

Summary
The transcriptional response to infection with the bacterium Listeria monocytogenes (Lm) requires cooperative signals of the type I interferon (IFN-I)-stimulated JAK-STAT and proinflammatory NF-κB pathways. Using ChIP-seq analysis, we define genes induced in Lm-infected macrophages through synergistic transcriptional activation by NF-κB and the IFN-I-activated transcription factor ISGF3. Using the Nos2 and IL6 genes as prime examples of this group, we show that NF-κB functions to recruit enzymes that establish histone marks of transcriptionally active genes. In addition, NF-κB regulates transcriptional elongation by employing the mediator kinase module for the recruitment of the pTEFb complex. ISGF3 has a major role in associating the core mediator with the transcription start as a prerequisite for TFIID and RNA polymerase II (Pol II) binding. Our data suggest that the functional cooperation between two major antimicrobial pathways is based on promoter priming by NF-κB and the engagement of the core mediator for Pol II binding by ISGF3.
Graphical Abstract
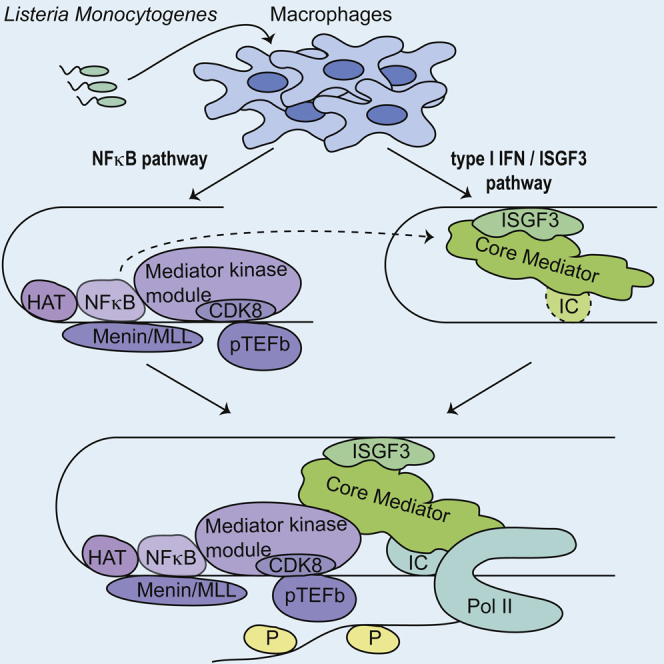
Highlights
-
•
STAT-NF-κB cooperativity shapes the transcriptional response to infection
-
•
NF-κB recruits RNA polymerase II kinase complexes and enzymes marking active chromatin
-
•
NF-κB attracts the mediator kinase module, whereas STATs contact the core mediator
-
•
STAT-NF-κB cooperation tethers a complete, functional mediator to antimicrobial genes
Innate immunity requires transcriptional activation of antimicrobial genes. Wienerroither et al. show that the mediator, a multisubunit complex, serves as a hub for signal integration and cooperative transcriptional activation by two major antimicrobial signaling avenues, the STAT and NF-κB pathways.
Introduction
Immune cells respond to microbial invaders such as the Gram-positive intracellular bacterium Listeria monocytogenes (Lm) with specialized gene expression profiles (Hamon et al., 2006; McCaffrey et al., 2004). Initial sensing of the microbe occurs by surface and endosomal Toll-like receptors (TLRs), whereas Lms escape from endosomal confinement to the cytoplasm causes the engagement of different cytoplasmic receptors to detect infection (Kawai and Akira, 2009; Mancuso et al., 2009; Sauer et al., 2011; Seki et al., 2002; Woodward et al., 2010). Collectively, these pattern recognition receptors (PRRs) activate an extensive network of signals, leading to NF-κB activation and the interferon regulatory factor (IRF)-mediated synthesis of mRNA for type I interferons (IFN-I). IFN-I synthesis takes place exclusively upon recognition of cytosolic bacteria (O’Riordan and Portnoy, 2002; Stockinger et al., 2002). When escape from the phagosome is impeded, the NF-κB pathway is activated without IFN-I synthesis (Farlik et al., 2010).
The IFN-I receptor complex causes the phosphorylation of signal transducers and activators of transcription 1 (STAT1) and STAT2 by the receptor-associated Janus tyrosine kinases (JAK). The tyrosine-phosphorylated STATs form heterodimers and associate with IRF9 to form a trimeric complex, interferon-stimulated gene factor 3 (ISGF3). Depending on the promoter, ISGF3 may be both necessary and sufficient for the transcription of IFN-stimulated genes, or it may require input from additional signaling pathways (Levy and Darnell, 2002). A prominent example of a gene whose expression is strongly enhanced upon stimulation by an additional pathway is Nos2, which encodes inducible nitric oxide synthase (iNOS), a critical component of antibacterial defense (Bogdan, 2001; Farlik et al., 2010). In macrophages infected with Lm, IFN-I/ISGF3 and NF-κB synergize to stimulate the production of iNOS. During infection, NF-κB binds first to the Nos2 promoter, converting the PRR signal into a transcriptional memory effect for the subsequent IFN-I-dependent deposition of ISGF3. NF-κB is necessary for the recruitment of TFIIH and pTEFb, the complexes containing the RNA polymerase II (Pol II) kinases CDK7 and CDK9, whereas ISGF3 is essential for binding of the general transcription factor TFIID and Pol II (Farlik et al., 2010; Wienerroither et al., 2014).
The transcriptionally active state of a gene requires chromatin remodeling and modification as well as the phosphorylation of serines (S) within the Pol II carboxy-terminal domain (CTD). S5 phosphorylation by CDK7 is a prerequisite for promoter clearance and mRNA 5′ end processing, whereas CDK9 phosphorylation of the CTD at S2 is essential for subsequent mRNA elongation. Many groups have reported that the bromo and extra terminal (BET) family member Brd4 is involved in pTEFb recruitment, tethering the complex to transcriptional activators or acetylated histones or acting in the context of superelongation complexes (SECs) (Brasier et al., 2011; Jang et al., 2005; Luo et al., 2012; Yang et al., 2005). pTEFb association with the Nos2 promoter is unaffected by BET inhibition (Wienerroither et al., 2014), so recruitment of pTEFb to the Nos2 promoter takes place by a different mechanism.
The kinase module of the mediator provides an alternative platform for pTEFb recruitment. The mediator is a multi-subunit protein complex that bridges transcription factors with Pol II and initiation and elongation factors (Conaway and Conaway, 2013; Malik and Roeder, 2010). Association with the kinase module containing the subunits MED12, MED13, cyclinC (CcnC), and CDK8 is dynamic and influenced by transcription factors interacting with the mediator core (Conaway and Conaway, 2013; Donner et al., 2010; Ebmeier and Taatjes, 2010; Malik and Roeder, 2010). The presence of the kinase module permits mediator association with transcriptional cofactors such as pTEFb (Donner et al., 2010; Ebmeier and Taatjes, 2010). The MED26 subunit has also been proposed to play a part in pTEFb binding. Takahashi and colleagues co-purified pTEFb with a complex containing MED26 and subunits shared with the SEC. The results suggest that the MED26-containing complex exchanges promoter-bound TFIID for pTEFb (Takahashi et al., 2011).
The interaction of the mediator and its kinase module with STATs has been little studied (Jamieson et al., 2012). CDK8 has recently been shown to regulate the activity of STAT1 dimers (Bancerek et al., 2013). Serrat et al. (2014) found LPS to enrich CDK8 at the Nos2 promoter, an effect enhanced by histone deacetylase (HDAC) inhibition. Because HDAC inhibitors suppress Nos2, the authors propose that CDK8 negatively regulates Nos2.
We used chromatin immunoprecipitation sequencing (ChIP-seq) analysis to identify other genes that are subject to regulation by both NF-κB and ISGF3 in Lm-infected cells. We examined the role of the STAT and NF-κB pathways in the modification of promoter chromatin and the modification of Pol II. We report essential roles for different portions of the mediator complex in the NF-κB-dependent deposition of pTEFb and in the ISGF3-dependent recruitment of Pol II. Our results provide a mechanistic explanation for the interaction between the STAT and NF-κB pathways in the response to infection.
Results
Genome-wide Search for Genes Co-regulated by the STAT and NF-κB Pathways
A recent microarray analysis indicated a significant number of genes synergistically induced by IFN-I/STAT and other pathways (Farlik et al., 2010). To further define this group of genes under the experimental conditions used throughout this study, bone marrow-derived macrophages (BMDMs) were treated with heat-killed Listeria (hkL, non-STAT pathways activated), IFN-I alone (only the IFN-I/STAT pathway activated), or both stimuli together. Genes induced under these conditions in microarrays were filtered for a synergy factor >2; i.e., a more than 2-fold higher induction with both stimuli over the addition of each stimulus alone. 42 genes were identified, fulfilling this criterion (Table S1), and examples of these were validated by qPCR (Figure 1B). To determine whether synergistic induction with IFN-I reflected functional cooperativity with the NF-κB pathway, BMDMs were subjected to the same treatment, and chromatin immunoprecipitates with STAT1, NF-κB p65, or Pol II antibodies were sequenced. Around 1,200 genes had STAT1 and NF-κB bound above background levels within 50 kB of their annotated transcription start sites (TSSs). A comparison with the microarray analysis is shown in Figure 1C. 127 “synergy genes” with NF-κB and STAT1 binding sites were defined by ≥2-fold Pol II binding within the 50-kb promoter region after hkL/IFN-I co-treatment compared with single treatment. 36 of these contained NF-κB and STAT1 peaks within 500 base pairs (bps) upstream of the TSS, such as the IL6 gene (Figure S1). 11 genes (blue segment in Figure 1C; Nos2, Il6, Ccl7, Ccrl2, Cd83, Edn1, Il10, Il1rn, Il27, Slamf1, and Slc28a2) showed a microarray synergy factor of >2, and 28 genes (orange segment) showed synergy factors between 1- and 2-fold. 22 genes showed no added value of the co-treatment (Figure 1C, green segment), and 11 responded to one stimulus only (white segment). Together, the data define a group of genes under the synergistic control of the STAT and NF-κB pathways.
Figure 1.

Genes Co-regulated by Type I Interferons and the NF-κB Pathway in Macrophages Exposed to Heat-Killed Listeria and Type I IFN
(A) Scheme depicting the scientific question.
(B) BMDMs were treated with hkL, IFNβ or a combination of both for 4 hr. Gene microarrays were performed, and selected genes showing synergistic induction by both stimuli were validated by qPCR.
(C) The distribution of 127 genes that were synergistically induced by hkL and IFN-I according to ChIP-seq Pol II signals with respect to fold change in microarray analysis. Values represent means ± SE of three or more independent biological replicates.
∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
A search for overrepresented short motifs (Bailey et al., 2006) in genomic regions enriched by ChIP with an NF-κB-reactive antibody revealed as best hit a motif corresponding closely to the NF-κB binding motif in the JASPAR database (Figure 2B; Sandelin et al., 2004). For the STAT1-reactive antibody, the best hit corresponded to the ISGF3 binding motif (interferon stimulation response element [ISRE]; Figure 2C). Among genes with significant STAT1 peaks in the 50-kb upstream region (including gene coordinates), we found slightly more γ interferon activation sites (GASs) than ISRE motifs (59% and 53%, respectively). Among genes with significant STAT2 peaks, we also found 59% with GAS sites but more with ISRE sites (65%).
Figure 2.

Global Analysis of STAT1 and NF-κB Binding and Detection of Novel Binding Sites in the Nos2 Upstream Region
(A) Scheme depicting the scientific question.
(B and C) Motif analysis of genomic sequences ChIPed (enriched) with (B) NF-κB p65 and (C) STAT1 antibodies. Shown at the bottom are the web logos of de novo computed motifs by DAVID from our ChIP-seq data. Shown at the top are the web logos of motifs present in the JASPAR database. The p value is the probability that the match between the two web logos occurred by random chance according to the null model, as computed by TOMTOM.
(D) Co-occurrence of STAT1 and NF-κB binding sites within 50 kB of TSSs of all genes (left), genes not synergistically induced by hkL/IFNβ cotreatment (center), or genes with synergistic cooperativity according to ChIP-seq pol II signals (right).
(E) BMDMs were treated with hkL, IFNβ, or a combination of both for 4 hr. For STAT1, ChIP treatment was with IFNβ for 2 hr. Cells were processed for ChIP with the indicated antibodies (Pol II, NF-κB p65, and STAT1), followed by next-generation sequencing of the precipitate. Genome browser views of the ChIP-seq results obtained for Nos2 are shown with a scale of mapped reads. Shaded areas indicate the promoter-proximal region and distal enhancer-containing NF-κB and IFN response elements. Asterisks mark ChIP binding sites with coinciding NF-κB or ISRE consensus sequences as identified by motif search algorithms. For detailed information, see text.
(F–H) BMDMs were treated for 4 hr with a combination of hkL and IFNβ (F) or for 2 hr with IFNβ (G and H). NF-κB p65 (n = 3) (F), STAT1 (n = 3) (G), and STAT2 (n = 5) binding (H) to proximal promoter regions containing the NF-κB site or the IFN-responsive regions or the shaded distal region indicated in Figure 2E was measured by ChIP. In the distal region, a 240-bp fragment (chr11: 78886608–78886847) containing ChIP peaks for both NF-κB and STAT1 was amplified.
Values represent mean and SE of at least three independent biological replicates. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ns, not significant.
A genome-wide analysis of STAT1 and NF-κB peaks in genes and the relationship to transcriptional activity is shown in Figure 2D and Table S2A. Venn diagrams indicate the individual or combined occurrence of STAT1 and NF-κB binding sites within 50 kB upstream of a TSS, either for all genes (Figure 2D, left), those that do not show synergy of Pol II binding after hkL + IFNβ treatment (center), and those that show synergistic Pol II association (right). Of the genes that are synergistic, 42% have NF-κB peaks. The majority of these (78%) also contain STAT1 peaks, a highly significant overrepresentation (p = 1.97e-6; Table S2B). The fraction of genes with STAT1 peaks in this group is larger (65%), in keeping with the dominance of the Jak-STAT pathway in the transcriptional response to IFN. Similarly, synergistic genes are significantly enriched for peaks of both NF-κB and STAT1 jointly compared with all other genes (p = 2.2e-16; Table S2C).
Surprisingly, binding of both NF-κB and STAT1 to the Nos 2 promoter sites mapped previously by transient transfection (Kleinert et al., 2004) and ChIP (Farlik et al., 2010) was very weak. This includes an NF-κB site at −86/−76 and an IFN response region with imperfect ISRE sequences at −952/−940 and −924/−911 with respect to the TSS (contained within the Chr11:78915 kb–78925 kb region in Figure 2E; the TSS corresponds to nucleotide 78920787). Stronger binding of both NF-κB and STAT1 occurred approximately 30 kb upstream of the Nos2 TSS (chr11:78870 kb–78890 kb). A search for NF-κB and STAT binding sites in the Nos2 gene body and 50 kB upstream sequence based on position weight matrices from the JASPAR database revealed consensus NF-κB sequences slightly downstream of the TSS and two peaks in the distal enhancer (marked with an asterisk in Figure 2E). Furthermore, this algorithm identified ISRE consensus sequences in two STAT1 peaks of the distal enhancer (see asterisks). At the stringency set for this search, neither the upstream NF-κB element nor the putative ISRE sequences at −952/−940 and −924/−911 showed up as consensus elements, consistent with weak STAT1 binding. Furthermore, two consensus ISREs corresponded to regions without any apparent STAT1 association. Likewise, NF-κB consensus elements without corresponding ChIP-seq peaks were found.
NF-κB, STAT1, and STAT2 binding to the distal enhancer was confirmed by site-directed ChIP and found to be stronger than binding to the promoter-proximal sites indicated above (Figures 2F–2H). The region amplified in the distal enhancer is within the shaded area of Figure 2E and contains both NF-κB and STAT1 ChIP-seq peaks (corresponding to chr11:78886608–78886847 or −34179/−33939 with respect to the TSS). Interestingly, the IL6 promoter also contained a far upstream enhancer with NF-κB and STAT1 binding sites (29940 kb–29950 kb in Figure S1), in agreement with a recent report (Qiao et al., 2013). The control ISG Mx2 showed no p65 binding but a strong STAT1 peak, whereas the NF-κB target control Nfκbia showed a prominent promoter-proximal p65 binding site but no binding of STAT1 (Figure S1).
Gene ontology (GO) analysis was performed using DAVID (Jiao et al., 2012) on the 127 STAT1/NF-κB-p65 synergy genes. Significantly enriched (p < 0.05) GO terms clustered into the following categories: innate immune response, adaptive immune response, apoptosis, regulation of immune response, part of the extracellular region, and cytokine/chemokine activity (Table S3). Analysis of the same gene group for Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways produced the Jak-STAT pathway as the second-most overrepresented pathway (Table S3), with many other genes being clearly linked to pathways relevant for innate immunity. In summary, these findings indicate that the interaction between ISGF3 and NF-κB in transcriptional control of functionally interlinked genes supports cell-based innate immunity to bacterial infection.
Histone 4 Acetylation at the Nos2 Promoter Occurs via NF-κB-Dependent HAT Recruitment
The Nos2 gene was further analyzed as a paradigm for the interplay of the IFN-I and NF-κB pathways in transcriptional regulation. Critical findings were confirmed with the IL6 gene, another member of the “synergy group.”
Our first aim was to analyze histone modifications of active promoters. First, global histone 4 acetylation (H4ac) after Lm infection in nucleosomes proximal to the TSS was examined (Figure 3B). Acetylation of histone 4 lysine 5 (H4K5), histone 4 lysine 8 (H4K8), histone 4 lysine 12 (H4K12), and histone 4 lysine 16 (H4K16) occurred after treatment of BMDMs with hkL alone (solid lines in Figure 3C) and was reduced by the simultaneous inhibition of NF-κB activation by the IKKβ inhibitor BI605906 (Figure 3C, dashed lines). To identify histone acetyltransferases (HATs) responsible for proximal promoter acetylation, hkL-induced binding of PCAF, CBP, and GCN5 was analyzed. PCAF and GCN5 showed very low enrichment, whereas robust, BI605906-sensitive recruitment was observed for CBP (Figure 3D). The data demonstrate NF-κB-dependent CBP recruitment at the Nos2 and Il6 (Figure S2A) genes. They favor the hypothesis that active chromatin at these genes in hkL-treated macrophages requires CBP activity, not ruling out a contribution of other HATs.
Figure 3.

Recruitment of Histone Modifiers and the Establishment of Histone Modifications by Heat-Killed Listeria and IFNβ
(A) Scheme depicting the scientific question. HMT, histone methyl transferase.
(B) BMDMs were infected with Lm for the indicated time, followed by ChIP with antibodies to acetyl-H4.
(C) BMDMs were treated with hkL for the indicated time in the presence (dashed line) or absence (solid line) of the IKKβ inhibitor BI605906 (10 μM), followed by ChIP with antibodies to the acetylated H4 lysines H4K5ac, H4K8ac, H4K12ac, and H4K16ac and amplification of the proximal Nos2 promoter, including the TSS.
(D) BMDMs were treated as described in (C) for 4 hr, followed by ChIP with antibodies to the histone acetyl transferases CBP, PCAF, and GCN5 (n = 3). untr., untreated.
(E) BMDMs were infected with Lm for the indicated time (n = 2). Deposition of the H3K4me3 mark at the distal and proximal Nos2 promoter was determined by ChIP.
(F and G) BMDMs were transfected with the indicated siRNAs to inhibit the expression of essential subunits of the Set1A-B, MLL1-2, or MLL3-4 methyltransferase complexes. Transfected cells were treated with hkL + IFNβ for 4 hr. The cells were assayed for synthesis of Nos2 mRNA by qPCR (n = 2) (F) and for the recruitment of Menin and Pol II or the establishment of the H3K4me3 mark by ChIP (n = 3) at the Nos2 promoter (G). The data in (G) represent percent binding compared with cells transfected with control (contr.) siRNA. H3 binding was measured as a negative control.
(H–J) BMDMs were treated with hkL or IFNβ for 4 hr. H3K4me3 (n = 2) (H), Menin (n = 2) recruitment (J), and H2B ubiquitination (n = 2) (K) were determined by ChIP. All ChIP experiments with antibodies to a histone modification were additionally normalized to total histone 3 levels.
Values represent mean and SE of at least three independent biological replicates. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
NF-κB Signaling Causes the Appearance of the H3K4me3 Chromatin Mark at the Proximal Nos2 Promoter
Another hallmark of transcriptionally active genes is the deposition of a histone 3 lysine 4 trimethylation (H3K4me3) mark at nucleosomes close to the TSS. Reportedly, the Taf3 subunit binds to H3K4me3 to aid TFIID binding (Lauberth et al., 2013; Vermeulen et al., 2007). Lm infection induced H3K4me3 proximal to the Nos2 TSS and, weaker, at the proximal IFN response region (Figure 3E). Mammalian H3K4me3 methylase complexes are distinguished by, respectively, WDR82 (Set1 A-B/COMPASS-like), Menin (MLL1-2/COMPASS-like), or PTIP subunits (MLL3-4/COMPASS-like; Shilatifard, 2012; Smith et al., 2011). Small interfering RNA (siRNA)-mediated knockdown of individual subunits revealed a prominent role of the Menin-containing complex for transcriptional induction of Nos2 and Il6 by hkL/IFNβ treatment (Figure 3F; Figure S2B). In contrast, expression of the NF-κB-independent ISGF3 target Mx2 was suppressed by WDR82 siRNA but not by Menin targeting (Figure S2C), suggesting activity of Set1 A-B/COMPASS-like methyl tranferases. Menin siRNA decreased both Menin binding and the H3K4me3 mark at the proximal Nos2 and IL6 promoters (Figure 3G; Figures S2D and S2E). Consistently, the appearance of Menin and H3K4me3 showed similar kinetics, and both were stimulated by hkL but not by IFNβ treatment (Figures 3H and 3I).
Monoubiquitination of H2B (H2Bub) is a frequent prerequisite for H3K4 trimethylation (Soares and Buratowski, 2013). Like Menin and H3K4me3, this modification was established by hkL treatment alone, suggesting that the NF-κB pathway mediates both H2Bub and H3K4me3 modifications (Figure 3J). The data therefore suggest that NF-κB recruits the machinery for Nos2 and Il6 H3K4 trimethylation.
Mediator Kinase Module Recruits CDK9 to the Nos2 Promoter
pTEFb/CDK9 association with the Nos2 promoter is Brd4-independent but requires NF-κB (Wienerroither et al., 2014). To identify the pTEFb/CDK9 recruitment mechanism, pTEFb interaction with CDK8 and the mediator kinase module was examined (Donner et al., 2010). In BMDMs, binding of CDK9 or of CDK8 and its regulatory subunit CcnC to a region encompassing the Nos2 (Figures 4B–4E) or IL6 (Figure S3) TSS followed the same pattern. It was stimulated by hkL, not by IFNβ, and both stimuli together produced a slight, statistically insignificant increase over hkL alone (Figures 4B–4D; Figure S3). CDK8 binding was sensitive to IKKβ inhibition (Figure 4E) and largely unperturbed by Stat1 deficiency (Figure 4F). CDK8 binding to the Il6 gene was less robust but occurred under the same conditions (Figure S4A). Consistent with the presence of these proteins in a common complex, CDK8 and CDK9 were co-immunoprecipitated from BMDM lysates, particularly after hkL/IFNβ treatment (Figure 4G). The lack of interaction between CDK7 and CDK8 is shown as a control.
Figure 4.

CDK9 Recruitment by the Mediator Kinase Module
(A) Scheme depicting the scientific question.
(B–D) BMDMs were treated with hkL or IFNβ or a combination of both, followed by ChIP analysis with antibodies to CDK9 (B), CDK8 (C), or CcnC (D).
(E and F) WT and Stat1−/− BMDMs were treated with hkL, IFNβ, or a combination of both, for 4 hr with and without 10 μM IKKβ inhibitor BI605906 as indicated, followed by ChIP analysis with antibody to CDK8 and qPCR for the proximal Nos2 promoter, including the TSS.
(G) BMDMs were treated for 2 or 4 hr with hkL + IFNβ, followed by cell lysis and precipitation for CDK8 protein. Samples were loaded on an SDS-PAGE gel, and antibodies to CDK7 and CDK9 were used in a western blot to analyze the interaction with CDK8.
(H–K) Knockdown of MED18 or CcnC was performed in BMDMs. (H) The cells were treated with hkL and IFNβ for 4 hr, followed by mRNA extraction and qPCR for Nos2 expression. Additionally, cells were analyzed by ChIP with antibodies to CDK9 (I), pS2Pol II (J), or Pol II (K). Enrichment fold values at the proximal Nos2 promoter for the control siRNA samples were as follows: CDK9, 2.7; pS2Pol II, 3.31; Pol II, 4.1.
Values represent means ± SE of three or more independent biological replicates. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
CcnC siRNA and, as a control, MED18 siRNA were used to verify the importance of the CDK8/CcnC pair for CDK9 recruitment. Both CcnC and MED18 siRNAs caused an approximately 50% reduction of Nos2 mRNA induction by combined hkL/IFNβ treatment (Figures 4H and S4B). Consistent with a role of CDK8/CcnC in CDK9 recruitment, CcnC siRNA, but not MED18 siRNA, reduced CDK9 association with Nos2 in hkL/IFNβ-treated BMDMs (Figure 4I). Accordingly, CcnC knockdown specifically abrogated S2 phosphorylation of the Pol II CTD (Figure 4J). By contrast, MED18 siRNA, but not CcnC siRNA, reduced the binding of Pol II to the Nos2 TSS (Figure 4K). The results obtained for the IL6 gene were in agreement with those for Nos2 (Figures S4C–S4E). Our data support the notion that the mediator kinase module is required for pTEFb association with the Nos2 and Il6 promoters. They are in line with recent findings showing a requirement of CDK8 for pTEFb recruitment, but not CDK7 (TFIIH) recruitment, at hypoxia-inducible genes (Galbraith et al., 2013).
Concerted Action of the STAT and NF-κB Pathways in Mediator Recruitment
Our results show that NF-κB recruits histone-modifying enzymes, Pol II kinases, and the mediator kinase module. By contrast, IFN-I and ISGF3 are necessary for TFIID and Pol II binding (Farlik et al., 2010). The STAT1 as well as STAT2 subunits of the ISGF3 complex interact functionally and physically with the mediator (Lau et al., 2003; Zakharova et al., 2003). Because the mediator bridges the initiation complex with transcriptional activators, we examined the contribution of the STAT and NF-κB pathways to its association with the Nos2 promoter.
We first determined the presence of the MED1 and MED26 subunits because these do not always co-purify with the core mediator (Malik and Roeder, 2010). Particularly MED26-containing mediators are suggested to function without the kinase module, possibly as an alternative recruitment mechanism for pTEFb (Conaway and Conaway, 2013; Donner et al., 2010; Takahashi et al., 2011). Both MED1 and MED26 associated with the Nos2 promoter in response to stimulation of BMDMs with hkL/IFNβ (Figures 5B and 5D). Less MED1 binding occurred after stimulation with hkL and none after IFNβ treatment alone. Binding was sensitive to IKKβ inhibition (Figure 5B) but resisted Stat1 deficiency (Figure 5C). The IL6 gene showed a similar behavior toward MED1, although binding was less robust (Figure S4F). MED26 bound weakly to the Nos2 TSS after IFNβ treatment alone, whereas induction by hkL was marginal. MED26 resisted IKKβ inhibition but was entirely lost in Stat−/− macrophages (Figures 5D and 5E). STAT1-dependent MED26 recruitment, therefore, differs strikingly not only from MED1 but also from the conditions of pTEFb/CDK9 promoter binding (Figure 4F), ruling out a role of MED26 in tethering pTEFb to the Nos2 promoter.
Figure 5.

Association of the Mediator Subunits MED1 and MED26 with the Nos2 Promoter
(A) Scheme depicting the scientific question.
(B–E) WT or Stat1−/− BMDMs were treated with hkL, IFNβ, or a combination of both with or without 10 μM IKKβ inhibitor for 4 hr. The cells were processed for ChIP with antibodies to MED1 (B and C) or MED26 (D and E). Enrichment at the proximal Nos2 promoter/TSS was analyzed by qPCR. Values represent means ± SE of four or more independent biological replicates. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
The data suggest that Lm signaling recruits a mediator, including MED1, MED26, and the kinase module, to the Nos2 promoter through NF-κB-ISGF3 cooperativity. To determine whether this extends to subunits of the core mediator, binding of the tail subunit MED5/24 was examined. As for MED26 recruitment, binding was lost in Stat1−/− cells but maximally induced with STAT and NF-κB signaling together (Figures 6B and 6C). Similarly, binding of MED18, a subunit of the mediator head, required STAT1 (Figures 6D and 6E). At the IL6 promoter, MED18 binding displayed a similar behavior although less dependence on NF-κB signaling. Again, enrichment was less robust as that seen for Nos2 (Figures S4G and S4H).
Figure 6.

Nos2 Promoter Binding of the Core Mediator
(A) Scheme depicting the scientific question.
(B–L) WT, Stat1−/−, or Stat2−/− BMDMs were treated with hkL, IFNβ, or a combination of both with or without 10 μM IKKβ inhibitor BI605906 for 4 hr. The cells were processed for ChIP with antibodies to MED5/24 (B, C, and K), MED18 (D, E, and J), MED4 (F and G), MED7 (H and I), or CDK8 (L). Enrichment at the proximal Nos2 promoter/TSS was analyzed by qPCR. Values represent means ± SE of four or more independent biological replicates. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
The subunits of the mediator middle domain, MED4 and MED7, differed from the head and tail domains by their stronger dependence on NF-κB for binding to the Nos2 TSS. Association was stimulated by hkL treatment alone and reduced by the IKKβ inhibitor (Figures 6F and 6H). However, input from IFN signaling was confirmed in Stat1−/− cells and by a low level of MED4 and MED7 association after IFNβ treatment alone (Figures 6F–6I).
Mediator core recruitment to the Nos2 promoter in Stat2−/− macrophages was essentially as in Stat1−/− cells. The MED18 and MED5/24 subunits were missing, but the kinase/CDK8 module was present after hkL/IFNβ treatment (Figures 6J–6L). The simultaneous presence of the mediator and ISGF3 at the Nos2 promoter was ascertained by ChIP-reChIP analysis (Farlik et al., 2010; Figure S5). Both STATs were re-precipitated from MED18/MED5/24 ChIPs and, vice versa, the MED subunits from STAT1/2 ChIPs.
Mx2 and IκBα were used, respectively, as IFNβ/ISGF3 and hkL/NF-κB control genes (Figure S6). Unlike Nos2 and IL6, all tested mediator subunits associated with their respective stimulus alone, although CDK8 at the Mx2 promoter was increased further by combined hkL treatment. Our data support the idea that genes showing NF-κB/ISGF3 cooperation require both transcription factors to associate with a complete mediator complex.
Discussion
Higher eukaryotes must be able to respond rapidly to changing environmental conditions or challenges such as invading microorganisms. Transcriptional gene activation is an indispensable component of the complex cellular response to microbial infection. We studied the case of Lm infection, focusing on transcriptional synergism by NF-κB and ISGF3, transcription factors at the endpoints of two major antimicrobial pathways. Our work reveals that different portions of the mediator complex are involved in the NF-κB-dependent binding of pTEFb and in the ISGF3-dependent recruitment of Pol II to gene promoters. We are therefore able to provide a mechanistic explanation for the interaction between the two pathways (Figure 7).
Figure 7.

Model Describing the Regulation of the Nos2 Gene in Listeria monocytogenes-Infected Macrophages by the STAT and NF-κB Pathways
NF-κB association with the Nos2 promoter is the initial step and delivers histone-modifying enzymes (CBP and MLL1/MLL2 COMPASS-like) that produce transcription-friendly chromatin. NF-κB also recruits TFIIH/CDK7 and pTEFb/CDK9, the kinases required for Pol II phosphorylation. NF-κB-dependent stable binding of TFIIH/CDK7 is achieved with the help of the BET protein Brd4. pTEFb/CDK9 is recruited to the promoter via the mediator kinase module, which, like MED1, associates in complete dependence on NF-κB. By contrast, ISGF3 binding is essential for the formation of a pre-initiation complex, particularly the binding of TFIID. ISGF3 also provides a critical contribution to the acquisition of the core mediator and Pol II. For a further explanation, see the text and accompanying references.
In the early phase of infection, before IFN-I is produced, NF-κB delivers histone-modifying enzymes that produce transcription-friendly chromatin. Although the strongest acetyl transferase signal was obtained for CBP binding, an involvement of other HATs cannot be excluded from these findings. NF-κB also manages the acquisition of the Pol II CTD kinases (Farlik et al., 2010), and Brd4 helps stabilize binding of TFIIH/CDK7 to the promoter of the iNOS gene (Wienerroither et al., 2014). pTEFb/CDK9 are brought in via the mediator kinase module (Figures 4G–4K). At a later stage, signaling by the IFN-I receptor causes ISGF3 binding, which is essential for the acquisition of the core mediator (Figure 6), TFIID (Farlik et al., 2010), and Pol II (Farlik et al., 2010). Interestingly, recruitment of pTEFb/CDK9 by NF-κB draws upon the mediator kinase module before the core is recruited with the help of ISGF3. Deposition of the kinases needed for both promoter clearance and elongation prepares Nos2-coregulated promoters for immediate firing in response to the second activating signal, which is provided by IFN-I/ISGF3. It seems unlikely that such a sophisticated mechanism would be unique to a single gene (Nos2). Indeed, we confirmed key results with the Nos2-like IL6 gene.
A detailed examination of the chromatin modifications associated with transcriptionally active chromatin revealed H4 acetylation and H3K4me3 at nucleosomes proximal to the TSS. The modifications are the result of the modifying enzyme CBP and the MLL1-2/COMPASS-like complexes, which associate with the Nos2 promoter in the absence of IFN-I. Menin/MLL1-2 methyltransferase complexes are known to regulate a number of cellular genes, including Hox genes and other genes important for development (Wang et al., 2009). We now show that MLL1-2 also has a role in the innate immune response.
The H2Bub and H3K4me3 marks at the Nos2 and Il6 genes appear prior to the ISGF3-dependent recruitment of Pol II and depend solely on the NF-κB pathway. In contrast, initial recruitment of the yeast complex containing the Set H3K4 methyltransferase occurs by the Pol II-associated PAF complex, and transcription is required for H2B ubiquitination as well as for H3K4me3 catalysis. Reportedly, H3K4 trimethylation enhances transcriptional initiation through interaction with the TFIID complex (Lauberth et al., 2013; Vermeulen et al., 2007). It is attractive to speculate that NF-κB-dependent H3K4 trimethylation supports the ISGF3-dependent recruitment of TFIID (Farlik et al., 2010), which would represent a further example of crosstalk between the NF-κB and IFN pathways.
Phosphorylation of the Pol II CTD is essential for gene transcription. In Lm-infected cells, the Nos2 promoter is occupied by the TFIIH and pTEFb complexes as a result of NF-κB signaling (Farlik et al., 2010; Wienerroither et al., 2014). CTD phosphorylation at both S5 and S2 occurs rapidly after promoter association of Pol II, which depends on ISGF3. Although TFIIH persistence requires the BET protein Brd4, pTEFb/CDK9 binding is independent of Brd4 (Wienerroither et al., 2014) because the mediator kinase module, particularly its CcnC/CDK8 subunits, is able to recruit pTEFb to the Nos2 and IL6 promoters. Our conclusion is based on the following three lines of evidence: the recruitment conditions and kinetics of CcnC/CDK8 and CDK9 are strikingly similar; antibody-mediated co-purification of CDK8 and CDK9 is possible in untreated macrophages and enhanced after stimulation with hkL/IFNβ; and siRNA-mediated knockdown of CcnC selectively suppresses CDK9 binding and S2 phosphorylation of the Pol II CTD. The data are consistent with observations of increased CDK8 association in LPS-treated macrophages (Serrat et al., 2014) and with previous findings on serum-responsive genes (Donner et al., 2010; Ebmeier and Taatjes, 2010).
The MED26 subunit of the mediator has been implicated in the recruitment of pTEFb to promoters (Takahashi et al., 2011). However, the conditions under which MED26 associated with the Nos2 promoter were different from those under which pTEFb associated. In addition, MED26 is believed to act by the exchange of TFIID and pTEFb, which is not compatible with the deposition of pTEFb by the NF-κB pathway independently of ISGF3-mediated TFIID association. It appears, therefore, that the kinase module alone suffices for pTEFb binding. Nevertheless, recruitment of MED26 by IFN-I may partially explain the enhancement of NF-κB-mediated CDK8 binding by the cytokine.
MED26 is not the only subunit of the core mediator to depend on IFN-I for recruitment to the Nos2 and Il6 promoters. Recruitment of all core subunits we investigated also requires IFN-I and is, therefore, generally affected by Stat1 and Stat2 deficiency. For example, MED5/24 and MED18 are not detected at the Nos2 promoter in Stat1-deficient macrophages treated with a combination of hkL and IFN-I, and a smaller amount of MED4 binds the Nos2 TSS under these conditions. Consistent with the requirement for STAT1/2, subunits of the mediator core were induced to various degrees to bind the Nos2 promoter after IFN-I treatment alone. The subunits of the middle portion of the mediator, MED4 and MED7, revealed a significant input of NF-κB signaling and were partially recruited by hkL treatment alone. The data show that ISGF3 and NF-κB interact to assemble a complete mediator at the Nos2 TSS, with recruitment of the kinase module and MED1 completely dependent on NF-κB and recruitment of other subunits requiring STAT1, with various degrees of input from NF-κB signaling.
Two mechanistic interpretations could account for the dual requirement for ISGF3 and NF-κB. A preassembled mediator might require tethering by both transcription factors, or the mediator might be only partially preformed and require assembly by concerted action in a promoter-specific context. The latter interpretation is supported by studies showing that the composition of the mediator is determined by the interacting transcription factors (Ebmeier and Taatjes, 2010; Poss et al., 2013) and that about 30% of CDK8 exists independent of the core mediator complex (Knuesel et al., 2009). It is further consistent with a recent study in yeast that suggested a modular character of the mediator complex (Paul et al., 2015).
Our mechanistic studies of the Nos2 and Il6 promoters are relevant to a large number of genes. ChIP-seq targeting STAT1, NF-κB, and Pol II has defined 127 genes that are bound by both ISGF3 and NF-κB and that are expressed as a result of synergy between the two pathways. The ChIP-seq data also reveal that the major binding site for NF-κB (and ISGF3) at the Nos2 promoter is not where we and others suspected (Farlik et al., 2010; Kleinert et al., 2004), so our understanding of transcription binding sites to other promoters may also require revision.
We describe here the mechanism for the interaction of ISGF3 and NF-κB at the Nos2 and Il6 promoters and present indications that the synergy between these transcription factors might also take place, with subtle differences in mechanism, at a number of other promoters. It is likely that variations of the mechanism apply also to interactions between other transcription factors. The ISGF3 complex is not the only transcription factor to interact extensively with NF-κB. An in-depth, ChIP-seq-based analysis of virus-infected cells has shown that IRF3 also interacts with NF-κB (Freaney et al., 2013). We suggest that the mechanism we uncovered may represent a widespread method for higher eukaryotes to coordinate the activation of a number of genes, not only in the innate immune response but also to enable rapid and precise responses to changing environmental conditions or to challenges posed by microorganisms.
Experimental Procedures
Reagents
Recombinant IFN-β was purchased from PBL and used at 250 U/ml. The inhibitor BI605906 (a gift from Phillip Cohen) was used at 10 μM.
Bacteria and Infection
Growth of Listeria monocytogenes strain LO28 and infection of cells at MOI 20 was as described previously (Stockinger et al., 2002). Heat-killed Listeria was generated by incubation for 20 min at 70°C.
Mice and Cells
Mice were housed under specific pathogen-free (SPF) conditions. C57BL/6, Stat1−/− (Durbin et al., 1996) and Stat2−/− (Park et al., 2000) mice in a C57BL/6 genetic background were sacrificed between 7–10 weeks of age. BMDMs were obtained by culture of bone marrow in L-cell-derived colony-stimulating factor 1 (Stockinger et al., 2002).
siRNA-Mediated Gene Knockdown
siRNAs (Menin, PTIP, WDR82, MED18, and CcnC; ON-TARGETplus, Dharmacon, 6 pmol) or non-target control siRNA were mixed with Opti-MEM I medium (Gibco) and transfected into BMDMs using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer’s instructions. 48 hr after transfection, the medium was replaced by fresh medium without siRNA. Another 24 hr later, cells were treated as described in the text and figure legends.
Chromatin Immunoprecipitation, Re-ChIP, and ChIP-Seq
ChIP re-ChIP experiments were performed as described previously (Wienerroither et al., 2014). Antibodies are listed in Table S4, and primers for precipitate amplification are listed in Table S5. Unless indicated otherwise, the proximal Nos2 promoter was analyzed by amplification of a 251-bp fragment (gene coordinates Chr11: 78920580–78920830). The proximal IL-6 promoter was analyzed by amplifying a 198-bp fragment encompassing gene coordinates Chr5: 30013385–30013582. 5–10 ng of DNA precipitate was used for the generation of sequencing libraries using the KAPA library preparation kit for Illumina systems. Libraries were quantified with a Bioanalyzer dsDNA 1000 assay kit (Agilent Technologies) and a qPCR next-generation sequencing (NGS) library quantification kit (KAPA). Cluster generation and sequencing was performed with a HiSeq 2000 system with a read length of 100 nucleotides according to the manufacturer’s guidelines (Illumina).
RNA Preparation and qPCR
RNA was isolated using a NucleoSpin RNA II kit (Macherey-Nagel) and quantified using the NanoDrop method (ND1000, Peq lab). Primers for quantitative real-time PCR are given in Table S5. mRNA expression data were normalized to the housekeeping Gapdh gene and calculated relative to the respective unstimulated controls.
Western Blot
Western blots with CDK7 (sc-529) and CDK9 (sc-484) antibodies at a dilution of 1:200 were performed as described previously (Kovarik et al., 1998). For detection, the fluorescence-labeled secondary antibodies IRDye800 and IRDye700 (LI-COR Biosciences) and an infrared imaging system (Odyssey, LI-COR Biosciences) were used.
Microarray Analysis
Total RNA from BMDMs treated with heat-killed Listeria, IFN-β, or both for 2, 4, and 8 hr was collected from three sets of independent experiments and purified with Trizol (Invitrogen) and an RNeasy mini kit (QIAGEN). Microarray analysis was performed by the Genomics Core of the Lerner Research Institute, Cleveland Clinic, using 1 μg of total RNA and Illumina Mouse Ref-8 v2 expression bead chips. The data were normalized by the quantile method. Expression levels from untreated and treated cells were compared, and genes were selected that increased >4-fold (differential p values, <0.05; average signals in treated samples, >50).
ChIP-Seq Analysis
Quality-based trimming was performed at the 3′ end of raw reads using the “trim-fastq.pl” script of the PoPoolation toolbox, where all trimmed reads with a length of less than 40 bp were discarded. Quality-controlled reads were mapped to the mouse genome (UCSC, mm10) using Bowtie (Langmead et al., 2009), where all non-unique alignments were discarded. For post-alignment filtering, reads mapped in a proper pair were selected, and PCR duplicates were removed using SAMtools (Li et al., 2009). Peak calling was performed using MACS (Zhang et al., 2008) version 2.0.10 with default parameters for NF-κB, STAT1, and STAT2 samples and the “–broad” parameter for pol2 samples. A threshold of q < 0.1 was set for calling significant peaks, which were annotated within the nearest 50 kb upstream of genes (including the gene itself) using PeakAnalyzer (Salmon-Divon et al., 2010).
DNA Motif Analysis
Overlapping areas of enrichment are merged by MACS, resulting into much wider peaks than the actual binding sites. With the PeakSplitter module of the PeakAnalyzer tool, these enriched areas were sub-divided by accurately partitioning enriched loci into finer resolution sets of individual binding sites. These sequences were used for de novo motif analysis of the peak summit regions where binding is most probable. De novo motif finding was performed by MEME (Bailey et al., 2006) on all of the significant ChIP-seq binding peaks for NF-κB (HkL and IFNβ treatment) and STAT1 (IFNβ treatment) samples. The parameter “minimum motif width” of MEME was set to 11 for NF-κB and 15 for STAT1 because this is their known motif width from the JASPAR (Sandelin et al., 2004) database. The most significant de novo computed motifs were compared with known motifs present in the JASPAR database with TOMTOM (Gupta et al., 2007). Additionally, we scanned the promoter (50 kb) and gene coordinates of Nos2 for the presence of NF-κB (relA), ISRE (STAT2:STAT1), and GAS (STAT1) motifs, by using their appropriate position weight matrices (PWM) from the JASPAR database. To the frequencies of each site of the PWMs we added one (to avoid zero probabilities) and calculated the probabilities of each of the four bases at each position. Sliding along each position of the DNA sequence of the iNOS gene from 50 kb upstream to the end of the gene, we calculated the fit of a position weight matrix starting at the respective position by summing the log probabilities of the bases from the beginning to the end of the PWM. We extracted fits exceeding a threshold that was specific for each transcription factor.
GO Analysis
Enrichment of GO terms in 127 genes that had STAT1 and NF-κB-p65 binding sites and greater than 2-fold enrichment of Pol II binding in the co-treated sample was computed using DAVID (Jiao et al., 2012). Among the significantly enriched GO terms (EASE score <0.05 according to a modified Fisher’s exact p value), similar terms were grouped with the “Functional Annotation Clustering” module of DAVID, which uses an algorithm to measure the relationship among the annotated GO terms based on the degrees of their co-association genes.
Cooperativity between the Transcription Factors
For the transcription factors NF-κB_STAT1, we computed 2x2 contingency tables according to the presence or absence of at least one peak within the upstream 50-kb region of the focal gene (including the gene coordinates). Furthermore, we divided this dataset into genes that show at least 2-fold synergistic enrichment in Pol II with double treatment versus single treatment samples and those that do not. Various hypotheses were addressed with 2x2 contingency tables and tested with chi-square tests.
Pathway Analysis
The same list of 127 genes was analyzed for overrepresented KEGG pathways using DAVID. Again, an EASE score of less than 0.05 was used as a threshold to filter significantly overrepresented pathways.
Statistical Analysis
Differences between mean values for qPCR results of either mRNA expression or ChIP experiments were analyzed by t test.
Acknowledgments
This paper is dedicated to Karl Decker on the occasion of his 90th birthday. We thank Gijs Versteeg for valuable comments on the manuscript. Christian Seiser and Anna Sawicka are thanked for critical discussions and providing antibodies to modified histones. We gratefully acknowledge Graham Tebb for editing and improving the clarity of our manuscript. Funding was provided by the Austrian Science Fund (FWF) through grant SFB-28 (to B.S., M.M., and T.D.). S.W. and A.M. were supported by the FWF through the doctoral program Molecular Mechanisms of Cell Signaling. Next-generation sequencing was performed at the Vienna Biocenter’s CSF NGS Unit (http://www.csf.ac.at/home/).
Published: July 2, 2015
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Supplemental Information includes six figures and five tables and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2015.06.021.
Accession Numbers
Microarray and ChIP-seq data reported in this paper have been deposited to the ArrayExpress database and are available under accession number ArrayExpress: E-MTAB-3015, E-MTAB-2972.
Supplemental Information
Untreated cells are compared with cells subjected to single IFNβ or hkL treatment or with cells treated with combined hkL + IFNβ.
References
- Bailey T.L., Williams N., Misleh C., Li W.W. MEME: discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res. 2006;34:W369–W373. doi: 10.1093/nar/gkl198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bancerek J., Poss Z.C., Steinparzer I., Sedlyarov V., Pfaffenwimmer T., Mikulic I., Dölken L., Strobl B., Müller M., Taatjes D.J., Kovarik P. CDK8 kinase phosphorylates transcription factor STAT1 to selectively regulate the interferon response. Immunity. 2013;38:250–262. doi: 10.1016/j.immuni.2012.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdan C. Nitric oxide and the immune response. Nat. Immunol. 2001;2:907–916. doi: 10.1038/ni1001-907. [DOI] [PubMed] [Google Scholar]
- Brasier A.R., Tian B., Jamaluddin M., Kalita M.K., Garofalo R.P., Lu M. RelA Ser276 phosphorylation-coupled Lys310 acetylation controls transcriptional elongation of inflammatory cytokines in respiratory syncytial virus infection. J. Virol. 2011;85:11752–11769. doi: 10.1128/JVI.05360-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conaway R.C., Conaway J.W. The Mediator complex and transcription elongation. Biochim. Biophys. Acta. 2013;1829:69–75. doi: 10.1016/j.bbagrm.2012.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donner A.J., Ebmeier C.C., Taatjes D.J., Espinosa J.M. CDK8 is a positive regulator of transcriptional elongation within the serum response network. Nat. Struct. Mol. Biol. 2010;17:194–201. doi: 10.1038/nsmb.1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbin J.E., Hackenmiller R., Simon M.C., Levy D.E. Targeted disruption of the mouse Stat1 gene results in compromised innate immunity to viral disease. Cell. 1996;84:443–450. doi: 10.1016/s0092-8674(00)81289-1. [DOI] [PubMed] [Google Scholar]
- Ebmeier C.C., Taatjes D.J. Activator-Mediator binding regulates Mediator-cofactor interactions. Proc. Natl. Acad. Sci. USA. 2010;107:11283–11288. doi: 10.1073/pnas.0914215107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farlik M., Reutterer B., Schindler C., Greten F., Vogl C., Müller M., Decker T. Nonconventional initiation complex assembly by STAT and NF-kappaB transcription factors regulates nitric oxide synthase expression. Immunity. 2010;33:25–34. doi: 10.1016/j.immuni.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freaney J.E., Kim R., Mandhana R., Horvath C.M. Extensive cooperation of immune master regulators IRF3 and NFκB in RNA Pol II recruitment and pause release in human innate antiviral transcription. Cell Rep. 2013;4:959–973. doi: 10.1016/j.celrep.2013.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galbraith M.D., Allen M.A., Bensard C.L., Wang X., Schwinn M.K., Qin B., Long H.W., Daniels D.L., Hahn W.C., Dowell R.D., Espinosa J.M. HIF1A employs CDK8-mediator to stimulate RNAPII elongation in response to hypoxia. Cell. 2013;153:1327–1339. doi: 10.1016/j.cell.2013.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S., Stamatoyannopoulos J.A., Bailey T.L., Noble W.S. Quantifying similarity between motifs. Genome Biol. 2007;8:R24. doi: 10.1186/gb-2007-8-2-r24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamon M., Bierne H., Cossart P. Listeria monocytogenes: a multifaceted model. Nat. Rev. Microbiol. 2006;4:423–434. doi: 10.1038/nrmicro1413. [DOI] [PubMed] [Google Scholar]
- Jamieson, A.M., Farlik, M., and Decker, T. (2012). How Stats Interact with the Molecular Machinery of Transcriptional Activation, T. Decker and M. Müller, eds. (Springer), pp. 65–89.
- Jang M.K., Mochizuki K., Zhou M., Jeong H.-S., Brady J.N., Ozato K. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol. Cell. 2005;19:523–534. doi: 10.1016/j.molcel.2005.06.027. [DOI] [PubMed] [Google Scholar]
- Jiao X., Sherman B.T., Huang W., Stephens R., Baseler M.W., Lane H.C., Lempicki R.A. DAVID-WS: a stateful web service to facilitate gene/protein list analysis. Bioinformatics. 2012;28:1805–1806. doi: 10.1093/bioinformatics/bts251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T., Akira S. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int. Immunol. 2009;21:317–337. doi: 10.1093/intimm/dxp017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinert H., Pautz A., Linker K., Schwarz P.M. Regulation of the expression of inducible nitric oxide synthase. Eur. J. Pharmacol. 2004;500:255–266. doi: 10.1016/j.ejphar.2004.07.030. [DOI] [PubMed] [Google Scholar]
- Knuesel M.T.M., Meyer K.D.K., Donner A.J.A., Espinosa J.M.J., Taatjes D.J.D. The human CDK8 subcomplex is a histone kinase that requires Med12 for activity and can function independently of mediator. Mol. Cell. Biol. 2009;29:650–661. doi: 10.1128/MCB.00993-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovarik P., Stoiber D., Novy M., Decker T. Stat1 combines signals derived from IFN-γ and LPS receptors during macrophage activation. EMBO J. 1998;17:3660–3668. doi: 10.1093/emboj/17.13.3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Trapnell C., Pop M., Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau J.F., Nusinzon I., Burakov D., Freedman L.P., Horvath C.M. Role of metazoan mediator proteins in interferon-responsive transcription. Mol. Cell. Biol. 2003;23:620–628. doi: 10.1128/MCB.23.2.620-628.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauberth S.M., Nakayama T., Wu X., Ferris A.L., Tang Z., Hughes S.H., Roeder R.G. H3K4me3 interactions with TAF3 regulate preinitiation complex assembly and selective gene activation. Cell. 2013;152:1021–1036. doi: 10.1016/j.cell.2013.01.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy D.E., Darnell J.E., Jr. Stats: transcriptional control and biological impact. Nat. Rev. Mol. Cell Biol. 2002;3:651–662. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Z., Lin C., Shilatifard A. The super elongation complex (SEC) family in transcriptional control. Nat. Rev. Mol. Cell Biol. 2012;13:543–547. doi: 10.1038/nrm3417. [DOI] [PubMed] [Google Scholar]
- Malik S., Roeder R.G. The metazoan Mediator co-activator complex as an integrative hub for transcriptional regulation. Nat. Rev. Genet. 2010;11:761–772. doi: 10.1038/nrg2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancuso G., Gambuzza M., Midiri A., Biondo C., Papasergi S., Akira S., Teti G., Beninati C. Bacterial recognition by TLR7 in the lysosomes of conventional dendritic cells. Nat. Immunol. 2009;10:587–594. doi: 10.1038/ni.1733. [DOI] [PubMed] [Google Scholar]
- McCaffrey R.L., Fawcett P., O’Riordan M., Lee K.-D., Havell E.A., Brown P.O., Portnoy D.A. A specific gene expression program triggered by Gram-positive bacteria in the cytosol. Proc. Natl. Acad. Sci. USA. 2004;101:11386–11391. doi: 10.1073/pnas.0403215101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Riordan M., Portnoy D.A. The host cytosol: front-line or home front? Trends Microbiol. 2002;10:361–364. doi: 10.1016/s0966-842x(02)02401-0. [DOI] [PubMed] [Google Scholar]
- Park C., Li S., Cha E., Schindler C. Immune response in Stat2 knockout mice. Immunity. 2000;13:795–804. doi: 10.1016/s1074-7613(00)00077-7. [DOI] [PubMed] [Google Scholar]
- Paul E., Zhu Z.I., Landsman D., Morse R.H. Genome-wide association of mediator and RNA polymerase II in wild-type and mediator mutant yeast. Mol. Cell. Biol. 2015;35:331–342. doi: 10.1128/MCB.00991-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poss Z.C., Ebmeier C.C., Taatjes D.J. The Mediator complex and transcription regulation. Crit. Rev. Biochem. Mol. Biol. 2013;48:575–608. doi: 10.3109/10409238.2013.840259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao Y., Giannopoulou E.G., Chan C.H., Park S.-H., Gong S., Chen J., Hu X., Elemento O., Ivashkiv L.B. Synergistic activation of inflammatory cytokine genes by interferon-γ-induced chromatin remodeling and toll-like receptor signaling. Immunity. 2013;39:454–469. doi: 10.1016/j.immuni.2013.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmon-Divon M., Dvinge H., Tammoja K., Bertone P. PeakAnalyzer: genome-wide annotation of chromatin binding and modification loci. BMC Bioinformatics. 2010;11:415–427. doi: 10.1186/1471-2105-11-415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandelin A., Alkema W., Engström P., Wasserman W.W., Lenhard B. JASPAR: an open-access database for eukaryotic transcription factor binding profiles. Nucleic Acids Res. 2004;32:D91–D94. doi: 10.1093/nar/gkh012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer J.-D., Sotelo-Troha K., von Moltke J., Monroe K.M., Rae C.S., Brubaker S.W., Hyodo M., Hayakawa Y., Woodward J.J., Portnoy D.A., Vance R.E. The N-ethyl-N-nitrosourea-induced Goldenticket mouse mutant reveals an essential function of Sting in the in vivo interferon response to Listeria monocytogenes and cyclic dinucleotides. Infect. Immun. 2011;79:688–694. doi: 10.1128/IAI.00999-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki E., Tsutsui H., Tsuji N.M., Hayashi N., Adachi K., Nakano H., Futatsugi-Yumikura S., Takeuchi O., Hoshino K., Akira S. Critical roles of myeloid differentiation factor 88-dependent proinflammatory cytokine release in early phase clearance of Listeria monocytogenes in mice. J. Immunol. 2002;169:3863–3868. doi: 10.4049/jimmunol.169.7.3863. [DOI] [PubMed] [Google Scholar]
- Serrat N., Sebastian C., Pereira-Lopes S., Valverde-Estrella L., Lloberas J., Celada A. The response of secondary genes to lipopolysaccharides in macrophages depends on histone deacetylase and phosphorylation of C/EBPβ. J. Immunol. 2014;192:418–426. doi: 10.4049/jimmunol.1203500. [DOI] [PubMed] [Google Scholar]
- Shilatifard A. The COMPASS family of histone H3K4 methylases: mechanisms of regulation in development and disease pathogenesis. Annu. Rev. Biochem. 2012;81:65–95. doi: 10.1146/annurev-biochem-051710-134100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith E., Lin C., Shilatifard A. The super elongation complex (SEC) and MLL in development and disease. Genes Dev. 2011;25:661–672. doi: 10.1101/gad.2015411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares L.M., Buratowski S. Histone Crosstalk: H2Bub and H3K4 Methylation. Mol. Cell. 2013;49:1019–1020. doi: 10.1016/j.molcel.2013.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockinger S., Materna T., Stoiber D., Bayr L., Steinborn R., Kolbe T., Unger H., Chakraborty T., Levy D.E., Müller M., Decker T. Production of type I IFN sensitizes macrophages to cell death induced by Listeria monocytogenes. J. Immunol. 2002;169:6522–6529. doi: 10.4049/jimmunol.169.11.6522. [DOI] [PubMed] [Google Scholar]
- Takahashi H., Parmely T.J., Sato S., Tomomori-Sato C., Banks C.A.S., Kong S.E., Szutorisz H., Swanson S.K., Martin-Brown S., Washburn M.P. Human mediator subunit MED26 functions as a docking site for transcription elongation factors. Cell. 2011;146:92–104. doi: 10.1016/j.cell.2011.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeulen M., Mulder K.W., Denissov S., Pijnappel W.W.M.P., van Schaik F.M.A., Varier R.A., Baltissen M.P.A., Stunnenberg H.G., Mann M., Timmers H.T.M. Selective anchoring of TFIID to nucleosomes by trimethylation of histone H3 lysine 4. Cell. 2007;131:58–69. doi: 10.1016/j.cell.2007.08.016. [DOI] [PubMed] [Google Scholar]
- Wang P., Lin C., Smith E.R., Guo H., Sanderson B.W., Wu M., Gogol M., Alexander T., Seidel C., Wiedemann L.M. Global analysis of H3K4 methylation defines MLL family member targets and points to a role for MLL1-mediated H3K4 methylation in the regulation of transcriptional initiation by RNA polymerase II. Mol. Cell. Biol. 2009;29:6074–6085. doi: 10.1128/MCB.00924-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wienerroither S., Rauch I., Rosebrock F., Jamieson A.M., Bradner J., Muhar M., Zuber J., Müller M., Decker T. Regulation of NO synthesis, local inflammation, and innate immunity to pathogens by BET family proteins. Mol. Cell. Biol. 2014;34:415–427. doi: 10.1128/MCB.01353-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodward J.J., Iavarone A.T., Portnoy D.A. c-di-AMP secreted by intracellular Listeria monocytogenes activates a host type I interferon response. Science. 2010;328:1703–1705. doi: 10.1126/science.1189801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z., Yik J.H.N., Chen R., He N., Jang M.K., Ozato K., Zhou Q. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol. Cell. 2005;19:535–545. doi: 10.1016/j.molcel.2005.06.029. [DOI] [PubMed] [Google Scholar]
- Zakharova N., Lymar E.S., Yang E., Malik S., Zhang J.J., Roeder R.G., Darnell J.E., Jr. Distinct transcriptional activation functions of STAT1alpha and STAT1beta on DNA and chromatin templates. J. Biol. Chem. 2003;278:43067–43073. doi: 10.1074/jbc.M308166200. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W., Liu X.S. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Untreated cells are compared with cells subjected to single IFNβ or hkL treatment or with cells treated with combined hkL + IFNβ.