

Abstract
Procaspase-Activating Compound 1 (PAC-1) is an ortho-hydroxy-N-acylhydrazone that induces apoptosis in cancer cells by chelation of labile inhibitory zinc from procaspase-3. PAC-1 has been assessed in a wide variety of cell culture experiments and in vivo models of cancer, with promising results, and a Phase 1 clinical trial in cancer patients has been initiated (NCT02355535). For certain applications, however, the in vivo half-life of PAC-1 could be limiting. Thus, with the goal of developing a compound with enhanced metabolic stability, a series of PAC-1 analogues was designed containing modifications that systematically block sites of metabolic vulnerability. Evaluation of the library of compounds identified four potentially superior candidates with comparable anticancer activity in cell culture, enhanced metabolic stability in liver microsomes, and improved tolerability in mice. In head-to-head experiments with PAC-1, pharmacokinetic evaluation in mice demonstrated extended elimination half-lives and greater AUC values for each of the four compounds, suggesting them as promising candidates for further development.
Graphical abstract
Introduction
The development of personalized therapeutics has emerged as a promising strategy in anticancer drug discovery. Translocation, mutation, and abnormal expression of genes can produce unique proteins that exist only in tumors, and the selective modulation of these proteins can kill cancer cells with little effect on healthy cells, minimizing adverse side effects.1 As evasion of apoptosis is a hallmark of cancer,2, 3 many recent efforts to develop new anticancer drugs have focused on inhibition of antiapoptotic proteins, including MDM2,4 Bcl-2,5, 6 and XIAP.7 Similarly, small molecules capable of enhancing the activity of proapoptotic proteins hold promise for the treatment of cancer. One target that has received considerable attention is procaspase-3,8–11 a member of the caspase family of proteases critical to apoptosis. Both the intrinsic and extrinsic pathways of apoptosis converge to activate executioner caspases-3, -6, and -7, from their proenzyme forms.12, 13 The low frequency of procaspase-3 mutations in cancer,14 its downstream location relative to apoptotic proteins that are frequently mutated,13 and the overexpression of procaspase-3 in a number of cancer types, including lymphoma,15, 16 leukemia,17, 18 melanoma,19, 20 glioblastoma,21, 22 pancreatic cancer,23 liver cancer,24 lung cancer,25–27 breast cancer,28–31 esophageal cancer,32 and colon cancer,8, 33–35 have made the small molecule-mediated activation of procaspase-3 an attractive strategy for personalized medicine.8–11
Procaspase-Activating Compound 1 (PAC-1, 1, Figure 1) was identified via a high-throughput screen for compounds that could enhance procaspase-3 enzymatic activity in vitro.8 PAC-1 has been shown to be cytotoxic against a diverse array of cancer cells in culture, including cell lines derived from hematopoietic tumors (lymphoma,8, 36–43 leukemia,8, 38, 42–47 and multiple myeloma47), carcinomas of diverse origin (breast,8, 38, 42–46, 48–50 renal,8 adrenal,8, 51 colon, 8, 42, 47, 49, 50, 52 lung,8, 42–50, 52–55 cervical,38, 47 gastric, 42, 43, 47, 49, 50, 52 ovarian,47 liver,42, 43, 47 prostate,42, 43 and gallbladder42, 43), and other solid tumor histologies (melanoma,8, 38, 42, 43 osteosarcoma,47 neuroblastoma,8, 47, 49, 50 and glioblastoma42, 43). PAC-1 and derivatives also induce apoptosis in patient-derived samples from colon cancer8 and chronic lymphocytic leukemia,18 and have anticancer efficacy in multiple murine tumor models8, 42, 43, 48, 54–56 and in pet dogs with cancer.38
Figure 1.

Structures of PAC-1 (1) and S-PAC-1 (2).
The ortho-hydroxy-N-acylhydrazone was identified as the key pharmacophore of PAC-1 through extensive studies of structure-activity relationships (SAR).36, 37 Several PAC-1 derivatives containing this motif have comparable activity in vitro and in cell culture, but modification of the core results in a loss of activity.37 The ortho-hydroxy-N-acylhydrazone is known to chelate metals, including iron,57 copper,58 and zinc,59 and many divalent metal cations are also known to inhibit procaspase36 and caspase60–63 enzymes. In particular, zinc from the labile zinc pool, which is bound loosely to certain proteins, co-localizes with procaspase-3 in cells64 and inhibits the enzymatic activity of both procaspase-336 and caspase-3,61 and a putative binding site on procaspase-3/caspase-3 for labile zinc ions has been identified.65 The mechanism of action of PAC-1 most likely involves the chelation of labile zinc from procaspase-3, relieving the zinc-mediated inhibition and allowing procaspase-3 to process itself to the active form.36, 37, 41 Using genetically-encoded zinc sensors, PAC-1 has been shown to mobilize the labile zinc pool in cancer cells.41 Providing further support to this direct procaspase-3 activation mechanism, cells treated with PAC-1 or a derivative show cleaved procaspase-3 and poly-ADP ribose polymerase-1 prior to release of cytochrome c from the mitochondria or cleavage of initiator procaspases-8 and -9,8, 42, 43, 66 and PAC-1-mediated apoptosis occurs regardless of the status of Bcl-2 family proteins.67, 68 Because of this unique mechanism, PAC-1 is increasingly being used as a tool to directly activate procaspase-3 in a variety of biological settings.66, 67, 69, 70 In addition, PAC-1 and derivatives have shown synergy with experimental therapeutics18, 48 and with the anticancer drug paclitaxel.55
Despite the potential for promiscuity and/or instability with certain ortho-hydroxy-N-acylhydrazones,71 PAC-1 shows minimal inhibitory activity towards zinc-dependent enzymes, including matrix metalloproteinases-9 and -14,72 and a derivative of PAC-1 showed minimal inhibition toward carboxypeptidase A and histone deacetylases.18 These results are consistent with the known modest affinity of PAC-1 for zinc,36 allowing for a high degree of selectivity for chelation of zinc ions from the labile pool over essential zinc ions in canonical zinc binding sites within metalloproteins. In addition, PAC-1 is stable in aqueous solution; degradation of PAC-1 is observed only when the compound is subjected to extremes in temperature and pH outside of relevant physiological ranges.73
Pharmacokinetic studies with PAC-1 in mice and dogs revealed that serum concentrations of approximately 10 μM can be achieved with few adverse events,39 with transient neuroexcitation observed only at elevated doses when PAC-1 is administered via IV or IP injection.38 A Phase 1 clinical trial of PAC-1 given orally to cancer patients has been initiated (NCT02355535). A sulfonamide-containing derivative of PAC-1, called S-PAC-1 (2, Figure 1), is well tolerated by mice at doses of 350 mg/kg or higher via IP injection, with peak plasma concentrations of 3.5 mM at this dose.38 It is likely that the improved safety profile is due in large part to the decreased ability of S-PAC-1 to cross the blood-brain barrier, as compared to PAC-1.41 In addition to the ability of S-PAC-1 to induce cell death to a variety of cancer cell lines in culture,38 recent efforts have demonstrated the potential for S-PAC-1 to sensitize cancer cells in culture to ionizing radiation.74 Encouragingly, S-PAC-1 was effective in reducing or stabilizing tumor growth in four out of six canine patients with spontaneously occurring lymphoma, and the compound was well tolerated in all six dogs.38 These results demonstrate the potential for procaspase-3 activation as a safe and promising anticancer strategy.
While studies with PAC-1 and S-PAC-1 have been encouraging, a challenge in using these compounds in animals is the relatively short in vivo half-lives of both PAC-1 (2.1 ± 0.3 h in dogs)39 and S-PAC-1 (1.09 ± 0.02 h in dogs)38 following IV administration. A study in rats identified three main pathways of metabolism for PAC-1, including oxidative N-debenzylation, olefin oxidation, and arene oxidation (Figure 2).75 While many of these metabolites may be active based on the predicted structure-activity relationships, the alcohols and secondary amines resulting from these metabolites provide sites for conjugation, including sulfation and glucuronidation; these conjugates are then cleared from circulation. The metabolic liabilities present in PAC-1 likely contribute to its pharmacokinetic profile, necessitating relatively large doses to achieve therapeutic levels in vivo. A PAC-1 analogue lacking some of these liabilities may allow for lower dosing, which could potentially reduce off-target toxicity. In this work, we describe the design, synthesis, and evaluation of a family of PAC-1 derivatives with the goal of enhancing metabolic stability, and we report on promising compounds with enhanced metabolic stability in vitro and in vivo.
Figure 2.

PAC-1 is susceptible to enzymatic oxidation in vitro and in vivo, giving metabolites that result from N-debenzylation, olefin oxidation, and arene oxidation.75
Results
Library Design
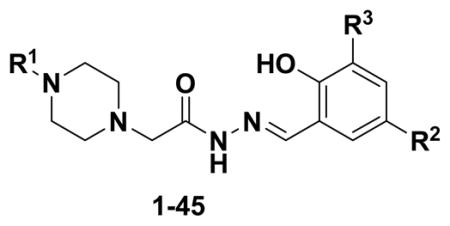
The structure-activity relationships of PAC-1 indicate that modifications to the aryl rings can be tolerated, as long as the core ortho-hydroxy-N-acylhydrazone remains intact.8, 36–38 The synthetic strategy that has been adopted to access these active compounds involves the late-stage condensation of a hydrazide and an aldehyde to form the key ortho-hydroxy-N-acylhydrazone.8, 37, 38, 40, 42–46, 49, 51, 53 This strategy was useful for the generation of a large combinatorial library of 837 diverse PAC-1 analogues.40 However, for this study, we sought a more focused library design, with an emphasis on the creation of derivatives with systematic removal of the metabolic liabilities. The library (Figure 3) consists of 45 PAC-1 analogues (1–45), constructed from nine hydrazides (46a-i) and five aldehydes (47a-e). In order to avoid oxidative N-debenzylation, the benzyl moiety was modified to a benzoyl (as in 46c, 46e, 46g, and 46i), hypothesized to be more resistant to oxidation.76 In order to avoid olefin oxidation, the allyl group was changed to a propyl group (as in 47b and 47e) or removed entirely (as in 47c). Finally, in order to block arene oxidation, building blocks were introduced containing nitrile (46d-e), fluorine (46f-g, 47c-e), and trifluoromethyl (46h-i) substituents. Multiple derivatives were synthesized containing only one modification to the PAC-1 core, so that the effect of individual changes could be systematically evaluated.
Figure 3.

Nine hydrazides and five aldehydes were used to construct a library of 45 PAC-1 derivatives designed to display enhanced metabolic stability by blocking oxidative N-debenzylation, olefin oxidation, and/or arene oxidation.
Compound Synthesis
Synthesis of the library involved the construction of the nine hydrazides and five aldehydes, followed by the condensation of each hydrazide with each aldehyde to give the PAC-1 derivatives. The hydrazides were synthesized according to Scheme 1a. The synthesis began with the alkylation of piperazine (48) with ethyl chloroacetate (49) to form monosubstituted piperazine 50. Compound 50 was then reacted with substituted benzyl or benzoyl halides to give disubstituted piperazines 51a-i in high yields. Reaction of the esters with hydrazine then gave hydrazides 46a-i.
Scheme 1.

Synthesis of PAC-1 analogues. (a) Synthesis of hydrazides 46a-i. (b) Synthesis of aldehydes 47a-e. (c) Condensation of hydrazides and aldehydes to form PAC-1 analogues 1–45.
Synthesis of the aldehydes is shown in Scheme 1b. Both salicylaldehyde (52) and 5-fluorosalicylaldehyde (47c) were alkylated with allyl bromide to give allyloxybenzaldehydes 53a-b in high yields. These compounds underwent Claisen rearrangements upon heating at 200°C, yielding aldehydes 47a and 47d in approximately 50% yield. Finally, hydrogenation with diphenyl sulfide as a catalyst poison allowed for chemoselective reduction of the olefins,77 giving aldehydes 47b and 47e in high yield. As shown in Scheme 1c, each of the hydrazides (46a-i) was condensed with each of the aldehydes (47a-e) in the presence of a catalytic amount of HCl to give PAC-1 derivatives 1–45, the structures of which are given in Table 1. Chromatographic purification of the library members yielded the PAC-1 analogues in high purity (97% average purity). All derivatives were at least 95% pure except compounds 20, 22, and 24; because this standard of purity was not met for these three compounds, they were excluded from further evaluation.
Table 1.
|
| ||||||
|---|---|---|---|---|---|---|
| compound | R1 | R2 | R3 | U-937 72h IC50 (μM) | RLM 3h % Stability | mouse toxicity IP 200 mg/kg (* = 100 mg/kg) |
| 1 (PAC-1) | Bn | H | All | 10.2 ± 0.3 | 38 ± 2 | severe |
| 2 (S-PAC-1) | 4-SO2NH2-Bn | H | All | 8.9 ± 0.6 | 84 ± 1 | none |
| 3 | Bz | H | All | 12.1 ± 1.3 | 89 ± 4 | - |
| 4 | 4-CN-Bn | H | All | 13.7 ± 0.9 | 48 ± 2 | - |
| 5 | 4-CN-Bz | H | All | 13.1 ± 3.7 | 90 ± 4 | - |
| 6 | 4-F-Bn | H | All | 11.1 ± 2.1 | 31 ± 1 | - |
| 7 | 4-F-Bz | H | All | 10.2 ± 1.7 | 86 ± 2 | moderate |
| 8 | 4-CF3-Bn | H | All | 15.3 ± 6.7 | 16 ± 1 | - |
| 9 | 4-CF3-Bz | H | All | 6.6 ± 1.9 | 85 ± 6 | lethal* |
| 10 | Bn | H | n-Pr | 9.6 ± 2.1 | 30 ± 1 | - |
| 11 | 4-SO2NH2-Bn | H | n-Pr | 4.9 ± 0.4 | 61 ± 2 | - |
| 12 | Bz | H | n-Pr | 9.4 ± 1.3 | 71 ± 3 | - |
| 13 | 4-CN-Bn | H | n-Pr | 9.0 ± 1.2 | 30 ± 2 | - |
| 14 | 4-CN-Bz | H | n-Pr | 12.8 ± 2.7 | 61 ± 3 | - |
| 15 | 4-F-Bn | H | n-Pr | 10.0 ± 1.7 | 24 ± 2 | - |
| 16 | 4-F-Bz | H | n-Pr | 7.3 ± 0.9 | 69 ± 4 | - |
| 17 | 4-CF3-Bn | H | n-Pr | 4.1 ± 0.4 | 15 ± 2 | - |
| 18 | 4-CF3-Bz | H | n-Pr | 4.8 ± 1.2 | 64 ± 1 | lethal* |
| 19 | Bn | F | H | 17.0 ± 1.4 | 64 ± 4 | - |
| 20 | 4-SO2NH2-Bn | F | H | - | - | - |
| 21 | Bz | F | H | 15.7 ± 2.6 | 88 ± 1 | - |
| 22 | 4-CN-Bn | F | H | - | - | - |
| 23 | 4-CN-Bz | F | H | 15.3 ± 1.3 | 88 ± 4 | - |
| 24 | 4-F-Bn | F | H | - | - | - |
| 25 | 4-F-Bz | F | H | 15.3 ± 0.8 | 86 ± 2 | - |
| 26 | 4-CF3-Bn | F | H | 4.7 ± 0.3 | 30 ± 5 | - |
| 27 | 4-CF3-Bz | F | H | 8.7 ± 0.5 | 87 ± 3 | moderate |
| 28 | Bn | F | All | 9.5 ± 0.9 | 56 ± 1 | - |
| 29 | 4-SO2NH2-Bn | F | All | 9.8 ± 1.3 | 89 ± 3 | lethal |
| 30 | Bz | F | All | 8.6 ± 2.0 | 93 ± 7 | moderate |
| 31 | 4-CN-Bn | F | All | 12.7 ± 2.0 | 65 ± 2 | - |
| 32 | 4-CN-Bz | F | All | 10.1 ± 2.0 | 95 ± 4 | mild |
| 33 | 4-F-Bn | F | All | 10.3 ± 4.1 | 57 ± 1 | - |
| 34 | 4-F-Bz | F | All | 8.5 ± 1.4 | 92 ± 3 | severe |
| 35 | 4-CF3-Bn | F | All | 3.4 ± 0.6 | 49 ± 3 | - |
| 36 | 4-CF3-Bz | F | All | 6.5 ± 0.6 | 90 ± 2 | moderate |
| 37 | Bn | F | n-Pr | 8.9 ± 1.2 | 49 ± 6 | - |
| 38 | 4-SO2NH2-Bn | F | n-Pr | 8.7 ± 0.4 | 62 ± 3 | - |
| 39 | Bz | F | n-Pr | 12.3 ± 1.0 | 86 ± 5 | - |
| 40 | 4-CN-Bn | F | n-Pr | 11.2 ± 0.9 | 49 ± 5 | - |
| 41 | 4-CN-Bz | F | n-Pr | 9.4 ± 1.2 | 66 ± 3 | mild |
| 42 | 4-F-Bn | F | n-Pr | 7.5 ± 0.7 | 48 ± 1 | - |
| 43 | 4-F-Bz | F | n-Pr | 7.5 ± 1.4 | 67 ± 3 | severe |
| 44 | 4-CF3-Bn | F | n-Pr | 3.9 ± 0.6 | 40 ± 1 | - |
| 45 | 4-CF3-Bz | F | n-Pr | 5.2 ± 0.6 | 64 ± 5 | lethal |
Cells treated with compounds for 72 hours. Biomass quantified by sulforhodamine B assay. IC50 values shown are mean ± s.e.m. (n = 3).
Rat liver microsomes treated with compounds (10 μM) for 3 hours. Percent stability values shown are mean ± s.e.m. (n = 3).
Mice dosed with compound via i.p. injection at 200 mg/kg (except *100 mg/kg). Dash indicates that the compound was not evaluated.
Evaluation of PAC-1 analogues
Upon completion of the synthesis of the 45 PAC-1 derivatives, the compounds were evaluated in biological assays. First, the ability of the compounds to induce cell death in U-937 (human lymphoma) cells in culture was determined (Table 1 and Supporting Information). Each of the compounds induced dose-dependent cell death under these conditions, and most of the compounds were approximately as potent as PAC-1 and S-PAC-1, confirming the previously determined SAR.8, 37, 38, 40
The metabolic stability of the compounds was then evaluated in rat liver microsomes. The compounds were incubated with liver microsomes for 3 hours at 10 μM, and the metabolites were observed by LC/MS, with (±)-propranolol hydrochloride as a positive control;78 approximately 20% of the control remained. The results of this assay are shown in Table 1. Compounds that contained benzoyl substituents were significantly more stable than analogous compounds containing benzyl groups; for example, compound 3 was more stable than PAC-1, and compound 32 was more stable than compound 31. The propyl-containing compounds were less stable than the allyl-containing compounds (e.g., PAC-1 was more stable than compound 10). In addition, S-PAC-1 was relatively stable in the liver microsomes, despite the short in vivo half-life of the compound.38 This suggests that clearance mechanisms other than oxidative metabolism are responsible for the elimination of S-PAC-1 from treated animals.
The results of selected liver microsome experiments are shown in Figure 4 (The full set of results is in the Supporting Information). PAC-1 (Figure 4A) was 38% stable in the assay, and several metabolites, including an N-dealkylated product, a dihydroxylated product, and multiple monooxygenated products, were observed. S-PAC-1 (Figure 4B) was found to be more stable than PAC-1, and fewer metabolites were observed. One of the modifications that improved stability to the greatest degree was the addition of the benzoyl in place of the benzyl substituent, as demonstrated with compound 3 (Figure 4C). This modification prevented the N-debenzylation completely and increased stability to 89% during the three-hour incubation. In addition, compounds with a benzoyl substituent had fewer monooxygenated species than PAC-1, as the amide likely acted to deactivate the aromatic ring towards oxidation. The addition of fluorine to the benzylidene ring, as in compound 19 (Figure 4D), was also successful in reducing the number of monooxygenated metabolites, and as expected, dihydroxylated metabolites were not formed from compounds lacking the allyl group. Finally, combining multiple modifications, as in compounds 7 (Figure 4E) and 32 (Figure 4F), led to highly stable compounds that gave significantly fewer metabolites in the liver microsome experiment.
Figure 4.

Metabolic stability of PAC-1 and derivatives was evaluated in rat liver microsomes at 10 μM for 3 hours. LC/MS results of liver microsome experiments for A. PAC-1, B. S-PAC-1, C. 3, D. 19, E. 7, and F. 32 are shown. Data shown are representative of three independent experiments.
Evaluation of toxicity in mice
Based on the comparable cytotoxicity and improved in vitro metabolic stability profile, several compounds were selected for further in vivo evaluation. In order to assess the in vivo tolerability of the compounds, they were administered to mice via intraperitoneal injection at a dose of 200 mg/kg, the maximum tolerated dose of PAC-1.48 Compounds were formulated at 5 mg/mL in a 200 mg/mL aqueous solution of 2-hydroxypropyl-β-cyclodextrin. The results of this experiment are shown in Table 1. Responses are graded as mild, moderate, or severe; compounds that were lethal to the mice at 200 mg/kg were also noted. Compounds 9 and 18 were lethal at a lower dose of 100 mg/kg, and compound 36 induced hemolysis in the animals, so the compounds containing the (trifluoromethyl)benzoyl substituent were not pursued further.
Secondary assays
Because of their high stability, comparable potency, and improved in vivo tolerability as compared to PAC-1, compounds 7, 30, 32, and 41 were selected for further investigation. In order to confirm that the hit compounds act similarly to PAC-1, the compounds were evaluated for their ability to chelate zinc in vitro, activate executioner caspases in whole cells, and induce apoptosis in cancer cells. Zinc binding was determined using an EGTA titration experiment.79 In this experiment, varying concentrations of Zn(OTf)2 were added to each well of a 96-well plate with a HEPES-buffered solution containing EGTA and PAC-1 derivative, and the fluorescence of the complex was analyzed, a slight variant of our previous protocol for assessment of zinc binding.37 As shown in Table 2, PAC-1 binds zinc with a Kd of 1.28 ± 0.03 nM, while S-PAC-1 binds zinc with a Kd of 2.72 ± 0.13 nM. Each of the four new compounds displays affinity for zinc in the range of 1–2 nM. PAC-1a, an inactive derivative of PAC-1 lacking the allyl and hydroxyl substituents,8, 36, 37 does not bind zinc.
Table 2.
| Zn2+ Kd (nM) | % Caspase-3/-7 Activity | |
|---|---|---|
|
|
||
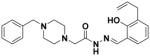 PAC-1 |
1.28 ± 0.03 | 87 ± 7 |
|
S-PAC-1 |
2.72 ± 0.13 | 64 ± 11 |
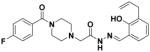 7 |
1.46 ± 0.07 | 64 ± 4 |
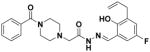 30 |
1.07 ± 0.09 | 67 ± 6 |
|
32 |
1.37 ± 0.10 | 83 ± 3 |
|
41 |
1.37 ± 0.03 | 81 ± 4 |
|
PAC-1a |
>106 | 3 ± 0.1 |
Increasing amounts of Zn(OTf)2 added to a buffered solution of EGTA (7.3 mM) and PAC-1 derivative (100 μM). Kd was determined by comparing fluorescence intensity (ex. 410 nm, em. 475 nm) and free zinc concentration.
U-937 cells treated with compounds (30 μM) for 16 hours, then lysed. Caspase-3/7 activity assessed by cleavage of fluorogenic substrate Ac-DEVD-AFC.
In addition, the ability of compounds to activate executioner caspases in whole cells was evaluated. Cells were treated with compound for 0 or 16 hours, then the cells were lysed, and cleavage of the fluorescent caspase-3/-7 substrate Ac-DEVD-AFC was analyzed via kinetic reads. The percent activity at 16 hours was normalized to the slope of each compound at 0 hours (0% activity) and the slope of the positive control compound staurosporine at 16 hours (100% activity). As shown in Table 2 and Figure 5, PAC-1 induces the highest degree of caspase activation, while each of the other active compounds induces greater than 60% activation of the executioner caspases. As expected, treatment with DMSO alone or PAC-1a induces minimal caspase activity in the cells.
Figure 5.

PAC-1 and active derivatives activate executioner caspases in cells. U-937 cells were treated with compounds (30 μM for PAC-1 derivatives, 1 μM for staurosporine) for 16 hours, and then lysed. Caspase-3/7 activity was assessed by cleavage of the fluorogenic substrate Ac-DEVD-AFC. Cells treated with vehicle alone or inactive derivative PAC-1a show minimal caspase activity after 16 hours. Values shown are mean ± s.e.m. (n = 3).
In order to determine the mode of cell death induced by the compounds, U-937 cells were treated with compounds at 50 μM for 12 hours, and viability was assessed by Annexin V-FITC/propidium iodide staining (Figure 6). Each of these compounds induced approximately 50% cell death under these conditions, and the presence of large populations in the lower right quadrants of the histograms (Annexin V-FITC positive, propidium iodide negative) confirms that the compounds induce apoptosis in these cancer cells.
Figure 6.

PAC-1 and derivatives induce apoptosis in U-937 cells. Cells were treated for 12 hours at 50 μM, and viability was assessed by Annexin V-FITC/propidium iodide staining. Data shown are representative of three independent experiments.
As many PAC-1 derivatives show activity against white blood cell cancer lines8, 37, 38, 40, 42–47 and patient-derived leukemic lymphocytes18 in culture, the compounds were evaluated for their ability to induce cell death in a panel of lymphoma and leukemia cell lines, including Jurkat (human leukemia), GL-1 (dog lymphoma), OSW (dog lymphoma), and EL4 (mouse lymphoma) cells, in order to complement the previously determined IC50 values in U-937 (human lymphoma) cells. As shown in Table 3, the compounds displayed comparable potency against each given cell line. These results provide further support to the previously determined structure-activity relationships, as the modifications to improve metabolic stability had minimal effect on the activity of the new compounds, and further suggest the potential of PAC-1 and derivatives for the treatment of white blood cell cancers.
Table 3.
PAC-1and derivatives are cytotoxic to white blood cell cancer lines in culture.a
| Cell line | Species | Origin | 72-hour IC50 (μM) | |||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| PAC-1 | S-PAC-1 | 7 | 30 | 32 | 41 | |||
| U-937 | human | lymphoma | 10.2 ± 0.3 | 8.9 ± 0.6 | 10.2 ± 1.7 | 8.6 ± 2.0 | 10.1 ± 2.0 | 9.4 ± 1.2 |
| Jurkat | human | leukemia | 4.4 ± 0.6 | 4.5 ± 1.2 | 4.0 ± 0.5 | 4.1 ± 0.7 | 3.5 ± 0.2 | 3.4 ± 0.6 |
| GL-1 | dog | lymphoma | 3.0 ± 0.1 | 3.2 ± 0.2 | 3.0 ± 0.1 | 3.4 ± 0.2 | 2.4 ± 0.4 | 2.2 ± 0.3 |
| OSW | dog | lymphoma | 10.0 ± 0.8 | 9.8 ± 0.1 | 9.3 ± 0.2 | 10.0 ± 0.6 | 9.5 ± 0.7 | 8.5 ± 0.7 |
| EL4 | mouse | lymphoma | 6.5 ± 0.5 | 7.9 ± 0.5 | 6.5 ± 0.8 | 7.3 ± 1.2 | 5.1 ± 0.4 | 4.7 ± 0.7 |
Cells treated with compounds for 72 hours. Biomass quantified by sulforhodamine B assay. IC50 values shown are mean ± s.e.m. (n = 3).
Pharmacokinetics
Because compounds 7, 30, 32, and 41 all chelate zinc, activate executioner caspases in whole cells, and induce apoptosis, all four of the hit compounds were studied further in vivo. The pharmacokinetics of the four compounds plus PAC-1 and S-PAC-1 were evaluated in mice at a dose of 25 mg/kg (IV injection), and the results are shown in Figure 7 and Table 4. PAC-1 and S-PAC-1 were cleared rapidly and were no longer detectable after 5 and 6 hours post-treatment, respectively. In contrast, detectable levels of each of the four new derivatives remained in circulation for at least 8 hours post-treatment.
Figure 7.

Pharmacokinetic profiles of PAC-1 and selected derivatives following 25 mg/kg intravenous dose (n = 2). Detectable levels of the novel derivatives are present in serum for at least 8 hours post-treatment, while PAC-1 and S-PAC-1 are no longer detectable after 5 and 6 hours post-treatment, respectively.
Table 4.
Pharmacokinetic parameters for PAC-1 and selected derivatives.a
| compound | t1/2 (min) | AUC (IV) (min*μg/mL) | AUC (PO) (min*μg/mL) | %Foral |
|---|---|---|---|---|
| PAC-1 | 24.6 ± 0.9 | 210.3 ± 9.3 | 31.6 ± 1.6 | 15.1 ± 1.4 |
| S-PAC-1 | 38.1 ± 3.3 | 446.0 ± 114.1 | 54.0 ± 12.4 | 12.9 ± 6.1 |
| 7 | 89.5 ± 19.3 | 362.3 ± 55.8 | 92.2 ± 6.0 | 25.6 ± 2.3 |
| 30 | 120.5 ± 16.3 | 291.0 ± 40.6 | 25.7 ± 18.3 | 8.5 ± 5.1 |
| 32 | 88.7 ± 3.3 | 364.9 ± 5.6 | 105.2 ± 24.2 | 28.8 ± 6.2 |
| 41 | 122.3 ± 1.4 | 313.4 ± 5.5 | 113.0 ± 3.7 | 36.1 ± 0.5 |
25 mg/kg dose was administered via intravenous injection or oral gavage. Values shown are mean ± standard deviation (n = 2).
The elimination half-life of PAC-1 was 24.6 ± 0.9 minutes, and the half-life of S-PAC-1 was 38.1 ± 3.3 minutes. Each of the four new derivatives displayed half-lives of at least 88 minutes, with compound 41 having the longest half-life at 122.3 ± 1.4 minutes. In addition, AUC values from intravenous administration for the four new derivatives were all significantly higher than that of PAC-1. Compounds 7, 32, and 41 were also found to display increased oral bioavailability as compared to PAC-1 and S-PAC-1.
Discussion
The introduction of substituents designed to block oxidative metabolism is among the most attractive methods to achieve a favorable pharmacokinetic profile, as the drug can pass through the liver without being modified and remain in circulation for longer periods of time. The knowledge of the metabolites formed from PAC-1 in vivo facilitated the design of a library of PAC-1 derivatives whose members lacked many of the metabolic liabilities present on the parent compound. The flexible, modular nature of the PAC-1 synthesis allowed for the rapid generation of 45 derivatives from nine hydrazides and five aldehydes.
Metal binders in medicine
Given the increased attention paid to so-called “PAINS” (pan-assay interference compounds), a further discussion of PAC-1 and derivatives in relationship to PAINS compounds is warranted. The metal-binding ability of ortho-hydroxy-N-acylhydrazones can cause members of this class of compounds to appear as hits in screening assays due to interference with the assay screening system, rather than via specific interactions with biological targets.71 In these cases, attempts to validate such hits will fail because the apparent activity of the screening hit is unrelated to the target. However, rather than interfering with the in vitro procaspase-3 enzymatic assay, the chelation of zinc from procaspase-3 in vitro by PAC-1 is highly biologically relevant: PAC-1 is directly modulating zinc, an endogenous inhibitor of procaspase-3, and the binding site on procaspase-3/caspase-3 for this inhibitory zinc has been identified.65 Through this “inhibiting the inhibitor” mechanism, PAC-1 is akin to compounds that bind to other endogenous apoptotic inhibitors and induce apoptosis, such as those binding MDM24 and XIAP.7
PAC-1 also chelates labile zinc in cancer cells in culture, as determined by detailed experiments with genetically encoded fluorescent sensors specific for zinc.41 This removal of zinc leads to the observed anticancer effect in cell culture. PAC-1 shows no activity toward several other enzymes as assessed by in vitro assays,80 PAC-1 and derivatives do not affect the activity of proteins that rely on tightly-bound zinc,18, 72 and PAC-1 derivatives that do not bind zinc in vitro are inactive in cells.37 As shown by multiple investigators, treatment of cells with PAC-1 or a derivative results in the cleavage of procaspase-3 prior to the cleavage of initiator procaspases-8 and -9,42, 43 and co-treatment of PAC-1 with a covalent inhibitor of caspase-9 does not prevent cleavage of procaspase-3,66 further supporting the hypothesis that the proapoptotic activity of PAC-1 is due to chelation of labile inhibitory zinc from procaspase-3, leading to the activation of procaspase-3 and apoptotic cell death.
While many metal chelators will interfere with in vitro enzyme assays, it would be inappropriate to disregard all metal chelators from consideration as drug candidates due to this in vitro artifact. Indeed, metal chelators have a rich history in drug discovery and have made a positive impact on many diseases through a diverse range of mechanisms and targets.81 The many examples of therapeutic metal-binding compounds include the marketed drugs vorinostat (Zn2+),82 penicillamine (Cu2+),81 and the entire class of bisphosphonates (Ca2+),83 as well as the experimental therapeutics elesclomol (Cu2+),81 ML-133/Apto-253 (Zn2+),84 and triapine (Fe3+/Fe2+),85 all of which rely on metal chelation in vivo for their mechanism of action. It is safe to say that many of these compounds would interfere with certain in vitro assays that are contingent upon metal-bound proteins. While it is reasonable for metal chelators and metal-chelating motifs to be structural alerts when examining screening hits, if the desired biological activity is metal chelation, then obviously metal chelation is the precise trait to look for in a screening hit.
Translational potential of PAC-1 derivatives
Cell culture evaluation of the compounds reported herein confirm previously determined structure-activity relationships, in that substituents can be introduced to the aromatic rings without abolishing activity if the core ortho-hydroxy-N-acylhydrazone remains intact. Removal of the allyl group leads to a diminution in cell culture potency, consistent with previous reports,8, 37 although reduction to the fully saturated propyl group was tolerated in the cell culture experiment. It is likely that the increased hydrophobicity of the alkyl chain contributes to increased cell permeability, as the allyl group does not affect the ability of PAC-1 to bind zinc.37
The benzoyl-containing compounds display similar cell culture activity to PAC-1. This substitution changes the electronics at both the arene and the piperazine nitrogen; the role of the benzylpiperazine in PAC-1 activity merits further evaluation. Evaluation of the metabolic stability of the library members in rat liver microsomes suggested that N-debenzylation was the main route of metabolism in vitro; the PAC-1 derivatives containing benzoyl substituents were more stable than those containing benzyl substituents. These substitutions also reduced the extent of arene oxidation, providing further support for advancement of these compounds.
Four compounds (7, 30, 32, and 41) were identified with favorable cell culture potency, in vitro metabolic stability, and in vivo tolerability. Each of these compounds contains the benzoyl substitution, as well as at least one arene substituent (fluorine and/or nitrile) not present on PAC-1. The introduction of fluorine is common in medicinal chemistry, especially the use of aryl fluorides to block undesired metabolic arene oxidation, as in the cholesterol-lowering drug ezetimibe.86 Aryl nitriles are also commonly employed to accomplish this goal, as nitriles typically pass through the body unmodified, and the electron-withdrawing nature of the group deactivates the arene towards oxidative metabolism at other sites.87 Trifluoromethyl groups can deactivate arenes similarly in certain cases,88 and in vitro results with the (trifluoromethyl)benzoyl-containing PAC-1 derivatives were encouraging. However, these compounds were not evaluated further due to unacceptable levels of toxicity in vivo.
Further evaluation of the four lead compounds demonstrates that they chelate zinc, activate executioner caspases in whole cells, and induce apoptosis similarly to PAC-1 and S-PAC-1. The four derivatives display three- to five-fold higher elimination half-lives and up to two-fold higher overall compound exposure compared to PAC-1. Results from the liver microsome experiment are mostly consistent with the observed in vivo pharmacokinetic profiles: fewer metabolites formed from the new derivatives than from PAC-1 in vitro, and the compounds remain in serum for longer periods of time than PAC-1. In contrast, S-PAC-1 is stable in the liver microsome assay but has a relatively short in vivo half-life. This suggests that the main mode of clearance for S-PAC-1 may not be via oxidative metabolism; instead, the compound may be excreted without modification. A more thorough understanding of this phenomenon may allow for the design of PAC-1 derivatives that improve upon the pharmacokinetics even further than those described in this report. The rapid clearance of PAC-1 and S-PAC-1 from circulation makes them challenging to evaluate in certain efficacy models in vivo; these studies typically require large doses of compound that increase the potential for toxicity. The four novel derivatives remain in circulation for longer than either PAC-1 or S-PAC-1, and thus offer promise as novel therapeutic agents for the treatment of cancer.
Experimental
Chemical Information
Materials and Methods
General
All reactions requiring anhydrous conditions were conducted under a positive atmosphere of nitrogen or argon in oven-dried glassware. Standard syringe techniques were used for anhydrous addition of liquids. Unless otherwise noted, all starting materials, solvents, and reagents were acquired from commercial suppliers and used without further purification. Flash chromatography was performed using 230–400 mesh silica gel. Compound syntheses are discussed in the order in which they appear in the text. Syntheses of 46a,8 46b,37 46h,40 47a,37 50,37 PAC-1 (1),8 S-PAC-1 (2),37 and PAC-1a8 have been described previously.
All NMR experiments were recorded either in CDCl3 (Sigma or Cambridge), CD3OD (Sigma), or (CD3)2CO (Sigma or Cambridge) on a Varian Unity 500 MHz spectrometer with residual undeuterated solvent as the internal reference for 1H-NMR and 13C-NMR, and C6F6 as the internal reference for 19F-NMR. Chemical shift, δ (ppm); coupling constants, J (Hz); multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, quint = quintet, sext = sextet, m = multiplet, br = broad); and integration are reported. High-resolution mass spectral data was recorded on a Micromass Q-Tof Ultima hybrid quadrupole/time-of-flight ESI mass spectrometer or a Micromass 70-VSE at the University of Illinois Mass Spectrometry Laboratory. Compound purity was assessed by analytical HPLC (monitoring at 254 nm) on a Waters Alliance e2695 HPLC with a Waters XBridge C18 column, 4.6 × 150 mm. Mobile phase A was 0.1% F3CCO2H in H2O, B was MeCN (solvent B). A gradient was run with 0% B for 1 min, then 0–100% B for 10 min, then constant 100% B for 5 min, then 100-0% B for 1 min, then constant 0% B for 5 min. All compounds evaluated in biological assays were ≥95% pure.
General Procedure A: Synthesis of dialkylated piperazines
To a round-bottom flask were added benzyl halide (1.0 equiv.), K2CO3 (3.0 equiv.), and acetone (0.2 M). The mixture was stirred, and 50 (1.5 equiv.) was added. The reaction mixture was stirred at reflux overnight. The reaction mixture was cooled to room temperature. The solid was filtered and washed with acetone. The filtrate was concentrated, and the product was purified by silica gel column chromatography.
General Procedure B: Synthesis of amides
To an oven-dried round-bottom flask were added 50 (1.0 equiv.), anhydrous tetrahydrofuran (0.2 M), and freshly distilled Et3N (2.0 equiv.). The solution was stirred at 0°C under N2, and the benzoyl chloride (1.0 equiv.) was added. The reaction mixture was stirred overnight at room temperature under N2. The reaction mixture was diluted with EtOAc and washed with sat. NaHCO3 (2x), H2O, and brine. The organic layer was dried over MgSO4, filtered, and concentrated. The product was purified by silica gel column chromatography.
General Procedure C: Synthesis of hydrazides
To a round-bottom flask were added ethyl ester (1.0 equiv.) and EtOH or 2:1 EtOH:MeOH (0.5 M). The solution was stirred, and anhydrous hydrazine (4.0 equiv.) was added dropwise. The reaction mixture was stirred at reflux overnight. The reaction mixture was cooled to room temperature and concentrated. The resulting residue was partitioned between CH2Cl2/1:1 brine:0.1 M KOH. The layers were separated, and the aqueous layer was extracted with CH2Cl2 (2x). The combined organic layers were dried over MgSO4, filtered, and concentrated. Purification by silica gel column chromatography or recrystallization yielded pure hydrazide.
Ethyl 4-Benzoyl-1-piperazineacetate (51c)
Synthesized according to General Procedure B: 50 (2.45 g, 14.2 mmol, 1.0 equiv.), anhydrous tetrahydrofuran (70 mL, 0.2 M), freshly distilled Et3N (4.0 mL, 28.4 mmol, 2.0 equiv.), benzoyl chloride (54c, 2.0 g, 1.7 mL, 1.0 equiv.). Purification by silica-gel column chromatography (50–100% EtOAc/hexanes) afforded 51c (2.87 g, 73.1%) as a pale yellow oil. 1H-NMR (500 MHz, CDCl3) δ 7.41-7.38 (m, 5H), 4.19 (q, 2H, J = 7.0 Hz), 3.85 (br s, 2H), 3.48 (br s, 2H), 3.25 (s, 2H), 2.68 (br s), 2.54 (br s, 2H), 1.27 (t, 3H, J = 7.0 Hz). 13C-NMR (125 MHz, CDCl3)δ 170.5, 170.2, 135.9, 129.9, 128.7, 127.3, 61.0, 59.4, 53.3 (br), 52.8 (br), 47.8 (br), 42.1 (br), 14.4. HRMS (ESI): 277.1552 (M+H)+; calcd. for C15H21N2O3: 277.1552.
4-Benzoyl-1-piperazineacetohydrazide (46c)
Synthesized according to General Procedure C: 51c (2.87 g, 10.4 mmol, 1.0 equiv.), anhydrous hydrazine (1.31 mL, 41.6 mmol, 4.0 equiv.), EtOH (20 mL, 0.5 M). 46c (1.41 g, 51.5%) was obtained as a white solid after extraction without further purification. 1H-NMR (500 MHz, CDCl3) δ 8.10 (s, 1H), 7.39-7.34 (m, 5H), 3.84 (br s, 2H), 3.77 (br s, 2H), 3.43 (br s, 2H), 3.08 (s, 2H), 2.56 (br s, 2H), 2.44 (br s, 2H). 13C-NMR (125 MHz, CDCl3) δ 170.5, 169.9, 135.5, 130.0, 128.7, 127.1, 60.6, 53.9 (br), 53.4 (br), 47.7 (br), 42.2 (br). HRMS (ESI): 263.1513 (M+H)+; calcd. for C13H19N4O2: 263.1508.
Ethyl 4-(4-Cyanophenylmethyl)-1-piperazineacetate (51d)
Synthesized according to General Procedure A: 4-(bromomethyl)benzonitrile (54d, 2.0 g, 10.2 mmol, 1.0 equiv.), 50 (2.64 g, 15.3 mmol, 1.5 equiv.), K2CO3 (4.22 g, 30.6 mmol, 3.0 equiv.), acetone (50 mL, 0.2 M). Purification by silica gel column chromatography (50–100% EtOAc/hexanes) afforded 51d (2.71 g, 92.3%) as a yellow solid. 1H-NMR (500 MHz, CDCl3) δ 7.60 (d, 2H, J = 8.0 Hz), 7.44 (d, 2H, J = 8.0 Hz), 4.18 (q, 2H, J = 7.0 Hz), 3.55 (s, 2H), 3.20 (s, 2H), 2.61 (br s, 4H), 2.51 (br s, 4H), 1.26 (t, 3H, J = 7.0 Hz). 13C-NMR (125 MHz, CDCl3) δ 170.4, 144.4, 132.3, 129.7, 119.2, 111.0, 62.5, 60.8, 59.6, 53.1, 53.1, 14.4. HRMS (ESI): 288.1718 (M+H)+; calcd. for C16H22N3O2: 288.1712.
4-(4-Cyanophenylmethyl)-1-piperazineacetohydrazide (46d)
Synthesized according to General Procedure C: 51d (2.71 g, 9.43 mmol, 1.0 equiv.), anhydrous hydrazine (1.18 mL, 37.7 mmol, 4.0 equiv.), EtOH (19 mL, 0.5 M). 46d (1.73 g, 67.1%) was obtained as an off-white solid after extraction without further purification. 1H-NMR (500 MHz, CDCl3) δ 8.10 (br s, 1H), 7.60 (d, 2H, J = 8.0 Hz), 7.43 (d, 2H, J = 8.5 Hz), 3.84 (br d, 2H, J = 5.0 Hz), 3.55 (s, 2H), 3.08 (s, 2H), 2.55 (br s, 4H), 2.46 (br s, 4H). 13C-NMR (125 MHz, CDCl3) δ 170.6, 144.1, 132.4, 129.6, 119.1, 111.2, 62.5, 60.8, 53.8, 53.3. HRMS (ESI): 274.1673 (M+H)+; calcd. for C14H20N5O: 274.1668.
Ethyl 4-(4-Cyanobenzoyl)-1-piperazineacetate (51e)
Synthesized according to General Procedure B: 50 (5.20 g, 30.2 mmol, 1.0 equiv.), anhydrous tetrahydrofuran (150 mL, 0.2 M), freshly distilled Et3N (8.4 mL, 60.4 mmol, 2.0 equiv.), 4-cyanobenzoyl chloride (54e, 5.0 g, 30.2 mmol, 1.0 equiv.). Purification by silica gel column chromatography (0–10% MeOH/EtOAc) afforded 51e (5.95 g, 65.4%) as a white solid. 1H-NMR (500 MHz, CDCl3) δ 7.68 (d, 2H, J = 8.0 Hz), 7.47 (d, 2H, J = 8.0 Hz), 4.14 (q, 2H, J = 7.0 Hz), 3.80 (br s, 2H), 3.37 (br s, 2H), 3.23 (s, 2H), 2.67 (br s, 2H), 2.53 (br s, 2H), 1.23 (t, 3H, J = 7.0 Hz). 13C-NMR (125 MHz, CDCl3) δ 170.0, 168.3, 140.1, 132.5, 127.9, 118.2, 113.6, 60.9, 59.0, 52.9, 52.3, 47.5, 42.1, 14.3. HRMS (ESI): 302.1501 (M+H)+; calcd. for C16H20N3O3: 302.1505.
4-(4-Cyanobenzoyl)-1-piperazineacetate (46e)
Synthesized according to General Procedure C with modification as noted: 51e (5.59 g, 18.6 mmol, 1.0 equiv.), anhydrous hydrazine (2.4 mL, 74.4 mmol, 4.0 equiv.), EtOH (35 mL, 0.5 M). After extraction with CH2Cl2, the aqueous layer was extracted with EtOAc (3x). 46e (2.98 g, 55.8%) was obtained as an off-white solid after extraction without further purification. 1H-NMR (500 MHz, CDCl3) δ 8.02 (br s, 1H), 7.70 (d, 2H, J = 8.5 Hz), 7.48 (d, 2H, J = 8.5 Hz), 3.86 (br d, 2H, J = 3.5 Hz), 3.78 (br s, 2H), 3.37 (br s, 2H), 3.11 (s, 2H), 2.60 (br s, 2H), 2.46 (br s, 2H). 13C-NMR (125 MHz, CDCl3) δ 169.8, 168.4, 139.8, 132.6, 127.9, 118.1, 113.8, 60.6, 53.8 (br), 53.3 (br), 47.6 (br), 42.2 (br). HRMS (ESI): 288.1464 (M+H)+; calcd. for C14H18N5O2: 288.1461.
Ethyl 4-(4-Fluorophenylmethyl)-1-piperazineacetate (51f)
Synthesized according to General Procedure A: 4-fluorobenzyl chloride (54f, 2.5 g, 2.1 mL, 17.3 mmol, 1.0 equiv.), 50 (4.48 g, 26.0 mmol, 1.5 equiv.), K2CO3 (7.19 g, 52.0 mmol, 3.0 equiv.), acetone (90 mL, 0.2 M). Purification by silica gel column chromatography (gradient, 50–100% EtOAc/hexanes) afforded 51f (3.66 g, 75.4%) as a yellow oil. 1H-NMR (500 MHz, CDCl3) δ 7.24-7.20 (m, 2H), 6.95-6.91 (m, 2H), 4.13 (q, 2H, J = 7.0 Hz), 3.42 (s, 2H), 3.15 (s, 2H), 2.55 (br s, 4H), 2.46 (br s, 4H), 1.21 (t, 3H, J = 7.0 Hz). 13C-NMR (125 MHz, CDCl3) δ 170.3, 162.0 (d, JC-F = 243.5 Hz), 133.9, 130.6 (d, JC-F = 8.0 Hz), 115.0 (d, JC-F = 21.0 Hz), 62.2, 60.6, 59.6, 53.1, 52.8, 14.3. 19F-NMR (470 MHz, CDCl3) δ −119.1. HRMS (ESI): 281.1659 (M+H)+; calcd. for C15H22FN2O2: 281.1665.
4-(4-Fluorophenylmethyl)-1-piperazineacetohydrazide (46f)
Synthesized according to General Procedure C: 51f (3.0 g, 10.7 mmol, 1.0 equiv.), anhydrous hydrazine (1.4 mL, 42.8 mmol, 4.0 equiv.), EtOH (20 mL, 0.5 M). 46f (2.59 g, 91.1%) was obtained as a white solid after extraction without further purification. 1H-NMR (500 MHz, CDCl3) δ 8.15 (br s, 1H), 7.22-7.19 (m, 2H), 6.95-6.91 (m, 2H), 3.84 (br s, 2H), 3.40 (s, 2H), 3.01 (s, 2H), 2.47 (br s, 4H), 2.39 (br s, 4H). 13C-NMR (125 MHz, CDCl3) δ 170.5, 162.0 (d, JC-F = 243.6 Hz), 133.7 (d, JC-F = 2.8 Hz), 130.6 (d, JC-F = 8.3 Hz), 115.1 (d, JC-F = 21.1 Hz), 62.0, 60.6, 53.7, 53.0. 19F-NMR (470 MHz, CDCl3) δ −118.9. HRMS (ESI): 267.1630 (M+H)+; calcd. for C13H20FN4O: 267.1621.
Ethyl 4-(4-Fluorobenzoyl)-1-piperazineacetate (51g)
Synthesized according to General Procedure B: 50 (2.58 g, 15.0 mmol, 1.0 equiv.), anhydrous tetrahydrofuran (30 mL, 0.5 M), freshly distilled Et3N (4.2 mL, 30.0 mmol, 2.0 equiv.), 4-fluorobenzoyl chloride (54g, 1.8 mL, 15.0 mmol, 1.0 equiv.). Purification by silica gel column chromatography (50–100% EtOAc/hexanes) afforded 51g (3.74 g, 84.7%) as a pale yellow oil. 1H-NMR (500 MHz, CDCl3) δ 1H-NMR (500 MHz, CDCl3) δ 7.38-7.34 (m, 2H), 7.06-7.01 (m, 2H), 4.13 (q, 2H, J = 7.0 Hz), 3.77 (br s, 2H), 3.43 (br s, 2H), 3.21 (s, 2H), 2.61 (br s, 2H), 2.52 (br s, 2H), 1.22 (t, 3H, J = 7.0 Hz). 13C-NMR (125 MHz, CDCl3) δ 170.0, 169.4, 163.5 (d, JC-F = 248.1 Hz), 131.8, 129.5 (d, JC-F = 8.3 Hz), 115.6 (d, JC-F = 22.0 Hz), 60.8, 59.1, 52.8 (br), 47.8 (br), 42.2 (br), 14.3. 19F-NMR (470 MHz, CDCl3) δ −113.4. HRMS (ESI): 295.1457 (M+H)+; calcd. for C15H20FN2O3: 295.1458.
4-(4-Fluorobenzoyl)-1-piperazineacetohydrazide (46g)
Synthesized according to General Procedure C: 51g (3.73 g, 12.7 mmol, 1.0 equiv.), anhydrous hydrazine (1.6 mL, 50.8 mmol, 4.0 equiv.), EtOH (25 mL, 0.5 M). 46g (2.28 g, 64.1%) was obtained as a white solid after extraction without further purification. 1H-NMR (500 MHz, CDCl3) δ 1H-NMR (500 MHz, CDCl3) δ 8.09 (br s, 1H), 7.37-7.33 (m, 2H), 7.06-7.02 (m, 2H), 3.85 (br s, 2H), 3.70 (br s, 2H), 3.42 (br s, 2H), 3.06 (s, 2H), 2.48 (br s, 4H). 13C-NMR (125 MHz, CDCl3) δ 169.8, 169.5, 163.5 (d, JC-F = 249.1 Hz), 131.5 (d, JC-F = 2.8 Hz), 129.4 (d, JC-F = 9.1 Hz), 115.7 (d, JC-F = 22.0 Hz), 60.6, 53.5 (br), 47.7 (br), 42.3 (br). 19F-NMR (470 MHz, CDCl3) δ −113.1. HRMS (ESI): 281.1409 (M+H)+; calcd. for C13H18FN4O2: 281.1414.
Ethyl 4-[4-(Trifluoromethyl)benzoyl]-1-piperazineacetate (51i)
Synthesized according to General Procedure B: 50 (2.58 g, 15.0 mmol, 1.0 equiv.), anhydrous tetrahydrofuran (30 mL, 0.5 M), freshly distilled Et3N (4.2 mL, 30.0 mmol, 2.0 equiv.), 4-(trifluoroemthyl)benzoyl chloride (54i, 2.2 mL, 15.0 mmol, 1.0 equiv.). Purification by silica-gel column chromatography (50–100% EtOAc/hexanes) afforded 51i (4.01 g, 77.5%) as a yellow oil. 1H-NMR (500 MHz, CDCl3) δ 7.65 (d, 2H, J = 8.0 Hz), 7.49 (d, 2H, J = 8.0 Hz), 4.16 (q, 2H, J = 7.0 Hz), 3.82 (br s, 2H), 3.41 (br s, 2H), 3.24 (s, 2H), 2.68 (br s, 2H), 2.54 (br s, 2H), 1.24 (t, 3H, J = 7.0 Hz). 13C-NMR (125 MHz, CDCl3)δ 170.1, 168.9, 139.4 131.8 (q, JC-F = 32.6 Hz), 127.6, 125.7 (q, JC-F = 3.8 Hz), 123.8 (q, JC-F = 271.0 Hz), 60.9, 59.1, 53.0 (br), 52.5 (br), 47.6 (br), 42.2 (br), 14.3. 19F-NMR (470 MHz, CDCl3) δ −66.0. HRMS (ESI): 345.1430 (M+H)+; calcd. for C16H20F3N2O3: 345.1426.
4-[4-(Trifluoromethyl)benzoyl]-1-piperazineacetohydrazide (46i)
Synthesized according to General Procedure C: 51i (4.00 g, 11.6 mmol, 1.0 equiv.), anhydrous hydrazine (1.5 mL, 46.4 mmol, 4.0 equiv.), EtOH (25 mL, 0.5 M). 46i (2.35 g, 61.4%) was obtained as a white solid after extraction without further purification. 1H-NMR (500 MHz, CDCl3) δ 8.09 (br s, 1H), 7.62 (d, 2H, J = 8.0 Hz), 7.45 (d, 2H, J = 8.0 Hz), 3.88 (br s, 2H) 3.75 (br s, 2H), 3.35 (br s, 2H), 3.07 (s, 2H), 2.56 (br s, 2H), 2.42 (br s, 2H). 13C-NMR (125 MHz, CDCl3)δ 169.8, 168.9, 139.1 131.8 (q, JC-F = 32.1 Hz), 127.5, 125.7 (q, JC-F = 3.6 Hz), 123.7 (q, JC-F = 271.1 Hz), 60.5, 53.7 (br), 53.2 (br), 47.6 (br), 42.2 (br). 19F-NMR (470 MHz, CDCl3) δ −66.0. HRMS (ESI): 331.1374 (M+H)+; calcd. for C14H18F3N4O2: 331.1382.
2-Hydroxy-3-propylbenzaldehyde (47b)
To a round-bottom flask were added aldehyde 47a (1.62 g, 10.0 mmol, 1.0 equiv.), 5% Pd/C (324 mg, 20 wt% of 47a), diphenyl sulfide (17 μL, 0.10 mmol, 0.010 equiv.), and EtOAc (40 mL, 0.25 M). The reaction mixture was stirred overnight at room temperature under an atmosphere of H2 (balloon pressure). The reaction mixture was filtered through Celite and washed thoroughly with EtOAc. The filtrate was concentrated to afford aldehyde 47b (1.50 g, 91.7%) as a yellow oil. 1H-NMR (500 MHz, CDCl3) δ 11.27 (s, 1H), 9.88 (s, 1H), 7.41-7.38 (m, 2H), 6.95 (t, 1H, J = 7.5 Hz), 2.64 (t, 2H, J = 7.5 Hz), 1.65 (sext, 2H, J = 7.5 Hz), 0.96 (t, 3H, J = 7.5 Hz). 13C-NMR (125 MHz, CDCl3) δ 197.0, 160.0, 137.4, 131.7, 131.4, 120.4, 119.6, 31.3, 22.7, 14.1. HRMS (EI): 164.08383 (M+); calcd. for C10H12O2: 164.08373.
5-Fluoro-2-(2-propenyloxy)benzaldehyde (53b)
To a round bottom flask were added 5-fluorosalicylaldehyde (47c, 4.0 g, 28.5 mmol, 1.0 equiv.), potassium carbonate (4.92 g, 35.6 mmol, 1.25 equiv.), and DMF (20 mL). Allyl bromide (3.7 mL, 42.8 mmol, 1.5 equiv.) was added slowly to the mixture. The reaction mixture was stirred overnight at room temperature. The reaction mixture was diluted with water (50 mL) and extracted with ethyl acetate (3 × 50 mL). The combined organic layers were washed with water (2 × 25 mL), 0.1M KOH (2 × 25 mL), water (2 × 25 mL), and brine (2 × 25 mL), dried over MgSO4, filtered, and concentrated to yield 53b (4.80 g, 93.3%) as a pale yellow liquid. 1H-NMR (500 MHz, CDCl3) δ 10.47 (d, 1H, J = 3.0 Hz), 7.50 (dd, 1H, J = 3.0, 8.0 Hz), 7.23 (ddd, 1H, J = 3.0, 7.5, 11.0 Hz), 6.95 (dd, 1H, J = 4.0, 9.0 Hz), 6.06 (tdd, 1H, J = 5.0, 10.5, 17.5 Hz), 5.44 (qd, 1H, J = 1.5, 17.0 Hz), 5.34 (ddd, 1H, J = 1.5, 2.5, 10.5 Hz), 4.64 (td, 2H, J = 1.5, 5.0 Hz). 13C-NMR (125 MHz, CDCl3) δ 188.8, 157.4 (d, JC-F = 1.9 Hz), 157.2 (d, JC-F = 240.5 Hz), 132.4, 126.1 (d, JC-F = 5.9 Hz), 122.6 (d, JC-F = 23.8 Hz), 118.5, 114.8 (d, JC-F = 7.1 Hz), 114.2 (d, JC-F = 23.1 Hz), 70.1. 19F-NMR (470 MHz, CDCl3) δ −125.5. HRMS (EI): 180.05789 (M+); calcd. for C10H9FO2: 180.05866.
5-Fluoro-2-hydroxy-3-(2-propenyl)benzaldehyde (47d)
53b (4.64 g, 25.8 mmol) was heated neat overnight at 200°C. The crude product was purified by silica gel column chromatography (gradient, 0–10% CH2Cl2/hexanes) to yield 47d (2.24 g, 48.3%) as a bright yellow oil. 1H-NMR (500 MHz, CDCl3) δ 11.10 (s, 1H), 9.83 (s, 1H), 7.17 (dd, 1H, J = 3.0, 9.0 Hz), 7.11 (dd, 1H, J = 3.0, 7.5 Hz) 5.96 (tdd, 1H, J = 6.5, 10.0, 17.0 Hz), 5.16-5.14 (m, 1H), 5.12 (qd, 1H, J = 1.5, 11.0 Hz), 3.42 (d, 2H, J = 6.5 Hz). 13C-NMR (125 MHz, CDCl3) δ 195.9 (d, JC-F = 2.5 Hz), 156.0 (d, JC-F = 1.0 Hz), 155.7 (d, JC-F = 238.8 Hz), 135.1, 131.6 (d, JC-F = 6.4 Hz), 124.8 (d, JC-F = 23.6 Hz), 119.8 (d, JC-F = 6.4 Hz), 117.3, 116.0 (d, JC-F = 22.3 Hz), 33.2. 19F-NMR (470 MHz, CDCl3) δ −126.9. HRMS (EI): 180.05761 (M+); calcd. for C10H9FO2: 180.05866.
5-Fluoro-2-hydroxy-3-propylbenzaldehyde (47e)
To a round-bottom flask were added aldehyde 47d (1.10 g, 6.11 mmol, 1.0 equiv.), 5% Pd/C (220 mg, 20 wt% of 47d), diphenyl sulfide (10 μL, 0.061 mmol, 0.010 equiv.), and EtOAc (25 mL, 0.25 M). The reaction mixture was stirred overnight at room temperature under an atmosphere of H2 (balloon pressure). The reaction mixture was filtered through Celite and washed thoroughly with EtOAc. The filtrate was concentrated to afford aldehyde 47e (991 mg, 89.3%) as a yellow oil. 1H-NMR (500 MHz, CDCl3) δ 11.06 (br s, 1H), 9.80 (s, 1H), 7.12 (dd, 1H, J = 3.0, 9.0 Hz), 7.06 (dd, 1H, J = 3.0, 7.5 Hz), 2.62 (t, 2H, J = 7.5 Hz), 1.63 (sext, 2H, J = 7.5 Hz), 0.96 (t, 3H, J = 7.5 Hz). 13C-NMR (125 MHz, CDCl3) δ 195.9 (d, JC-F = 2.5 Hz), 156.3 (d, JC-F = 1.0 Hz), 155.5 (d, JC-F = 238.1 Hz), 133.9 (d, JC-F = 6.3 Hz), 124.7 (d, JC-F = 23.1 Hz), 119.6 (d, JC-F = 6.5 Hz), 115.4 (d, JC-F = 22.4 Hz), 31.2, 22.4, 14.0. 19F-NMR (470 MHz, CDCl3) δ −127.3. HRMS (EI): 182.07392 (M+); calcd. for C10H11FO2: 182.07431.
General Procedure D: Synthesis of PAC-1 analogues
To a 16 × 150 mm test tube were added hydrazide (1.0 equiv.), aldehyde (1.0 equiv.), EtOH or 2:1 MeOH:MeCN (0.15 M), and 1.2 M HCl (7 mol%). The reaction mixture was shaken overnight at reflux on a Büchi Syncore parallel synthesizer. The reaction mixture was cooled to room temperature, concentrated, and purified by silica gel column chromatography or recrystallization to yield pure PAC-1 analogue.
N′-[2-hydroxy-3-(2-propenyl)phenylmethylene]-4-benzoyl-1-piperazineacetohydrazide (3)
Synthesized according to General Procedure D, but in a round-bottom flask: 46c (262 mg, 1.0 mmol, 1.0 equiv.), 47a (162 mg, 1.0 mmol, 1.0 equiv.), 1.2 M HCl (58 μL, 0.070 mmol, 0.070 equiv.), EtOH (7 mL, 0.15 M). Purification by silica gel column chromatography (gradient, 0–10% MeOH/EtOAc) yielded 3 (284 mg, 69.8%) as a white solid. 1H-NMR (500 MHz, CDCl3) δ 11.19 (s, 1H), 9.94 (br s, 1H), 8.45 (s, 1H), 7.46-7.41 (m, 5H), 7.20 (d, 1H, J = 6.5 Hz), 7.08 (dd, 1H, J = 1.5, 7.5 Hz), 6.85 (t, 1H, J = 7.0 Hz), 6.03 (tdd, 1H, J = 6.5, 10.0, 16.5 Hz), 5.10-5.05 (m, 2H), 3.88 (br s, 2H), 3.58 (s, 2H), 3.52 (br s, 2H), 3.45 (d, 2H, J = 6.5 Hz), 3.25 (s, 2H), 2.68 (br s, 2H), 2.61 (br s, 2H). 13C-NMR (125 MHz, CDCl3) δ 170.6, 165.4, 156.4, 151.6, 136.5, 135.4, 132.5, 130.2, 129.3, 128.8, 128.3, 127.1, 119.2, 116.9, 115.8, 61.0, 53.7, 53.1, 47.6, 42.1, 33.9. HRMS (ESI): 407.2077 (M+H)+; calcd. for C23H27N4O3: 407.2083. HPLC purity: 95%.
N′-[2-Hydroxy-3-(2-propenyl)phenylmethylene]-4-(4-cyanophenylmethyl)-1-piperazineacetohydrazide (4)
Synthesized according to General Procedure D: 46d (273 mg, 1.0 mmol, 1.0 equiv.), 47a (162 mg, 1.0 mmol, 1.0 equiv.), 1.2 M HCl (58 μL, 0.070 mmol, 0.070 equiv.), EtOH (7 mL, 0.15 M). Purification by silica gel column chromatography (gradient, 0–15% MeOH/EtOAc) yielded 4 (367 mg, 87.7%) as a white solid. 1H-NMR (500 MHz, CDCl3) δ 11.25 (br s, 1H), 9.99 (br s, 1H), 8.40 (s, 1H), 7.61 (d, 2H, J = 8.0 Hz), 7.44 (d, 2H, J = 8.0 Hz), 7.18 (dd, 1H, J = 1.5, 7.5 Hz), 7.07 (dd, 1H, J = 1.5, 7.5 Hz), 6.84 (t, 1H, J = 7.5 Hz), 6.02 (tdd, 1H, J = 6.5, 10.0, 16.5 Hz), 5.11-5.04 (m, 2H), 3.58 (s, 2H), 3.44 (d, 2H, J = 7.0 Hz), 3.19 (s, 2H), 2.63 (br s, 4H), 2.53 (br s, 4H). 13C-NMR (125 MHz, CDCl3) δ 165.9, 156.5, 151.5, 144.0, 136.6, 132.5, 132.4, 129.6, 129.3, 128.4, 119.2, 119.1, 117.0, 115.8, 111.2, 62.4, 61.1, 53.8, 53.2, 34.0. HRMS (ESI): 418.2242 (M+H)+; calcd. for C24H28N5O2: 418.2243. HPLC purity: 96%.
N′-[2-Hydroxy-3-(2-propenyl)phenylmethylene]-4-(4-cyanobenzoyl)-1-piperazineacetohydrazide (5)
Synthesized according to General Procedure D: 46e (287 mg, 1.0 mmol, 1.0 equiv.), 47a (162 mg, 1.0 mmol, 1.0 equiv.), 1.2 M HCl (58 μL, 0.070 mmol, 0.070 equiv.), EtOH (7 mL, 0.15 M). Purification by silica gel column chromatography (gradient, 0–10% MeOH/EtOAc) yielded 5 (378 mg, 87.6%) as a light yellow solid. 1H-NMR (500 MHz, CDCl3) δ 11.23 (br s, 1H), 9.98 (br s, 1H), 8.32 (s, 1H), 7.69 (d, 2H, J = 8.5 Hz), 7.48 (d, 2H, J = 8.0 Hz), 7.17 (d, 1H, J = 7.0 Hz), 6.99 (dd, 1H, J = 1.5, 8.0 Hz), 6.81 (t, 1H, J = 7.5 Hz), 5.98 (tdd, 1H, J = 6.5, 10.0, 17.0), 5.08-5.02 (m, 2H), 3.85 (br s, 2H), 3.42-3.39 (m, 4H), 3.23 (s, 2H), 2.68 (br s, 2H), 2.86 (br s, 4H). 13C-NMR (125 MHz, CDCl3) δ 168.4, 165.3, 156.4, 151.7, 139.7, 136.5, 132.6, 132.6, 129.4, 128.2, 127.9, 119.3, 118.1, 116.8, 115.9, 113.8, 60.9, 53.7 (br), 53.3 (br), 47.5 (br), 42.1 (br), 33.9. HRMS (ESI): 432.2034 (M+H)+; calcd. for C24H26N5O3: 432.2036. HPLC purity: 97%.
N′-[2-Hydroxy-3-(2-propenyl)phenylmethylene]-4-(4-fluorophenylmethyl)-1-piperazineacetohydrazide (6)
Synthesized according to General Procedure D: 46f (133 mg, 0.50 mmol, 1.0 equiv.), 47a (81 mg, 0.50 mmol, 1.0 equiv.), 1.2 M HCl (29 μL, 0.035 mmol, 0.070 equiv.), EtOH (3 mL, 0.15 M). Purification by silica gel column chromatography (gradient, 0–10% MeOH/EtOAc) followed by precipitation from Et2O yielded 6 (182 mg, 89.0%) as a white solid. 1H-NMR (500 MHz, CDCl3) δ 11.26 (br s, 1H), 10.02 (br s, 1H), 8.41 (s, 1H), 7.29-7.26 (m, 2H), 7.19 (dd, 1H, J = 1.5, 7.5 Hz), 7.08 (dd, 1H, J = 1.5, 8.0 Hz), 7.02-6.99 (m, 2H), 6.85 (t, 1H, J = 7.5 Hz), 6.03 (tdd, 1H, J = 6.5, 10.0, 16.5 Hz), 5.11-5.04 (m, 2H), 3.50 (s, 2H), 3.45 (d, 2H, J = 6.5 Hz), 3.19 (s, 2H), 2.62 (br s, 4H), 2.51 (br s, 4H). 13C-NMR (125 MHz, CDCl3) δ 166.0, 162.2 (d, JC-F = 243.9 Hz), 156.6, 151.5, 136.7, 133.7 (d, JC-F = 3.1 Hz), 132.5, 130.7 (d, JC-F = 7.8 Hz), 129.3, 128.4, 119.2, 117.0, 115.8, 115.3 (d, JC-F = 21.0 Hz), 62.2, 61.2, 53.9, 53.1, 34.0. 19F-NMR (470 MHz, CDCl3) δ −118.8. HRMS (ESI): 411.2203 (M+H)+; calcd. for C23H28FN4O2: 411.2196. HPLC purity: 98%.
N′-[2-Hydroxy-3-(2-propenyl)phenylmethylene]-4-(4-fluorobenzoyl)-1-piperazineacetohydrazide (7)
Synthesized according to General Procedure D: 46g (140 mg, 0.50 mmol, 1.0 equiv.), 47a (81 mg, 0.50 mmol, 1.0 equiv.), 1.2 M HCl (29 μL, 0.035 mmol, 0.070 equiv.), EtOH (3 mL, 0.15 M). Purification by silica gel column chromatography (gradient, 0–10% MeOH/EtOAc) yielded 7 (171 mg, 80.5%) as a white solid. 1H-NMR (500 MHz, CDCl3) δ 11.19 (br s, 1H), 9.91 (br s, 1H), 8.43 (s, 1H), 7.43-7.40 (m, 2H), 7.19 (dd, 1H, J = 1.0, 7.5 Hz), 7.12-7.09 (m, 2H), 7.06 (dd, 1H, J = 1.5, 8.0 Hz), 6.85 (t, 1H, J = 7.5 Hz), 6.02 (tdd, 1H, J = 6.5, 10.0, 16.5 Hz), 5.10-5.04 (m, 2H), 3.69 (br s, 4H), 3.44 (d, 2H, J = 6.5 Hz), 3.24 (s, 2H), 2.64 (br s, 4H). 13C-NMR (125 MHz, CDCl3) δ 169.6, 165.4, 163.6 (d, JC-F = 249.1 Hz), 156.4, 151.6, 136.5, 132.5, 131.4 (d, JC-F = 3.4 Hz), 129.5 (d, JC-F = 8.4 Hz), 129.3, 128.2, 119.2, 116.8, 115.8, 115.8 (d, JC-F = 21.5 Hz), 60.9, 53.6 (br), 47.7 (br), 42.3 (br), 33.9. 19F-NMR (470 MHz, CDCl3) δ −112.8. HRMS (ESI): 425.1989 (M+H)+; calcd. for C23H26FN4O3: 425.1989. HPLC purity: 97%.
N′-[2-Hydroxy-3-(2-propenyl)phenylmethylene]-4-[4-(trifluoromethyl)phenylmethyl]-1-piperazineacetohydrazide (8)
Synthesized according to General Procedure D: 46h (158 mg, 0.50 mmol, 1.0 equiv.), 47a (81 mg, 0.50 mmol, 1.0 equiv.), 1.2 M HCl (29 μL, 0.035 mmol, 0.070 equiv.), EtOH (3 mL, 0.15 M). Purification by silica gel column chromatography (gradient, 0–15% MeOH/EtOAc) yielded 8 (125 mg, 54.4%) as a white solid. 1H-NMR (500 MHz, CDCl3) δ 11.32 (br s, 1H), 10.11 (br s, 1H), 8.33 (s, 1H), 7.56 (d, 2H, J = 8.5 Hz), 7.43 (d, 2H, J = 8.0 Hz), 7.17 (dd, 1H, J = 1.5, 7.5 Hz), 7.04 (dd, 1H, J = 1.5, 8.0 Hz), 6.83 (t, 1H, J = 7.5 Hz), 6.02 (tdd, 1H, J = 6.5, 10.0, 16.5 Hz), 5.10-5.04 (m, 2H), 3.57 (s, 2H), 3.44 (d, 2H, J = 7.0 Hz), 3.19 (s, 2H), 2.63 (br s, 4H), 2.53 (br s, 4H). 13C-NMR (125 MHz, CDCl3) δ 166.0, 156.4, 151.2, 142.4, 136.6, 132.4, 129.5 (q, JC-F = 32.0 Hz), 129.3, 129.3, 128.3, 125.3 (q, JC-F = 3.8 Hz), 123.9 (q, JC-F = 270.6 Hz), 119.2, 117.0, 115.8, 62.3, 61.0, 53.7, 53.1, 34.0. 19F-NMR (470 MHz, CDCl3) δ −65.4. HRMS (ESI): 461.2160 (M+H)+; calcd. for C24H28F3N4O2: 461.2164. HPLC purity: 95%.
N′-[2-Hydroxy-3-(2-propenyl)phenylmethylene]-4-[4-(trifluoromethyl)benzoyl]-1-piperazineacetohydrazide (9)
Synthesized according to General Procedure D: 46i (165 mg, 0.50 mmol, 1.0 equiv.), 47a (81 mg, 0.50 mmol, 1.0 equiv.), 1.2 M HCl (29 μL, 0.035 mmol, 0.070 equiv.), EtOH (3 mL, 0.15 M). Purification by silica gel column chromatography (gradient, 0–15% MeOH/EtOAc) yielded 9 (211 mg, 89.1%) as a white solid. 1H-NMR (500 MHz, CDCl3) δ 11.28 (br s, 1H), 10.13 (br s, 1H), 8.27 (s, 1H), 7.65 (d, 2H, J = 8.0 Hz), 7.48 (d, 2H, J = 8.0 Hz), 7.15 (d, 1H, J = 8.0 Hz), 6.94 (d, 2H, J = 7.0 Hz), 6.79 (t, 1H, J = 7.5 Hz), 5.97 (tdd, 1H, J = 6.5, 10.0, 17.0 Hz), 5.05-5.00 (m, 2H), 3.86 (br s, 2H), 3.43 (br s, 2H), 3.39 (d, 2H, J = 6.5 Hz), 3.21 (s, 2H), 2.66 (br s, 2H), 2.58 (br s, 2H). 13C-NMR (125 MHz, CDCl3) δ 169.0, 165.4, 156.3, 151.5, 139.0, 136.4, 132.5, 131.9 (q, JC-F = 32.6 Hz), 129.3, 128.2, 127.5, 125.8 (q, JC-F = 3.5 Hz), 123.7 (q, JC-F = 271.1 Hz), 119.3, 116.8, 115.8, 60.8, 53.5 (br), 47.5 (br), 42.0 (br), 33.8. 19F-NMR (470 MHz, CDCl3) δ −66.0. HRMS (ESI): 475.1964 (M+H)+; calcd. for C24H26F3N4O3: 475.1957. HPLC purity: 97%.
N′-(2-Hydroxy-3-propylphenylmethylene)-4-phenylmethyl-1-piperazineacetohydrazide (10)
Synthesized according to General Procedure D, but in a round-bottom flask: 46a (248 mg, 1.0 mmol, 1.0 equiv.), 47b (164 mg, 1.0 mmol, 1.0 equiv.), 1.2 M HCl (58 μL, 0.070 mmol, 0.070 equiv.), EtOH (7 mL, 0.15 M). Purification by silica gel column chromatography (gradient, 0–20% MeOH/EtOAc) yielded 10 (345 mg, 87.3%) as an off-white solid. 1H-NMR (500 MHz, CDCl3) δ 11.30 (s, 1H), 10.12 (br s, 1H), 8.31 (s, 1H), 7.35-7.30 (m, 4H), 7.30-7.25 (m, 1H), 7.17 (d, 1H, J = 7.5 Hz), 7.03 (d, 1H, J = 7.5 Hz), 6.82 (t, 1H, J = 7.5 Hz), 3.54 (s, 2H), 3.19 (s, 2H), 2.67 (t, 2H, J = 7.5 Hz), 2.62 (br s, 4H), 2.54 (br s, 4H), 1.67 (sext, 2H, J = 7.5 Hz), 0.97 (t, 3H, J = 7.5 Hz). 13C-NMR (125 MHz, CDCl3) δ 165.9, 156.7, 151.2, 137.9, 132.5, 130.7, 129.2, 128.8, 128.4, 127.3, 118.9, 116.8, 62.9, 61.0, 53.7, 53.0, 32.0, 22.7, 14.2. HRMS (ESI): 395.2436 (M+H)+; calcd. for C23H31N4O2: 395.2447. HPLC purity: 98%.
4-{4-[N′-(2-Hydroxy-3-propylphenylmethylene)hydrazinecarbonylmethyl]-1-piperazinylmethyl}benzenesulfonamide (11)
Synthesized according to General Procedure D: 46b (164 mg, 0.50 mmol, 1.0 equiv.), 47b (82 mg, 0.50 mmol, 1.0 equiv.), 1.2 M HCl (29 μL, 0.035 mmol, 0.070 equiv.), 2:1 MeOH:MeCN (3 mL, 0.15 M). Purification by silica gel column chromatography (gradient, 0–20% MeOH/EtOAc) yielded 11 (211 mg, 89.0%) as a white solid. 1H-NMR (500 MHz, (CD3)2CO) δ 11.78 (s, 1H), 10.76 (br s, 1H), 8.48 (s, 1H), 7.84 (d, 2H, J = 8.5 Hz), 7.51 (d, 2H, J = 8.5 Hz), 7.17 (d, 1H, J = 7.0 Hz), 7.14 (dd, 1H, J = 1.5, 8.0 Hz), 6.82 (t, 1H, J = 7.5 Hz), 6.54 (br s, 2H), 3.59 (s, 2H), 3.17 (s, 2H), 2.64-2.59 (m, 6H), 2.52 (br s, 4H), 1.63 (sext, 2H, J = 7.5 Hz), 0.93 (t, 3H, J = 7.5 Hz). 13C-NMR (125 MHz, (CD3)2CO) δ 166.3, 157.3, 150.9, 144.0, 143.8, 132.7, 130.8, 129.9, 129.6, 126.9, 119.6, 118.3, 62.6, 61.7, 54.3, 53.6, 32.5, 23.4, 14.2. HRMS (ESI): 474.2175 (M+H)+; calcd. for C23H32N5O4S: 474.2175. HPLC purity: 95%.
N′-(2-Hydroxy-3-propylphenylmethylene)-4-benzoyl-1-piperazineacetohydrazide (12)
Synthesized according to General Procedure D: 46c (131 mg, 0.50 mmol, 1.0 equiv.), 47b (82 mg, 0.50 mmol, 1.0 equiv.), 1.2 M HCl (29 μL, 0.035 mmol, 0.070 equiv.), EtOH (3 mL, 0.15 M). Purification by silica gel column chromatography (gradient, 50–100% EtOAc/hexanes, then 5% MeOH/EtOAc) yielded 12 (174 mg, 85.5%) as a white solid. 1H-NMR (500 MHz, CDCl3) δ 11.29 (s, 1H), 10.29 (br s, 1H), 8.23 (s, 1H), 7.41-7.34 (m, 5H), 7.13 (dd, 1H, J = 1.5, 7.5 Hz), 6.90 (dd, 1H, J = 1.5, 7.5 Hz), 6.76 (t, 1H, J = 7.5 Hz), 3.80 (br s, 2H), 3.47 (br s, 2H), 3.18 (s, 2H), 2.71-2.52 (m, 6H, Ar-CH2-CH2), 1.61 (sext, 2H, J = 7.5 Hz), 0.91 (t, 3H, J = 7.5 Hz). 13C-NMR (125 MHz, CDCl3) δ 170.5, 165.5, 156.6, 151.5, 135.3, 132.5, 130.6, 130.1, 128.8, 128.7, 127.0, 118.9, 116.7, 60.8, 53.6 (br), 53.0 (br), 47.5 (br), 42.0 (br), 31.9, 22.7, 14.1. HRMS (ESI): 409.2238 (M+H)+; calcd. for C23H29N4O3: 409.2240. HPLC purity: 98%.
N′-(2-Hydroxy-3-propylphenylmethylene)-4-(4-cyanophenylmethyl)-1-piperazineacetohydrazide (13)
Synthesized according to General Procedure D: 46d (273 mg, 1.0 mmol, 1.0 equiv.), 47b (164 mg, 1.0 mmol, 1.0 equiv.), 1.2 M HCl (58 μL, 0.070 mmol, 0.070 equiv.), EtOH (7 mL, 0.15 M). Purification by silica gel column chromatography (gradient, 0–15% MeOH/EtOAc) yielded 13 (373 mg, 88.8%) as a white solid. 1H-NMR (500 MHz, CDCl3) δ 11.19 (br s, 1H), 9.99 (br s, 1H), 8.37 (s, 1H), 7.60 (d, 2H, J = 8.0 Hz), 7.44 (d, 2H, J = 7.5 Hz), 7.16 (dd, 1H, J = 1.5, 7.5 Hz), 7.04 (dd, 1H, J = 1.5, 7.5 Hz), 6.82 (t, 1H, J = 7.5 Hz), 3.58 (s, 2H), 3.19 (s, 2H), 2.66-2.63 (m, 6H), 2.52 (br s, 4H), 1.64 (sext, 2H, J = 7.5 Hz), 0.95 (t, 3H, J = 7.5 Hz). 13C-NMR (125 MHz, CDCl3) δ 165.8, 156.8, 151.6, 144.0, 132.7, 132.3, 130.8, 129.6, 128.9, 119.1, 119.0, 116.8, 111.2, 62.4, 61.1, 53.8, 53.2, 32.0, 22.8, 14.2. HRMS (ESI): 420.2396 (M+H)+; calcd. for C24H30N5O2: 420.2400. HPLC purity: 97%.
N′-(2-Hydroxy-3-propylphenylmethylene)-4-(4-cyanobenzoyl)-1-piperazineacetohydrazide (14)
Synthesized according to General Procedure D: 46e (287 mg, 1.0 mmol, 1.0 equiv.), 47b (164 mg, 1.0 mmol, 1.0 equiv.), 1.2 M HCl (58 μL, 0.070 mmol, 0.070 equiv.), EtOH (7 mL, 0.15 M). Purification by silica gel column chromatography (gradient, 0-15% MeOH/EtOAc) yielded 14 (377 mg, 86.9%) as a light yellow solid. 1H-NMR (500 MHz, CDCl3) δ 11.15 (br s, 1H), 9.92 (br s, 1H), 8.32 (s, 1H), 7.71 (d, 2H, J = 8.0 Hz), 7.49 (d, 2H, J = 7.5 Hz), 7.16 (d, 1H, J = 7.0 Hz), 6.98 (dd, 1H, J = 1.5, 7.5 Hz), 6.80 (t, 1H, J = 7.5 Hz), 3.86 (br s, 2H), 3.44 (br s, 2H), 3.24 (s, 2H), 2.70 (br s, 2H), 2.64-2.57 (m, 4H), 1.62 (sext, 2H, J = 7.5 Hz), 0.93 (t, 3H, J = 7.5 Hz). 13C-NMR (125 MHz, CDCl3) δ 168.5, 165.2, 156.7, 152.0, 139.8, 132.9, 132.7, 130.8, 129.0, 127.9, 119.1, 118.1, 116.7, 113.9, 61.0, 53.5 (br), 47.5 (br), 42.1 (br), 32.0, 22.8, 14.2. HRMS (ESI): 434.2188 (M+H)+; calcd. for C24H28N5O3: 434.2192. HPLC purity: 98%.
N′-(2-Hydroxy-3-propylphenylmethylene)-4-(4-fluorophenylmethyl)-1-piperazineacetohydrazide (15)
Synthesized according to General Procedure D: 46f (133 mg, 0.50 mmol, 1.0 equiv.), 47b (82 mg, 0.50 mmol, 1.0 equiv.), 1.2 M HCl (29 μL, 0.035 mmol, 0.070 equiv.), EtOH (3 mL, 0.15 M). Purification by silica gel column chromatography (gradient, 0–20% MeOH/EtOAc) followed by precipitation from Et2O yielded 15 (137 mg, 66.4%) as a white solid. 1H-NMR (500 MHz, CDCl3) δ 11.26 (br s, 1H), 10.09 (br s, 1H), 8.31 (s, 1H), 7.26 (dd, 2H, J = 6.0, 8.0 Hz), 7.16 (dd, 1H, J = 1.5, 6.5 Hz), 7.02-6.97 (m, 3H), 6.80 (t, 1H, J = 7.5 Hz), 3.48 (s, 2H), 3.18 (s, 2H), 2.65 (t, 2H, J = 7.5 Hz), 2.61 (br s, 4H), 2.50 (br s, 4H), 1.65 (sext, 2H, J = 7.5 Hz), 0.95 (t, 3H, J = 7.5 Hz). 13C-NMR (125 MHz, CDCl3) δ 165.9, 162.1 (d, JC-F = 243.6 Hz), 156.7, 151.3, 133.7 (d, JC-F = 3.0 Hz), 132.5, 130.7, 130.6 (d, JC-F = 7.8 Hz), 128.9, 118.9, 116.8, 115.2 (d, JC-F = 21.0 Hz), 62.1, 61.0, 53.8, 53.0, 32.0, 22.8, 14.2. 19F-NMR (470 MHz, CDCl3) δ −118.8. HRMS (ESI): 413.2361 (M+H)+; calcd. for C23H30FN4O2: 413.2353. HPLC purity: 97%.
N′-(2-Hydroxy-3-propylphenylmethylene)-4-(4-fluorobenzoyl)-1-piperazineacetohydrazide (16)
Synthesized according to General Procedure D: 46g (140 mg, 0.50 mmol, 1.0 equiv.), 47b (82 mg, 0.50 mmol, 1.0 equiv.), 1.2 M HCl (29 μL, 0.035 mmol, 0.070 equiv.), EtOH (3 mL, 0.15 M). Purification by silica gel column chromatography (gradient, 0–15% MeOH/EtOAc) yielded 16 (133 mg, 62.4%) as a white solid. 1H-NMR (500 MHz, CDCl3) δ 11.24 (br s, 1H), 10.17 (br s, 1H), 8.25 (s, 1H), 7.37 (dd, 2H, J = 5.5, 8.5 Hz), 7.13 (dd, 1H, J = 1.5, 7.5 Hz), 7.06 (t, 2H, J = 8.5 Hz), 6.91 (dd, 1H, J = 1.5, 7.5 Hz), 6.77 (t, 1H, J = 7.5 Hz), 3.83 (br s, 2H), 3.49 (br s, 2H), 3.20 (s, 2H), 2.62-2.58 (m, 6H), 1.60 (sext, 2H, J = 7.5 Hz), 0.91 (t, 3H, J = 7.5 Hz). 13C-NMR (125 MHz, CDCl3) δ 169.6, 165.4, 163.6 (d, JC-F = 249.3 Hz), 156.6, 151.7, 132.6, 131.4 (d, JC-F = 3.4 Hz), 129.5 (d, JC-F = 8.5 Hz), 128.9, 119.0, 116.7, 115.8 (d, JC-F = 21.8 Hz), 60.9, 53.5 (br), 47.7 (br), 42.2 (br), 31.9, 22.7, 14.1. 19F-NMR (470 MHz, CDCl3) δ −112.8. HRMS (ESI): 427.2141 (M+H)+; calcd. for C23H28FN4O3: 427.2145. HPLC purity: 96%.
N′-(2-Hydroxy-3-propylphenylmethylene)-4-[4-(trifluoromethyl)phenylmethyl]-1-piperazineacetohydrazide (17)
Synthesized according to General Procedure D: 46h (158 mg, 0.50 mmol, 1.0 equiv.), 47b (82 mg, 0.50 mmol, 1.0 equiv.), 1.2 M HCl (29 μL, 0.035 mmol, 0.070 equiv.), EtOH (3 mL, 0.15 M). Purification by silica gel column chromatography (gradient, 0–15% MeOH/EtOAc) yielded 17 (93.9 mg, 40.6%) as a yellow solid. 1H-NMR (500 MHz, CDCl3) δ 11.23 (br s, 1H), 10.05 (br s, 1H), 8.33 (s, 1H), 7.57 (d, 2H, J = 8.0 Hz), 7.44 (d, 2H, J = 8.0 Hz), 7.16 (dd, 1H, J = 1.5, 7.5 Hz), 7.02 (dd, 2H, J = 1.5, 7.5 Hz), 6.81 (t, 1H, J = 7.5 Hz), 3.58 (s, 2H), 3.19 (s, 2H), 2.67-2.62 (m, 6H), 2.53 (br s, 4H), 1.65 (sext, 2H, J = 7.5 Hz), 0.95 (t, 3H, J = 7.5 Hz). 13C-NMR (125 MHz, CDCl3) δ 165.9, 156.8, 151.5, 142.4, 132.6, 130.8, 129.6 (q, JC-F = 32.0 Hz), 129.3, 128.9, 125.4 (q, JC-F = 3.6 Hz), 124.4 (q, JC-F = 270.5 Hz), 119.0, 116.9, 62.4, 61.1, 53.8, 53.2, 32.0, 22.8, 14.2. 19F-NMR (470 MHz, CDCl3) δ −65.5. HRMS (ESI): 463.2321 (M+H)+; calcd. for C24H30F3N4O2: 463.2321. HPLC purity: 98%.
N′-(2-Hydroxy-3-propylphenylmethylene)-4-[4-(trifluoromethyl)benzoyl]-1-piperazineacetohydrazide (18)
Synthesized according to General Procedure D: 46i (165 mg, 0.50 mmol, 1.0 equiv.), 47b (82 mg, 0.50 mmol, 1.0 equiv.), 1.2 M HCl (29 μL, 0.035 mmol, 0.070 equiv.), EtOH (3 mL, 0.15 M). Purification by silica gel column chromatography (gradient, 0–15% MeOH/EtOAc) yielded 18 (216 mg, 90.8%) as a white solid. 1H-NMR (500 MHz, CDCl3) δ 11.24 (br s, 1H), 10.14 (br s, 1H), 8.23 (s, 1H), 7.64 (d, 2H, J = 8.0 Hz), 7.47 (d, 2H, J = 8.0 Hz), 7.13 (d, 1H, J = 8.0 Hz), 6.89 (d, 1H, J = 7.5 Hz), 6.76 (t, 1H, J = 7.5 Hz), 3.85 (br s, 2H), 3.43 (br s, 2H), 3.21 (s, 2H), 2.73-2.58 (m, 6H), 1.60 (sext, 2H, J = 7.5 Hz), 0.90 (t, 3H, J = 7.5 Hz). 13C-NMR (125 MHz, CDCl3) δ 169.0, 165.4, 156.5, 151.6, 139.0, 132.7, 131.9 (q, JC-F = 32.5 Hz), 130.6, 128.8, 127.5, 125.8 (q, JC-F = 3.5 Hz), 123.7 (q, JC-F = 271.3 Hz), 119.0, 116.7, 60.8, 53.5 (br), 47.5 (br), 42.0 (br), 31.9, 22.7, 14.1. 19F-NMR (470 MHz, CDCl3) δ −66.0. HRMS (ESI): 477.2108 (M+H)+; calcd. for C24H28F3N4O3: 477.2114. HPLC purity: 99%.
N′-(5-Fluoro-2-hydroxyphenylmethylene)-4-phenylmethyl-1-piperazineacetohydrazide (19)
Synthesized according to General Procedure D: 46a (124 mg, 0.50 mmol, 1.0 equiv.), 47c (70 mg, 0.50 mmol, 1.0 equiv.), 1.2 M HCl (29 μL, 0.035 mmol, 0.070 equiv.), EtOH (3 mL, 0.15 M). Purification by silica gel column chromatography (gradient, 0–20% MeOH/EtOAc) yielded 19 (173 mg, 93.7%) as a white solid. 1H-NMR (500 MHz, CDCl3) δ 10.81 (br s, 1H), 10.13 (br s, 1H), 8.39 (s, 1H), 7.33-7.30 (m, 4H), 7.28-7.25 (m, 1H), 7.00 (dt, 1H, J = 3.0, 9.0 Hz), 6.94-6.89 (m, 2H), 3.55 (s, 2H), 3.19 (s, 2H), 2.63 (br s, 4H), 2.53 (br s, 4H). 13C-NMR (125 MHz, CDCl3) δ 166.3, 155.9 (d, JC-F = 235.8 Hz), 154.8, 150.0, 137.9, 129.3, 128.5, 127.4, 118.9 (d, JC-F = 23.1 Hz), 118.4 (d, JC-F = 7.6 Hz), 117.6 (d, JC-F = 7.5 Hz), 116.1 (d, JC-F = 23.8 Hz), 63.0, 61.1, 53.9, 53.1. 19F-NMR (470 MHz, CDCl3) δ −128.5. HRMS (ESI): 371.1877 (M+H)+; calcd. for C20H24FN4O2: 371.1883. HPLC purity: 95%.
4-{4-[N′-(5-Fluoro-2-hydroxyphenylmethylene)hydrazinecarbonylmethyl]-1-piperazinylmethyl}benzenesulfonamide (20)
Synthesized according to General Procedure D: 46b (164 mg, 0.50 mmol, 1.0 equiv.), 47c (70 mg, 0.50 mmol, 1.0 equiv.), 1.2 M HCl (29 μL, 0.035 mmol, 0.070 equiv.), 2:1 MeOH:MeCN (3 mL, 0.15 M). Purification by silica gel column chromatography (gradient, 0–20% MeOH/EtOAc) yielded 20 (172 mg, 82.1%) as a yellow solid. 1H-NMR (500 MHz, (CD3)2CO) δ 11.33 (br s, 1H), 10.95 (br s, 1H), 8.49 (s, 1H), 7.85 (d, 2H, J = 8.0 Hz), 7.49 (d, 2H, J = 8.0 Hz), 7.12 (dd, 1H, J = 3.0, 9.0 Hz), 7.07 (dt, 1H, J = 3.0, 8.5 Hz), 6.91 (dd, 1H, J = 5.0, 9.0 Hz), 6.62 (br s, 2H), 3.55 (s, 2H), 3.19 (s, 2H), 2.59 (br s, 4H), 2.49 (br s, 4H). 13C-NMR (125 MHz, (CD3)2CO) δ 166.8, 156.4 (d, JC-F = 233.5 Hz), 155.4, 155.1 (d, JC-F = 2.8 Hz), 143.9, 143.6, 129.9, 126.7, 119.2 (d, J C-F = 7.6 Hz), 118.7 (d, JC-F = 17.8 Hz), 118.6, 116.6 (d, JC-F = 23.9 Hz), 62.5, 61.5, 54.1, 53.4. 19F-NMR (470 MHz, (CD3)2CO) δ −127.3. HRMS (ESI): 450.1609 (M+H)+; calcd. for C20H25FN5O4S: 450.1611. HPLC purity: 92%.
N′-(5-Fluoro-2-hydroxyphenylmethylene)-4-benzoyl-1-piperazineacetohydrazide (21)
Synthesized according to General Procedure D: 46c (131 mg, 0.50 mmol, 1.0 equiv.), 47c (70 mg, 0.50 mmol, 1.0 equiv.), 1.2 M HCl (29 μL, 0.035 mmol, 0.070 equiv.), EtOH (3 mL, 0.15 M). Purification by silica gel column chromatography (gradient, 0–20% MeOH/EtOAc) yielded 21 (157 mg, 81.6%) as a white solid. 1H-NMR (500 MHz, CDCl3) δ 10.89 (br s, 1H), 10.50 (br s, 1H), 8.21 (s, 1H), 7.40-7.33 (m, 5H), 6.93 (dt, 1H, J = 2.5, 8.5 Hz), 6.84 (dd, 1H, J = 4.5, 9.0 Hz), 6.74 (dd, 1H, J = 2.5, 8.5 Hz), 3.79 (br s, 2H), 3.48 (br s, 2H), 3.17 (s, 2H), 2.60 (br s, 2H), 2.53 (br s, 2H). 13C-NMR (125 MHz, CDCl3) δ 170.5, 165.8, 155.7 (d, JC-F = 235.8 Hz), 154.5, 149.7, 135.3, 130.1, 128.7, 127.0, 118.8 (d, JC-F = 23.1 Hz), 118.2 (d, JC-F = 7.5 Hz), 117.6 (d, JC-F = 7.4 Hz), 116.0 (d, JC-F = 23.6 Hz), 60.7, 53.6 (br), 53.4 (br), 47.6 (br), 42.0 (br). 19F-NMR (470 MHz, CDCl3) δ −128.3. HRMS (ESI): 385.1674 (M+H)+; calcd. for C20H22FN4O3: 385.1676. HPLC purity: 97%.
N′-(5-Fluoro-2-hydroxyphenylmethylene)-4-(4-cyanophenylmethyl)-1-piperazineacetohydrazide (22)
Synthesized according to General Procedure D: 46d (137 mg, 0.50 mmol, 1.0 equiv.), 47c (70 mg, 0.50 mmol, 1.0 equiv.), 1.2 M HCl (29 μL, 0.035 mmol, 0.070 equiv.), EtOH (3 mL, 0.15 M). Purification by silica gel column chromatography (gradient, 0–20% MeOH/EtOAc) yielded 22 (169 mg, 85.5%) as a white solid. 1H-NMR (500 MHz, CDCl3) δ 10.84 (br s, 1H), 10.20 (br s, 1H), 8.31 (s, 1H), 7.56 (d, 2H, J = 8.5 Hz), 7.41 (d, 1H, J = 8.0 Hz), 6.95 (dt, 1H, J = 3.0, 9.0 Hz), 6.88-6.85 (m, 2H), 3.54 (s, 2H), 3.18 (s, 2H), 2.60 (br s, 4H), 2.50 (br s, 4H). 13C-NMR (125 MHz, CDCl3) δ 166.2, 155.7 (d, JC-F = 235.9 Hz), 154.6, 149.6, 144.0, 132.2, 129.5, 118.9 (d, JC-F = 22.6 Hz), 118.6, 118.2 (d, JC-F = 7.6 Hz), 117.5 (d, JC-F = 7.5 Hz), 116.0 (d, JC-F = 23.8 Hz), 110.9, 62.2, 61.0, 53.6, 53.0. 19F-NMR (470 MHz, CDCl3) δ −128.4. HRMS (ESI): 396.1838 (M+H)+; calcd. for C21H23FN5O2: 396.1836. HPLC purity: 94%.
N′-(5-Fluoro-2-hydroxyphenylmethylene)-4-(4-cyanobenzoyl)-1-piperazineacetohydrazide (23)
Synthesized according to General Procedure D: 46e (144 mg, 0.50 mmol, 1.0 equiv.), 47c (70 mg, 0.50 mmol, 1.0 equiv.), 1.2 M HCl (29 μL, 0.035 mmol, 0.070 equiv.), EtOH (3 mL, 0.15 M). Purification by silica gel column chromatography (gradient, 0–20% MeOH/EtOAc) yielded 23 (144 mg, 70.1%) as a white solid. 1H-NMR (500 MHz, CDCl3) δ 10.79 (br s, 1H), 10.16 (br s, 1H), 8.28 (s, 1H), 7.67 (d, 2H, J = 8.0 Hz), 7.47 (d, 2H, J = 8.5 Hz), 6.95 (dt, 1H, J = 3.0, 8.0 Hz), 6.85 (dd, 1H, J = 4.5, 9.0 Hz), 6.79 (dd, 1H, J = 3.0, 8.5 Hz), 3.82 (br s, 2H), 3.41 (br s, 2H), 3.22 (s, 2H), 2.67 (br s, 2H), 2.54 (br s, 2H). 13C-NMR (125 MHz, CDCl3) δ 168.4, 165.5, 155.7 (d, JC-F = 236.1 Hz), 154.5, 149.9, 139.7, 132.6, 127.8, 119.0 (d, JC-F = 23.1 Hz), 118.2 (d, JC-F = 7.6 Hz), 118.1, 117.4 (d, JC-F = 7.5 Hz), 116.0 (d, JC-F = 23.6 Hz), 113.7, 60.8, 53.4 (br), 52.7 (br), 47.4 (br), 42.0 (br). 19F-NMR (470 MHz, CDCl3) δ −128.1. HRMS (ESI): 410.1623 (M+H)+; calcd. for C21H21FN5O3: 410.1628. HPLC purity: 96%.
N′-(5-Fluoro-2-hydroxyphenylmethylene)-4-(4-fluorophenylmethyl)-1-piperazineacetohydrazide (24)
Synthesized according to General Procedure D: 46f (133 mg, 0.50 mmol, 1.0 equiv.), 47c (70 mg, 0.50 mmol, 1.0 equiv.), 1.2 M HCl (29 μL, 0.035 mmol, 0.070 equiv.), EtOH (3 mL, 0.15 M). Purification by silica gel column chromatography (gradient, 0–20% MeOH/EtOAc) yielded 24 (152 mg, 78.2%) as a pale yellow solid. 1H-NMR (500 MHz, CDCl3) δ 10.83 (br s, 1H), 10.19 (br s, 1H), 8.33 (s, 1H), 7.25 (dd, 2H, J = 5.5, 8.5 Hz), 6.99-6.95 (m, 3H), 6.90-6.86 (m, 2H), 3.47 (s, 2H), 3.18 (s, 2H), 2.60 (br s, 4H), 2.49 (br s, 4H). 13C-NMR (125 MHz, CDCl3) δ 166.3, 162.1 (d, JC-F = 243.9 Hz), 155.8 (d, JC-F = 236.0 Hz), 154.7 (d, JC-F = 1.4 Hz), 149.7 (d, JC-F = 2.6 Hz), 133.7 (d, JC-F = 3.0 Hz), 130.6 (d, JC-F = 7.9 Hz), 118.8 (d, JC-F = 23.3 Hz), 118.3 (d, JC-F = 7.6 Hz), 117.6 (d, JC-F = 7.5 Hz), 116.0 (d, JC-F = 23.8 Hz), 115.2 (d, JC-F = 21.1 Hz), 62.1, 61.0, 53.8, 52.9. 19F-NMR (470 MHz, CDCl3) δ −118.8, −128.4. HRMS (ESI): 389.1787 (M+H)+; calcd. for C20H23F2N4O2: 389.1789. HPLC purity: 94%.
N′-(5-Fluoro-2-hydroxyphenylmethylene)-4-(4-fluorobenzoyl)-1-piperazineacetohydrazide (25)
Synthesized according to General Procedure D: 46g (140 mg, 0.50 mmol, 1.0 equiv.), 47c (70 mg, 0.50 mmol, 1.0 equiv.), 1.2 M HCl (29 μL, 0.035 mmol, 0.070 equiv.), EtOH (3 mL, 0.15 M). Purification by silica gel column chromatography (gradient, 0–20% MeOH/EtOAc) yielded 25 (101 mg, 50.1%) as a white solid. 1H-NMR (500 MHz, CDCl3) δ 10.81 (br s, 1H), 10.25 (br s, 1H), 8.29 (s, 1H), 7.38 (dd, 2H, J = 5.5, 8.5 Hz), 7.07 (t, 2H, J = 8.5 Hz), 6.98-6.94 (m, 1H), 6.86 (dd, 1H, J = 4.0, 9.0 Hz), 6.79 (dd, 1H, J = 2.0, 8.0 Hz), 3.63 (br s, 4H), 3.21 (s, 2H), 2.59 (br s, 4H). 13C-NMR (125 MHz, CDCl3) δ 169.7, 165.7, 163.6 (d, JC-F = 249.4 Hz), 155.8 (d, JC-F = 236.3 Hz), 154.6 (d, JC-F = 0.9 Hz), 150.0 (d, JC-F = 2.3 Hz), 131.3 (d, JC-F = 3.4 Hz), 129.5 (d, JC-F = 8.4 Hz), 119.0 (d, JC-F = 23.1 Hz), 118.3 (d, JC-F = 7.5 Hz), 117.5 (d, JC-F = 7.3 Hz), 116.0 (d, JC-F = 24.6 Hz), 115.8 (d, JC-F = 21.9 Hz), 60.9, 53.5, 47.7, 42.2. 19F-NMR (470 MHz, CDCl3) δ −112.6, −128.2. HRMS (ESI): 403.1573 (M+H)+; calcd. for C20H21F2N4O3: 403.1582. HPLC purity: 96%.
N′-(5-Fluoro-2-hydroxyphenylmethylene)-4-[4-(trifluoromethyl)phenylmethyl]-1-piperazineacetohydrazide (26)
Synthesized according to General Procedure D: 46h (158 mg, 0.50 mmol, 1.0 equiv.), 47c (70 mg, 0.50 mmol, 1.0 equiv.), 1.2 M HCl (29 μL, 0.035 mmol, 0.070 equiv.), EtOH (3 mL, 0.15 M). Purification by silica gel column chromatography (gradient, 0–20% MeOH/EtOAc) yielded 26 (194 mg, 88.6%) as a pale yellow solid. 1H-NMR (500 MHz, CDCl3) δ 10.83 (br s, 1H), 10.17 (br s, 1H), 8.34 (s, 1H), 7.56 (d, 2H, J = 8.0 Hz), 7.43 (d, 2H, J = 8.0 Hz), 6.98 (dt, 1H, J = 3.0, 8.0 Hz), 6.91-6.86 (m, 2H), 3.57 (s, 2H), 3.19 (s, 2H), 2.62 (br s, 4H), 2.52 (br s, 4H). 13C-NMR (125 MHz, CDCl3) δ 166.3, 155.8 (d, JC-F = 236.0 Hz), 154.7 (d, JC-F = 1.5 Hz), 149.8 (d, JC-F = 2.4 Hz), 142.4 (d, JC-F = 0.8 Hz), 129.5 (q, JC-F = 32.1 Hz), 129.3, 125.3 (q, JC-F = 3.8 Hz), 124.4 (q, JC-F = 270.6 Hz), 118.9 (d, JC-F = 23.0 Hz), 118.3 (d, JC-F = 7.6 Hz), 117.6 (d, JC-F = 7.5 Hz), 116.1 (d, JC-F = 23.6 Hz), 62.3, 61.0, 53.8, 53.1. 19F-NMR (470 MHz, CDCl3) δ −65.4, −128.4. HRMS (ESI): 439.1765 (M+H)+; calcd. for C21H23F4N4O2: 439.1757. HPLC purity: 96%.
N′-(5-Fluoro-2-hydroxyphenylmethylene)-4-[4-(trifluoromethyl)benzoyl]-1-piperazineacetohydrazide (27)
Synthesized according to General Procedure D: 46i (165 mg, 0.50 mmol, 1.0 equiv.), 47c (70 mg, 0.50 mmol, 1.0 equiv.), 1.2 M HCl (29 μL, 0.035 mmol, 0.070 equiv.), EtOH (3 mL, 0.15 M). Purification by silica gel column chromatography (gradient, 0–20% MeOH/EtOAc) yielded 27 (173 mg, 76.5%) as a white solid. 1H-NMR (500 MHz, CDCl3) δ 10.80 (br s, 1H), 10.20 (br s, 1H), 8.28 (s, 1H), 7.65 (d, 2H, J = 8.0 Hz), 7.48 (d, 2H, J = 7.5 Hz), 6.96 (dt, 1H, J = 2.5, 8.0 Hz), 6.87 (dd, 1H, J = 4.5, 8.5 Hz), 6.79 (dd, 1H, J = 2.5, 8.0 Hz), 3.84 (br s, 2H), 3.44 (br s, 2H), 3.22 (s, 2H), 2.70 (br s, 2H), 2.55 (br s, 2H). 13C-NMR (125 MHz, CDCl3) δ 169.1, 165.6, 155.8 (d, JC-F = 236.3 Hz), 154.6 (d, JC-F = 1.3 Hz), 150.0 (d, JC-F = 1.9 Hz), 138.9, 132.0 (q, JC-F = 32.6 Hz), 127.5, 125.8 (q, JC-F = 3.6 Hz), 123.7 (q, JC-F = 271.3 Hz), 119.0 (d, JC-F = 23.1 Hz), 118.3 (d, JC-F = 7.6 Hz), 117.4 (d, JC-F = 7.5 Hz), 116.0 (d, JC-F = 23.8 Hz), 60.8, 53.5 (br), 47.5 (br), 42.0 (br). 19F-NMR (470 MHz, CDCl3) δ −66.0, −128.1. HRMS (ESI): 453.1552 (M+H)+; calcd. for C21H21F4N4O3: 453.1550. HPLC purity: 97%.
N′-[5-Fluoro-2-hydroxy-3-(2-propenyl)phenylmethylene]-4-phenylmethyl-1-piperazineacetohydrazide (28)
Synthesized according to General Procedure D: 46a (124 mg, 0.50 mmol, 1.0 equiv.), 47d (90 mg, 0.50 mmol, 1.0 equiv.), 1.2 M HCl (29 μL, 0.035 mmol, 0.070 equiv.), EtOH (3 mL, 0.15 M). Purification by silica gel column chromatography (gradient, 0–20% MeOH/EtOAc) yielded 28 (186 mg, 90.8%) as a white solid. 1H-NMR (500 MHz, CDCl3) δ 11.16 (s, 1H), 10.20 (br s, 1H), 8.28 (s, 1H), 7.34-7.30 (m, 4H), 7.28-7.25 (m, 1H), 6.91 (dd, 1H, J = 3.0, 9.0 Hz), 6.74 (dd, 1H, J = 3.0, 8.0 Hz), 5.98 (tdd, 1H, J = 6.5, 10.0, 16.5 Hz), 5.13-5.08 (m, 2H), 3.54 (s, 2H), 3.42 (d, 2H, J = 7.0 Hz), 3.20 (s, 2H), 2.63 (br s, 4H), 2.53 (br s, 4H). 13C-NMR (125 MHz, CDCl3) δ 166.2, 155.5 (d, JC-F = 235.8 Hz), 152.5 (d, JC-F = 1.4 Hz), 149.8 (d, JC-F = 2.5 Hz), 137.9, 135.7, 130.2 (d, JC-F = 6.8 Hz), 129.2, 128.4, 127.3, 119.0 (d, JC-F = 23.1 Hz), 116.8 (d, JC-F = 7.9 Hz), 116.6, 113.9 (d, JC-F = 23.5 Hz), 62.9, 61.0, 53.8, 53.0, 33.8. 19F-NMR (470 MHz, CDCl3) δ −128.6. HRMS (ESI): 411.2191 (M+H)+; calcd. for C23H28FN4O2: 411.2196. HPLC purity: 99%.
4-{4-[N′-(5-Fluoro-2-hydroxy-3-(2-propenyl)phenylmethylene)hydrazinecarbonylmethyl]-1-piperazinylmethyl}benzensulfonamide (29)
Synthesized according to General Procedure D: 46b (164 mg, 0.50 mmol, 1.0 equiv.), 47d (90 mg, 0.50 mmol, 1.0 equiv.), 1.2 M HCl (29 μL, 0.035 mmol, 0.070 equiv.), EtOH (3 mL, 0.15 M). Purification by silica gel column chromatography (gradient, 0–20% MeOH/EtOAc) yielded 29 (214 mg, 87.2%) as a white solid. 1H-NMR (500 MHz, (CD3)2CO) δ 11.73 (br s, 1H), 10.94 (br s, 1H), 8.46 (s, 1H), 7.85 (d, 2H, J = 8.5 Hz), 7.48 (d, 2H, J = 8.5 Hz), 6.96 (d, 2H, J = 9.0 Hz), 6.62 (br s, 2H), 5.99 (tdd, 1H, J = 6.5, 10.0, 16.5 Hz), 5.10 (qd, 1H, J = 1.5, 17.0 Hz), 5.04 (qd, 2H, J = 1.5, 10.0 Hz), 3.55 (s, 2H), 3.40 (d, 2H, J = 7.0 Hz), 3.19 (s, 2H), 2.59 (br s, 4H), 2.49 (br s, 4H). 13C-NMR (125 MHz, (CD3)2CO) δ 166.8, 156.1 (d, JC-F = 233.6 Hz), 153.1 (d, JC-F = 1.3 Hz), 149.6, 143.9, 143.5, 136.6, 130.5 (d, JC-F = 7.0 Hz), 129.8, 126.7, 118.7 (d, JC-F = 23.1 Hz), 118.4 (d, JC-F = 8.0 Hz), 116.5, 114.6 (d, JC-F = 23.6 Hz), 62.5, 61.5, 54.1, 53.4, 34.2. 19F-NMR (470 MHz, (CD3)2CO) δ −127.4. HRMS (ESI): 490.1930 (M+H)+; calcd. for C23H29FN5O4S: 490.1924. HPLC purity: 98%.
N′-[5-Fluoro-2-hydroxy-3-(2-propenyl)phenylmethylene]-4-benzoyl-1-piperazineacetohydrazide (30)
Synthesized according to General Procedure D: 46c (131 mg, 0.50 mmol, 1.0 equiv.), 47d (90 mg, 0.50 mmol, 1.0 equiv.), 1.2 M HCl (29 μL, 0.035 mmol, 0.070 equiv.), EtOH (3 mL, 0.15 M). Purification by silica gel column chromatography (gradient, 0–15% MeOH/EtOAc) yielded 30 (186 mg, 87.8%) as a white solid. 1H-NMR (500 MHz, CDCl3) δ 11.17 (s, 1H), 10.45 (br s, 1H), 8.19 (s, 1H), 7.40-7.33 (m, 5H), 6.86 (dd, 1H, J = 3.0, 9.0 Hz), 6.60 (dd, 1H, J = 3.0, 8.5 Hz), 5.91 (tdd, 1H, J = 6.5, 9.5, 18.0 Hz), 5.09-5.03 (m, 2H), 3.80 (br s, 2H), 3.47 (br s, 2H), 3.35 (d, 2H, J = 6.5 Hz), 3.19 (s, 2H), 2.56 (br s, 4H). 13C-NMR (125 MHz, CDCl3) δ 170.5, 165.6, 155.4 (d, JC-F = 235.5 Hz), 152.4 (d, JC-F = 1.4 Hz), 150.1 (d, JC-F = 1.8 Hz), 135.6, 135.3, 130.1, 130.1, 128.7, 127.0, 119.0 (d, JC-F = 23.0 Hz), 116.8 (d, JC-F = 7.9 Hz), 116.5, 113.9 (d, JC-F = 23.5 Hz), 60.7, 53.6 (br), 47.6 (br), 42.0 (br), 33.7. 19F-NMR (470 MHz, CDCl3) δ −128.5. HRMS (ESI): 425.1991 (M+H)+; calcd. for C23H26FN4O3: 425.1989. HPLC purity: 97%.
N′-[5-Fluoro-2-hydroxy-3-(2-propenyl)phenylmethylene]-4-(4-cyanophenylmethyl)-1-piperazineacetohydrazide (31)
Synthesized according to General Procedure D: 46d (137 mg, 0.50 mmol, 1.0 equiv.), 47d (90 mg, 0.50 mmol, 1.0 equiv.), 1.2 M HCl (29 μL, 0.035 mmol, 0.070 equiv.), EtOH (3 mL, 0.15 M). Purification by silica gel column chromatography (gradient, 0–15% MeOH/EtOAc) yielded 31 (164 mg, 75.3%) as a white solid. 1H-NMR (500 MHz, CDCl3) δ 11.12 (br s, 1H), 10.16 (br s, 1H), 8.28 (s, 1H), 7.57 (d, 2H, J = 8.0 Hz), 7.42 (d, 2H, J = 8.0 Hz), 6.88 (dd, 1H, J = 3.0, 9.0 Hz), 6.72 (dd, 1H, J = 3.0, 8.0 Hz), 5.94 (tdd, 1H, J = 6.5, 10.0, 17.0 Hz), 5.09-5.05 (m, 2H), 3.55 (s, 2H), 3.38 (d, 2H, J = 7.0 Hz), 3.19 (s, 2H), 2.62 (br s, 4H), 2.51 (br s, 4H). 13C-NMR (125 MHz, CDCl3) δ 166.0, 155.5 (d, JC-F = 235.5 Hz), 152.5 (d, JC-F = 1.4 Hz), 149.9, 144.0, 135.7, 132.2, 130.1 (d, JC-F = 6.8 Hz), 129.5, 119.0 (d, JC-F = 23.0 Hz), 119.0, 116.8 (d, JC-F = 7.8 Hz), 116.5, 113.9 (d, JC-F = 23.5 Hz), 111.0, 62.3, 60.9, 53.8, 53.1, 33.8. 19F-NMR (470 MHz, CDCl3) δ −128.6. HRMS (M+H)+; 436.2144 (M+1); calcd. for C24H27FN5O2: 436.2149. HPLC purity: 95%.
N′-[5-Fluoro-2-hydroxy-3-(2-propenyl)phenylmethylene]-4-(4-cyanobenzoyl)-1-piperazineacetohydrazide (32)
Synthesized according to General Procedure D: 46e (144 mg, 0.50 mmol, 1.0 equiv.), 47d (90 mg, 0.50 mmol, 1.0 equiv.), 1.2 M HCl (29 μL, 0.035 mmol, 0.070 equiv.), EtOH (3 mL, 0.15 M). Purification by silica gel column chromatography (gradient, 0–15% MeOH/EtOAc) yielded 32 (196 mg, 87.2%) as a white solid. 1H-NMR (500 MHz, CDCl3) δ 11.10 (br s, 1H), 10.21 (br s, 1H), 8.20 (s, 1H), 7.65 (d, 2H, J = 8.0 Hz), 7.45 (d, 2H, J = 8.5 Hz), 6.84 (dd, 1H, J = 3.0, 9.0 Hz), 6.61 (dd, 1H, J = 3.0, 8.0 Hz), 5.88 (tdd, 1H, J = 7.0, 10.0, 16.5 Hz), 5.03-5.00 (m, 2H), 3.81 (br s, 2H), 3.40 (br s, 2H), 3.31 (d, 2H, J = 6.5 Hz), 3.21 (s, 2H), 2.66 (br s, 2H), 2.54 (br s, 2H). 13C-NMR (125 MHz, CDCl3) δ 168.3, 165.4, 155.4 (d, JC-F = 235.9 Hz), 152.3, 150.1, 139.7, 135.4, 132.5, 130.0 (d, JC-F = 6.8 Hz), 127.7, 119.1 (d, JC-F = 23.1 Hz), 118.0, 116.6 (d, JC-F = 7.9 Hz), 116.5, 113.8 (d, JC-F = 23.5 Hz), 113.6, 60.7, 53.3 (br), 47.3 (br), 42.0 (br), 33.6. 19F-NMR (470 MHz, CDCl3) δ −128.3. HRMS (ESI): 450.1931 (M+H)+; calcd. for C24H25FN5O3: 450.1941. HPLC purity: 97%.
N′-[5-Fluoro-2-hydroxy-3-(2-propenyl)phenylmethylene]-4-(4-fluorophenylmethyl)-1-piperazineacetohydrazide (33)
Synthesized according to General Procedure D: 46f (133 mg, 0.50 mmol, 1.0 equiv.), 47d (90 mg, 0.50 mmol, 1.0 equiv.), 1.2 M HCl (29 μL, 0.035 mmol, 0.070 equiv.), EtOH (3 mL, 0.15 M). Purification by silica gel column chromatography (gradient, 0–20% MeOH/EtOAc) yielded 33 (176 mg, 82.0%) as a yellow solid. 1H-NMR (500 MHz, CDCl3) δ 11.15 (br s, 1H), 10.22 (br s, 1H), 8.26 (s, 1H), 7.25 (dd, 2H, J = 5.5, 8.5 Hz), 6.98 (t, 2H, J = 8.5 Hz), 6.89 (dd, 1H, J = 3.0, 9.0 Hz), 6.72 (dd, 1H, J = 3.0, 8.0 Hz), 5.95 (tdd, 1H, J = 6.5, 10.0, 17.0 Hz), 5.10-5.06 (m, 2H), 3.47 (s, 2H), 3.39 (d, 2H, J = 6.5 Hz), 3.18 (s, 2H), 2.61 (br s, 4H), 2.49 (br s, 4H). 13C-NMR (125 MHz, CDCl3) δ 166.2, 162.1 (d, JC-F = 243.8 Hz), 155.5 (d, JC-F = 235.5 Hz), 152.5 (d, JC-F = 0.9 Hz), 149.8, 135.7, 133.6 (d, JC-F = 3.0 Hz), 130.6 (d, JC-F = 7.8 Hz), 130.1 (d, JC-F = 6.8 Hz), 119.0 (d, JC-F = 23.0 Hz), 116.8 (d, JC-F = 7.8 Hz), 116.5, 115.1 (d, JC-F = 21.0 Hz), 113.9 (d, JC-F = 23.5 Hz), 62.1, 61.0, 53.7, 52.9, 33.8. 19F-NMR (470 MHz, CDCl3) δ −118.8, −128.6. HRMS (ESI): 429.2095 (M+H)+; calcd. for C23H27F2N4O2: 429.2102. HPLC purity: 98%.
N′-[5-Fluoro-2-hydroxy-3-(2-propenyl)phenylmethylene]-4-(4-fluorobenzoyl)-1-piperazineacetohydrazide (34)
Synthesized according to General Procedure D: 46g (140 mg, 0.50 mmol, 1.0 equiv.), 47d (90 mg, 0.50 mmol, 1.0 equiv.), 1.2 M HCl (29 μL, 0.035 mmol, 0.070 equiv.), EtOH (3 mL, 0.15 M). Purification by silica gel column chromatography (gradient, 0–15% MeOH/EtOAc) yielded 34 (163 mg, 73.8%) as a white solid. 1H-NMR (500 MHz, CDCl3) δ 11.10 (br s, 1H), 10.23 (br s, 1H), 8.25 (s, 1H), 7.38 (dd, 2H, J = 5.5, 8.5 Hz), 7.07 (t, 2H, J = 8.5 Hz), 6.88 (dd, 1H, J = 3.0, 9.0 Hz), 6.65 (dd, 1H, J = 3.0, 8.5 Hz), 5.92 (tdd, 1H, J = 6.5, 9.5, 17.0 Hz), 5.10-5.04 (m, 2H), 3.82 (br s, 2H), 3.50 (br s, 2H), 3.36 (d, 2H, J = 6.5 Hz), 3.21 (s, 2H), 2.59 (br s, 4H). 13C-NMR (125 MHz, CDCl3) δ 169.7, 165.6, 163.6 (d, JC-F = 249.8 Hz), 155.5 (d, JC-F = 235.6 Hz), 152.5 (d, JC-F = 1.4 Hz), 150.3 (d, JC-F = 2.5 Hz), 135.6, 131.3 (d, JC-F = 3.5 Hz), 130.2 (d, JC-F = 6.9 Hz), 129.5 (d, JC-F = 8.4 Hz), 119.2 (d, JC-F = 23.1 Hz), 116.7 (d, JC-F = 7.8 Hz), 116.6, 115.9 (d, JC-F = 21.8 Hz), 113.9 (d, JC-F = 23.5 Hz), 60.9, 53.6 (br), 47.7 (br), 42.2 (br), 33.8. 19F-NMR (470 MHz, CDCl3) δ −112.6, −128.4. HRMS (ESI): 443.1886 (M+H)+; calcd. for C23H25F2N4O3: 443.1895. HPLC purity: 98%.
N′-[5-Fluoro-2-hydroxy-3-(2-propenyl)phenylmethylene]-4-[4(trifluoromethyl)phenylmethyl]-1-piperazineacetohydrazide (35)
Synthesized according to General Procedure D: 46h (158 mg, 0.50 mmol, 1.0 equiv.), 47d (90 mg, 0.50 mmol, 1.0 equiv.), 1.2 M HCl (29 μL, 0.035 mmol, 0.070 equiv.), EtOH (3 mL, 0.15 M). Purification by silica gel column chromatography (gradient, 0–20% MeOH/EtOAc) yielded 35 (176 mg, 73.7%) as a white solid. 1H-NMR (500 MHz, CDCl3) δ 11.14 (br s, 1H), 10.19 (br s, 1H), 8.28 (s, 1H), 7.56 (d, 2H, J = 8.0 Hz), 7.43 (d, 2H, J = 8.0 Hz), 6.90 (dd, 1H, J = 3.0, 9.0 Hz), 6.72 (dd, 2H, J = 3.0, 8.0 Hz), 5.96 (tdd, 1H, J = 6.5, 10.0, 17.0 Hz), 5.11-5.08 (m, 2H), 3.57 (s, 2H), 3.40 (d, 2H, J = 6.5 Hz), 3.20 (s, 2H), 2.63 (br s, 4H), 2.53 (br s, 4H). 13C-NMR (125 MHz, CDCl3) δ 166.1, 155.6 (d, JC-F = 235.5 Hz), 152.5 (d, JC-F = 1.3 Hz), 149.9 (d, JC-F = 2.4 Hz), 142.4, 135.7, 130.2 (d, JC-F = 6.8 Hz), 129.5 (q, JC-F = 32.0 Hz), 129.3, 125.3 (q, JC-F = 3.8 Hz), 124.4 (q, JC-F = 270.5 Hz), 119.1 (d, JC-F = 23.1 Hz), 116.9 (d, JC-F = 7.8 Hz), 116.6, 114.0 (d, JC-F = 23.5 Hz), 62.3, 61.0, 53.8, 53.1, 33.8. 19F-NMR (470 MHz, CDCl3) δ −65.4, −128.5. HRMS (ESI): 479.2066 (M+H)+; calcd. for C24H27F4N4O2: 479.2070. HPLC purity: 96%.
N′-[5-Fluoro-2-hydroxy-3-(2-propenyl)phenylmethylene]-4-[4-(trifluoromethyl)benzoyl]-1-piperazineacetohydrazide (36)
Synthesized according to General Procedure D: 46i (165 mg, 0.50 mmol, 1.0 equiv.), 47d (90 mg, 0.50 mmol, 1.0 equiv.), 1.2 M HCl (29 μL, 0.035 mmol, 0.070 equiv.), EtOH (3 mL, 0.15 M). Purification by silica gel column chromatography (gradient, 0–15% MeOH/EtOAc) yielded 36 (139 mg, 56.3%) as a white solid. 1H-NMR (500 MHz, CDCl3) δ 11.07 (br s, 1H), 10.14 (br s, 1H), 8.26 (s, 1H), 7.66 (d, 2H, J = 8.5 Hz), 7.50 (d, 2H, J = 8.0 Hz), 6.89 (dd, 1H, J = 3.0, 9.0 Hz), 6.66 (dd, 1H, J = 3.0, 8.5 Hz), 5.93 (tdd, 1H, J = 7.0, 10.0, 16.5 Hz), 5.10-5.04 (m, 2H), 3.86 (br s, 2H), 3.45 (br s, 2H), 3.37 (d, 2H, J = 6.5 Hz), 3.23 (s, 2H), 2.69 (br s, 2H), 2.57 (br s, 2H). 13C-NMR (125 MHz, CDCl3) δ 169.1, 165.5, 155.6 (d, JC-F = 235.9 Hz), 152.5, 150.4 (d, JC-F = 2.1 Hz), 138.9, 135.6, 132.0 (q, JC-F = 32.6 Hz), 130.3 (d, JC-F = 6.8 Hz), 127.5, 125.9 (q, JC-F = 3.8 Hz), 123.7 (q, JC-F = 271.1 Hz), 119.3 (d, JC-F = 23.0 Hz), 116.7 (d, JC-F = 4.6 Hz), 116.7, 114.0 (d, JC-F = 23.5 Hz), 60.9, 53.6 (br), 47.5 (br), 42.2 (br), 33.8. 19F-NMR (470 MHz, CDCl3) δ −66.0, −128.4. HRMS (ESI): 493.1868 (M+H)+; calcd. for C24H25F4N4O3: 493.1863. HPLC purity: 97%.
N′-(5-Fluoro-2-hydroxy-3-propylphenylmethylene)-4-phenylmethyl-1-piperazineacetohydrazide (37)
Synthesized according to General Procedure D: 46a (124 mg, 0.50 mmol, 1.0 equiv.), 47e (91 mg, 0.50 mmol, 1.0 equiv.), 1.2 M HCl (29 μL, 0.035 mmol, 0.070 equiv.), EtOH (3 mL, 0.15 M). Purification by silica gel column chromatography (gradient, 0–15% MeOH/EtOAc) yielded 37 (174 mg, 84.6%) as an off-white solid. 1H-NMR (500 MHz, CDCl3) δ 11.10 (s, 1H), 10.19 (br s, 1H), 8.26 (s, 1H), 7.32-7.30 (m, 4H), 7.28-7.25 (m, 1H), 6.89 (dd, 1H, J = 3.0, 9.0 Hz), 6.71 (dd, 1H, J = 3.0, 8.5 Hz), 3.54 (s, 2H), 3.20 (s, 2H), 2.66-2.59 (m, 6H), 2.53 (br s, 4H), 1.64 (sext, 2H, J = 7.5 Hz), 0.95 (t, 3H, J = 7.5 Hz). 13C-NMR (125 MHz, CDCl3) δ 166.1, 155.4 (d, JC-F = 235.1 Hz), 152.8, 150.0, 137.9, 132.7 (d, JC-F = 6.6 Hz), 129.2, 128.4, 127.3, 119.1 (d, JC-F = 22.5 Hz), 116.7 (d, JC-F = 7.9 Hz), 113.4 (d, JC-F = 23.4 Hz), 63.0, 61.0, 53.8, 53.1, 31.9, 22.5, 14.0. 19F-NMR (470 MHz, CDCl3) δ −129.1. HRMS (ESI): 413.2345 (M+H)+; calcd. for C23H30FN4O2: 413.2353. HPLC purity: 99%.
4-{4-[N′-(5-Fluoro-2-hydrxy-3-propylphenylmethylene)hydrazinecarbonylmethyl]-1-piperazinylmethyl}benzenesulfonamide (38)
Synthesized according to General Procedure D: 46b (164 mg, 0.50 mmol, 1.0 equiv.), 47e (91 mg, 0.50 mmol, 1.0 equiv.), 1.2 M HCl (29 μL, 0.035 mmol, 0.070 equiv.), EtOH (3 mL, 0.15 M). Purification by silica gel column chromatography (gradient, 0–20% MeOH/EtOAc) yielded 38 (221 mg, 89.9%) as a white solid. 1H-NMR (500 MHz, (CD3)2CO) δ 11.68 (br s, 1H), 10.92 (br s, 1H), 8.46 (s, 1H), 7.84 (d, 2H, J = 8.0 Hz), 7.49 (d, 2H, J = 8.0 Hz), 6.98 (dd, 1H, J = 3.0, 9.5 Hz), 6.93 (dd, 1H, J = 3.0, 8.5 Hz), 6.59 (br s, 2H), 3.57 (s, 2H), 3.18 (s, 2H), 2.64-2.59 (m, 6H), 2.50 (br s, 4H), 1.63 (sext, 2H, J = 7.5 Hz), 0.93 (t, 3H, J = 7.5 Hz). 13C-NMR (125 MHz, (CD3)2CO) δ 166.6, 156.1 (d, JC-F = 233.1 Hz), 153.4 (d, JC-F = 1.4 Hz), 149.7 (d, JC-F = 2.9 Hz), 143.9, 143.6, 132.8 (d, JC-F = 6.9 Hz), 129.9, 126.8, 119.0 (d, JC-F = 22.8 Hz), 118.3 (d, JC-F = 8.1 Hz), 114.2 (d, JC-F = 23.6 Hz), 62.5, 61.6, 54.2, 53.4, 32.3, 23.1, 14.1. 19F-NMR (470 MHz, (CD3)2CO) δ −127.7. HRMS (ESI): 492.2074 (M+H)+; calcd. for C23H31FN5O4S: 492.2081. HPLC purity: 96%.
N′-(5-Fluoro-2-hydroxy-3-propylphenylmethylene)-4-benzoyl-1-piperazineacetohydrazide (39)
Synthesized according to General Procedure D: 46c (131 mg, 0.50 mmol, 1.0 equiv.), 47e (91 mg, 0.50 mmol, 1.0 equiv.), 1.2 M HCl (29 μL, 0.035 mmol, 0.070 equiv.), EtOH (3 mL, 0.15 M). Purification by silica gel column chromatography (gradient, 0–10% MeOH/EtOAc) yielded 39 (189 mg, 88.9%) as a white solid. 1H-NMR (500 MHz, CDCl3) δ 11.13 (s, 1H), 10.48 (br s, 1H), 8.15 (s, 1H), 7.38-7.32 (m, 5H), 6.83 (dd, 1H, J = 3.0, 9.0 Hz), 6.55 (dd, 1H, J = 3.0, 8.5 Hz), 3.80 (br s, 2H), 3.46 (br s, 2H), 3.18 (s, 2H), 2.59-2.54 (m, 6H), 1.57 (sext, 2H, J = 7.5 Hz), 0.88 (t, 3H, J = 7.5 Hz). 13C-NMR (125 MHz, CDCl3) δ 170.5, 165.6, 155.3 (d, JC-F = 235.1 Hz), 152.6 (d, JC-F = 1.4 Hz), 150.2 (d, JC-F = 2.5 Hz), 135.3, 132.5 (d, JC-F = 6.8 Hz), 130.1, 128.7, 126.9, 119.1 (d, JC-F = 22.6 Hz), 116.6 (d, JC-F = 7.9 Hz), 113.4 (d, JC-F = 23.4 Hz), 60.7, 53.5 (br), 47.5 (br), 42.0 (br), 31.8, 22.4, 13.9. 19F-NMR (470 MHz, CDCl3) δ −129.0. HRMS (ESI): 427.2144 (M+H)+; calcd. for C23H28FN4O3: 427.2145. HPLC purity: 99%.
N′-(5-Fluoro-2-hydroxy-3-propylphenylmethylene)-4-(4-cyanopheylmehtyl)-1-piperazineacetohydrazide (40)
Synthesized according to General Procedure D: 46d (137 mg, 0.50 mmol, 1.0 equiv.), 47e (91 mg, 0.50 mmol, 1.0 equiv.), 1.2 M HCl (29 μL, 0.035 mmol, 0.070 equiv.), EtOH (3 mL, 0.15 M). Purification by silica gel column chromatography (gradient, 0–15% MeOH/EtOAc) yielded 40 (180 mg, 82.1%) as a white solid. 1H-NMR (500 MHz, CDCl3) δ 11.07 (br s, 1H), 10.16 (br s, 1H), 8.25 (s, 1H), 7.57 (d, 2H, J = 8.5 Hz), 7.42 (d, 2H, J = 8.0 Hz), 6.86 (dd, 1H, J = 3.0, 9.0 Hz), 6.68 (dd, 1H, J = 3.0, 8.0 Hz), 3.55 (s, 2H), 3.18 (s, 2H), 2.70-2.57 (m, 6H), 2.51 (br s, 4H). 13C-NMR (125 MHz, CDCl3) δ 166.0, 155.4 (d, JC-F = 235.1 Hz), 152.7 (d, JC-F = 1.4 Hz), 150.0 (d, JC-F = 2.5 Hz), 144.0, 132.6 (d, JC-F = 6.8 Hz), 132.2, 129.5, 119.1 (d, JC-F = 22.6 Hz), 119.0, 116.6 (d, JC-F = 8.0 Hz), 113.4 (d, JC-F = 23.5 Hz), 110.9, 62.3, 60.9, 53.7, 53.1, 31.9, 22.4, 14.0. 19F-NMR (470 MHz, CDCl3) δ −129.0. HRMS (ESI): 438.2301 (M+H)+; calcd. for C24H29FN5O2: 438.2305. HPLC purity: 96%.
N′-(5-Fluoro-2-hydroxy-3-propylphenylmethylene)-4-(4-cyanobenzoyl)-1-piperazineacetohydrazide (41)
Synthesized according to General Procedure D: 46e (144 mg, 0.50 mmol, 1.0 equiv.), 47e (91 mg, 0.50 mmol, 1.0 equiv.), 1.2 M HCl (29 μL, 0.035 mmol, 0.070 equiv.), EtOH (3 mL, 0.15 M). Purification by silica gel column chromatography (gradient, 0–10% MeOH/EtOAc) yielded 41 (196 mg, 86.5%) as a white solid. 1H-NMR (500 MHz, CDCl3) δ 11.03 (br s, 1H), 10.17 (br s, 1H), 8.19 (s, 1H), 7.66 (d, 2H, J = 8.0 Hz), 7.46 (d, 2H, J = 8.0 Hz), 6.84 (dd, 1H, J = 3.0, 9.0 Hz), 6.59 (dd, 1H, J = 3.0, 8.5 Hz), 3.82 (br s, 2H), 3.41 (br s, 2H), 3.22 (s, 2H), 2.66 (br s, 2H), 2.57-2.52 (m, 2H), 1.55 (sext, 2H, J = 7.5 Hz), 0.87 (t, 3H, J = 7.5 Hz). 13C-NMR (125 MHz, CDCl3) δ 168.3, 165.4, 155.3 (d, JC-F = 235.5 Hz), 152.6 (d, JC-F = 1.1 Hz), 150.3 (d, JC-F = 2.0 Hz), 139.7, 132.6 (d, JC-F = 6.8 Hz), 132.5, 127.7, 119.2 (d, JC-F = 22.6 Hz), 118.0, 116.4 (d, JC-F = 7.9 Hz), 113.6, 113.4 (d, JC-F = 23.3 Hz), 60.7, 53.4 (br), 47.4 (br), 42.0 (br), 31.7, 22.3, 13.9. 19F-NMR (470 MHz, CDCl3) δ −128.7. HRMS (ESI): 452.2098 (M+H)+; calcd. for C24H27FN5O3: 452.2098. HPLC purity: 99%.
N′-(5-Fluoro-2-hydroxy-3-propylphenylmethylene)-4-(4-fluoropheylmethyl)-1-piperazineacetohydrazide (42)
Synthesized according to General Procedure D: 46f (133 mg, 0.50 mmol, 1.0 equiv.), 47e (91 mg, 0.50 mmol, 1.0 equiv.), 1.2 M HCl (29 μL, 0.035 mmol, 0.070 equiv.), EtOH (3 mL, 0.15 M). Purification by silica gel column chromatography (gradient, 0–20% MeOH/EtOAc) yielded 42 (202 mg, 93.8%) as a yellow solid. 1H-NMR (500 MHz, CDCl3) δ 11.07 (br s, 1H), 10.16 (br s, 1H), 8.26 (s, 1H), 7.26 (dd, 2H, J = 5.5, 8.5 Hz), 6.99 (t, 2H, J = 8.5 Hz), 6.88 (dd, 1H, J = 3.0, 9.0 Hz), 6.70 (dd, 1H, J = 3.0, 8.0 Hz), 3.48 (s, 2H), 3.18 (s, 2H), 2.66-2.58 (m, 6H), 2.50 (br s, 4H), 1.62 (sext, 2H, J = 7.5 Hz), 0.94 (t, 3H, J = 7.5 Hz). 13C-NMR (125 MHz, CDCl3) δ 166.1, 162.1 (d, JC-F = 243.8 Hz), 155.5 (d, JC-F = 235.0 Hz), 152.8 (d, JC-F = 0.9 Hz), 150.1 (d, JC-F = 2.0 Hz), 133.7, 132.7 (d, JC-F = 6.8 Hz), 130.7 (d, JC-F = 7.9 Hz), 119.2 (d, JC-F = 22.5 Hz), 116.7 (d, JC-F = 7.9 Hz), 115.2 (d, JC-F = 21.0 Hz), 113.5 (d, JC-F = 23.4 Hz), 62.1, 61.0, 53.8, 53.0, 31.9, 22.5, 14.1. 19F-NMR (470 MHz, CDCl3) δ −118.8, −129.0. HRMS (ESI): 431.2250 (M+H)+; calcd. for C23H29F2N4O2: 431.2259. HPLC purity: 98%.
N′-(5-Fluoro-2-hydroxy-3-propylphenylmethylene)-4-(4-fluorobenzoyl)-1-piperazineacetohydrazide (43)
Synthesized according to General Procedure D: 46g (140 mg, 0.50 mmol, 1.0 equiv.), 47e (91 mg, 0.50 mmol, 1.0 equiv.), 1.2 M HCl (29 μL, 0.035 mmol, 0.070 equiv.), EtOH (3 mL, 0.15 M). Purification by silica gel column chromatography (gradient, 0–10% MeOH/EtOAc) yielded 43 (195 mg, 87.7%) as a white solid. 1H-NMR (500 MHz, CDCl3) δ 11.07 (br s, 1H), 10.32 (br s, 1H), 8.18 (s, 1H), 7.36 (dd, 2H, J = 5.0, 8.5 Hz), 7.05 (t, 2H, J = 8.5 Hz), 6.84 (dd, 1H, J = 3.0, 9.0 Hz), 6.57 (dd, 1H, J = 3.0, 8.5 Hz), 3.81 (br s, 2H), 3.48 (br s, 2H), 3.20 (s, 2H), 2.62-2.50 (m, 6H), 1.56 (sext, 2H, J = 7.5 Hz), 0.88 (t, 3H, J = 7.5 Hz). 13C-NMR (125 MHz, CDCl3) δ 169.6, 165.6, 163.5 (d, JC-F = 249.3 Hz), 155.4 (d, JC-F = 235.4 Hz), 152.7 (d, JC-F = 0.9 Hz), 150.3, 132.6 (d, JC-F = 6.8 Hz), 131.3 (d, JC-F = 3.4 Hz), 129.4 (d, JC-F = 8.4 Hz), 119.2 (d, JC-F = 22.6 Hz), 116.5 (d, JC-F = 7.9 Hz), 115.8 (d, JC-F = 21.6 Hz), 113.4 (d, JC-F = 23.4 Hz), 60.7, 53.5 (br), 47.7 (br), 42.1 (br), 31.8, 22.4, 13.9. 19F-NMR (470 MHz, CDCl3) δ −112.6, −128.8. HRMS (ESI): 445.2049 (M+H)+; calcd. for C23H27F2N4O3: 445.2051. HPLC purity: 98%.
N′-(5-Fluoro-2-hydroxy-3-propylphenylmethylene)-4-[4-(trifluoromethyl)phenylmethyl]-1-piperazineacetohydrazide (44)
Synthesized according to General Procedure D: 46h (158 mg, 0.50 mmol, 1.0 equiv.), 47e (91 mg, 0.50 mmol, 1.0 equiv.), 1.2 M HCl (29 μL, 0.035 mmol, 0.070 equiv.), EtOH (3 mL, 0.15 M). Purification by silica gel column chromatography (gradient, 0–20% MeOH/EtOAc) yielded 44 (184 mg, 76.5%) as a light yellow solid. 1H-NMR (500 MHz, CDCl3) δ 11.08 (br s, 1H), 10.18 (br s, 1H), 8.25 (s, 1H), 7.56 (d, 2H, J = 8.0 Hz), 7.43 (d, 2H, J = 8.0 Hz), 6.88 (dd, 1H, J = 3.0, 9.0 Hz), 6.69 (dd, 2H, J = 3.0, 8.5 Hz), 3.56 (s, 2H), 3.20 (s, 2H), 2.64-2.58 (m, 6H), 2.52 (br s, 4H), 1.62 (sext, 2H, J = 7.5 Hz), 0.93 (t, 3H, J = 7.5 Hz). 13C-NMR (125 MHz, CDCl3) δ 166.1, 155.5 (d, JC-F = 235.1 Hz), 152.8 (d, JC-F = 1.2 Hz), 150.0 (d, JC-F = 2.6 Hz), 142.4, 132.7 (d, JC-F = 6.8 Hz), 129.5 (q, JC-F = 32.1 Hz), 129.3, 125.3 (q, JC-F = 3.8 Hz), 124.4 (q, JC-F = 270.5 Hz), 119.2 (d, JC-F = 22.6 Hz), 116.7 (d, JC-F = 7.9 Hz), 113.5 (d, JC-F = 23.5 Hz), 62.3, 61.0, 53.8, 53.1, 31.9, 22.5, 14.0. 19F-NMR (470 MHz, CDCl3) δ −65.4, −128.9. HRMS (ESI): 481.2216 (M+H)+; calcd. for C24H29F4N4O2: 481.2227. HPLC purity: 97%.
N′-(5-Fluoro-2-hydroxy-3-propylphenylmethylene)-4-[4-(trifluoromethyl)benzoyl]-1-piperazineacetohydrazide (45)
Synthesized according to General Procedure D: 46i (165 mg, 0.50 mmol, 1.0 equiv.), 47e (91 mg, 0.50 mmol, 1.0 equiv.), 1.2 M HCl (29 μL, 0.035 mmol, 0.070 equiv.), EtOH (3 mL, 0.15 M). Purification by silica gel column chromatography (gradient, 0–10% MeOH/EtOAc) yielded 45 (140 mg, 56.7%) as a white solid. 1H-NMR (500 MHz, CDCl3) δ 11.04 (br s, 1H), 10.19 (br s, 1H), 8.21 (s, 1H), 7.65 (d, 2H, J = 8.5 Hz), 7.48 (d, 2H, J = 8.0 Hz), 6.86 (dd, 1H, J = 3.0, 9.0 Hz), 6.60 (dd, 1H, J = 3.0, 8.0 Hz), 3.85 (br s, 2H), 3.44 (br s, 2H), 3.22 (s, 2H), 2.67 (br s, 2H), 2.60-2.51 (m, 4H), 1.58 (sext, 2H, J = 7.5 Hz), 0.90 (t, 3H, J = 7.5 Hz). 13C-NMR (125 MHz, CDCl3) δ 169.0, 165.5, 155.4 (d, JC-F = 235.4 Hz), 152.7 (d, JC-F = 0.9 Hz), 150.4 (d, JC-F = 2.4 Hz), 138.9, 132.7 (d, JC-F = 6.8 Hz), 132.0 (q, JC-F = 32.6 Hz), 127.5, 125.8 (q, JC-F = 3.6 Hz), 123.7 (q, JC-F = 271.0 Hz), 119.3 (d, JC-F = 22.6 Hz), 116.5 (d, JC-F = 7.9 Hz), 113.4 (d, JC-F = 23.4 Hz), 60.8, 53.5 (br), 47.5 (br), 42.0 (br), 31.8, 22.4, 14.0. 19F-NMR (470 MHz, CDCl3) δ −66.0, −128.7. HRMS (ESI): 495.2008 (M+H)+; calcd. for C24H27F4N4O3: 495.2019. HPLC purity: 98%.
Supplementary Material
Acknowledgments
We are grateful to the National Institutes of Health (R01-CA120439) and University of Illinois for support of this work. H.S.R. was partially supported by the Richard B. Silverman Predoctoral Fellowship from the American Chemical Society Division of Medicinal Chemistry. R.C.B. is a National Science Foundation predoctoral fellow, a Robert C. and Carolyn J. Springborn graduate fellow, and a member of the NIH Chemistry-Biology Interface Training Grant (NRSA 1-T32-GM070421).
Abbreviation Used
- PAC-1
Procaspase-Activating Compound 1
Footnotes
Supporting Information
Full biological protocols, copies of NMR spectra, LC traces for liver microsome experiments, formation curves for IC50 determination and zinc binding determination. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Schilsky RL. Personalized medicine in oncology: the future is now. Nat Rev Drug Discov. 2010;9:363–366. doi: 10.1038/nrd3181. [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 3.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 4.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 5.Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, Joseph MK, Kitada S, Korsmeyer SJ, Kunzer AR, Letai A, Li C, Mitten MJ, Nettesheim DG, Ng S, Nimmer PM, O’Connor JM, Oleksijew A, Petros AM, Reed JC, Shen W, Tahir SK, Thompson CB, Tomaselli KJ, Wang BL, Wendt MD, Zhang HC, Fesik SW, Rosenberg SH. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 6.Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, Dayton BD, Ding H, Enschede SH, Fairbrother WJ, Huang DC, Hymowitz SG, Jin S, Khaw SL, Kovar PJ, Lam LT, Lee J, Maecker HL, Marsh KC, Mason KD, Mitten MJ, Nimmer PM, Oleksijew A, Park CH, Park CM, Phillips DC, Roberts AW, Sampath D, Seymour JF, Smith ML, Sullivan GM, Tahir SK, Tse C, Wendt MD, Xiao Y, Xue JC, Zhang H, Humerickhouse RA, Rosenberg SH, Elmore SW. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013;19:202–208. doi: 10.1038/nm.3048. [DOI] [PubMed] [Google Scholar]
- 7.Lu J, Bai L, Sun H, Nikolovska-Coleska Z, McEachern D, Qiu S, Miller RS, Yi H, Shangary S, Sun Y, Meagher JL, Stuckey JA, Wang S. SM-164: a novel, bivalent Smac mimetic that induces apoptosis and tumor regression by concurrent removal of the blockade of cIAP-1/2 and XIAP. Cancer Res. 2008;68:9384–9393. doi: 10.1158/0008-5472.CAN-08-2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Putt KS, Chen GW, Pearson JM, Sandhorst JS, Hoagland MS, Kwon JT, Hwang SK, Jin H, Churchwell MI, Cho MH, Doerge DR, Helferich WG, Hergenrother PJ. Small-molecule activation of procaspase-3 to caspase-3 as a personalized anticancer strategy. Nat Chem Biol. 2006;2:543–550. doi: 10.1038/nchembio814. [DOI] [PubMed] [Google Scholar]
- 9.Wolan DW, Zorn JA, Gray DC, Wells JA. Small-molecule activators of a proenzyme. Science. 2009;326:853–858. doi: 10.1126/science.1177585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schipper JL, MacKenzie SH, Sharma A, Clark AC. A bifunctional allosteric site in the dimer interface of procaspase-3. Biophys Chem. 2011;159:100–109. doi: 10.1016/j.bpc.2011.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vickers CJ, Gonzalez-Paez GE, Umotoy JC, Cayanan-Garrett C, Brown SJ, Wolan DW. Small-molecule procaspase activators identified using fluorescence polarization. ChemBioChem. 2013;14:1419–1422. doi: 10.1002/cbic.201300315. [DOI] [PubMed] [Google Scholar]
- 12.Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407:770–776. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- 13.Elmore S. Apoptosis: A review of programmed cell death. Toxicol Pathol. 2007;35:495–516. doi: 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ghavami S, Hashemi M, Ande SR, Yeganeh B, Xiao W, Eshraghi M, Bus CJ, Kadkhoda K, Wiechec E, Halayko AJ, Los M. Apoptosis and cancer: mutations within caspase genes. J Med Genet. 2009;46:497–510. doi: 10.1136/jmg.2009.066944. [DOI] [PubMed] [Google Scholar]
- 15.Soini Y, Paakko P. Apoptosis and expression of caspases 3, 6 and 8 in malignant non-Hodgkin’s lymphomas. APMIS. 1999;107:1043–1050. doi: 10.1111/j.1699-0463.1999.tb01508.x. [DOI] [PubMed] [Google Scholar]
- 16.Wrobel G, Maldyk J, Kazanowska B, Rapala M, Maciejka-Kapuscinska L, Chaber R. Immunohistochemical expression of procaspase-3 and its clinical significance in childhood non-Hodgkin lymphomas. Pediatr Dev Pathol. 2011;14:173–179. doi: 10.2350/10-01-0779-OA.1. [DOI] [PubMed] [Google Scholar]
- 17.Estrov Z, Thall PF, Talpaz M, Estey EH, Kantarjian HM, Andreeff M, Harris D, Van Q, Walterscheid M, Kornblau SM. Caspase 2 and caspase 3 protein levels as predictors of survival in acute myelogenous leukemia. Blood. 1998;92:3090–3097. [PubMed] [Google Scholar]
- 18.Patel V, Balakrishnan K, Keating MJ, Wierda WG, Gandhi V. Expression of executioner procaspases and their activation by a procaspase activating compound in chronic lymphocytic leukemia cells. Blood. 2015;125:1126–1136. doi: 10.1182/blood-2014-01-546796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fink D, Schlagbauer-Wadl H, Selzer E, Lucas T, Wolff K, Pehamberger H, Eichler HG, Jansen B. Elevated procaspase levels in human melanoma. Melanoma Res. 2001;11:385–393. doi: 10.1097/00008390-200108000-00009. [DOI] [PubMed] [Google Scholar]
- 20.Chen N, Gong J, Chen X, Meng W, Huang Y, Zhao F, Wang L, Zhou Q. Caspases and inhibitor of apoptosis proteins in cutaneous and mucosal melanoma: expression profile and clinicopathologic significance. Hum Pathol. 2009;40:950–956. doi: 10.1016/j.humpath.2008.12.001. [DOI] [PubMed] [Google Scholar]
- 21.Gdynia G, Grund K, Eckert A, Bock BC, Funke B, Macher-Goeppinger S, Sieber S, Herold-Mende C, Wiestler B, Wiestler OD, Roth W. Basal caspase activity promotes migration and invasiveness in glioblastoma cells. Mol Cancer Res. 2007;5:1232–1240. doi: 10.1158/1541-7786.MCR-07-0343. [DOI] [PubMed] [Google Scholar]
- 22.Murphy AC, Weyhenmeyer B, Schmid J, Kilbride SM, Rehm M, Huber HJ, Senft C, Weissenberger J, Seifert V, Dunst M, Mittelbronn M, Kogel D, Prehn JH, Murphy BM. Activation of executioner caspases is a predictor of progression-free survival in glioblastoma patients: a systems medicine approach. Cell Death Dis. 2013;4:e629. doi: 10.1038/cddis.2013.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Virkajarvi N, Paakko P, Soini Y. Apoptotic index and apoptosis influencing proteins bcl-2, mcl-1, bax and caspases 3, 6 and 8 in pancreatic carcinoma. Histopathology. 1998;33:432–439. doi: 10.1046/j.1365-2559.1998.00553.x. [DOI] [PubMed] [Google Scholar]
- 24.Persad R, Liu C, Wu TT, Houlihan PS, Hamilton SR, Diehl AM, Rashid A. Overexpression of caspase-3 in hepatocellular carcinomas. Mod Pathol. 2004;17:861–867. doi: 10.1038/modpathol.3800146. [DOI] [PubMed] [Google Scholar]
- 25.Tormanen-Napankangas U, Soini Y, Kahlos K, Kinnula V, Paakko P. Expression of caspases-3, -6 and -8 and their relation to apoptosis in non-small cell lung carcinoma. Int J Cancer. 2001;93:192–198. doi: 10.1002/ijc.1315. [DOI] [PubMed] [Google Scholar]
- 26.Krepela E, Prochazka J, Liul X, Fiala P, Kinkor Z. Increased expression of Apaf-1 and procaspase-3 and the functionality of intrinsic apoptosis apparatus in non-small cell lung carcinoma. Biol Chem. 2004;385:153–168. doi: 10.1515/BC.2004.034. [DOI] [PubMed] [Google Scholar]
- 27.Krepela E, Prochazka J, Fiala P, Zatloukal P, Selinger P. Expression of apoptosome pathway-related transcripts in non-small cell lung cancer. J Cancer Res Clin Oncol. 2006;132:57–68. doi: 10.1007/s00432-005-0048-6. [DOI] [PubMed] [Google Scholar]
- 28.O’Donovan N, Crown J, Stunell H, Hill AD, McDermott E, O’Higgins N, Duffy MJ. Caspase 3 in breast cancer. Clin Cancer Res. 2003;9:738–742. [PubMed] [Google Scholar]
- 29.Zapata JM, Krajewska M, Krajewski S, Huang RP, Takayama S, Wang HG, Adamson E, Reed JC. Expression of multiple apoptosis-regulatory genes in human breast cancer cell lines and primary tumors. Breast Cancer Res Treat. 1998;47:129–140. doi: 10.1023/a:1005940832123. [DOI] [PubMed] [Google Scholar]
- 30.Krajewski S, Krajewska M, Turner BC, Pratt C, Howard B, Zapata JM, Frenkel V, Robertson S, Ionov Y, Yamamoto H, Perucho M, Takayama S, Reed JC. Prognostic significance of apoptosis regulators in breast cancer. Endocr Relat Cancer. 1999;6:29–40. doi: 10.1677/erc.0.0060029. [DOI] [PubMed] [Google Scholar]
- 31.Nakopoulou L, Alexandrou P, Stefanaki K, Panayotopoulou E, Lazaris AC, Davaris PS. Immunohistochemical expression of caspase-3 as an adverse indicator of the clinical outcome in human breast cancer. Pathobiology. 2001;69:266–273. doi: 10.1159/000064337. [DOI] [PubMed] [Google Scholar]
- 32.Jiang H, Gong M, Cui Y, Ma K, Chang D, Wang TY. Upregulation of caspase-3 expression in esophageal cancer correlates with favorable prognosis: an immunohistochemical study from a high incidence area in northern China. Dis Esophagus. 2010;23:487–492. doi: 10.1111/j.1442-2050.2009.01043.x. [DOI] [PubMed] [Google Scholar]
- 33.Roy S, Bayly CI, Gareau Y, Houtzager VM, Kargman S, Keen SL, Rowland K, Seiden IM, Thornberry NA, Nicholson DW. Maintenance of caspase-3 proenzyme dormancy by an intrinsic “safety catch” regulatory tripeptide. Proc Natl Acad Sci US A. 2001;98:6132–6137. doi: 10.1073/pnas.111085198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sadowska A, Car H, Pryczynicz A, Guzinska-Ustymowicz K, Kowal KW, Cepowicz D, Kedra B. Expression of apoptotic proteins in human colorectal cancer and metastatic lymph nodes. Pathol Res Pract. 2014;210:576–581. doi: 10.1016/j.prp.2014.04.023. [DOI] [PubMed] [Google Scholar]
- 35.Hector S, Conlon S, Schmid J, Dicker P, Cummins RJ, Concannon CG, Johnston PG, Kay EW, Prehn JH. Apoptosome-dependent caspase activation proteins as prognostic markers in Stage II and III colorectal cancer. Br J Cancer. 2012;106:1499–1505. doi: 10.1038/bjc.2012.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Peterson QP, Goode DR, West DC, Ramsey KN, Lee JJY, Hergenrother PJ. PAC-1 activates procaspase-3 in vitro through relief of zinc-mediated inhibition. J Mol Biol. 2009;388:144–158. doi: 10.1016/j.jmb.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peterson QP, Hsu DC, Goode DR, Novotny CJ, Totten RK, Hergenrother PJ. Procaspase-3 activation as an anti-cancer strategy: structure-activity relationship of Procaspase-Activating Compound 1 (PAC-1) and its cellular co-localization with caspase-3. J Med Chem. 2009;52:5721–5731. doi: 10.1021/jm900722z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peterson QP, Hsu DC, Novotny CJ, West DC, Kim D, Schmit JM, Dirikolu L, Hergenrother PJ, Fan TM. Discovery and canine preclinical assessment of a nontoxic procaspase-3-activating compound. Cancer Res. 2010;70:7232–7241. doi: 10.1158/0008-5472.CAN-10-0766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lucas PW, Schmit JM, Peterson QP, West DC, Hsu DC, Novotny CJ, Dirikolu L, Churchwell MI, Doerge DR, Garrett LD, Hergenrother PJ, Fan TM. Pharmacokinetics and derivation of an anticancer dosing regimen for PAC-1, a preferential small molecule activator of procaspase-3, in healthy dogs. Invest New Drugs. 2011;29:901–911. doi: 10.1007/s10637-010-9445-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hsu DC, Roth HS, West DC, Botham RC, Novotny CJ, Schmid SC, Hergenrother PJ. Parallel synthesis and biological evaluation of 837 analogues of Procaspase-Activating Compound 1 (PAC-1) ACS Comb Sci. 2012;14:44–50. doi: 10.1021/co2001372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.West DC, Qin Y, Peterson QP, Thomas DL, Palchaudhuri R, Morrison KC, Lucas PW, Palmer AE, Fan TM, Hergenrother PJ. Differential effects of procaspase-3 activating compounds in the induction of cancer cell death. Mol Pharmaceutics. 2012;9:1425–1434. doi: 10.1021/mp200673n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang FY, Wang LH, Zhao YF, Li Y, Ping GF, Xiao S, Chen K, Zhu WF, Gong P, Yang JY, Wu CF. A novel small-molecule activator of procaspase-3 induces apoptosis in cancer cells and reduces tumor growth in human breast, liver and gallbladder cancer xenografts. Mol Oncol. 2014;8:1640–1652. doi: 10.1016/j.molonc.2014.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang F, Liu Y, Wang L, Yang J, Zhao Y, Wang N, Cao Q, Gong P, Wu C. Targeting procaspase-3 with WF-208, a novel PAC-1 derivative, causes selective cancer cell apoptosis. J Cell Mol Med. doi: 10.1111/jcmm.12566. Online early access. Published Online: March 8: 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang B, Zhao YF, Zhai X, Fan WJ, Ren JL, Wu CF, Gong P. Design, synthesis and antiproliferative activities of diaryl urea derivatives bearing N-acylhydrazone moiety. Chin Chem Lett. 2012;23:915–918. doi: 10.1248/cpb.c12-00234. [DOI] [PubMed] [Google Scholar]
- 45.Zhang B, Zhao YF, Zhai X, Wang LH, Yang JY, Tan ZH, Gong P. Design, synthesis and anticancer activities of diaryl urea derivatives bearing N-acylhydrazone moiety. Chem Pharm Bull. 2012;60:1046–1054. doi: 10.1248/cpb.c12-00234. [DOI] [PubMed] [Google Scholar]
- 46.Zhai X, Huang Q, Jiang N, Wu D, Zhou HY, Gong P. Discovery of hybrid dual N-acylhydrazone and diaryl urea derivatives as potent antitumor agents: design, synthesis and cytotoxicity evaluation. Molecules. 2013;18:2904–2923. doi: 10.3390/molecules18032904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huang Q, Fu Q, Liu Y, Bai J, Wang Q, Liao H, Gong P. Design, synthesis and anticancer activity of novel 6-(aminophenyl)-2,4-bismorpholino-1,3,5-triazine derivatives bearing arylmethylene hydrazine moiety. Chem Res Chin Univ. 2014;30:257–265. [Google Scholar]
- 48.Botham RC, Fan TM, Im I, Borst LB, Dirikolu L, Hergenrother PJ. Dual small-molecule targeting of procaspase-3 dramatically enhances zymogen activation and anticancer activity. J Am Chem Soc. 2014;136:1312–1319. doi: 10.1021/ja4124303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ma J, Zhang G, Han X, Bao G, Wang L, Zhai X, Gong P. Synthesis and biological evaluation of benzothiazole derivatives bearing the ortho-hydroxy-N-acylhydrazone moiety as potent antitumor agents. Arch Pharm Chem Life Sci. 2014;347:936–949. doi: 10.1002/ardp.201400230. [DOI] [PubMed] [Google Scholar]
- 50.Ma JJ, Chen D, Lu K, Wang LH, Han XQ, Zhao YF, Gong P. Design, synthesis, and structure-activity relationships of novel benzothiazole derivatives bearing the ortho-hydroxy N-carbamoylhydrazone moiety as potent antitumor agents. Eur J Med Chem. 2014;86:257–269. doi: 10.1016/j.ejmech.2014.08.058. [DOI] [PubMed] [Google Scholar]
- 51.Astrand OAH, Aziz G, Ali SF, Paulsen RE, Hansen TV, Rongved P. Synthesis and initial in vitro biological evaluation of two new zinc-chelating compounds: comparison with TPEN and PAC-1. Bioorg Med Chem. 2013;21:5175–5181. doi: 10.1016/j.bmc.2013.06.037. [DOI] [PubMed] [Google Scholar]
- 52.Qin MZ, Liao WK, Xu C, Fu BL, Ren JG, Gu YC, Gong P. Synthesis and biological evaluation of novel 4-(2-fluorophenoxy)-2-(1H-tetrazol-1-yl)pyridines bearing semicarbazone moieties as potent antitumor agents. Arch Pharm Chem Life Sci. 2013;346:840–850. doi: 10.1002/ardp.201300188. [DOI] [PubMed] [Google Scholar]
- 53.Hao-ming L, Chun-ling Y, Xiao-ying Z, Ming-ming Z, Dan J, Jun-hai X, Xiao-hong Y, Song L. Design, synthesis, and antitumor activity of a novel series of PAC-1 analogues. Chem Res Chin Univ. 2013;29:906–910. [Google Scholar]
- 54.Razi SS, Schwartz G, Boone D, Li X, Belsley S, Todd G, Connery CP, Bhora FY. Small molecule activation of procaspase-3 induces apoptosis in human lung adenocarcinoma: a tailored anti-cancer strategy. J Surg Res. 2010;158:402–403. [Google Scholar]
- 55.Razi SS, Rehmani S, Li X, Park K, Schwartz GS, Latif MJ, Bhora FY. Antitumor activity of paclitaxel is significantly enhanced by a novel proapoptotic agent in non–small cell lung cancer. J Surg Res. 2015;194:622–630. doi: 10.1016/j.jss.2014.11.004. [DOI] [PubMed] [Google Scholar]
- 56.Razi SS, Schwartz G, Li XG, Boone D, Belsley S, Todd G, Connery CP, Bhora FY. Direct activation of procaspase-3 inhibits human lung adenocarcinoma in a murine model. J Am Coll Surgeons. 2010;211:S36–S36. [Google Scholar]
- 57.Charkoudian LK, Pham DM, Franz KJ. A pro-chelator triggered by hydrogen peroxide inhibits iron-promoted hydroxyl radical formation. J Am Chem Soc. 2006;128:12424–12425. doi: 10.1021/ja064806w. [DOI] [PubMed] [Google Scholar]
- 58.Johnson DK, Murphy TB, Rose NJ, Goodwin WH, Pickart L. Cytotoxic chelators and chelates. 1. Inhibition of DNA synthesis in cultured rodent and human cells by aroylhydrazones and by a copper(II) complex of salicylaldehyde benzoyl hydrazone. Inorg Chim Acta. 1982;67:159–165. [Google Scholar]
- 59.Peng X, Tang X, Qin W, Dou W, Guo Y, Zheng J, Liu W, Wang D. Aroylhydrazone derivative as fluorescent sensor for highly selective recognition of Zn2+ ions: syntheses, characterization, crystal structures and spectroscopic properties. Dalton Trans. 2011;40:5271–5277. doi: 10.1039/c0dt01590c. [DOI] [PubMed] [Google Scholar]
- 60.Muzio M, Salvesen GS, Dixit VM. FLICE induced apoptosis in a cell-free system. Cleavage of caspase zymogens. J Biol Chem. 1997;272:2952–2956. doi: 10.1074/jbc.272.5.2952. [DOI] [PubMed] [Google Scholar]
- 61.Perry DK, Smyth MJ, Stennicke HR, Salvesen GS, Duriez P, Poirier GG, Hannun YA. Zinc is a potent inhibitor of the apoptotic protease, caspase-3. A novel target for zinc in the inhibition of apoptosis. J Biol Chem. 1997;272:18530–18533. doi: 10.1074/jbc.272.30.18530. [DOI] [PubMed] [Google Scholar]
- 62.Huber KL, Hardy JA. Mechanism of zinc-mediated inhibition of caspase-9. Protein Sci. 2012;21:1056–1065. doi: 10.1002/pro.2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Velazquez-Delgado EM, Hardy JA. Zinc-mediated allosteric inhibition of caspase-6. J Biol Chem. 2012;287:36000–36011. doi: 10.1074/jbc.M112.397752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Truong-Tran AQ, Grosser D, Ruffin RE, Murgia C, Zalewski PD. Apoptosis in the normal and inflamed airway epithelium: role of zinc in epithelial protection and procaspase-3 regulation. Biochem Pharmacol. 2003;66:1459–1468. doi: 10.1016/s0006-2952(03)00498-2. [DOI] [PubMed] [Google Scholar]
- 65.Daniel AG, Peterson EJ, Farrell NP. The bioinorganic chemistry of apoptosis: potential inhibitory zinc binding sites in caspase-3. Angew Chem Int Ed Engl. 2014;53:4098–4101. doi: 10.1002/anie.201311114. [DOI] [PubMed] [Google Scholar]
- 66.Putinski C, Abdul-Ghani M, Stiles R, Brunette S, Dick SA, Fernando P, Megeney LA. Intrinsic-mediated caspase activation is essential for cardiomyocyte hypertrophy. Proc Natl Acad Sci US A. 2013;110:E4079–E4087. doi: 10.1073/pnas.1315587110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Monaco G, Decrock E, Akl H, Ponsaerts R, Vervliet T, Luyten T, De Maeyer M, Missiaen L, Distelhorst CW, De Smedt H, Parys JB, Leybaert L, Bultynck G. Selective regulation of IP3-receptor-mediated Ca2+ signaling and apoptosis by the BH4 domain of Bcl-2 versus Bcl-Xl. Cell Death Differ. 2012;19:295–309. doi: 10.1038/cdd.2011.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Seervi M, Joseph J, Sobhan PK, Bhavya BC, Santhoshkumar TR. Essential requirement of cytochrome c release for caspase activation by procaspase-activating compound defined by cellular models. Cell Death Dis. 2011;2:e207. doi: 10.1038/cddis.2011.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Guo WT, Wang XW, Yan YL, Li YP, Yin X, Zhang Q, Melton C, Shenoy A, Reyes NA, Oakes SA, Blelloch R, Wang Y. Suppression of epithelial-mesenchymal transition and apoptotic pathways by miR-294/302 family synergistically blocks let-7-induced silencing of self-renewal in embryonic stem cells. Cell Death Differ. doi: 10.1038/cdd.2014.205. Online early access. Published Online: December 12, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Campbell DS, Okamoto H. Local caspase activation interacts with Slit-Robo signaling to restrict axonal arborization. J Cell Biol. 2013;203:657–672. doi: 10.1083/jcb.201303072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Baell JB, Holloway GA. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem. 2010;53:2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- 72.Sjoli S, Solli AI, Akselsen O, Jiang Y, Berg E, Hansen TV, Sylte I, Winberg JO. PAC-1 and isatin derivatives are weak matrix metalloproteinase inhibitors. Biochim Biophys Acta. 2014;1840:3162–3169. doi: 10.1016/j.bbagen.2014.07.011. [DOI] [PubMed] [Google Scholar]
- 73.Song Z, Chen XH, Zhang D, Ren L, Fang LN, Cheng WM, Gong P, Bi KS. Kinetic study of the degradation of PAC-1 and identification of a degradation product in alkaline condition. Chromatographia. 2009;70:1575–1580. [Google Scholar]
- 74.Abbineni G, Bisht K, Roth H, Hergenrother P, Haglund K. S-PAC-1, a small molecule activator of procaspase 3, sensitizes human breast cancer and other cell lines to ionizing radiation. Int J Radiat Oncol Biol Phys. 2012;84:S698. [Google Scholar]
- 75.Ren L, Bi K, Gong P, Cheng W, Song Z, Fang L, Chen X. Characterization of the in vivo and in vitro metabolic profile of PAC-1 using liquid chromatography–mass spectrometry. J Chromatogr B. 2008;876:47–53. doi: 10.1016/j.jchromb.2008.10.006. [DOI] [PubMed] [Google Scholar]
- 76.Nassar AEF, Kamel AM, Clarimont C. Improving the decision-making process in the structural modification of drug candidates: enhancing metabolic stability. Drug Discov Today. 2004;9:1020–1028. doi: 10.1016/S1359-6446(04)03280-5. [DOI] [PubMed] [Google Scholar]
- 77.Mori A, Miyakawa Y, Ohashi E, Haga T, Maegawa T, Sajiki H. Pd/C-catalyzed chemoselective hydrogenation in the presence of diphenylsulfide. Org Lett. 2006;8:3279–3281. doi: 10.1021/ol061147j. [DOI] [PubMed] [Google Scholar]
- 78.Soars MG, Gelboin HV, Krausz KW, Riley RJ. A comparison of relative abundance, activity factor and inhibitory monoclonal antibody approaches in the characterization of human CYP enzymology. Br J Clin Pharmacol. 2003;55:175–181. doi: 10.1046/j.1365-2125.2003.01721.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Huang S, Clark RJ, Zhu L. Highly sensitive fluorescent probes for zinc ion based on triazolyl-containing tetradentate coordination motifs. Org Lett. 2007;9:4999–5002. doi: 10.1021/ol702208y. [DOI] [PubMed] [Google Scholar]
- 80.Goode DR, Totten RK, Heeres JT, Hergenrother PJ. Identification of promiscuous small molecule activators in high-throughput enzyme activation screens. J Med Chem. 2008;51:2346–2349. doi: 10.1021/jm701583b. [DOI] [PubMed] [Google Scholar]
- 81.Franz KJ. Clawing back: broadening the notion of metal chelators in medicine. Curr Opin Chem Biol. 2013;17:143–149. doi: 10.1016/j.cbpa.2012.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Richon VM. Cancer biology: mechanism of antitumour action of vorinostat (suberoylanilide hydroxamic acid), a novel histone deacetylase inhibitor. Br J Cancer. 2006;95:S2–S6. [Google Scholar]
- 83.Russell RG, Watts NB, Ebetino FH, Rogers MJ. Mechanisms of action of bisphosphonates: similarities and differences and their potential influence on clinical efficacy. Osteoporosis Int. 2008;19:733–759. doi: 10.1007/s00198-007-0540-8. [DOI] [PubMed] [Google Scholar]
- 84.Huesca M, Lock LS, Khine AA, Viau S, Peralta R, Cukier IH, Jin H, Al-Qawasmeh RA, Lee Y, Wright J, Young A. A novel small molecule with potent anticancer activity inhibits cell growth by modulating intracellular labile zinc homeostasis. Mol Cancer Ther. 2009;8:2586–2596. doi: 10.1158/1535-7163.MCT-08-1104. [DOI] [PubMed] [Google Scholar]
- 85.Yu Y, Wong J, Lovejoy DB, Kalinowski DS, Richardson DR. Chelators at the cancer coalface: desferrioxamine to triapine and beyond. Clin Cancer Res. 2006;12:6876–6883. doi: 10.1158/1078-0432.CCR-06-1954. [DOI] [PubMed] [Google Scholar]
- 86.Purser S, Moore PR, Swallow S, Gouverneur V. Fluorine in medicinal chemistry. Chem Soc Rev. 2008;37:320–330. doi: 10.1039/b610213c. [DOI] [PubMed] [Google Scholar]
- 87.Fleming FF, Yao L, Ravikumar PC, Funk L, Shook BC. Nitrile-containing pharmaceuticals: efficacious roles of the nitrile pharmacophore. J Med Chem. 2010;53:7902–7917. doi: 10.1021/jm100762r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Park BK, Kitteringham NR, O’Neill PM. Metabolism of fluorine-containing drugs. Annu Rev Pharmacol Toxicol. 2001;41:443–470. doi: 10.1146/annurev.pharmtox.41.1.443. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.