Histochem Cytochem. 1998;46:911–34. doi: 10.1177/002215549804600806. [DOI] [PubMed] [Google Scholar]
- 20.Iwata T, Yamakoshi Y, Hu JC-C, Ishikawa I, Bartlett JD, Krebsbach PH, et al. Processing of ameloblastin by MMP-20. J Dent Res. 2007;86:153–7. doi: 10.1177/154405910708600209. [DOI] [PubMed] [Google Scholar]
- 21.Fukumoto S, Kiba T, Hall B, Iehara N, Nakamura T, Longenecker G, et al. Ameloblastin is a cell adhesion molecule required for maintaining the differentiation state of ameloblasts. J Cell Biol. 2004;167:973–83. doi: 10.1083/jcb.200409077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sonoda A, Iwamoto T, Nakamura T, Fukumoto E, Yoshizaki K, Yamada A, et al. Critical role of heparin binding domains of ameloblastin for dental epithelium cell adhesion and ameloblastoma proliferation. J Biol Chem. 2009;284:27176–84. doi: 10.1074/jbc.M109.033464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Y, Zhang X, Lu X, Atsawasuwan P, Luan X. Ameloblastin regulates cell attachment and proliferation through RhoA and p27. Eur J Oral Sci. 2011;119:280–5. doi: 10.1111/j.1600-0722.2011.00887.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zeichner-David M, Chen L-S, Hsu Z, Reyna J, Caton J, Bringas P. Amelogenin and ameloblastin show growth-factor like activity in periodontal ligament cells. Eur J Oral Sci. 2006;114:244–53. doi: 10.1111/j.1600-0722.2006.00322.x. [DOI] [PubMed] [Google Scholar]
- 25.Bartlett JD, Ganss B, Goldberg M, Moradian-Oldak J, Paine ML, Snead ML, et al. Protein–protein interactions of the developing enamel matrix. In: Schatten GP editor. Current Topics in Developmental Biology. Volume 74. Academic Press, Waltham; 2006:57–115. [DOI] [PMC free article] [PubMed]
- 26.Nakamura Y, Slaby I, Spahr A, Pezeshki G, Matsumoto K, Lyngstadaas SP. Ameloblastin fusion protein enhances pulpal healing and dentin formation in porcine teeth. Calcif Tissue Int. 2006;78:278–84. doi: 10.1007/s00223-005-0144-2. [DOI] [PubMed] [Google Scholar]
- 27.Iizuka S, Kudo Y, Yoshida M, Tsunematsu T, Yoshiko Y, Uchida T, et al. Ameloblastin regulates osteogenic differentiation by inhibiting Src kinase via cross talk between Integrin β1 and CD63. Mol Cell Biol. 2011;31:783–92. doi: 10.1128/MCB.00912-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Spahr A, Lyngstadaas SP, Slaby I, Haller B, Boeckh C, Tsoulfidou F, et al. Expression of amelin and trauma-induced dentin formation. Clin Oral Investig. 2002;6:51–7. doi: 10.1007/s00784-001-0139-y. [DOI] [PubMed] [Google Scholar]
- 29.Yamakoshi Y, Tanabe T, Oida S, Hu C-C, Simmer JP, Fukae M. Calcium binding of enamel proteins and their derivatives with emphasis on the calcium-binding domain of porcine sheathlin. Arch Oral Biol. 2001;46:1005–14. doi: 10.1016/s0003-9969(01)00070-x. [DOI] [PubMed] [Google Scholar]
- 30.Vymětal J, Slabý I, Spahr A, Vondrášek J, Lyngstadaas SP. Bioinformatic analysis and molecular modelling of human ameloblastin suggest a two-domain intrinsically unstructured calcium-binding protein. Eur J Oral Sci. 2008;116:124–34. doi: 10.1111/j.1600-0722.2008.00526.x. [DOI] [PubMed] [Google Scholar]
- 31.Zhang X, Diekwisch TGH, Luan X. Structure and function of ameloblastin as an extracellular matrix protein: adhesion, calcium binding, and CD63 interaction in human and mouse. Eur J Oral Sci. 2011;119:270–9. doi: 10.1111/j.1600-0722.2011.00889.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dhamija S, Liu Y, Yamada Y, Snead ML, Krebsbach PH. Cloning and characterization of the murine ameloblastin promoter. J Biol Chem. 1999;274:20738–43. doi: 10.1074/jbc.274.29.20738. [DOI] [PubMed] [Google Scholar]
- 33.Tamburstuen MV, Snead ML, Reseland JE, Paine ML, Lyngstadaas SP. Ameloblastin upstream region contains structural elements regulating transcriptional activity in a stromal cell line derived from bone marrow. Eur J Oral Sci. 2011;119:286–92. doi: 10.1111/j.1600-0722.2011.00910.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dhamija S, Krebsbach PH. Role of Cbfa1 in ameloblastin gene transcription. J Biol Chem. 2001;276:35159–64. doi: 10.1074/jbc.M010719200. [DOI] [PubMed] [Google Scholar]
- 35.Spahr A, Lyngstadaas SP, Slaby I, Pezeshki G. Ameloblastin expression during craniofacial bone formation in rats. Eur J Oral Sci. 2006;114:504–11. doi: 10.1111/j.1600-0722.2006.00403.x. [DOI] [PubMed] [Google Scholar]
- 36.Tamburstuen MV, Reseland JE, Spahr A, Brookes SJ, Kvalheim G, Slaby I, et al. Ameloblastin expression and putative autoregulation in mesenchymal cells suggest a role in early bone formation and repair. Bone. 2011;48:406–13. doi: 10.1016/j.bone.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tamburstuen MV, Reppe S, Spahr A, Sabetrasekh R, Kvalheim G, Slaby I, et al. Ameloblastin promotes bone growth by enhancing proliferation of progenitor cells and by stimulating immunoregulators. Eur J Oral Sci. 2010;118:451–9. doi: 10.1111/j.1600-0722.2010.00760.x. [DOI] [PubMed] [Google Scholar]
- 38.Kuroda S, Wazen R, Sellin K, Tanaka E, Moffatt P, Nanci A. Ameloblastin is not implicated in bone remodelling and repair. Eur Cell Mater. 2011;22:56–66. doi: 10.22203/ecm.v022a05. [DOI] [PubMed] [Google Scholar]
- 39.Sire J-Y, Delgado SC, Girondot M. Hen’s teeth with enamel cap: from dream to impossibility. BMC Evol Biol. 2008;8:246. doi: 10.1186/1471-2148-8-246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Deméré TA, McGowen MR, Berta A, Gatesy J. Morphological and molecular evidence for a stepwise evolutionary transition from teeth to baleen in mysticete whales. Syst Biol. 2008;57:15–37. doi: 10.1080/10635150701884632. [DOI] [PubMed] [Google Scholar]
- 41.Meredith RW, Gatesy J, Springer MS. Molecular decay of enamel matrix protein genes in turtles and other edentulous amniotes. BMC Evol Biol. 2013;13:20. doi: 10.1186/1471-2148-13-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kärrman Mårdh C, Bäckman B, Simmons D, Golovleva I, Gu TT, Holmgren G, et al. Human ameloblastin gene: genomic organization and mutation analysis in amelogenesis imperfecta patients. Eur J Oral Sci. 2001;109:8–13. doi: 10.1034/j.1600-0722.2001.00979.x. [DOI] [PubMed] [Google Scholar]
- 43.Hart PS, Wright JT, Savage M, Kang G, Bensen JT, Gorry MC, et al. Exclusion of candidate genes in two families with autosomal dominant hypocalcified amelogenesis imperfecta. Eur J Oral Sci. 2003;111:326–31. doi: 10.1034/j.1600-0722.2003.00046.x. [DOI] [PubMed] [Google Scholar]
- 44.Santos MC, Hart PS, Ramaswami M, Kanno CM, Hart TC, Line SR. Exclusion of known gene for enamel development in two Brazilian families with amelogenesis imperfecta. Head Face Med. 2007;3:8. doi: 10.1186/1746-160X-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chan H-C, Estrella NMRP, Milkovich RN, Kim J-W, Simmer JP, Hu JC-C. Target gene analyses of 39 amelogenesis imperfecta kindreds. Eur J Oral Sci. 2011;119:311–23. doi: 10.1111/j.1600-0722.2011.00857.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.MacDougall M. Dental structural diseases mapping to human chromosome 4q21. Connect Tissue Res. 2003;44(Suppl 1):285–91. [PubMed] [Google Scholar]
- 47.MacDougall M, DuPont BR, Simmons D, Reus B, Krebsbach P, Kärrman C, et al. Ameloblastin gene (AMBN) maps within the critical region for autosomal dominant amelogenesis imperfecta at chromosome 4q21. Genomics. 1997;41:115–8. doi: 10.1006/geno.1997.4643. [DOI] [PubMed] [Google Scholar]
- 48.Poulter JA, Murillo G, Brookes SJ, Smith CEL, Parry DA, Silva S, et al. Deletion of ameloblastin exon 6 is associated with amelogenesis imperfecta. Hum Mol Genet. 2014;23:5317–24. doi: 10.1093/hmg/ddu247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fukumoto S, Yamada A, Nonaka K, Yamada Y. Essential roles of ameloblastin in maintaining ameloblast differentiation and enamel formation. Cells Tissues Organs. 2005;181:189–95. doi: 10.1159/000091380. [DOI] [PubMed] [Google Scholar]
- 50.Perdigão PF, Gomez RS, Pimenta FJGS, De Marco L. Ameloblastin gene (AMBN) mutations associated with epithelial odontogenic tumors. Oral Oncol. 2004;40:841–6. doi: 10.1016/j.oraloncology.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 51.Perdigão PF, Carvalho VM, Marco LD, Gomez RS. Mutation of ameloblastin gene in calcifying epithelial odontogenic tumor. Anticancer Res. 2009;29:3065–7. [PubMed] [Google Scholar]
- 52.Hirayama K, Miyasho T, Ohmachi T, Watanabe T, Yokota H, Taniyama H. Biochemical and immunohistochemical characterization of the amyloid in canine amyloid-producing odontogenic tumor. Vet Pathol Online. 2010;47:915–22. doi: 10.1177/0300985810375047. [DOI] [PubMed] [Google Scholar]
- 53.Delgado S, Girondot M, Sire J-Y. Molecular evolution of amelogenin in mammals. J Mol Evol. 2005;60:12–30. doi: 10.1007/s00239-003-0070-8. [DOI] [PubMed] [Google Scholar]
- 54.Al-Hashimi N, Sire J-Y, Delgado S. Evolutionary analysis of mammalian enamelin, the largest enamel protein, supports a crucial role for the 32-kDa peptide and reveals selective adaptation in rodents and primates. J Mol Evol. 2009;69:635–56. doi: 10.1007/s00239-009-9302-x. [DOI] [PubMed] [Google Scholar]
- 55.Bardet C, Delgado S, Sire J-Y. MEPE evolution in mammals reveals regions and residues of prime functional importance. Cell Mol Life Sci. 2010;67:305–20. doi: 10.1007/s00018-009-0185-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gasse B, Silvent J, Sire J-Y. Evolutionary analysis suggests that AMTN is enamel-specific and a candidate for AI. J Dent Res. 2012;91:1085–9. doi: 10.1177/0022034512460551. [DOI] [PubMed] [Google Scholar]
- 57.Silvent J, Sire J-Y, Delgado S. The dentin matrix acidic phosphoprotein 1 (DMP1) in the light of mammalian evolution. J Mol Evol. 2013;76:59–70. doi: 10.1007/s00239-013-9539-2. [DOI] [PubMed] [Google Scholar]
- 58.Kumar S, Dudley JT, Filipski A, Liu L. Phylomedicine: an evolutionary telescope to explore and diagnose the universe of disease mutations. Trends Genet. 2011;27:377–86. doi: 10.1016/j.tig.2011.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.National Center for Biotechnology Information. [http://www.ncbi.nlm.nih.gov]
- 60.Ensembl. [http://www.ensembl.org]
- 61.Davit-Béal T, Tucker AS, Sire J-Y. Loss of teeth and enamel in tetrapods: fossil record, genetic data and morphological adaptations. J Anat. 2009;214:477–501. doi: 10.1111/j.1469-7580.2009.01060.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Higgins DG, Thompson JD, Gibson TJ. Using CLUSTAL for multiple sequence alignments. In: Doolittle RF, editor. Methods in Enzymology. Volume 266. Academic Press, Waltham; 1996:383–402. [Computer Methods for Macromolecular Sequence Analysis] [DOI] [PubMed]
- 63.Rambaut A. Se-Al: Sequence Alignement Editor. Oxford: Department of Zoology. University of Oxford; 1996. [Google Scholar]
- 64.Se-Al software. [http://tree.bio.ed.ac.uk/software/seal/]
- 65.DNA Functional Site Miner. [http://dnafsminer.bic.nus.edu.sg]
- 66.Kozak M. Compilation and analysis of sequences upstream from the translational start site in eukaryotic mRNAs. Nucleic Acids Res. 1984;12:857–72. doi: 10.1093/nar/12.2.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kozak M. Point mutations define a sequence flanking the AUG initiator codon that modulates translation by eukaryotic ribosomes. Cell. 1986;44:283–92. doi: 10.1016/0092-8674(86)90762-2. [DOI] [PubMed] [Google Scholar]
- 68.SignalP Server. [www.cbs.dtu.dk/services/SignalP]
- 69.Dyrløv Bendtsen J, Nielsen H, von Heijne G, Brunak S. Improved prediction of signal peptides: SignalP 3.0. J Mol Biol. 2004;340:783–95. doi: 10.1016/j.jmb.2004.05.028. [DOI] [PubMed] [Google Scholar]
- 70.Katoh K, Misawa K, Kuma K, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30:3059–66. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28:1647–9. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ranwez V, Harispe S, Delsuc F, Douzery EJP. MACSE: Multiple Alignment of Coding SEquences accounting for frameshifts and stop codons. PLoS ONE. 2011;6:e22594. doi: 10.1371/journal.pone.0022594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000;17:540–52. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]
- 74.Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–90. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- 75.Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–91. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 76.Meredith RW, Janečka JE, Gatesy J, Ryder OA, Fisher CA, Teeling EC, et al. Impacts of the Cretaceous terrestrial revolution and KPg extinction on mammal diversification. Science. 2011;334:521–4. doi: 10.1126/science.1211028. [DOI] [PubMed] [Google Scholar]
- 77.Romiguier J, Ranwez V, Delsuc F, Galtier N, Douzery EJP. Less is more in mammalian phylogenomics: AT-rich genes minimize tree conflicts and unravel the root of placental mammals. Mol Biol Evol. 2013;30:2134–44. doi: 10.1093/molbev/mst116. [DOI] [PubMed] [Google Scholar]
- 78.Nielsen R, Yang Z. Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics. 1998;148:929–36. doi: 10.1093/genetics/148.3.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yang Z, Nielsen R, Goldman N, Pedersen A-MK. Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics. 2000;155:431–49. doi: 10.1093/genetics/155.1.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yang Z. Likelihood ratio tests for detecting positive selection and application to primate lysozyme evolution. Mol Biol Evol. 1998;15:568–73. doi: 10.1093/oxfordjournals.molbev.a025957. [DOI] [PubMed] [Google Scholar]
- 81.Burk-Herrick A, Scally M, Amrine-Madsen H, Stanhope MJ, Springer MS. Natural selection and mammalian BRCA1 sequences: elucidating functionally important sites relevant to breast cancer susceptibility in humans. Mamm Genome. 2006;17:257–70. doi: 10.1007/s00335-005-0067-2. [DOI] [PubMed] [Google Scholar]
- 82.Kirwan JD, Bekaert M, Commins JM, Davies KTJ, Rossiter SJ, Teeling EC. A phylomedicine approach to understanding the evolution of auditory sensory perception and disease in mammals. Evol Appl. 2013;6:412–22. doi: 10.1111/eva.12047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lartillot N, Delsuc F. Joint reconstruction of divergence times and life-history evolution in placental mammals using a phylogenetic covariance model. Evolution. 2012;66:1773–87. doi: 10.1111/j.1558-5646.2011.01558.x. [DOI] [PubMed] [Google Scholar]
- 84.Lartillot N, Poujol R. A phylogenetic model for investigating correlated evolution of substitution rates and continuous phenotypic characters. Mol Biol Evol. 2011;28:729–44. doi: 10.1093/molbev/msq244. [DOI] [PubMed] [Google Scholar]
- 85.Benton MJ, Donoghue PCJ, Asher RJ. Calibrating and constraining molecular clocks. In: SB Hedges and S Kumar, editors. The Timetree of Life. Oxford University Press, Oxford; 2009.
- 86.Kumar S, Hedges SB. TimeTree2: species divergence times on the iPhone. Bioinformatics. 2011;27:2023–4. doi: 10.1093/bioinformatics/btr315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jones KE, Bielby J, Cardillo M, Fritz SA, O’Dell J, Orme CDL, et al. PanTHERIA: a species-level database of life history, ecology, and geography of extant and recently extinct mammals. Ecology. 2009;90:2648–8.
- 88.Yang Z, Nielsen R. Synonymous and nonsynonymous rate variation in nuclear genes of mammals. J Mol Evol. 1998;46:409–18. doi: 10.1007/pl00006320. [DOI] [PubMed] [Google Scholar]
- 89.Pupko T, Pe’er I, Hasegawa M, Graur D, Friedman N. A branch-and-bound algorithm for the inference of ancestral amino-acid sequences when the replacement rate varies among sites: Application to the evolution of five gene families. Bioinformatics. 2002;18:1116–23. doi: 10.1093/bioinformatics/18.8.1116. [DOI] [PubMed] [Google Scholar]
- 90.Ashkenazy H, Penn O, Doron-Faigenboim A, Cohen O, Cannarozzi G, Zomer O, et al. FastML: a web server for probabilistic reconstruction of ancestral sequences. Nucleic Acids Res. 2012;40:W580–4. doi: 10.1093/nar/gks498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hassanin A, Delsuc F, Ropiquet A, Hammer C, van Vuuren BJ, Matthee C, et al. Pattern and timing of diversification of Cetartiodactyla (Mammalia, Laurasiatheria), as revealed by a comprehensive analysis of mitochondrial genomes. C R Biol. 2012;335:32–50. doi: 10.1016/j.crvi.2011.11.002. [DOI] [PubMed] [Google Scholar]
- 92.Kawasaki K, Weiss KM. SCPP gene evolution and the dental mineralization continuum. J Dent Res. 2008;87:520–31. doi: 10.1177/154405910808700608. [DOI] [PubMed] [Google Scholar]
- 93.Davis MJ, Hanson KA, Clark F, Fink JL, Zhang F, Kasukawa T, et al. Differential use of signal peptides and membrane domains is a common occurrence in the protein output of transcriptional units. PLoS Genet. 2006;2:e46. doi: 10.1371/journal.pgen.0020046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Al-Hashimi N, Lafont A-G, Delgado S, Kawasaki K, Sire J-Y. The enamelin genes in lizard, crocodile, and frog and the pseudogene in the chicken provide new insights on enamelin evolution in tetrapods. Mol Biol Evol. 2010;27:2078–94. doi: 10.1093/molbev/msq098. [DOI] [PubMed] [Google Scholar]
- 95.Jin T, Ito Y, Luan X, Dangaria S, Walker C, Allen M, et al. Elongated polyproline motifs facilitate enamel evolution through matrix subunit compaction. PLoS Biol. 2009;7:e1000262. doi: 10.1371/journal.pbio.1000262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Conne B, Stutz A, Vassalli JD. The 3′ untranslated region of messenger RNA: a molecular “hotspot” for pathology? Nat Med. 2000;6:637–41. doi: 10.1038/76211. [DOI] [PubMed] [Google Scholar]
- 97.Beaudoing E, Freier S, Wyatt JR, Claverie J-M, Gautheret D. Patterns of variant polyadenylation signal usage in human genes. Genome Res. 2000;10:1001–10. doi: 10.1101/gr.10.7.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Edwalds-Gilbert G, Veraldi KL, Milcarek C. Alternative poly(A) site selection in complex transcription units: means to an end? Nucleic Acids Res. 1997;25:2547–61. doi: 10.1093/nar/25.13.2547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gautheret D, Poirot O, Lopez F, Audic S, Claverie J-M. Alternate polyadenylation in human mRNAs: a large-scale analysis by EST clustering. Genome Res. 1998;8:524–30. doi: 10.1101/gr.8.5.524. [DOI] [PubMed] [Google Scholar]
- 100.Sehic A, Risnes S, Khuu C, Khan Q-E-S, Osmundsen H. Effects of in vivo transfection with anti-miR-214 on gene expression in murine molar tooth germ. Physiol Genomics. 2011;43:488–98. doi: 10.1152/physiolgenomics.00248.2010. [DOI] [PubMed] [Google Scholar]
- 101.Fukae M, Kanazashi M, Nagano T, Tanabe T, Oida S, Gomi K. Porcine sheath proteins show periodontal ligament regeneration activity. Eur J Oral Sci. 2006;114:212–8. doi: 10.1111/j.1600-0722.2006.00309.x. [DOI] [PubMed] [Google Scholar]
- 102.Yamakoshi Y, Hu JC-C, Zhang H, Iwata T, Yamakoshi F, Simmer JP. Proteomic analysis of enamel matrix using a two-dimensional protein fractionation system. Eur J Oral Sci. 2006;114:266–71. doi: 10.1111/j.1600-0722.2006.00279.x. [DOI] [PubMed] [Google Scholar]
- 103.Uchida T, Murakami C, Wakida K, Satoda T, Dohi N, Takahashi O. Synthesis, secretion, degradation, and fate of ameloblastin during the matrix formation stage of the rat incisor as shown by immunocytochemistry and immunochemistry using region-specific antibodies. J Histochem Cytochem. 1997;45:1329–40. doi: 10.1177/002215549704501002. [DOI] [PubMed] [Google Scholar]
- 104.Hu JC-C, Yamakoshi Y, Yamakoshi F, Krebsbach PH, Simmer JP. Proteomics and genetics of dental enamel. Cells Tissues Organs. 2005;181:219–31. doi: 10.1159/000091383. [DOI] [PubMed] [Google Scholar]
- 105.Kobayashi K, Yamakoshi Y, Hu JC-C, Gomi K, Arai T, Fukae M, et al. Splicing determines the glycosylation state of ameloblastin. J Dent Res. 2007;86:962–7. doi: 10.1177/154405910708601009. [DOI] [PubMed] [Google Scholar]
- 106.Bohley P. Surface hydrophobicity and intracellular degradation of proteins. Biol Chem. 1996;377:425–35. [PubMed] [Google Scholar]
- 107.Mayer BJ. SH3 domains: complexity in moderation. J Cell Sci. 2001;114:1253–63. doi: 10.1242/jcs.114.7.1253. [DOI] [PubMed] [Google Scholar]
- 108.Ingham RJ, Colwill K, Howard C, Dettwiler S, Lim CSH, Yu J, et al. WW domains provide a platform for the assembly of multiprotein networks. Mol Cell Biol. 2005;25:7092–106. doi: 10.1128/MCB.25.16.7092-7106.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Fukumoto S, Yamada A, Iwamoto T, Nakamura T. Dental epithelium proliferation and differentiation regulated by ameloblastin. In: Sasano T et al, editors. Interface Oral Health Science. Springer Japan, Tokyo; 2010:33–8.
- 110.Yoo SY, Kobayashi M, Lee PP, Lee S-W. Early osteogenic differentiation of mouse preosteoblasts induced by collagen-derived DGEA-peptide on nanofibrous phage tissue matrices. Biomacromolecules. 2011;12:987–96. doi: 10.1021/bm1013475. [DOI] [PubMed] [Google Scholar]
- 111.Wazen RM, Moffatt P, Zalzal SF, Yamada Y, Nanci A. A mouse model expressing a truncated form of ameloblastin exhibits dental and junctional epithelium defects. Matrix Biol. 2009;28:292–303. doi: 10.1016/j.matbio.2009.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sire J-Y, Delgado S, Fromentin D, Girondot M. Amelogenin: lessons from evolution. Arch Oral Biol. 2005;50:205–12. doi: 10.1016/j.archoralbio.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 113.Shellis RP, Beynon AD, Reid DJ, Hiiemae KM. Variations in molar enamel thickness among primates. J Hum Evol. 1998;35:507–22. doi: 10.1006/jhev.1998.0238. [DOI] [PubMed] [Google Scholar]
- 114.Dumont ER. Enamel thickness and dietary adaptation among extant primates and chiropterans. J Mammal. 1995;76:1127–36. [Google Scholar]
- 115.Pampush JD, Duque AC, Burrows BR, Daegling DJ, Kenney WF, McGraw WS. Homoplasy and thick enamel in primates. J Hum Evol. 2013;64:216–24. doi: 10.1016/j.jhevol.2013.01.009. [DOI] [PubMed] [Google Scholar]
- 116.Kelley JL, Swanson WJ. Dietary change and adaptive evolution of enamelin in humans and among primates. Genetics. 2008;178:1595–603. doi: 10.1534/genetics.107.077123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Meredith RW, Gatesy J, Murphy WJ, Ryder OA, Springer MS. Molecular decay of the tooth gene enamelin (ENAM) mirrors the loss of enamel in the fossil record of placental mammals. PLoS Genet. 2009;5:e1000634. doi: 10.1371/journal.pgen.1000634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Meredith RW, Zhang G, Gilbert MTP, Jarvis ED, Springer MS. Evidence for a single loss of mineralized teeth in the common avian ancestor. Science. 2014;346:1254390. doi: 10.1126/science.1254390. [DOI] [PubMed] [Google Scholar]
- 119.Chun Y-HP, Yamakoshi Y, Yamakoshi F, Fukae M, Hu JC-C, Bartlett JD, et al. Cleavage site specificity of MMP-20 for secretory-stage ameloblastin. J Dent Res. 2010;89:785–90. doi: 10.1177/0022034510366903. [DOI] [PMC free article] [PubMed] [Google Scholar]