

Abstract
The alteration of cellular functions by anchoring macromolecules to specified organelles may reveal a new area of therapeutic potential and clinical treatment. In this work, a unique phenotype was evoked by influencing cellular behavior through the modification of subcellular structures with genetically targetable macromolecules. These fluorogen-functionalized polymers, prepared via controlled radical polymerization, were capable of exclusively decorating actin, cytoplasmic, or nuclear compartments of living cells expressing localized fluorgen-activating proteins. The macromolecular fluorogens were optimized by establishing critical polymer architecture-biophysical property relationships which impacted binding rates, binding affinities, and the level of internalization. Specific labeling of subcellular structures was realized at nanomolar concentrations of polymer, in the absence of membrane permeabilization or transduction domains, and fluorogen-modified polymers were found to bind to protein intact after delivery to the cytosol. Cellular motility was found to be dependent on binding of macromolecular fluorogens to actin structures causing rapid cellular ruffling without migration.
Introduction
The modification of cells and proteins with macromolecules has shown significant clinical impact and therapeutic potential.1 Poly(ethylene glycol) modifications, i.e. PEGylation, can inhibit the immune response to foreign proteins and cells, improve biomolecule solubility, increase protein stability and circulation lifetime, and prevent rejection of heterologous transplanted cells.1c Protein-polymer hybrids have been used in numerous applications, including uses as siRNA delivery vehicles, sensitive tags for fluorescence or mass-based detection, and as high-dose drug and radionuclide carriers.1d,2 The effectiveness of these approaches and their ability to alter biological function in living systems is contingent upon the degree of specificity to which these hybrid molecules can be delivered to their target organism or subcellular structure.
Targeting and anchoring of macromolecules onto subcellular structures and organelles has been an ongoing scientific challenge and represents a new frontier in polymer-based engineering of biological systems.3 To date, many effective approaches exist to target macromolecules within the body and to enhance their uptake by specific cell types using various “passive” and “active” targeting ligands.3a Macromolecule conjugated proteins, including antibodies and fragments,4 affibodies,5 nanobodies,6 and recombinant endogenous proteins7 have been effectively used for cellular targeting within tumors and brain tissues. Macromolecules coupled to peptides, sugars, and folate molecules,8 as well as nucleic acid-type targeting ligands (e.g. aptamers)9 have enabled receptor mediated targeting and uptake in specific cells in-vivo. In spite of these accomplishments, successful navigation of complex in vivo environments remains elusive, and targeting of macromolecular structures has been limited to cell-specific targets and cytosolic internalization. To our knowledge, targeting of polymeric materials to specific intracellular structures or subcellular organelles is not yet possible.3c
Fluorogen-activating proteins (FAPs) and their cognate small molecule fluorogens provide a powerful imaging tool uniquely capable of homing small molecule probes to programmed locations within living cells. Fluorogen molecules, when in the presence of FAPs, specifically and non-covalently bind with strong host-guest binding affinities of ca. < 1 nM accompanied by an incipient fluorescent activity.10 In their unbound state, fluorogen molecules are fluorescently inactive which circumvents non-specific background signals and simplifies labeling procedures. A variety of fluorogen molecules have been developed based on unrigidized dyes including thiazole orange,11 dimethylindole red,12 and malachite green (MG).10 Fluorogen molecules can also be coupled with other augmenting molecules to provide supplemental imaging elements (i.e. “tandem-dyes”),13 stimuli-responsive diagnostic properties,14 or to enhance probe performance.15 Among the aforementioned fluorogens, MG based fluorogens have been demonstrated to have excellent binding affinities to FAPs in the pM concentration range and to function within living cells.16 To extend the functionality of this fluoromodule technology, we envisioned that this FAP-fluorogen tool could serve as a self-reporting molecular anchoring system to position macromolecular structures within cells (Figure 1A-B). Similar to polymer-modified proteins in vitro and in vivo, the properties of cellular proteins fused with FAPs could be altered upon polymer binding thereby providing dials to influence cellular behavior (Figure 1C).
Figure 1.
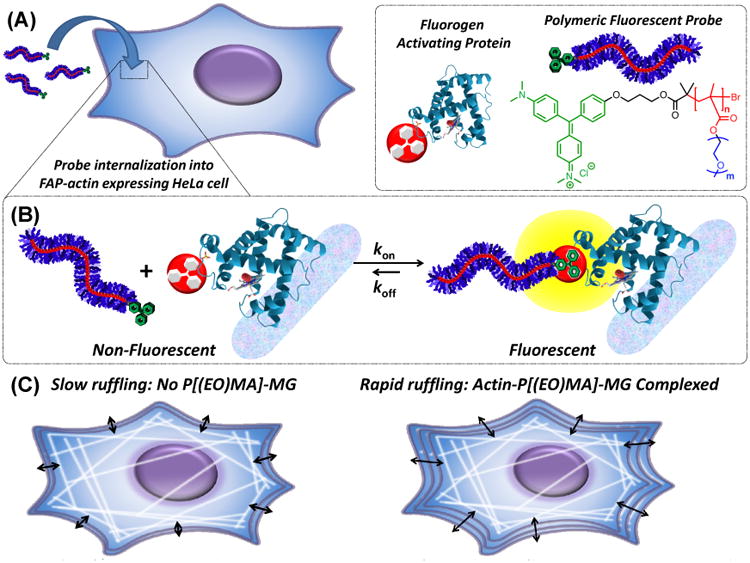
(A) Illustration depicting internalization of a polymer fluorogen into a genetically targetable actin-modified HeLa cell. Legend and schematic breakdown of the polymer fluorogen consisting of MG (green), methacrylate backbone (red) with a DP of “n” repeat units, and ethylene oxide (EO) side-chains (blue) with “m” repeat units. Subsequent data in Figure 3 and 4 are color-coded for clarity, where red figures indicated backbone DP variations and blue EO side-chain variations. (B) Polymeric fluorescent probe disassociated and complexed with a FAP representing its non-fluorescent and fluorescent states, respectively. (C) Illustration of binding polymer fluorogens to FAP-fused actin and their effect on cellular ruffling behavior.
Here, we present the use of a genetically encoded FAP combined with a new macromolecular platform to achieve specific and pre-defined targeting of the same polymeric materials to distinct subcellular structures within living cells (Figure 1). Site-specific targeting was unequivocally demonstrated via live-cell imaging using stable cell lines expressing FAP-fusion constructs within actin, cytoplasmic, and nuclear compartments. Genetically targetable polymers, based on a fluorogen functionalized initiator and a controlled radical polymerization process, have nanomolar FAP binding affinities and cell membrane permeability under physiological conditions (Figure 1A-B). Modification of the actin cytoskeleton via polymer binding shows pronounced effects on macroscopic cellular ruffling (Figure 1C). To our knowledge, this work represents the first instance of a genetically targetable macromolecule that can be specifically delivered to subcellular compartments, visualized, and which perturbs cellular behavior. This approach can reveal details about subcellular targeting of polymeric materials and drug carriers, improve targeting and design of diagnostic and therapeutic biomaterials, and can provide a means to manipulate cellular behavior and function for applications in fundamental and synthetic biology.
Results and Discussion
Prior to evaluating whether the FAP-fluorogen platform could be utilized as a macromolecular homing device, imaging agent, and cellular modification tool; the influence of polymer architecture on fluoromodule properties was first investigated. A series of narrow dispersity fluorogen-functionalized polymers was prepared to determine the effect of side-chain length and polymer backbone length on their biophysical properties when complexed with dL5** protein. In order to establish these critical polymer structure-biophysical property relationships, controlled radical polymerization was employed to produce well-defined macromolecules from an assortment of monomers having pre-determined number-average molecular weights (Mn) with narrow molar-mass dispersity values (ĐM ≤ 1.3).17 In other literature accounts, step-growth polymerization was used to synthesize conjugated polymers having broad dispersity values (ĐM ≥ 1.8) for live cell imaging; however, these systems lacked the required uniformity to extract fundamental polymer structure-biophysical property relationships for improving their performance and were not targeted to specific subcellular components.18
Atom transfer radical polymerization (ATRP) was employed to synthesize well-defined polymeric fluorogens comprised of oligo(ethylene oxide)methacrylate (OEOMA) monomer and a custom MG functionalized ATRP initiator (Malachite Green-Propyl Bromo isoButyrate: MG-PBiB). MG-PBiB was synthesized according to Scheme S1 in the supporting information (SI) to contain a latent MG moiety10 for imaging and an alkyl halide functionality for ATRP. The bromoisobutyrate functionality is known to be an effective initiator for ATRP.19 Furthermore, MG specifically binds with FAPs, in this case dL5**, to generate highly fluorescent complexes.10,15 Ethylene oxide based monomers were selected based upon its polymers inherent water solubility, biocompatibility,20 and effectiveness in penetrating living cells.21
Monomer, initiator, solvent, and a copper-based catalyst (CuII/TPMA, TPMA = Tris(2-pyridylmethyl)amine) were charged into a reactor producing a dark green solution and then polymerization was initiated by injecting SnII(Oct)2 reducing agent into the reaction. All polymerizations were carried out in anisole containing a small amount of DMSO for solubility purposes. Figure 2A shows that ∼75 % of the monomer was consumed within 5 hours and that first-order behavior existed with respect to monomer. Linearity in the first-order kinetic plot confirmed a constant concentration of propagating chain-ends was present throughout the polymerization. Gel permeation chromatography multi-angle laser light scattering (GPC-MALLS, SI Figure S1) was used to characterize the polymer's Mn and ĐM during the progression of polymerization. The linear relationship between the Mn and monomer conversion in Figure 2B confirmed that this was a controlled polymerization process. These results verified that predetermined molecular weight polymers could be synthesized with narrow dispersity values (ĐM ≤ 1.3). After purification, the resulting polymers were observed to be a dark green tacky material as shown in Figure 2A (inset). Each polymer chain was assumed to be uniform owing to the controlled nature of the polymerization and to contain a single end-capped fluorogen moiety because MG was an integral part of the initiator.
Figure 2.
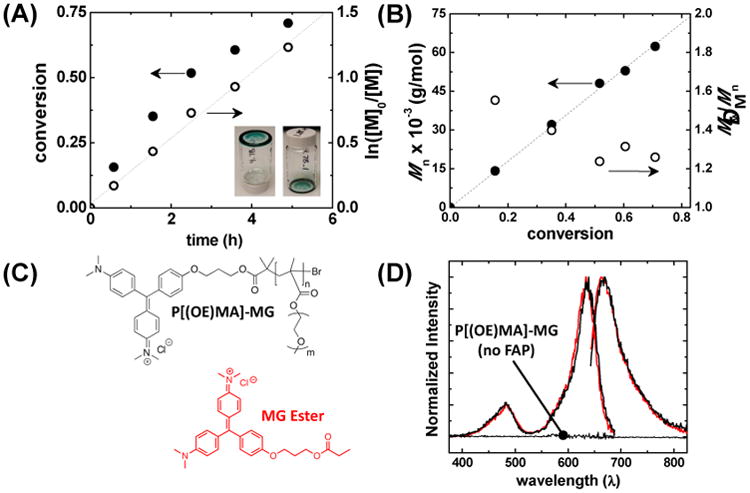
ATRP of OEOMA monomer with MG-PBiB initiator, and spectral properties of MG functionalized P[(EO)8.5MA]372 (black) and MG-Ester (red) in PBS+. (A) Monomer conversion and first-order plot with respect to monomer (M) versus time, and (B) evolution of number-averaged molecular weight (Mn) and ĐM versus monomer conversion. (C) Structures of MG functionalized polymer and MG-Ester. (D) Normalized excitation scans observed at λ = 730 nm and emission scans excited at 636 nm using P[(EO)8.5MA]372-MG in the presence and absence of FAP. Excitation and emission scans of MG-Ester in the presence of FAP are shown in red for comparison. Polymerization conditions: [OEOMA475]0 = 0.339 M in anisole at 60 °C and a total volume of 4 mL. [OEOMA475]:[MG-PBiB]:[TPMA]:[CuIIBr2]:[SnII(Oct)2] = 150:1:0.81:0.27:2.7.
Once polymerization control was established, a range of MG functionalized polymers with varying side-chain lengths and backbone lengths were synthesized, as summarized in Table 1. For clarity, all polymers described in the following discussion will be identified as P[(EO)mMA]n-MG; where subscript “m” represents the number-average of ethylene oxide (EO) side-chain repeat units and subscript “n” represents the number-average of methacrylate backbone repeat units, i.e. the degree of polymerization (DP). A structural depiction of the polymer fluorogen can be found in Figure 1A. Two main categories of P[(EO)mMA]n-MG were synthesized to investigate the influence of (1) overall backbone length (Entry 1-4: DP or n = 66-372) and (2) side-chain length, which were prepared from monomers having varying ethylene oxide (EO) repeat unit lengths (Entry 1,5-8: EO = 2-19.3). Two of the polymers synthesized for the side-chain length study were disregarded from further analysis because of either broad dispersity values (Entry 7: EO = 19.3, Đ = 1.82) or limited solubility in PBS+ buffer (Entry 8: EO = 2). A broad dispersity would reduce the accuracy of subsequent dissociation constant and binding-rate constant measurements owing to the diverse population of polymers, whereas the limited water solubility from short side-chains created analytical difficulties. One additional polymer (Entry 9) was polymerized using a unique MG amide initiator, synthesized according to SI Scheme S2, containing a longer molecular spacer between the MG functionality and the attached P[(EO)8.5MA]204 polymer.
Table 1. Characterization Results of Synthesized Polymeric Fluorogens.
| Entry | Mn | Đ | DP | EO | KD | kON | τdc | Brightness |
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| (#) | (g/mole) | (n) | (m) | (nM) | (M-1*s-1) | (μs) | (kHz) | |
| 1 | 177,400 | 1.26 | 372 | 8.5 | 33 | 1.36E4 | 611 | 6.499 |
| 2 | 71,820 | 1.20 | 150 | 8.5 | 42 | 1.62E4 | 490 | 6.728 |
| 3 | 43,560 | 1.20 | 90 | 8.5 | 29 | 1.89E4 | 363 | 7.052 |
| 4 | 31,970 | 1.40 | 66 | 8.5 | 10 | 3.98E4 | 353 | 7.323 |
| 5 | 49,890 | 1.31 | 164 | 4.5 | 4.7 | 3.14E4 | 404 | 7.573 |
| 6 | 38,750 | 1.33 | 164 | 3.0 | 6.0 | 3.36E4 | 377 | 9.230 |
| 7 | 180,200 | 1.82 | 202 | 19.3 | -- | -- | 532 | 7.678 |
| 8 | 23,130 | 1.44 | 120 | 2.0 | -- | -- | -- | -- |
| 9a | 97,960 | 1.84 | 204 | 8.5 | 1.9 | 3.46E4 | -- | 6.855 |
| MG-Esterb | -- | -- | -- | -- | 0.15 | 8.2E6 | 217 | 7.720 |
MG amide initiator containing a longer linker was used instead of PBiB-MG.
KD and kON values were obtained from J. Mol. Biol. 2013, 425, 4595.
Diffusion correlation time from FCS measurements by fitting autocorrelation curves using a one component 3-D diffusion model.
Initial spectroscopic analysis of P[(EO)8.5MA]372-MG complexed with dL5** FAP revealed nearly identical behavior to the small molecule MG-ester (Figure 2C-D). UV-Vis absorption spectra in PBS+ buffer (SI Figure S2) displayed maximum absorption bands at 463 nm and 616 nm for the MG functional polymer, and 463 nm and 608 nm for the small molecule analog. The slight bathochromic shift of the polymer fluorogen was attributed to its modification of the solvent environment surrounding the dye.22 Fluorescence excitation and emission spectra showed that the polymeric fluorogen was efficiently quenched unless bound to the FAP, whereas when complexed, it became activated at similar levels to that of the small molecule fluorogen (Figure 2D). Molecular brightness values determined by fluorescence correlation spectroscopy (FCS) were similar (+/- 15 %) for P[(EO)mMA]n-MG and MG-ester fluorogens (Table 1). The observed spectroscopic behavior confirmed these small molecule and macromolecular fluorogens were analogous, and that they would function as protein-targeted and activated fluorogens in physiological solution.
Polymers bound to small ligands may perturb their binding to proteins.23 To assess the functional consequence of these macromolecules on MG's binding affinity to FAPs, KD and kON values were determined for each polymer (Method and Figures, SI - Fig S3-S4). While the small molecule KD with dL5** protein is 18 pM,16 KD's of these macromolecular fluorogens were considerably higher, i.e. weaker binding constants of ca. 2-40 nM depending on the specific macromolecular architecture. At high molecular weights, KD values showed little dependence on polymer length (Figure 3A, DP ≈ 150-400), whereas short polymers bound with lower KD values of ca. 10 nM at a DP = 66. These results were bolstered by kinetic measurements (Figure 3A – y2 axis) demonstrating the largest kON values at low molecular weights (DP = 66), and smaller but fairly constant values at higher molecular weights (DP ≈ 100-400). Larger kON values can be in part rationalized by faster diffusion of smaller polymer chains, inferred from smaller diffusion times (τd) (Figure 3C), thereby reducing the KD values (i.e. KD = kOFF/kON). The molecular weight dependence of polymer diffusion coefficients are known to obey power-law functions resembling the curves in in Figure 3A.24 It should be stressed that the observed kON values of these polymeric fluorogens are as much as 1000-fold lower than the kON values measured for MG-ester binding to the same protein in solution, suggesting the polymer's inherent bulk provides a considerable steric barrier to association.
Figure 3.
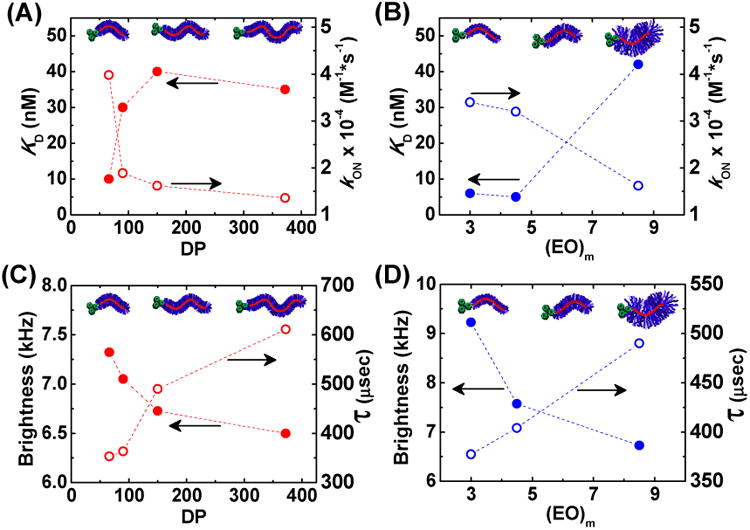
KD and kON as a function of (A) polymer backbone length (DP) displayed in red and (B) ethylene oxide (EO) side chain length displayed in blue. KD values were determined with a constant [FAP] typically between 1-10 nM in PBS+ using a λex = 636 nm and λem = 664 nm. For each kon [MG-PEO] = 4, 2, and 1 uM and [dL5**] = 50 nM in PBS+ with λex = 636/20 nm and λem = 700/20 nm. Brightness (solid circle) and diffusion time (hollow circle) from FCS as a function of (C) polymer backbone DP and (D) EO side chain length. For Figures 3A and 3C, all polymers had EO side chain lengths of ∼8.5 (Table 1, Entries 1-4), whereas polymers in Figures 3B and 3D had similar backbone DPs ≈ 150-170 (Table 1, Entries 2, 5-6).
Considering the polymer's steric bulk, it is reasonable to anticipate that there may be more pronounced effects from side-chain length in comparison to polymer backbone length. A series of polymers having similar DPs (Entry 2, 5-6: DP ≈ 150-170), but with different EO side-chain lengths (Figure 3B), was evaluated for their biophysical properties. Shorter EO side chains reduced the determined KD values to as low as 5 nM, whereas their larger counterpart had a KD value of 45 nM. kON values reinforced these results with shorter side-chains showing faster association (3×104 M-1 s-1) compared to longer side-chains (1.5×104 M-1 s-1). However, a 9-fold change in KD cannot be explained by a two-fold change in kON, suggesting longer side-chains may also facilitate faster dissociation from the protein complex. This behavior is rationalized to be a response of two main phenomena: shorter side-chain polymers have (1) smaller hydrodynamic radii increasing their diffusion rate inferred from respective τd values (Figure 3D), i.e. from diminished side-chain bulk and reduced solubility in PBS+, and (2) minimal steric hindrance directly adjacent to the MG functionality improving its propensity to remain bound, i.e. smaller dissociation-rate coefficient.
To further investigate the influence of side-chain sterics on MG binding, an additional initiator (MG-Amide) was synthesized containing a longer spacer between the MG functionality and the attached polymer. Synthetic procedures and characterization for this initiator can be found in the SI. The polymer fluorogen synthesized from this initiator (Entry 9, KD = 1.9 nM) was observed to have a 20-fold reduction in the KD compared to an analogous MG-PBiB polymer (Entry 2, KD = 42 nM). This result suggests that providing an adequate space for proper FAP-MG association mitigates the steric hindrance generated from polymer side-chains. Although this polymer containing the extended linker had a broader molecular weight distribution (ĐM = 1.84) than similar MG-PBiB derived polymers, the measured KD is lower than the shortest MG-PBiB polymer, which implies this is a direct effect of the extended linker and not a consequence of rapidly binding low molecular weight polymers.
Optimal biochemical properties may not necessary translate into ideal properties for intracellular labeling. Intracellular labeling requires that the macromolecular probe is capable of cellular internalization (Figure 1A) and potentially subcellular transmembrane penetration prior to binding (e.g. when labeling nuclear-localized FAPs, the polymer must cross the nuclear envelope). The steady-state labeling of intracellular FAP-actin fusion proteins in live HeLa cells was determined by flow cytometry for each polymer after 12 hours incubation in DMEM at 37° C, while using parental HeLa cells lacking FAP expression as controls. The observed trends were similar to those seen with regard to the biochemical properties, where the polymer backbone length imparted a minimal effect while side-chain length exerted a dramatic effect, i.e. the shortest side-chains produced up to a 5-fold higher signal with no change in the associated background (Figure 4A-B). This intensity enhancement is attributed to a mixture of biochemical and macromolecular properties, including slightly enhanced brightness values, smaller KD values, and improved transport across the plasma membrane from lipophilicity differences between compositionally disparate polymers.25
Figure 4.
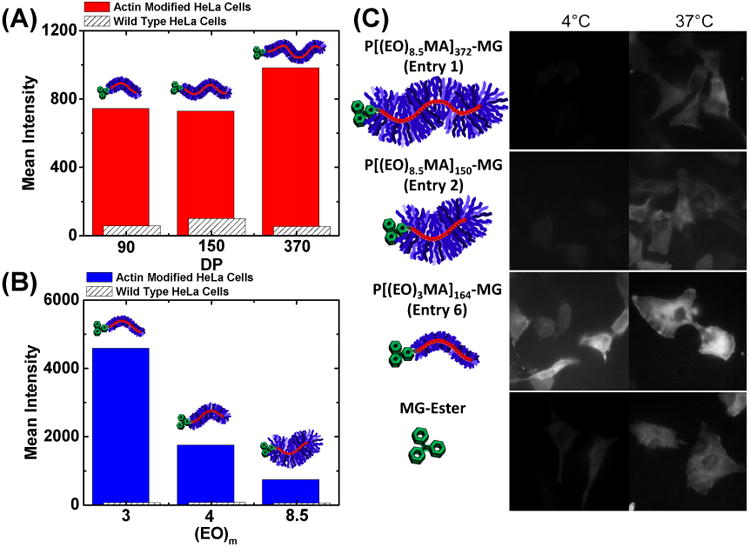
Polymeric probe internalization via flow cytometry and fluorescence microscopy. All cells were incubated in DMEM + 10% fetal bovine serum (FBS) containing 500 nM polymeric dye for 12 hours. (A) Mean fluorescent intensity as a function of polymer backbone DP with all polymers having EO side chain lengths of ∼8.5 (Table 1, Entries 1-3). Red bars correspond to FAP-actin modified HeLa cells, whereas white bars with black hashes correspond to control wild-type HeLa cells. (B) Mean fluorescent intensity as a function of varying EO side-chain length (integer corresponds to the number of EO repeat units) with all polymers having backbone DPs ≈ 150-170 (Table 1, Entries 2, 5-6). Red and blue bars correspond to FAP-actin modified HeLa cells, whereas white bars with black hashes correspond to control wild-type HeLa cells. A red laser was used to excite MG at 635 nm and emissions of MG bound to FAP were recorded between 650-710 nm. (C) Confocal images of FAP-actin modified HeLa cells incubated with 500nM P[(EO)mMA]n-MG and MG ester in DMEM + 10% FBS at 4°C and 37°C for 6 hours. The scale bar in confocal images represents 20 μm.
If solely an endocytosis process is in place, the size of these macromolecules should have minimal impact on internalization. To gain insight into the mechanism of internalization, labeling experiments were conducted using confocal microscopy at 4 and 37 °C (Figure 4C) with various polymer fluorogens. Polymers with long and short backbone lengths, P[(EO)8.5MA]372-MG (entry 1) and P[(EO)8.5MA]150-MG (entry 2) respectively, were inhibited from take-up by cells at 4 °C indicating internalization through endocytosis. In contrast, labeling experiments conducted at 4 °C for the same amount of time revealed a surprising side-chain dependent uptake mechanism. The shortest side-chain polymer, P[(EO)3MA]164-MG (entry 6), was internalized equally at 4 and 37 °C showing an endocytosis independent mechanism, likely passive insertion into the cell membrane as a result of its smaller thickness and increased lipophilicity. Based upon these and previous results, short side-chain polymer fluorogens provided superior biophysical properties and ability to internalize into living systems.
Genetically encoded targeting proteins can be expressed in any biological compartment of interest. Actin, cytoplasmic, and nuclear localized FAPs were expressed in various cell lines prior to labeling experiments using our optimized polymer fluorogen (P[(EO)3MA]164-MG, Entry 6). Successful targeting of intracellular structures was observed, however, with signal levels 4 fold lower than those seen with the small molecule MG fluorogen. Confocal images (Figure 5A) unequivocally illustrate specific binding and activation of our macromolecular probe to the actin, cytoplasm, and nuclear compartments of the cell. In FAP-actin cells, stress fibers are plainly visible and homogenously distributed throughout the cell, showing these stress fibers are a consequence of the fluorogen FAP-actin fusion and not a result of off-target nonspecific interactions. Additional high-resolution FAP-actin images can be found in SI Figure S5. The presence of the cytoplasmic FAP in the nucleus is consistent with previous reports for size selectivity of the nuclear pore.26 When expressed with a nuclear localization signal, the FAP-fluorogen signal is found exclusively in the nucleus (Figure 5A).
Figure 5.
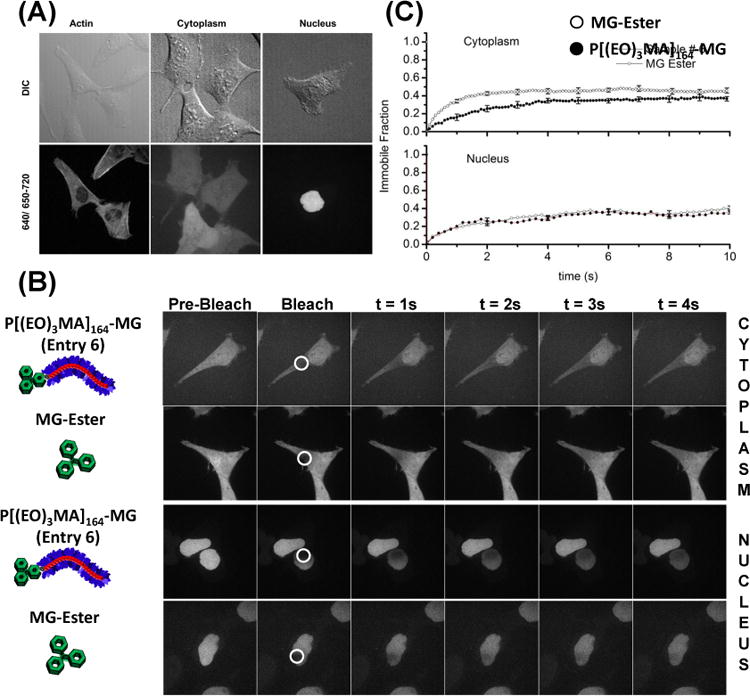
Live-cell confocal microscopy images of cells stably expressing targeted FAP proteins. (A) FAP-Actin (HeLa), Cytoplasm (HEK-293), and Nuclear targeted proteins all show proper localization when labeled with the polymeric fluorogen P[(EO)3MA]164-MG (Table 1, Entry 6). Cells were incubated in DMEM with a 500 nM polymeric dye for 12 h. Emission of MG bound to FAPs was observed between 650-710 nm and excited with a 633 nm laser. Additional Actin images are supplied in the supporting information (Figure S5). (B) Fluorescence recovery after photobleaching (FRAP) images for polymeric dye and MG ester in the cytoplasm and nucleus. The cells were imaged for a single frame at an exposure of a 100 ms followed by bleaching in the region shown using a circle with a white border. The subsequent images show the recovery of the signal in a 4s duration. (C) Plots of average intensity as a function of time in the circular regions shown in B, in nucleus and cytoplasm for polymeric dye and MG ester. Scalebar in confocal images is 20 μm.
Fluorescence does not necessarily ensure that the polymeric fluorogen has reached the site of signal intact. The integrity of any artificial cargo or probe within cellular environments may be compromised because of bond-cleavage by esterases, reductive conditions, or reactive oxygen. Therefore, the integrity of these polymeric fluorogens was investigated in the cytoplasm and nucleus of living cells using fluorescence recovery after photobleaching (FRAP). Previous diffusion measurements by FCS (Table 1) revealed marked differences in hydrodynamic diameter of the polymer-FAP complex compared to the MG-FAP complex. If the MG functionality was cleaved from the polymer, FRAP diffusion measurements of polymer-FAP complex would be equivalent to small molecule MG-FAP complex. Cytoplasmic FRAP measurements revealed slower recovery of the polymer-FAP complex compared to the MG-FAP complex, confirming the polymer fluorogen remained intact. These recovery rates were consistent with measured differences in τd from FCS, where FRAP had a 2.5 fold difference and FCS had a 1.7 fold difference. The suitable difference between these analytical techniques is attributed to sub-Brownian diffusion of larger molecules in the crowded cellular environment.27 Within the nucleus, the recovery rate and immobile fraction of fluorophores were identical for MG-ester and the polymer-fluorogen complexes. Similar size independent diffusion within the nucleus was also observed by Bancaud and coworkers when using inert molecules and was ascribed to the fractal organization of nuclear chromatin.28
Targeting macromolecules to intracellular proteins, especially those integral to cell movement may allow alteration of cellular motility. There are two cell motion related phenomena of particular interest here: membrane ruffling and cell migration. Membrane ruffling is a repeated protrusion and retraction of cell-substrate contact surfaces, while migration is polarized protrusion and retraction at the leading and trailing edges of the cell. In preliminary experimentation, we observed that FAP-actin expressing HeLa cells labeled with P[(EO)8.5MA]372-MG (Entry 1), P[(EO)8.5MA]150-MG (Entry 2), and P[(EO)3MA]164-MG (Entry 6) showed statistically faster ruffling rates compared to cells without polymeric fluorogen. In order to further enhance this effect, a knockdown of β-actin was performed using siRNA (Figure 6A) to reduce the native β-actin without affecting the FAP-actin concentration in the cells; thus ensuring a high degree of polymer could be bound to the actin cytoskeleton upon labeling. HeLa cells were incubated with polymeric fluorogen, MG-ester, and MG-ester plus a 0.2% w/v P[(EO)3MA150] without MG functionality. Incubation media, containing the fluorogens, was replaced with imaging media and then the cells were monitored and recorded using an epi-fluorescence setup as described in the SI. From each resulting movie, supplied in the SI, several kymographs were generated for each system in regions that showed high ruffling rates (Figure 6B). These kymographs were segmented and speeds were calculated from selected points on the cell's surface. Figure 6C shows the distribution of these speeds using various controls and polymer fluorogen systems. Statistics of this speed analysis are available in SI Table S1. In all cases where polymer fluorogens were bound to the FAP-actin cytoskeleton, two-fold larger mean-instantaneous speed values (ῡ ≈ 45-50 nm/s) were observed compared to the unlabeled and labeled control systems (ῡ ≈ 19-22 nm/s) using MG-ester and unfunctionalized polymer. Furthermore, while ruffling rates were higher compared to control systems minimal cellular migration occurred. Although a detailed analysis of the mechanism underlying these observations is beyond the scope of this paper, it is possible that polymer modification of actin prevents engagement of actin bundles with the focal adhesions; allowing ruffling, but reducing traction forces and decreasing cellular motility. This work demonstrates that polymer-modified cellular structures can influence cellular behavior to display unique engineered phenotypes, which may grant access to a new area of therapeutic and clinical treatments.
Figure 6.
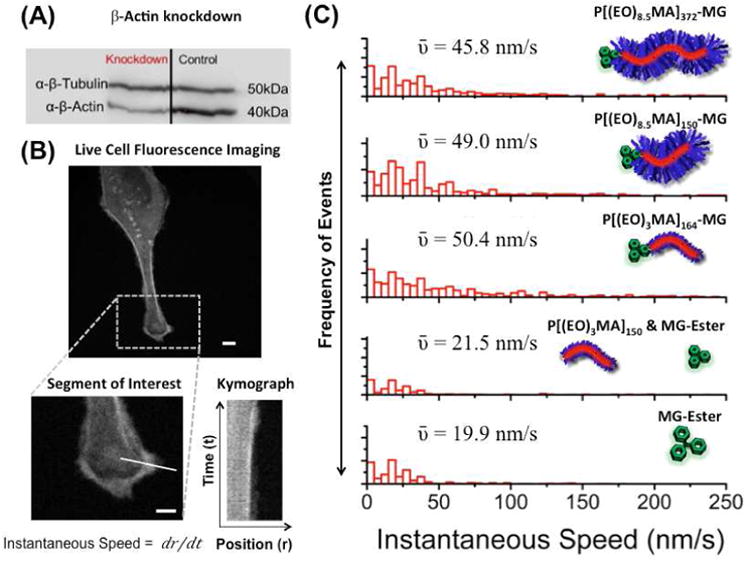
Effect of actin bound polymer fluorogen on membrane ruffling. (A). Western blot showing the knockdown of β-actin by siRNA in HeLa cells expressing modified actin. (B) Fluorescence images of HeLa cells after knockdown and incubation with 500 nM polymeric dye or MG ester in DMEM + 10% FBS for 6 hours. A segment of interest was chosen along the cell membrane and a kymograph was generated for the selected segment. Scalebars in confocal images represents 10 μm. From the kymograph, instantaneous speeds were obtained at selected segment for various time points. (C) Histograms of the instantaneous speed are shown for the knockdown cells with various polymer fluorogens and control systems with their corresponding mean-instantaneous speed values (ῡ).
Conclusion
Here, genetically encoded FAPs combined with a new macromolecular platform have achieved specific targeting of polymeric materials to distinct subcellular structures within living cells. Site-specific targeting was demonstrated via live-cell imaging using stable cell lines expressing FAP-fusion constructs within actin, cytoplasmic, and nuclear compartments. Genetically targetable polymers, based on a fluorogen functionalized initiator and a controlled radical polymerization process, have nanomolar FAP binding affinities and cell membrane permeability under physiological conditions. Modification of the actin cytoskeleton via polymer binding shows pronounced effects on macroscopic cellular ruffling and migration. To our knowledge, this work represents the first instance of a genetically targetable macromolecule that can be specifically delivered to subcellular compartments, visualized, and perturb cellular behavior. This approach may reveal details of subcellular targeting of polymeric materials and drug carriers, improve targeting and design of diagnostic and therapeutic biomaterials, and can provide a means to manipulate cellular behavior for applications in fundamental and synthetic biology.
Supplementary Material
Figure S1: GPC-MALLS refractive index traces as a function of monomer conversion during the polymerization of OEOMA475 in anisole.
Figure S2: UV-Vis absorbance scans of P[(EO)8.5MA]372-MG (black) and MG-Ester (red) in PBS+.
Figure S3: Example determination of kon for P[(EO)8.5MA]372-MG under pseudo-first order kinetics. (A) Raw fluorescence intensity versus time as a function of time using four P[(OE)8.5MA)]372-MG (Table 1, entry 1) concentrations. Experiments were performed using 50 nM of FAP in PBS+, and excitation and emission wavelengths were set to 636 and 700 nm, respectively. (B) Plot of apparent rate coefficients (kapp) versus P[(EO)8.5MA]372-MG concentration.
Figure S4: Example titration curve for the determination of KD for P[(EO)8.5MA]372-MG using 1 nM FAP (Table 1, entry 1). Emission intensity as a function of P[(OE)8.5MA)]372-MG concentration. KD was determined to be 34 nM. In all experiments, excitation and emission wavelengths were set to 636 and 664 nm, respectively.
Figure S5. Images from live-cell confocal microscopy using genetically engineered HeLa cells expressing FAP proteins fused with actin filaments. HeLa cells were incubated in DMEM using 500 nM polymer fluorogen for 12 hours. Emission signals P[(EO)3MA]164-MG bound to FAPs were observed between 650-710 nm and excited with a 633 nm laser. Images were taken using P[(EO)3MA]164-MG.
Scheme S1: Synthesis of Malachite Green Functional Initiator (5).
Scheme S2: Synthesis of Amide Malechite Green Functional Initiator (12)
Table S0. Primers used for constructing the plasmids for FAP, mCerulean3 expression in HEK cells.
Table S1. Statistical values for the ruffling analyses showing the total number of instantaneous speed points used to calculate mean speeds and standard deviations in Figure 6C. Each segment on a cell consisted of ten speed points. The speed points are the number of times the instantaneous speed was measured for each segments.
Acknowledgments
This work was supported by the NIH, through the Technology Centers for Networks and Pathways program (U54GM103529). SS was supported in part by the McWilliams Graduate Fellowship.
Footnotes
Supporting Information: Detailed descriptions of all materials, equipment, and experimental procedures are provided in the supporting information including chemical reagents, instrumentation, analytical techniques, synthetic procedures for small molecules and polymerizations, FAP cell line descriptions, DNA constructs, and additional supporting references.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.(a) Roberts MJ, Bentley MD, Harris JM. Adv Drug Delivery Rev. 2002;54:459. doi: 10.1016/s0169-409x(02)00022-4. [DOI] [PubMed] [Google Scholar]; (b) Rossi NAA, Constantinescu I, Brooks DE, Scott MD, Kizhakkedathu JN. J Am Chem Soc. 2010;132:3423. doi: 10.1021/ja909174x. [DOI] [PubMed] [Google Scholar]; (c) Scott MD, Murad KL, Koumpouras F, Talbot M, Eaton JW. Proc Natl Acad Sci U S A. 1997;94:7566. doi: 10.1073/pnas.94.14.7566. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Cummings C, Murata H, Koepsel R, Russell AJ. Biomaterials. 2013;34:7437. doi: 10.1016/j.biomaterials.2013.06.027. [DOI] [PubMed] [Google Scholar]
- 2.Haag R, Kratz F. Angew Chem, Int Ed. 2006;45:1198. doi: 10.1002/anie.200502113. [DOI] [PubMed] [Google Scholar]
- 3.(a) Kamaly N, Xiao Z, Valencia PM, Radovic-Moreno AF, Farokhzad OC. Chem Soc Rev. 2012;41:2971. doi: 10.1039/c2cs15344k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lim CS. Adv Drug Delivery Rev. 2007;59:697. [Google Scholar]; (c) Chou LYT, Ming K, Chan WCW. Chem Soc Rev. 2011;40:233. doi: 10.1039/c0cs00003e. [DOI] [PubMed] [Google Scholar]
- 4.(a) Bartlett DW, Su H, Hildebrandt IJ, Weber WA, Davis ME. Proc Natl Acad Sci U S A. 2007;104:15549. doi: 10.1073/pnas.0707461104. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kirpotin DB, Drummond DC, Shao Y, Shalaby MR, Hong K, Nielsen UB, Marks JD, Benz CC, Park JW. Cancer Res. 2006;66:6732. doi: 10.1158/0008-5472.CAN-05-4199. [DOI] [PubMed] [Google Scholar]; (c) Nobs L, Buchegger F, Gurny R, Allemann E. Bioconjugate Chem. 2006;17:139. doi: 10.1021/bc050137k. [DOI] [PubMed] [Google Scholar]
- 5.Orlova A, Tolmachev V, Pehrson R, Lindborg M, Tran T, Sandstroem M, Nilsson FY, Wennborg A, Abrahmsen L, Feldwisch J. Cancer Res. 2007;67:2178. doi: 10.1158/0008-5472.CAN-06-2887. [DOI] [PubMed] [Google Scholar]
- 6.Cortez-Retamozo V, Backmann N, Senter PD, Wernery U, De BP, Muyldermans S, Revets H. Cancer Res. 2004;64:2853. doi: 10.1158/0008-5472.can-03-3935. [DOI] [PubMed] [Google Scholar]
- 7.(a) Choi CHJ, Alabi CA, Websterb P, Davis ME. Proc Natl Acad Sci U S A. 2010;107:1235. doi: 10.1073/pnas.0914140107. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Cui B, Wu C, Chen L, Ramirez A, Bearer EL, Li WP, Mobley WC, Chu S. Proc Natl Acad Sci U S A. 2007;104:13666. doi: 10.1073/pnas.0706192104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Quintana A, Raczka E, Piehler L, Lee I, Myc A, Majoros I, Patri AK, Thomas T, Mule J, Baker JR., Jr Pharm Res. 2002;19:1310. doi: 10.1023/a:1020398624602. [DOI] [PubMed] [Google Scholar]
- 9.Bagalkot V, Farokhzad OC, Langer R, Jon S. Angew Chem, Int Ed. 2006;45:8149. doi: 10.1002/anie.200602251. [DOI] [PubMed] [Google Scholar]
- 10.Szent-Gyorgyi C, Schmidt BA, Creeger Y, Fisher GW, Zakel KL, Adler S, Fitzpatrick JAJ, Woolford CA, Yan Q, Vasilev KV, Berget PB, Bruchez MP, Jarvik JW, Waggoner A. Nat Biotechnol. 2008;26:235. doi: 10.1038/nbt1368. [DOI] [PubMed] [Google Scholar]
- 11.Shank NI, Pham HH, Waggoner AS, Armitage BA. J Am Chem Soc. 2013;135:242. doi: 10.1021/ja308629w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Senutovitch N, Stanfield RL, Bhattacharyya S, Rule GS, Wilson IA, Armitage BA, Waggoner AS, Berget PB. Biochemistry. 2012;51:2471. doi: 10.1021/bi201422g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yushchenko DA, Zhang M, Yan Q, Waggoner AS, Bruchez MP. ChemBioChem. 2012;13:1564. doi: 10.1002/cbic.201200334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grover A, Schmidt BF, Salter RD, Watkins SC, Waggoner AS, Bruchez MP. Angew Chem, Int Ed. 2012;51:4838. doi: 10.1002/anie.201108107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Szent-Gyorgyi C, Schmidt BF, Fitzpatrick JAJ, Bruchez MP. J Am Chem Soc. 2010;132:11103. doi: 10.1021/ja9099328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Szent-Gyorgyi C, Stanfield RL, Andreko S, Dempsey A, Ahmed M, Capek S, Waggoner AS, Wilson IA, Bruchez MP. J Mol Biol. 2013;425:4595. doi: 10.1016/j.jmb.2013.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.(a) Patten TE, Xia J, Abernathy T, Matyjaszewski K. Science (Washington, D C) 1996;272:866. doi: 10.1126/science.272.5263.866. [DOI] [PubMed] [Google Scholar]; (b) Georges MK, Veregin RPN, Kazmaier PM, Hamer GK. Macromolecules. 1993;26:2987. [Google Scholar]; (c) Chiefari J, Chong YK, Ercole F, Krstina J, Jeffery J, Le TPT, Mayadunne RTA, Meijs GF, Moad CL, Moad G, Rizzardo E, Thang SH. Macromolecules. 1998;31:5559. [Google Scholar]
- 18.(a) McRae RL, Phillips RL, Kim IB, Bunz UHF, Fahrni CJ. J Am Chem Soc. 2008;130:7851. doi: 10.1021/ja8007402. [DOI] [PubMed] [Google Scholar]; (b) Moon JH, McDaniel W, MacLean P, Hancock LF. Angew Chem, Int Ed. 2007;46:8223. doi: 10.1002/anie.200701991. [DOI] [PubMed] [Google Scholar]
- 19.Matyjaszewski K, Xia J. Chem Rev. 2001;101:2921. doi: 10.1021/cr940534g. [DOI] [PubMed] [Google Scholar]
- 20.(a) Chang CW, Bays E, Tao L, Alconcel SNS, Maynard HD. Chem Commun (Cambridge, U K) 2009:3580. doi: 10.1039/b904456f. [DOI] [PubMed] [Google Scholar]; (b) Lutz JF, Andrieu J, Uezguen S, Rudolph C, Agarwal S. Macromolecules. 2007;40:8540. [Google Scholar]
- 21.(a) Zhang Y, Kohler N, Zhang M. Biomaterials. 2002;23:1553. doi: 10.1016/s0142-9612(01)00267-8. [DOI] [PubMed] [Google Scholar]; (b) van VLE, Vyas TK, Amiji MM. Pharm Res. 2007;24:1405. doi: 10.1007/s11095-007-9284-6. [DOI] [PubMed] [Google Scholar]
- 22.Korppitommola J, Yip RW. Can J Chem-Rev Can Chim. 1981;59:191. [Google Scholar]
- 23.(a) Kramer RH, Karpen JW. Nature. 1998;395:710. doi: 10.1038/27227. [DOI] [PubMed] [Google Scholar]; (b) Mecinović J, Snyder PW, Mirica KA, Bai S, Mack ET, Kwant RL, Moustakas DT, Héroux A, Whitesides GM. J Am Chem Soc. 2011;133:14017. doi: 10.1021/ja2045293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.(a) Shimada K, Kato H, Saito T, Matsuyama S, Kinugasa S. J Chem Phys. 2005;122:244914/1. doi: 10.1063/1.1948378. [DOI] [PubMed] [Google Scholar]; (b) Duda JL, Vrentas JS, Ju ST, Liu HT. AIChE J. 1982;28:279. [Google Scholar]
- 25.(a) Dong H, Matyjaszewski K. Macromolecules. 2010;43:4623. [Google Scholar]; (b) Lutz JF. Adv Mater. 2011;23:2237. [Google Scholar]
- 26.Solmaz SR, Blobel G, Melčák I. Proc Natl Acad Sci U S A. 2013 doi: 10.1073/pnas.1302655110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weiss M, Elsner M, Kartberg F, Nilsson T. Biophys J. 2004;87:3518. doi: 10.1529/biophysj.104.044263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.(a) Bancaud A, Huet S, Daigle N, Mozziconacci J, Beaudouin J, Ellenberg J. EMBO J. 2009;28:3785. doi: 10.1038/emboj.2009.340. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Recamier V, Izeddin I, Bosanac L, Dahan M, Proux F, Darzacq X. Nucleus. 2014;5:75. doi: 10.4161/nucl.28227. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: GPC-MALLS refractive index traces as a function of monomer conversion during the polymerization of OEOMA475 in anisole.
Figure S2: UV-Vis absorbance scans of P[(EO)8.5MA]372-MG (black) and MG-Ester (red) in PBS+.
Figure S3: Example determination of kon for P[(EO)8.5MA]372-MG under pseudo-first order kinetics. (A) Raw fluorescence intensity versus time as a function of time using four P[(OE)8.5MA)]372-MG (Table 1, entry 1) concentrations. Experiments were performed using 50 nM of FAP in PBS+, and excitation and emission wavelengths were set to 636 and 700 nm, respectively. (B) Plot of apparent rate coefficients (kapp) versus P[(EO)8.5MA]372-MG concentration.
Figure S4: Example titration curve for the determination of KD for P[(EO)8.5MA]372-MG using 1 nM FAP (Table 1, entry 1). Emission intensity as a function of P[(OE)8.5MA)]372-MG concentration. KD was determined to be 34 nM. In all experiments, excitation and emission wavelengths were set to 636 and 664 nm, respectively.
Figure S5. Images from live-cell confocal microscopy using genetically engineered HeLa cells expressing FAP proteins fused with actin filaments. HeLa cells were incubated in DMEM using 500 nM polymer fluorogen for 12 hours. Emission signals P[(EO)3MA]164-MG bound to FAPs were observed between 650-710 nm and excited with a 633 nm laser. Images were taken using P[(EO)3MA]164-MG.
Scheme S1: Synthesis of Malachite Green Functional Initiator (5).
Scheme S2: Synthesis of Amide Malechite Green Functional Initiator (12)
Table S0. Primers used for constructing the plasmids for FAP, mCerulean3 expression in HEK cells.
Table S1. Statistical values for the ruffling analyses showing the total number of instantaneous speed points used to calculate mean speeds and standard deviations in Figure 6C. Each segment on a cell consisted of ten speed points. The speed points are the number of times the instantaneous speed was measured for each segments.