

Abstract
The congenital myasthenic syndromes are diverse disorders linked by abnormal signal transmission at the motor endplate that stem from defects in single or multiple proteins. Multiple endplate proteins are affected by mutations of single enzymes required for protein glycosylation, and deletion of PREPL exerts its effect by activating adaptor protein 1. Finally, neuromuscular transmission is also impaired in some congenital myopathies. The specific diagnosis of some syndromes is facilitated by clinical clues pointing to a disease gene. In absence of such clues, exome sequencing is a useful tool for finding the disease gene. Deeper understanding of disease mechanisms come from structural and in vitro electrophysiologic studies of the patient endplate, and from engineering the mutant and wild-type gene into a suitable expression system that can be interrogated by appropriate electrophysiologic and biochemical studies. Most CMS are treatable. Importantly, however, some medication beneficial in one syndrome can be detrimental in another.
Introduction
The congenital myasthenic syndromes (CMS) are inherited disorders in which the safety margin of neuromuscular transmission is impaired by one or more specific mechanisms. The CMS have been recognized as distinct clinical entities since the 1970s, after the autoimmune origin of myasthenia gravis and of the Lambert-Eaton myasthenic syndromes had been established. Initially, the CMS were delineated by combined clinical, in vitro electrophysiologic, and structural studies. The study of the CMS gained further impetus when sequences of genes coding for EP-associated proteins were determined and with the advent of Sanger sequencing. In the past three years, whole exome sequencing facilitated discovery of novel CMS at an accelerated pace and by now no fewer than 20 CMS disease gene have been identified. Figure 1 shows the distribution of identified CMS disease proteins at the EP. In this review we consider the factors that affect the safety margin of neuromuscular transmission, classify the CMS identified to date, describe their distinguishing features and pathogenesis, and consider available therapies.
Figure 1.
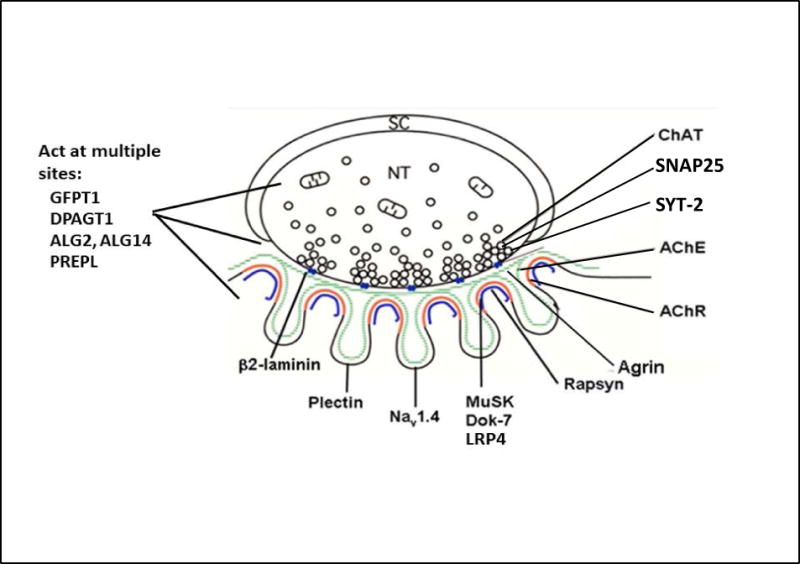
Schematic diagram of an EP with locations of pre-synaptic, synaptic and postsynaptic CMS disease proteins. Green line, synaptic basal lamina; red line, AChR on crests of the junctional folds; blue line, LRP4, MuSK, Dok-7, and rapsyn closely associated with AChR. SC, Schwann cell; NT, nerve terminal. (Modified from figure 1, Neuromuscul Disord 2012;22:99–111).
The safety margin of neuromuscular transmission
The safety margin of neuromuscular transmission (NMT) is a function of the difference between the depolarization caused by the EP potential (EPP) and the depolarization required to activate the voltage gated Nav1.4 channels deployed on the postsynaptic membrane.1 The amplitude of the EPP is a function of the number of acetylcholine (ACh) molecules per synaptic vesicle, the number of synaptic vesicles released by nerve impulse, and the efficacy of the released quanta. Quantal efficacy is determined by the EP geometry, the density and functional state of AChE in the synaptic space, the density, affinity for ACh, and kinetic properties of AChR, and the density and kinetic properties of Nav1.4.1,2
Classification of the CMS
Table 1 shows a classification of the currently recognized CMS based on 356 index patients investigated at the Mayo Clinic. Initially, the CMS were classified according to the location of the mutant protein as presynaptic, synaptic basal lamina-associated, and postsynaptic. The current classification takes into account the CMS caused by defects in protein glycosylation where the abnormal proteins are located at any EP site, and assigns a separate group to the CMS caused by defects of EP development and maintenance. Of interest, in another series of 680 CMS patients, frequencies of mutations in ChAT, ColQ, AChR subunits, rapsyn, and Dok7 were identical to those investigated at the Mayo Clinic.3
Panel 2.
Phenotypic clues to CMS disease proteins or syndromes
|
Diagnosis
Generic diagnosis
A generic diagnosis of a CMS can be made on the basis of onset at birth to early childhood, fatigable weakness affecting especially the ocular and other cranial muscles, a positive family history, and a decremental EMG response or an abnormal single-fiber EMG. However, some CMS present later in life, the weakness can affect proximal and torso rather than cranial muscles, and the decremental EMG response may be detected only after prolonged subtetanic stimulation. Tests for anti-AChR and anti-MuSK antibodies are indicated in sporadic patients after the age of 1 year and in arthrogrypotic infants even if the mother has no myasthenic symptoms. Panel 1 lists the differential diagnosis of the CMS.
Panel 1.
The differential diagnosis of congenital myasthenic syndromes
Neonatal period, infancy, childhood
|
Older patients
|
Not reported in the first year of life.
This diagnosis was suspected in some cases of the slow-channel congenital myasthenic syndrome.
Genetic diagnosis
The genetic diagnosis of a specific CMS is greatly facilitated when clinical, and EMG studies point to a candidate gene (Panel 2). If a sufficient number of affected and unaffected relatives is available, linkage analysis can point to a candidate chromosomal locus. This approach works best in inbred populations or multiplex families.4
Testing for CMS mutations in previously identified CMS genes is now commercially available but is best used in a targeted manner based on specific clinical clues. In recent years, whole exome sequencing has been used to identify CMS mutations. This approach presently captures ~97% of the entire exome but reads only 75% of the exome with more than 20× coverage. Analysis is facilitated if DNA from both parents and more than one affected family member is tested. Exome sequencing with the bioinformatics analysis is still expensive and the putative mutations must be confirmed by Sanger sequencing. The analysis can miss pathogenic noncoding variants and large deletions or duplications but the latter can be identified by array based comparative genomic hybridization.5 Finally, synonymous variants that can cause exon skipping are often filtered out. If a novel CMS disease gene is discovered, then expression studies with the genetically engineered mutant protein can be used to confirm its pathogenicity.6 Deeper insights into disease mechanisms can be obtained by in vitro analysis of neuromuscular transmission and structural studies of the neuromuscular junction (Panel 3). Although available at only few medical centers, they are important for identifying direct effects of the mutations on neuromuscular transmission, characterizing novel CMS, and providing clues for therapy.
Panel 3.
Detailed Investigation of Congenital Myasthenic Syndromes
MORPHOLOGY STUDIES
|
ENDPLATE-SPECIFIC 125I-α-BUNGAROTOXIN BINDING SITES IN VITRO ELECTROPHYSIOLOGY STUDIES
|
MOLECULAR GENETIC STUDIES
|
| EXPRESSION STUDIES IF MUTATION IDENTIFIED |
Abbreviations: AChE = acetylcholinesterase; AChR = acetylcholine receptor; MEPP = miniature endplate potential; MEPC = miniature endplate current; m = number of ACh quanta released by nerve impulse; n = number of ACh quanta available for release; p= probability of quantal release.
Currently available therapies
Current therapies for the CMS include cholinergic agonists, namely pyridostigmine and 3,4-diaminopyridine (3,4-DAP), long-lived open-channel blockers of the AChR ion channel, and adrenergic agonists.
Pyridostigmine acts by inhibiting AChE in the synaptic basal lamina which increases the number of AChRs activated by a single quantum. 3,4-Diaminopyridine (3,4-DAP) increases the number of ACh quanta released by nerve impulse. Alone, and especially in combination, they increase the amplitude of the EP potential and thereby the safety margin of neuromuscular transmission. Thus they are beneficial in patients with EP AChR deficiency and also in the fast-channel syndromes in which the safety margin of transmission is compromised by a decreased synaptic response to ACh and the abnormally fast decay of the synaptic current.
Fluoxetine and quinidine are long-lived open-channel blockers of AChR used in the treatment of the slow-channel syndrome. By curtailing the duration of the prolonged synaptic currents, they prevent a depolarization block and desensitization of AChR at physiologic rates of stimulation and mitigate the cationic overloading of the postsynaptic region that causes degeneration of the junctional folds and alters the EP geometry.
The adrenergic agonists albuterol and ephedrine were empirically found to be effective in the CMS caused by mutations in the ColQ component of AChE, Dok-7, and laminin-β2 as well as in some patients harboring low-expressor mutations of the AChR. The mechanisms by which these agents improve neuromuscular transmission is not understood.
Finally, it is important to note that agents that benefit one type of CMS can be ineffective or harmful in another type. For example, patients harboring low-expressor or fast-channel mutation in AChR are improved by cholinergic agonists whereas patients with slow-channel mutations in AChR are worsened by these medications. Patients harboring mutations in Dok-7 are rapidly worsened by cholinergic agonists but are improved by adrenergic agonists. Therefore it is essential that a molecular diagnosis should inform the choice of therapy. Finally, the cholinergic agonists pyridostigmine and 3,4-DAP exert their effect as soon as the medication is absorbed whereas the adrenergic agonists and the AChR channel blockers act more slowly over days, weeks, or months. Table 2 summarizes the pharmacotherapy of the currently recognized CMS.
Presynaptic syndromes
Choline acetyltransferase (ChAT) deficiency
ChAT catalyzes the synthesis of ACh from acetyl-CoA and choline in cholinergic neurons. The diagnosis is suggested by sudden episodes of apnea provoked by stress or without apparent cause occurring in patients with few or no myasthenic symptoms. Some patients are apneic and hypotonic at birth; others are normal at birth and develop apneic attacks during infancy or childhood.7–11 However, apneic episodes also can occur in patients with Na-channel myasthenia2 or with mutations in rapsyn.12,13 In some children an apneic attack is followed by ventilatory failure for weeks.14 Few patients never breathe spontaneously, and some develop cerebral atrophy from hypoxemia.11 A clue to the identity of the disease gene came from the observation that subtetanic stimulation reduced the amplitude of the compound muscle action potential (CMAP) and of EPP to 50% below the baseline (normal <30%) followed by slow recovery over 5–10 minutes. A marked decrease of the CMAP amplitude after subtetanic stimulation also occurs in other CMS but is followed by recovery in less than 5 minutes. The slow recovery implicated a defect in ACh resynthesis and provided the clue to discovery of mutations in ChAT15 (figure 2A).
Figure 2.
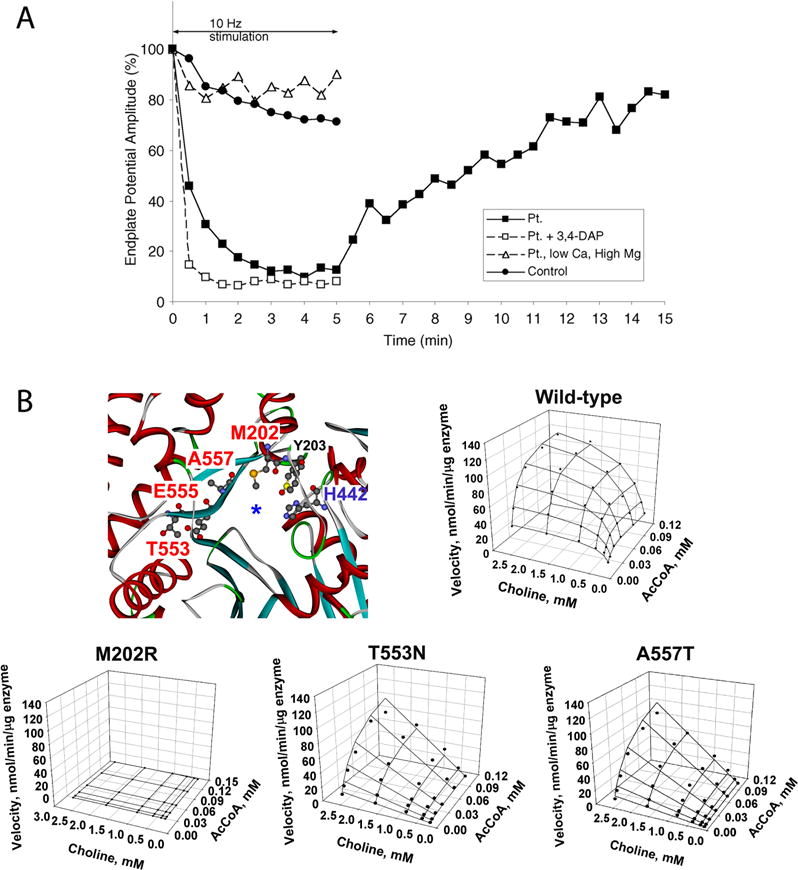
ChAT deficiency. (A) Subtetanic stimulation rapidly decreases the endplate potential (EPP) which returns to the baseline slowly over more than 10 min. 3,4-diaminopyridine (3,4-DAP) which increases quantal release accelerates the decline of the EPP, whereas a low Ca2+/high Mg2+ solution which reduces quantal release prevents the abnormal decline of the EPP. (B) Positions of mutated residues in active site tunnel of ChAT, kinetic landscapes of wild-type and mutant enzymes obtained from reaction velocities over a range and AcCoA concentrations, and normalized kinetic parameters of wild-type and mutant ChAT. The catalytic efficiencies of the enzymes with AcCoA (kcat/Kma), choline (kcat/Kmb), and both substrates (kcat/KiaKmb), were calculated as described in Reference 11. The catalytic efficiencies of the mutant enzymes are <0.1% of wild-type. Kcat = maximal reaction velocity/enzyme concentration; KmA and KmB = Michelis-Menten constants for AcCoA and choline; Kia = dissociation constant for the enzyme-AcCoA complex. (Panel A is reproduced from Nature Rev Neurosci, 2003;330–352; Panel B is reproduced from Hum Mutat 2011;32:1259–1267.)
The severity of the CMS has been correlated with position of the mutant residues in the atomic structural model of ChAT, and with the level of expression, catalytic activity, and structural stability of recombinant mutant enzymes.11 The most severe kinetic abnormalities are caused by mutations in the active-site tunnel of the enzyme or close to the substrate binding site, or exert their effect allosterically (figure 2B). The safety margin of NMT is compromised by incomplete filling of the synaptic vesicles with ACh.
Therapy consists of AChE inhibitors that must be continued even in asymptomatic patients but the most severely affected patients may fail to respond. The parents must be instructed in use of a portable respirator, an apnea monitor at home, and intramuscular injection of neostigmine methylsulfate.7
Synaptic basal lamina associated syndromes
EP AChE deficiency due to mutations in COLQ
The EP species of AChE is an asymmetric enzyme composed of 1, 2, or 3 homotetramers of globular catalytic subunits (AChET) attached to a triple-stranded collagenic tail (ColQ) that anchors the enzyme in the synaptic basal lamina16 (figure 3). Conserved domains of ColQ include an N-terminal proline-rich attachment domain (PRAD) that associates each ColQ strand with an AChET tetramer, a central collagen domain, and a C-terminal region required for assembly of the ColQ strands in a triple helix (figure 3A and B). ColQ is anchored in the synaptic space by binding to the extracellular domain of MuSK17 and by its cationic residues to perlecan. The synaptic currents and potentials are prolonged owing to the extended lifetime of ACh in the synaptic space. The nerve terminals are abnormally small and curtail the number of quanta available for release, and some junctional folds are degenerating. Mutations have now been identified in each ColQ domain (figure 3A). Depending on their location, they prevent the collagen domain from associating with the catalytic subunits, truncate and prevent assembly of the triple-helical collagen domain, or reduce expression of the assembled enzyme.
Figure 3.
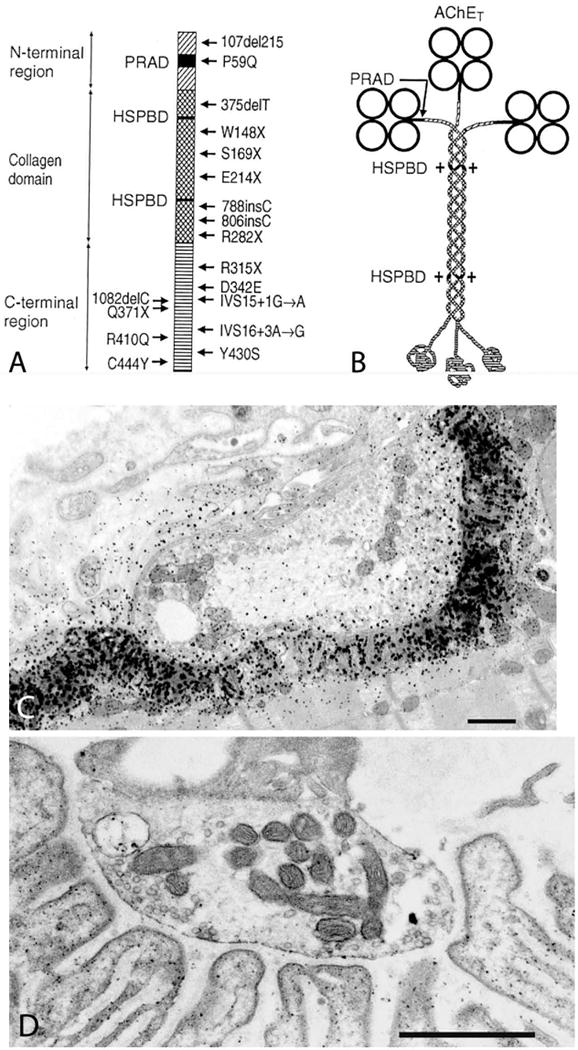
EP AChE deficiency. (A) Schematic diagram showing domains of a ColQ strand with 24 identified ColQ mutations. (B) Schematic diagram of the A12 species of asymmetric AChE. PRAD = proline rich attachment domain; HSPBD, heparan sulfate proteoglycan binding domain. (C) Electron cytochemical localization of AChE in a control subject (C) and in an AChE-deficient patient (D). There is no reaction for AChE at the patient EP. Bars in (C) and (D) = 1 μm. (Panel A is reproduced by from Neuromuscul Disord 22:99–111, 2012)
Severely affected patients present in infancy.18,19,20 Less severely affected patients present in childhood and become disabled later in life. The weakness can affect all voluntary muscles but may spare the ocular muscles, and in few patients it has a limb-girdle distribution. Treatment with ephedrine21,22 or albuterol,23,24 is beneficial but the effects develop gradually, over several months or longer.24 The mechanism by which adrenergic agonists improve the disease is not understood.
CMS associated with laminin-β2 deficiency
Laminin β2, encoded by LAMB2, is highly expressed in basal lamina of the eye, kidney, and -EP. At the EP, laminin β2 aligns the nerve terminal with the postsynaptic region and regulates their trophic interactions. A single report describes a patient with renal and ocular malformations (Pierson syndrome) and a CMS due to heteroallelic missense and frameshift mutations in LAMB2.25 NMT was compromised by decreased quantal content of the EPP and low-amplitude MEPPs. The nerve terminals were small, as in EP AChE deficiency, accounting for the decreased quantal release. The reduced MEPP amplitude was attributed to simplification of the postsynaptic region. The patient was worsened by pyridostigmine but was improved by ephedrine.
Defects in AChR
The muscle form of nicotinic AChR
Most CMS stem from molecular defects in the muscle nicotinic AChR, an integral membrane protein with the subunit composition α2βδɛ at the adult EP or α2βδγ at the fetal EP and extrajunctional regions. The genes encoding the α (CHRNA1), δ (CHRND), and γ (CHRNG) subunits are at different loci on chromosome 2q and those encoding the β (CHRNB) and ɛ (CHRNE) subunits are at different loci on chromosome 17p. At the normal EP, AChR is concentrated on the crests of the junctional folds.
The five subunits of AChR are organized like barrel staves around a central cation-permeable channel. Each subunit has a large N-terminal extracellular ligand-binding domain composed mostly of β strands, an extracellular coupling domain, a pore forming domain composed of four transmembrane α-helices (M1–M4) and a short C-terminal extracellular domain (figure 4). The M2 α-helices from each subunit align to form the ion channel, while the C-terminus of each M1 domain may also contribute to the channel in the resting state.26 The extracellular M2/M3 linker and intracellular M1/M2 and M3/M4 linkers join the four transmembrane α-helices of each subunit. The cytoplasmic M3/M4 linker is unusually long. It is important for attachment to the cytoskeleton, contains phosphorylation sites that may be important for desensitization, and in the ɛ subunit it harbors residues that contribute to the kinetics and the fidelity of channel gating.27,28 Each AChR has two solvent accessible ACh-binding pockets located at the α/ɛ (or α/γ) and α/δ subunit interfaces. Residues from seven distinct regions of the primary sequences of each subunit pair converge to form the ACh binding pocket: three regions originate from the α subunit and four from the δ, ɛ, or γ subunits.29
Figure 4.
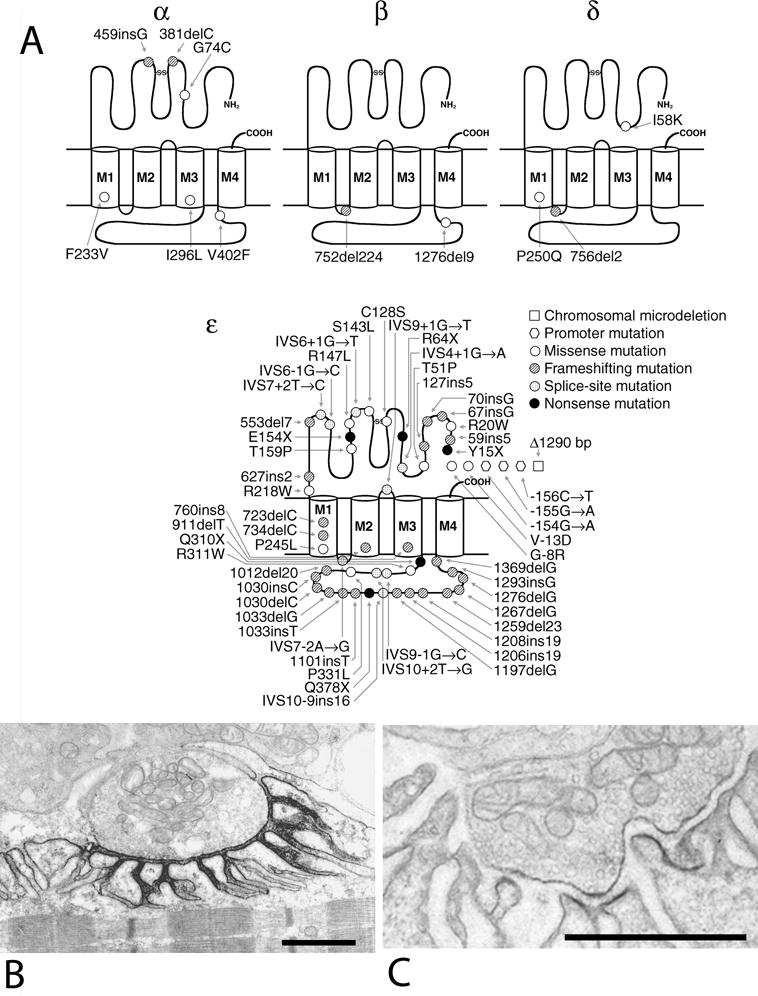
EP AChR deficiency. (A) Schematic diagram of low-expressor and null mutations reported in the AChR α, β, δ, and ɛ subunits. Most mutations occur in the ɛ subunit. Splice-site mutations point to the N-terminal codons of the predicted skipped exons. Panels (B) and (C) show ultrastructural localization of AChR with peroxidase-labeled α-bungartoxin at an EP from a control subject and from a patient homozygous for c.ɛ553del7. Bars in (B) and (C) = 1 μm. (Panel A is reproduced from Muscle Nerve, 27:4–25, 2003)
Following its release from the nerve terminal, ACh binds to the two binding sites on the receptor with rate constants that approach the limit of diffusion, thus rapidly forming a doubly-liganded complex with the receptor in which the ion channel remains closed. After a very short latency, the channel opens and it remains open for a millisecond or so. After the channel returns to the doubly-liganded closed state, ACh rapidly dissociates from the binding sites and is cleared from the synaptic cleft by diffusion and hydrolysis by AChE. The combination of rapid dissociation and rapid clearance of ACh ensures that the synaptic response is brief, and that a succeeding response can be elicited in quick succession. Because the two ACh binding sites contain different non-α subunits, the rates of ACh association and dissociation differ at each site, resulting in distinct agonist binding affinities.30 The extent to which the binding affinities differ depends on animal species; for the adult human AChR the affinities differ by approximately 5-fold, with the α/δ site exhibiting high affinity and the α/ɛ site low affinity.31 The mechanisms of AChR activation are described and discussed in online Appendix 1 and online Panel 1.
CMS caused by primary AChR deficiency
These CMS result from recessive missense, nonsense, or splice site and promoter region mutations in each AChR subunit, but most occur in the ɛ subunit.32 Preference for the ɛ subunit has been attributed to phenotypic rescue by substitution of the fetal γ subunit for the defective ɛ subunit27,33,34 (figure 5A). Despite this, the EP AChR content is reduced to approximately 10 percent of normal (figure 5B and C), and the synaptic response to ACh, reflected by the low amplitude of the MEPP and MEPC, is markedly decreased. Evoked quantal release is increased and partially compensates for the AChR deficiency.35 Single-channel patch-clamp recordings from patient EPs reveal low-amplitude, long-duration channel openings typical of the fetal AChR. The synaptic contacts consist of multiple small regions over an extended length of the fiber surface, but the structural integrity of the pre- and postsynaptic regions is preserved. Nearly all patients have eyelid ptosis, oculoparesis that often becomes fixed, and variable but frequently moderate to severe limb muscle weakness. Patients harboring null mutations in both alleles of CHRNA1, CHRNB, or CHRND cannot be rescued because no substituting subunits exist and hence are likely embryonic lethal. Patients with heterozygous or homozygous low-expressor mutations in the non-ɛ subunits are severely affected and have a high mortality in infancy or early childhood. The first line of therapy is an AChE inhibitor, such as pyridostigmine, which increases the number of AChRs activated by each quantum. Patients failing to respond adequately often derive additional benefit from 3,4-diaminopyridine (3,4-DAP), a potassium channel blocker that enhances quantal release by prolonging the duration of the presynaptic action potential, and hence Ca2+ entry into the nerve terminal.36 A beneficial effect of albuterol was reported in two patients with low expressor ɛ subunit mutations who failed to respond to pyridostigmine and 3,4-DAP.37
Figure 5.
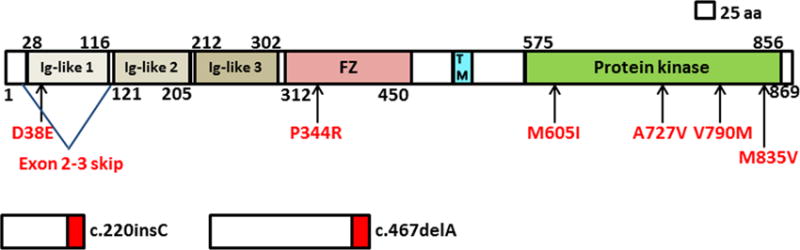
Scaled linear model of MUSK with its domains and observed mutations. Exon 2–3 skip predicts in-frame deletion. Red boxes in the frameshifting mutations represent missense amino acids. Transmembrane domain includes amino acids 496–516. FZ, frizzled; TM, transmembrane.
CMS caused by kinetic defects in AChR: Slow-channel syndrome
This CMS is caused by dominant mutations in the ligand binding or pore domains of AChR (see green symbols in figure 4 and online Panel 2) and result in prolonged synaptic currents and potentials. Mutations in the ligand binding domain enhance affinity for ACh by slowing its dissociation rate from the doubly-liganded receptor, whereas most mutations in the pore domain enhance steps in the process of receptor activation subsequent to agonist binding.32 Either mechanism prolongs the single-channel currents (figure 6B), EP currents, and EPPs, while mutations in the pore domain also cause spontaneous channel openings. As in EP AChE deficiency, the prolonged EPP triggers a repetitive CMAP (figure 6A), but in this syndrome edrophonium enhances the repetitive response. Also, during activity, successive prolonged EPPs progressively depolarize the postsynaptic membrane causing a depolarization block. The prolonged as well as spontaneous openings of the AChR channel result in Ca2+ accumulation in the postsynaptic region38 (figure 6D and E) and an EP myopathy (figure 6F). The safety margin of NMT is compromised by depolarization block of the action potential, desensitization of AChR at physiologic rates of neuromuscular activity, and by the EP myopathy that causes loss of AChR from the junctional folds and alters the EP geometry.
Figure 6.
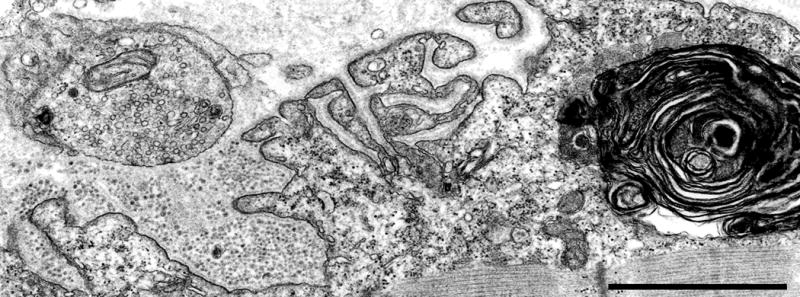
Degenerating EP in Dok7 myasthenia. Note replacement of degenerate junctional folds by debris, and large myeloid structure in junctional sarcoplasm. Bar = 1 μm.
The slow channel syndrome usually presents in the first decade but severely affected patients present in the neonatal period. There is selectively severe involvement of the cervical, scapular, and dorsal forearm muscles. The ocular muscles are usually spared. Some patients have mild asymmetric ptosis. Pyridostigmine worsens the disease by increasing the EP myopathy but long-lived open-channel blockers of the AChR channel, such as quinidine (figure 6G and H),39 quinine, or fluoxetine40–42 gradually improve and even remove the symptoms.
CMS caused by kinetic defects in AChR: Fast-channel syndrome
This syndrome is caused by a recessive fast-channel mutation of one allele accompanied by a null or low-expressor mutation of the second allele, or rarely by another fast-channel mutation on the second allele32 (see red symbols in figure 4, and online Panel 3). It is the physiological opposite of the slow-channel syndrome in that the EP currents decay abnormally fast, the channel openings are abnormally brief (figure 7B), and the amplitude of the synaptic currents and potentials is reduced owing to a reduced probability that the AChR channel is opened by physiologic concentrations of ACh (figure 7C).
Figure 7.
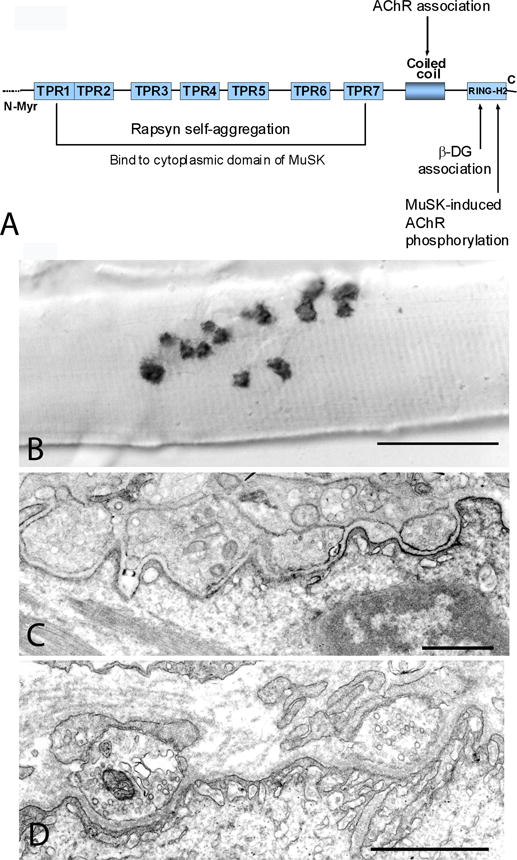
CMS due to mutations in rapsyn. (A) Rapsyn domains and interactions. (B) Small cholinesterase reactive EP regions are distributed over an extended length of the muscle fiber. (C) and (D) Multiple small nerve terminals appear over poorly developed junctional folds. In (C), the distribution of AChR on the postsynaptic membrane, visualized with peroxidase-labeled α-bungarotoxin, is patchy. Bars 50 μm in (A) and 1 μm in (B) and (C).(Reproduced from Neurology 2009;73:228–235.)
The mutations reside in different domains and subunits of AChR (figure 4) and exert their effects by different mechanisms. In the extracellular domains, ɛP121L,43 and ɛW55R44 decrease affinity for ACh,43 whereas αV132L,45 αV188M,46 and δL42P47 interfere with coupling of agonist binding to channel gating. In the M3 transmembrane domain αV285I reduces gating efficiency.48 In the long cytoplasmic loop of the ɛ subunit, N436del shortens channel openings by reducing the stability of the diliganded receptor,49 and c.1254ins18, A411P produce a wide range of channel gating kinetics resulting in an increased proportion of brief channel openings27,28 Finally, some mutations are pathogenic by a combination of these factors.45,50,46 No clinical clues point to the diagnosis of a fast-channel syndrome. Consequently, in vitro microelectrode studies of the neuromuscular junction and single-channel patch-clamp studies are required to detect and define the kinetic abnormality. The clinical consequences are severe when the pathogenic mutation appears at or near the agonist binding site44 or affects coupling of agonist binding to gating,45 or is positioned in a transmembrane domain,48 and less severe when it is in the long cytoplasmic loop of the ɛ subunit.28 Therapy consists of 3,4-DAP and pyridostigmine.
Defects in EP development and maintenance
To date, mutations in proteins essential for EP development and maintenance have been detected in MuSK (MUSK), neural agrin (AGRN), LRP4 (LRP4), Dok-7 (DOK7), and rapsyn (RAPSN) which form a signaling network essential for EP development and maintenance. Agrin, secreted into the synaptic space by the nerve terminal, binds to the lipoprotein-related protein LRP4 in the postsynaptic membrane. The Agrin-LRP4 complex binds to and activates MuSK. This enhances MuSK phosphorylation and leads to clustering of LRP4 and MuSK. Activated MuSK in concert with postsynaptic Dok-7 and other postsynaptic proteins acts on rapsyn to concentrate AChR in the postsynaptic membrane, enhances synapse specific gene expression by postsynaptic nuclei, and promotes postsynaptic differentiation. Clustered LRP4, in turn, promotes differentiation of motor axons. The agrin-LRP4-MuSK-Dok-7 signaling system is also essential for maintaining the structure of the adult neuromuscular junction (reviewed in Ref. 51).
Agrin deficiency
Only three reports describe a CMS caused by defects in agrin. In one report, two siblings had eyelid ptosis and mild weakness of the facial and limb-girdle muscles.52 They carry a homozygous missense mutation (G1709R) in the laminin G-like 2 domain required for MuSK activation. The EPs showed no AChR and agrin deficiency but some were misshaped or regenerating, and some postsynaptic gutters lacked nerve endings. The postsynaptic regions were preserved. Expression studies revealed no effect on MuSK activation by agrin. This mutation perturbs EP maintenance without preventing postsynaptic differentiation. Pyridostigmine and 3,4-DAP were ineffective.
The second report describes a severe CMS caused by two heteroallelic mutations: Q353X in the N-terminal region and V1727F located in the second laminin G-like domain of agrin.{Maselli, 2012 7822/id} The nonsense mutation abolishes agrin expression and the missense mutation markedly reduces agrin-induced AChR clustering in C2 muscle cells. The synaptic contacts were dispersed and fragmented, the postsynaptic regions simplified, the nerve terminals small, and the junctional cytoplasm displayed degenerative changes. The patient did not respond to pyridostigmine or 3,4-DAP but responded partially to ephedrine.
LRP4 deficiency
A recent study identified heteroallelic E1233K and R1277H mutations at the edge of the third beta-propeller domain of LRP4 in a 14-year-old girl. Both mutations decrease binding affinity of LRP4 for both MuSK and agrin in a position specific-manner. The patient had a respiratory arrest at birth, delayed motor development, and fatigable weakness of the limb-girdle muscles. Therapy with pyridostigmine worsened her weakness. In an intercostal muscle, the synaptic contacts were disarrayed and the pre- and post-synaptic regions were hypoplastic. Despite these structural changes, the AChR content of the EPs, quantal release by nerve impulse, and the synaptic response to ACh were unaffected.54
MuSK deficiency
In the past decade, 6 reports have described CMS caused by mutations in MUSK (figure 8). The disease presents at birth or early life with eyelid ptosis or respiratory distress.55,56,57,58 Subsequently, it involves the ocular, facial and proximal limb muscles, and in some kinships the bulbar muscles as well. Structural studies in humans55,58 and transgenic mice59 reveal extensive remodeling of the EPs due to recurrent cycles of focal denervation and reinnervation. Pyridostigmine therapy is ineffective55,57,58 or worsens the disease.55,56 3,4-DAP therapy improved symptoms in one patient.55 Importantly, a recent report indicates that therapy with albuterol has been highly effective in two brothers.60 No clear genotype-phenotype correlations have been observed.
Dok-7 deficiency
The observation that Dok-7 is a muscle-intrinsic activator of MuSK61 prompted a search for CMS caused by mutations DOK7. This resulted in detection of numerous mutations in DOK762–66,67 and c.1124_1127dupTGCC in exon 7 emerged as a common variant. Other mutations prevent Dok-7 to associate with and become activated by Crk1/Crk1L. No consistent phenotype-genotype correlations have emerged.
All affected patients have limb-girdle weakness with lesser facial and cervical muscle involvement but a few have severe bulbar weakness and significant oculoparesis.64 Some were initially misdiagnosed as having limb-girdle muscular dystrophy. The clinical course is mild to severe. Type 1 fiber preponderance, type 2 fiber atrophy, mild myopathic changes and target formations are associated features. All EPs consist of one to multiple small synaptic contacts. Impaired maintenance of the EPs is evidenced by ongoing destruction and remodeling of the EPs (figure 9). NMT is compromised by the decreased quantal content of the EPP and the reduced MEPP amplitude.64 Importantly, this CMS is rapidly worsened by cholinergic agonists but responds well over a period of time to ephedrine68 or albuterol.64
Rapsyn deficiency
Rapsyn concentrates and anchors AChR in the postsynaptic membrane69 and is required for development of the junctional folds.12 Distinct regions of rapsyn subserve different functions: a myristoylated N-terminal region links rapsyn to the postsynaptic membrane; 7 tetratricopeptide repeats are important for rapsyn self-aggregation and to bind to the cytoplasmic domain of MuSK; a coiled-coil domain interacts with long-cytoplasmic loops of AChR subunits; and a C-terminal domain binds to β-dystroglycan which links rapsyn to the actin cytoskeleton69,70 (figure 10A). Recent cryoelectron tomography studies indicate that rapsyn and AChR form a network with up to 3 rapsyn dimers contacting the cytoplasmic domain of each AChR.71
Most patients present in the first year and a few in later life.13 Arthrogryposis at birth and other congenital malformations occur in nearly one-third of the patients.12,13,72 Intercurrent infections or fever can trigger respiratory crises resulting in anoxic encephalopathy.12,13,73,74 The clinical features can suggest autoimmune myasthenia gravis but the ocular ductions are intact in most patients.13,72 Multiple synaptic contacts appear on single muscle fibers (figure 10B). The EP AChR deficiency is milder than in primary AChR deficiency72 and the junctional folds are not well differentiated (figure 10C and D). Most patients respond well to AChE inhibitors; some derive additional benefit from 3,4-DAP, and some benefit from the added use of ephedrine or albuterol.73
Indo-Europeans harbor a common N88K mutation in RAPSN.75 Other mutations appear in the promoter region and in the entire open reading frame of RAPSN. Different mutations hinder rapsyn self-association or binding to AChR, impede agrin-MuSK-LRP4-mediated clustering of AChR, or decrease rapsyn expression.12,76 There are no genotype-phenotype correlations except that Near-Eastern Jewish patients with a homozygous E-box mutation (c.-38A>G) have a mild phenotype with ptosis, prognathism, severe masticatory and facial muscle weakness, and hypernasal speech.77
Congenital defects of glycosylation
Glycosylation increases solubility, folding, stability, assembly, and intracellular transport of nascent peptides. O-glycosylation occurs in the Golgi apparatus with addition of sugar residues to hydroxyl groups of serine and threonine; N-glycosylation occurs in the endoplasmic reticulum in sequential reactions that decorate the amino group of asparagine with a core glycan composed of 2 glucose, 9 mannose and 2 N-acetylglucosamine (GlcNAc)78,79 To date, defects in four enzymes subserving glycosylation cause a CMS: GFPT1 (glutamine fructose-6-phosphate transaminase),4,80 DPAGT1 (dolichyl-phosphate [UDP-N-acetylglucosamine] N-acetylglucosaminephosphotransferase 1),81,82 ALG2 (alpha-1,3-mannosyl transferase), and ALG14 (UDP-N-acetylglucosaminyltransferase subunit).83 Tubular aggregates of the sarcoplasmic reticulum (SR) in muscle fibers are a phenotypic clue to the diagnosis but are not present in all patients. Because glycosylated proteins are present at all EP sites, NMT is compromised by a combination of pre- and postsynaptic abnormalities.
Defects in GFPT1
GFPT1 controls glucose entry into the hexosamine pathway, and hence formation of precursors for N- and O-linked protein glycosylation. A CMS caused by defects in GFPT1 causing limb-girdle muscle weakness responsive to pyridostigmine was reported in 16 patients in 2011.4 A subsequent study of 11 patients revealed slowly progressive weakness in 10, but one patient with mutations disrupting the muscle-specific exon of GFPT1 never moved in utero, was arthrogrypotic at birth, and remains bedfast and tube-fed at age 8 years. She has a severe myopathy with numerous dilated and degenerating vesicular profiles, autophagic vacuoles, and bizarre apoptotic nuclei.80 Muscle specimens in 9 less affected patients revealed tubular aggregates in 6. Electron microscopy demonstrated abnormally small EP regions and poorly developed junctional folds. In vitro electrophysiology studies revealed a reduced synaptic response to ACh and decreased quantal release in the most severely affected patient. That many EPs in this CMS are underdeveloped likely stems from hypoglycosylation and altered function of EP-associated glycoproteins, such as MuSK, agrin, and dystroglycans.
Defects in DPAGT1
DPAGT1 catalyzes the first committed step of N-linked protein glycosylation. DPAGT1 deficiency predicts impaired asparagine glycosylation of multiple proteins distributed throughout the organism, but in the first 5 patients harboring DPAGT1 mutations only neuromuscular transmission was adversely affected; this was attributed to reduced AChR expression at the EP but the patient EPs were not examined.81 A subsequent study of two siblings and of a third patient with DPAGT1 deficiency extended the phenotypic spectrum of the disease.82 These patients have moderately severe to severe weakness and are intellectually disabled. The siblings respond poorly to pyridostigmine and 3,4-DAP; the third patient responds partially to pyridostigmine and albuterol. Whole exome sequencing revealed that the siblings harbored an M1L mutation that reduces protein expression, and H375Y which decreases enzyme activity. The third patient carries V264M that abolishes enzyme activity and a synonymous L120L mutation that markedly augments exon skipping resulting in many skipped and few nonskipped alleles.
Intercostal muscle studies showed fiber type disproportion, small tubular aggregates, and an autophagic vacuolar myopathy. Electron microscopy revealed few degenerating EPs and small pre- and postsynaptic regions. Evoked quantal release, postsynaptic response to ACh, and the EP AChR content were all reduced to ~50% of normal. Immunoblots of muscle extracts with two different antibodies indicated decreased to absent glycosylation of different proteins, including that of STIM1, an SR-associated calcium sensor that operates in concert with Orai1 on the plasma membrane to homeostatically regulate the SR calcium content.84 Because mutations in STIM1 cause a tubular aggregate myopathy,85 STIM1 hypoglycosylation is a likely cause of the tubular aggregates in muscle in N-glycosylation disorders.
Defects in ALG2 and ALG14
ALG2 catalyzes the second and third committed steps of N-glycosylation. In one kinship four affected siblings were homozygous for an insertion/deletion mutation, and another patient was homozygous for a low-expressor V68G mutation. ALG14 forms a multiglycosyltransferase complex with ALG13 and DPAGT1 and thus contributes to the first committed step of N-glycosylation. In one family two affected siblings carried heteroallelic P65L and V68G mutations. EP ultrastructure and parameters of neuromuscular transmission were not investigated.83
Other myasthenic syndromes
PREPL deletion syndrome
The hypotonia-cystinuria syndrome is caused by recessive deletions involving the SLC3A1 and PREPL genes at chromosome 2p21. The major clinical features are type A cystinuria, growth hormone deficiency, muscle weakness, ptosis, and feeding problems. A patient with isolated PREPL deficiency had myasthenic symptoms since birth, a positive edrophonium test and growth hormone deficiency but no cystinuria, and responded transiently to pyridostigmine during infancy.86 She harbors a paternally inherited nonsense mutation in PREPL and a maternally inherited deletion involving both PREPL and SLC3A1; therefore the PREPL deficiency determines the phenotype. PREPL expression was absent from the patient’s muscle and EPs. EP studies revealed decreased evoked quantal release and small MEPPs despite robust EP AChR expression.86 Because PREPL is an essential activator of the clathrin associated adaptor protein 1 (AP1),87 and AP1 is required by the vesicular ACh transporter to fill the synaptic vesicles with ACh,88 the small MEPP is attributed to a decreased vesicular content of ACh.
Plectin deficiency
Organelle and tissue specific isoforms of plectin, encoded by PLEC, crosslink intermediate filaments to their targets in different tissues and thereby assure their cytoskeletal support. Defects in plectin can cause epidermolysis bullosa simplex (EBS),89 muscular dystrophy,90,91 and a myasthenic syndrome.92,93 The muscular dystrophy is caused by loss of cytoskeletal support of the muscle fiber organelles, which become dislocated, and of the sarcolemma which displays multiple small defects allowing calcium ingress into the fibers.93 The two patients investigated by us respectively harbor Q2057X and R2319X, and a shared c.12043dupG mutation92,93 Both patients had EBS since infancy and later developed a progressive myopathy and a CMS refractory to pyridostigmine. Both had a decremental EMG response on repetitive nerve stimulation, and half-normal MEPPs attributed to degeneration of the junctional folds with loss of AChR and altered EP geometry.93
Defect in Nav1.4
This CMS was detected in a single patient with brief abrupt attacks of muscle weakness and respiratory arrest that caused an anoxic encephalopathy. The synaptic response to ACh and quantal release by nerve impulse were normal, but normal amplitude EPPs failed to generate muscle action potentials, pointing to a defect in action potential generation. Mutation analysis of SCN4A, the gene encoding Nav1.4, revealed two mutations (S246L in the S4/S5 linker in domain I and V1442E in S4/S5 linker in domain IV). Nav1.4 expression at the EPs was normal. Expression studies in HEK cells of the V1442E-sodium channels revealed marked enhancement of fast inactivation near the resting membrane potential and enhanced use-dependent inactivation on high frequency stimulation. S246L had only minor kinetic effects and is likely a benign mutation. The safety margin of NMT is compromised because most Nav1.4 channels are inexcitable in the resting state.2 The patient responded partially to therapy with pyridostigmine and acetazolamide.
Myasthenias associated with congenital myopathies
Eyelid ptosis, ophthalmoparesis, weakness of facial muscles, exercise intolerance, a decremental EMG study, and response to pyridostigmine have been documented in patients with centronuclear myopathies (CNM) caused by mutations in amphiphysin (BIN1),94 myotubularin (MTM1),95 and dynamin 2 (DNM2)96 as well as in other CNM patients with no identified mutations.97 Importantly, knockdown of MTM1 or DNM2 in zebrafish causes reduced spontaneous and touch-evoked movements that respond dramatically to edrophonium.95,96 Detailed investigation of neuromuscular transmission in an adult CNM patient with myasthenic symptoms and no identified mutations revealed EP remodeling, mild EP AChR deficiency, simplified postsynaptic regions, 60% reduction of the MEPP amplitude, and a 40% decrease of evoked quantal release attributed to a decreased number of synaptic vesicles available for release.97 Interestingly, two patients with a congenital myopathy caused by tropomyosin 3 deficiency had signs and symptoms mimicking a CMS.98
Conclusions and future directions
Initially, molecular bases for the CMS were approached with in-depth clinical evaluation combined with electrophysiologic and morphometric analyses of patient endplates. With the introduction of gene cloning and sequencing of complementary DNAs, the candidate gene approach provided a powerful complement to these analyses. After a variant in a candidate gene was identified, the goal was to demonstrate its pathogenicity, which could be achieved by incorporating the wild type and mutant genes into a heterologous expression system and then assessing the level of expression of the mutant protein and its functional properties by appropriate methods that included the patch-clamp studies to dissect the kinetic effects of AChR mutants, and enzyme assays of the wild-type and mutant species of ChAT, DPAGT1 and AChE. The net result was demonstration that the CMS were caused by a diversity of disease targets and molecular mechanisms, which together guided an individualized therapy.
Despite the power of the candidate gene approach, in some CMS the disease gene has remained elusive. For these CMS, whole exome sequencing, available in the past 3 years, proved to be a lodestone to discovery of unsuspected defects in genes subserving protein glycosylation, and more are surely in the pipeline. The power of this approach is enhanced if multiple family members, and especially trios are analyzed, and interpretation of the results is greatly facilitated by phenotypic clues. Although whole exome sequencing fails to identify large scale duplications or deletions, these can now be detected by microarray based comparative gene hybridization. Another future approach will be the use of custom made microarrays designed to detect mutations in heretofore identified CMS disease genes. A drawback of this method will be that it cannot identify newly emerging CMS disease genes. However, in many patients it will obviate the need for more expensive whole exome sequencing.
It is highly likely that more CMS disease genes will be discovered but demonstration of pathogenicity associated with individual mutations will remain a necessity. This will take advantage of tried and true methods for gene expression and functional comparison of the wild type and mutant gene products. Clinicians armed with knowledge of the molecular mechanism behind the aberrant gene product will be in an advantageous position to promote rational and individualized therapy of each form of CMS.
Supplementary Material
Table 1.
Classification of the CMSa
| Index cases | Percent | |
|---|---|---|
| Presynaptic (6.2%) | ||
| Choline acetyltransferase deficiency | 18 | 5.1 |
| Paucity of synaptic vesicles and reduced quantal releaseb | 1 | 0.3 |
| Other presynaptic defectsb | 3 | 0.8 |
| Synaptic basal lamina-associated (12.9%) | ||
| Endplate AChE deficiency | 45 | 12.6 |
| Laminin β2 deficiency | 1 | 0.3 |
| Defects in AChR (50.6%) | ||
| Primary AChR deficiency ± minor kinetic abnormality | 118 | 33.1 |
| Primary kinetic defect ± minor AChR deficiency | 62 | 17.4 |
| Defects in EP development and maintenance (25.3%) | ||
| Agrin deficiency | 1 | 0.3 |
| LRP4 myasthenia | 2 | 0.6 |
| MuSK deficiency | 1 | 0.3 |
| Dok-7 myasthenia | 35 | 9.8 |
| Rapsyn deficiency | 51 | 14.3 |
| Congenital defect of glycosylation (3.6%) | ||
| GFPT1 myasthenia | 11 | 3.1 |
| DPAGT1 myasthenia | 2 | 0.6 |
| ALG2c | 0 | 0 |
| ALG14c | 0 | 0 |
| Other myasthenic syndromes (1.4%) | ||
| PREPL deletion syndrome | 1 | 0.3 |
| Kinetic defect in Nav 1.4 | 1 | 0.3 |
| Plectin deficiency | 2 | 0.6 |
| CMS associated with centronuclear myopathies | 1 | 0.3 |
| Total | 356 | 100 |
Classification based on cohort of congenital myasthenic syndrome patients investigated at the Mayo Clinic between 1988 and 2014
No gene defect identified
Defects in ALG2 and ALG14 were identified at another medical center.83
Table 2.
Pharmacotherapy of the CMS
| Syndrome | Therapy | Caveats |
|---|---|---|
| ChAT deficiency | Pyridostigmine; parenteral neostigmine methyl sulfate for acute apneic episodes. | |
| AChE deficiency | Albuterol or ephedrine | Avoid pyridostigmine and 3,4-DAP |
| Laminin-β2 deficiency | Ephedrine | Avoid pyridostigmine |
| Simple AChR deficiency | Pyridostigmine; 3,4-DAP also helps. Albuterol may help if refractory to above | |
| Slow-channel CMS | Quinine, quinidine, or fluoxetine | Avoid pyridostigmine and 3,4-DAP |
| Fast-channel CMS | Pyridostigmine and 3,4-DAP | Avoid quinine, quinidine, or fluoxetine |
| Rapsyn deficiency | Pyridostigmine, 3,4-DAP, albuterol | |
| Musk deficiency | Variable response to 3,4-DAP. Good response to albuterol in one patient | Conventional doses of pyridostigmine can worsen symptoms |
| Dok-7 myasthenia | Albuterol or ephedrine | Avoid pyridostigmine |
| Agrin deficiency | No response to pyridostigmine or 3,4-DAP in one pt; partial response to ephedrine in a second pt. | Use pyridostigmine with caution |
| LRP4 deficiency | Albuterol or ephedrine not tested | Avoid pyridostigmine |
| GFPT1, DAPGT1, ALG2 and ALG14 | Pyridostigmine; 3,4-DAP may confer additional benefit. | |
| Na-channel myasthenia | Pyridostigmine and acetazolamide | |
| PREPL deficiency | Pyridostigmine beneficial in infancy | Older patients are refractory to pyridostigmine |
Search strategy for the article.
PubMed was searched for articles published on congenital myasthenic syndromes between January 2004 and April 2014. The primary search term was: Congenital myasthenic syndrome. The secondary search terms were: acetylcholine receptor; choline acetyltransferase; ColQ; laminin beta-2; agrin; LRP4; MuSK; Dok-7; rapsyn; GFPT1; DPAGT1; ALG2, ALG14; Prepl; plectin; centronuclear myopathies; Nav1.4 channel. The authors also searched their own reprint files, clinical histories of their patients, and data files of their own research studies.
Acknowledgments
Sources of support: Drs. Andrew G Engel and Xin-Ming Shen were supported by NIH Grant NS06277. SMS was supported by NIH grant NS031744.
Footnotes
Contributors
All authors contributed equally to the literature search, figures, study design, data analysis, data interpretation, and manuscript preparation.
Conflicts of Interest
None of the authors reports a conflict of interest.
References
- 1.Wood SJ, Slater CP. Safety factor at the neuromuscular junction. Prog Neurobiol. 2001;64:393–429. doi: 10.1016/s0301-0082(00)00055-1. [DOI] [PubMed] [Google Scholar]
- 2.Tsujino A, Maertens C, Ohno K, et al. Myasthenic syndrome caused by mutation of the SCN4A sodium channel. Proc Natl Acad Sci USA. 2003;100:7377–7382. doi: 10.1073/pnas.1230273100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abicht A, Dusl M, Guergueltcheva V, et al. Congenital myasthenic syndromes: achievements and limitations of phenotype-guided gene-after-gene sequencing in diagnostic practice: a study of 680 patients. Hum Mutat. 2014;33:1474–1484. doi: 10.1002/humu.22130. [DOI] [PubMed] [Google Scholar]
- 4.Senderek J, Muller JS, Dusl M, et al. Hexosamine biosynthetic pathway mutations cause neuromuscular transmission defect. Am J Hum Genet. 2011;88:162–172. doi: 10.1016/j.ajhg.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shinawi M, Cheung SW. The array CGH and its clinical applications. Drug Discov Today. 2008;13:760–770. doi: 10.1016/j.drudis.2008.06.007. [DOI] [PubMed] [Google Scholar]
- 6.Engel AG. The investigation of congenital myasthenic syndromes. Ann N Y Acad Sci. 1993;681:425–434. doi: 10.1111/j.1749-6632.1993.tb22927.x. [DOI] [PubMed] [Google Scholar]
- 7.Byring RF, Pihko H, Shen X-M, et al. Congenital myasthenic syndrome associated with episodic apnea and sudden infant death. Neuromuscul Disord. 2002;12:548–553. doi: 10.1016/s0960-8966(01)00336-4. [DOI] [PubMed] [Google Scholar]
- 8.Maselli RA, Chen D, Mo D, et al. Choline acetyltransferase mutations in myasthenic syndrome due to deficient acetylcholine resynthesis. Muscle Nerve. 2003;27:180–187. doi: 10.1002/mus.10300. [DOI] [PubMed] [Google Scholar]
- 9.Mallory LA, Shaw JG, Burgess SL, et al. Congenital myasthenic syndrome with episodic apnea. Pediatr Neurol. 2009;41:42–45. doi: 10.1016/j.pediatrneurol.2009.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schara U, Christen H-J, Durmus H, et al. Long-term follow-up in patients with congenital myasthenic syndrome due to CHAT mutations. Eur J Paediatr Neurol. 2010;14:326–333. doi: 10.1016/j.ejpn.2009.09.009. [DOI] [PubMed] [Google Scholar]
- 11.Shen X-M, Crawford TO, Brengman J, et al. Functional consequences and structural interpretation of mutations in human choline acetyltransferase. Hum Mutat. 2011;32:1259–1267. doi: 10.1002/humu.21560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ohno K, Engel AG, Shen X-M, et al. Rapsyn mutations in humans cause endplate acetylcholine receptor deficiency and myasthenic syndrome. Am J Hum Genet. 2002;70:875–885. doi: 10.1086/339465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burke G, Cossins J, Maxwell S, et al. Rapsyn mutations in hereditary myasthenia. Distinct early- and late-onset phenotypes. Neurology. 2003;61:826–828. doi: 10.1212/01.wnl.0000085865.55513.ae. [DOI] [PubMed] [Google Scholar]
- 14.Kraner S, Lufenberg I, Strassburg HM, Sieb JP, Steinlein OK. Congenital myasthenic syndrome with episodic apnea in patients homozygous for a CHAT missense mutation. Arch Neurol. 2003;60:761–763. doi: 10.1001/archneur.60.5.761. [DOI] [PubMed] [Google Scholar]
- 15.Ohno K, Tsujino A, Shen XM, et al. Choline acetyltransferase mutations cause myasthenic syndrome associated with episodic apnea in humans. Proc Natl Acad Sci USA. 2001;98:2017–2022. doi: 10.1073/pnas.98.4.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Massoulié J, Pezzementi L, Bon S, Krejci E, Valette F-M. Molecular and cellular biology of cholinesterases. Prog Neurobiol. 1993;41:31–91. doi: 10.1016/0301-0082(93)90040-y. [DOI] [PubMed] [Google Scholar]
- 17.Cartaud A, Strochlic L, Guerra M, et al. MuSK is required for anchoring acetylcholinesterase at the neuromuscular junction. J Cell Biol. 2004;165:505–515. doi: 10.1083/jcb.200307164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Engel AG, Lambert EH, Gomez MR. A new myasthenic syndrome with end-plate acetylcholinesterase deficiency, small nerve terminals, and reduced acetylcholine release. Ann Neurol. 1977;1:315–330. doi: 10.1002/ana.410010403. [DOI] [PubMed] [Google Scholar]
- 19.Hutchinson DO, Walls TJ, Nakano S, et al. Congenital endplate acetylcholinesterase deficiency. Brain. 1993;116:633–653. doi: 10.1093/brain/116.3.633. [DOI] [PubMed] [Google Scholar]
- 20.Ohno K, Brengman JM, Tsujino A, Engel AG. Human endplate acetylcholinesterase deficiency caused by mutations in the collagen-like tail subunit (ColQ) of the asymmetric enzyme. Proc Natl Acad Sci USA. 1998;95:9654–9659. doi: 10.1073/pnas.95.16.9654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bestue-Cardiel M, de-Cabazon-Alvarez AS, Capablo-Liesa JL, et al. Congenital endplate acetylcholinesterase deficiency responsive to ephedrine. Neurology. 2005;65:144–146. doi: 10.1212/01.wnl.0000167132.35865.31. [DOI] [PubMed] [Google Scholar]
- 22.Mihaylova V, Muller JS, Vilchez JJ, Salih MA, et al. Clinical and molecular genetic findings in COLQ-mutant congenital myasthenic syndromes. Brain. 2008;131:747–759. doi: 10.1093/brain/awm325. [DOI] [PubMed] [Google Scholar]
- 23.Liewluck T, Selcen D, Engel AG. Beneficial effects of albuterol in congenital endplate acetylcholinesterase deficiency and DOK-7 myasthenia. Muscle Nerve. 2011;44:789–794. doi: 10.1002/mus.22176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chan SH, Wong VC, Engel AG. Congenital myasthenia syndrome with a novel COLQ mutation responsive to albuterol. Pediatr Neurol. 2012;47:137–140. doi: 10.1016/j.pediatrneurol.2012.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maselli RA, Ng JJ, Andreson JA, et al. Mutations in LAMB2 causing a severe form of synaptic congenital myasthenic syndrome. J Med Genet. 2009;46:203–208. doi: 10.1136/jmg.2008.063693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Unwin N. Refined structure of the nicotinic acetylcholine receptor at 4 Å resolution. J Mol Biol. 2005;346:967–989. doi: 10.1016/j.jmb.2004.12.031. [DOI] [PubMed] [Google Scholar]
- 27.Milone M, Wang H-L, Ohno K, et al. Mode switching kinetics produced by a naturally occurring mutation in the cytoplasmic loop of the human acetylcholine receptor ɛ subunit. Neuron. 1998;20:575–588. doi: 10.1016/s0896-6273(00)80996-4. [DOI] [PubMed] [Google Scholar]
- 28.Wang H-L, Ohno K, Milone M, et al. Fundamental gating mechanism of nicotinic receptor channel revealed by mutation causing a congenital myasthenic syndrome. J Gen Physiol. 2000;116:449–460. doi: 10.1085/jgp.116.3.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brejc K, van Dijk WV, Schuurmans M, et al. Crystal structure of ACh-binding protein reveals the ligand-binding domain of nicotinic receptors. NAT. 2001;411:269–276. doi: 10.1038/35077011. [DOI] [PubMed] [Google Scholar]
- 30.Sine SM, Claudio T, Sigworth FJ. Activation of Torpedo acetylcholine receptors expressed in mouse fibroblasts: single-channel current kinetics reveal distinct agonist binding affinities. J Gen Physiol. 1990;96:395–437. doi: 10.1085/jgp.96.2.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sine SM. The nicotinic receptor ligand binding domain. J Neurobiol. 2002;53(4):431–446. doi: 10.1002/neu.10139. [DOI] [PubMed] [Google Scholar]
- 32.Engel AG, Ohno K, Sine SM. Sleuthing molecular targets for neurological diseases at the neuromuscular junction. Nature Rev Neurosci. 2003;4:339–352. doi: 10.1038/nrn1101. [DOI] [PubMed] [Google Scholar]
- 33.Engel AG, Ohno K, Bouzat C, Sine SM, Griggs RG. End-plate acetylcholine receptor deficiency due to nonsense mutations in the ɛ subunit. Ann Neurol. 1996;40:810–817. doi: 10.1002/ana.410400521. [DOI] [PubMed] [Google Scholar]
- 34.Ohno K, Quiram P, Milone M, et al. Congenital myasthenic syndromes due to heteroallelic nonsense/missense mutations in the acetylcholine receptor ɛ subunit gene: identification and functional characterization of six new mutations. Hum Mol Genet. 1997;6:753–766. doi: 10.1093/hmg/6.5.753. [DOI] [PubMed] [Google Scholar]
- 35.Plomp JJ, van Kempen GThH, De Baets MB, et al. Acetylcholine release in myasthenia gravis: Regulation at single end-plate level. Ann Neurol. 1995;37:627–636. doi: 10.1002/ana.410370513. [DOI] [PubMed] [Google Scholar]
- 36.Engel AG. The therapy of congenital myasthenic syndromes. Neurotherapeutics. 2007;4:252–257. doi: 10.1016/j.nurt.2007.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sadeh M, Shen X-M, Engel AG. Beneficial effect of albuterol in congenital myasthenic syndrome with ɛ subunit mutations. Muscle Nerve. 2011;44:289–291. doi: 10.1002/mus.22153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ohno K, Hutchinson DO, Milone M, et al. Congenital myasthenic syndrome caused by prolonged acetylcholine receptor channel openings due to a mutation in the M2 domain of the ɛ subunit. Proc Natl Acad Sci USA. 1995;92:758–762. doi: 10.1073/pnas.92.3.758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Harper CM, Engel AG. Quinidine sulfate therapy for the slow-channel congenital myasthenic syndrome. Ann Neurol. 1998;43:480–484. doi: 10.1002/ana.410430411. [DOI] [PubMed] [Google Scholar]
- 40.Harper CM, Fukudome T, Engel AG. Treatment of slow channel congenital myasthenic syndrome with fluoxetine. Neurology. 2003;60:170–173. doi: 10.1212/01.wnl.0000061483.11417.1b. [DOI] [PubMed] [Google Scholar]
- 41.Colomer J, Muller JS, Vernet A, et al. Long-term improvement of slow-channel myasthenic syndrome with fluoxetine. Neuromuscul Disord. 2006;16:329–333. doi: 10.1016/j.nmd.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 42.Chaouch A, Muller JS, Guergueltcheva V, et al. A retrospective clinical study of the slow-channel congenital myasthenic syndrome. J Neurol. 2012;259:474–481. doi: 10.1007/s00415-011-6204-9. [DOI] [PubMed] [Google Scholar]
- 43.Ohno K, Wang H-L, Milone M, et al. Congenital myasthenic syndrome caused by decreased agonist binding affinity due to a mutation in the acetylcholine receptor ɛ subunit. Neuron. 1996;17:157–170. doi: 10.1016/s0896-6273(00)80289-5. [DOI] [PubMed] [Google Scholar]
- 44.Shen XM, Brengman J, Edvardson S, Sine SM, Engel AG. Highly fatal fast-channel congenital syndrome caused by AChR ɛ subunit mutation at the agonist binding site. Neurology. 2012;79:449–454. doi: 10.1212/WNL.0b013e31825b5bda. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shen X-M, Ohno K, Tsujino A, et al. Mutation causing severe myasthenia reveals functional asymmetry of AChR signature Cys-loops in agonist binding and gating. J Clin Invest. 2003;111:497–505. doi: 10.1172/JCI16997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shen X-M, Brengman J, Sine SM, Engel AG. Myasthenic syndrome AChRα C-loop mutant disrupts initiation of channel gating. J Clin Invest. 2012;122:2613–2621. doi: 10.1172/JCI63415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shen X-M, Fukuda T, Ohno K, Sine SM, Engel AG. Congenital myasthenia-related AChR δ subunit mutation interferes with intersubunit communication essential for channel gating. J Clin Invest. 2008;118:1867–1876. doi: 10.1172/JCI34527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang H-L, Milone M, Ohno K, et al. Acetylcholine receptor M3 domain: Stereochemical and volume contributions to channel gating. Nature Neurosci. 1999;2:226–233. doi: 10.1038/6326. [DOI] [PubMed] [Google Scholar]
- 49.Shen X-M, Ohno K, Sine SM, Engel AG. Subunit-specific contribution to agonist binding and channel gating revealed by inherited mutation in muscle AChR M3-M4 linker. Brain. 2005;128:345–355. doi: 10.1093/brain/awh364. [DOI] [PubMed] [Google Scholar]
- 50.Sine SM, Shen X-M, Wang H-L, et al. Naturally occurring mutations at the acetylcholine receptor binding site independently alter ACh binding and channel gating. J Gen Physiol. 2002;120:483–496. doi: 10.1085/jgp.20028568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Burden SJ, Yumoto N, Zhang W. The role of MuSK in synapse formation and neuromuscular disease. Cold Spring Harb Perspect Biol. 2013;5:a009167. doi: 10.1101/cshperspect.a009167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huze C, Bauche S, Richard P, et al. Identification of an agrin mutation that causes congenital myasthenia and affects synapse function. Am J Hum Genet. 2009;85:155–167. doi: 10.1016/j.ajhg.2009.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Maselli RA, Fernandez JM, Arredondo J, et al. LG2 agrin mutation causing severe congenital myasthenic syndrome mimics functional characteristics of non-neural agrin (z-) agrin. Hum Genet. 2012;131:1123–1135. doi: 10.1007/s00439-011-1132-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ohkawara B, Cabrera-Serrano M, Nakat T, et al. LRP4 third β-propeller domain mutations cause novel congenital myasthenic syndrome by compromising agrin-mediated MuSK signalling in a position-specific manner. Hum Mol Genet. 2014;23:1856–1868. doi: 10.1093/hmg/ddt578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chevessier F, Faraut B, Ravel-Chapuis A, et al. MUSK, a new target for mutations causing congenital myasthenic syndrome. Hum Mol Genet. 2004;13:3229–3240. doi: 10.1093/hmg/ddh333. [DOI] [PubMed] [Google Scholar]
- 56.Mihaylova V, Salih MA, Mukhtar MM, et al. Refinement of the clinical phenotype in MUSK-related congenital myasthenic syndromes. Neurology. 2009;73:1926–1928. doi: 10.1212/WNL.0b013e3181c3fce9. [DOI] [PubMed] [Google Scholar]
- 57.Maselli R, Arredondo J, Cagney O, et al. Mutations in MUSK causing congenital myasthenic syndrome impair MuSK-Dok-7 interaction. Hum Mol Genet. 2010;19:2370–2379. doi: 10.1093/hmg/ddq110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ben Ammar A, Soltanzadeh P, Bauchê S, et al. A mutation causes MuSK reduced sensitivity to agrin and congenital myasthenia. PLoS One. 2013 Jan 9;8:e53826. doi: 10.1371/journal.pone.0053826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chevessier F, Girard E, Molgo J, et al. A mouse model for congenital myasthenic syndrome due to MuSK mutations reveals defects in structure and function of neuromuscular junctions. Hum Mol Genet. 2008;17:3577–3595. doi: 10.1093/hmg/ddn251. [DOI] [PubMed] [Google Scholar]
- 60.Gallenmuller C, Muller-Felber W, Dusl M, et al. Salbutamol-responsive limb-girdle congenital myasthenic syndrome due to a novel missese mutaion and heteroallelic deletion in MUSK. Neuromuscul Disord. 2014;24:31–35. doi: 10.1016/j.nmd.2013.08.002. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Okada K, Inoue A, Okada M, et al. The muscle protein Dok-7 is essential for neuromuscular synaptogenesis. Science. 2006;312:1802–1805. doi: 10.1126/science.1127142. [DOI] [PubMed] [Google Scholar]
- 62.Beeson D, Higuchi O, Palace J, et al. Dok-7 mutations underlie a neuromuscular junction synaptopathy. Science. 2006;313:1975–1978. doi: 10.1126/science.1130837. [DOI] [PubMed] [Google Scholar]
- 63.Muller JS, Herczegfalvi A, Vilchez JJ, et al. Phenotypical spectrum of DOK7 mutations in congenital myasthenic syndromes. Brain. 2007;130:1497–1506. doi: 10.1093/brain/awm068. [DOI] [PubMed] [Google Scholar]
- 64.Selcen D, Milone M, Shen X-M, et al. Dok-7 myasthenia: phenotypic and molecular genetic studies in 16 patients. Ann Neurol. 2008;64:71–87. doi: 10.1002/ana.21408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Anderson JA, Ng JJ, Bowe C, et al. Variable phenotypes associated with mutations in DOK7. Muscle Nerve. 2008;37:448–456. doi: 10.1002/mus.20944. [DOI] [PubMed] [Google Scholar]
- 66.Ammar AB, Petit F, Alexandri K, et al. Phenotype-genotype analysis in 15 patients presenting a congenital myasthenic syndrome due to mutations in DOK7. J Neurol. 2010;257:754–766. doi: 10.1007/s00415-009-5405-y. [DOI] [PubMed] [Google Scholar]
- 67.Slater CR, Fawcett PRW, Walls TJ, et al. Pre- and postsynaptic abnormalities associated with impaired neuromuscular transmission in a group of patients with ‘limb-girdle myasthenia’. Brain. 2006;127:2061–2076. doi: 10.1093/brain/awl200. [DOI] [PubMed] [Google Scholar]
- 68.Schara U, Barisic N, Deschauer M, et al. Ephedrine therapy in eight patients with congenital myasthenic syndrome due to DOK7 mutations. Neuromuscul Disord. 2010;19:828–832. doi: 10.1016/j.nmd.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 69.Ramarao MK, Cohen JB. Mechanism of nicotinic acetylcholine receptor cluster formation by rapsyn. Proc Natl Acad Sci USA. 1998;95:4007–4012. doi: 10.1073/pnas.95.7.4007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ramarao MK, Bianchetta MJ, Lanken J, Cohen JB. Role of rapsyn tetratricopeptide repeat and coiled-coil domains in self-association and nicotinic acetylcholine receptor clustering. J Biol Chem. 2001;276:7475–7483. doi: 10.1074/jbc.M009888200. [DOI] [PubMed] [Google Scholar]
- 71.Zuber B, Unwin N. Structure and superorganization of the acetylcholine receptor-rapsyn complex. Proc Natl Acad Sci U S A. 2013;110:10622–10627. doi: 10.1073/pnas.1301277110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Milone M, Shen XM, Selcen D, et al. Myasthenic syndrome due to defects in rapsyn: Clinical and molecular findings in 39 patients. Neurology. 2009;73:228–235. doi: 10.1212/WNL.0b013e3181ae7cbc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Banwell BL, Ohno K, Sieb JP, Engel AG. Novel truncating RAPSN mutation causing congenital myasthenic syndrome responsive to 3,4-diaminopyridine. Neuromuscul Disord. 2004;14:202–207. doi: 10.1016/j.nmd.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 74.Skeie GO, Aurlien H, Müller JS, Norgard G, Bindoff LA. Unusual features in a boy with rapsyn N88K mutation. Neurology. 2006;67:2262–2263. doi: 10.1212/01.wnl.0000249184.09369.c2. [DOI] [PubMed] [Google Scholar]
- 75.Müller JS, Mildner G, Müller-Felber W, et al. Rapsyn N88K is a frequent cause of CMS in European patients. Neurology. 2003;60:1805–1811. doi: 10.1212/01.wnl.0000072262.14931.80. [DOI] [PubMed] [Google Scholar]
- 76.Cossins J, Burke G, Maxwell S, et al. Diverse molecular mechanisms involved in AChR deficiency due to rapsyn mutations. Brain. 2006;129:2773–2783. doi: 10.1093/brain/awl219. [DOI] [PubMed] [Google Scholar]
- 77.Ohno K, Sadeh M, Blatt I, Brengman JM, Engel AG. E-box mutations in RAPSN promoter region in eight cases with congenital myasthenic syndrome. Hum Mol Genet. 2003;12:739–748. doi: 10.1093/hmg/ddg089. [DOI] [PubMed] [Google Scholar]
- 78.Haeuptle MA, Hennet T. Congenital disorders of glycosylation: An update on defects affecting the biosynthesis of dolichol-linked oligosaccharides. Hum Mutat. 2009;30:1628–1641. doi: 10.1002/humu.21126. [DOI] [PubMed] [Google Scholar]
- 79.Freeze HH, Chong JX, Bamshad MJ, Ng BG. Solving glycosylation disorders: Fundamental approaches reveal complicated pathways. Am J Hum Genet. 2014;94:161–165. doi: 10.1016/j.ajhg.2013.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Selcen D, Shen X-M, Milone M, et al. GFPT1-myasthenia: Clinical, structural, and electrophysiologic heterogeneity. Neurology. 2013;23:370–378. doi: 10.1212/WNL.0b013e31829c5e9c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Belaya K, Finlayson S, Slater C, et al. Mutations in DPAGT1 cause a limb-girdle congenital myasthenic syndrome with tubular aggregates. Am J Hum Genet. 2012;91:1–9. doi: 10.1016/j.ajhg.2012.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Selcen D, Shen X-M, Li Y, et al. DPAGT1 myasthenia and myopathy. Genetic, phenotypic, and expression studies. Neurology. 2014;82:1822–1830. doi: 10.1212/WNL.0000000000000435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cossins J, Belaya K, Hicks D, et al. Congenital myasthenic syndromes due to mutations in ALG2 and ALG14. Brain. 2013;136:944–956. doi: 10.1093/brain/awt010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Soboloff J, Rothberg BS, Madesh M, Gill DL. STIM proteins: dynamic calcium signal transducers. Nat Rev Mol Cell Biol. 2012;13:549–565. doi: 10.1038/nrm3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bohm J, Chevessier F, De Paula AM, et al. Constitutive activation of STIM1 causes tubular aggregate myopathy. Am J Hum Genet. 2013;92:271–278. doi: 10.1016/j.ajhg.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Regal L, Shen XM, Selcen D, et al. PREPL deficiency with or without cystinuria causes a novel myasthenic syndrome. Neurology. 2014;82:1254–1260. doi: 10.1212/WNL.0000000000000295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Radhakrishnan K, Baltes J, Creemers JWM, Schu P. Trans-Golgi network morphology and sorting is regulated by prolyl-oligopeptidase-like protein PREPL and AP-1 complex subunit μ1A. J Cell Sci. 2013;126:1155–1163. doi: 10.1242/jcs.116079. [DOI] [PubMed] [Google Scholar]
- 88.Kim M-H, Hersh LB. The vesicular acetylcholine transporter interacts with clathrin-associated adaptor complexes AP-1 and AP-2. J Biol Chem. 2004;279:12580–12587. doi: 10.1074/jbc.M310681200. [DOI] [PubMed] [Google Scholar]
- 89.Rezniczeck GA, Walko G, Wiche G. Plectin defects lead to various forms of epidermolysis bullosa simplex. Dermatol Clin. 2009;28:33–41. doi: 10.1016/j.det.2009.10.004. [DOI] [PubMed] [Google Scholar]
- 90.Smith FJD, Eady RAJ, Leigh IM, et al. Plectin deficiency results in muscular dystrophy with epidermolysis bullosa. Nat Genet. 1996;13:450–457. doi: 10.1038/ng0896-450. [DOI] [PubMed] [Google Scholar]
- 91.McMillan JR, Akiyama M, Rouan F, et al. Plectin defects in epidermolysis bullosa simplex with muscular dystrophy. Muscle Nerve. 2007;35:24–35. doi: 10.1002/mus.20655. [DOI] [PubMed] [Google Scholar]
- 92.Banwell BL, Russel J, Fukudome T, et al. Myopathy, myasthenic syndrome, and epidermolysis bullosa simplex due to plectin deficiency. J Neuropathol Exp Neurol. 1999;58:832–846. doi: 10.1097/00005072-199908000-00006. [DOI] [PubMed] [Google Scholar]
- 93.Selcen D, Juel VC, Hobson-Webb LD, et al. Myasthenic syndrome caused by plectinopathy. Neurology. 2011;76:327–336. doi: 10.1212/WNL.0b013e31820882bd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Claeys KG, Maisonobe T, Bohm J, et al. Phenotype of a patient with recessive centronuclear myopathy and a novel BIN1 mutation. Neurology. 2010;74:519–521. doi: 10.1212/WNL.0b013e3181cef7f9. [DOI] [PubMed] [Google Scholar]
- 95.Robb SA, Sewry CA, Dowling JJ, et al. Impaired neuromuscular transmission and response to aceylcholinesterase inhibitors in centronuclear myopathy. Neuromuscul Disord. 2011;21:379–386. doi: 10.1016/j.nmd.2011.02.012. [DOI] [PubMed] [Google Scholar]
- 96.Gibbs EM, Clarke NF, Rose K, et al. Neuromuscular junction abnrormalities in DNM2-related centronuclear myopathy. J Mol Med (Berl) 2013;91:727–737. doi: 10.1007/s00109-013-0994-4. [DOI] [PubMed] [Google Scholar]
- 97.Liewluck T, Shen X-M, Milone M, Engel AG. Endplate structure and parameters of neuromuscular transmission in sporadic centronuclear myopathy associated with myasthenia. Neuromuscul Disord. 2011;21:387–395. doi: 10.1016/j.nmd.2011.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Munot P, Lashley D, Jungbluth H, et al. Congenital fibre type disproportion associated with mutations in the tropomyosin 3 (TPM3) gene mimicking congenital myasthenia. Neuromuscul Disord. 2010;20:796–800. doi: 10.1016/j.nmd.2010.07.274. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.