

Abstract
Ribonucleic acid (RNA) is one of the two nucleic acids used by extant biochemistry and plays a central role as the intermediary carrier of genetic information in transcription and translation. If RNA was involved in the origin of life, it should have a facile prebiotic synthesis. A wide variety of such syntheses have been explored. However, to date no one-pot reaction has been shown capable of yielding RNA monomers from likely prebiotically abundant starting materials, though this does not rule out the possibility that simpler, more easily prebiotically accessible nucleic acids may have preceded RNA. Given structural constraints, such as the ability to form complementary base pairs and a linear covalent polymer, a variety of structural isomers of RNA could potentially function as genetic platforms. By using structure-generation software, all the potential structural isomers of the ribosides (BC5H9O4, where B is nucleobase), as well as a set of simpler minimal analogues derived from them, that can potentially serve as monomeric building blocks of nucleic acid–like molecules are enumerated. Molecules are selected based on their likely stability under biochemically relevant conditions (e.g., moderate pH and temperature) and the presence of at least two functional groups allowing the monomers to be incorporated into linear polymers. The resulting structures are then evaluated by using molecular descriptors typically applied in quantitative structure–property relationship (QSPR) studies and predicted physicochemical properties. Several databases have been queried to determine whether any of the computed isomers had been synthesized previously. Very few of the molecules that emerge from this structure set have been previously described. We conclude that ribonucleosides may have competed with a multitude of alternative structures whose potential proto-biochemical roles and abiotic syntheses remain to be explored. Key Words: Evolution—Chemical evolution—Exobiology—Prebiotic chemistry—RNA world. Astrobiology 15, 538–558.
1. Introduction
The molecular solutions life has arrived at for information storage, in the form of DNA and RNA (Fig. 1), are likely evolutionarily optimized with regard to various constraints, including stability, ability to encode information, and ability to compact it in small spaces, such as cells. These requirements can likely only be met by certain molecules given the rules of organic chemistry, though the set of possible molecules could be very large. If there were alternative molecules that could better fulfill these criteria, then extant genetic systems could be considered suboptimal. It is of interest to understand whether biology's solution to these various problems is optimal, suboptimal, or arbitrary. One way to explore this is with structure generation software and in silico property screening.
FIG. 1.

The molecular structures of RNA and DNA and their components. (A) The sugars ribose and deoxyribose and their atom-numbering conventions. Note the stereochemistry of the bonds between the ring and its substituents. (B) The nitrogen heterocycles used in RNA [adenine (A), uracil (U), guanine (G), and cytosine (C)] and DNA [A, G, C, and thymine (T)] and their ring-atom-numbering conventions. (C and D) The structures of phosphate-linked RNA and DNA. (E) A simplified generic nucleoside analog structure.
RNA plays a central role in biochemistry as the transcriptional intermediary of genetic information as well as the mediator of the translation of mRNA messages into peptides and proteins. It has been suggested that RNA preceded DNA in biochemical evolution based on several lines of evidence: the central role of RNA in the flow of information within the cell (Woese, 1967; Crick, 1968; Orgel, 1968), the fact that deoxyribonucleotides are often biosynthesized from ribonucleotides (Benner et al., 1989), and because many enzyme cofactors are ribonucleotide derivatives (White, 1976; King, 1980).
The antiquity and central importance of RNA in biochemistry suggests that RNA is not only ancient but also primordial (Gesteland et al., 2006). Consequently, the quest for a prebiotic synthesis of RNA has been actively pursued over the last 50 years (Sanchez and Orgel, 1970; Fuller et al., 1972a, 1972b; Powner and Sutherland, 2008; Powner et al., 2010; Benner et al., 2012) (Fig. 2).
FIG. 2.

Some previously explored prebiotic syntheses of nucleosides and nucleotides.
To date, no one-pot reaction has yielded either the purine or pyrimidine ribonucleosides directly from likely prevalent prebiotic starting materials, though various steps along the way have been shown to be chemically feasible (Fuller et al., 1972a, 1972b; Ricardo et al., 2004). Several synthetic strategies have been tried, and all lead to some degree to mixtures of isomers. For example, the direct condensation of purines with ribose gives mixtures of α- and β-isomers, as well as various exocyclic amine-substituted molecules (Fuller et al., 1972a, 1972b). The condensation reactions of thymine, cytosine, and uracil with ribose are known to be nonproductive, although the use of alternative pyrimidines has been shown to yield small amounts of pyrimidine ribosides (Bean et al., 2007; Sheng et al., 2009).
Doubts remain regarding the prebiotic plausibility of some of the schemes that have been investigated to date for RNA monomer synthesis (Shapiro, 1984, 1988, 1995; McCollom, 2013). It has thus been suggested that, since RNA is not unique in its ability to carry out its biological functions such as base-pairing and information transfer, it may merely be a frozen accident, perhaps the result of some combination of structural requirements for carrying out a cybernetic function and requirements of the complex set of reinforcing reactions that comprised metabolism during evolution (Braakman and Smith, 2013).
According to this view, RNA is an exceptional solution to the problem of molecular information storage but was not the first solution to this problem in a continuum of molecular systems (Cairns-Smith, 1977; Joyce et al., 1987). Nevertheless, the apparent difficulty of RNA's prebiotic synthesis has led relatively few of those interested in the origin of life to explore the prebiotic synthesis of other molecules that are perhaps more robust (e.g., Nelson et al., 2000).
“Structure space” represents the number of molecular structures that could exist given specific defining parameters (Oprea and Gottfries, 2001; Kirkpatrick and Ellis, 2004; Drew et al., 2012), for example, the total organic structure space, the druglike structure space (Lipinski and Hopkins, 2004; Eberhardt et al., 2011), the amino acid structure space (Meringer et al., 2013), and so on. Many of these chemical spaces are very large. For example, the total number of possible stable druglike organic molecules may be on the order of 1033 to 10180 (Gorse, 2006; Polishchuk et al., 2013). Limiting the diversity and range of properties culls these sets significantly; for example, the number of druglike compounds estimated to be synthesizable by current techniques is on the order of 1020 to 1024 (Ertl, 2003). For comparison, the number of neuro-active compounds, which may be comparable to the size of nucleic acid chemistry-space due to their size and number of potential host-protein recognition surface interaction-point requirements, has been estimated to be in the range of 1015 molecules (Weaver and Weaver, 2011).
In contrast, the number of known naturally occurring or synthetic molecules is much smaller. As of July 2009, there were 49,037,297 unique organic and inorganic chemical substances registered with the Chemical Abstracts Service (Ruddigkeit et al., 2012). The Dictionary of Natural Products as of March 2009 (Ji et al., 2009) listed ∼214,000 compounds, though the “common core” of modern metabolism comprises a mere ∼500 compounds (Dobson, 2004; Smith and Morowitz, 2004), and there are perhaps a few thousand total low-molecular-weight compounds found in the human body (Goto et al., 2002). As a final comparison, a recent exploration of the organic contents of methanol extracts of the Murchison meteorite using high-resolution mass spectrometry revealed a complex though relatively small set of compounds ranging from 105 to perhaps 107 (Schmitt-Kopplin et al., 2010). Clearly, nature is constrained in its exploration of the vastness of chemical space by the reaction mechanisms available to it at any given point in time and the physicochemical stability of the resulting structures in their environmental context.
Antiviral and antisense research has shown that a wide variety of nonnatural nucleotide analogues have biological activity (Périgaud et al., 1992). The recognition and replication features of DNA and RNA are also not unique to those molecules but can also be engendered by a range of related structures (Egholm et al., 1992; Eschenmoser and Loewenthal, 1992; Schöning et al., 2000; Eschenmoser, 2005; Zhang et al., 2010; Pinheiro et al., 2012) (Fig. 3).
FIG. 3.

(A) Some commercial antiviral nucleoside analogues. (B) Antisense nucleoside-analog-based polymers that have found use in biotechnology or that have been found to be capable of Watson-Crick-type base-pairing.
A nucleic acid can be viewed as a polymer of tri-functional monomers, with the three functional moieties being a recognition surface and two points capable of being joined via covalent linkages (Fig. 1E) (Cleaves and Bada, 2012). In naturally occurring nucleic acids, the “recognition surface” is a nitrogenous base, while we term the sugar and its attendant functional groups the “core.” Ribonucleosides are thus tetra-functional (containing a recognition surface and three alcohol groups: 2′, 3′, and 5′), but their reactivity and regiochemistries are enzymatically controlled, avoiding the less stable 2′,5′-linkages (Usher and McHale, 1976; Yin and Steitz, 2002). Natural nucleic acid monomers also include a “linker” molecule, in this case a phosphate moiety. Many other functionalities have been demonstrated to function as unnatural internucleoside linkages, including ethers, esters, sulfonates, amines, phosphonates, and amides (Schneider and Benner, 1990a; Li et al., 2002; Bean et al., 2006; Cleaves and Bada, 2012).
The number of molecules that could fulfill the minimal requirements of being “nucleic acid–like” is remarkably large and in principle limitless, though reasonable arguments could probably be made as to why monomers cannot contain more than some given number of carbon atoms, which might depend on the chemical behavior of such molecules or might be impractical with respect to a dynamic and material-stingy evolving metabolism (Cleaves, 2010). It is in principle difficult to ascertain how common such molecules are in structure space. One way to address this question is to limit exploration of the structural complexity allowed to a specific range of molecular formulas. Presently, there are known nonnatural nucleic acid analogues that have core formulas containing three carbon atoms [e.g., GNA, BC3H7O2 (Ueda et al., 1971)] to eight carbon atoms [e.g., tricyclo-DNA analogues BC8H11O3 (Dugovic et al., 2014)]. Molecules that contain N, S, or other heteroatoms in the linker, recognition elements, or core, while retaining their base-mediated hydrogen-bonding functionality, are also known in both natural and nonnatural systems, and the existence of a few known exemplars makes consideration of nucleic acid–like structure space truly daunting. The number of valid, likely-to-be-stable molecular formulas that contain potentially base-pairing-compatible isomers and include cores containing between three and eight carbon atoms is undoubtedly very large. BC5H9O4 was thus chosen for this study merely for the fact that it is the formula of the extant biological isomer.
Enumeration of the riboside BC5H9O4 space gives some appreciation of the size and dimensionality of nucleic acid–like molecule space and allows some consideration of the optimality or arbitrariness of biology's choice of this particular isomer. Enumeration of the BC5H9O4 chemical space should thus merely be considered an exploratory transect in the very complex formula space of nucleic-acid compatible building blocks.
With respect to the atom choice explored here (using only C, H, and O), we note first that C, H, and O are among the most cosmo- and geochemically abundant elements and that CHO isomers are in principle derivable from formose-type chemistry, which allows an obvious linkage to abiotic geochemistry. S (kept at the −2 oxidation state) could be swapped for O in any of the structures presented. A one-to-one mapping would give an equal number of structures, but there could be plausible molecules containing combinations of S and O (barring chemical incompatibilities that would bear further consideration). There could be as many as 17 times as many possible core isomers where S could entirely, or partially, substitute for O (yielding ∼3859 total isomers for BC5H9OxSy, where x+y=4 and y≥1).
Inclusion of N in the core (restricting it to the −3 oxidation state of amine functionalities) would complicate this analysis considerably. This again underscores the question of why biology restricts itself to this molecular formula for this most fundamental molecule type and whether the functions of RNA could equally well have been carried out by structures not otherwise explored by biology. We are currently calculating the number of plausible isomers over a much broader and more inclusive formula range, with results to be reported shortly. The evaluation of the BC5H9O4 isomer space must thus be viewed as a first practical example of an exploration of what is a much larger chemical space.
We present here the results of a simple exploration of the constitutional space of the ribosides with respect to fundamental functional and chemical constraints imposed by bonding and reactivity rules. Limiting the search to structural isomers with the molecular formula of the core sugar of RNA (BC5H9O4, where B=a nitrogenous base), the range and variety of possible structures is enumerated precisely with structure generation software. The resulting structural isomers are further categorized according to a number of computed descriptors. This gives a glimpse of what abiotic chemistry and evolving biochemistry could produce and provides a host of interesting synthesis targets for medicinal chemistry and basic research.
2. Materials and Methods
2.1. Structure generation
The isomers of the natural ribosides were generated with MOLGEN 5.0 (Gugisch et al., 2009, 2014). MOLGEN 5.0 allows for the calculation of all constitutional isomers of a given molecular formula. In this case, we chose to explore all the isomers of BC5H9O4, the furanosyl ribosides' formula, leaving the phosphate moiety off, which could in any event potentially be replaced by another linker such as glyoxylate (Bean et al., 2006). We coded the base moiety as a “dummy” univalent chlorine (Cl) atom to give a final formula of C5H9O4Cl. This allows enumeration of the isomers of all the A, U, G, C, and T ribosides simultaneously with less computing time. The structure generation is performed iteratively with the concomitant development of a restrictive “bad” list, which precludes the output of structures deemed structurally unstable. In addition, a previously compiled bad list for organic compounds in aqueous solution (Meringer et al., 2013) and a preloaded bad list that excludes highly strained ring systems are used, rings of size 3 and 4 are forbidden, and Cl being connected to a carbon atom is prescribed. Numbers of functional groups such as -OH can be specified by MOLGEN's ability to define the number of hydrogen atoms as part of an atom state. The output was restricted to include molecules with at least two -OH groups. Certain structural constraints could only be formulated as substructures with substructure restrictions, for example, the property of certain atoms lying on rings. Such substructure restrictions are not offered by MOLGEN 5, but they are accessible via MOLGEN–QSPR (Kerber et al., 2004) and were applied as a post-generation filtering step. To generate the complete ribosides, the final output sd file then had the chlorine atom replaced with AUGC or T via an N9 linkage for the purines or an N1 linkage for the pyrimidines. For this, MOLGEN–COMB's ability to virtually execute user-defined reactions on multiple reactants was used (Gugisch et al., 2000; Kerber et al., 2007).
The output files were then processed with ChemAxon's reactor stereoisomer program (http://www.chemaxon.com). These files were then energy-minimized by MOLGEN–QSPR and their descriptors computed. A complete list of the descriptors computable with MOLGEN–QSPR may be found at http://molgen.de/documents/molgenqspr-descriptors/MOLGEN_Descriptors.html.
2.2. Gibbs free energy of formation estimation
The free energies of formation of the adenine-substituted isomers were computed with Group Contribution Method software developed previously (Jankowski et al., 2008).
2.3. Synthetic accessibility scoring
Some organic compounds are more difficult to make, in the laboratory or abiotically, than others. This is due to several factors, among them the ready environmental, commercial, or synthetic availability of precursors; the difficulty of various organic couplings; and the difficulty of introducing and/or maintaining stereocenters during synthesis (Boda et al., 2007). Ease of synthesis can be computed against databases of known synthesized compounds. The commercially available SYLVIA (Estimation of the Synthetic Accessibility of Organic Compounds) software (https://www.molecular-networks.com/products/sylvia) was used to assess the ease of laboratory synthesis of the compounds discussed herein. SYLVIA assigns a synthetic accessibility score on a scale from 1 (facile synthesis) to 10 (complex and challenging synthesis).
SYLVIA's method for calculating synthetic accessibility takes into account a variety of criteria such as the complexity of the target structure, the complexity of the target structure's ring systems, the number of stereo centers in the target, the similarity to commercially available compounds, and the applicability of known synthesis reactions to the target's formation. These criteria are individually weighted and combined to give a single value for synthetic accessibility. Results of a survey of several medicinal chemists are also considered in the weighting process. The synthetic accessibility estimation consists of five components. The molecular graph complexity score is based on graph and information theory and takes into account the size, symmetry, branching, rings, multiple bonds, and heteroatoms of the target molecule. The ring complexity score penalizes bridged and fused ring systems that might be difficult to synthesize. The stereochemical complexity score counts the number of tetrahedral stereo centers in the target. The starting material similarity score takes into account the fact that structures with complex structural motifs can still easily be synthesized if the parts are available starting materials. More similar compounds are identified, and the higher the coverage of the target molecule, the easier it is to synthesize a given target compound.
Synthesis design programs perform retrosynthetic analysis to transform the target structure to a sequence of progressively simpler structures along a retrosynthetic pathway, which ultimately leads to simple starting materials. The overall synthetic accessibility score of a target structure is calculated by summing the five weighted individual components.
The isomer sets were uploaded directly into this software and evaluated using SYLVIA's algorithm.
2.4. Comparison with structure databases
The resulting output files containing the enumerated and filtered structures were parsed against a number of public [PubChem (https://pubchem.ncbi.nlm.nih.gov) and ChEMBL (https://www.ebi.ac.uk/chembl)], publicly available [GDB11 (Fink and Reymond, 2007), GDB13 (Blum and Reymond, 2009), and GDB17 (Ruddigkeit et al., 2012)], and private [Reaxys (www.reaxys.com)] databases to determine whether any of the enumerated molecules had been described previously, either synthetically or computationally. The complete set of U and C riboside formula isomers from the GDB17 database were kindly provided by its authors as SMILES strings and converted to InChi format with OpenBabel software (www.openbabel.org). These were compared against our U and C isomer files for overlap.
The workflow of the structure generation process is shown in Fig. 4.
FIG. 4.
A general work flow for the enumeration of the riboside isomers. The enumeration of good structures can be iterated to produce increasingly “good” sets of output.
3. Results
3.1. Isomer space
Our initial output comprised 32,710 molecular graphs. Excluding disallowed “bad” list elements gave 854 structures, which contained numerous orthoesters and hemiacetals. These were further refined by additions to the bad list or via postprocessing to give a final set of 227 structures shown in Fig. 5.
FIG. 5.

The final enumerated set of riboside isomers. Structures are ordered from right to left and top to bottom according to the location of the double bond equivalent (DBE), beginning with aldehydes, then ketones, esters, BC(=O)C linkages, BC(= O)O linkages, carboxylic acids, and finally rings. The structure corresponding to the natural ribosides is highlighted by a black cartouche.
3.2. Structural properties
The 227 structures in the output library include both acyclic and cyclic species (Table 1) and are available as a supplementary sd file (Supplementary Data are available online at www.liebertonline.com/ast).
Table 1.
Some Pairwise Structural Motifs Found in the Enumerated Isomer Space
| Structure | Aldehyde | Ketone | COOH | B(C=O)C | B(C=O)O | 0°B | 1°B | 2°B | 3°B | 5 Ring | 6 Ring | 7 Ring | Diol | Triol | Tetrol | cis-Diol | Ester-linkable | Diol-linkable | Both linkages possible |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Aldehyde | 28 | 0 | 0 | 0 | 0 | 5 | 12 | 9 | 2 | 0 | 0 | 0 | 16 | 11 | 0 | 7 | 0 | 28 | 0 |
| Ketone | 0 | 17 | 0 | 0 | 0 | 3 | 9 | 3 | 2 | 0 | 0 | 0 | 8 | 9 | 0 | 2 | 0 | 17 | 0 |
| COOH | 0 | 0 | 79 | 0 | 0 | 10 | 39 | 27 | 17 | 0 | 0 | 0 | 29 | 0 | 0 | 14 | 79 | 33 | 33 |
| B(C=O)C | 0 | 0 | 0 | 21 | 9 | 0 | 21 | 0 | 0 | 0 | 0 | 0 | 9 | 12 | 0 | 7 | 0 | 21 | 0 |
| B(C=O)O | 0 | 0 | 0 | 9 | 9 | 9 | 0 | 0 | 0 | 0 | 0 | 0 | 9 | 0 | 0 | 1 | 0 | 9 | 0 |
| 0°B | 5 | 3 | 10 | 0 | 9 | 40 | 0 | 0 | 0 | 6 | 2 | 1 | 32 | 0 | 0 | 5 | 10 | 32 | 0 |
| 1°B | 12 | 9 | 39 | 21 | 9 | 0 | 114 | 0 | 0 | 15 | 5 | 1 | 62 | 27 | 0 | 46 | 39 | 89 | 13 |
| 2°B | 9 | 3 | 27 | 0 | 0 | 0 | 0 | 61 | 0 | 11 | 4 | 0 | 30 | 14 | 1 | 22 | 27 | 22 | 13 |
| 3°B | 2 | 2 | 17 | 0 | 0 | 0 | 0 | 0 | 12 | 2 | 0 | 0 | 11 | 4 | 0 | 2 | 6 | 11 | 5 |
| 5 Ring | 0 | 0 | 0 | 0 | 0 | 6 | 15 | 11 | 2 | 34 | 0 | 0 | 21 | 3 | 1 | 11 | 0 | 34 | 0 |
| 6 Ring | 0 | 0 | 0 | 0 | 0 | 2 | 5 | 4 | 0 | 0 | 11 | 0 | 7 | 4 | 0 | 4 | 0 | 11 | 0 |
| 7 Ring | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 2 | 2 | 0 | 0 | 2 | 0 | 2 | 0 |
| Diol | 16 | 8 | 29 | 9 | 9 | 32 | 62 | 30 | 11 | 21 | 7 | 2 | 129 | 44 | 0 | 50 | 33 | 129 | 31 |
| Triol | 11 | 9 | 0 | 12 | 0 | 0 | 27 | 14 | 4 | 3 | 4 | 0 | 44 | 44 | 0 | 33 | 0 | 44 | 0 |
| Tetrol | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 1 | 0 |
| cis-Diol | 7 | 2 | 14 | 7 | 1 | 5 | 46 | 22 | 2 | 11 | 4 | 2 | 50 | 33 | 1 | 87 | 7 | 87 | 14 |
| Ester-linkable | 0 | 0 | 79 | 0 | 0 | 10 | 39 | 27 | 6 | 0 | 0 | 0 | 33 | 0 | 0 | 7 | 79 | 33 | 33 |
| Diol-linkable | 28 | 17 | 33 | 21 | 9 | 32 | 89 | 22 | 11 | 34 | 11 | 2 | 129 | 44 | 1 | 87 | 33 | 181 | 33 |
| Both linkages possible | 0 | 0 | 33 | 0 | 0 | 0 | 13 | 13 | 5 | 0 | 0 | 0 | 31 | 0 | 0 | 14 | 33 | 33 | 33 |
Categorical definitions: Aldehyde, the molecule contains a terminal RCHO group; Ketone, the molecule contains a C(C=O)C motif; COOH, the molecule contains a free carboxyl group; B(C=O)C, the molecule contains the B moiety attached to a carboxyl-hybridized carbon atom attached to another carbon atom; B(C=O)O, the molecule contains the B moiety attached to a carboxyl-hybridized carbon atom attached to an oxygen atom; 0° to 3° B, the B moiety is attached to a C atom that is attached to 0–3 other C atoms; 5–7 Ring, the molecule contains a ring of this size; Diol–Tetrol, the molecule contains 2–4 hydroxyl groups that are not part of COOH groups; cis-Diol, the molecule contains a 1,2-diol motif; Ester-linkable, the molecule can be incorporated as a polyester directly as it contains both terminal -OH and -COOH groups; Diol-linkable, the molecule can be linked into a polymer either directly as an ether or via the incorporation of an intermediary linker moiety such as phosphate; Both linkages possible, the molecule can be linked in either of the two fashions described above.
In a few cases, these were still permutational isomers representing unique ring closure of the same connectivity isomer, for example, ribofuranose versus ribopyranose isomers; however, these represented a small percentage of the overall enumerated isomers.
It should be noted that the frequency of a given structural motif in this structure space does not imply that the relative frequency of structural motifs would be retained in neighboring formulas with different numbers of atoms. For example, a neighboring formula with a different number of double bond equivalents (DBEs) may increase or lower the output structures that can contain any given set of motifs in an almost unpredictable fashion.
3.3. Aldehyde series
There are 28 aldehyde-containing structures in this set. All but eight of them can in principle cyclize to give 5- and/or 6-membered hemiacetals.
3.4. Ketone series
There are 17 ketone-containing structures, of which only four could potentially cyclize to give 5- or 6-membered hemiketals. In some cases, cyclization could mask hydroxyl groups that could otherwise be employed in polymerization.
3.5. Carboxylic acid series
There are 79 molecules containing free carboxylic acid (i.e., those not involved in ester or other functionalities). All of these could be oligomerized as esters, and 34 of them could cyclize to give 5- or 6-membered lactones. The cyclic lactone products could possibly facilitate polymerization by a ring-opening mechanism (Brunelle, 1993).
3.6. B(C=O)C (glycosyl) acyl-linked nucleobase series
There are 30 isomers containing this motif in the computed set. These appear inherently unstable. It is difficult, however, to know what biochemical systems might accomplish, and such molecules are known in the literature (Dutta et al., 1977), though their decomposition half-lives under physiological or other conditions have not been rigorously determined.
3.7. B(C=O)O (glycosyl) carbamate-linked nucleobase series
There are nine isomers containing this motif, which should be even less stable than the acyl-linked analogues. Given known exemplars from the literature (Dyer and Minnier, 1968), they were retained in this analysis.
3.8. Nucleobase-backbone attachment point
Carbon atoms can be described as unique (herein designated 0°), primary (1°), secondary (2°), or tertiary (3°) based on the number of additional carbon atoms attached to them, here 0, 1, 2, or 3, respectively. The nucleobase moiety could be linked to a 0–3° carbon center. The 1°-C linkage, like the one found in the natural ribosides was observed in 50.2% molecules, with the remainder 0° (17.6%), 2° (26.9%), or 3° (5.2%).
3.9. Ring series
The ring series includes the furanose ribosides (such as the arabinosides), as well as their pyranose forms. A total of 47 (∼21%) out of the 227 output structures include rings. The presence of a ring may limit the “floppiness” of the core and thus reduce the entropic cost of base-pairing in duplex assemblies, although the resulting impact on base-pairing geometries and stabilities is difficult to predict a priori.
These ring systems include thirty-four 5-membered rings, eleven 6-membered rings, and two 7-membered rings, and include one cyclopentane, nineteen furans, fourteen dioxolanes, four pyrans, seven 1,3-dioxanes, and two 1,3-dioxepanes. All of these ring system types are known to occur in natural products (Buckingham, 1993) and thus are in principle derivable from biocatalysis.
3.10. Diol, triol, tetrol, and cis-diol motifs
Simply due to the number of DBEs and the starting formula, a large number of the output structures contain two or more hydroxyl groups. Thus, 56.8% are diols, 19.4% are triols, and one structure (0.4% of the set) is a tetrol. A good deal of RNA's ability to act as a catalyst stems from the cis-diol motif present across the C2′-C3′ portion of the monomer (Ward et al., 2014). In fact, in the set of isomers, this motif occurs 87 (38.3%) times and is thus not especially rare. Of course, some of the utility of the cis-diol lies in the fact that only one of the two hydroxyl groups is involved in backbone formation, while the other is free. The cis-diol motif occurs in molecules that are also triols in 33 instances (14.5% of the set).
3.11. Possible backbone linkages
A total of 181 of the 227 output backbone structures could be linked via two hydroxyl groups (for example, by a phosphate diester or directly as ethers), 79 could be linked directly as esters, and 33 could be linked both ways.
3.12. Ester-linkable molecules
There are 79 structural isomers in this set that could be oligomerized as polyesters, and 46 of these can only be linked as esters. Due to the single DBE in the input formula, all the ester-linkable molecules are acyclic.
3.13. Diol-linkable molecules
A total of 181 of the 227 structural isomers in this set could be oligomerized via two hydroxyl groups (see section 3.11 above). These include acyclic molecules and ring-containing structures.
3.14. Monomers linkable in more than one fashion
Thirty-three of the output structures could be linked by using either OH/OH-mediated linkage chemistry or ester linkage chemistry. In all these cases, there are three possible variations on the mixed linkage; that is, the molecules could be linked via OH/OH linkages and then by either of two ester linkages.
3.15. Stereoisomer space
The 227 generated structural isomers yield 962 total stereoisomers. The ability of each to enable base-pairing bears consideration. The molecules in the final set each contain between zero and five stereocenters. The distribution of stereocenters is shown in Fig. 6, along with the cumulative number of stereoisomers.
FIG. 6.
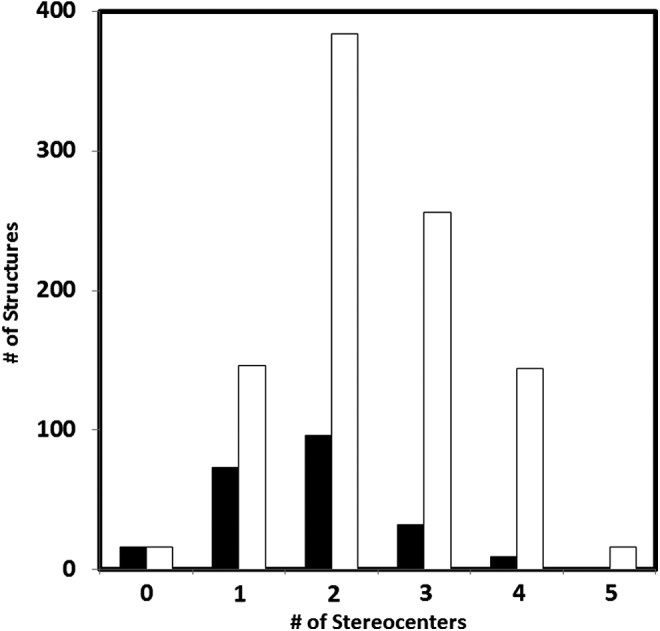
The number of structures with a given number of stereocenters of the output set. Black bars: number of connectivity isomers. White bars: number of total stereoisomers.
Even the lowest molecular weight CHO nucleoside structure (BC3H7O2) has two structural isomers (GNA and iso-GNA), as well as two stereoisomers for GNA (the R and S 2,3-dihydroxypropyl derivatives, iso-GNA is prochiral), while the furanosyl riboside connectivity isomers include α- and β-anomers of eight stereoisomers, for a total of 16 stereoisomers. Biological β-D-ribosides, with four stereocenters (at the C2′, C3′, C4′, and anomeric C1′ atoms), thus are at the high end of the stereoisomerism distribution for this isomer space. Only one of the 227 output isomers has more stereocenters (5) than the biological ribosides, though this isomer has only 16 enantiomers due to meso symmetries that arise. Fourteen of the output structures are achiral, or technically prochiral, as a stereocenter would be introduced upon the formation of a polymer.
Of the 79 ester-linkable molecules, all but two contain stereocenters, and these exceptions would again be prochiral. Of the remaining 77, 34 have one stereocenter (68 enantiomers total), 41 have two (164 enantiomers), and 2 have three each (16 enantiomers).
The majority (∼93%) of the structures in the total output set are chiral. This suggests that a chiral bias may be highly likely in the development of linear genetic polymers of this level of structural complexity. This could be considered as a metric of the synthetic complexity of these molecules or a measure of the functional utility of having many stereocenters in precise orientations in a given molecule.
3.16. Overlap with existing compound databases
The initial Cl-containing and nucleobase-substituted libraries were compared to the Reaxys database (www.reaxys.com), a database of 24,442,243 compounds as of April 11, 2014. The stereochemistry was suppressed in this search; thus the overlap is representative of structural isomerism and not stereoisomerism. We further compared these libraries to the GDB11 [2.64×106 compounds (Fink and Reymond, 2007)], GDB13 [9.7×108 structures (Blum and Reymond, 2009)], and GDB17 [1.66×1011 structures (Ruddigkeit et al., 2012)] libraries, which were computationally generated data sets designed to contain large numbers of druglike molecules. The overlap is shown in Table 2.
Table 2.
Overlap of the Generated Structural Isomer Library with Existing Synthetic and Computational Databases
| Database\Derivative | Cl | A | U | G | C | T |
|---|---|---|---|---|---|---|
| ChEMBL | 0 | 5 | 1 | 1 | 1 | 1 |
| PubMed | nd | 4 | 1 | 1 | 2 | 3 |
| Reaxys | 9 | 16 | 8 | 3 | 7 | 8 |
| GDB11 | 0/33* | X | X | X | X | X |
| GDB13 | 0/0* | X | X | X | X | X |
| GDB17 | 0/0* | X | 54 | X | 54 | X |
Computed with F substituting for Cl.
nd=not determined. X=not possible given the library constraints.
A search through the ChEMBL database (https://www.ebi.ac.uk/chembl), containing 1,359,508 distinct compounds as of April 10, 2014, revealed very few hits (<1%), mainly stereoisomers of the natural nucleosides in both furanosyl and pyranosyl configurations (Table 2). There was no overlap with the Cl-containing library. The result was similar for PubChem (https://pubchem.ncbi.nlm.nih.gov), which contained 11 out of 1135 potential structures (the AUGCT-substituted isomers, also <1%). The Reaxys database, on the other hand, contained a total of 51 out of 1362 possible isomers or 3.7%. It is evident that chemistry space has not been well explored with respect to this structural class.
Finally, the output library was compared to the GDB11, GDB13, and GDB17 databases, both before and after the addition of nucleobases. There was no particular reason to expect that these structures should be present in these libraries; this is simply a measure of the overlap of the two and in a sense the completeness of the libraries. It is worth noting that these databases are limited by the numbers of heavy atoms they can contain (11, 13, or 17, respectively), among other structural restrictions. Our search formula already contains 10 heavy atoms (C5O4Cl), and the addition of a nitrogenous base adds from 8 to 11 heavy atoms (using U or C as replacements for Cl, with 8 heavy atoms each: C4N2O2 or C4N3O, or G, with 11 heavy atoms: C5N5O). Thus, only GDB17 can contain our base-modified output isomers, and then only of the U and C derivatives. GDB17 contains 56,683,397 isomers of cytidine and 26,419,868 isomers of uridine. Among these, there were 133 each C- and U-containing stereoisomers that overlapped with the 962 for each nucleobase derivative in our enumerated sets (∼13.8% overlap), representing 54 of the basic 227 potentially polymer-incorporable stable riboside isomers enumerated here (∼23.8% overlap).
Considering the sizes of these databases, it is somewhat surprising that the output libraries contain so little overlap for molecules containing more than 10 heavy atoms. Some of this is simply due to the truly enormous size of chemical space, in the case of the computed libraries, and some of it is due to the logic and methodologies that drive exploration of real-world chemical space.
4. Comparison with RNA Monomers
RNA is normally linked by 3′→5′ linkages; however, as has been shown by Szostak and coworkers (Engelhart et al., 2013), 2′→5′ linked polymers are also capable of base-pairing and expressing catalytic function. Thus, analogous to the alternative linkages investigated by Eschenmoser (2005), of the 100 output structural isomers that can only be linked via OH/OH-mediated linkages (such as phosphodiesters), 62 are limited to a single isomeric intermolecular linkage. However, another 27 isomers within the enumerated set, including the natural ribosides, can be intermolecularly coupled via three distinct regiochemistries, with the additional complexity of head-to-tail, head-to-head, and tail-to-tail linkage directionality. One remaining isomer lends itself to six possible linkage regiochemistries.
This regioisomeric ambiguity has a functional utility in the linkage of nucleic acids and peptides in translation. Acyl-activated amino acids may jump from the 2′ and 3′ hydroxyl groups of the donor tRNA, or seen in another light, one of the -OH groups makes the other more reactive. A cis-diol motif may thus be a structural requirement, and if encountered in a ring system, it may be required that the two -OH groups be vicinal and syn-oriented.
4.1. Linkage distances
While native nucleic acid oligomers manifest a 6-atom repeating structure along the backbone, as do many analogues such as LNA, HNA, and PNA, this is not an absolute requirement to enable base-pairing. For example, TNA and GNA have 5-atom-unit repeating backbones, and base-pairing systems with 3-, 4-, 5-, and 7-atom backbone repeats are also known (Diederichsen and Schmitt, 1998; Eschenmoser, 2004; Zhang et al., 2005; Kashida et al., 2011).
The distribution of backbone atom repeats for both sets is shown in Fig. 7.
FIG. 7.
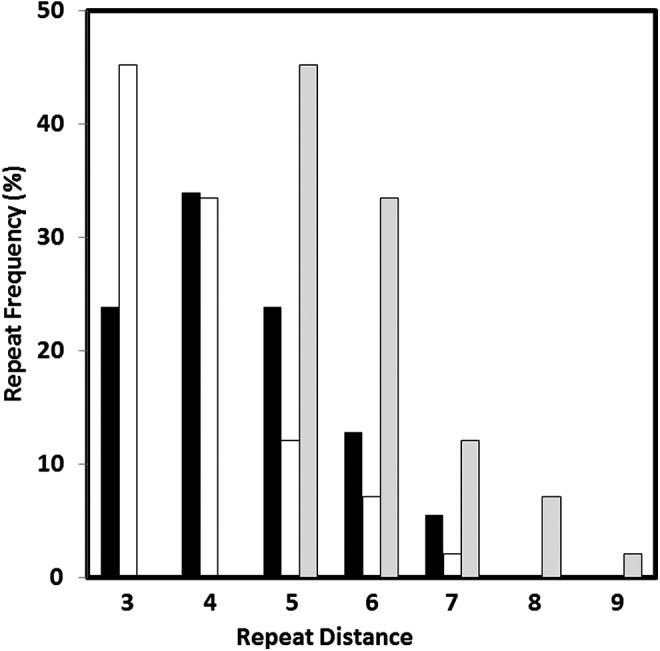
Distribution of possible backbone distance monomer repeats from the enumerated set. Black bars: ester linkages. White bars: diol-mediated linkages connected directly as ethers. Light gray bars: diol linkages assuming incorporation of a phosphate linker. Linkage distance repeat frequencies are summed over the total number of occurrences in the set, i.e., a single structure that can be linked three unique ways is counted three times.
For the enumerated output isomer set, 34% of the ester-linkable structures would give 6-atom repeats, while 33% of the diol-linkage structures would do similarly if linked by a phosphate moiety. The 6-atom repeat is thus not an especially rare motif within this structure space.
The ribosides include more than two hydroxyl groups and can thus be linked in multiple ways, for example as 3′, 5′ linkages or 2′, 5′ linkages. Excluding the combinatorics involved in addition of multiple types of nitrogenous bases [which could be further complicated by the use of other noncanonical bases and other glycosidic or pseudo-glycosidic linkages such as C-glycosides (Benner et al., 1998)], we estimate there are 390 potential regular connectivity polymer systems (ignoring stereochemistry) using only direct ester or diol-phosphate-linkages derivable from even this very restricted set of constitutional isomers of the ribosides.
4.2. Structural comparison with RNA
The presence of the extra, unmasked hydroxyl or COOH group in many of these enumerated riboside isomers potentiates a variety of emergent chemical and catalytic competencies. Eighty-six (∼37.9%) of the enumerated isomers computed here could also retain a free hydroxyl group following macromolecular synthesis.
Among the many properties computed that do not appear to be restricted to the riboside structures, some properties appear to be relatively special in the case of the ribosides. A comparison of the computed moments of inertia (MOI) and molecular shadows of the adenosine isomers along the three principle axes of each molecule is shown in Fig. 8. The MOI is a measure of the distribution of mass about the center of gravity and thus to some degree a measure of the relative “compactness” of the density distribution of a molecule, while the shadow is similarly, but in a sense inversely, a measure of the relative “expansiveness” of the molecule.
FIG. 8.

Computed MOI (top) and cross-sectional molecular shadows (bottom) of the 962 energy-minimized output adenosine-analog stereoisomers, shown in descending order from left to right of the pairwise metrics for the three major axes (i.e., X+Y, Y+Z, Z+X). The stereoisomers of the ribofuranosides are shown as open red circles. D and L β-ribofuranosyladenosine are represented as a solid red circle. Color graphics are available at www.liebertonline.com/ast.
As can be seen, β-ribofuranosyladenosine falls relatively close to the center of the MOI distributions but almost at the extreme of the shadow distributions for the enumerated set. This hints at there being some utility in presenting the largest possible cross section, which may maximize the potential for recognition interactions between the molecules and elements such as nucleic acid–binding peptides.
The rigidity of a nucleoside is also important, as this limits the number of ways the base-pairing elements are presented to complements. More rigid molecules tend to have less freely rotatable bonds. A comparison of optimal compaction and rigidity as measured by computed van der Waals volume and the number of freely rotatable bonds is shown in Fig. 9.
FIG. 9.
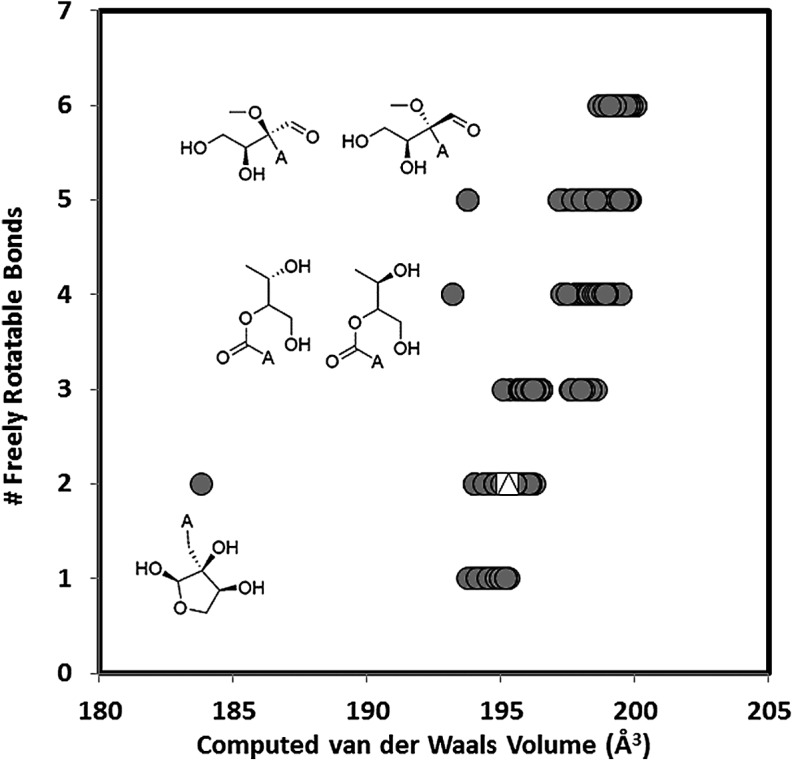
Plot of the number of free rotatable bonds to the computed van der Waals volume. The DL-β-ribofuranosyl- and DL-β-ribopyranosyladenosine structures are shown as a white triangle and square, respectively, superposed over the remaining 958 stereoisomers. The structures associated with the three outlying left-most points are also shown for comparison.
In Fig. 9, more compact and more rigid isomers would fall toward the lower left of the plot. Clearly, among the various possible isomers, the β-ribofuranosylribosides belong to the most compact and rigid set.
4.3. Estimated standard molar Gibbs free energies of formation
The standard molar Gibbs free energies of formation 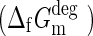 at 298.15 K were computed for the adenosine isomers with group contribution methods previously reported in the literature (Jankowski et al., 2008) (Fig. 10).
at 298.15 K were computed for the adenosine isomers with group contribution methods previously reported in the literature (Jankowski et al., 2008) (Fig. 10).
FIG. 10.
Computed standard molar Gibbs free energies of formation 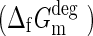 of the 227 A-substituted isomers. The calculated and measured values for adenosine are shown as vertical red and blue lines, respectively. The shaded area represents the estimated uncertainty of±13.4% in the calculated value. Color graphics are available at www.liebertonline.com/ast.
of the 227 A-substituted isomers. The calculated and measured values for adenosine are shown as vertical red and blue lines, respectively. The shaded area represents the estimated uncertainty of±13.4% in the calculated value. Color graphics are available at www.liebertonline.com/ast.
The computed values ranged from −28.43 to −71.12 kcal mol−1, with uncertainties estimated between 8% and 20%. Adenosine fell roughly in the middle of the range with a ΔG of −43.1 kcal mol−1 [literature value −46.5 kcal mol−1 (Boerio-Goates et al., 2001)] and an estimated uncertainty of ±13.4%. For reference, the average value for the entire set was −47.4 kcal mol−1.
4.4. Functionally and structurally minimal core structures
Ribosides are in many senses structurally minimal. There is likely a selective utility for each of their observed functionalities. For example, the atomic repeat of the polymer optimizes the base-stacking interactions, the rigidity of the ring structure optimizes base presentation for base-pairing, the 2′, 3′ cis-diol motif optimizes both metal chelation and strand instability, and so on. However, with respect to the minimal nucleoside motif described in Fig. 1, in principle only the attachment points for incorporation into a linear polymer and the recognition surface are required.
Many of the generated isomers can be reduced to simpler structures based on their core connectivity patterns. This reduction is arbitrary with respect to the functional utility each chemical moiety affords the final structure. For example, various side-chains that are not directly required for forming the core scaffold can be removed to give minimal functional cores. The groups removed could of course confer functional advantages, such as solubility, to the original structures.
Such minimal nucleosides have found considerable use as antiviral compounds, and a few have also been incorporated into strongly base-pairing nucleic acid analogues. The functionally minimal derivatives of this set were created by removing any atoms that would not be directly involved in the formation of a polymer, as shown in Fig. 11.
FIG. 11.

Method for extracting functionally minimal nucleoside motifs from the enumerated riboside isomers.
This allowed the definition of a set containing 34 minimal acyclic riboside analog structures (Fig. 12).
FIG. 12.

The 68 functionally minimal acyclic substructures derived from the computed library, presented from left to right by the number of carbon atoms they contain.
Six of the minimal structures are prochiral, and the remaining 62 have one stereocenter. A total of 37 of the 68 are linkable as esters; the remainder are diol-linkable.
Although the 227 riboside isomers include some useful antivirals (Périgaud et al., 1992), this set also includes many structures that would not be economically reasonable to explore in the quest for useful drugs. The minimal acyclic set, however, has been more widely explored and includes several potent antivirals such as ganciclovir and penciclovir. The 68 structures in the minimal acyclic set were searched against the Reaxys database as the A, U, G, C, and T derivatives. There were 21 hits against N9-G-containing structures, 19 against N9-A-containing structures, 18 against N1-T-containing structures, 16 against N1-U-containing structures, and 9 against N1-C-containing structures. In total, these represent 31 of the 68 structures, including five containing carboxylic acid functional groups. Only four structural motifs represent all five derivatives, and only five motifs (including the previously mentioned four) represent complete potentially “coding sets” (including AUGC or ATGC). The earliest synthesized analogue was made in 1965. Thus, this enterprise has been explored for 50 years already, but little effort has been made to extend it into the alternative nucleic acid space. In this light, it is significant that only three of the 68 structures (GNA, isoGNA, and FNA) (Zhang et al., 2005, 2010; Karri et al., 2013) have been explored as base-pairing systems. It should be noted that the criteria for deciding whether structures are worthy of further exploration has hinged on their measurable effects over the timescales of biological systems, which are already based on another coding structure. Proto-biological systems using other motifs would almost necessarily be more inefficient relative to these and not worthy of exploration from the standpoint of biomedical science. They might be perfectly viable, though, in carrying out information transfer in less robust systems.
The manner in which exploration of this structure space has been carried out is explicable by the fact that the main goal of nucleoside analog research has been antiviral therapy. It has been found that many of the modifications of the riboside structure appeared to predictably not yield virus-inhibitory drugs. For example, none of the seco-nucleosides proved to be good antivirals (Agrofoglio and Challand, 1998), and a few early studies showed that most acyclonucleosides were not good base-pairing systems (Schneider and Benner, 1990a). However, some of the unexplored motifs might have interesting properties in polymeric contexts, and this structure space cannot as yet be considered exhaustively explored. In any event, we provide here a fairly large list of structures that might be explored by intrepid “chemonauts” curious about the fundamental properties of chemical space.
4.5. Synthetic accessibility
The riboside isomers and minimal structures were evaluated with SYLVIA. Molecules are scored according to a database of reactions and known commercially available starting materials, and according to the number of disconnections necessary for synthesis and the number of stereocenters. The SYLVIA-generated overall synthetic accessibility and stereochemical complexity scores are shown in Fig. 13.
FIG. 13.
SYLVIA-generated synthetic accessibility scores for the riboside isomers and minimal structures. The riboside isomers are shown as open black circles, the minimal structures as open red circles. Individual isomers are annotated. Color graphics are available at www.liebertonline.com/ast.
As can be seen, the β-ribofuranosyl structure is not the simplest within this enumerated chemistry space based on this metric, though it is simpler than many of its isomers. Of course this is evaluated from the standpoint of laboratory synthesis, and the starting materials and transformational chemistry are well known and readily available in the case of the β-ribofuranosyl structure. An equivalent calculation cannot as yet readily be carried out to evaluate prebiotic synthesizability.
The stereochemical score, which simply determines how many stereocenters are present in the target, highlights that the ribosides are considerably more complex than many of the riboside isomers and all the minimal structures. Interestingly, isoGNA, likely by virtue of being prochiral, scores among the simplest of molecules. FNA is similarly scored as very simple. GNA, likely by virtue of having a stereocenter, scores as moderately complex.
5. Discussion
The structural space explored here is restricted to the molecular formula of the core RNA riboside but nonetheless includes a large number of possible isomers. In the formula range from BC3H7O2 (GNA's formula) to BC5H9O4 (RNA's) there are likely scores of valid formulas. These could collectively produce many thousands of structurally sound isomers. In turn, each of these isomers could yield many stereo- and macromolecular linkage-isomers, leading ultimately to perhaps billions of nucleic acid polymer types potentially capable of supporting base-pairing.
It is likely that only a subset of these structural and stereoisomers would lead to stable base-pairing systems, and we presently have no guiding algorithm to predict the location or population density of base-pairing systems in molecular formula or structure space, which undoubtedly depends on more subtle questions of molecular geometry and stereoelectronics. A more detailed view into these structure spaces will likely require continued experimental efforts as well as molecular dynamics simulations. There are only two connectivity isomers of GNA (GNA and iso-GNA), which together have three stereoisomers, which fulfill the two OH criteria. Only one (50% in structural space or 67% in enantiomer space) of these has generally proven to be a good base-pairing system using canonical nucleobases as recognition surfaces and phosphate as a linker (Seita et al., 1972; Meggers and Zhang, 2010; Karri et al., 2013). Eschenmoser and colleagues explored many of the 16 riboside stereoisomers (Eschenmoser, 1999a, 2005) and found that a good number were effective base-pairing systems, though our analysis suggests many isomers remain to be explored. Thus, the best we can say is that the potential number of base-pairing systems is likely less than 50% in isomer space. Given that so few of these molecules have been synthesized, this still leaves a vast number of polymer systems to be explored experimentally.
Freier and Altmann described some 200 experimental modifications of DNA structure, including modifications of the base, sugar, and backbone, and their impact on hybridization to complementary RNA strands as measured by change in melting temperature (Tm) (Freier and Altmann, 1997). The large majority of modifications resulted in duplex destabilization. One important reason these authors suggested some structures performed better as complements is that the modified strands were able to “pre-organize” to adopt conformations that were compatible with those of their RNA complements. Thus, it may be reasonable to conclude that the vast majority of the structures generated here would not be good RNA-antisense molecules.
However, as Freier and Altmann also noted, there are exceptions, for example, PNA (Egholm et al., 1992) and morpholino-linked analogues (Summerton and Weller, 1997), which are very good duplex-forming molecules. These authors also noted an even more significant exception in the context of the notion of alternative informational systems, as opposed to antisense systems, in the 2,3-dideoxy-D-glucopyanosyl homo-DNA described by Eschenmoser and colleagues, duplexes of which have a higher Tm than cognate DNA/DNA duplexes, which do not form stable duplexes themselves with complementary DNA strands and would not be expected to do so based on their geometry (Eschenmoser and Dobler, 1992). Thus, there is good reason to expect that few of the molecules enumerated here might be good RNA-binders in a pharmaceutical context, though whether they could be passable in an evolutionary context remains to be determined. Many more, however, could be capable of binding complements with their own composition. This is unfortunately the crucial question, and more unfortunately this can presently only be determined empirically. The development of computational methods for rapid screening of these questions would be of enormous interest.
It is of course possible that there is a literature bias toward structures that exhibit stable base-pairing due to the tendency of researchers to not report structures that fail to do so. We cannot reasonably speculate what fraction of experiments has not been reported in the literature. It may be that most of the isomeric structures in the final set of 227 (and their much larger coterie of enantiomers) have been synthesized and failed to show base-pairing in the laboratory; however, this may be unlikely. There are many structures that have required complicated synthesis, have failed to show stable base-pairing (though not necessarily isomers of the single formula considered herein), and nevertheless have been reported in the literature, dating back at least 40 years. Some examples include FNA (Schneider and Benner, 1990b), which has since been found to be a more stable base-pairing structure than was originally believed (Zhang et al., 2010); various S-linked riboside polymers, which were found to be poor base-pairing systems (Schneider and Benner, 1990a); amino acid backbone systems (Buttrey et al., 1975); and systems using unusual bases (Mittapalli et al., 2007).
It remains a matter of speculation whether the molecular structures involved in contemporary biochemistry are fundamental or were derived during evolution (Pace, 2001; Davila and McKay, 2014). Many biochemicals, such as various amino acids, are robustly made via abiotic synthesis (Oró and Kimball, 1961; Miller and Orgel, 1974; Eschenmoser and Loewenthal, 1992); however, this must be considered carefully in the context of structure space. Evolving biological systems must have devised ways to reproduce them reliably, which decoupled them from dependence on environmentally supplied organics.
Although some nucleic acid components, such as adenine and guanine, are also easily derived from abiotic chemistry (Borquez et al., 2005; Callahan et al., 2011), nucleosides themselves are more complex and may require biochemical systems for their construction (Bywater, 2012). It is not known what percentage of the riboside analogues described here could easily be accessed by prebiotic chemistry. Although the synthetic space of the extant nucleotides has been explored, and this has given at best ambiguous results, there are likely other structures worthy of exploration in the search for primordial informational genetic polymers.
5.1. Charge and COSMIC “inter”-LOPERs
It has been suggested that proteins and nucleic acids explore fundamentally different structure-property spaces (Benner, 1999); proteins often fold into compact globular structures in which molecular surface properties counterbalance propensities for precipitation. Biological nucleic acids have the useful property that their sequences can be completely modified without almost ever compromising the molecule's overall solubility, as the invariant portion of the molecule, the backbone linker and core, compensate for any changes introduced by modification of the recognition elements. This requires approximately one permanent charge per ∼300 Da of mass. Thus, genetic molecules must be, as Benner (1999) termed them, “COSMIC LOPERs” (Capable Of Searching Mutation-space Independent of Concern over Loss Of Properties Essential for Replication), which may be one of the reasons for the inclusion of phosphate in biological nucleic acids (Westheimer, 1987). The inclusion of this criterion would severely limit the number of structures compatible with open-ended evolution. Of course, at earlier points in biological evolution, smaller genomes could have tolerated a greater variety of component monomers, “COSMIC interLOPERs,” with properties less optimal than those of ribonucleotides.
The enumerated set includes molecules that can be linked into polymers in a variety of ways. Though OH/OH-linked molecules can, for example, be linked via ether linkages, this would provide no net charge on the resulting polymer. Alternatively, such molecules could be joined by any number of uncharged or ionizable linkers, such as phosphate, as in DNA and RNA. Ester-linked polymers, though the linkage is in some ways simpler as no linker is required, would not provide solubilizing charges.
5.2. Retrosynthetic analysis
There are many viable, though synthetically challenging, structures in this library, the majority of which have not yet been explored. Some may have unusual properties, including facile abiotic syntheses, suggesting that there may be an evolutionary explorable landscape of structures from which early life could have scaffolded itself into more optimal structures. Further, there appear to be many structures that could have been arrived at from various formose reaction products (Shigemasa et al., 1977; Schwartz and Degraaf, 1993) or prebiotic Michael acceptors (Cleaves, 2002). For example, structures reminiscent of the pentaerythrityl isomers suggested by Joyce et al. (1987) or the atactic glycerol-like structures studied by Schwartz et al. (Visscher and Schwartz, 1988; Zhang et al., 2010) are represented here.
Our initial structure-generation task generated molecules that could contain up to four hydroxyl groups, but -OH groups are also a component of carboxyl groups. Thus, since the input formula contains one DBE, molecules containing one carboxyl group and an additional two hydroxyl groups were allowed output. While our initial structure-generation task was to generate molecules that could be used as part of phosphate-linked backbone structures, molecules containing one hydroxyl group and one carboxyl group could also be joined together in polyester polymers.
5.3. Prebiotic synthesis
Relatively few precursors are likely responsible or necessary for the molecular diversity observed in prebiotic chemistry, for example that observed in carbonaceous chondrites (Nagy et al., 1963; Peltzer et al., 1984; Callahan et al., 2011; Cooper et al., 2011), Miller-Urey-type reactions (Miller, 1957), and various other prebiotic chemical-diversity-generating systems (Schwartz and Degraaf, 1993; Neish et al., 2010; He et al., 2012).
Collectively, the origins-of-life research community has directed much effort into understanding how the ribosides could have arisen de novo on primitive Earth, while relatively little research has gone into exploring the prebiotic synthesis of nucleotide analogues that may have considerably simpler syntheses and comparable properties (Joyce et al., 1987; Nielsen, 1993; Cleaves and Bada, 2012). Thanks to the achievements of synthetic organic chemists, scores of alternative molecules are now known, along with the large number described in this article, including both the riboside isomers and the structurally minimal nucleoside analogues. For reasons that are likely guided by the logic of Occam's razor, and the so-called continuity principle (Mulkidjanian et al., 2012), together suggesting that only extant biological molecules are valid synthetic targets for prebiotic chemistry, the prebiotic syntheses of very few of these has yet been explored (Nelson et al., 2000).
5.4. Biosynthesis
Analysis of the Gibbs free energies of formation of the various isomers shows that β-adenosine is not thermodynamically unusual within this structure space, and there are both thermodynamically more and less costly structures available. This suggests that thermodynamics was not the principle criterion for nucleoside selection during evolution but rather provided only a weak constraint acting in concert with other more stringent filters such as stability and concordance of structural properties with functional needs.
6. Conclusions
The isomer space of the biological ribosides has been exhaustively computed here. A total of 227 structures were deemed to be structurally reasonable from the standpoint of organic chemistry in water, representing 962 enantiomers. These isomers can potentially be incorporated into polymers that are either linked by a double condensation-based linkage using phosphate or another linker, or a single condensation reaction as esters. Prebiotic chemistry, which has been explored to introduce linkers, such as phosphate (Beck et al., 1967; Rabinowitz et al., 1968; Schwartz and Ponnampe, 1968; Lohrmann and Orgel, 1971; Slabaugh et al., 1974; Schoffstall, 1976; Pasek, 2008) and activating groups into such molecules, should be in many cases equally applicable to many of these isomers.
This search was constrained by the use of the riboside formula, which limits both atom types and ratios, by consideration of the likely chemical stability of the resulting analogues and by the overlay of structural requirements allowing incorporation of the analogues into a polymer. Greater structural diversity could be obtained by using a less restrictive formula search and by including other elements (such as nitrogen), which is the subject of a forthcoming study. Nevertheless, the present work, even within its restricted scope, indicates the remarkable number of alternative genetic platforms that nature could have explored during biochemical evolution.
There are likely many tens of thousands of possible side chain scaffolds containing only C, H, and O that would allow for the presentation of two hydroxyl groups and the stable attachment of a nitrogenous base in the numerous valid molecular formulas extending up to that of the ribosides. Known examples outside of this formula range include GNA (Ueda et al., 1971; Seita et al., 1972; Zhang et al., 2005) and iso-GNA (3-carbon side-chain analogues), TNA [a 4-carbon side-chain analogue (Eschenmoser, 2004; Herdewijn, 2001)], and various five-carbon isomers, including β-D pyranose riboside isomers (Pitsch et al., 1993; Eschenmoser, 1999b). Six-carbon isomers that enable effective supramolecular base-pairing are also known (see Fig. 3).
Many known antiviral and antimicrobial drugs are nucleoside analogues (Périgaud et al., 1992; Gallant et al., 2003), and there is a very poor overlap of the generated library with drugs explored to date. Some of the as-yet-unexplored isomers generated here may very well be useful as therapeutics. That so many of these have not been explored points to there possibly being activities that remain to be clinically studied. The requirement of having two attachment points is of course antithetical to the exploration of chain-terminating nucleoside analogues, such as 2′,3′-dideoxynucleoside analogues; thus there is a bias against finding these types of therapeutics in this set.
Examination of these alternative nucleoside structures raises several interesting questions. How might we detect other biologies that use these or other alternative genetic molecules? Is RNA a frozen accident of the evolutionary process that gave rise to terrestrial biochemistry, or is it highly predetermined by its inherent chemical properties?
The results presented here can be interpreted in multiple ways. First, laboratory synthesis has demonstrated that RNA is not unique in being able to carry out functions considered fundamental to genetic inheritance (Schwartz and Orgel, 1985; Schmidt et al., 1997; Kozlov et al., 1999a, 1999b; Chen et al., 2009; Pinheiro et al., 2012). Second, as we have shown here the structure space of molecules that could carry out these functions is very sparsely explored computationally or synthetically. Third, the prebiotic synthesis of molecules that are capable of carrying out these functions is extremely poorly explored, and prebiotic syntheses of the vast majority remain uninvestigated. Given this enormous structural space, the notion of an easily prebiotically accessible alternative genetic polymer or as a possible alternative evolutionary outcome should not be discounted too quickly.
Supplementary Material
Abbreviations Used
- DBE
double bond equivalent
- MOI
moments of inertia
- QSPR
quantitative structure–property relationship
Acknowledgments
H.J.C. would like to thank the Center for Chemical Evolution for funding support for the software used in this study. J.T.G. and M.M. would like to thank the Earth-Life Science Institute at the Tokyo Institute of Technology for travel and research support during the preparation of this manuscript. J.T.G. would also like to acknowledge individual support through a 2013–15 AAAS Science & Technology Policy Fellowship. We also thank Professor Linda Broadbent and Dr. Di Wu of Northwestern University for generously supplying us with their software package and assisting us in its use. We are also indebted to Professor Jean-Louis Reymond and Dr. Ricardo Visini of the University of Berne for providing us with the C and U isomer output of the GDB17.
Author Disclosure Statement
No competing financial interests exist.
References
- Agrofoglio L., and Challand S.R. (1998) Acyclic, Carbocyclic and L-Nucleosides, Springer, Dordrecht, the Netherlands [Google Scholar]
- Bean H.D., Anet F.A., Gould I.R., and Hud N.V. (2006) Glyoxylate as a backbone linkage for a prebiotic ancestor of RNA. Orig Life Evol Biosph 36:39–63 [DOI] [PubMed] [Google Scholar]
- Bean H.D., Sheng Y., Collins J.P., Anet F.A., Leszczynski J., and Hud N.V. (2007) Formation of a beta-pyrimidine nucleoside by a free pyrimidine base and ribose in a plausible prebiotic reaction. J Am Chem Soc 129:9556–9557 [DOI] [PubMed] [Google Scholar]
- Beck A., Lohrmann R., and Orgel L.E. (1967) Phosphorylation with inorganic phosphates at moderate temperatures. Science 157:952. [DOI] [PubMed] [Google Scholar]
- Benner S.A. (1999) How small can a microorganism be? In Size Limits of Very Small Microorganisms: Proceedings of a Workshop, Steering Group for the Workshop on Size Limits of Very Small Microorganisms, Space Studies Board, National Academies Press, Washington, DC, pp 126–138 [Google Scholar]
- Benner S.A., Ellington A.D., and Tauer A. (1989) Modern metabolism as a palimpsest of the RNA world. Proc Natl Acad Sci USA 86:7054–7058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benner S.A., Battersby T.R., Eschgfaller B., Hutter D., Kodra J.T., Lutz S., Arslan T., Baschlin D.K., Blattler M., Egli M., Hammer C., Held H.A., Horlacher J., Huang Z., Hyrup B., Jenny T.F., Jurczyk S.C., Konig M., von Krosigk U., Lutz M.J., MacPherson L.J., Moroney S.E., Muller E., Nambiar K.P., Piccirilli J.A., Switzer C.Y., Vogel J.J., Richert C., Roughton A.L., Schmidt J., Schneider K.C., and Stackhouse J. (1998) Redesigning nucleic acids. Pure Appl Chem 70:263–266 [DOI] [PubMed] [Google Scholar]
- Benner S.A., Kim H.-J., and Carrigan M.A. (2012) Asphalt, water, and the prebiotic synthesis of ribose, ribonucleosides, and RNA. Acc Chem Res 45:2025–2034 [DOI] [PubMed] [Google Scholar]
- Blum L.C. and Reymond J.-L. (2009) 970 million druglike small molecules for virtual screening in the chemical universe database GDB-13. J Am Chem Soc 131:8732–8733 [DOI] [PubMed] [Google Scholar]
- Boda K., Seidel T., and Gasteiger J. (2007) Structure and reaction based evaluation of synthetic accessibility. J Comput Aided Mol Des 21:311–325 [DOI] [PubMed] [Google Scholar]
- Boerio-Goates J., Francis M.R., Goldberg R.N., da Silva R., Manuel A., Maria D., and Tewari Y.B. (2001) Thermochemistry of adenosine. J Chem Thermodyn 33:929–947 [Google Scholar]
- Borquez E., Cleaves H.J., Lazcano A., and Miller S.L. (2005) An investigation of prebiotic purine synthesis from the hydrolysis of HCN polymers. Orig Life Evol Biosph 35:79–90 [DOI] [PubMed] [Google Scholar]
- Braakman R. and Smith E. (2013) The compositional and evolutionary logic of metabolism. Phys Biol 10, doi: 10.1088/1478-3975/10/1/011001 [DOI] [PubMed] [Google Scholar]
- Brunelle D.J. (1993) Ring-Opening Polymerization: Mechanisms, Catalysis, Structure, Utility, Oxford University Press, Oxford, UK [Google Scholar]
- Buckingham J. (1993) Dictionary of Natural Products, Chapman & Hall, London [Google Scholar]
- Buttrey J.D., Jones A.S., and Walker R.T. (1975) Synthetic analogues of polynucleotides—XIII: the resolution of DL-β-(thymin-1-yl)alanine and polymerisation of the β-(thymin-1-yl)alanines. Tetrahedron 31:73–75 [Google Scholar]
- Bywater R.P. (2012) On dating stages in prebiotic chemical evolution. Naturwissenschaften 99:167–176 [DOI] [PubMed] [Google Scholar]
- Cairns-Smith A.G. (1977) Takeover mechanisms and early biochemical evolution. Biosystems 9:105–109 [DOI] [PubMed] [Google Scholar]
- Callahan M.P., Smith K.E., Cleaves H.J., Ruzicka J., Stern J.C., Glavin D.P., House C.H., and Dworkin J.P. (2011) Carbonaceous meteorites contain a wide range of extraterrestrial nucleobases. Proc Natl Acad Sci USA 108:13995–13998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.J., Cai X., and Szostak J.W. (2009) N2′→P3′ Phosphoramidate glycerol nucleic acid as a potential alternative genetic system. J Am Chem Soc 131:2119–2121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleaves H.J., II (2002) The reactions of nitrogen heterocycles with acrolein: scope and prebiotic significance. Astrobiology 2:403–415 [DOI] [PubMed] [Google Scholar]
- Cleaves H.J., II (2010) The origin of the biologically coded amino acids. J Theor Biol 263:490–498 [DOI] [PubMed] [Google Scholar]
- Cleaves H.J., II, and Bada J. (2012) The prebiotic chemistry of alternative nucleic acids. In Genesis—In The Beginning: Precursors of Life, Chemical Models and Early Biological Evolution, edited by Seckbachs J., Springer, Dordrecht, the Netherlands, pp 3–33 [Google Scholar]
- Cooper G., Reed C., Nguyen D., Carter M., and Wang Y. (2011) Detection and formation scenario of citric acid, pyruvic acid, and other possible metabolism precursors in carbonaceous meteorites. Proc Natl Acad Sci USA 108:14015–14020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crick F.H.C. (1968) The origin of the genetic code. J Mol Biol 38:367–379 [DOI] [PubMed] [Google Scholar]
- Davila A.F. and McKay C.P. (2014) Chance and necessity in biochemistry: implications for the search for extraterrestrial biomarkers in Earth-like environments. Astrobiology 14:534–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diederichsen U. and Schmitt H.W. (1998) β-Homoalanyl PNAs: synthesis and indication of higher ordered structures. Angew Chem Int Ed Engl 37:302–305 [DOI] [PubMed] [Google Scholar]
- Dobson C.M. (2004) Chemical space and biology. Nature 432:824–828 [DOI] [PubMed] [Google Scholar]
- Drew K.L., Baiman H., Khwaounjoo P., Yu B., and Reynisson J. (2012) Size estimation of chemical space: how big is it? J Pharm Pharmacol 64:490–495 [DOI] [PubMed] [Google Scholar]
- Dugovic B., Wagner M., and Leumann C.J. (2014) Structure/affinity studies in the bicyclo-DNA series: synthesis and properties of oligonucleotides containing bcen-T and iso-tricyclo-T nucleosides. Beilstein J Org Chem 10:1840–1847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta S.P., Hong C.I., Tritsch G.L., Cox C., Parthasarthy R., and Chheda G.B. (1977) Synthesis and biological activities of some N6- and N9-carbamoyladenines and related ribonucleosides. J Med Chem 20:1598–1607 [DOI] [PubMed] [Google Scholar]
- Dyer E. and Minnier C.E. (1968) Acylations of some 2-amino-6-halo- and 2-amino-6-alkylthiopurines. J Med Chem 11:1232–1234 [DOI] [PubMed] [Google Scholar]
- Eberhardt L., Kumar K., and Waldmann H. (2011) Exploring and exploiting biologically relevant chemical space. Curr Drug Targets 12:1531–1546 [DOI] [PubMed] [Google Scholar]
- Egholm M., Buchardt O., Nielsen P.E., and Berg R.H. (1992) Peptide nucleic acids (PNA). Oligonucleotide analogs with an achiral peptide backbone. J Am Chem Soc 114:1895–1897 [Google Scholar]
- Engelhart A.E., Powner M.W., and Szostak J.W. (2013) Functional RNAs exhibit tolerance for non-heritable 2′–5′ versus 3′–5′ backbone heterogeneity. Nat Chem 5:390–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ertl P. (2003) Cheminformatics analysis of organic substituents: identification of the most common substituents, calculation of substituent properties, and automatic identification of drug-like bioisosteric groups. J Chem Inf Comput Sci 43:374–380 [DOI] [PubMed] [Google Scholar]
- Eschenmoser A. (1999a) Chemical etiology of nucleic acid structure. Science 284:2118–2124 [DOI] [PubMed] [Google Scholar]
- Eschenmoser A. (1999b) Pyranosyl-oligonucleotides. Nucleosides Nucleotides Nucleic Acids 18:1363–1364 [Google Scholar]
- Eschenmoser A. (2004) The TNA-family of nucleic acid systems: properties and prospects. Orig Life Evol Biosph 34:277–306 [DOI] [PubMed] [Google Scholar]
- Eschenmoser A. (2005) Searching for nucleic acid alternatives. Chimia 59:836–850 [Google Scholar]
- Eschenmoser A. and Dobler M. (1992) Warum Pentose- und nicht Hexose-Nucleinsäuren? Teil I. Einleitung und Problemstellung, Konformationsanalyse für Oligonucleotid-Ketten aus 2′,3′-Dideoxyglucopyranosyl-Bausteinen (‘Homo-DNS’) sowie Betrachtungen zur Konformation von A- und B-DNS. Helv Chim Acta 75:218–259 [Google Scholar]
- Eschenmoser A. and Loewenthal E. (1992) Chemistry of potentially prebiological natural products. Chem Soc Rev 21:1–16 [Google Scholar]
- Fink T. and Reymond J.-L. (2007) Virtual exploration of the chemical universe up to 11 atoms of C, N, O, F: assembly of 26.4 million structures (110.9 million stereoisomers) and analysis for new ring systems, stereochemistry, physicochemical properties, compound classes, and drug discovery. J Chem Inf Model 47:342–353 [DOI] [PubMed] [Google Scholar]
- Freier S.M. and Altmann K.-H. (1997) The ups and downs of nucleic acid duplex stability: structure-stability studies on chemically-modified DNA: RNA duplexes. Nucleic Acids Res 25:4429–4443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller W.D., Sanchez R.A., and Orgel L.E. (1972a) Studies in prebiotic synthesis. VI. Synthesis of purine nucleosides. J Mol Biol 67:25–33 [DOI] [PubMed] [Google Scholar]
- Fuller W.D., Sanchez R.A., and Orgel L.E. (1972b) Studies in prebiotic synthesis: VII. Solid-state synthesis of purine nucleosides. J Mol Evol 1:249–257 [DOI] [PubMed] [Google Scholar]
- Gallant J.E., Gerondelis P.Z., Wainberg M.A., Shulman N.S., Haubrich R.H., St Clair M., Lanier E.R., Hellmann N.S., and Richman D.D. (2003) Nucleoside and nucleotide analogue reverse transcriptase inhibitors: a clinical review of antiretroviral resistance. Antivir Ther 8:489–506 [PubMed] [Google Scholar]
- Gesteland R.F., Cech T., and Atkins J.F. (2006) The RNA World: The Nature of Modern RNA Suggests a Prebiotic RNA World, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- Gorse A.-D. (2006) Diversity in medicinal chemistry space. Curr Top Med Chem 6:3–18 [DOI] [PubMed] [Google Scholar]
- Goto S., Okuno Y., Hattori M., Nishioka T., and Kanehisa M. (2002) LIGAND: database of chemical compounds and reactions in biological pathways. Nucleic Acids Res 30:402–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gugisch R., Kerber A., Laue R., Meringer M., and Weidinger J. (2000) MOLGEN–COMB, a software package for combinatorial chemistry. Match: Communications in Mathematical and in Computer Chemistry 41:189–203 [Google Scholar]
- Gugisch R., Kerber A., Kohnert A., Laue R., Meringer M., Rücker C., and Wassermann A. (2009) MOLGEN Structure Elucidation, Reference Guide, version 5.0. Available online at http://molgen.de/documents/manual50.pdf
- Gugisch R., Kerber A., Kohnert A., Laue R., Meringer M., Rücker C., and Wassermann A. (2014) MOLGEN 5.0, a molecular structure generator. In Advances in Mathematical Chemistry and Applications, edited by Basak S.C., Restrepo G., and Villavecess J.L., Bentham Science Publishers Ltd, Bussum, the Netherlands, p 113–138 [Google Scholar]
- He C., Lin G., and Smith M.A. (2012) NMR identification of hexamethylenetetramine and its precursor in Titan tholins: implications for Titan prebiotic chemistry. Icarus 220:627–634 [Google Scholar]
- Herdewijn P. (2001) TNA as a potential alternative to natural nucleic acids. Angew Chem Int Ed Engl 40:2249–2251 [DOI] [PubMed] [Google Scholar]
- Jankowski M.D., Henry C.S., Broadbelt L.J., and Hatzimanikatis V. (2008) Group contribution method for thermodynamic analysis of complex metabolic networks. Biophys J 95:1487–1499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji H.F., Li X.J., and Zhang H.Y. (2009) Natural products and drug discovery. EMBO Rep 10:194–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce G.F., Schwartz A.W., Miller S.L., and Orgel L.E. (1987) The case for an ancestral genetic system involving simple analogues of the nucleotides. Proc Natl Acad Sci USA 84:4398–4402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karri P., Punna V., Kim K., and Krishnamurthy R. (2013) Base-pairing properties of a structural isomer of glycerol nucleic acid. Angewandte Chemie 125:5952–5956 [DOI] [PubMed] [Google Scholar]
- Kashida H., Murayama K., Toda T., and Asanuma H. (2011) Control of the chirality and helicity of oligomers of serinol nucleic acid (SNA) by sequence design. Angewandte Chemie 123:1321–1324 [DOI] [PubMed] [Google Scholar]
- Kerber A., Laue R., Meringer M., and Rücker C. (2004) MOLGEN–QSPR, a software package for the search of quantitative structure property relationships. Match: Communications in Mathematical and in Computer Chemistry 51:187–204 [Google Scholar]
- Kerber A., Laue R., Meringer M., and Rücker C. (2007) Molecules in silico: a graph description of chemical reactions. J Chem Inf Model 47:805–817 [DOI] [PubMed] [Google Scholar]
- King G. (1980) Evolution of the coenzymes. Biosystems 13:23–45 [DOI] [PubMed] [Google Scholar]
- Kirkpatrick P. and Ellis C. (2004) Chemical space. Nature 432:823 [Google Scholar]
- Kozlov I.A., Politis P.K., Pitsch S., Herdewijn P., and Orgel L.E. (1999a) A highly enantio-selective hexitol nucleic acid template for nonenzymatic oligoguanylate synthesis. J Am Chem Soc 121:1108–1109 [DOI] [PubMed] [Google Scholar]
- Kozlov I.A., Politis P.K., Van Aerschot A., Busson R., Herdewijn P., and Orgel L.E. (1999b) Nonenzymatic synthesis of RNA and DNA oligomers on hexitol nucleic acid templates: the importance of the A structure. J Am Chem Soc 121:2653–2656 [DOI] [PubMed] [Google Scholar]
- Li X., Zhan Z.-Y.J., Knipe R., and Lynn D.G. (2002) DNA-catalyzed polymerization. J Am Chem Soc 124:746–747 [DOI] [PubMed] [Google Scholar]
- Lipinski C. and Hopkins A. (2004) Navigating chemical space for biology and medicine. Nature 432:855–861 [DOI] [PubMed] [Google Scholar]
- Lohrmann R. and Orgel L.E. (1971) Urea-inorganic phosphate mixtures as prebiotic phosphorylating agents. Science 171:490–494 [DOI] [PubMed] [Google Scholar]
- McCollom T.M. (2013) Miller-Urey and beyond: what have we learned about prebiotic organic synthesis reactions in the past 60 years? Annu Rev Earth Planet Sci 41:207–229 [Google Scholar]
- Meggers E. and Zhang L. (2010) Synthesis and properties of the simplified nucleic acid glycol nucleic acid. Acc Chem Res 43:1092–1102 [DOI] [PubMed] [Google Scholar]
- Meringer M., Cleaves H.J., and Freeland S.J. (2013) Beyond terrestrial biology: charting the chemical universe of α-amino acid structures. J Chem Inf Model 35:2851–2862 [DOI] [PubMed] [Google Scholar]
- Miller S.L. (1957) The mechanism of synthesis of amino acids by electric discharges. Biochim Biophys Acta 23:480–489 [DOI] [PubMed] [Google Scholar]
- Miller S.L. and Orgel L.E. (1974) The Origins of Life on the Earth, Prentice-Hall, Englewood Cliffs, NJ [Google Scholar]
- Mittapalli G.K., Reddy K.R., Xiong H., Munoz O., Han B., De Riccardis F., Krishnamurthy R., and Eschenmoser A. (2007) Mapping the landscape of potentially primordial informational oligomers: oligodipeptides and oligodipeptoids tagged with triazines as recognition elements. Angew Chem Int Ed Engl 46:2470–2477 [DOI] [PubMed] [Google Scholar]
- Mulkidjanian A.Y., Bychkov A.Y., Dibrova D.V., Galperin M.Y., and Koonin E.V. (2012) Origin of first cells at terrestrial, anoxic geothermal fields. Proc Natl Acad Sci USA 109:E821–E830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy B., Meinschein W.G., and Hennessy D.J. (1963) Aqueous, low temperature environment of the Orgueil meteorite parent body. Ann NY Acad Sci 108:534–552 [DOI] [PubMed] [Google Scholar]
- Neish C.D., Somogyi Á., and Smith M.A. (2010) Titan's primordial soup: formation of amino acids via low-temperature hydrolysis of tholins. Astrobiology 10:337–347 [DOI] [PubMed] [Google Scholar]
- Nelson K.E., Levy M., and Miller S.L. (2000) Peptide nucleic acids rather than RNA may have been the first genetic molecule. Proc Natl Acad Sci USA 97:3868–3871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen P.E. (1993) Peptide nucleic acid (PNA): a model structure for the primordial genetic material? Orig Life Evol Biosph 23:323–327 [DOI] [PubMed] [Google Scholar]
- Oprea T.I. and Gottfries J. (2001) Chemography: the art of navigating in chemical space. J Comb Chem 3:157–166 [DOI] [PubMed] [Google Scholar]
- Orgel L.E. (1968) Evolution of the genetic apparatus. J Mol Biol 38:381–393 [DOI] [PubMed] [Google Scholar]
- Oró J. and Kimball A.P. (1961) Synthesis of purines under possible primitive Earth conditions. I. Adenine from hydrogen cyanide. Arch Biochem Biophys 94:217–227 [DOI] [PubMed] [Google Scholar]
- Pace N.R. (2001) The universal nature of biochemistry. Proc Natl Acad Sci USA 98:805–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasek M.A. (2008) Rethinking early Earth phosphorus geochemistry. Proc Natl Acad Sci USA 105:853–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peltzer E.T., Bada J.L., Schlesinger G., and Miller S.L. (1984) The chemical conditions on the parent body of the Murchison meteorite: some conclusions based on amino, hydroxy and dicarboxylic acids. Adv Space Res 4:69–74 [DOI] [PubMed] [Google Scholar]
- Périgaud C., Gosselin G., and Imbach J.L. (1992) Nucleoside analogues as chemotherapeutic agents: a review. Nucleosides Nucleotides 11:903–945 [Google Scholar]
- Pinheiro V.B., Taylor A.I., Cozens C., Abramov M., Renders M., Zhang S., Chaput J.C., Wengel J., Peak-Chew S.-Y., McLaughlin S.H., Herdewijn P., and Holliger P. (2012) Synthetic genetic polymers capable of heredity and evolution. Science 336:341–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitsch S., Wendeborn S., Jaun B., and Eschenmoser A. (1993) Why pentose- and not hexose-nucleic acids? Part VII. Pyranosyl-RNA (‘p-RNA’). Helv Chim Acta 76:2161–2183 [Google Scholar]
- Polishchuk P., Madzhidov T., and Varnek A. (2013) Estimation of the size of drug-like chemical space based on GDB-17 data. J Comput Aided Mol Des 27:675–679 [DOI] [PubMed] [Google Scholar]
- Powner M.W. and Sutherland J.D. (2008) Potentially prebiotic synthesis of pyrimidine beta-D-ribonucleotides by photoanomerization/hydrolysis of alpha-D-cytidine-2′-phosphate. Chembiochem 9:2386–2387 [DOI] [PubMed] [Google Scholar]
- Powner M.W., Sutherland J.D., and Szostak J.W. (2010) Chemoselective multicomponent one-pot assembly of purine precursors in water. J Am Chem Soc 132:16677–16688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinowitz J., Chang S., and Ponnamperuma C. (1968) Phosphorylation by way of inorganic phosphate as a potential prebiotic process. Nature 218:442–443 [DOI] [PubMed] [Google Scholar]
- Ricardo A., Carrigan M.A., Olcott A.N., and Benner S.A. (2004) Borate minerals stabilize ribose. Science 303:196. [DOI] [PubMed] [Google Scholar]
- Ruddigkeit L., Van Deursen R., Blum L.C., and Reymond J.-L. (2012) Enumeration of 166 billion organic small molecules in the chemical universe database GDB-17. J Chem Inf Model 52:2864–2875 [DOI] [PubMed] [Google Scholar]
- Sanchez R.A. and Orgel L.E. (1970) Studies in prebiotic synthesis. V. Synthesis and photoanomerization of pyrimidine nucleosides. J Mol Biol 47:531–543 [DOI] [PubMed] [Google Scholar]
- Schmidt J.G., Christensen L., Nielsen P.E., and Orgel L.E. (1997) Information transfer from DNA to peptide nucleic acids by template-directed syntheses. Nucleic Acids Res 25:4792–4796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt-Kopplin P., Gabelica Z., Gougeon R.D., Fekete A., Kanawati B., Harir M., Gebefuegi I., Eckel G., and Hertkorn N. (2010) High molecular diversity of extraterrestrial organic matter in Murchison meteorite revealed 40 years after its fall. Proc Natl Acad Sci USA 107:2763–2768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider K. and Benner S. (1990a) Building-blocks for oligonucleotide analogs with dimethylene-sulfide, dimethylene-sulfoxide, and dimethylene-sulfone groups replacing phosphodiester linkages. Tetrahedron Lett 31:335–338 [Google Scholar]
- Schneider K. and Benner S. (1990b) Oligonucleotides containing flexible nucleoside analogs. J Am Chem Soc 112:453–455 [Google Scholar]
- Schoffstall A.M. (1976) Prebiotic phosphorylation of nucleosides in formamide. Orig Life 7:399–412 [DOI] [PubMed] [Google Scholar]
- Schöning K.-U., Scholz P., Guntha S., Wu X., Krishnamurthy R., and Eschenmoser A. (2000) Chemical etiology of nucleic acid structure: the α-threofuranosyl-(3′→2′) oligonucleotide system. Science 290:1347–1351 [DOI] [PubMed] [Google Scholar]
- Schwartz A. and Ponnamperuma C. (1968) Phosphorylation of adenosine with linear polyphosphate salts in aqueous solution. Nature 218:443. [DOI] [PubMed] [Google Scholar]
- Schwartz A.W. and Degraaf R.M. (1993) The prebiotic synthesis of carbohydrates—a reassessment. J Mol Evol 36:101–106 [Google Scholar]
- Schwartz A.W. and Orgel L.E. (1985) Template-directed synthesis of novel, nucleic acid-like structures. Science 228:585–587 [DOI] [PubMed] [Google Scholar]
- Seita T., Yamauchi K., Kinoshita M., and Imoto M. (1972) Condensation polymerization of nucleotide analogues. Die Makromolekulare Chemie 154:255–261 [Google Scholar]
- Shapiro R. (1984) The improbability of prebiotic nucleic acid synthesis. Orig Life Evol Biosph 14:565–570 [DOI] [PubMed] [Google Scholar]
- Shapiro R. (1988) Prebiotic ribose synthesis: a critical analysis. Orig Life Evol Biosph 18:71–85 [DOI] [PubMed] [Google Scholar]
- Shapiro R. (1995) The prebiotic role of adenine: a critical analysis. Orig Life Evol Biosph 25:83–98 [DOI] [PubMed] [Google Scholar]
- Sheng Y., Bean H.D., Mamajanov I., Hud N.V., and Leszczynski J. (2009) Comprehensive investigation of the energetics of pyrimidine nucleoside formation in a model prebiotic reaction. J Am Chem Soc 131:16088–16095 [DOI] [PubMed] [Google Scholar]
- Shigemasa Y., Fujitani T., Sakazawa C., and Matsuura T. (1977) Formose reactions. III. Evaluation of various factors affecting the formose reaction. Bull Chem Soc Jpn 50:1527–1531 [Google Scholar]
- Slabaugh M.R., Harvey A.J., and Nagyvary J. (1974) The possible role of inorganic thiophosphate as a prebiotic phosphorylating agent. J Mol Evol 3:317–321 [DOI] [PubMed] [Google Scholar]
- Smith E. and Morowitz H.J. (2004) Universality in intermediary metabolism. Proc Natl Acad Sci USA 101:13168–13173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summerton J. and Weller D. (1997) Morpholino antisense oligomers: design, preparation, and properties. Antisense Nucleic Acid Drug Dev 7:187–195 [DOI] [PubMed] [Google Scholar]
- Ueda N., Kawabata T., and Takemoto K. (1971) Synthesis of N-(2,3-dihydroxypropyl) derivatives of nucleic bases. J Heterocycl Chem 8:827–829 [Google Scholar]
- Usher D. and McHale A. (1976) Hydrolytic stability of helical RNA: a selective advantage for the natural 3′,5′-bond. Proc Natl Acad Sci USA 73:1149–1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visscher J. and Schwartz A.W. (1988) Template-directed synthesis of acyclic oligonucleotide analogues. J Mol Evol 28:3–6 [DOI] [PubMed] [Google Scholar]
- Ward W.L., Plakos K., and DeRose V.J. (2014) Nucleic acid catalysis: metals, nucleobases, and other cofactors. Chem Rev 114:4318–4342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver D.F. and Weaver C.A. (2011) Exploring neurotherapeutic space: how many neurological drugs exist (or could exist)? J Pharm Pharmacol 63:136–139 [DOI] [PubMed] [Google Scholar]
- Westheimer F.H. (1987) Why nature chose phosphates. Science 235:1173–1178 [DOI] [PubMed] [Google Scholar]
- White H.B., III (1976) Coenzymes as fossils of an earlier metabolic state. J Mol Evol 7:101–104 [DOI] [PubMed] [Google Scholar]
- Woese C.R. (1967) The Genetic Code: The Molecular Basis for Genetic Expression, Harper & Row, New York [Google Scholar]
- Yin Y.W. and Steitz T.A. (2002) Structural basis for the transition from initiation to elongation transcription in T7 RNA polymerase. Science 298:1387–1395 [DOI] [PubMed] [Google Scholar]
- Zhang L., Peritz A., and Meggers E. (2005) A simple glycol nucleic acid. J Am Chem Soc 127:4174–4175 [DOI] [PubMed] [Google Scholar]
- Zhang S., Switzer C., and Chaput J.C. (2010) The resurgence of acyclic nucleic acids. Chem Biodivers 7:245–258 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.