

Abstract
Objective:
We aimed to decipher the molecular genetic basis of disease in a cohort of children with a uniform clinical presentation of neonatal irritability, spastic or dystonic quadriplegia, virtually absent psychomotor development, axonal neuropathy, and elevated blood/CSF lactate.
Methods:
We performed whole-exome sequencing of blood DNA from the index patients. Detected compound heterozygous mutations were confirmed by Sanger sequencing. Structural predictions and a bacterial activity assay were performed to evaluate the functional consequences of the mutations. Mass spectrometry, Western blotting, and protein oxidation detection were used to analyze the effects of selenoprotein deficiency.
Results:
Neuropathology indicated laminar necrosis and severe loss of myelin, with neuron loss and astrogliosis. In 3 families, we identified a missense (p.Thr325Ser) and a nonsense (p.Tyr429*) mutation in SEPSECS, encoding the O-phosphoseryl-tRNA:selenocysteinyl-tRNA synthase, which was previously associated with progressive cerebellocerebral atrophy. We show that the mutations do not completely abolish the activity of SEPSECS, but lead to decreased selenoprotein levels, with demonstrated increase in oxidative protein damage in the patient brain.
Conclusions:
These results extend the phenotypes caused by defective selenocysteine biosynthesis, and suggest SEPSECS as a candidate gene for progressive encephalopathies with lactate elevation.
Mitochondrial dysfunction is a frequent cause of childhood encephalopathy. Besides the typical multisystemic disorders, an increasing number of mitochondrial defects are shown to cause a CNS-specific phenotype.1–5 Lactate elevation raises suspicion of mitochondrial involvement and may be observed even in encephalopathies in which muscle biopsies show normal mitochondrial respiratory chain (RC) function.1–3,6 Within our cohort of pediatric patients, we identified patients with an undefined cause of cerebellocerebral atrophy, seizures, severe spasticity, and axonal neuropathy with lactate elevation. We report that despite many of the clinical and neuropathologic signs pointing toward mitochondrial impairment, the patients had novel mutations in the SEPSECS gene, which functions in cytoplasmic transfer RNA (tRNA)-charging in the selenoprotein biosynthesis pathway. We describe the uniform clinical, neuroradiologic, and neuropathologic features of this entity and a detailed mutation characterization. Moreover, our results indicate oxidative damage in the brain as part of the pathogenic mechanism resulting from selenoprotein deficiency.
METHODS
Standard protocol approvals, registrations, and patient consents.
All patient and control samples were taken according to the Declaration of Helsinki, with informed consent. The study was approved by the review board of the Helsinki University Central Hospital.
The patients were identified within a cohort of 64 clinically similar patients. One patient (patient 3) was part of the original PEHO (progressive encephalopathy with edema, hypsarrhythmia, and optic atrophy) syndrome patient series7 and was included in the neuroradiologic (group A) and ophthalmologic study (patient 11) of that series.
A detailed neuropathologic examination was available for 3 patients (patients 1, 2, and 3), including the spinal cord from patient 1; from patient 4, records pertaining to cerebellum, brainstem, and cerebral hemispheres were available. General autopsy records were available from 3 patients (patients 1, 2, and 4). Fresh-frozen tissue samples of patient 3 were available for the study as well as fibroblasts of patients 1 and 2 and myoblasts of patient 2.
DNA sequencing.
For whole-exome sequencing, the exome targets of the patients' DNA were captured with the NimbleGen Sequence Capture 2.1M Human Exome v2.0 array (NimbleGen, Basel, Switzerland) followed by sequencing with the Illumina Genome Analyzer-IIx platform (Illumina, Inc., San Diego, CA) with 2 × 82 base pair paired-end reads. The variant calling pipeline of the Finnish Institute for Molecular Medicine was used for the reference genome alignment and variant calling.8 The coding exons of SEPSECS were sequenced by Sanger sequencing.
Structural analysis of the mutations.
Structural analysis was based on the crystal structure of the human SEPSECS-tRNASec binary complex (PDB ID: 3HL2). The SEPSECS mutants p.Thr325Ser and p.Tyr429* were generated in silico and analyzed in Coot.9 All figures were produced in PyMOL (The PyMOL Molecular Graphics System, version 1.5.0.4, Schrödinger, LLC).
Oxyblot.
The brain protein lysates were extracted using RIPA buffer, and Oxyblot method was performed using an OxyBlot Protein Oxidation Detection Kit (Millipore Corp., Billerica, MA) according to the manufacturer's instructions. The e-Methods on the Neurology® Web site at Neurology.org include full descriptions of haplotype analysis, in vivo activity assay, and protein analysis methods.
RESULTS
Clinical data.
We investigated 4 children from 3 unrelated Finnish families. Clinical features of the patients are summarized in table 1. These children were born after uncomplicated pregnancies at term to healthy nonconsanguineous parents. Two patients were microcephalic at birth. The children were irritable from birth and presented by the age of 1 to 2 months with opisthotonus posturing, absent head control, tremors, and myoclonic jerks. Severe spastic or dystonic quadriplegia with absent psychomotor development became evident during the first few months. Three patients had epileptic seizures, including infantile spasms. As a sign of peripheral neuropathy, the deep tendon reflexes attenuated or vanished by the age of 2.5 years. The optic discs were pale but not atrophic. All patients had edema of hands, feet, and face, as well as narrow forehead, tapering fingers, and high palate.
Table 1.
Summary of clinical and laboratory examinations of patients
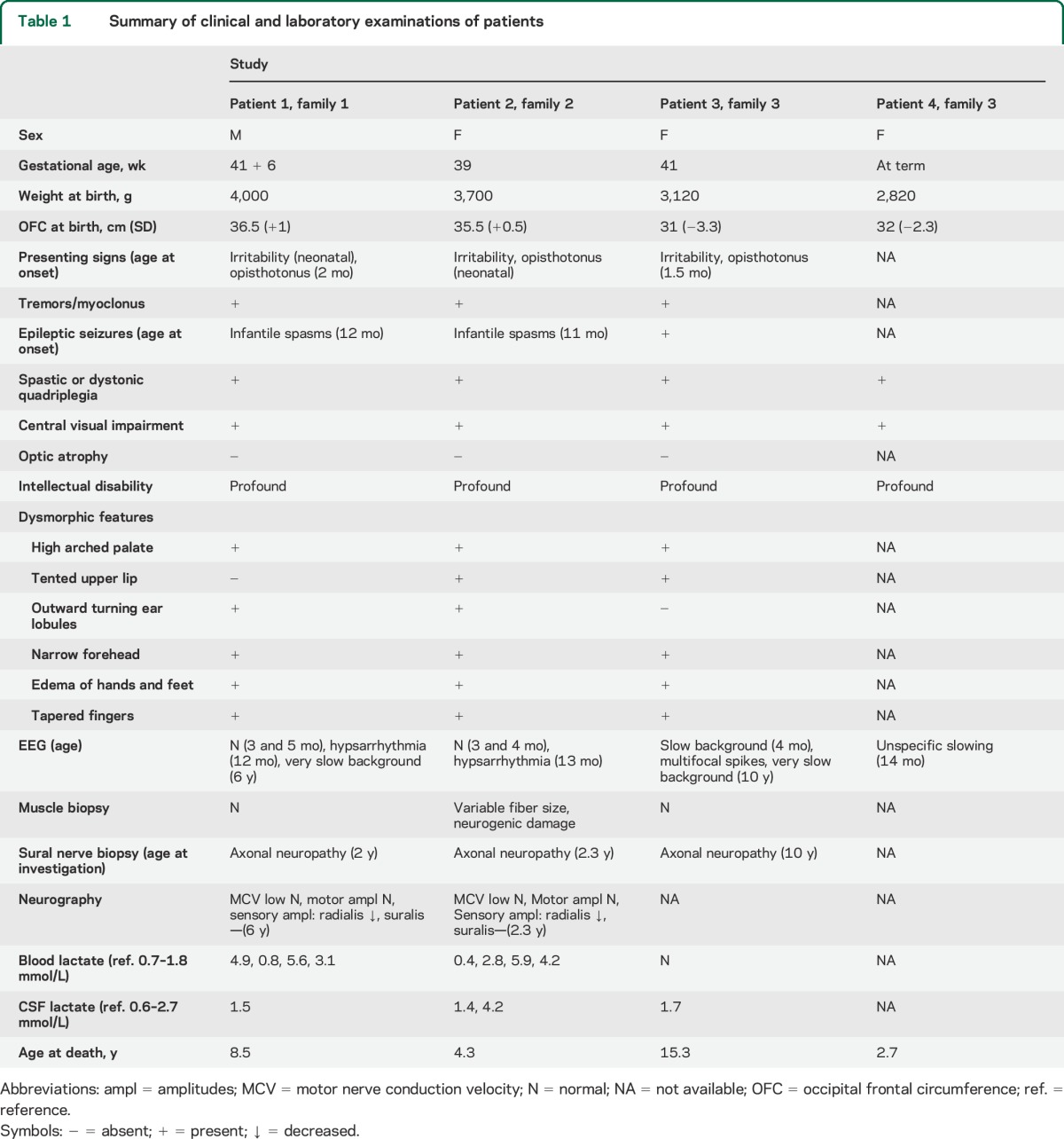
In 2 patients, early EEG studies (younger than 6 months of age) were normal, and later, hypsarrhythmia with infantile spasms was documented. Later EEG recordings showed severe slowing of background activity. Sensory axonal neuropathy was verified by sural nerve biopsy and electroneuromyography (table 1).
Two patients had elevated blood lactate levels and one of them also had elevated lactate in the CSF. Patients 1 and 2 showed mild elevation of thyroid-stimulating hormone (TSH) with normal levels of thyroxine (T4) (table 1). Triiodothyronine (T3) levels were not available. Other laboratory evaluations, including liver transaminases, were unremarkable.
Neuroradiology and neuropathology showed neuron and myelin loss.
Neuroradiologic examinations showed progressive cerebellar atrophy and less pronounced cerebral atrophy (table e-1). Myelination was delayed in the early MRIs and subsequently arrested. Cerebral white matter showed pronounced and progressive volume loss (figure 1).
Figure 1. Brain imaging findings of SEPSECS deficiency.

T2 and fluid-attenuated inversion recovery images of patient 1 at age 5 months (A) and 2 years, 10 months (B, C) and patient 2 at age 6 months (D) and 1 year, 8 months (E, F). The early images (A, D) show loss of periventricular white matter volume and almost complete lack of myelin (arrows). Upon disease progression (B, C, E, F), widening of both central and cortical CSF spaces and cerebellar atrophy are present, with no signal for myelin.
Neuropathologic analysis revealed that all 4 patients shared features of progressive neuronal degeneration: laminar subtotal necrosis of the neocortex, which was especially pronounced in the parietooccipital regions (figure 2A), with relative sparing of the hippocampi. The white matter showed myelin loss and pallor with gliosis, consistent with degeneration secondary to the neuronal loss. Patient 1 had subtotal striatal degeneration (figure 2B) and also some thalamic atrophy. All cases shared a quite severe degeneration and atrophy of the brainstem and cerebellar cortex, creating an olivopontocerebellar atrophy–like appearance: basis pontis, inferior olives, and tegmentum of the medulla oblongata were severely atrophic; the cerebellar cortex was severely atrophic as well (figure 2, C and D). Here, the molecular layer was thin, accompanied by a subtotal loss of Purkinje cells and a very thin granule cell layer (figure 2E). The spinal cord, available from one case, showed atrophy and degeneration especially in the posterior columns. Finally, the general autopsy revealed mild to moderate mostly microvacuolar fatty degeneration of the liver parenchyma (figure 2F).
Figure 2. Histologic findings of SEPSECS deficiency.

(A) Parietooccipital cortex displaying edemic transcortical laminar necrosis (ln) with a subtotal neuronal loss (arrows). (B) Putamen (put) is severely neuron-depleted and gliotic (arrows). (C) Pontine basis (pb) is narrow, both neurons and transverse fibers are reduced. (D) Atrophy of the inferior olives (io) and hili with olivocerebellar fibers and narrowed tegmentum seen in low magnification of the medulla oblongata. (E) Atrophic cerebellar cortex, the molecular and granular layers are thin, with practically total loss of Purkinje cells (arrows). (F) Liver parenchyma exhibiting a moderate microvacuolar fatty degeneration (fat visible as white droplets). Paraffin sections, hematoxylin & eosin (A, C, D, F), Luxol fast blue–cresyl violet (B, E); original magnification ×40 (A, E), ×100 (B), ×10 (C, D), ×400 (F).
SEPSECS mutations identified as the genetic cause.
The genetic cause of the disease was identified by whole-exome sequencing of DNA samples of 2 patients (patients 1 and 2) from 2 unrelated families. The identified variants were first filtered to exclude nongenic variants and those common in populations. On the basis that both families were of Finnish ancestry and the clinical manifestation of both patients closely resembled each other, we searched for homozygous or compound heterozygous variants that the patients shared. Two novel heterozygous variants in SEPSECS were subsequently identified in both patients, c.974C>G in exon 8 leading to a missense mutation p.Thr325Ser and c.1287C>A in exon 11 leading to a nonsense mutation p.Tyr429* (RefSeq NM_016955.3). The variants were validated by Sanger sequencing (figure 3A). The parents of patient 1 and the mother of patient 2 were heterozygous carriers; the DNA sample of the father of patient 2 was not available. The variants were not present in the 1000 Genomes database (www.1000genomes.org) or in the NHLBI GO ESP Exome Variant Server (evs.gs.washington.edu/EVS/) or in approximately 230 screened Finnish control samples. However, a recent database of 3,323 exome sequences of Finnish individuals provided a heterozygous carrier frequency of 1:277 for the c.1287C>A, p.Tyr429* variant (Sequencing Initiative Suomi, sisu.fimm.fi), suggesting enrichment of the nonsense variant in Finland, but without homozygous occurrence.
Figure 3. SEPSECS gene defect with structural and functional consequences of the mutations.

(A) SEPSECS mutation sequences. The arrow indicates the mutation site. (B) Haplotypes on the chromosome 4 region containing the SEPSECS gene in 3 patients from 3 unrelated families. Parent samples were used to construct the haplotypes when available. (C) Alignment of the sequence region containing Thr325 (arrow) in eukaryotic SEPSECS proteins. (D) Thr325 is located in the middle of helix α12 in the C-terminal domain of the protein. Helix α12 provides support for the major elements that constitute the active site of SEPSECS by 2 direct interactions: (1) helix α12 (orange) interacts with helix α10 (from monomer 1, in gray); (2) helix α10 carries a catalytic residue Lys284 (orange sticks) to which the cofactor PLP (orange sticks) is covalently attached and forms the floor of the active site. The α10–α12 interaction places Thr325 (orange sticks) behind the active-site pocket and approximately 15 Å away from PLP (shown with up-and-down arrow). In addition, helix α12 interacts with helix α3 (blue) from the second SEPSECS monomer (monomer 2, in light blue), which is flanked by loop 70 and P-loop (blue) that form the clefts of the active site. The CCA end of tRNASec (beige) binds in the proximity of the P-loop, which is also implicated in binding of the phosphoseryl group. (E) Sodium dodecyl sulfate–polyacrylamide gel electrophoresis from brain samples; SEPSECS patient (P) and control (C). The full-length SEPSECS of approximately 55 kDa was readily detected, but no signal of the size of the truncated protein (∼47 kDa) was present. (F) In vivo dilution series assay of the ability of human SEPSECS variants to restore the benzyl viologen–reducing activity of the selenoprotein formate dehydrogenase H in the Escherichia coli ΔselA deletion strain. GAPDH = glyceraldehyde 3-phosphate dehydrogenase; PLP = pyridoxal phosphate; tRNA = transfer RNA.
Next we screened 11 SEPSECS exons for mutations in additional patients with mitochondrial encephalopathy and/or other shared features. One patient (patient 3) from an affected sib-pair with similar clinical findings was found to be compound heterozygous for the same SEPSECS mutations c.974C>G (p.Thr325Ser) and c.1287C>A (p.Tyr429*). The DNA sample of the affected sib was not available for the study. Investigation of the family histories of the 3 patients with shared SEPSECS mutations revealed that they all originated from a restricted area in eastern Finland. Haplotype analysis of the nearby microsatellite markers indicated shared ancestral haplotypes, further supporting the distant common origin of the mutations in our patients (figure 3B).
Predicted effects of the mutations onto the SEPSECS protein structure.
SEPSECS codes for O-phosphoseryl-tRNA:selenocysteinyl-tRNA synthase, the key enzyme in the sole biosynthetic route to selenocysteine (Sec) in eukaryotes and archaea.10,11 Residue Thr325, which is affected by a missense mutation in our patients, is a highly conserved amino acid in eukaryotic SEPSECS (figure 3C). In the SEPSECS structure, Thr325 is located in the middle of helix α12 in the C-terminal domain of the protein (figure 3D). Helix α12 provides support for the major elements that constitute the active site of SEPSECS. Thr325 interacts only with the backbone and side-chain atoms of helix α12, and its replacement with serine may destabilize the structure of α-helix. This could lead to altered positioning of the cofactor pyridoxal phosphate, and the floor (helix α10) and the clefts (loop 70 and P-loop) of the active site. These structural rearrangements in the catalytic pocket would ultimately yield an enzyme with reduced catalytic power.
The p.Tyr429* mutant messenger RNA escapes nonsense-mediated decay, and is predicted to lead to truncation of the 73 C-terminal amino acids of SEPSECS, the region critical both for tRNA binding and enzyme activity (figure e-1). The full-length SEPSECS of approximately 55 kDa was readily detected by Western blotting—even in a higher amount than in the control sample (figure 3E)—in the brain autopsy sample of patient 3, but no evidence of a truncated protein (∼47 kDa) was found, indicating that it was either not produced or rapidly degraded.
To functionally confirm the mutation effects, we showed that both mutations severely affected SEPSECS activity in vivo in an anaerobic Escherichia coli assay.12 For the assay, we utilized the ΔselA strain and inspected the ability of human SEPSECS to restore the benzyl viologen–reducing activity of an E coli selenoprotein, the formate dehydrogenase H. As predicted, p.Tyr429* mutant was completely inactive in this assay, while the p.Thr325Ser mutant was active albeit at the decreased level (figure 3F).
Functional consequences of the SEPSECS mutations.
Inactivating SEPSECS mutations presumably inhibit synthesis of 25 selenoproteins (the human selenoproteome), which participate in diverse biological processes.13 We measured selenoprotein levels in the lysate obtained from the autopsy brain material of patient 3 using selected reaction monitoring–mass spectrometry (SRM-MS). Protein levels were normalized against glyceraldehyde 3-phosphate dehydrogenase and phosphoglycerate kinase 1. To validate the method, the levels of the glial fibrillary acidic protein, which resides in astrocytes, were measured and shown to be increased, thus indicating astrogliosis (figure 4, A, D). In contrast, levels of myelin basic protein and neurofilament medium were clearly reduced (figure 4A), which is consistent with the observed myelin and neuron loss in the patients. Three selenoproteins, thioredoxin reductase TXNRD1 and glutathione peroxidases GPX1 and GPX4, were abundant enough to be reliably detected. Their levels were decreased by 15% to 40% in the patient brain sample compared with controls (figure 4B). Consistent with a partial defect in selenoprotein production, levels of TXNRD1 and TXNRD2 were decreased in the patient's brain as shown by Western blotting (figure 4D). However, the steady-state level of tRNASec was not altered (figure 4F). Of note, the observed defects were tissue-specific, as the patient's fibroblasts, myoblasts, or differentiated myotubes did not show reduced TXNRD levels (data not shown).
Figure 4. Selenoprotein and respiratory chain protein amounts in SEPSECS deficiency.

(A–C) Selected reaction monitoring–mass spectrometry; autopsy brain sample of patient 3 and 3 controls. The results are shown for (A) glial fibrillary acidic protein (GFAP), myelin basic protein (MBP), and neurofilament medium (NFM), markers of brain cells, for (B) selenoproteins glutathione peroxidases 1 and 4 (GPX1 and GPX4) and thioredoxin reductase 1 (TRXR1) and for (C) mitochondrial respiratory chain complex subunits: NADH dehydrogenase (ubiquinone) Fe-S protein 1 (NDUFS1), NADH dehydrogenase (ubiquinone) Fe-S protein 8 (NDUFS8), ubiquinol-cytochrome c reductase core protein II (QCR2), cytochrome c oxidase subunit Va (COX5A), mitochondrially encoded ATP synthase 6 (ATPA), and citrate synthase (CISY). (D) Western blotting of the brain samples for GFAP and 2 selenoproteins (P, patient 3; C, controls 1–3). (E) Blue native electrophoresis of mitochondrial respiratory chain complexes in the patient (P) and control (C) brain samples. (F) Steady-state levels of tRNASec compared with mitochondrial tRNAAla in the brain sample of patient 3 (P) and controls (C1–C4). (G) Oxyblot shows increased amounts of oxidized proteins in patient brain (P) compared with controls (C1–C3). GAPDH = glyceraldehyde 3-phosphate dehydrogenase; mt-tRNA = mitochondrial transfer RNA; tRNASec = selenocysteine-specific transfer RNA.
Because of the lactate elevation, we analyzed the amounts of mitochondrial RC complexes I–IV in the autopsy brain samples of patient 3 by blue native electrophoresis, but found them to be similar to controls (figure 4E). In addition, when we quantified the amounts of RC complex subunits by SRM-MS, they were shown to be mostly unaffected (figure 4C).
Because the decreased selenoprotein synthesis affected glutathione peroxidases and thioredoxin reductases—enzymes of antioxidant defense—we analyzed protein carbonylation in the patient's brain, and found protein oxidation to be clearly increased (figure 4G).
DISCUSSION
We report here that pathogenic mutations in SEPSECS lead to severe cerebellocerebral atrophy by attenuating the synthesis of selenoproteins, which leads to considerable oxidative damage in the patient brains.
SEPSECS mutations were previously described in a single report, underlying progressive cerebellocerebral atrophy with profound mental retardation, progressive microcephaly, severe spasticity, and myoclonic or generalized tonic-clonic seizures, later classified as pontocerebellar hypoplasia type 2D (PCH2D) (MIM 613811).12,14 The MRI findings of progressive cerebellar atrophy, followed by cerebral atrophy involving both white and gray matter, mimicked the findings in our patients. In contrast to a normal metabolic profile of patients with PCH2D,12 our patients had lactacidemia, and they also presented with axonal neuropathy. Similarities between the patients with PCH2D and the patients described here support the disease-causing role of the identified mutations, thus extending the phenotypes caused by defective selenocysteine biosynthesis.
The neuropathologic changes of SEPSECS deficiency, such as progressive neuronal degeneration, more pronounced laterally than midline, are reminiscent of Alpers syndrome,15 which is caused by defects in mitochondrial proteins, most commonly in polymerase gamma, but also in Twinkle helicase and the phenylalanyl-tRNA synthetase.4,16,17 The topography of the lesions corresponds to highly energy-dependent regions of the CNS; in case of SEPSECS deficiency, this is particularly the parietooccipital region. The patients with SEPSECS mutations described here also showed moderate degeneration of the liver postmortem. These findings, together with elevated lactate, suggest that encephalopathy due to SEPSECS deficiency and mitochondrial encephalopathies, such as Alpers syndrome, could share some common pathogenic mechanisms. However, we did not identify significant alterations in the mitochondrial RC complexes in the patient's brain sample that would explain changes typical for RC deficiencies.
Similar manifestations between mitochondrial and selenoprotein disorders could be explained by the role selenoproteins have in maintaining the cellular redox potential and H2O2 detoxification.13 In other words, selenoproteins are an important part of the antioxidant defense. We showed remarkable increase of protein carbonylation as a sign of oxidative stress in the brain of patients with SEPSECS deficiency. As mitochondria are one of the main sources of cellular reactive oxygen species, SEPSECS deficiency could especially damage cells with high mitochondrial activity. Future work on mouse models is needed to clarify this aspect of selenoprotein pathology.
Progressive cerebellar atrophy of our patients together with infantile spasms, dysmorphic features, and edema of hands, feet, and face resembled the findings typically seen in PEHO syndrome.7,18 However, unlike patients with PEHO, our patients did not have optic atrophy and were spastic rather than hypotonic. Previously, a connection between pontocerebellar hypoplasias, mitochondrial encephalopathies, and PEHO-like features has been proposed in PCH6 that is caused by RARS2 mutations.19 RARS2 encodes the mitochondrial aminoacyl-tRNA synthetase essential for charging tRNAArg for protein synthesis of mitochondrial RC complexes.6 Of note, SEPSECS also affects cellular functions through tRNA, but in a different biological pathway, by catalyzing the conversion of the phosphoseryl-tRNASec intermediate into selenocysteinyl-tRNASec.10,11 Furthermore, PCH2 types A, B, and C are caused by mutations in genes encoding for subunits of the tRNA-splicing endonuclease complex, TSEN54, TSEN2 and TSEN34, respectively.20 This complex performs the splicing of intron-containing tRNAs, which comprise approximately 6% of human tRNAs needed for cytoplasmic translation.21 Whether a common mechanism exists by which the defects of the different cellular protein synthesis processes lead to similar neurodegenerative phenotypes or whether tRNA involvement in all these entities is purely coincidental remains to be established.
SEPSECS is a tetramer composed of 2 dimers, where each dimer contains 2 active sites formed at the dimer interface.10,11 The novel compound heterozygous mutations identified in this study were predicted to have differential effects on the function of the tetrameric SEPSECS: p.Thr325Ser introduced Ser in the middle of an α-helix predicting destabilization of the helix,22 thereby modifying the catalytic pocket of the enzyme, whereas p.Tyr429* resulted in a total loss of function. Our dilution series of the bacterial in vivo assay verified these predictions. The previously reported SEPSECS mutations of patients with PCH2D displayed no detectable activity in an anaerobic in vivo assay for SEPSECS activity.12 Thus, the differences in the phenotypes of our patients and those reported previously may be caused by differences in the residual SEPSECS activity. The fact that the fibroblasts, myoblasts, and myotubes of our patients did not display decreased selenoprotein levels (not shown) suggests that the residual SEPSECS activity was sufficient to maintain selenoprotein synthesis in these cell types but not in the brain.
The 25 human selenoproteins function in remarkably diverse processes. Besides SEPSECS deficiency, the only known human disease affecting selenoprotein synthesis is caused by mutations in SECISBP2, which encodes a protein that recognizes the specific insertion sequence in the selenoprotein messenger RNAs. SECISBP2 mutations cause either a multisystem disorder23 or abnormal thyroid hormone metabolism with elevated TSH, T4, and reverse T3 and reduced T3 levels.24 Our patients presented with increased TSH and normal T4 levels, as measured at ages 2 and 6 years. Thus, although affecting the common metabolic pathway, mutations in SECISBP2 and SEPSECS yield distinct phenotypes.
The selenoproteome is essential for mammals as shown by the early embryonic lethality of the tRNASec gene (Trsp) knockout mouse.25 The selenoprotein knockout mice have further clarified their importance for brain function.26 The neuron-specific knockout mice of GPx4,27 Txnrd1,28 and Trsp29 are characterized by neurodegeneration, in general, and by cerebellar hypoplasia, in particular.30 In the Txnrd1 mice, the effect was attributed to decreased proliferation of granule cell precursors within the external granular layer.28 Furthermore, the full knockout of SelP,31 which encodes a plasma protein SELP that transports selenium from the liver to peripheral tissues, was shown to have a neurologic phenotype with spasticity and seizures.32 It is likely that the lack of several selenoproteins contributes to the neuronal loss in the selenoprotein biosynthesis defects. Our SRM-MS and Western blotting results derived from the analysis of an autopsy brain sample from a patient with SEPSECS mutations showed that selenoproteins were present albeit at significantly reduced levels. However, because of the severe neuronal loss and astrogliosis in the patient brain, a comparative analysis has limitations. For instance, neurons may have had more severe reductions of selenoproteins before their death. Also, the measurement of selenoenzyme activities was not feasible with this material. Regardless of these shortcomings, our result of SEPSECS mutations causing a selenoprotein deficiency in human brain implicates a specific requirement of selenoproteins in postnatal brain development.
Based on the results of our study, we suggest SEPSECS sequencing in progressive early childhood brain atrophies of unknown cause, especially when patients present with sensory axonal neuropathy and elevated lactate.
Supplementary Material
ACKNOWLEDGMENT
Anu Harju and Riitta Lehtinen are thanked for technical help. The authors acknowledge the exome capture, sequencing, and variant calling pipeline analysis performed by the Institute for Molecular Medicine Finland FIMM, Technology Centre, and University of Helsinki. Proteome analysis was made possible through use of the National Proteomics and Metabolomics infrastructure of Biocenter Finland.
GLOSSARY
- PCH2D
pontocerebellar hypoplasia type 2D
- PEHO
progressive encephalopathy with edema, hypsarrhythmia, and optic atrophy
- RC
respiratory chain
- SRM-MS
selected reaction monitoring–mass spectrometry
- T4
thyroxine
- tRNA
transfer RNA
- TSH
thyroid-stimulating hormone
- T3
triiodothyronine
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
A.-K.A. collected and analyzed patient data and drafted the manuscript. T.H., A.L., and E.Y. sequenced patients and performed protein and cell culture experiments. T. Linnankivi and P.I. contributed to the clinical analysis. R.L.F. and M. Simonović performed the structural analysis. Y.L. and D.S. performed the in vivo bacterial assay. D.M.-P. and G.L.C. performed the mass spectrometry analysis. M. Somer, M.L., T. Lönnqvist, and H.P. contributed to the clinical analysis. L.V. performed neuroradiology. A.P. performed neuropathology. H.T. performed the exome sequencing analysis. A.-E.L., A.S., and H.T. designed the study and drafted the manuscript. All authors revised the manuscript.
STUDY FUNDING
The authors thank the Sigrid Jusélius Foundation, Academy of Finland and University of Helsinki (to H.T. and A.S.), Jane and Aatos Erkko Foundation (to A.S.), Folkhälsan Research Foundation (to A.-E.L.), the US NIH grant GM097042 (to M. Simonović), the US National Institute of General Medical Sciences GM22854 (to D.S.), Arvo and Lea Ylppö Foundation (to A.-K.A. and H.T.), Orion Farmos Research Foundation (to H.T.), Helsinki University Central Hospital Research Fund (to A.-K.A.), and Foundation for Pediatric Research (to P.I.) for funding support.
DISCLOSURE
A. Anttonen has received research support from Arvo and Lea Ylppö Foundation and Helsinki University Central Hospital Research Fund. T. Hilander and T. Linnankivi report no disclosures relevant to the manuscript. P. Isohanni has received research support from Foundation for Pediatric Research. R. French and Y. Liu report no disclosures relevant to the manuscript. M. Simonović has received research support from the US NIH grant GM097042. D. Söll received research support from the US National Institute of General Medical Sciences (GM22854). M. Somer, D. Muth-Pawlak, G. Corthals, A. Laari, E. Ylikallio, M. Lähde, L. Valanne, T. Lönnqvist, H. Pihko, and A. Paetau report no disclosures relevant to the manuscript. A. Lehesjoki has received research support from the Folkhälsan Research Foundation. A. Suomalainen has received research support from Sigrid Jusélius Foundation, Academy of Finland, University of Helsinki, and Jane and Aatos Erkko Foundation. H. Tyynismaa has received research support from Arvo and Lea Ylppö Foundation, Orion Farmos Research Foundation, Sigrid Jusélius Foundation, Academy of Finland, and University of Helsinki. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Koskinen T, Santavuori P, Sainio K, Lappi M, Kallio AK, Pihko H. Infantile onset spinocerebellar ataxia with sensory neuropathy: a new inherited disease. J Neurol Sci 1994;121:50–56. [DOI] [PubMed] [Google Scholar]
- 2.Nikali K, Suomalainen A, Saharinen J, et al. Infantile onset spinocerebellar ataxia is caused by recessive mutations in mitochondrial proteins Twinkle and Twinky. Hum Mol Genet 2005;14:2981–2990. [DOI] [PubMed] [Google Scholar]
- 3.Scheper GC, van der Klok T, van Andel RJ, et al. Mitochondrial aspartyl-tRNA synthetase deficiency causes leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation. Nat Genet 2007;39:534–539. [DOI] [PubMed] [Google Scholar]
- 4.Elo JM, Yadavalli SS, Euro L, et al. Mitochondrial phenylalanyl-tRNA synthetase mutations underlie fatal infantile Alpers encephalopathy. Hum Mol Genet 2012;21:4521–4529. [DOI] [PubMed] [Google Scholar]
- 5.Ylikallio E, Suomalainen A. Mechanisms of mitochondrial diseases. Ann Med 2012;44:41–59. [DOI] [PubMed] [Google Scholar]
- 6.Edvardson S, Shaag A, Kolesnikova O, et al. Deleterious mutation in the mitochondrial arginyl-transfer RNA synthetase gene is associated with pontocerebellar hypoplasia. Am J Hum Genet 2007;81:857–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Somer M. Diagnostic criteria and genetics of the PEHO syndrome. J Med Genet 1993;30:932–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sulonen AM, Ellonen P, Almusa H, et al. Comparison of solution-based exome capture methods for next generation sequencing. Genome Biol 2011;12:R94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of coot. Acta Crystallogr D Biol Crystallogr 2010;66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Palioura S, Sherrer RL, Steitz TA, Soll D, Simonovic M. The human SepSecS-tRNASec complex reveals the mechanism of selenocysteine formation. Science 2009;325:321–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yuan J, Palioura S, Salazar JC, et al. RNA-dependent conversion of phosphoserine forms selenocysteine in eukaryotes and archaea. Proc Natl Acad Sci USA 2006;103:18923–18927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Agamy O, Ben Zeev B, Lev D, et al. Mutations disrupting selenocysteine formation cause progressive cerebello-cerebral atrophy. Am J Hum Genet 2010;87:538–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bellinger FP, Raman AV, Reeves MA, Berry MJ. Regulation and function of selenoproteins in human disease. Biochem J 2009;422:11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ben-Zeev B, Hoffman C, Lev D, et al. Progressive cerebellocerebral atrophy: a new syndrome with microcephaly, mental retardation, and spastic quadriplegia. J Med Genet 2003;40:e96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harding BN. Progressive neuronal degeneration of childhood with liver disease (Alpers-Huttenlocher syndrome): a personal review. J Child Neurol 1990;5:273–287. [DOI] [PubMed] [Google Scholar]
- 16.Hakonen AH, Isohanni P, Paetau A, Herva R, Suomalainen A, Lonnqvist T. Recessive Twinkle mutations in early onset encephalopathy with mtDNA depletion. Brain 2007;130:3032–3040. [DOI] [PubMed] [Google Scholar]
- 17.Naviaux RK, Nguyen KV. POLG mutations associated with Alpers' syndrome and mitochondrial DNA depletion. Ann Neurol 2004;55:706–712. [DOI] [PubMed] [Google Scholar]
- 18.Salonen R, Somer M, Haltia M, Lorentz M, Norio R. Progressive encephalopathy with edema, hypsarrhythmia, and optic atrophy (PEHO syndrome). Clin Genet 1991;39:287–293. [DOI] [PubMed] [Google Scholar]
- 19.Rankin J, Brown R, Dobyns WB, et al. Pontocerebellar hypoplasia type 6: a British case with PEHO-like features. Am J Med Genet A 2010;152A:2079–2084. [DOI] [PubMed] [Google Scholar]
- 20.Budde BS, Namavar Y, Barth PG, et al. tRNA splicing endonuclease mutations cause pontocerebellar hypoplasia. Nat Genet 2008;40:1113–1118. [DOI] [PubMed] [Google Scholar]
- 21.Kasher PR, Namavar Y, van Tijn P, et al. Impairment of the tRNA-splicing endonuclease subunit 54 (tsen54) gene causes neurological abnormalities and larval death in zebrafish models of pontocerebellar hypoplasia. Hum Mol Genet 2011;20:1574–1584. [DOI] [PubMed] [Google Scholar]
- 22.Serrano L, Fersht AR. Capping and alpha-helix stability. Nature 1989;342:296–299. [DOI] [PubMed] [Google Scholar]
- 23.Schoenmakers E, Agostini M, Mitchell C, et al. Mutations in the selenocysteine insertion sequence-binding protein 2 gene lead to a multisystem selenoprotein deficiency disorder in humans. J Clin Invest 2010;120:4220–4235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dumitrescu AM, Liao XH, Abdullah MS, et al. Mutations in SECISBP2 result in abnormal thyroid hormone metabolism. Nat Genet 2005;37:1247–1252. [DOI] [PubMed] [Google Scholar]
- 25.Bosl MR, Takaku K, Oshima M, Nishimura S, Taketo MM. Early embryonic lethality caused by targeted disruption of the mouse selenocysteine tRNA gene (trsp). Proc Natl Acad Sci USA 1997;94:5531–5534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kasaikina MV, Hatfield DL, Gladyshev VN. Understanding selenoprotein function and regulation through the use of rodent models. Biochim Biophys Acta 2012;1823:1633–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seiler A, Schneider M, Forster H, et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab 2008;8:237–248. [DOI] [PubMed] [Google Scholar]
- 28.Soerensen J, Jakupoglu C, Beck H, et al. The role of thioredoxin reductases in brain development. PLoS One 2008;3:e1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wirth EK, Conrad M, Winterer J, et al. Neuronal selenoprotein expression is required for interneuron development and prevents seizures and neurodegeneration. FASEB J 2010;24:844–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wirth EK, Bharathi BS, Hatfield D, Conrad M, Brielmeier M, Schweizer U. Cerebellar hypoplasia in mice lacking selenoprotein biosynthesis in neurons. Biol Trace Elem Res 2014;158:203–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schomburg L, Schweizer U, Holtmann B, Flohe L, Sendtner M, Kohrle J. Gene disruption discloses role of selenoprotein P in selenium delivery to target tissues. Biochem J 2003;370:397–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hill KE, Zhou J, McMahan WJ, Motley AK, Burk RF. Neurological dysfunction occurs in mice with targeted deletion of the selenoprotein P gene. J Nutr 2004;134:157–161. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.