

Abstract
The caged complex between orotidine 5′-monophosphate decarboxylase (ScOMPDC) and 5-fluoroorotidine 5′-monophosphate (FOMP) undergoes decarboxylation ∼300 times faster than the caged complex between ScOMPDC and the physiological substrate, orotidine 5′-monophosphate (OMP). Consequently, the enzyme conformational changes required to lock FOMP at a protein cage and release product 5-fluorouridine 5′-monophosphate (FUMP) are kinetically significant steps. The caged form of ScOMPDC is stabilized by interactions between the side chains from Gln215, Tyr217, and Arg235 and the substrate phosphodianion. The control of these interactions over the barrier to the binding of FOMP and the release of FUMP was probed by determining the effect of all combinations of single, double, and triple Q215A, Y217F, and R235A mutations on kcat/Km and kcat for turnover of FOMP by wild-type ScOMPDC; its values are limited by the rates of substrate binding and product release, respectively. The Q215A and Y217F mutations each result in an increase in kcat and a decrease in kcat/Km, due to a weakening of the protein–phosphodianion interactions that favor fast product release and slow substrate binding. The Q215A/R235A mutation causes a large decrease in the kinetic parameters for ScOMPDC-catalyzed decarboxylation of OMP, which are limited by the rate of the decarboxylation step, but much smaller decreases in the kinetic parameters for ScOMPDC-catalyzed decarboxylation of FOMP, which are limited by the rate of enzyme conformational changes. By contrast, the Y217A mutation results in large decreases in kcat/Km for ScOMPDC-catalyzed decarboxylation of both OMP and FOMP, because of the comparable effects of this mutation on rate-determining decarboxylation of enzyme-bound OMP and on the rate-determining enzyme conformational change for decarboxylation of FOMP. We propose that kcat = 8.2 s–1 for decarboxylation of FOMP by the Y217A mutant is equal to the rate constant for cage formation from the complex between FOMP and the open enzyme, that the tyrosyl phenol group stabilizes the closed form of ScOMPDC by hydrogen bonding to the substrate phosphodianion, and that the phenyl group of Y217 and F217 facilitates formation of the transition state for the rate-limiting conformational change. An analysis of kinetic data for mutant enzyme-catalyzed decarboxylation of OMP and FOMP provides estimates for the rate and equilibrium constants for the conformational change that traps FOMP at the enzyme active site.
Orotidine 5′-monophosphate decarboxylase from Saccharomyces cerevisiae (ScOMPDC) provides a large 31 kcal/mol stabilization of the transition state for decarboxylation of OMP to form uridine 5′-monophosphate [UMP (Scheme 1)],1,2 through a UMP carbanion intermediate,3−8 and shows an extraordinary specificity in binding this transition state with an affinity much higher than that of OMP, whose ground-state Michaelis complex is stabilized by only 8 kcal/mol.9,10 This rate acceleration is achieved by sequestering OMP in a structured protein cage,11−15 which provides for optimal stabilizing interactions with the decarboxylation transition state.11,16 The protein cage is formed by an energetically demanding conformational change, driven by the binding interactions with the substrate phosphodianion and other nonreacting substrate fragments,10,11 which function to mold two flexible protein loops into an active enzyme (Figure 1).17
Scheme 1.

Figure 1.
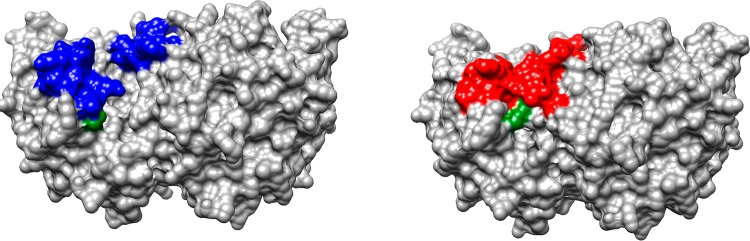
Space filling models of ScOMPDC from yeast.18 The structure on the left is the open unliganded form of ScOMPDC (PDB entry 1DQW), and the structure on the right shows the complex with 6-hydroxyuridine 5′-monophosphate (PDB entry 1DQX). Two colored flexible loops close to form the active site cage: Pro202–Val220, on the left-hand side of each structure, interact with the substrate dianion, and Glu152–Thr165, on the right-hand side, interact with the pyrimidine ring. The guanidine side chain of R235, at the base of the left-hand loop, is colored green.
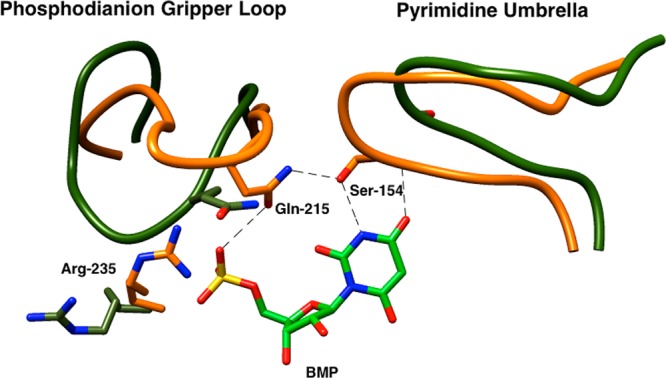
Figure 2 shows interactions of the ligand phosphodianion with three side chains of ScOMPDC: (i) the amide side chain of Gln215, (ii) the phenol side chain of Tyr217, and (iii) the guanidine side chain of Arg235.18 The side chain of Arg235 sits on the protein surface adjacent to a phosphodianion gripper loop [Pro202–Val220 (Figure 1)]. The role of these gripper side chains was probed by preparing all combinations of single (Q215A, R235A, and Y217F), double (Q215A/Y217F, Q215A/R235A, and R235A/Y217F), and triple (Q215A/R235A/Y217F) mutants and determining the effect of these mutations on the kinetic parameters for ScOMPDC-catalyzed decarboxylation of OMP and the phosphodianion-truncated substrate 1-(β-d-erythrofuranosyl)orotic acid (EO).4,19 The single mutations result in ≤2.5-fold decreases in kcat/Km for the catalyzed reaction of EO.4 By contrast, these mutations result in similar large decreases in kcat/Km for ScOMPDC-catalyzed decarboxylation of OMP,19 and in the third-order rate constant for activation of ScOMPDC-catalyzed decarboxylation of EO by phosphite dianion.4 These results show that interactions of ScOMPDC with the phosphodianion of OMP, or with phosphite dianion have the sole function of activating the enzyme for catalysis of decarboxylation at the distant orotate ring, through stabilization of a catalytically active caged substrate complex.4,10,11,19 This model also provides a simple rationalization for the activation by phosphite dianion of triosephosphate isomerase20−23 and l-glycerol 3-phosphate dehydrogenase-catalyzed reactions24−26 of their phosphodianion-truncated substrate.
Figure 2.
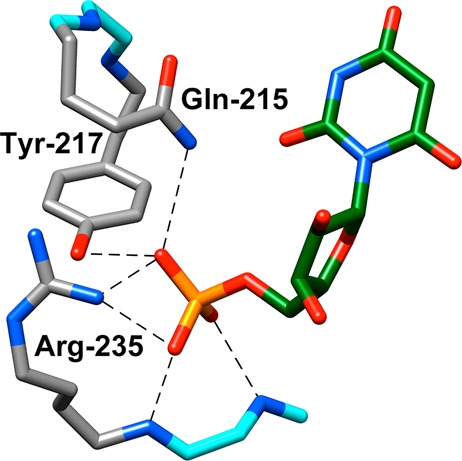
X-ray crystal structure (PDB entry 1DQX) of yeast ScOMPDC in a complex with 6-hydroxyuridine 5′-monophosphate.18 This structure shows the important interactions of Gln215, Tyr217, and Arg235 side chains from the phosphodianion gripper loop with the ligand phosphodianion. Reproduced from ref (4). Copyright 2012 American Chemical Society.
In this paper, we consider the kinetic barriers for conversion of the open form of ScOMPDC to the caged complex to substrate (Figure 1), and for conversion of the caged product complex to the open enzyme. Rate constants for conformational changes that convert enzymes from an inactive to an active form may be determined directly as the appropriate enzyme kinetic parameter, when the conformational change is rate-determining for catalysis. Substrate binding is already partly rate-determining for ScOMPDC-catalyzed decarboxylation of OMP,9,27 while the 5-F substituent of 5-fluorouridine 5′-monophosphate (FOMP) is exceptional in providing strong stabilization of transition states for ScOMPDC-catalyzed reactions.27,28 For example, the 5-F results in a 3400-fold increase in kcat for the ScOMPDC-catalyzed deuterium exchange reaction of enzyme-bound UMP that proceeds through the C-6 UMP carbanion intermediate3,6 and a 390-fold increase in the second-order rate constant kcat/Km for ScOMPDC-catalyzed decarboxylation of truncated substrate EO (Scheme 2).29,30 Now, if enzyme conformational changes are partly rate-determining for ScOMPDC-catalyzed decarboxylation of OMP, then these steps should be strongly rate-determining for chemically rapid ScOMPDC-catalyzed decarboxylation of FOMP.27,28
Scheme 2.
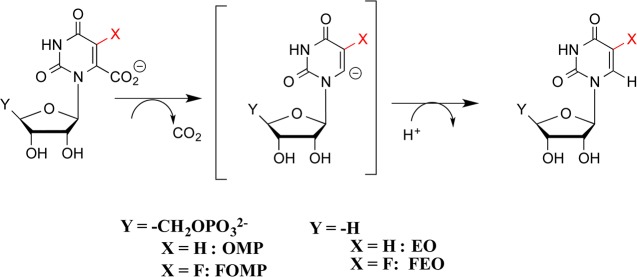
The 5-F at FOMP results in only small 1.1- and 6-fold increases in kcat/Km and kcat, respectively, for decarboxylation of OMP (Scheme 2),29 so that the 5-F substituent effect is only weakly expressed at the virtual transition states that govern the values of these kinetic parameters.31 Instead, the value of kcat/Km is mainly limited by the rate of formation of the Michaelis complex, and the value of kcat is mainly limited by the rate of product release.27 The values of the kinetic parameter for decarboxylation of FOMP are not strongly affected by changes in solvent viscosity η, so that substrate binding and product release are not strictly diffusion-controlled reactions.27 It was proposed that kcat/Km is instead limited by the rate of the protein conformational changes that follow substrate binding.27 Roughly, a 390/1.1 = 350-fold increase (Scheme 2) in the observed rate constant of kcat/Km for decarboxylation of FOMP, relative to that for decarboxylation of OMP, is required to effect similar rate-determining decarboxylation steps for the catalyzed reactions of both OMP and FOMP.
A comparison of the effects of site-directed mutations on the kinetic parameters for ScOMPDC-catalyzed decarboxylation of OMP, which is limited by chemical decarboxylation, and on ScOMPDC-catalyzed decarboxylation of FOMP, which is limited by the rate of formation and breakdown of Michaelis complexes, provides insight into the effect of the mutations on the relative barriers to the chemical steps and the enzyme conformational changes.32 We previously reported the effect of single (Q215A, R235F, and Y217F), double (Q215A/Y217F, Q215A/R235A, and R235A/Y217F), and triple (Q215A/R235A/Y217F) mutations of amino acid residues that interact with the phosphodianion of OMP (Figure 2) on the kinetic parameters for ScOMPDC-catalyzed decarboxylation of OMP.4,19 We report here the effect of these same mutations on the kinetic parameters for ScOMPDC-catalyzed decarboxylation of FOMP. We note that these mutations generally cause larger changes in the values of the kinetic parameters for decarboxylation of OMP than of FOMP. This is because of their larger effects on the barrier to the decarboxylation reaction, which limits the rate of decarboxylation OMP, compared with their effect on the barrier to the enzyme conformational change, which limits the rate of decarboxylation of FOMP.27 We also note interesting exceptions to this trend, which show that mutations of gripper side chains cause significant changes in the barriers to the protein conformational changes, which control the rate constants for formation and breakdown of complexes between ScOMPDC and FOMP or FUMP.
Experimental Procedures
Materials
OMP28,29 and FOMP29,33 were prepared enzymatically by literature procedures. 3-(N-Morpholino)propanesulfonic acid (MOPS, ≥99.5%) was purchased from Fluka. All other chemicals were reagent grade or better and were used without further purification.
Wild-Type and Mutant Forms of ScOMPDC
Plasmid pScODC-15b containing the gene encoding ScOMPDC from S. cerevisiae with an N-terminal His6 or His10 tag was available from previous studies.27,34 The procedures for the preparation of the Q215A,34 Y217F,4 Y217A,5 Q215A/Y217F,4 Q215A/R235A,4 Y217F/R235A,4 Q215A/Y217F/R235A,4 S154A,34 and Q215A/S154A34 mutant enzymes were described in earlier work. In all cases, the N-terminal His6 or His10 tag was removed by the action of thrombin (1 unit/mg of mutant ScOMPDC) at room temperature for ∼16 h, as described in the Supporting Information of ref (34). Wild-type and mutant forms of ScOMPDC were stored at −80 °C. Prior to use, frozen ScOMPDC was defrosted and dialyzed at 4 °C against 10 mM MOPS (50% free base) and 100 mM NaCl (pH 7.1).
Kinetic Parameters for Decarboxylation of FOMP
The following conditions were used for all enzyme assays: 30 mM MOPS (50% free base) at pH 7.1 (30 mM MOPS), 25 °C, and I = 0.105 (NaCl). The concentration of ScOMPDC in stock enzyme solutions was determined from the absorbance at 280 nm using values of 29900 M–1 cm–1 for wild-type and most mutant enzymes or 28400 M–1 cm–1 for the Y217F and Y217A mutants that were calculated using the ProtParam tool available on the ExPASy server.35,36 Initial velocities, v0 (M s–1), for decarboxylation of FOMP (0.01–3.0 mM) catalyzed by mutants of yeast ScOMPDC were determined spectrophotometrically by following the decrease in absorbance at 282, 290, 295, or 300 nm. The following values for the difference in the extinction coefficient of FOMP and FUMP were determined from the initial absorbance of a solution that contains a known concentration of FOMP, and the final absorbance after quantitative enzyme-catalyzed decarboxylation to form FUMP: −1380, −1090, −805, and −490 M–1 cm–1 for FOMP at 282, 290, 295, and 300 nm, respectively. The following wavelengths were generally monitored for enzyme-catalyzed decarboxylation reactions at different [FOMP]:[FOMP] ≤ 0.2 mM at 282 nm, [FOMP] = 0.2–0.4 mM at 290 nm, [FOMP] = 0.3–0.8 mM at 295 nm, and [OMP] ≥ 0.8 mM at 300 nm. Enzyme-catalyzed decarboxylation of FOMP, in a total volume of 1 mL, was initiated by the addition of 1–10 μL of a stock solution of mutant ScOMPDC to give the following enzyme concentrations: 5–10 nM, Q215A and Y217F mutants; 200 nM, Q215A/R235A mutant; 1.8 μM, Y217F/R235A mutant; 50–100 nM, S154A and S154A/Q215A mutants; 200 nM, Y217A mutant. Initial reaction velocity v0 was determined for the decarboxylation of the first ∼5% of FOMP. Values of kcat and Km for the mutant enzyme-catalyzed reactions were obtained from the nonlinear least-squares fits of the values of v0/[E] (s–1) to the Michaelis–Menten equation.
The decarboxylation of FOMP (50 μM) catalyzed by the Q215A/Y217F/R235A triple mutant of ScOMPDC was monitored over 8–10 reaction half times. Reactions in a volume of 1 mL were initiated by addition of 100 μL of a stock solution of the mutant enzyme to give a final concentration of 25 μM and then monitored spectrophotometrically at 295 nm. The rate constants kobs (s–1) for first-order decay of FOMP to FUMP were obtained by fitting the absorbance date to the equation for an exponential decay. The value of kcat/Km (M–1s–1) was then calculated from the relationship kcat/Km = kobsd/[E].
Results
Panels A–C of Figure 3 show the fits of plots of v0/[E] versus [FOMP] to the Michaelis–Menten equation for decarboxylation of FOMP catalyzed by site-directed mutants of ScOMPDC in solutions that contain 30 mM MOPS (50% free base) at pH 7.1, 25 °C, and I = 0.105 (NaCl). These fits gave the values for kcat and Km for the mutant enzyme decarboxylation of FOMP reported in Table 1, with the exception of data for the reaction catalyzed by the Y217F/R235A mutant, which were fit to a linear equation to give the value for kcat/Km. The value of kcat/Km for the Q215A/Y217F/R235A triple mutant ScOMPDC-catalyzed reaction was determined by monitoring the entire time course for the decarboxylation of 50 μM FOMP (Km ≫ 50 μM) as described in Experimental Procedures. Table 1 reports the kinetic parameters kcat, Km, and kcat/Km for wild-type and mutant ScOMPDC-catalyzed decarboxylation of FOMP determined in this study. The corresponding kinetic parameters for decarboxylation of OMP were reported previously.19,34,37 We note a kcat of 4.7 s–1, a Km of 0.0019 M–1, and a kcat/Km of 2500 M–1 s–1 reported for decarboxylation of OMP catalyzed by the Y217A mutant of ScOMPDC in solutions that contain 30 mM MOPS (50% free base) at pH 7.1, 25 °C, and I = 0.105 (NaCl).37 By comparison, a kcat of 2.0 s–1, a Km of 9.4 × 10–5 M–1, and a kcat/Km of 21000 M–1 s–1 were reported for the reaction catalyzed by this mutant at an undefined low ionic strength.38 A decrease in ionic strength from I = 0.105 to I = 0.020 was shown in earlier work to result in a 9-fold decrease in Km and kcat/Km for the decarboxylation of OMP catalyzed by the R235A mutant of ScOMPDC.29
Figure 3.

Dependence of v/[E] for decarboxylation of FOMP catalyzed by mutant forms of ScOMPDC on the concentration of FOMP for reactions at 25 °C, pH 7.1 (30 mM MOPS), and I = 0.105 (NaCl): (A) (▲) Y217F mutant, (●) Q215A/Y217F mutant, and (inset) Q215A mutant; (B) (▲) Q215A/R235A mutant and (●) Y217F/R235A mutant; and (C) (●) S154A mutant and (○) Q215A/S154A mutant.
Table 1. Effect of Mutations of Phosphodianion Gripper Amino Acid Residues on the Kinetic Parameters for ScOMPDC-Catalyzed Decarboxylation of FOMPa.
| ScOMPDC | kcat (s–1)b | (kcat)F/(kcat)Hh | Km (M)b | kcat/Km (M–1 s–1) | (kcat/Km)F/(kcat/Km)Hh |
|---|---|---|---|---|---|
| wild-typec | 95 | 6 | 8 × 10–6 | 1.2 × 107 | 1.1 |
| Q215Ad | 190 ± 10 | 8 | (9.6 ± 1) × 10–5 | 2.0 × 106 | 8 |
| Y217Fd | 430 ± 30 | 21 | (4.2 ± 0.6) × 10–4 | 1.1 × 106 | 6 |
| R235c | 92 | 92 | 5.8 × 10–4 | 1.6 × 105 | 180 |
| Q215A/Y217Fd | 49 ± 5 | 10 | (9.2 ± 1.7) × 10–4 | 5.3 × 104 | 16 |
| Q215A/R235Ae | 4.7 ± 0.3 | 240 | (6.5 ± 0.8) × 10–4 | 7200 | 500 |
| Y217F/R235Ae | 820 | 200 | |||
| triple mutant | 28 | 760 | |||
| S154Af | 16 ± 0.5 | 200i | (5.5 ± 0.6) × 10–5 | 2.9 × 105 | 460i |
| S154A/Q215Af | 6.6 ± 0.3 | 160i | (8.6 ± 1.3) × 10–5 | 7.7 × 104 | 200i |
| Y217Ag | 8.2 ± 0.5 | 2g | (6.2 ± 0.6) × 10–4 | 1.3 × 104 | 5g |
For reactions at pH 7.1 (30 mM MOPS), 25 °C, and I = 0.105 (NaCl).
The quoted errors are the standard deviations obtained from the nonlinear least-squares fits of data from panels A–C of Figure 3 to the Michaelis–Menten equation.
From ref (29).
Kinetic parameters for reactions of OMP from ref (37).
Kinetic parameters for reactions of OMP from ref (19), unless indicated otherwise.
Kinetic parameters for reactions of OMP from ref (34).
Discussion
An enzyme conformational change at the ternary E·FEO·HPO32– complex is rate-determining for phosphite dianion activation of ScOMPDC-catalyzed decarboxylation of the fluorinated–truncated substrate FEO to form FEU.30 The 1.1-fold effect of the 5-F at the whole substrate FOMP on kcat/Km for wild-type ScOMPDC-catalyzed decarboxylation of OMP shows that there is minimal stabilization of this rate-determining transition state by polar/electrostatic interactions between the electronegative -F and transition-state negative charge at C-6. The simplest interpretation is that kcat/Km is limited by the rate of diffusion-controlled formation of the Michaelis complex between ScOMPDC and FOMP. However, a study of the effect of changes in solvent viscosity (ηo) on kinetic parameter (kcat/Km)o for decarboxylation in water gave slopes of 0.37 and 0.64 for plots of (kcat/Km)o/(kcat/Km)obs for decarboxylation of OMP and FOMP, respectively, against η/ηo,27 which are smaller than the slope of 1.0 expected for a diffusion-limited reaction.39 This shows that the rate of formation of the Michaelis complex is partly limited by a second step, which we propose is the conformational change that traps FOMP in a protein cage [kc (Scheme 3)].27
Scheme 3.

The small value of (kcat)F/(kcat)H = 6 for ScOMPDC-catalyzed decarboxylation of OMP and FOMP, compared to ratios observed when the entire 5-F substituent effect on carbanion stability is expressed at the rate-determining transition state (Scheme 2), shows that decarboxylation of FOMP is not limited by rate constant kchem (Scheme 3) for decarboxylation of bound substrate. Slopes of 0.39 and 1.0, respectively, were determined for plots of (kcat)obs/(kcat)o for decarboxylation of OMP and FOMP, respectively, versus η/ηo.27 These are consistent with the conclusion that k′–c and/or k′–d are partly rate-determining for ScOMPDC-catalyzed decarboxylation of OMP (Scheme 3) and fully rate-determining for decarboxylation of FOMP.
Simple Conclusions about the Effect of Mutations on 5-F Substituent Effects
The effect of the 5-F at FOMP on the kinetic parameters kcat/Km and kcat for ScOMPDC-catalyzed decarboxylation of OMP, when chemical decarboxylation is clearly rate-determining, should be similar to the 390-fold effect of 5-F on kcat/Km for ScOMPDC-catalyzed decarboxylation of the phosphodianion-truncated substrate EO (Scheme 2). Table 1 shows that the values of (kcat/Km)F/(kcat/Km)H increase as ScOMPDC is crippled by mutations of gripper side chains to 760 for the Q215A/Y217A/R235A triple mutant, consistent with rate-determining decarboxylation for the two substrates. The largest observed ratio of (kcat)F/(kcat)H is 240 for the Q215A/R235A mutant enzyme-catalyzed reactions. The more severely crippled mutants bind OMP and FOMP too weakly to approach saturation.
Single mutations of gripper side chains result in increases in kF/kH for kinetic parameters kcat and kcat/Km, because of the stronger effect of the mutations on kcat and kcat/Km for enzyme-catalyzed decarboxylation of OMP. The following effects of Q215A and Y217F single mutations on kF/kH for kinetic parameters kcat/Km and kcat provide strong evidence that the mutations affect the rate constants for binding and release of FOMP.
(1) The Q215A and Y217F mutations of ScOMPDC cause (kcat/Km)F/(kcat/Km)H to increase to 8 and 6, respectively. These are still much smaller than the ratio of ≈400 for rate-determining decarboxylation. Therefore, formation of the EC·FOMP complex remains rate-determining for the mutant enzyme-catalyzed reactions. The mutations result in surprising 6- and 10-fold decreases in kcat/Km for Q215A and Y217F mutant enzyme-catalyzed decarboxylation of FOMP, respectively (Table 1), which can reflect only decreases in kc for rate-determining conversion of EO·FOMP to EC·FOMP (Scheme 3). This shows that a weakening of the protein–phosphodianion interactions by these mutations results in a decrease in the rate of enzyme conformational change, which allows expression of these interactions (Figure 2).
(2) The Q215A and Y217F mutations of ScOMPDC cause an increase in (kcat)F/(kcat)H, which is due mainly to a surprising increase in (kcat)F for decarboxylation of FOMP by rate-determining breakdown of EC·FUMP (Scheme 3). We conclude that kcat ≈ k′–c = 95 s–1 (Scheme 3) for the wild-type enzyme-catalyzed reaction and that weakening of the protein–dianion interactions results in an increase in k′–c (Scheme 3) for the rate-determining enzyme conformational change.
The S154A mutation results in large 180- and 15000-fold decreases in kinetic parameters kcat and (kcat/Km), respectively, for decarboxylation of OMP34 but much smaller 6- and 41-fold decreases in kcat and (kcat/Km), respectively, for decarboxylation of FOMP (Table 1). Consequently, the S154A mutant shows a high reactivity toward catalysis of decarboxylation of FOMP: (kcat/Km)F/(kcat/Km)H = 460, and (kcat)F/(kcat)H = 200. These 5-F effects are similar to the 400-fold effect of the 5-F on kcat/Km for decarboxylation of the phosphodianion-truncated substrate EO (Scheme 2). We conclude that the stabilizing interactions between the 5-F and neighboring C-6 carbanion are strongly expressed at the rate-determining transition state for decarboxylation of FOMP catalyzed by S154A mutant ScOMPDC. We suggest that the ∼2-fold larger 5-F substituent effect on kcat/Km compared with that on kcat reflects the small stabilization of the Michaelis complex to FOMP by an interaction between the protein and the 5-F substituent.
The CH2OH side chain of Ser154 accepts a hydrogen bond from the pyrimidine NH group and acts as a hydrogen bond donor to the oxygen of the amide side chain of Gln215 (Figure 4).18,34 The second H-bond acts to clamp two enzyme loops over the pyrimidine ring and the phosphodianion of OMP. The effect of these individual mutations on the kinetic parameters for decarboxylation of OMP depends upon the order of the amino acid substitutions.34 The 6 kcal/mol total side chain interactions can be partitioned into an ∼2 kcal/mol interaction between the amide side chain of Gln215 and the phosphodianion group of OMP, observed for the Q215A mutation of wild-type ScOMPDC, and an ∼4 kcal/mol interaction between the -CH2OH side chain of Ser154 and the pyrimidine ring of OMP, observed for the S154A mutation of Q215A mutant ScOMPDC. The opposite order of these mutations leads to expression of nearly the entire 6 kcal/mol effect observed for the S154A/Q215A double mutation at the S154A single mutant, with only a small 0.3 kcal/mol additional effect of the second Q215A mutation.34 These results reflect cooperativity in the interactions of these side chains with the transition state for decarboxylation, due to the requirement for a hydrogen bond between the -CH2OH group of Ser154 and the amide side chain of Gln215 to position the latter side chain to interact with the phosphodianion group of OMP. It was proposed that the S154A single mutation leads to loss of the transition-state stabilization contributed by the side chains of both Ser154 and Gln215. These trends are hardly discernible in the effects of the same mutations on the kinetic parameters for ScOMPDC-catalyzed decarboxylation of FOMP, because the large effects on the rate of chemical decarboxylation are weakly expressed as changes in the enzyme kinetic parameters.
Figure 4.
Partial X-ray crystal structure of ScOMPDC complexed with 6-hydroxyuridine 5′-monophosphate (PDB entry 1DQX) superimposed over the structure for unliganded ScOMPDC (PDB entry 1DQW). The movement of the phosphodianion gripper loop (Pro202–Val220) toward the hydrophobic pyrimidine umbrella (Glu152–Thr165) is shown for the unliganded (olive green) and liganded (orange) enzymes. The closure of these loops is cooperative and driven by the formation of a hydrogen bond between the side chains of Gln215 from the “gripper” loop and of Ser 154 from the “umbrella”. Reproduced from ref (29). Copyright 2013 American Chemical Society.
Eliminating Protein–Phosphodianion Interactions
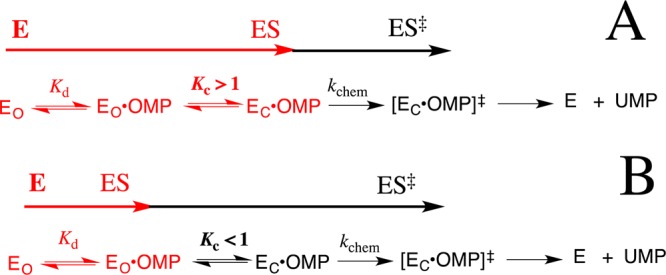
The results from a recent study on the effect of mutations of gripper side chains on the kinetic parameters for ScOMPDC-catalyzed decarboxylation of EO and OMP provide strong evidence that the effects of these mutations on the enzyme kinetic parameters for decarboxylation of OMP are due essentially entirely to changes in the relative stability of the inactive open and active closed forms of ScOMPDC (Scheme 4).4,19,29 We will summarize the experimental evidence that supports the model shown in Scheme 4(4,19,29) and then use this model in the interpretation of the effects of these mutations on the kinetic parameters for ScOMPDC-catalyzed decarboxylation of FOMP.
Scheme 4.
(1) The Q215A, Y217A, and R235A single mutations result in ≥2.5-fold decreases in kcat/Km for decarboxylation of the phosphodianion-truncated substrate EO,4 but in much larger decreases in the third-order rate constant for phosphite dianion activation of ScOMPDC-catalyzed decarboxylation of EO,4 and in the second-order rate constant kcat/Km for decarboxylation of OMP.19 The first result shows that there is little or no direct interaction between the loop side chains and the site of chemical decarboxylation of the orotate ring. It was concluded that the large effect of these mutations on the kinetic parameters for dianion activation of decarboxylation of EO and for decarboxylation of OMP is entirely due to the loss of stabilizing interactions between the deleted side chains and the enzyme-bound dianion.4,19
(2) The model in Scheme 4 was used to rationalize the effects of mutations of gripper side chains on the kinetic parameters for ScOMPDC-catalyzed decarboxylation of OMP. The complex between wild-type ScOMPDC and OMP is proposed to exist mainly in the closed form [Kc ≫ 1 (Scheme 4A)]. Consequently, the first mutations of gripper side chains, which result in up to 4 kcal/mol destabilization of EC, are entirely expressed as a decrease in the Michaelis constant to the limiting value of (Km)obs = Kd = 1 mM for dissociation of OMP from EO·OMP. Mutations that result in further decreases to Kc values of <1 then lead to a decrease in kcat, which falls below the invariant value of kchem for ScOMPDC-catalyzed decarboxylation of OMP (Scheme 3).19 This is because the loop dianion interactions now only develop on proceeding from the dominant Michaelis complex (EO·OMP) to the transition state for decarboxylation at EC·OMP [ES⧧ (Scheme 4B)]. We estimate a Kc value of ≈1000 (Table 2) for the reaction catalyzed by wild-type ScOMPDC from the difference in Kd = 1 μM for decarboxylation of OMP9 and estimated Kd ≈ 1 mM for dissociation of OMP from EO·OMP.19
Table 2. Microscopic Rate Constants for Decarboxylation of FOMP (Scheme 5) by Wild-Type and Mutant Forms of ScOMPDC at pH 7.1 (30 mM MOPS), 25 °C, and I = 0.105 (NaCl), Calculated As Described in the Text.
| ScOMPDC | kcat (s–1)a | kchem (s–1)b | [(kcat)wt/(kcat)mut]OMPc | Kcd | kc (s–1)e | k–c (s–1)f | Λg (kcal/mol) | k0 (s–1)h |
|---|---|---|---|---|---|---|---|---|
| wild-type | 95 | 5000 | 1000 | 104 | 10 | 13.9 | 350 | |
| R235A | 92 | 5000 | 15 | 0.071 | 120 | 1800 | 13.7 | 500 |
| Q215A/R235A | 4.7 | 5000 | 750 | 0.0013 | 17 | 13000 | 13.7 | 560 |
| Q215A/Y217F | 49 | 5000 | 3.1 | 0.48 | 50 | 100 | 14.9 | 70 |
| Y217A | 8.2 | 5000 | 3.2 | 0.45 | 8.2 | 18 | 15.9 | 12 |
Calculated from kcat = 15 s–1 for decarboxylation of OMP19 and the ratios kchem/kcat = 1.8 for decarboxylation of OMP,9 where kcat is the true decarboxylation rate constant, and (kcat)F/(kcat)H ≈ 200 (Table 1) when chemistry is rate-determining for ScOMPDC-catalyzed decarboxylation of OMP and FOMP (Table 1).
The ratio of observed rate constants kcat for decarboxylation of OMP catalyzed by wild-type [(kcat)wt] and mutant [(kcat)mut] forms of ScOMPDC.19
Estimated equilibrium constants for conversion of EO·OMP to EC·OMP (Scheme 5), calculated for mutants of ScOMPDC using eq 1.
Rate constant for conversion of EC·OMP to EO·OMP.
Microscopic Rate Constants
Wild-Type ScOMPDC
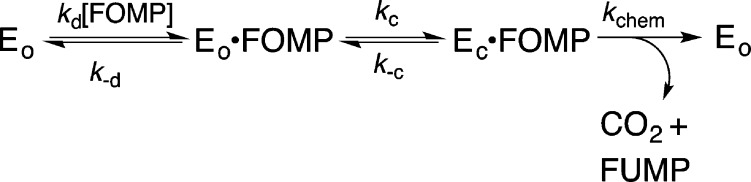
Substrate binding (kd) and enzyme conformational change kc are each partly rate-determining for wild-type ScOMPDC-catalyzed decarboxylation of FOMP at [FOMP] ≪ Km, so that k–d ≈ kc (Scheme 5).27 Combining the values of (kcat/Km) ≈ kd = 1.2 × 107 M–1 s–1 for encounter-limited decarboxylation of FOMP determined for wild-type OMPDC and Kd = (k–d/kd) ≈ 10–3 M [1 mM (see above)] for breakdown of the EO·FOMP complex gives kc ≈ k–d = 104 s–1 for the enzyme conformational change (Table 2). The value of kchem = 5000 s–1 for decarboxylation at EC·FOMP by wild-type ScOMPDC (Table 2) was calculated from kcat = 15 s–1 for decarboxylation of OMP,19 and the ratios kchem/kcat = 1.8 for decarboxylation of OMP,9 where kchem is the true decarboxylation rate constant, and (kcat)F/(kcat)H ≈ 200 when chemistry (Table 1) is rate-determining for OMPDC-catalyzed reactions of both substrates.
Scheme 5.
Mutants of ScOMPDC
The Q215A and Y217F mutations each cause kcat for turnover of FOMP to increase above the value of kcat = 95 s–1 for wild-type ScOMPDC, because of an increase in the rate of release of product to water (Table 1). By contrast, the more severe R235A, Q215A/Y217F, and Q215A/R235A mutations cause kcat to decrease to a value as small as 4.7 s–1 for the Q215A/R235A mutant (Table 1). The value of kchem for the reaction of EC·FOMP is not affected by mutations of the gripper residues, because there are only weak interactions between these side chains and the orotate ring at the transition state for ScOMPDC-catalyzed decarboxylation of enzyme-bound substrate.4,19 Rather, the decreases in kcat, as the protein dianion interactions are weakened and product release becomes much faster than turnover, show that the values of rate constants kc, k–c, and kchem (Scheme 5, which omits the step for product release) begin to control the observed value of kcat.
Equation 1 relates the equilibrium constant Kc for loop closure at mutant ScOMPDCs to the effect of the individual mutation on (kcat)wt for wild-type ScOMPDC-catalyzed decarboxylation of OMP. This equation was derived by assuming that these mutations do not affect microscopic rate constant kchem for decarboxylation of EC·OMP, and that the entire decrease in (kcat)mut compared to (kcat)wt is due to the decrease, from the value of fEC = 1.0 for wild-type ScOMPDC, in the fraction of the Michaelis complex present in the reactive EC·OMP form during turnover [(kcat)mut = (kcat)wtfEC]. The estimated values of Kc reported in Table 2 for mutant ScOMPDC-catalyzed decarboxylation of FOMP were calculated using eq 1 (Scheme 3), and assuming identical values of Kc for loop closure over OMP and FOMP.
| 1 |
Table 2 reports estimated rate constants (Scheme 5) for mutants of ScOMPDC, which were calculated from kchem = 5000 s–1, the values of (kcat)mut for decarboxylation of FOMP (Table 1) and of Kc for the enzyme conformational change (see above), and using eqs 2 and 3 derived for Scheme 5. We note again this treatment assumes that mutations of gripper side chains do not affect the value of kchem (Scheme 5) for decarboxylation of OMP and FOMP. The observation that this treatment provides a rationalization for the following otherwise confusing effects of the mutations of gripper side chains on the kinetic parameters for decarboxylation of OMP and FOMP provides strong justification for the simplifying assumptions used in the calculation of the rate constants from Table 2.
| 2 |
| 3 |
(1) The 5-fold larger value of kcat for decarboxylation of OMP catalyzed by the Q215A/Y217F mutant (4.8 s–1) compared with that for the R235A mutant (1.0 s–1) is due to the larger effect of the R235A mutation on the equilibrium constant Kc (Table 2).19 By contrast, the Q215A/Y217F mutant shows a 2-fold smaller value of kcat for decarboxylation of FOMP (Table 1). We propose that the kcat values of 49 and 92 s–1 for the Q215A/Y217F and R235A mutant ScOMPDC-catalyzed decarboxylations of FOMP, respectively, are limited by the kc values of 50 and 120 s–1 for loop closure to form EC·FOMP (Table 2). In other words, rate-determining loop closure during R235A mutant enzyme-catalyzed decarboxylation of FOMP is faster than for the Q215A/Y217F mutant enzyme-catalyzed reaction, in spite of the 5-fold more unfavorable equilibrium constant for loop closure at the R235A mutant.
(2) The large value of (kcat)F/(kcat)H = 240 for decarboxylation catalyzed by Q215A/R235A mutant ScOMPDC shows that the step for kchem for decarboxylation of EC·FOMP limits the value of kcat for turnover (Scheme 3). The kcat value of 4.7 s–1 for decarboxylation of FOMP catalyzed by this mutant is much smaller than the kchem of ≈5000 s–1 and the kc of =17 s–1 (Table 2), because the barrier to decarboxylation kcat includes the ∼4 kcal/mol barrier for conversion of EO·FOMP to EC·FOMP [Kc = 1/750, and k–c = 13000 s–1 (Table 2)]. In other words, loop closure over enzyme-bound FOMP is partly reversible (k–c > kchem) so that decarboxylation at EC·FOMP (kchem) is the rate-determining step.
(3) The Y217A mutation results in similar large decreases in kcat and kcat/Km for wild-type ScOMPDC-catalyzed decarboxylation of both OMP and FOMP, so that there is only a small effect of the 5-F substituent on the kinetic parameters for this mutant enzyme-catalyzed reaction: (kcat)F/(kcat)H = 2, and (kcat/Km)F/(kcat/Km)H = 5 (Table 1). We conclude that the Y217A mutation results in a similar increase in the barriers to decarboxylation of OMP, where chemistry is rate-determining, and for decarboxylation of FOMP, where the enzyme conformational change is rate-determining. This is reflected by the large decrease in rate constant kc for the enzyme conformational change (Scheme 5) to form the loop-closed ScOMPDC (Table 2).
| 4 |
(4) Equations 2 and 4 are derived for mutants (Table 2) whose reaction is described by Scheme 5. These equations predict that kinetic parameters kcat and kcat/Km for decarboxylation of FOMP are related by 1/Kd for formation of EO·FOMP. This provides a rationalization for the observation that the kinetic data from Table 1 can be used to calculate a nearly constant ratio of kcat/(kcat/Km) ≈ Kd = 6–9 × 10–4 M for decarboxylation of FOMP catalyzed by the different mutants from Table 2, which is similar to Kd ≈ 10–3 M estimated in earlier work for the release of substrate from EO·OMP to form EO.19
Free Energy Reaction Profiles
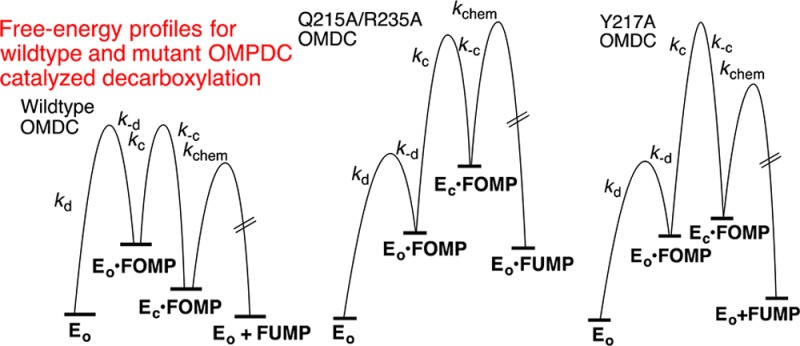
Figure 5 shows free energy profiles for ScOMPDC-catalyzed decarboxylation of FOMP catalyzed by wild-type OMPDC (Figure 5A), the Q215A/R235A (Figure 5B), and Y217A (Figure 5C) mutant enzymes, drawn using the kinetic parameters from Table 2 for reactions at [S] ≪ Km. These profiles rationalize the two very different effects of the 5-F substituent on mutant-enzyme catalyzed decarboxylation of OMP.
Figure 5.

Free energy profiles for ScOMPDC-catalyzed decarboxylation of FOMP (Scheme 5), drawn for reactions at [S] ≪ Km using the kinetic parameters from Table 2. (A) Decarboxylation of FOMP catalyzed by wild-type OMPDC, which shows (a) the similar barriers to partitioning of EO·FOMP between dissociation of OMP and the enzyme conformational change to form EC·FOMP (k–d ≈ kc),27 (b) thermodynamically favorable conversion of EO·FOMP to the EC·FOMP caged complex,19 and (c) the ≈3.5 kcal/mol difference [RT ln(390/1.1) (Scheme 2)] between the barriers for formation of EC·FOMP and decarboxylation of EC·FOMP. (B) Decarboxylation of FOMP catalyzed by the Q215A/R235A mutant of ScOMPDC. This mutation results in a 106-fold decrease in Kc for loop closure compared with that of wild-type ScOMPDC, but in little change in intrinsic barrier Λ for loop closure (Table 2). The decarboxylation step is rate-determining because k–c > kchem. (C) Decarboxylation of FOMP catalyzed by the Y217A mutant of ScOMPDC. This mutation results in a 2000-fold decrease in Kc for loop closure and a large increase in intrinsic barrier Λ for slow loop closure (Table 2), so that loop closure is rate-determining for this decarboxylation reaction.
(1) The large 240- and 500-fold 5-F substituent effects on kcat and kcat/Km for Q215A/R235A ScOMPDC-catalyzed decarboxylation show that the step for chemical decarboxylation, kchem (Scheme 5), limits the values of these kinetic parameters. This reflects the small barrier to k–c for fast loop opening at the crippled enzyme, relative to the barrier for kchem for decarboxylation, which is not affected by these mutations. Figure 5B shows that the overall barrier to kcat/Km is the sum of the barriers to (i) formation of the Michaelis complex ([S] ≪ Km), (ii) Kc for the enzyme conformational change, and (iii) the activation barrier to kchem.
(2) By comparison, the small 2- and 5-fold effects of 5-F on kcat and kcat/Km for Y217A mutant ScOMPDC-catalyzed decarboxylation of OMP (Table 2) show that a slow enzyme conformational change is rate-determining for the mutant enzyme-catalyzed decarboxylation reaction (Figure 5C). Now, the expected increase in k–c for loop opening for the mutant enzyme, due to the effect of the mutation on Kc (Table 2), has been approximately balanced by a second effect of this mutation that results in a decrease in k–c.
Intrinsic Reaction Barriers
Similar rate constants (kc) are estimated for the Q215A/R235A [100 s–1 (Table 2)] and Y217A (18 s–1) mutant enzyme-catalyzed reactions; however, the conformational change for the Q215A/R235A mutant is relatively slow because of the large unfavorable thermodynamic driving force (KC = 1.3 × 10–3), while this conformational change for the Y217A mutant is slow because of the large intrinsic kinetic reaction barrier. These intrinsic kinetic barriers Λ (kilocalories per mole) may be defined by the Marcus equation (eq 5) as the reaction barrier in the absence of a thermodynamic driving force (ΔG° = 0).40−45 Intrinsic barriers Λ for loop closure, with different ScOMPDC mutants, were estimated from rate constants kc and corresponding equilibrium constants Kc reported in Table 2, using eq 5, derived for a reaction at 298 K.46,47 The Marcus intrinsic rate constants for hypothetical thermoneutral loop closure reactions at the different mutant enzymes (Table 2) were then estimated from Λ using eq 6, also derived at 298 K.46,47
| 5 |
| 6 |
Table 2 shows that the R235A/Q215A double mutation results in an ≈106-fold decrease in Kc for loop closure but causes little change in intrinsic barrier Λ for the enzyme conformational change. By contrast, the Q215A/Y217F double mutation results in a 1.2 kcal/mol increase in Λ, which we attribute mainly to the effect of the conservative Y217F substitution, while the less conservative Y217A mutation results in a larger 2.2 kcal/mol increase in Λ. We propose that the phenol group at position 217 functions both to stabilize the closed form of ScOMPDC, through formation of a hydrogen bond to the substrate phosphodianion,18,19,34 and to reduce the intrinsic kinetic barrier to this enzyme conformational change. We suggest that the effect of these mutations of Y217 on Λ is related to their effect on the rate of formation of the highly organized intraloop clamping interaction between the -CH2OH side chain of Ser154 and the amide side chain of Gln215, which sits close to the phenol side chain of Y217 (Figure 4).18,34
Concluding Remark
The simplest interpretation of these results is that the large conformational change of OMPDC is necessary to lock OMP and FOMP into catalytically active caged Michaelis complexes. We have no evidence that this conformational change is coupled in any way to the decarboxylation step of the enzyme-bound substrate. The steady-state kinetic parameters from Table 1 were obtained by the direct determination of decarboxylation reaction velocities. In several cases, the barrier to a nonchemical step, which we propose is an enzyme conformational change, was shown to control the observed reaction barrier. In these cases, the steady-state kinetic parameters were used to estimate the rate and equilibrium constants for this conformational change (Table 2). It would be interesting to examine the changes in the nuclear magnetic resonance48−50 or fluorescence51,52 spectral properties of ScOMPDC during turnover of FOMP, to obtain an independent estimate of the kinetic and thermodynamic barriers to this enzyme conformational change.
Glossary
Abbreviations
- ScOMPDC
orotidine 5′-monophosphate decarboxylase from S. cerevisiae
- OMP
orotidine 5′-monophosphate
- UMP
uridine 5′-monophosphate
- FOMP
5-fluoroorotidine 5′-monophosphate
- FUMP
5-fluorouridine 5′-monophosphate
- BMP
6-hydroxyuridine 5′-monophosphate
- EO
1-(β-d-erythrofuranosyl)orotic acid
- EU
1-(β-d-erythrofuranosyl)uracil
- FEO
1-(β-d-erythrofuranosyl)-5-fluoroorotic acid
- FEU
1-(β-d-erythrofuranosyl)-5-fluorouracil
- MOPS
3-(N-morpholino)propanesulfonic acid
- PDB
Protein Data Bank.
This work was funded by National Institutes of Health Grant GM39754 (to J.P.R.) and Grant GM65155 (to J.A.G.).
The authors declare no competing financial interest.
References
- Miller B. G.; Wolfenden R. (2002) Catalytic proficiency: the unusual case of OMP decarboxylase. Annu. Rev. Biochem. 71, 847–885 10.1146/annurev.biochem.71.110601.135446. [DOI] [PubMed] [Google Scholar]
- Radzicka A.; Wolfenden R. (1995) A proficient enzyme. Science 267, 90–93 10.1126/science.7809611. [DOI] [PubMed] [Google Scholar]
- Tsang W.-Y.; Wood B. M.; Wong F. M.; Wu W.; Gerlt J. A.; Amyes T. L.; Richard J. P. (2012) Proton Transfer from C-6 of Uridine 5′-Monophosphate Catalyzed by Orotidine 5′-Monophosphate Decarboxylase: Formation and Stability of a Vinyl Carbanion Intermediate and the Effect of a 5-Fluoro Substituent. J. Am. Chem. Soc. 134, 14580–14594 10.1021/ja3058474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amyes T. L.; Ming S. A.; Goldman L. M.; Wood B. M.; Desai B. J.; Gerlt J. A.; Richard J. P. (2012) Orotidine 5′-monophosphate decarboxylase: Transition state stabilization from remote protein-phosphodianion interactions. Biochemistry 51, 4630–4632 10.1021/bi300585e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth K.; Amyes T. L.; Wood B. M.; Chan K.; Gerlt J. A.; Richard J. P. (2010) Product Deuterium Isotope Effects for Orotidine 5′-Monophosphate Decarboxylase: Effect of Changing Substrate and Enzyme Structure on the Partitioning of the Vinyl Carbanion Reaction Intermediate. J. Am. Chem. Soc. 132, 7018–7024 10.1021/ja102408k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amyes T. L.; Wood B. M.; Chan K.; Gerlt J. A.; Richard J. P. (2008) Formation and Stability of a Vinyl Carbanion at the Active Site of Orotidine 5′-Monophosphate Decarboxylase: pKa of the C-6 Proton of Enzyme-Bound UMP. J. Am. Chem. Soc. 130, 1574–1575 10.1021/ja710384t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth K.; Amyes T. L.; Wood B. M.; Chan K.; Gerlt J. A.; Richard J. P. (2007) Product Deuterium Isotope Effect for Orotidine 5′-Monophosphate Decarboxylase: Evidence for the Existence of a Short-Lived Carbanion Intermediate. J. Am. Chem. Soc. 129, 12946–12947 10.1021/ja076222f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goryanova B.; Amyes T. L.; Gerlt J. A.; Richard J. P. (2011) OMP Decarboxylase: Phosphodianion Binding Energy Is Used To Stabilize a Vinyl Carbanion Intermediate. J. Am. Chem. Soc. 133, 6545–6548 10.1021/ja201734z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter D. J. T.; Short S. A. (2000) Yeast Orotidine-5′-Phosphate Decarboxylase: Steady-State and Pre-Steady-State Analysis of the Kinetic Mechanism of Substrate Decarboxylation. Biochemistry 39, 11788–11800 10.1021/bi001199v. [DOI] [PubMed] [Google Scholar]
- Amyes T. L.; Richard J. P. (2013) Specificity in transition state binding: The Pauling model revisited. Biochemistry 52, 2021–2035 10.1021/bi301491r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard J. P.; Amyes T. L.; Goryanova B.; Zhai X. (2014) Enzyme architecture: on the importance of being in a protein cage. Curr. Opin. Chem. Biol. 21, 1–10 10.1016/j.cbpa.2014.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malabanan M. M.; Amyes T. L.; Richard J. P. (2010) A role for flexible loops in enzyme catalysis. Curr. Opin. Struct. Biol. 20, 702–710 10.1016/j.sbi.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai B. J.; Wood B. M.; Fedorov A. A.; Fedorov E. V.; Goryanova B.; Amyes T. L.; Richard J. P.; Almo S. C.; Gerlt J. A. (2012) Conformational changes in orotidine 5′-monophosphate decarboxylase: A structure-based explanation for how the 5′-phosphate group activates the enzyme. Biochemistry 51, 8665–8678 10.1021/bi301188k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood B. M.; Amyes T. L.; Fedorov A. A.; Fedorov E. V.; Shabila A.; Almo S. C.; Richard J. P.; Gerlt J. A. (2010) Conformational changes in orotidine 5′-monophosphate decarboxylase: ″Remote″ residues that stabilize the active conformation. Biochemistry 49, 3514–3516 10.1021/bi100443a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth K.; Amyes T. L.; Wood B. M.; Chan K. K.; Gerlt J. A.; Richard J. P. (2009) An Examination of the Relationship between Active Site Loop Size and Thermodynamic Activation Parameters for Orotidine 5′-Monophosphate Decarboxylase from Mesophilic and Thermophilic Organisms. Biochemistry 48, 8006–8013 10.1021/bi901064k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfenden R. (1974) Enzyme catalysis. Conflicting requirements of substrate access and transition state affinity. Mol. Cell. Biochem. 3, 207–211 10.1007/BF01686645. [DOI] [PubMed] [Google Scholar]
- Richard J. P.; Zhai X.; Malabanan M. M. (2014) Reflections on the catalytic power of a TIM-barrel. Bioorg. Chem. 57, 206–212 10.1016/j.bioorg.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller B. G.; Hassell A. M.; Wolfenden R.; Milburn M. V.; Short S. A. (2000) Anatomy of a proficient enzyme: the structure of orotidine 5′-monophosphate decarboxylase in the presence and absence of a potential transition state analog. Proc. Natl. Acad. Sci. U. S. A. 97, 2011–2016 10.1073/pnas.030409797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman L. M.; Amyes T. L.; Goryanova B.; Gerlt J. A.; Richard J. P. (2014) Enzyme Architecture: Deconstruction of the Enzyme-Activating Phosphodianion Interactions of Orotidine 5′-Monophosphate Decarboxylase. J. Am. Chem. Soc. 136, 10156–10165 10.1021/ja505037v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai X.; Amyes T. L.; Richard J. P. (2014) Enzyme Architecture: Remarkably Similar Transition States for Triosephosphate Isomerase-Catalyzed Reactions of the Whole Substrate and the Substrate in Pieces. J. Am. Chem. Soc. 136, 4145–4148 10.1021/ja501103b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go M. K.; Amyes T. L.; Richard J. P. (2009) Hydron Transfer Catalyzed by Triosephosphate Isomerase. Products of the Direct and Phosphite-Activated Isomerization of [1-13C]-Glycolaldehyde in D2O. Biochemistry 48, 5769–5778 10.1021/bi900636c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amyes T. L.; Richard J. P. (2007) Enzymatic catalysis of proton transfer at carbon: activation of triosephosphate isomerase by phosphite dianion. Biochemistry 46, 5841–5854 10.1021/bi700409b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard J. P. (2012) A Paradigm for Enzyme-Catalyzed Proton Transfer at Carbon: Triosephosphate Isomerase. Biochemistry 51, 2652–2661 10.1021/bi300195b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang W.-Y.; Amyes T. L.; Richard J. P. (2008) A Substrate in Pieces: Allosteric Activation of Glycerol 3-Phosphate Dehydrogenase (NAD+) by Phosphite Dianion. Biochemistry 47, 4575–4582 10.1021/bi8001743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes A. C.; Zhai X.; Morgan K. T.; Reinhardt C. J.; Amyes T. L.; Richard J. P. (2015) The Activating Oxydianion Binding Domain for Enzyme-Catalyzed Proton Transfer, Hydride Transfer and Decarboxylation: Specificity and Enzyme Architecture. J. Am. Chem. Soc. 137, 1372–1382 10.1021/ja5123842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes A. C.; Koudelka A. P.; Amyes T. L.; Richard J. P. (2015) Enzyme Architecture: Optimization of Transition State Stabilization from a Cation–Phosphodianion Pair. J. Am. Chem. Soc. 137, 5312–5315 10.1021/jacs.5b02202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood B. M.; Chan K. K.; Amyes T. L.; Richard J. P.; Gerlt J. A. (2009) Mechanism of the Orotidine 5′-Monophosphate Decarboxylase-Catalyzed Reaction: Effect of Solvent Viscosity on Kinetic Constants. Biochemistry 48, 5510–5517 10.1021/bi9006226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Vleet J. L.; Reinhardt L. A.; Miller B. G.; Sievers A.; Cleland W. W. (2008) Carbon isotope effect study on orotidine 5′-monophosphate decarboxylase: support for an anionic intermediate. Biochemistry 47, 798–803 10.1021/bi701664n. [DOI] [PubMed] [Google Scholar]
- Goryanova B.; Goldman L. M.; Amyes T. L.; Gerlt J. A.; Richard J. P. (2013) Role of a Guanidinium Cation–Phosphodianion Pair in Stabilizing the Vinyl Carbanion Intermediate of Orotidine 5′-Phosphate Decarboxylase-Catalyzed Reactions. Biochemistry 52, 7500–7511 10.1021/bi401117y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goryanova B.; Spong K.; Amyes T. L.; Richard J. P. (2013) Catalysis by Orotidine 5′-Monophosphate Decarboxylase: Effect of 5-Fluoro and 4′-Substituents on the Decarboxylation of Two-Part Substrates. Biochemistry 52, 537–546 10.1021/bi301650d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein R. L.; Fujihara H.; Quinn D. M.; Fischer G.; Kuellertz G.; Barth A.; Schowen R. L. (1984) Transition-state structural features for anilide hydrolysis from β-deuterium isotope effects. J. Am. Chem. Soc. 106, 1457–1461 10.1021/ja00317a045. [DOI] [Google Scholar]
- Chan K. K.; Wood B. M.; Fedorov A. A.; Fedorov E. V.; Imker H. J.; Amyes T. L.; Richard J. P.; Almo S. C.; Gerlt J. A. (2009) Mechanism of the Orotidine 5′-Monophosphate Decarboxylase-Catalyzed Reaction: Evidence for Substrate Destabilization. Biochemistry 48, 5518–5531 10.1021/bi900623r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross A.; Abril O.; Lewis J. M.; Geresh S.; Whitesides G. M. (1983) Practical synthesis of 5-phospho-D-ribosyl α-1-pyrophosphate (PRPP): enzymatic routes from ribose 5-phosphate or ribose. J. Am. Chem. Soc. 105, 7428–7435 10.1021/ja00363a037. [DOI] [Google Scholar]
- Barnett S. A.; Amyes T. L.; Wood B. M.; Gerlt J. A.; Richard J. P. (2008) Dissecting the Total Transition State Stabilization Provided by Amino Acid Side Chains at Orotidine 5′-Monophosphate Decarboxylase: A Two-Part Substrate Approach. Biochemistry 47, 7785–7787 10.1021/bi800939k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasteiger E., Hoogland C., Gattiker A., Duvaud S., Wilkins M. R., Appel R. D., and Bairoch A., Eds. (2005) Protein Identification and Analysis Tools on the ExPASy Server, Humana Press Inc., Totowa, NJ. [DOI] [PubMed] [Google Scholar]
- Gasteiger E.; Gattiker A.; Hoogland C.; Ivanyi I.; Appel R. D.; Bairoch A. (2003) ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 31, 3784–3788 10.1093/nar/gkg563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett S. A. (2009) Studies on the Mechanism of Action of Orotidine 5′-Monophosphate Decarboxylase. Ph.D. Thesis, University at Buffalo, Buffalo, NY. [Google Scholar]
- Miller B. G.; Butterfoss G. L.; Short S. A.; Wolfenden R. (2001) Role of Enzyme-Ribofuranosyl Contacts in the Ground State and Transition State for Orotidine 5′-Phosphate Decarboxylase: A Role for Substrate Destabilization?. Biochemistry 40, 6227–6232 10.1021/bi0028993. [DOI] [PubMed] [Google Scholar]
- Blacklow S. C.; Raines R. T.; Lim W. A.; Zamore P. D.; Knowles J. R. (1988) Triosephosphate isomerase catalysis is diffusion controlled. Biochemistry 27, 1158–1165 10.1021/bi00404a013. [DOI] [PubMed] [Google Scholar]
- Marcus R. A. (1969) Unusual Slopes of Free Energy Plots in Kinetics. J. Am. Chem. Soc. 91, 7224–7225 10.1021/ja01054a003. [DOI] [Google Scholar]
- Marcus R. A. (1968) Theoretical Relations among Rate Constants, Barriers, and Brønsted Slopes of Chemical Reactions. J. Phys. Chem. 72, 891–899 10.1021/j100849a019. [DOI] [Google Scholar]
- Richard J. P.; Williams K. B. (2007) A Marcus Treatment of Rate Constants for Protonation of Ring-Substituted α-Methoxystyrenes: Intrinsic Reaction Barriers and the Shape of the Reaction Coordinate. J. Am. Chem. Soc. 129, 6952–6961 10.1021/ja071007k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard J. P.; Amyes T. L.; Williams K. B. (1998) Intrinsic barriers to the formation and reaction of carbocations. Pure Appl. Chem. 70, 2007–2014 10.1351/pac199870102007. [DOI] [Google Scholar]
- Richard J. P.; Amyes T. L.; Toteva M. M. (2001) Formation and Stability of Carbocations and Carbanions in Water and Intrinsic Barriers to Their Reactions. Acc. Chem. Res. 34, 981–988 10.1021/ar0000556. [DOI] [PubMed] [Google Scholar]
- Richard J. P.; Nagorski R. W. (1999) Mechanistic Imperatives for Catalysis of Aldol Addition Reactions: Partitioning of the Enolate Intermediate between Reaction with Bronsted Acids and the Carbonyl Group. J. Am. Chem. Soc. 121, 4763–4770 10.1021/ja9900297. [DOI] [Google Scholar]
- Guthrie J. P. (1991) Rate-equilibrium correlations for the aldol condensation: an analysis in terms of Marcus theory. J. Am. Chem. Soc. 113, 7249–7255 10.1021/ja00019a024. [DOI] [Google Scholar]
- Guthrie J. P.; Guo J. (1996) Intramolecular Aldol Condensations: Rate And Equilibrium Constants. J. Am. Chem. Soc. 118, 11472–11487 10.1021/ja954247l. [DOI] [Google Scholar]
- Massi F.; Wang C.; Palmer A. G. III. (2006) Solution NMR and computer simulation studies of active site loop motion in triosephosphate isomerase. Biochemistry 45, 10787–10794 10.1021/bi060764c. [DOI] [PubMed] [Google Scholar]
- Loria J. P.; Berlow R. B.; Watt E. D. (2008) Characterization of enzyme motions by solution NMR relaxation dispersion. Acc. Chem. Res. 41, 214–221 10.1021/ar700132n. [DOI] [PubMed] [Google Scholar]
- Rozovsky S.; Jogl G.; Tong L.; McDermott A. E. (2001) Solution-state NMR investigations of triosephosphate isomerase active site loop motion: Ligand release in relation to active site loop dynamics. J. Mol. Biol. 310, 271–280 10.1006/jmbi.2001.4673. [DOI] [PubMed] [Google Scholar]
- Hernandez-Alcantara G.; Rodriguez-Romero A.; Reyes-Vivas H.; Peon J.; Cabrera N.; Ortiz C.; Enriquez-Flores S.; De la Mora-De la Mora I.; Lopez-Velazquez G. (2008) Unraveling the mechanisms of tryptophan fluorescence quenching in the triosephosphate isomerase from Giardia lamblia. Biochim. Biophys. Acta, Proteins Proteomics 1784, 1493–1500 10.1016/j.bbapap.2008.05.016. [DOI] [PubMed] [Google Scholar]
- Desamero R.; Rozovsky S.; Zhadin N.; McDermott A.; Callender R. (2003) Active Site Loop Motion in Triosephosphate Isomerase: T-Jump Relaxation Spectroscopy of Thermal Activation. Biochemistry 42, 2941–2951 10.1021/bi026994i. [DOI] [PubMed] [Google Scholar]