

Abstract
Purpose
Vascular endothelial growth factor is a key regulator of physiological and pathologic angiogenesis. Although much is known about the major upstream signaling pathways of VEGF in endothelial cells, less is known about key transcription factors involved in VEGF action. The transcription factor myocyte enhancer factor (MEF)-2C is thought to play an important role in vasculogenesis and angiogenesis during vascular development. The purpose of this study was to investigate the regulation of MEF2C expression and MEF2-dependent activity in endothelial cells by VEGF.
Methods
Expression of MEF2C in human retinal endothelial cells and human umbilical vein endothelial cells was assayed by real-time PCR and Western blot. VEGF regulation of MEF2-dependent transcription was studied using an MEF2-luciferase reporter construct containing three copies of a high-affinity MEF2 binding site. The effect of MEF2 on endothelial cell migration was evaluated using a dominant-negative MEF2C mutant.
Results
VEGF induced MEF2C expression in a dose- and time-dependent fashion. This induction was completely abrogated by the inhibition of protein kinase C and was partially blocked by the inhibition of PKC-β and PKC-δ. In addition to regulating MEF2C expression, VEGF stimulated transcription from an MEF2-dependent promoter. VEGF stimulation of transcription was significantly reduced by the inhibition of calcineurin, CaMKII, p38 MAPK, and PKC, but not by the inhibition of ERK1/2 or BMK1/ERK5. Transfection of a dominant-negative mutant of MEF2C significantly inhibited VEGF-stimulated endothelial cell migration.
Conclusions
These results implicate VEGF as a key regulator of MEF2C and suggest that MEF2 may be an important mediator of VEGF in endothelial cells.
Vascular endothelial growth factor (VEGF, also referred to as VEGF-A) is a key regulator in physiological angiogenesis and in pathologic angiogenic processes, such as those associated with tumor growth and retinal neovascularization.1 The proangiogenic activity of VEGF derives from its ability to regulate many steps in the angiogenic process, including proliferation, migration, survival, and permeability. Much is known about the upstream events mediating the effect of VEGF on endothelial cells, including the major signaling pathways that are activated.2–4 Relatively less is known about the key transcription factors involved in the angiogenic effects of VEGF.
The myocyte enhancer factor (MEF)-2 family of transcription factors consists of a group of transcriptional activators (MEF2A, -B, -C, and -D) that show homology in a MADS (MCM1, Agamous, Deficiens, serum response factor) box and an adjacent motif known as the MEF2 domain.5 MEF2 factors play a pivotal role in the morphogenesis and myogenesis of skeletal, cardiac, and smooth muscle cells.6
Of the MEF2 family members, MEF2C appears to have a particularly important role in vasculogenesis and angiogenesis during vascular development. MEF2C is expressed in endothelial cells in vivo7 and in vitro.8 Targeted deletion of the MEF2C gene in mice results in embryonic lethality between embryonic day (E) 9.5 and E10,7,9 with significant cardiovascular defects.7,10 These mice exhibit severe vascular abnormalities, including the absence of yolk sac vascularization.7 In these mice, endothelial cells are able to differentiate but are unable to become organized into a vascular network.7 The overall phenotype of MEF2C mutant mice is similar to that of mice lacking VEGF or Flt-1.7
MEF2C appears to be a direct transcriptional target of Ets transcription factors in the developing and the adult vasculature. A transcriptional enhancer from the MEF2C gene containing four consensus Ets transcription factor binding sites is sufficient to direct gene expression to the vascular endothelium in developing and adult vasculature in transgenic LacZ reporter mice.11 The regulation of MEF2C gene expression by Ets transcription factors is of particular interest because VEGF is known to upregulate Ets-1 in endothelial cells.12
In light of the important role of MEF2C in vasculogenesis and angiogenesis, we were interested in characterizing the regulation of MEF2C by VEGF and the potential role of MEF2 in endothelial cells. We found that VEGF induces MEF2C gene expression in a protein kinase C–dependent manner. In addition, VEGF activates MEF2-dependent transcription, and this activation is regulated by several signaling pathways. Finally, MEF2C may play an important role in endothelial cell migration.
Methods
Reagents
Human recombinant VEGF, placental growth factor (PlGF), FGF-2, and erythropoietin were purchased from R&D Systems (Minneapolis, MN). Calphostin C, wortmannin, PD98059, SB203580, rottlerin, cyclosporin A, and KN93 were purchased from Calbiochem (San Diego, CA). LY333531 was purchased from A.G. Scientific, Inc. (San Diego, CA). Other chemicals and reagents were obtained from Sigma Chemical Company (St. Louis, MO) unless otherwise indicated.
Plasmids
A minimal MEF2-luciferase reporter construct (3X-MEF2-luc), in which firefly luciferase expression is driven by a thymidine kinase promoter with three upstream tandem copies of a high-affinity MEF2 binding site from the desmin gene (termed desMEF213,14), was kindly provided by Eric Olson. Plasmids encoding MEF2C mutants also were kindly provided by Eric Olson. pcDNA1-MEF2C 1 to 105 Flag encodes an MEF2C mutant consisting of amino acids 1 to 105 of MEF2C tagged with a flag sequence.15 This truncated version of MEF2C (including its DNA-binding domain but not its transcriptional activation domain) acts as a dominant-negative mutant. The dominant-negative MEF2C mutant cDNA was released from the parent plasmid by EcoRV restriction digest and was cloned into the corresponding site of pcDNA 3.1+. pcDNA1-MEF2C 1–117/VP16 encodes a chimeric protein comprising amino acids 1 to 117 of MEF2C (which includes the DNA-binding domain of MEF2C), fused to the transcriptional activation domain of the viral coactivator VP16.15 This constitutively active MEF2C is even more potent than wild-type MEF2C in activating transcription.15 The constitutively active MEF2C mutant cDNA was released from the parent plasmid by HindIII/NotI restriction digest and cloned into the corresponding site of pcDNA 3.1+.
A plasmid encoding FLAG-p38α (AF), a dominant-negative kinase inactive mutant of p38α, was a gift of Jiahuai Han.16,17 Plasmid pSRa-T286D CaMKII (constitutively active CaMKII) and pSRa-K42M/ T286D (kinase-dead CaMKII) were gifts of Leslie Griffith and Susan Birren.18–20 A cDNA encoding RCAN1/DSCR1 isoform 4 was obtained by PCR amplification and subcloned as previously described.21 Plasmid pCEFL encoding dominant-negative BMK1/ERK5 (ERK5-NH2 terminus) was kindly provided by J. Silvio Gutkind.22 The cDNAs for DN CaMK II and RCAN1/DSCR1 were liberated from the original plasmid and cloned into pCEFL, a gift of J. Silvio Gutkind.
Cell Culture
Human retinal endothelial cells (HRECs; Cell Systems, Kirkland, WA) and human umbilical vein endothelial cells (HUVECs; Clonetics, San Diego, CA) were cultured in medium (EGM2-MV; Clonetics) and used between passages 4 and 12. HRECs were cultured in fibronectin-coated dishes. Cells were grown in 5% CO2 at 37°C, and media were changed every 2 to 3 days.
Real-Time PCR Analysis
Real-time PCR was performed as previously described.23,24 Cells were harvested in reagent (Trizol; Invitrogen, Carlsbad, CA), and total RNA was isolated according to the manufacturer’s instructions. Single-stranded cDNA was synthesized from 1 μg total RNA using an oligo (dT) 18-mer as primer, and MMLV reverse transcriptase (Invitrogen) in a final reaction volume of 20 μL real-time PCR was performed (Quantitect SYBR Green PCR Kit [Qiagen, Valencia, CA] and LightCycler [Roche Diagnostics, GmbH, Germany]). The primers were hMEF2C sense 5′-GCCCTGAGTCTGAGGACAAG-3′ and antisense 5′-AGT-GAGCTGACAGGGTTGCT-3′. GAPDH was used as the reference for normalization, using hGAPDH sense primer 5′-GAGTCAACGGATTTG-GTCGT-3′ and antisense 5′-GACAAGCT TCCCGTTCTCAG-3′.
Western Blot Analysis
HRECs and HUVECs were grown to confluence in EGM2-MV medium and then starved in reduced-serum medium (OptiMEM; Invitrogen) supplemented with 0.5% FBS overnight. After treatment with VEGF for the indicated times, cells were washed with PBS and lysed in Laemmli sample buffer (Bio-Rad Laboratories, Hercules, CA). Proteins were subjected to SDS-PAGE on 10% gels and were transferred to nitrocellulose membrane (Sigma). The membranes were probed with polyclonal anti-MEF2C (Santa Cruz Biotechnology, Santa Cruz, CA), and monoclonal anti-GAPDH (Abcam, Cambridge, MA). The blots were detected with a chemiluminescent substrate (SuperSignal West Pico; Pierce Biotechnology, Rockford, IL).
Transient Transfection (Lipofection)
Cells (1.0 × 105 per well) were passaged into 12-well plates 24 hours before transfection. HRECs were cotransfected with three plasmids (0.3375 μg MEF2C mutant expression vector or control pCEFL vector, 0.1125 μg 3X-MEF2-firefly luciferase, and 50 ng pRL-SV40 containing the Renilla luciferase gene to allow normalization of firefly luciferase reporter activity) using 1.5 μL multi-component lipid-based transfection reagent (FuGENE 6; Roche Diagnostics, Indianapolis, IN) for each transfection, according to the manufacturer’s instructions. Cells were incubated with the DNA transfection reagent complex for 18 hours in growth medium and were then subjected to low serum conditions (reduced-serum medium [OptiMEM; Invitrogen] containing 0.5% FBS) for 8 hours before treatment with VEGF (25 ng/mL) for another 16 hours.
Luciferase Assay
Transfected cells were washed with PBS and lysed in 1× lysis buffer. Activities of firefly luciferase (from the 3X-MEF2-luciferase plasmid) and Renilla luciferase (from the pRL-SV40 plasmid) were measured with a reporter assay system (Dual-Luciferase; Promega, Madison, WI) according to the manufacturer’s instructions. All transfection experiments were performed in triplicate. Representative results of at least two independent experiments are shown in all cases.
Transient Transfection (Nucleofection)
For high-efficiency transfection, HUVECs were transfected by a nucleofection technique (Nucleofector Solution; Amaxa Inc., Gaithersburg, MD). HUVECs at passages 4 to 10 were used. Cells (0.5–1 × 106) at 90% confluence were trypsinized, pelleted, and resuspended in 100 μL solution for human microvascular endothelial cells (Amaxa Inc.). Cell suspensions were mixed with 5 μg highly purified plasmid DNA (EndoFree Plasmid Maxi Kit; Qiagen), and the entire suspension was transferred to an Amaxa cuvette. The cells were subjected to electroporation using the nucleofector program A-34 (Nucleofector I Device; Amaxa). Medium (EGM2-MV; Clonetics) was then added, and the cells were plated and incubated in 5% CO2 at 37°C. Transfection efficiency was confirmed after transfection by visualization of GFP (in cells transfected with a GFP-encoding plasmid) and found to be 75% to 84% efficient.
Cell Migration Assay
A modified Boyden chamber assay25,26 was used for studies of the role of MEF2C in endothelial cell migration. HUVECs were used because of their ability to achieve very high transfection efficiencies in this cell type. HUVECs were transfected by the nucleofection technique with a plasmid encoding dominant-negative MEF2C or corresponding control vector and were incubated for 24 hours Transfected cells were serum starved with 0.1% BSA in DMEM overnight, trypsinized, and seeded in DMEM 0.1% BSA at a density of 1 × 105 in 100 μL into the upper chamber of 8-μm pore size transwell membranes (Corning Costar, Cambridge, MA) precoated with type I collagen (INAMED, New York, NY) solution at a concentration of 10 μg/mL in PBS for 2 hours at room temperature. The lower chamber contained medium with or without VEGF at 25 ng/mL. After incubation at 37°C for 4 hours, the stationary cells on the top of the membrane were removed by a cotton swab, and the migrated cells on the bottom of the membrane were stained with DAPI. Eight photographs for each well were randomly taken under a microscope (Axiovert 200; Carl Zeiss, Thornwood, NY) at 20× objective, and the cells were counted using the ImageJ program (developed by Wayne Rasband, National Institutes of Health, Bethesda, MD; available at http://rsb.info.nih.gov/ij/index.html).
Statistical Analysis
All experiments were conducted at least two to three times. Results were reported as mean ± SD. An unpaired Student’s t-test was used to determine statistical significance. P < 0.05 was considered significant.
Results
VEGF Upregulates MEF2C mRNA Expression in Retinal Endothelial Cells
MEF2C is expressed in endothelial cells in vivo7 and in vitro.8 Given the important role of MEF2C in vasculogenesis and angiogenesis during vascular development,7 we were interested in determining whether MEF2C gene expression is regulated by angiogenic growth factors. For each growth factor, a concentration of 25 ng/mL—a dose at which endothelial cells exhibit a strong response to the various growth factors—was chosen. We performed real-time PCR of HRECs in the presence or absence of various growth factors. As shown in Figure 1, MEF2C mRNA expression was significantly upregulated by VEGF. In contrast, MEF2C expression was not increased by the angiogenic growth factors PlGF or FGF-2 or by EGF.
Figure 1.

Effects of different factors on MEF2C mRNA expression in endothelial cells. HRECs were treated with VEGF, PlGF, FGF-2, and EGF, each at a concentration of 25 ng/mL, for 4 hours. Total RNA was isolated, and reverse transcription was performed with 1 μg RNA. Quantitative real-time PCR was used to detect the expression of MEF2C mRNA. Values were normalized to GAPDH gene expression. Results are representative of three independent experiments, each performed with duplicate samples. CN, control. *P < 0.05 versus control.
VEGF Upregulates MEF2C mRNA and Protein Expression in a Time- and Dose-Dependent Fashion
Because VEGF displayed a dramatic upregulation of MEF2C expression, we further characterized its induction of MEF2C. As demonstrated in Figure 2A, VEGF upregulated MEF2C mRNA expression in HRECs and HUVECs in a dose-dependent manner. Upregulation was evident at a dose of 2.5 ng/mL and peaked at a dose of approximately 25 to 50 ng/mL. A time-course experiment (Fig. 2B) demonstrated that VEGF induces MEF2C RNA expression in HRECs as early as 1 hour after, with a peak effect by 4 hours of treatment. VEGF upregulation of MEF2C RNA expression persisted 24 hours after treatment. Similar results were observed with HUVECs, with peak expression within 4 hours of VEGF treatment and with a persistence of stimulation at 24 hours after treatment (data not shown). VEGF also upregulated MEF2C at the protein level. MEF2C protein expression in HRECs and HUVECs was significantly upregulated by VEGF at 4 hours and maximal at 8 hours (Fig. 2C). Of note, the immunoblot for MEF2C yielded two bands, possibly reflecting alternative splice variants of MEF2C27 or a biochemical modification such as phosphorylation28 or sumoylation,29 though this remains to be determined.
Figure 2.
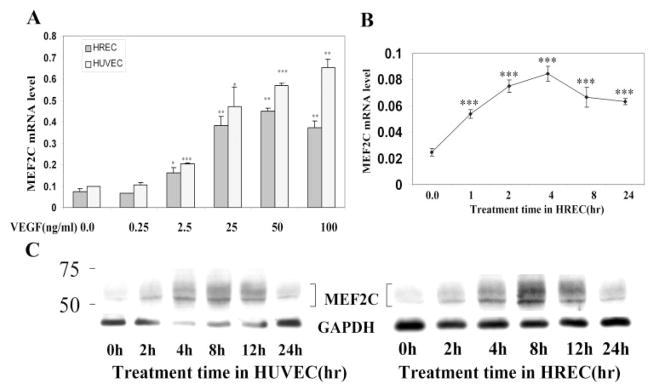
VEGF induction of MEF2C RNA and protein expression in a time- and dose-dependent manner in human endothelial cells. (A) HRECs and HUVECs were treated with VEGF at the indicated doses for 4 hours. Total RNA was isolated, and reverse transcription and quantitative real-time PCR were performed as in Figure 1. Values were normalized to GAPDH gene expression. (B) HRECs were treated with VEGF (25 ng/mL) for the indicated amount of time. Total RNA was isolated, and MEF2C expression was assessed by quantitative real-time PCR. Results are representative of three independent experiments, each performed with duplicate samples. CN, control. (C) HRECs and HUVECs were treated with VEGF (25 ng/mL) for the indicated amount of time. Cells were harvested and probed with antibody to MEF2C. The same membrane was stripped and blotted with antibody to GAPDH for normalization. Results shown are representative of three independent experiments. *P < 0.05, **P < 0.01, and ***P < 0.001 versus control.
VEGF Induction of MEF2C Expression Is Dependent on PKC
We next investigated the role of possible signaling pathways mediating VEGF induction of MEF2C. As shown in Figure 3A, the protein kinase C inhibitor calphostin C completely abrogated VEGF-induced upregulation of MEF2C RNA in HRECs. Calphostin C also significantly reduced the basal expression of MEF2C. In contrast, the phosphatidylinositol 3-kinase inhibitor wortmannin and the MEK inhibitor PD98059 had no significant effect on basal or VEGF-induced expression of MEF2C. Pretreatment of endothelial cells with PD98059 completely blocked VEGF-induced phosphorylation of ERK1/2, and pretreatment with wortmannin inhibited basal and VEGF-induced phosphorylation of Akt on immunoblot analysis (data not shown). These results demonstrate that VEGF-induced MEF2C expression in endothelial cells is dependent on protein kinase C signaling. Pretreatment of HRECs with the PKC-β selective inhibitor LY333531 or the PKC-δ selective inhibitor rottlerin partially inhibited VEGF induction of MEF2C RNA (Fig. 3A). Western blot analysis also demonstrated complete inhibition of VEGF induction of MEF2C protein expression by calphostin C, whereas PD98059 had no effect (Fig. 3B). In addition, selective inhibition of PKC-β and of PKC-δ partially inhibited this induction of MEF2C protein (Fig. 3C).
Figure 3.

Induction of MEF2C expression by VEGF is dependent on protein kinase C. (A) HRECs were pretreated with 100 nM wortmannin, 1 μM calphostin C, 2 μM LY333531, 5 μM rottlerin, and 50 μM PD98059 for 30 minutes, followed by treatment with VEGF (25 ng/mL) for 4 hours. Total RNA was isolated, and quantitative real-time PCR was used to measure the expression of MEF2C mRNA. Values were normalized to GAPDH gene expression. Results are representative of at least three independent experiments, each performed with duplicate samples. (B) HRECs were pretreated with calphostin C (1 μM) and PD98059 (50 μM) for 30 minutes, followed by treatment with VEGF (25 ng/mL) for 8 hours. Cells were harvested and probed with antibody to MEF2C. The same membrane was stripped and blotted with antibody to GAPDH for normalization. Results shown are representative of three independent experiments. (C) HRECs were pretreated with calphostin C, LY333531 (2 μM), and rottlerin (5 μM) for 30 minutes, then treated with VEGF (25 ng/mL) for 8 hours. Cells were then harvested for Western blot, as described. *P < 0.05, **P < 0.01, and ***P < 0.001 versus control.
VEGF Stimulates MEF2-Driven Gene Expression in Retinal Endothelial Cells
We next examined whether VEGF can activate MEF2C-dependent transcription in endothelial cells. To explore this issue, we used a minimal MEF2-luciferase reporter construct, in which firefly luciferase gene expression is driven by a thymidine kinase promoter with three upstream tandem copies of a canonical MEF2 binding site (3X-MEF2-luc). We evaluated the induction of MEF2-driven luciferase by various growth factors using a dose at which endothelial cells exhibit a strong response to the respective growth factor. As shown in Figure 4A, VEGF treatment significantly increased MEF2-driven luciferase activity in HRECs. In contrast, MEF2-driven luciferase activity was not stimulated by placental growth factor, fibroblast growth factor-2, or erythropoietin (Fig. 4A). Cotransfection of an expression plasmid encoding a constitutively active form of MEF2C independently increased luciferase activity in this system (Fig. 4B), as expected. Cotransfection of an expression plasmid encoding a dominant-negative mutant of MEF2C decreased basal luciferase activity and abrogated VEGF induction of MEF2-driven luciferase activity in this system (Fig. 4B). VEGF induced MEF2-driven luciferase activity in a dose-dependent manner, with a significant effect at 2.5 ng/mL and a maximal effect by approximately 25 ng/mL (Fig. 4C). Interestingly, the induction of MEF2-driven luciferase occurred quickly after VEGF treatment, with a statistically significant increase by 4 hours after VEGF treatment (Fig. 4D).
Figure 4.

VEGF stimulates MEF2C-dependent gene expression. (A) HRECs were cotransfected with a minimal 3X-MEF2-firefly Luc-reporter and internal control pRL-SV40 containing the Renilla luciferase gene. The 3×-MEF2-firefly luciferase contains three copies of a high-affinity MEF2-binding site from the desmin promoter (desMEF2). After overnight incubation, cells were treated with VEGF (25 ng/mL), PlGF (25 ng/mL), FGF-2 (25 ng/mL), or erythropoietin (10 U/mL), then lysed for measurements of firefly and Renilla luciferase activity. All results are corrected for variations in transfection efficiency by normalization to expression of the cotransfected pRL-SV40 plasmid. Data are expressed relative to the luciferase activity observed in the control state (no VEGF treatment). (B) HRECs were cotransfected with 3X-MEF2-Luc reporter, internal control pRL-SV40 containing the Renilla luciferase gene, and MEF2C mutant expressing plasmid or control vector. After overnight incubation, cells were treated with VEGF (25 ng/mL) for 16 hours, then lysed for measurement of luciferase activity. *P < 0.05 compared with control pcDNA3.1. **P < 0.01 compared with control pcDNA3.1 in the presence of VEGF. (C) HRECs were cotransfected, as described, with 3X-MEF2-Luc and pRL-SV40. Cells were treated with the indicated doses of VEGF for 16 hours and then assayed for luciferase activity. (D) HRECs were cotransfected with 3X-MEF2-Luc and pRL-SV40. Cells were treated with VEGF (25 ng/mL) for the indicated times and then assayed for luciferase activity. *P < 0.05, **P < 0.01, and ***P < 0.001 versus control for (A), (C), and (D).
VEGF Stimulation of MEF2-Driven Transcription Is Dependent on Multiple Pathways
We were interested in determining the signaling pathways that play important roles in mediating VEGF induction of MEF2-driven luciferase expression in endothelial cells. We investigated in particular those pathways known to be involved in regulating MEF2C in other cell types, including p38 MAPK,30 BMK1/ERK5,31 calcineurin,14,32 and calmodulin-dependent protein kinase.14,33 We first evaluated the possible roles of these pathways using pharmacologic inhibitors (Fig. 5). Endothelial cells were treated with inhibitors targeting calcineurin (cyclosporin A), p38 MAPK (SB203580), calmodulin-dependent kinase (KN93), protein kinase C (calphostin C), and MEK (PD98059) for 30 minutes, followed by treatment with VEGF for 16 hours. The addition of cyclosporin, SB203580, and KN93 all significantly inhibited VEGF stimulation of MEF2-driven transcription (Fig. 5A), and cyclosporin in particular nearly abrogated this VEGF stimulation. In addition, calphostin treatment abrogated VEGF stimulation of MEF2-driven transcription, whereas PD98059 had no effect (Fig. 5B). Pretreatment of endothelial cells with PD98059 completely abrogated VEGF-induced phosphorylation of ERK1/2 (data not shown).
Figure 5.

Pharmacologic inhibition of multiple signaling pathways inhibits VEGF induction of MEF2-dependent transcription. (A) HRECs were cotransfected with 3X-MEF2-Luc and pRL-SV40, as in Figure 4A. Thirty minutes before VEGF addition, cells were treated with cyclosporin (1 μM), KN93 (3 μM), or SB203580 (20 μM). Cells were harvested 16 hours after VEGF treatment and assayed for luciferase activity. Data are presented as in Figure 4. There was no statistically significant difference in relative luciferase activity between control and cyclosporin, KN-93, or SB203580. (B) Thirty minutes before VEGF addition, cells were treated with calphostin C (1 μM) or PD98059 (20 μ M). Luciferase activity was assayed 16 hours after VEGF treatment. *P < 0.05 and ***P < 0.001 versus control.
As an additional approach to delineate the involvement of specific signaling pathways, we performed transfection experiments expressing dominant-negative mutants or molecular regulators of various signaling molecules. Of the signaling molecules known to regulate MEF2, VEGF is known to activate p38,34 BMK1/ERK 5,35 and calcineurin.36,37 Transfection of a dominant-negative mutant of p38 MAPK partially inhibited VEGF activation of MEF2-luciferase (Fig. 6A). Dominant-negative BMK1/ERK5 had no effect on VEGF stimulation (Figs. 6A, 6B).
Figure 6.

VEGF induction of MEF2-dependent transcription is inhibited by modulation of calcineurin, CaMKII, and p38 MAPK. (A) HRECs were cotransfected with 3X-MEF2-Luc reporter, internal control pRL-SV40 containing the Renilla luciferase gene, and plasmid encoding DN-BMK1/ERK5 (ERK5-NH2 terminus), DN-p38α (kinase-inactive), RCAN1/DSCR1, or empty vector pCEFL. After overnight incubation, cells were treated with VEGF (25 ng/mL) for 16 hours, then lysed for measurement of luciferase activity. Data are presented as in Figure 4. (B) HRECs were cotransfected with 3X-MEF2-Luc, pRL-SV40, and plasmid encoding DN-CaMKII (kinase-dead), DN-BMK1, or empty vector pCEFL. Sixteen hours after VEGF treatment, cells were assayed for luciferase activity. *P < 0.05 and **P < 0.01 versus control.
In addition to p38 MAPK, we investigated the modulation of calcineurin because pharmacologic blockage with cyclosporin inhibited VEGF activation of MEF2-luciferase (Fig. 5A). Therefore, we transfected an expression plasmid for RCAN1/ DSCR1,38 which is known to function as an endogenous inhibitor of calcineurin in various cell types,39–41 including endothelial cells.21,42,43 Transfection of RCAN1/DSCR1 significantly inhibited VEGF activation of MEF2-luciferase (Fig. 6A).
In light of this result confirming the role of calcineurin in the VEGF regulation of MEF2, we investigated another calcium transducer, calcium-dependent calmodulin-dependent kinase (CaMK). Transfection of a plasmid encoding dominant-negative CaMKII significantly inhibited VEGF activation of MEF2-luciferase (Fig. 6B). Transfection of constitutive-active CaMKII resulted in a twofold increase in luciferase activity compared with control (data not shown).
MEF2 Modulates Endothelial Cell Migration
MEF2C is thought to play a major role in vascularization, especially during development.7 We were interested in examining whether MEF2C might play a role in VEGF-induced angiogenesis using endothelial cell migration as an end point. For these studies, we used HUVECs because of their ability to achieve high transfection efficiencies (greater than 80% as assessed by transfection with a GFP-expressing plasmid) in this cell type. Endothelial cells were transfected with a plasmid encoding dominant-negative MEF2C (Fig. 7A) or a corresponding control vector. After transfection, cells were seeded for modified Boyden chamber assay. VEGF was used as a chemotactic agent for 4 hours and significantly stimulated endothelial cell migration, consistent with previous findings25,26 (Fig. 7). Transfection with dominant-negative MEF2C (DN-MEF2C) significantly reduced the VEGF-stimulated migration of endothelial cells.
Figure 7.
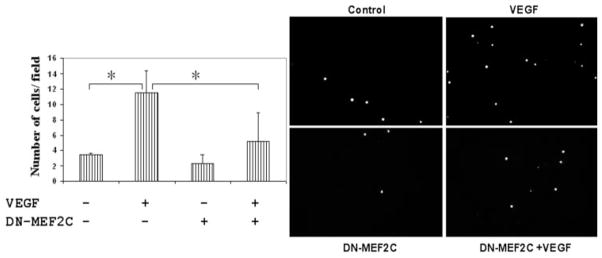
Expression of a dominant-negative MEF2C mutant inhibits VEGF-stimulated endothelial cell migration. (A) A modified Boyden chamber assay25,26 was used to study VEGF stimulation of endothelial cell migration. HUVECs were transfected with a plasmid encoding dominant-negative MEF2C or corresponding control vector. Transfected cells were seeded into the upper chamber of 8-μm pore size transwells. The lower chamber contained medium with or without VEGF at 25 ng/mL. After incubation at 37°C for 4 hours, migrated cells were counted in eight different fields (20× objective). Data are presented as mean ± SD (B) Representative figures for modified Boyden chamber migration assay. *P < 0.05 versus control plasmid pcDNA 3.1 + VEGF. **P < 0.001 versus control plasmid without VEGF. Results shown are representative of five independent experiments.
Discussion
Although the major upstream signaling pathways mediating VEGF proangiogenic effects on endothelial cells have been well characterized, relatively less is known regarding the transcription factors that mediate these effects. MEF2C has been demonstrated to play a critical role in vasculogenesis and angiogenesis during vascular development. Mice with targeted deletion of the MEF2C gene exhibit severe vascular abnormalities7,10 and a phenotype similar to that of mice lacking VEGF or Flt-1.7 Importantly, transcripts for VEGF and its receptors, Flt-1 and Flk-1, were detected at similar levels in wild-type and MEF2C mutant embryos, suggesting that the vascular defects in MEF2C mutant embryos did not arise from downregulation of these molecules.7
We were, therefore, interested in the concept that MEF2C might mediate, at least in part, the angiogenic effects of VEGF on endothelial cells. We initially looked for evidence that VEGF might regulate the expression of MEF2C. We found that VEGF strongly induces the expression of MEF2C RNA and protein, and this induction is completely abrogated by the inhibition of PKC. We were interested in testing the possible effects of specific PKC isoforms, particularly PKC-β given its known effects in mediating VEGF action in retinal endothelial cells and its effects on retinal neovascularization.44 We found that VEGF induction of MEF2C expression was significantly reduced by the inhibition of PKC-β and PKC-δ. To our knowledge, this is the first example of a cell-extrinsic signal or growth factor that induces MEF2C expression in any cell type. In contrast, the cell-intrinsic regulation of MEF2C expression has received greater attention, especially in the context of skeletal muscle development. The basic helix-loop-helix transcription factor myogenin has been shown to upregulate MEF2C expression in vitro,45,46 and myogenic basic helix-loop-helix proteins have been shown to activate MEF2C expression during skeletal muscle development.47,48 MEF2C, in turn, participates in the developmental program for myogenesis. It is conceivable that VEGF induction of MEF2C expression in endothelial cells plays an analogous role in angiogenesis.
MEF2C is known to be regulated at the protein level in multiple cell types by several signaling pathways. One important mechanism of regulation is activation of its transcriptional activation domain, notably by p38 MAPK and BMK1/ERK5 signaling. p38 MAPK is thought to activate MEF2C through phosphorylation of the Thr293, Thr300, and Ser387 sites within the MEF2C transactivation domain,30 whereas BMK1/ ERK5 enhances the transactivating activity of MEF2C by phosphorylating Ser 387.31 Because VEGF is known to activate the phosphorylation of p38 MAPK34 and BMK1/ERK5,35 we investigated whether these pathways play a role in VEGF stimulation of MEF2-driven transcription. For our experiments investigating MEF2-driven transcription, we used a 3×-MEF2 reporter approach that is widely used as a readout of cellular MEF2 activity both in vitro14 and in vivo.13 This reporter construct allows the investigation of transcription specifically driven by MEF2, in isolation from other transcription factors that might also be induced by VEGF. We found that the modulation of p38 either by pharmacologic inhibition or by transfection of dominant-negative p38 reduced VEGF stimulation of MEF2-driven transcription, whereas the inhibition of BMK1/ERK5 using a dominant-negative mutant did not.
In addition to p38 MAPK, we found that VEGF activation of MEF2-driven transcription is strongly dependent on the calcium-dependent protein phosphatase calcineurin because this activation was strongly inhibited by cyclosporin and by the calcineurin regulator, RCAN1/DSCR1. With regard to calcineurin regulation of transcription factors, the NFAT family is a well-known target of calcineurin, and NFAT transcription factors translocate to the nucleus in response to dephosphorylation by calcineurin.49 Of note, VEGF induces NFAT dephosphorylation and nuclear translocation in endothelial cells, and this induction is blocked by the calcineurin inhibitor cyclosporin.50 Furthermore, VEGF activation of NFAT in endothelial cells results in the induction of gene expression of DSCR1 (recently renamed RCAN138), which acts as a feedback inhibitor of calcineurin.21,42,43 In nonendothelial cells, calcineurin activates MEF2 using several potential mechanisms.28 Calcineurin dephosphorylates MEF2 in neurons51 and skeletal muscle.52 Calcineurin-mediated dephosphorylation of MEF2 transcription factors (specifically, Ser396 in MEF2C) inhibits sumoylation of a nearby lysine residue,53,54 thereby inhibiting repression of MEF2C.29 An additional mechanism by which calcineurin can activate MEF2 is the recruitment of NFAT, which associates with MEF2 and recruits the p300 coactivator.32 It will be of great interest to determine the specific mechanism(s) by which calcineurin activates MEF2C in endothelial cells.
The role of calcineurin in mediating VEGF activation of MEF2 prompted us to investigate the calcium-dependent protein kinase, calmodulin-dependent protein kinase (CaMK). CaMK has been demonstrated to activate MEF2 in nonendothelial cells by releasing it from class 2 histone deacetylases (HDACs), well-known inhibitors of MEF2.55 Although VEGF has not been reported to regulate CaMK in endothelial cells, CaMKII expression has been detected in endothelial cells,56 and CaMKII has been found to play a regulatory role in eNOS expression57 and nitric oxide synthesis58 in endothelial cells. Our results demonstrated that VEGF activation of MEF2-driven transcription is indeed partially dependent on CaMK because this activation was inhibited by the CaMK inhibitor KN93 and by a dominant-negative mutant of CaMKII. It will be interesting to determine whether CaMKII activates MEF2C in endothelial cells through the regulation of HDACs or through an alternative mechanism.
Because our studies demonstrate strong evidence that VEGF induces MEF2C expression and MEF2-dependent transcription, we sought to determine the potential functional effect on endothelial cells. Endothelial cell migration is an important facet of the angiogenic process. Our experiments showed that a dominant-negative mutant of MEF2C significantly inhibited VEGF-stimulated retinal endothelial cell migration (Fig. 7). We consider this to be particularly significant given the relatively short period (4 hours) of VEGF treatment in this assay. In this experiment, there was also a trend toward a reduction in basal migration (i.e., in the absence of VEGF) in endothelial cells transfected with dominant-negative MEF2C compared with control plasmid, though this did not reach statistical significance. In any event, these results indicate that MEF2 had a significant effect in modulating endothelial cell migration in this assay system. It will be of great interest to investigate other functional end points, including endothelial cell proliferation, survival, and tube formation.
VEGF has pleiotropic effects on endothelial cells resulting from its ability to activate a complex network of signaling pathways.4 In the context of this study, VEGF activation of MEF2C appears to be dependent on several different signaling pathways, suggesting a role for MEF2C as an important downstream target of these pathways. p38 MAPK and calcineurin have been proposed as important mediators of VEGF-induced migration,34,59 and our results suggest that MEF2C might serve as one of the downstream effectors of these two signaling molecules. Clearly, further studies will be necessary to fully dissect the role and importance of MEF2C in modulating the effects of these pathways.
Based on the current understanding of MEF2C and our results in this study, we propose a schematic profile of VEGF regulation of MEF2C in endothelial cells (Fig. 8). We hypothesize that VEGF induces MEF2C in endothelial cells at two levels. First, VEGF induces the expression of MEF2C RNA (and, therefore, of protein) in a PKC-dependent fashion and possibly by stimulation of transcription factors including Ets-1. In addition, VEGF increases the activity of MEF2C protein by activating signaling molecules, including p38 MAPK, calcineurin, and calmodulin-dependent protein kinase II. Activated MEF2C then plays a role in modulating the effects of VEGF on endothelial cells (including such activities as migration).
Figure 8.

Hypothesized schematic profile of MEF2C and its regulation by VEGF in endothelial cells. PKC, protein kinase C; p38, p38 MAPK; CaN, calcineurin; CaMK II, calmodulin-dependent protein kinase II; EC, endothelial cell.
Although our results suggest that MEF2C in particular may be important for the effects of VEGF, we cannot rule out the possible involvement of other MEF2 family members. In addition, it is highly likely that other transcription factors also play critical roles in the angiogenic actions of VEGF. VEGF has been shown to induce Egr-1 expression in endothelial cells,60 and this transcription factor plays a major role in VEGF induction of tissue factor.61 VEGF induction of Ets-1 expression is important for retinal angiogenesis.12 VEGF activates NFAT through dephosphorylation and nuclear translocation,50 and NFAT appears to have a role in VEGF-induced angiogenesis through Cox-2 induction.62
Nevertheless, VEGF regulation of MEF2C expression and MEF2-dependent transcription suggest an important role for this transcription factor in VEGF action. The stimulation of MEF2C expression and MEF2-driven transcription was specific to VEGF because we did not find evidence of stimulation by other proangiogenic growth factors. It will be important to delineate the spectrum of MEF2C functional roles in VEGF-induced angiogenesis and to elucidate its transcriptional program and critical target genes in endothelial cells.
Acknowledgments
Supported by National Institutes of Health Grants EY00398 and EY018138, the Juvenile Diabetes Research Foundation, and Richard and Sandra Forsythe/Forsythe Technology, Inc. EJD is a recipient of an RPB Career Development Award.
The authors thank Eric Olson, Jiahuai Han, Leslie Griffith, Susan Birren, and J. Silvio Gutkind for providing plasmids, and Chaitali Sarkar for technical assistance.
Footnotes
Disclosure: D. Maiti, None; Z. Xu, None; E.J. Duh, None
References
- 1.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–676. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 2.Caldwell RB, Bartoli M, Behzadian MA, et al. Vascular endothelial growth factor and diabetic retinopathy: pathophysiological mechanisms and treatment perspectives. Diabetes Metab Res Rev. 2003;19:442–455. doi: 10.1002/dmrr.415. [DOI] [PubMed] [Google Scholar]
- 3.Cross MJ, Dixelius J, Matsumoto T, Claesson-Welsh L. VEGF-receptor signal transduction. Trends Biochem Sci. 2003;28:488–494. doi: 10.1016/S0968-0004(03)00193-2. [DOI] [PubMed] [Google Scholar]
- 4.Zachary I. VEGF signalling: integration and multi-tasking in endothelial cell biology. Biochem Soc Trans. 2003;31:1171–1177. doi: 10.1042/bst0311171. [DOI] [PubMed] [Google Scholar]
- 5.Shore P, Sharrocks AD. The MADS-box family of transcription factors. Eur J Biochem. 1995;229:1–13. doi: 10.1111/j.1432-1033.1995.tb20430.x. [DOI] [PubMed] [Google Scholar]
- 6.Olson EN, Perry M, Schulz RA. Regulation of muscle differentiation by the MEF2 family of MADS box transcription factors. Dev Biol. 1995;172:2–14. doi: 10.1006/dbio.1995.0002. [DOI] [PubMed] [Google Scholar]
- 7.Lin Q, Lu J, Yanagisawa H, et al. Requirement of the MADS-box transcription factor MEF2C for vascular development. Development. 1998;125:4565–4574. doi: 10.1242/dev.125.22.4565. [DOI] [PubMed] [Google Scholar]
- 8.Hosking BM, Wang SC, Chen SL, Penning S, Koopman P, Muscat GE. SOX18 directly interacts with MEF2C in endothelial cells. Biochem Biophys Res Commun. 2001;287:493–500. doi: 10.1006/bbrc.2001.5589. [DOI] [PubMed] [Google Scholar]
- 9.Lin Q, Schwarz J, Bucana C, Olson EN. Control of mouse cardiac morphogenesis and myogenesis by transcription factor MEF2C. Science. 1997;276:1404–1407. doi: 10.1126/science.276.5317.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bi W, Drake CJ, Schwarz JJ. The transcription factor MEF2C-null mouse exhibits complex vascular malformations and reduced cardiac expression of angiopoietin 1 and VEGF. Dev Biol. 1999;211:255–267. doi: 10.1006/dbio.1999.9307. [DOI] [PubMed] [Google Scholar]
- 11.De Val S, Anderson JP, Heidt AB, Khiem D, Xu SM, Black BL. MEF2C is activated directly by Ets transcription factors through an evolutionarily conserved endothelial cell-specific enhancer. Dev Biol. 2004;275:424–434. doi: 10.1016/j.ydbio.2004.08.016. [DOI] [PubMed] [Google Scholar]
- 12.Watanabe D, Takagi H, Suzuma K, et al. Transcription factor Ets-1 mediates ischemia- and vascular endothelial growth factor-dependent retinal neovascularization. Am J Pathol. 2004;164:1827–1835. doi: 10.1016/S0002-9440(10)63741-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Naya FJ, Wu C, Richardson JA, Overbeek P, Olson EN. Transcriptional activity of MEF2 during mouse embryogenesis monitored with a MEF2-dependent transgene. Development. 1999;126:2045–2052. doi: 10.1242/dev.126.10.2045. [DOI] [PubMed] [Google Scholar]
- 14.Wu H, Naya FJ, McKinsey TA, et al. MEF2 responds to multiple calcium-regulated signals in the control of skeletal muscle fiber type. EMBO J. 2000;19:1963–1973. doi: 10.1093/emboj/19.9.1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Molkentin JD, Black BL, Martin JF, Olson EN. Mutational analysis of the DNA binding, dimerization, and transcriptional activation domains of MEF2C. Mol Cell Biol. 1996;16:2627–2636. doi: 10.1128/mcb.16.6.2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao M, New L, Kravchenko VV, et al. Regulation of the MEF2 family of transcription factors by p38. Mol Cell Biol. 1999;19:21–30. doi: 10.1128/mcb.19.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ge B, Gram H, Di Padova F, et al. MAPKK-independent activation of p38α mediated by TAB1-dependent autophosphorylation of p38α. Science. 2002;295:1291–1294. doi: 10.1126/science.1067289. [DOI] [PubMed] [Google Scholar]
- 18.Waldmann R, Hanson PI, Schulman H. Multifunctional Ca2+/cal-modulin-dependent protein kinase made Ca2+ independent for functional studies. Biochemistry. 1990;29:1679–1684. doi: 10.1021/bi00459a002. [DOI] [PubMed] [Google Scholar]
- 19.Hanson PI, Meyer T, Stryer L, Schulman H. Dual role of calmodulin in autophosphorylation of multifunctional CaM kinase may underlie decoding of calcium signals. Neuron. 1994;12:943–956. doi: 10.1016/0896-6273(94)90306-9. [DOI] [PubMed] [Google Scholar]
- 20.Slonimsky JD, Mattaliano MD, Moon JI, Griffith LC, Birren SJ. Role for calcium/calmodulin-dependent protein kinase II in the p75-mediated regulation of sympathetic cholinergic transmission. Proc Natl Acad Sci U S A. 2006;103:2915–2919. doi: 10.1073/pnas.0511276103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yao YG, Duh EJ. VEGF selectively induces Down syndrome critical region 1 gene expression in endothelial cells: a mechanism for feedback regulation of angiogenesis? Biochem Biophys Res Commun. 2004;321:648–656. doi: 10.1016/j.bbrc.2004.06.176. [DOI] [PubMed] [Google Scholar]
- 22.Chayama K, Papst PJ, Garrington TP, et al. Role of MEKK2-MEK5 in the regulation of TNF-α gene expression and MEKK2-MKK7 in the activation of c-Jun N-terminal kinase in mast cells. Proc Natl Acad Sci U S A. 2001;98:4599–4604. doi: 10.1073/pnas.081021898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yao YG, Yang HS, Cao Z, Danielsson J, Duh EJ. Upregulation of placental growth factor by vascular endothelial growth factor via a post-transcriptional mechanism. FEBS Lett. 2005;579:1227–1234. doi: 10.1016/j.febslet.2005.01.017. [DOI] [PubMed] [Google Scholar]
- 24.Xu Z, Yu Y, Duh EJ. Vascular endothelial growth factor upregulates expression of ADAMTS1 in endothelial cells through protein kinase C signaling. Invest Ophthalmol Vis Sci. 2006;47:4059–4066. doi: 10.1167/iovs.05-1528. [DOI] [PubMed] [Google Scholar]
- 25.Meadows KN, Bryant P, Pumiglia K. Vascular endothelial growth factor induction of the angiogenic phenotype requires Ras activation. J Biol Chem. 2001;276:49289–49298. doi: 10.1074/jbc.M108069200. [DOI] [PubMed] [Google Scholar]
- 26.Xu Z, Maiti D, Kisiel W, Duh EJ. Tissue factor pathway inhibitor-2 is upregulated by vascular endothelial growth factor and suppresses growth factor-induced proliferation of endothelial cells. Arterioscler Thromb Vasc Biol. 2006;26:2819–2825. doi: 10.1161/01.ATV.0000248731.55781.87. [DOI] [PubMed] [Google Scholar]
- 27.Zhu B, Gulick T. Phosphorylation and alternative pre-mRNA splicing converge to regulate myocyte enhancer factor 2C activity. Mol Cell Biol. 2004;24:8264–8275. doi: 10.1128/MCB.24.18.8264-8275.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McKinsey TA, Zhang CL, Olson EN. MEF2: a calcium-dependent regulator of cell division, differentiation and death. Trends Biochem Sci. 2002;27:40–47. doi: 10.1016/s0968-0004(01)02031-x. [DOI] [PubMed] [Google Scholar]
- 29.Kang J, Gocke CB, Yu H. Phosphorylation-facilitated sumoylation of MEF2C negatively regulates its transcriptional activity. BMC Biochem. 2006;7:5. doi: 10.1186/1471-2091-7-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Han J, Jiang Y, Li Z, Kravchenko VV, Ulevitch RJ. Activation of the transcription factor MEF2C by the MAP kinase p38 in inflammation. Nature. 1997;386:296–299. doi: 10.1038/386296a0. [DOI] [PubMed] [Google Scholar]
- 31.Kato Y, Kravchenko VV, Tapping RI, Han J, Ulevitch RJ, Lee JD. BMK1/ERK5 regulates serum-induced early gene expression through transcription factor MEF2C. EMBO J. 1997;16:7054–7066. doi: 10.1093/emboj/16.23.7054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Youn HD, Chatila TA, Liu JO. Integration of calcineurin and MEF2 signals by the coactivator p300 during T-cell apoptosis. EMBO J. 2000;19:4323–4331. doi: 10.1093/emboj/19.16.4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lu J, McKinsey TA, Nicol RL, Olson EN. Signal-dependent activation of the MEF2 transcription factor by dissociation from histone deacetylases. Proc Natl Acad Sci U S A. 2000;97:4070–4075. doi: 10.1073/pnas.080064097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rousseau S, Houle F, Landry J, Huot J. p38 MAP kinase activation by vascular endothelial growth factor mediates actin reorganization and cell migration in human endothelial cells. Oncogene. 1997;15:2169–2177. doi: 10.1038/sj.onc.1201380. [DOI] [PubMed] [Google Scholar]
- 35.Hayashi M, Kim SW, Imanaka-Yoshida K, et al. Targeted deletion of BMK1/ERK5 in adult mice perturbs vascular integrity and leads to endothelial failure. J Clin Invest. 2004;113:1138–1148. doi: 10.1172/JCI19890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu D, Jia H, Holmes DI, Stannard A, Zachary I. Vascular endothelial growth factor-regulated gene expression in endothelial cells: KDR-mediated induction of Egr3 and the related nuclear receptors Nur77, Nurr1, and Nor1. Arterioscler Thromb Vasc Biol. 2003;23:2002–2007. doi: 10.1161/01.ATV.0000098644.03153.6F. [DOI] [PubMed] [Google Scholar]
- 37.Johnson EN, Lee YM, Sander TL, et al. NFATc1 mediates vascular endothelial growth factor-induced proliferation of human pulmonary valve endothelial cells. J Biol Chem. 2003;278:1686–1692. doi: 10.1074/jbc.M210250200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Davies KJ, Ermak G, Rothermel BA, et al. Renaming the DSCR1/ Adapt78 gene family as RCAN: regulators of calcineurin. FASEB J. 2007;21:3023–3028. doi: 10.1096/fj.06-7246com. [DOI] [PubMed] [Google Scholar]
- 39.Rothermel B, Vega RB, Yang J, Wu H, Bassel-Duby R, Williams RS. A protein encoded within the Down syndrome critical region is enriched in striated muscles and inhibits calcineurin signaling. J Biol Chem. 2000;275:8719–8725. doi: 10.1074/jbc.275.12.8719. [DOI] [PubMed] [Google Scholar]
- 40.Rothermel BA, McKinsey TA, Vega RB, et al. Myocyte-enriched calcineurin-interacting protein, MCIP1, inhibits cardiac hypertrophy in vivo. Proc Natl Acad Sci U S A. 2001;98:3328–3333. doi: 10.1073/pnas.041614798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang J, Rothermel B, Vega RB, et al. Independent signals control expression of the calcineurin inhibitory proteins MCIP1 and MCIP2 in striated muscles. Circ Res. 2000;87:E61–E68. doi: 10.1161/01.res.87.12.e61. [DOI] [PubMed] [Google Scholar]
- 42.Hesser BA, Liang XH, Camenisch G, et al. Down syndrome critical region protein 1 (DSCR1), a novel VEGF target gene that regulates expression of inflammatory markers on activated endothelial cells. Blood. 2004;104:149–158. doi: 10.1182/blood-2004-01-0273. [DOI] [PubMed] [Google Scholar]
- 43.Minami T, Horiuchi K, Miura M, et al. Vascular endothelial growth factor- and thrombin-induced termination factor, Down syndrome critical region-1, attenuates endothelial cell proliferation and angiogenesis. J Biol Chem. 2004;279:50537–50554. doi: 10.1074/jbc.M406454200. [DOI] [PubMed] [Google Scholar]
- 44.Suzuma K, Takahara N, Suzuma I, et al. Characterization of protein kinase C beta isoform’s action on retinoblastoma protein phosphorylation, vascular endothelial growth factor-induced endothelial cell proliferation, and retinal neovascularization. Proc Natl Acad Sci U S A. 2002;99:721–726. doi: 10.1073/pnas.022644499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ridgeway AG, Wilton S, Skerjanc IS. Myocyte enhancer factor 2C and myogenin upregulate each other’s expression and induce the development of skeletal muscle in P19 cells. J Biol Chem. 2000;275:41–46. doi: 10.1074/jbc.275.1.41. [DOI] [PubMed] [Google Scholar]
- 46.Rogerson PJ, Jamali M, Skerjanc IS. The C-terminus of myogenin, but not MyoD, targets upregulation of MEF2C expression. FEBS Lett. 2002;524:134–138. doi: 10.1016/s0014-5793(02)03024-7. [DOI] [PubMed] [Google Scholar]
- 47.Wang DZ, Valdez MR, McAnally J, Richardson J, Olson EN. The Mef2c gene is a direct transcriptional target of myogenic bHLH and MEF2 proteins during skeletal muscle development. Development. 2001;128:4623–4633. doi: 10.1242/dev.128.22.4623. [DOI] [PubMed] [Google Scholar]
- 48.Dodou E, Xu SM, Black BL. mef2c is activated directly by myogenic basic helix-loop-helix proteins during skeletal muscle development in vivo. Mech Dev. 2003;120:1021–1032. doi: 10.1016/s0925-4773(03)00178-3. [DOI] [PubMed] [Google Scholar]
- 49.Crabtree GR. Generic signals and specific outcomes: signaling through Ca2+, calcineurin, and NF-AT. Cell. 1999;96:611–614. doi: 10.1016/s0092-8674(00)80571-1. [DOI] [PubMed] [Google Scholar]
- 50.Armesilla AL, Lorenzo E, Gomez del Arco P, Martinez-Martinez S, Alfranca A, Redondo JM. Vascular endothelial growth factor activates nuclear factor of activated T cells in human endothelial cells: a role for tissue factor gene expression. Mol Cell Biol. 1999;19:2032–2043. doi: 10.1128/mcb.19.3.2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mao Z, Wiedmann M. Calcineurin enhances MEF2 DNA binding activity in calcium-dependent survival of cerebellar granule neurons. J Biol Chem. 1999;274:31102–31107. doi: 10.1074/jbc.274.43.31102. [DOI] [PubMed] [Google Scholar]
- 52.Wu H, Rothermel B, Kanatous S, et al. Activation of MEF2 by muscle activity is mediated through a calcineurin-dependent pathway. EMBO J. 2001;20:6414–6423. doi: 10.1093/emboj/20.22.6414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gregoire S, Tremblay AM, Xiao L, et al. Control of MEF2 transcriptional activity by coordinated phosphorylation and sumoylation. J Biol Chem. 2006;281:4423–4433. doi: 10.1074/jbc.M509471200. [DOI] [PubMed] [Google Scholar]
- 54.Shalizi A, Gaudilliere B, Yuan Z, et al. A calcium-regulated MEF2 sumoylation switch controls postsynaptic differentiation. Science. 2006;311:1012–1017. doi: 10.1126/science.1122513. [DOI] [PubMed] [Google Scholar]
- 55.McKinsey TA, Zhang CL, Lu J, Olson EN. Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature. 2000;408:106–111. doi: 10.1038/35040593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Krizbai I, Deli M, Lengyel I, et al. In situ hybridization with digoxigenin labeled oligonucleotide probes: detection of CAMK-II gene expression in primary cultures of cerebral endothelial cells. Neurobiology (Bp) 1993;1:235–240. [PubMed] [Google Scholar]
- 57.Li H, Burkhardt C, Heinrich UR, Brausch I, Xia N, Forstermann U. Histamine upregulates gene expression of endothelial nitric oxide synthase in human vascular endothelial cells. Circulation. 2003;107:2348–2354. doi: 10.1161/01.CIR.0000066697.19571.AF. [DOI] [PubMed] [Google Scholar]
- 58.Schneider JC, El Kebir D, Chereau C, et al. Involvement of Ca2+/ calmodulin-dependent protein kinase II in endothelial NO production and endothelium-dependent relaxation. Am J Physiol Heart Circ Physiol. 2003;284:H2311–H2319. doi: 10.1152/ajpheart.00932.2001. [DOI] [PubMed] [Google Scholar]
- 59.Rafiee P, Heidemann J, Ogawa H, et al. Cyclosporin A differentially inhibits multiple steps in VEGF induced angiogenesis in human microvascular endothelial cells through altered intracellular signaling. Cell Commun Signal. 2004;2:3. doi: 10.1186/1478-811X-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lucerna M, Mechtcheriakova D, Kadl A, et al. NAB2, a corepressor of EGR-1, inhibits vascular endothelial growth factor-mediated gene induction and angiogenic responses of endothelial cells. J Biol Chem. 2003;278:11433–11440. doi: 10.1074/jbc.M204937200. [DOI] [PubMed] [Google Scholar]
- 61.Mechtcheriakova D, Wlachos A, Holzmuller H, Binder BR, Hofer E. Vascular endothelial cell growth factor-induced tissue factor expression in endothelial cells is mediated by EGR-1. Blood. 1999;93:3811–3823. [PubMed] [Google Scholar]
- 62.Hernandez GL, Volpert OV, Iniguez MA, et al. Selective inhibition of vascular endothelial growth factor-mediated angiogenesis by cyclosporin A: roles of the nuclear factor of activated T cells and cyclooxygenase 2. J Exp Med. 2001;193:607–620. doi: 10.1084/jem.193.5.607. [DOI] [PMC free article] [PubMed] [Google Scholar]