

Significance
Pex1 and Pex6 are members of the AAA family of ATPases, which contain two ATPase domains in a single polypeptide chain and form hexameric double rings. These two Pex proteins are involved in the biogenesis of peroxisomes, and mutations in them frequently cause diseases. Here, we determined structures of the Pex1/Pex6 complex by cryo-electron microscopy. Novel computational modeling methods allowed placement of Pex1/Pex6 domains into subnanometer density maps. Our results show that the peroxisomal Pex1/Pex6 ATPases form a unique double-ring structure in which the two proteins alternate around the ring. Our data shed light on the mechanism and function of this ATPase and suggest a role in peroxisomal protein import similar to that of p97 in ER-associated protein degradation.
Keywords: Pex1, Pex6, AAA ATPase, cryo-electron microscopy, peroxisome
Abstract
Members of the AAA family of ATPases assemble into hexameric double rings and perform vital functions, yet their molecular mechanisms remain poorly understood. Here, we report structures of the Pex1/Pex6 complex; mutations in these proteins frequently cause peroxisomal diseases. The structures were determined in the presence of different nucleotides by cryo-electron microscopy. Models were generated using a computational approach that combines Monte Carlo placement of structurally homologous domains into density maps with energy minimization and refinement protocols. Pex1 and Pex6 alternate in an unprecedented hexameric double ring. Each protein has two N-terminal domains, N1 and N2, structurally related to the single N domains in p97 and N-ethylmaleimide sensitive factor (NSF); N1 of Pex1 is mobile, but the others are packed against the double ring. The N-terminal ATPase domains are inactive, forming a symmetric D1 ring, whereas the C-terminal domains are active, likely in different nucleotide states, and form an asymmetric D2 ring. These results suggest how subunit activity is coordinated and indicate striking similarities between Pex1/Pex6 and p97, supporting the hypothesis that the Pex1/Pex6 complex has a role in peroxisomal protein import analogous to p97 in ER-associated protein degradation.
The AAA (ATPases associated with diverse cellular activities) family of ATPases contains a large number of proteins that perform important functions in cells (1). Many members of this family form hexamers. These hexamers consist either of a single ring of ATPase domains or of stacked rings formed by tandem ATPase domains in a single polypeptide chain (type I and II ATPases, respectively). Both classes of AAA ATPases often use the energy of ATP hydrolysis to move macromolecules. Examples of single-ring ATPases include the F1 ATPase (2), which rotates a polypeptide inside its central pore, ClpX (3), which moves polypeptides into the proteolytic chamber of ClpP, and the helicase gp4 of T7 phage (4), which pulls a DNA strand through its center. Double-ring ATPases generally act on polypeptides. Prominent members of this family include N-ethylmaleimide sensitive factor (NSF), p97/VCP (valosin-containing protein), and Hsp104 in eukaryotes and ClpB in bacteria (5–7). NSF dissociates SNARE complexes generated during membrane fusion, p97/VCP plays a role in many processes, including ER-associated protein degradation (ERAD), and Hsp104 and ClpB disassemble protein aggregates.
The molecular mechanism of many hexameric AAA ATPases is only poorly understood, particularly for those that move polypeptide chains. In addition, the mechanism appears to differ among the known examples. For the F1 ATPase, it has been established that ATPase domains hydrolyze ATP continuously in a strictly consecutive manner around the ring (2). In contrast, in another single-ring ATPase, ClpX, several ATPase domains hydrolyze ATP in sporadic bursts, either simultaneously or in short succession (3, 8–10). For double-ring ATPases, the coordination between ATPase subunits is even more complex, as there may be communication both within a given ring and between the two rings. It seems that generally most of the ATP hydrolysis occurs in only one ring. For example, the N-terminal ATPase domains of NSF, which form the D1 ring, hydrolyze ATP much more rapidly than the subunits in the D2 ring (11). The reverse is true for p97, in which the D2 ring performs the bulk of ATP hydrolysis (12, 13). Although single-ring AAA ATPases seem to move polypeptides through their central pore, it is not clear whether this model applies to NSF or p97 (14). More generally, it is unknown why some ATPases even have a second ring. In fact, some members of the double-ring ATPase family perform functions similar to their single ring counterparts (e.g., ClpAP vs. ClpXP, although ClpAP appears to grip substrates more tightly than ClpXP) (15).
Double-ring ATPases pose particular challenges. For single-ring ATPases, it was possible to covalently link three or even all six of the ATPase subunits (16). This linkage enabled mutagenesis of individual subunits, which in turn allowed testing the effect of one subunit on the activity of adjacent subunits in the ring. A similar analysis is possible in single-ring hexameric ATPases that consist of different subunits, such as the six Rpt subunits that form an ATPase ring in the 19S regulatory subunit of the proteasome (17) or the mitochondrial AAA proteases composed of two alternating subunits (18). For double-ring ATPases, covalent linkage of the subunits is impractical. However, double-ring ATPases consisting of different subunits are conceivable and would offer the opportunity to mutate specific subunits.
A deeper understanding of the mechanism of polypeptide-moving hexameric ATPases requires structural information. Unfortunately, only a few full-length structures are available at a resolution that allows fitting of the polypeptide chain (10, 19–21). Crystallography has proven difficult, particularly because crystallization is favored by symmetric structures in which all subunits are in the same nucleotide state. In addition, some domains in ATPases seem to be rather flexible. In such cases, electron microscopy (EM) analysis offers significant advantages over X-ray crystallography, as flexible particles can be analyzed and asymmetry is not limiting. Until recently, EM structures, including those of p97, NSF, and ClpB, were of relatively low resolution (>15 Å) (20, 22–25). Recent developments in cryo-electron microscopy (cryo-EM) single-particle analysis, including the use of direct electron detectors and novel image analysis algorithms, have paved the way to obtain higher resolution. Indeed, while this manuscript was under review, subnanometer-resolution structures of NSF were reported (26).
We are interested in understanding the structure and function of the Pex1/Pex6 complex, as this complex is important for human health and is a potential example of a double-ring ATPase consisting of two different polypeptide chains. Both proteins are predicted to contain two ATPase domains (D1 and D2; Fig. 1A) and are known to associate with one another (27, 28). Pex1 and Pex6 are peroxisome-associated proteins that are conserved in all eukaryotic cells. They were identified in genetic screens for peroxisome function in various organisms. Mutations in human Pex1 and Pex6 are by far the most frequent cause of peroxisome biogenesis disorders, such as Zellweger syndrome (29, 30). In these diseases, proteins fail to be imported from the cytosol into peroxisomes. Affected children have neurodevelopmental defects and multisystemic symptoms, including hepatic dysfunction, and they often die before puberty (31). Pex1 and Pex6 have also been implicated in the fusion of peroxisome precursor vesicles (32–34). Interestingly, the proposed role for Pex1 and Pex6 in peroxisomal protein import is similar to that of p97 in ERAD (35). In ERAD, p97 cooperates with the cofactors Ufd1 and Npl4 to extract ubiquitinated proteins from the ER membrane (36, 37), and similarly, Pex1 and Pex6 are thought to recycle ubiquitinated import receptor Pex5 by extracting it from the peroxisomal membrane (35).
Fig. 1.

Structure determination of the Pex1/Pex6 complex. (A) Domain structures of Pex1, Pex6, p97, and NSF. Shown are the N-terminal domains (N1, N2, N) and the ATPase domains (D1 and D2). (B) The Pex1/Pex6 complex was expressed in yeast cells and purified in different steps. Samples were analyzed by SDS/PAGE and Coomassie blue staining. Shown are samples from the crude lysate, after elution from Ni-NTA beads, after elution from streptavidin beads, and after gel filtration. MW, molecular weight markers. (C) Negative-stain EM image of the purified Pex1/Pex6 complex in the presence of ATPγS. (D) The Pex1/Pex6 complex in ATPγS was analyzed by cryo-EM. Shown is the density map in different views, calculated without imposition of symmetry. (E) Different views showing the three visible N-terminal domains of the Pex1/Pex6 complex. The numbers in the circles indicate different N domains.
Here, we used single-particle cryo-EM to provide a subnanometer-resolution EM structure of a double-ring AAA ATPase, the structure of the Saccharomyces cerevisiae Pex1/Pex6 complex. We were able to derive models for the Pex1/Pex6 complex in different nucleotide states, using a hybrid method that combines the placement of domains from structural homologs into the density map with model-building and refinement protocols implemented in Rosetta (38). Pex1 and Pex6 alternate around a double ring, forming an unprecedented arrangement. Each of the two proteins has tandem N-terminal domains, N1 and N2, which are structurally related to the single N domains in p97 and NSF. Both N2 domains and the N1 domain of Pex6 are packed against the double ring, whereas the N1 domain of Pex1 is mobile. Our data also indicate that the D1 subunits are inactive and form a symmetric ring, whereas the D2 subunits hydrolyze ATP and form an asymmetric ring, in which the subunits are likely in different nucleotide states. The structural similarity to p97 supports a role for the Pex1/Pex6 complex in peroxisomal protein import that is analogous to that of p97 in ERAD.
Results
Cryo-EM Density Map of the Pex1/Pex6 Complex in the Presence of ATPγS.
To obtain purified S. cerevisiae Pex1/Pex6 complex for structural analysis, we coexpressed both proteins in yeast cells. Pex1 was expressed under the GAL1 promoter with an N-terminal streptavidin-binding peptide (SBP) tag. Pex6 was also expressed under the GAL1 promoter with an N-terminal His14 tag. The protein complex was purified from the soluble fraction by consecutive affinity purification on Ni-NTA and streptavidin resins (Fig. 1B). The complex was finally subjected to gel-filtration chromatography in the presence of ATPγS (Fig. 1B), a nucleotide that is only poorly hydrolyzed. The purified complex contained only Pex1 and Pex6. They were present in stoichiometric amounts, as judged from the intensities of the bands in Coomassie blue-stained SDS gels. When Pex1 or Pex6 were overexpressed individually, the proteins were insoluble, suggesting that they need each other for proper folding.
In negative-stain EM images, the Pex1/Pex6 sample had a distinctly triangular shape (Fig. 1C), suggesting that Pex1 and Pex6 alternate around the ring. Essentially no side views were visible, indicating that the particles preferentially adsorbed to the grid in a face-on orientation.
To obtain higher resolution, samples were vitrified by plunge-freezing in liquid ethane for imaging by cryo-EM. The images (SI Appendix, Fig. S1A) and subsequent 2D class averages (SI Appendix, Fig. S1B), calculated with the iterative stable alignment and clustering (ISAC) procedure (39), showed that the complex adopted the same face-on view in vitrified ice as in negative-stain EM images. The preferred orientation in vitrified ice is likely due to the complexes aligning at the air/water interface. To increase the number of orientations of the Pex1/Pex6 complex, we reduced the surface tension by adding a low concentration of detergent immediately before freezing. In these images, additional views of the complex were present; this was confirmed by 2D class averages (SI Appendix, Fig. S1C). These class averages were used to calculate an initial 3D model using the stochastic hill-climbing (SHC) algorithm implemented in SPARX (40). This model was then used for 3D clustering and refinement of ∼47,000 particle images, either with or without imposition of threefold symmetry. The final symmetrized and unsymmetrized maps had resolutions of 6.2 and 7.3 Å, respectively (the unsymmetrized map is shown in Fig. 1D, a comparison of the two maps in SI Appendix, Fig. S1 D and E, and Fourier shell correlation curves in SI Appendix, Fig. S1F).
The Pex1/Pex6 complex consists of two stacked hexameric rings, D1 and D2, in which Pex1 and Pex6 alternate around the ring (Fig. 1 D and E). Three distinct regions of density corresponding to the N-terminal domains are visible. They allow the identification of the D1 ring and give the molecule a unique triangular shape with side lengths of ∼165 Å. Each of the D1 and D2 rings has a height of ∼35 Å and a diameter of ∼120 Å, similar to the dimensions of the ATPase rings in p97. The D1 ring has a relatively narrow triangular pore (side length of ∼20 Å), whereas the D2 ring has a wider pore with an annular diameter of ∼30 Å. Continuous density is seen for the D1 ring even at higher threshold levels, whereas the density of the D2 ring becomes fragmented. Even without symmetry imposed, the N domains and D1 ring show a near-perfect threefold symmetry, whereas the D2 ring is asymmetric. With symmetry imposed, the density in the D2 ring worsened, further supporting its native asymmetry. Density connecting the two rings is visible only at lower threshold levels. In the unsymmetrized map, these connections are asymmetric; they are strong on one side of the double ring and weak elsewhere (see below).
Model Building into the EM Density Maps.
The HHsearch algorithm (41) predicted that both Pex1 and Pex6 have two N-terminal domains, designated N1 and N2, which have a double Ψ-β barrel fold, followed by tandem AAA ATPase domains, D1 and D2. The D2 domains of Pex1 and Pex6 have structural homologs of high sequence identity (∼40%), whereas the N and D1 domains have only low sequence identity templates (<20%). However, even for the least conserved domain by sequence identity, the N2 of Pex1, the identified structural homologs all share the same overall fold (SI Appendix, Fig. S6), although these templates are as different from one another by sequence identity as they are from the Pex1 domain. HHsearch identified the crystal structures of p97 and NSF as the top-ranked templates for the domains of Pex1 and Pex6 (SI Appendix, Figs. S2–S5 and Fig. 1A).
To place the domains of Pex1 and Pex6 into the density map, we developed a method for the assembly of protein complex models. We first used a known representative Ψ-β barrel and a known AAA domain structure from the Protein Data Bank (PDB) to identify the location of these domains in the density map. To this end, a full rotation and translation search in the symmetrized cryo-EM map was performed, identifying candidate placements for each fold (see SI Appendix for further details). This preliminary positioning into the cryo-EM density map resulted in an average real-space correlation of 0.7, calculated per domain with the “fit to density” tool in UCSF Chimera. The agreement to the density map suggests that the domain fold identification results from HHsearch are reliable.
Next, we replaced these representative placements with the actual, unique domains of Pex1 and Pex6. We used the RosettaCM pipeline (38) to generate ∼25 structural homology models for each domain. In each case, nonaligned residues were excluded, resulting in partial models. We then used combinations of these models to identify mutually compatible domain placements. Compatibility was assessed with a scoring function that evaluates the quality of the fit into the density map, the degree of clashes between domains, and consistency with known linker lengths between adjacent domains in a polypeptide chain. Four possible domain assignments emerged. To distinguish among these, we added the unaligned residues originally omitted in the partial models and used RosettaCM to build and refine each domain to better fit the EM density map. After further optimization of domain compatibility, a single solution emerged in which the N1 domain of Pex1 was excluded from the map. Although the same domain placement emerged as the best solution using partial and complete amino acid sequences, the score difference from alternative solutions was significantly increased (SI Appendix, Table S1). This domain placement is distinguished from the next best solutions by a large score gap (SI Appendix, Fig. S6 and Tables S1 and S2). Furthermore, the top solution fits the density map better than the next best (SI Appendix, Fig. S7). Even if we consider fit-to-density or geometry criteria individually, the identified model has the best score (SI Appendix, Fig. S8). When the fit to the experimental density map was used as the sole criterion, the two best alternative solutions have the polypeptide chain termini in consecutive domains so far apart that they cannot be connected by the corresponding linker; they are therefore highly disfavored by the geometry terms (SI Appendix, Fig. S9). Taken together, these results show that the domains can be unambiguously placed into the density map.
We completed the structure by building the linkers between domains using RosettaCM and performing density-guided all-atom refinement in the context of the full assembly (42). The resulting symmetric model of the Pex1/Pex6 complex showed good agreement with the density map, particularly in the N and the D1 domains. To improve the model for the D2 ring, we used the unsymmetrized density map, which contains better resolved density in the D2 ring than the symmetrized map. Further refinement of individual D2 domains, followed by reassembly into the full-length proteins and refinement of the full-length proteins, resulted in a model for the entire complex. This final model fits well into the entire unsymmetrized EM density map (Fig. 2). Although we have refined all-atom models, we have refrained from drawing conclusions based on the modeled positions of amino acid side chains.
Fig. 2.
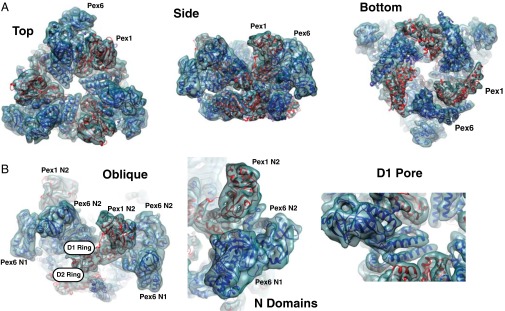
Fit of molecular models for Pex1 and Pex6 into the density map. (A) Models of Pex1 and Pex6 (red and dark blue, respectively) were fitted into the unsymmetrized density map (light blue envelope) determined in the presence of ATPγS. The model was derived as described in the text. Shown are different views. (B) Magnified views illustrating the quality of the fit. The different domains of Pex1 and Pex6 are indicated.
Confirmation of the Assignment of Domains in Pex1 and Pex6.
Given the sequence similarities of Pex1 and Pex6 and the resolution of the density map, it was important to experimentally validate the assignment of the polypeptide chains. We noticed that treatment of the Pex1/Pex6 complex with elastase or trypsin truncated Pex1, but left Pex6 intact (Fig. 3A). Mass spectrometry (MS) of fragments generated with these proteases showed that Pex1 was truncated at the N terminus, before residue Ile173 (SI Appendix, Fig. S10 A–C), resulting in a fragment that lacks only the N1 domain. We therefore purified the elastase-treated Pex1/Pex6 complex by gel filtration (Fig. 3B) and used it for cryo-EM analysis. A low-resolution map, generated from 5,000 particles, was completely superimposable onto a map of the full-length complex low-pass filtered to the same resolution (Fig. 3C). The similarity of the structures of the proteolyzed and nonproteolyzed samples provides strong evidence for the correct assignment of the domains of Pex1 and Pex6 in our model.
Fig. 3.

The N1 domain of Pex6 is not visible in the cryo-EM density map of the Pex1/Pex6 complex. (A) Purified Pex1/Pex6 complex was incubated with increasing concentrations of elastase or trypsin (given in μg/mL) for 20 min at 25 °C. The samples were analyzed by SDS/PAGE and Coomassie blue staining. The arrows indicate a protected fragment of Pex1 lacking the N1 domain, as determined by MS. MW, molecular weight markers. (B) Purification of a Pex1/Pex6 complex lacking the N1 domain of Pex1. The purified complex was treated with elastase and subjected to gel filtration. (C) Comparison of the density maps of the nonproteolyzed (blue) and proteolyzed (purple) Pex1/Pex6 complex, both determined in ATPγS. The map for the nonproteolyzed complex was filtered to about the same resolution as that for the proteolyzed complex (∼14 Å). The maps are shown individually in different views as well as after superposition. (D) Selected 2D class averages of negatively stained full-length Pex1/Pex6 complex. The brackets indicate a mobile domain peripheral to the triangular complex. (E) As in D, but class averages showing multiple mobile domains simultaneously.
Both the proteolysis and the modeling suggest that the N1 domain of Pex1 is connected to the rest of the protein through a flexible linker. The N1 domain per se is likely folded, as a crystal structure of the isolated domain has been reported (43). To confirm the presence of a flexibly attached domain in the full-length Pex1/Pex6 complex, we analyzed negative-stain EM images in more detail. 2D clustering with ISAC was performed using a relatively large window around each of the particles (SI Appendix, Fig. S10D). Consistent with the presence of a flexible linker to the N1 domain of Pex1, many of the classes displayed additional density at varying locations on the side of the triangular complex (Fig. 3D). Although flexibility likely precluded visualization of all three Pex1 N1 domains simultaneously, a number of 2D class averages showed two domains (Fig. 3E). The position of the domain is consistent with the expected location of the linker to the N2 domain of Pex1. These results, the cryo-EM structures of the nonproteolyzed and proteolyzed Pex1/Pex6 complex, and the modeling all support the conclusion that the N1 domain of Pex1 is attached through a flexible linker to the rest of the complex. Our assignment of Pex1 and Pex6 is also in agreement with a recent low-resolution structure determined by negative-stain EM, in which the position of Pex6 was determined by fusion to the maltose-binding protein (44). Finally, the experimental data provide strong validation for the computational domain-placement procedure.
Structural Features of the Pex1/Pex6 Complex.
The density map shows the N domains of Pex1 and Pex6 in positions above and at the side of the D1 ring (Fig. 2). In our model, the N2 domain of Pex1 is positioned above the plane of the D1 ring, whereas the N1 domain of Pex6 is located at the side of the D1 ring and reaches down to the D2 ring, without actually contacting it. The N2 domain of Pex6 is at an intermediate position with respect to the plane of the D1 ring. The N1 domain of Pex6 is larger than the N2 domains, as it contains two additional short helices. It is in direct contact with the N2 domain of Pex6.
Pex1 and Pex6 are structurally quite similar to each other. The N1 domain of Pex6 is readily superimposed on that of Pex1 (SI Appendix, Fig. S11A), the structure of which was previously determined by X-ray crystallography as an isolated domain (43). The N2 domains of the two proteins also have the same fold, but the differences are slightly more pronounced than for the N1 domains (SI Appendix, Fig. S11B). The individual D1 and D2 domains of Pex1 and Pex6 can also easily be superimposed (SI Appendix, Fig. S11 C and D).
The six D1 ATPase domains form a symmetric ring with all subunits in the same plane (Fig. 4 A and B). Thus, in the D1 ring, Pex1 and Pex6 are essentially equivalent, arranged as in other hexameric AAA ATPase complexes that consist of only one polypeptide chain (19, 45, 46). Nevertheless, there are subtle differences between Pex1 and Pex6. For example, each subunit contributes three helices that project slightly differently into the central pore (Fig. 2 A, Left and B, Right).
Fig. 4.

Subunit arrangement in the ATPase rings of the Pex1/Pex6 complex. (A) Slice through the D1 ring. Note that the ATPase subunits are arranged in a symmetric manner. (B) To demonstrate the planar character of the D1 ring, the same helix of each subunit is displayed. The rest of the protein was masked. (C) Slice through the D2 ring. Note that the subunits are unequally spaced with Pex1 and Pex6 arranged in three pairs. The approximate distances between the subunits are indicated. The distances across the Pex6→Pex1 interfaces are shown in red. Distances were determined between the Arg finger in one subunit and the conserved residues of the Walker A motif in the other. The arrows indicate the sites of strong density connecting the D1 and D2 rings. (D) As in B, but for the D2 ring. The arrows indicate the helices that are most tilted toward the central pore. They belong to the subunits indicated with an arrow in C. (E, Left) Strong connections between the D1 and D2 rings at the sites indicated by arrows in C and D. (Right) Equivalent view demonstrating weak connections at another point of the ring.
The D2 ring is asymmetric, but all six subunits are approximately in the same plane (Fig. 4 C and D). Within the ring, three pairs of closely apposed Pex1/Pex6 ATPase domains are clearly visible. At these Pex1→Pex6 interfaces, the second region of homology (SRH), containing conserved Arg residues of Pex1, comes close to residues in the nucleotide-binding pocket of Pex6. In contrast, the distances between the Pex6→Pex1 interfaces are different around the ring. At one interface, there is close contact (∼8–9 Å distance between the second conserved Arg in the SRH and the Walker A Lys in the nucleotide-binding pocket), whereas there are significant gaps at the other two (∼15 and ∼22 Å; Fig. 4C). At least at the widest junctions, strong binding of the nucleotide between neighboring ATPase subunits seems unlikely. These results suggest that not all subunits in the D2 ring are in the same nucleotide-bound state. Because the Pex1/Pex6 complex was saturated with ATPγS during purification, it is likely that some sites are in the ATP-bound state.
Interestingly, the D1 and D2 rings interact most strongly close to the location of the most widely spaced ATPase subunits in the D2 ring (Fig. 4 C, arrow, and E, Left). At the contact sites, the D2 subunits of both Pex1 and Pex6 are rotated within the ring plane such that the helical fingers pointing into the pore are moved downward (Fig. 4D, arrows). Little contact between D1 and D2 is seen on the other side of the D2 ring, where the subunits contact each other (Fig. 4E, Right).
Structure of the Pex1/Pex6 Complex in the Presence of ADP.
We next determined a structure of the Pex1/Pex6 complex in the presence of ADP. The complex was purified as before, except that ADP was present during gel filtration. The purified complex again consisted of stoichiometric quantities of Pex1 and Pex6, but it was less stable than in ATPγS, as assessed by negative-stain EM and as reported previously (47). However, the complex was stable at lower salt and higher nucleotide concentrations. We used cryo-EM analysis to generate density maps with and without threefold symmetry imposed at resolutions of 7.4 and 8.8 Å, respectively (SI Appendix, Fig. S1G). The unsymmetrized map (Fig. 5A) was used to fit a model of the Pex1/Pex6 complex, using the structure in ATPγS as a starting point for refinement in RosettaCM.
Fig. 5.

Comparison of the structures of the Pex1/Pex6 complex determined in ATPγS and ADP. (A) Density map of the Pex1/Pex6 complex obtained in ADP, calculated without imposing symmetry. Shown are different views. (B) Comparison of models of the Pex1/Pex6 complex in ATPγS and ADP (blue and orange, respectively). (C) As in B, but with magnified views of the individual domains.
The density maps and models were surprisingly similar for the structures in ATPγS and ADP (Fig. 5B). The N and D1 domain regions were completely superimposable, and only small, if any, changes were seen in the D2 ring (Fig. 5C). The D2 ring was again asymmetric, suggesting that the differences between the ATPase subunits are caused by nucleotide occupancy rather than by the presence of the γ-phosphate of the nucleotide.
Nucleotide Hydrolysis by the ATPase Domains of Pex1 and Pex6.
Sequence analysis of Pex1 and Pex6 shows that the D1 domains lack residues required for ATP hydrolysis in related ATPases. Specifically, the conserved Walker B motif residues Asp-Glu are changed to Asp-Gln in Pex1 and to Ala-His in Pex6 (Fig. 6A). Both mutations are known to severely affect ATP hydrolysis in related ATPases (48). In addition, the SRH domains of both proteins lack the Arg residues (Arg finger) that are required for ATP hydrolysis in the neighboring ATPase subunit. These features indicate that the ATPase subunits in the D1 ring can bind, but not hydrolyze, ATP. The tight spacing of the subunits and the rotational symmetry of the D1 ring suggest that all six sites are occupied by nucleotide. Because the Pex1/Pex6 complex disassembles in the absence of added nucleotide (47), it seems likely that nucleotide binding to the D1 ATPase subunits is required for the stability of the double-ring structure. Similar conclusions have been reached for p97 and NSF, in which the ring that is less active in ATP hydrolysis (D1 and D2, respectively) is thought to stabilize the overall complex (11–13).
Fig. 6.

ATPase activity of Pex1 and Pex6 mutants. (A) Sequences of the Walker A and B motifs and of the relevant segment of the SRH in the ATPase domains of Pex1 and Pex6. The first line shows the canonical sequences seen in AAA ATPases. Amino acids highlighted in red in the D1 and D2 domains of the Pex proteins are identical to the canonical residues. Residues in blue are different. Mutations made in the Walker A and B motifs are indicated. (B) ATPase activity of WT and mutant Pex1/Pex6 complex. Complexes containing either WT Pex1 or Pex6, or proteins with mutations in the D2 Walker A or B motifs, were purified, and equal concentrations were tested for ATPase activity. The activity is given relative to that of a complex in which both proteins are WT. Shown are means and SDs of three experiments.
The D2 subunits of both Pex1 and Pex6 contain the typical Walker A and B motifs, the two conserved Arg residues in the SRH, and other features characteristic of active ATPases (Fig. 6A). To test whether the D2 subunits in Pex1 and Pex6 are equivalent for overall ATP hydrolysis, we generated mutants with inactivated Walker A and B motifs and expressed and purified various combinations of these proteins. As expected, a complex in which the Walker B motifs of both Pex1 and Pex6 were mutated had essentially no ATPase activity (Fig. 6B). Similarly, complete inactivation was observed when only Pex6 lacked a functional Walker B motif, whereas in the reverse situation, ATPase activity was only reduced by 40%. Thus, the D2 ATPase domains of Pex1 and Pex6 are not equivalent. Interestingly, the essential Walker B motif of Pex6 is located at the tight Pex1→Pex6 interface in our structure (Fig. 4C), whereas the nonessential Walker B motif of Pex1 is located at the variably spaced Pex6→Pex1 interfaces. The correlation between ATPase activity and structural features also supports the assignment of Pex1 and Pex6 in our model. Mutations in the Walker A motifs did not completely eliminate ATP hydrolysis, even when present in both proteins (Fig. 6B), presumably because they did not entirely abolish ATP binding.
Discussion
Here we report EM structures of the peroxisomal AAA ATPase Pex1/Pex6. Our results show that the ATPase forms a unique double-ring structure in which the two proteins alternate around the ring. Both Pex1 and Pex6 have previously unrecognized tandem N-terminal domains that are structurally related to the single N domains in p97 and NSF. The D1 ring is symmetric, in contrast to the D2 ring. Like in p97, the D2 ring subunits conduct the bulk of ATP hydrolysis. These results are based on sequence homology and experimental data alone. However, a novel computational method was required to assign Pex1 and Pex6 into the density map and to realize that the N1 domain of Pex1 is flexibly attached to the complex. These model predictions could be experimentally confirmed by proteolysis and negative-stain EM experiments and are in agreement with the positions of Pex1 and Pex6 reported by others (44). Although the computational approach resulted in a molecular model of the Pex1/Pex6 complex, we have been careful not to interpret the data beyond the constraints of the density map resolution. Nevertheless, our results give insight into the mechanism and function of this double-ring ATPase.
The division of labor between the D1 and D2 rings of Pex1/Pex6 is similar to that in p97. However, the ATPases in the D1 ring of p97 have the canonical Walker motifs and retain a low ATPase activity (12), whereas the ones in the Pex1/Pex6 complex lack the typical motifs and seem to be inactive. The D2 ring is almost symmetric in all p97 structures, regardless of the nucleotide present, but the symmetry may be caused by crystallization. Asymmetry in an ATP-hydrolyzing ring, as in the D2 ring of the Pex1/Pex6 complex, has also been observed for other polypeptide-moving ATPases, such as ClpX, MecA/ClpC, and F1 ATPase (10, 21, 49). As demonstrated for ClpX and F1 ATPase, at any given moment, these ATPases may have only a subset of their subunits occupied with nucleotide. The variation in distances between D2 subunits of the Pex1/Pex6 complex is more pronounced than seen with F1 ATPase or MecA/ClpC and is equivalent to that of ClpX. Because the distances across the Pex1→Pex6 interfaces are small and constant, it seems that either the occupancy of the Pex6 subunits does not affect subunit spacing or these subunits are mostly in a nucleotide-occupied state. In contrast, two of the Pex6→Pex1 distances are too large to coordinate nucleotide by the neighboring subunits (Fig. 4C). We therefore assume that these two sites are unoccupied and that nucleotide occupancy of the Pex1 subunits has a major effect on subunit spacing. A correlation between subunit spacing and nucleotide occupancy has also been observed in recent structures of dynein (50). In our ATPγS structure, some of the occupied sites might contain ADP, as the differences from the ADP structure are only small and ATPγS can slowly be hydrolyzed or be contaminated with ADP.
Our Walker B mutations show that all subunits in the D2 ring hydrolyze ATP, but one interface between Pex1 and Pex6 is more important for overall ATP hydrolysis than the other. Specifically, Pex1 subunits cannot hydrolyze ATP when Pex6 subunits are in the ATP-bound state, whereas the reverse is not true (Fig. 6B). In vivo experiments with Walker B mutants show that ATP hydrolysis at both sites is needed for cell viability (51, 52). Thus, even the less important sites may hydrolyze ATP. We speculate that ATP hydrolysis occurs in a unidirectional manner around the ring, with one subunit primarily affecting ATP hydrolysis of only one of the neighboring subunits. ATP binding would be initiated on one side of the ring at the nucleotide-free sites where the subunits are most widely spaced, a model reminiscent of the rotary mechanism proposed for the F1 ATPase (49). Interestingly, in the case of ClpX, there may also be one or two nucleotide-free sites in the ring (10).
There might also be communication between the D1 and D2 rings during the functional cycle of the Pex1/Pex6 ATPase. Such communication has been suggested for other double-ring ATPases (53), but the density between the D1 and D2 rings observed on one side of the Pex1/Pex6 complex is stronger than previously observed. Interestingly, this strong connection between the rings occurs at the point of biggest gaps between D2 ring subunits. In addition, these subunits undergo a rotation toward the center of the D2 ring. The intriguing coincidence of these features could mean that either the D1 ring interaction affects the nucleotide state of subunits in the D2 ring or vice versa.
The differences between the structures determined in ATPγS and ADP are surprisingly small and may not even be significant at the resolution of our cryo-EM maps. However, similarly small differences have been seen in the F1 ATPase (54) or in p97 (55) crystallized with different nucleotides. In contrast, surprisingly large differences have recently been reported for NSF (26).
It is interesting to compare the position of the two N domains of Pex1 and Pex6 with that of the single N domain of p97. The N domain of p97 is in a “down conformation” relative to the plane of the D1 ring, regardless of the nucleotide state (19) (SI Appendix, Fig. S12). It displays an “up conformation” in p97 disease mutants crystallized with ATPγS (45) (SI Appendix, Fig. S12). In the structures of the Pex1/Pex6 complex, the N domains vary substantially in their positions relative to the D1 ring. In our model, the N2 domain of Pex1 is in the most extreme up conformation, even more above the D1 ring than the N domain in the p97 disease mutants. On the other hand, the N1 domain of Pex6 is in a down conformation similar to the situation in WT p97 (SI Appendix, Fig. S12).
The exact function and the substrates of the Pex1/Pex6 complex are currently unknown. However, the structural similarity of the Pex1/Pex6 complex to p97 supports a model in which the complex functions in peroxisomal protein import (35). In this model, the import receptor Pex5 delivers the substrate to the peroxisomal membrane, is ubiquitinated, and is then returned to the cytosol by the Pex1/Pex6 complex. Thus, the Pex1/Pex6 complex would function analogously to p97 in ERAD, in which the ATPase extracts poly-ubiquitinated proteins from the ER membrane. As proposed for p97, the Pex1/Pex6 complex may move substrate using the ATPase activity of the D2 ring domains. In ERAD, poly-ubiquitin chains are recognized by p97 and its cofactor Ufd1/Npl4 (13). Interestingly, these chains bind to N domains in both p97 and Ufd1. These domains share the same fold as the tandem N domains in Pex1 and Pex6 (19, 56). It is thus possible that the Pex1/Pex6 complex functionally corresponds to the p97/Ufd1/Npl4 complex, such that the peroxisomal ATPase would contain built-in cofactor domains. It is, however, conceivable that an additional unidentified cofactor is required.
It should be noted that the Pex1/Pex6 complex has also been proposed to function in the fusion of peroxisomal precursor vesicles (32–34), a role reminiscent of NSF in vesicular transport. However, no substrate or cofactor analogous to SNAREs or α-SNAP has been identified for the Pex1/Pex6 complex, and the activities of the ATPase domains D1 and D2 are reversed relative to NSF.
Our work establishes a precedent for generating a molecular model of a protein complex on the basis of an EM map of intermediate resolution. Whereas previous modeling approaches placed known crystal structures into the density map and used flexible fitting to improve the fit (57), our methodology uses density-guided comparative modeling of putative domain placements, followed by Monte Carlo sampling to identify the correct configuration of domains. This procedure allows modeling in cases where only structures of distantly related proteins are known or where domains of similar, but nonidentical, structure need to be placed into the density map. We anticipate that this approach will also be applicable to many other cases in which atomic resolution has been impossible to achieve.
Materials and Methods
Expanded details of experimental procedures are described in the SI Appendix, SI Materials and Methods.
Expression and Purification of the Pex1/Pex6 Complex.
S. cerevisiae Pex1 and Pex6 were coexpressed in yeast cells with an N-terminal SBP tag on Pex1 and an N-terminal His14 tag on Pex6, and purified by sequential affinity steps on Ni-NTA and streptavidin agarose resins. The protein complex was then subjected to gel filtration on a Superose 6 column in the presence of 0.3 mM ATPγS or 4 mM ADP.
Proteolysis Experiments.
Purified Pex1/Pex6 complex (0.9 mg/mL) was incubated with 0–100 μg/mL elastase or trypsin for 20 min at 25°C. For EM experiments, 1.7 mg/mL Pex1/Pex6 complex was incubated with 35 μg/mL elastase at 4°C for 10 min. The reaction was quenched with diisopropyl fluorophosphate before isolation by gel filtration.
ATPase Assays.
Pex1/Pex6 complex was incubated with 1 mM ATP at 25°C. Phosphate release was followed over time using the EnzChek assay (Life Technologies). All mutants were stable and assembled into hexamers, as assessed by negative-stain EM.
Electron Microscopy and Image Processing.
Specimens were negatively stained with uranyl formate as previously described (58). Samples were imaged using a 4K × 4K CCD camera (Gatan) on a Tecnai 12 electron microscope (FEI) operated at 120 kV. The particles were interactively selected with Boxer (59), windowed with SPIDER (60), and subjected to ISAC (39) implemented in SPARX (40). The final stable classes include ∼51% of the entire dataset.
For cryo-EM, vitrified specimens were prepared on a glow-discharged holey carbon grid by plunge-freezing in liquid ethane. In some cases, 0.018% Triton (wt/vol) was added to the protein solution just before freezing. Cryo-EM data were collected at liquid nitrogen temperature using a K2 Summit direct electron detector camera (Gatan) on a Tecnai F20 electron microscope operated at 200 kV. Dose-fractionated image stacks were recorded with UCSF Image 4 (61) in superresolution counting mode.
Dose-fractionated image stacks of vitrified Pex1/Pex6 complex were 2× binned and motion corrected, as previously described (61). The defocus was determined with CTFFIND3 (62). Particles were selected interactively (59). The particle images were subjected to ISAC (39), and resulting 2D class averages were used to calculate an initial 3D model with SPARX for 3D particle classification in RELION (63). Particles were then subjected to 3D auto-refinement in RELION. Where indicated, threefold symmetry was imposed during 3D classification and refinement. The resolutions of the final density maps were estimated by refining two half datasets independently and using the Fourier shell correlation (FSC) = 0.143 criterion (SI Appendix, Fig. S1 F and G). The maps were postprocessed in RELION and are shown after B-factor sharpening. Figures were prepared with UCSF Chimera (64).
Modeling Polypeptide Chains into Density Maps.
Sequence alignment with several different programs identified the domain organization of Pex1 and Pex6, with the D1 and D2 domains having AAA ATPase folds and the N1 and N2 domains having Ψ-β barrel folds. Rotational and translational searches with representative models for these domains resulted in a number of high-confidence placements. Next, individual homology models for the domains of Pex1 and Pex6 were placed into the preliminary positions of the Ψ-β barrel or AAA ATPase domain folds. The RosettaCM comparative modeling pipeline (38) was used to generate threaded templates (partial threads that include only the aligned residues for each template) for each of the domains of Pex1 and Pex6. Each of the partial threads was superimposed on the corresponding topology placements from the previous step. Each placement was further refined using rigid body minimization into the local density. Next, we placed all eight domains of Pex1 and Pex6 into the density map simultaneously. Placement was done with a simulated annealing Monte Carlo (MC) sampling procedure, in which each combination of putative domain placements is assigned a score (SI Appendix, SI Materials and Methods). This score assesses (i) the agreement of an assignment to the density data, (ii) whether neighboring domains clash with one another, and (iii) whether they can be connected with the known linker lengths. The first iteration of MC assembly was carried out using partial threads of individual domains as input and converged to four possible domain assignments. Next, we used the full sequences of the individual domains (not just the partial threads) to rebuild unaligned residues and to refine each domain into the density map (modeling was done per domain for tractability). The resulting models were then used as input for a second iteration of MC assembly. This second iteration converged at a single solution of domain placements, in which the N1 domain of Pex1 is unassigned into the map. The score of this solution was much better separated from the next best solution than after the first round of MC assembly (SI Appendix, Table S1). The best domain placement also gave a much better fit into the density map than the top incorrect solution (SI Appendix, Fig. S7).
Finally, full-length models were constructed in RosettaCM by rebuilding the linkers connecting individual domains and refining the symmetric assembly into the symmetrized density map. Although this gave good agreement over most of the structure, the D2 domains poorly fit the symmetrized map. We then further refined the D2 domains into the unsymmetrized map of the Pex1/Pex6 complex in ATPγS, using the Rosetta relax protocol augmented with a term assessing agreement to density. The linkers connecting the D1 and D2 domains were rebuilt, and the entire structure was refined against the unsymmetrized map. This model was used as a starting point for refinement of the Pex1/Pex6 structure in ADP, using the Rosetta relax protocol.
Supplementary Material
Acknowledgments
We thank Nicholas Bodnar, Ryan Baldridge, and Kelly Kim for critical reading of the manuscript. T.A.R., P.A.P., and D.B. are supported by National Institutes of Health (NIH) Grants GM05586, GM60635, and GM092802, respectively. N.B.B. was supported by NIH Grant GM007753. D.B., T.W., and T.A.R. are Howard Hughes Medical Institute Investigators.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The EM 3D maps of Pex1/Pex6 without imposed symmetry have been deposited in the EMDatabank, www.emdatabank.org (accession codes EMD-6359 and EMD-6360, for maps of complexes with ATPγS or ADP, respectively).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1500257112/-/DCSupplemental.
References
- 1.Hanson PI, Whiteheart SW. AAA+ proteins: Have engine, will work. Nat Rev Mol Cell Biol. 2005;6(7):519–529. doi: 10.1038/nrm1684. [DOI] [PubMed] [Google Scholar]
- 2.Leslie AG, Walker JE. Structural model of F1-ATPase and the implications for rotary catalysis. Philos Trans R Soc Lond B Biol Sci. 2000;355(1396):465–471. doi: 10.1098/rstb.2000.0588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baker TA, Sauer RT. ClpXP, an ATP-powered unfolding and protein-degradation machine. Biochim Biophys Acta. 2012;1823(1):15–28. doi: 10.1016/j.bbamcr.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Singleton MR, Dillingham MS, Wigley DB. Structure and mechanism of helicases and nucleic acid translocases. Annu Rev Biochem. 2007;76:23–50. doi: 10.1146/annurev.biochem.76.052305.115300. [DOI] [PubMed] [Google Scholar]
- 5.Bösl B, Grimminger V, Walter S. The molecular chaperone Hsp104—a molecular machine for protein disaggregation. J Struct Biol. 2006;156(1):139–148. doi: 10.1016/j.jsb.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 6.Meyer H, Bug M, Bremer S. Emerging functions of the VCP/p97 AAA-ATPase in the ubiquitin system. Nat Cell Biol. 2012;14(2):117–123. doi: 10.1038/ncb2407. [DOI] [PubMed] [Google Scholar]
- 7.Zhao C, Smith EC, Whiteheart SW. Requirements for the catalytic cycle of the N-ethylmaleimide-Sensitive Factor (NSF) Biochim Biophys Acta. 2012;1823(1):159–171. doi: 10.1016/j.bbamcr.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cordova JC, et al. Stochastic but highly coordinated protein unfolding and translocation by the ClpXP proteolytic machine. Cell. 2014;158(3):647–658. doi: 10.1016/j.cell.2014.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sen M, et al. The ClpXP protease unfolds substrates using a constant rate of pulling but different gears. Cell. 2013;155(3):636–646. doi: 10.1016/j.cell.2013.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stinson BM, et al. Nucleotide binding and conformational switching in the hexameric ring of a AAA+ machine. Cell. 2013;153(3):628–639. doi: 10.1016/j.cell.2013.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Whiteheart SW, et al. N-ethylmaleimide-sensitive fusion protein: A trimeric ATPase whose hydrolysis of ATP is required for membrane fusion. J Cell Biol. 1994;126(4):945–954. doi: 10.1083/jcb.126.4.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Song C, Wang Q, Li CC. ATPase activity of p97-valosin-containing protein (VCP). D2 mediates the major enzyme activity, and D1 contributes to the heat-induced activity. J Biol Chem. 2003;278(6):3648–3655. doi: 10.1074/jbc.M208422200. [DOI] [PubMed] [Google Scholar]
- 13.Ye Y, Meyer HH, Rapoport TA. Function of the p97-Ufd1-Npl4 complex in retrotranslocation from the ER to the cytosol: Dual recognition of nonubiquitinated polypeptide segments and polyubiquitin chains. J Cell Biol. 2003;162(1):71–84. doi: 10.1083/jcb.200302169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DeLaBarre B, Christianson JC, Kopito RR, Brunger AT. Central pore residues mediate the p97/VCP activity required for ERAD. Mol Cell. 2006;22(4):451–462. doi: 10.1016/j.molcel.2006.03.036. [DOI] [PubMed] [Google Scholar]
- 15.Olivares AO, Nager AR, Iosefson O, Sauer RT, Baker TA. Mechanochemical basis of protein degradation by a double-ring AAA+ machine. Nat Struct Mol Biol. 2014;21(10):871–875. doi: 10.1038/nsmb.2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martin A, Baker TA, Sauer RT. Rebuilt AAA + motors reveal operating principles for ATP-fuelled machines. Nature. 2005;437(7062):1115–1120. doi: 10.1038/nature04031. [DOI] [PubMed] [Google Scholar]
- 17.Beckwith R, Estrin E, Worden EJ, Martin A. Reconstitution of the 26S proteasome reveals functional asymmetries in its AAA+ unfoldase. Nat Struct Mol Biol. 2013;20(10):1164–1172. doi: 10.1038/nsmb.2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Augustin S, et al. An intersubunit signaling network coordinates ATP hydrolysis by m-AAA proteases. Mol Cell. 2009;35(5):574–585. doi: 10.1016/j.molcel.2009.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davies JM, Brunger AT, Weis WI. Improved structures of full-length p97, an AAA ATPase: Implications for mechanisms of nucleotide-dependent conformational change. Structure. 2008;16(5):715–726. doi: 10.1016/j.str.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 20.Lee S, et al. The structure of ClpB: A molecular chaperone that rescues proteins from an aggregated state. Cell. 2003;115(2):229–240. doi: 10.1016/s0092-8674(03)00807-9. [DOI] [PubMed] [Google Scholar]
- 21.Wang F, et al. Structure and mechanism of the hexameric MecA-ClpC molecular machine. Nature. 2011;471(7338):331–335. doi: 10.1038/nature09780. [DOI] [PubMed] [Google Scholar]
- 22.Carroni M, et al. Head-to-tail interactions of the coiled-coil domains regulate ClpB activity and cooperation with Hsp70 in protein disaggregation. eLife. 2014;3:e02481. doi: 10.7554/eLife.02481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chang LF, et al. Structural characterization of full-length NSF and 20S particles. Nat Struct Mol Biol. 2012;19(3):268–275. doi: 10.1038/nsmb.2237. [DOI] [PubMed] [Google Scholar]
- 24.Lee S, Sielaff B, Lee J, Tsai FT. CryoEM structure of Hsp104 and its mechanistic implication for protein disaggregation. Proc Natl Acad Sci USA. 2010;107(18):8135–8140. doi: 10.1073/pnas.1003572107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yeung HO, et al. Inter-ring rotations of AAA ATPase p97 revealed by electron cryomicroscopy. Open Biol. 2014;4:130142. doi: 10.1098/rsob.130142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao M, et al. Mechanistic insights into the recycling machine of the SNARE complex. Nature. 2015;518(7537):61–67. doi: 10.1038/nature14148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grimm I, Saffian D, Platta HW, Erdmann R. The AAA-type ATPases Pex1p and Pex6p and their role in peroxisomal matrix protein import in Saccharomyces cerevisiae. Biochim Biophys Acta. 2012;1823(1):150–158. doi: 10.1016/j.bbamcr.2011.09.005. [DOI] [PubMed] [Google Scholar]
- 28.Liu X, Ma C, Subramani S. Recent advances in peroxisomal matrix protein import. Curr Opin Cell Biol. 2012;24(4):484–489. doi: 10.1016/j.ceb.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Geisbrecht BV, Collins CS, Reuber BE, Gould SJ. Disruption of a PEX1-PEX6 interaction is the most common cause of the neurologic disorders Zellweger syndrome, neonatal adrenoleukodystrophy, and infantile Refsum disease. Proc Natl Acad Sci USA. 1998;95(15):8630–8635. doi: 10.1073/pnas.95.15.8630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reuber BE, et al. Mutations in PEX1 are the most common cause of peroxisome biogenesis disorders. Nat Genet. 1997;17(4):445–448. doi: 10.1038/ng1297-445. [DOI] [PubMed] [Google Scholar]
- 31.Braverman NE, D’Agostino MD, Maclean GE. Peroxisome biogenesis disorders: Biological, clinical and pathophysiological perspectives. Dev Disabil Res Rev. 2013;17(3):187–196. doi: 10.1002/ddrr.1113. [DOI] [PubMed] [Google Scholar]
- 32.Faber KN, Heyman JA, Subramani S. Two AAA family peroxins, PpPex1p and PpPex6p, interact with each other in an ATP-dependent manner and are associated with different subcellular membranous structures distinct from peroxisomes. Mol Cell Biol. 1998;18(2):936–943. doi: 10.1128/mcb.18.2.936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Titorenko VI, Rachubinski RA. Peroxisomal membrane fusion requires two AAA family ATPases, Pex1p and Pex6p. J Cell Biol. 2000;150(4):881–886. doi: 10.1083/jcb.150.4.881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van der Zand A, Gent J, Braakman I, Tabak HF. Biochemically distinct vesicles from the endoplasmic reticulum fuse to form peroxisomes. Cell. 2012;149(2):397–409. doi: 10.1016/j.cell.2012.01.054. [DOI] [PubMed] [Google Scholar]
- 35.Schliebs W, Girzalsky W, Erdmann R. Peroxisomal protein import and ERAD: Variations on a common theme. Nat Rev Mol Cell Biol. 2010;11(12):885–890. doi: 10.1038/nrm3008. [DOI] [PubMed] [Google Scholar]
- 36.Bagola K, Mehnert M, Jarosch E, Sommer T. Protein dislocation from the ER. Biochim Biophys Acta. 2011;1808(3):925–936. doi: 10.1016/j.bbamem.2010.06.025. [DOI] [PubMed] [Google Scholar]
- 37.Christianson JC, Ye Y. Cleaning up in the endoplasmic reticulum: Ubiquitin in charge. Nat Struct Mol Biol. 2014;21(4):325–335. doi: 10.1038/nsmb.2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Song Y, et al. High-resolution comparative modeling with RosettaCM. Structure. 2013;21(10):1735–1742. doi: 10.1016/j.str.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang Z, Fang J, Chittuluru J, Asturias FJ, Penczek PA. Iterative stable alignment and clustering of 2D transmission electron microscope images. Structure. 2012;20(2):237–247. doi: 10.1016/j.str.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hohn M, et al. SPARX, a new environment for Cryo-EM image processing. J Struct Biol. 2007;157(1):47–55. doi: 10.1016/j.jsb.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 41.Söding J. Protein homology detection by HMM-HMM comparison. Bioinformatics. 2005;21(7):951–960. doi: 10.1093/bioinformatics/bti125. [DOI] [PubMed] [Google Scholar]
- 42.DiMaio F, et al. Atomic accuracy models from 4.5 Å cryo-electron microscopy data with density-guided iterative local rebuilding and refinement. Nat Methods. 2015 doi: 10.1038/nmeth.3286. 12(4):361–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shiozawa K, et al. Structure of the N-terminal domain of PEX1 AAA-ATPase. Characterization of a putative adaptor-binding domain. J Biol Chem. 2004;279(48):50060–50068. doi: 10.1074/jbc.M407837200. [DOI] [PubMed] [Google Scholar]
- 44.Gardner BM, Chowdhury S, Lander GC, Martin A. The Pex1/Pex6 complex is a heterohexameric AAA+ motor with alternating and highly coordinated subunits. J Mol Biol. 2015;427(6 Pt B):1375–1388. doi: 10.1016/j.jmb.2015.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tang WK, et al. A novel ATP-dependent conformation in p97 N-D1 fragment revealed by crystal structures of disease-related mutants. EMBO J. 2010;29(13):2217–2229. doi: 10.1038/emboj.2010.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yu RC, Hanson PI, Jahn R, Brünger AT. Structure of the ATP-dependent oligomerization domain of N-ethylmaleimide sensitive factor complexed with ATP. Nat Struct Biol. 1998;5(9):803–811. doi: 10.1038/1843. [DOI] [PubMed] [Google Scholar]
- 47.Saffian D, Grimm I, Girzalsky W, Erdmann R. ATP-dependent assembly of the heteromeric Pex1p-Pex6p-complex of the peroxisomal matrix protein import machinery. J Struct Biol. 2012;179(2):126–132. doi: 10.1016/j.jsb.2012.06.002. [DOI] [PubMed] [Google Scholar]
- 48.Wendler P, Ciniawsky S, Kock M, Kube S. Structure and function of the AAA+ nucleotide binding pocket. Biochim Biophys Acta. 2012;1823(1):2–14. doi: 10.1016/j.bbamcr.2011.06.014. [DOI] [PubMed] [Google Scholar]
- 49.Abrahams JP, Leslie AG, Lutter R, Walker JE. Structure at 2.8 A resolution of F1-ATPase from bovine heart mitochondria. Nature. 1994;370(6491):621–628. doi: 10.1038/370621a0. [DOI] [PubMed] [Google Scholar]
- 50.Bhabha G, et al. Allosteric communication in the dynein motor domain. Cell. 2014;159(4):857–868. doi: 10.1016/j.cell.2014.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Birschmann I, Rosenkranz K, Erdmann R, Kunau WH. Structural and functional analysis of the interaction of the AAA-peroxins Pex1p and Pex6p. FEBS J. 2005;272(1):47–58. doi: 10.1111/j.1432-1033.2004.04393.x. [DOI] [PubMed] [Google Scholar]
- 52.Birschmann I, et al. Pex15p of Saccharomyces cerevisiae provides a molecular basis for recruitment of the AAA peroxin Pex6p to peroxisomal membranes. Mol Biol Cell. 2003;14(6):2226–2236. doi: 10.1091/mbc.E02-11-0752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tang WK, Xia D. Altered intersubunit communication is the molecular basis for functional defects of pathogenic p97 mutants. J Biol Chem. 2013;288(51):36624–36635. doi: 10.1074/jbc.M113.488924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Okazaki K, Takada S. Structural comparison of F1-ATPase: Interplay among enzyme structures, catalysis, and rotations. Structure. 2011;19(4):588–598. doi: 10.1016/j.str.2011.01.013. [DOI] [PubMed] [Google Scholar]
- 55.DeLaBarre B, Brunger AT. Nucleotide dependent motion and mechanism of action of p97/VCP. J Mol Biol. 2005;347(2):437–452. doi: 10.1016/j.jmb.2005.01.060. [DOI] [PubMed] [Google Scholar]
- 56.Park S, Isaacson R, Kim HT, Silver PA, Wagner G. Ufd1 exhibits the AAA-ATPase fold with two distinct ubiquitin interaction sites. Structure. 2005;13(7):995–1005. doi: 10.1016/j.str.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 57.Malet H, et al. Assembly principles of a unique cage formed by hexameric and decameric E. coli proteins. eLife. 2014;3:e03653. doi: 10.7554/eLife.03653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ohi M, Li Y, Cheng Y, Walz T. Negative staining and image classification - powerful tools in modern electron microscopy. Biol Proc Online. 2004;6:23–34. doi: 10.1251/bpo70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ludtke SJ, Baldwin PR, Chiu W. EMAN: Semiautomated software for high-resolution single-particle reconstructions. J Struct Biol. 1999;128(1):82–97. doi: 10.1006/jsbi.1999.4174. [DOI] [PubMed] [Google Scholar]
- 60.Frank J, et al. SPIDER and WEB: Processing and visualization of images in 3D electron microscopy and related fields. J Struct Biol. 1996;116(1):190–199. doi: 10.1006/jsbi.1996.0030. [DOI] [PubMed] [Google Scholar]
- 61.Li X, et al. Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat Methods. 2013;10(6):584–590. doi: 10.1038/nmeth.2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mindell JA, Grigorieff N. Accurate determination of local defocus and specimen tilt in electron microscopy. J Struct Biol. 2003;142(3):334–347. doi: 10.1016/s1047-8477(03)00069-8. [DOI] [PubMed] [Google Scholar]
- 63.Scheres SH. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol. 2012;180(3):519–530. doi: 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pettersen EF, et al. UCSF Chimera—A visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.