

Abstract
Exposure to improvised explosive devices can result in a unique form of traumatic brain injury—blast-induced traumatic brain injury (bTBI). At the mild end of the spectrum (mild bTBI [mbTBI]), there are cognitive and mood disturbances. Similar symptoms have been observed in post-traumatic stress disorder caused by exposure to extreme psychological stress without physical injury. A role of the monoaminergic system in mood regulation and stress is well established but its involvement in mbTBI is not well understood. To address this gap, we used a rodent model of mbTBI and detected a decrease in immobility behavior in the forced swim test at 1 d post-exposure, coupled with an increase in climbing behavior, but not after 14 d or later, possibly indicating a transient increase in anxiety-like behavior. Using in situ hybridization, we found elevated messenger ribonucleic acid levels of both tyrosine hydroxylase and tryptophan hydroxylase 2 in the locus coeruleus and the dorsal raphe nucleus, respectively, as early as 2 h post-exposure. High-performance liquid chromatography analysis 1 d post-exposure primarily showed elevated noradrenaline levels in several forebrain regions. Taken together, we report that exposure to mild blast results in transient changes in both anxiety-like behavior and brain region–specific molecular changes, implicating the monoaminergic system in the pathobiology of mbTBI.
Key words: : anxiety, mood disorder, noradrenaline, PTSD, stress
Introduction
Blast-induced traumatic brain injury (bTBI), and post-traumatic stress disorder (PTSD) have emerged as leading military health issues, with major long-term neuropsychiatric consequences.1–5 Blast-induced TBI is a spectrum disorder6 and at its mild end (mild bTBI [mbTBI]), the neurobehavioral symptoms substantially overlap with those observed in PTSD. PTSD is characterized by emotional instability, increased anxiety, and hyperarousal.7–11 The sudden and violent nature of the blast, even without detectable physical injury, can result in an initial elevation in anxiety and lead to sustained emotional impairment.2
In the majority of instances, these symptoms are acute; however, for some, they can develop into a chronic state.12,13 The lack of obvious or easily detectable structural damage and overlapping symptomology with PTSD have made the pathomechanism(s) of mbTBI difficult to identify and has hindered correct diagnosis.11 One, therefore, has to rely on the patient's symptoms, which are underreported and often biased and/or based on subjective neurobehavioral testing.14
A link between stress/mood disorders and distinct changes in the monoamine neurotransmitters have been extensively reported in the literature and include both noradrenaline (NA)15–25 and serotonin (5-hydroxytryptamine; 5-HT).26–33 Importantly, elevated NA levels are implicated in PTSD symptomology, associated with sleep disturbance and increased nightmare frequency, a hallmark of PTSD.34
NA is synthesized in the locus coeruleus (LC)35 involving the rate limiting enzyme tyrosine hydroxylase (TH).36,37 The LC gives rise to projections to virtually all regions of the central nervous system, including the prefrontal cortex (PFC), the dorsal and ventral hippocampal formation (dHiFo and vHiFo, respectively), and the hypothalamus.38–40 The dorsal raphe nucleus (DRN) and the median raphe nucleus (MRN) harbor the neuronal cell bodies, giving rise to the extensive serotonergic forebrain projections.35,41,42 The 5-HT neurones express the rate-limiting enzyme for the 5-HT synthesis, (brain) tryptophan hydroxylase 2 (TPH2).43
The role of the monoaminergic system in mbTBI is not well understood. One way to address this issue is to apply animal models, which have been developed to replicate the effects of bTBI.44,45 Here, we have used a metal blast tube as previously described.46 We tested exposed and sham animals for anxiety-/depression-like behavior using the forced swim test (FST)47–50; assessed the expression of the monoamine biosynthetic enzymes TH and TPH2 in the LC and DRN, respectively, using quantitative in situ hybridization (ISH); and quantified NA, dopamine (DA), and 5-HT, as well as some of their metabolites using high-performance liquid chromatography (HPLC).
Methods
Animals and housing conditions
A total of 78 male Sprague-Dawley rats (Taconic, Ry, Denmark), 10–12 weeks old and weighing 290–320 g at the beginning of the experiments, were used. They were housed in groups of four in Type IV MakrolonR plastic cages under standardized conditions (12 h light/dark cycle, lights on at 07:00; temperature of 22±0.5°C; and 40 – 50% relative humidity). Food and water were provided ad libitum to the animals.
Experimental groups and manipulations
This study is made up of three separate experiments, and for each experiment, different animals were used. All animals were handled and given a week to habituate to their environment prior to the experiments, then randomly assigned to one of three groups: control, sham, and exposed. Control rats were kept in the animal facility without any experimental manipulations for the duration of the study. Sham rats were anesthetized by isoflurane inhalation, then injected intraperitoneally with 2.4 mL/kg of a mixture of 1 mL Dormicum® (5 mg/mL midazolam; Roche, Stockholm, Sweden), 1 mL Hypnorm® (Fentanyl/fluaniscane; Janssen, Stockholm, Sweden) and 2 mL of distilled water. Exposed rats were treated identically to shams in addition to being exposed to a single blast wave.
Experiment One was a behavior test—the FST. Animals were divided into three groups: control (n=6), sham (n=7), and exposed (n=8). Experiment Two consisted of in situ hybridization and immunohistochemistry analysis. A total of 33 animals were terminated at four post-exposure time-points: 2 h (n=5), 1 d (n=5), 3 d (n=4) and 7 d (n=8), and a total of 11 shams were used. Experiment Three consisted of HPLC analysis. Twenty-four animals were sacrificed at 1 d=6, 6, and 7 d=6, 6, exposed and sham, respectively.
Exposure conditions
A validated blast tube46,51–53 designed by Clemedson in 1955 was used for exposure.54 Swedish army plastic explosive containing explosive m/46, 86% pentaerythritol tetranitrate and mineral oil was used with a Nonel ignition (Dyno Nobel Sweden, Nora, Sweden). Anesthetized animals were placed in a rigid metallic holder that protects all parts of the body except the head. This holder protects from lung injuries that otherwise would occur with the employed amount of explosive. The holder also prevents acceleration movements of the head relative to the rest of the body. The holder was subsequently mounted into the 1.5 m metal shock tube, with the rat placed in a transverse prone position at a distance of 1 m from the charge. Five grams of the explosive were then detonated, triggering a simple blast Friedlander-type wave at the surface of the animal with a peak pressure of 550 kPa and a duration of 0.2 msec.46 The rat's left side faced the charge. Post-exposure, animals were monitored for 1 h, after which time they awoke, as did the shams.
Forced swim test
Anxiety-/depression-like behavior was measured using the FST47 at 1 d pre-exposure (baseline) and 1 d, 14 d, and 35 d post-exposure. Animals were individually placed in a vertical plastic cylinder (50 cm height, 18 cm diameter) containing water to a height of 30 cm, at a temperature of 25±0.5°C. Animals were exposed to two swimming sessions, 24 h apart: a 10-min pre-test, and a 5-min test. The total duration of immobility and climbing behavior were recorded during the second, test day.49 Immobility was defined as floating passively in an upright position in water, with only small movements necessary to keep the head above the water surface, typically only one paw movement at a time. Climbing was defined as vigorous forepaw movements directed toward the walls of the cylinder.
In situ hybridization (ISH)
Animals were anesthetized with isoflurane, injected with 1.5–2.0 mL of pentobarbital, and then decapitated. The brains were rapidly removed and placed on dry ice and, once frozen, stored at −70°C until use. Serial coronal sections were cut at a thickness of 14 μm at the level of the LC (bregma −10.52 – −9.16 mm), and DRN (−8.30 – −7.30 mm; coordinates according to Paxinos and Watson55) using Cryo-Star HM 560M (MICROM International GmbH, Heidelberg, Germany), and two sections were thaw-mounted per super frost slide (Thermo Scientific, Stockholm, Sweden). Three slides per animal were processed for ISH. Method of selection of slides was random.
Oligonucleotides complementary to rat TPH2 nucleotides43: TCC TCC GTC CAA ATG TTG TCA GGT GGA TTC AGC GTC ACA ATG GTG GTC [GENBANK ID NM_017139], and TH37: GCG CTG GAT ACG AGA GGC ATA GTT CCT GAG CTT GTC [GENBANK ID NM_012740] (CyberGene AB, Solna, Sweden), were labeled with deoxyadenosine 5′triphosphate α–P32 at the 3′-end using terminal deoxynucleotidyltransferase (Thermo Scientific, Waltham, MA).
The labeled oligoprobes were purified using ProbeQuant G-50 Micro Columns (Amersham Pharmacia Biotech, Piscataway, NJ). Sections were air-dried and incubated with the oligonucleotide probe for 18 h at 42°C. Post-hybridization, sections were rinsed in 1×SSC, 4×30 min at 55°C followed by 1 h at room temperature (RT), dehydrated with 70%, followed by 90% and finally 100% ethanol, air-dried and dipped in liquid photo emulsion NTB2 at RT (Kodak, Rochester, NY).
The optimal exposure time was determined by exposing the slides to imaging plates (BAS-SR Fujifilm, Tokyo, Japan) for 24 h at RT, which were scanned using a phosphoimager (Fuji BAS 3000, Tokyo, Japan). Based on this data, TPH2 slides were developed after 72 h and TH after a week using D19 developer (Kodak) and AL-4 fixative (Kodak) and mounted in glycerol-phosphate.56
Dark field photomicrographs were captured in a microscope (Nikon Eclipse E-600; Nikon, Tokyo, Japan), connected to a digital camera (Digital Sight, U1; Nikon). The images were analyzed according to the mean gray density (MGD) of the messenger ribonucleic acid (mRNA) signal in the regions of interest (ROIs), using ImageJ 1.48 (National Institutes of Health, Bethesda, MD).
Histo- and immunohistochemistry (IHC)
Sections not used for ISH were used to evaluate nerve cell death (using Fluoro-Jade B [FJ] staining; Merck Millipore AG310, Darmstadt, Germany), β-amyloid precursor protein accumulation (APP; IHC) and leakages of blood vessels (using a secondary rat antibody, IHC). As positive control, sections from experiments with a focal penetrating injury model were used.57
For FJ staining, sections were air dried for 1 h before being fixed in 4% formaldehyde for 10 min, and rinsed in 0.01 M phosphate-buffered saline (PBS) 3×3 min. The sections were subsequently dipped in dH2O for 2 min, followed by 10 min in 0.06% potassium permanganate (KMnO4), then rinsed with dH2O for 2 min. Slides were then soaked in FJ solution for 30 min, washed in dH2O 3×1 min, and finally placed on a hot plate 50°C for 5 min before a quick dip in xylene and thereafter mounted with Entellan (Merck).
For all other staining, slides were fixed in ice cold methanol for 5 min, then shortly dipped in ice cold acetone, and rinsed in 0.01 M PBS 2×10 min. One group was incubated in a humid chamber at 4°C for 18 h with a solution of 0.3% Triton, 5% bovine serum albumin, and 0.1% sodium azide in 0.01M PBS and a rabbit poly-clonal antibody against APP (51-2700, dilution 1:400; Life Technologies, Stockholm, Sweden). The other group was not exposed to any primary antibody.
The following day, all sections were rinsed in 0.01M PBS 2×10 min and incubated for 1 h at RT with 0.01 M PBS, 0.1% sodium azide, 0.3% Triton. The first group then processed with an Alexa Fluor 488 conjugated anti-rabbit immunoglobulin G (dilution 1:400; Jackson ImmunoResearch, Suffolk, UK). The second group was incubated with CY2- conjugated donkey anti-rat antibody (Jackson ImmunoResearch, 712-225-153, dilution 1:100) to visualize blood vessel leakage.
Sections were rinsed in 0.01M PBS 2×10 min, and mounted in a mixture of glycerol and PBS (1:3), then cover-slipped. Slides were viewed in a microscope equipped with epifluorescence (Eclipse E600; Nikon).
High-performance liquid chromatography
Chemicals required for HPLC included: NA hydrochloride, DA hydrochloride, 5-HT hydrochloride, 3,4-dihydroxyphenylacetic acid (DOPAC), homovanillic acid (HVA), 5-hydroxyindoleacetic acid (5-HIAA), potassium chloride, phosphate monobasic monohydrate, sodium phosphate dibasic, sodium acetate, citric acid, octanesulfonic acid sodium salt and methanol, which were obtained from Sigma-Aldrich (St. Louis, MO). EDTA-2Na was purchased from Dojindo (Kumamoto, Japan).
The following regions were dissected using the appropriate landmarks as detailed by Paxinos and Watson55: PFC, hypothalamus, the dHiFo and vHiFo formation, occipital cortex (CX) and entorhinal cortex (ERC). Both ipsi-lateral (left)- and contra-lateral (right) regions were dissected, except the hypothalamus. The isolated brain regions were put into marked plastic vials and placed on dry ice. Dissected brain tissue samples (1–10 mg) were mixed at a ratio of 1:10 (weight/volume) with 0.2 M perchloric acid including 100 μM EDTA-2Na, and homogenized at 0°C in a glass-pestle micro-homogenizer. Following standing for 3 min on ice, the homogenates were centrifuged for 15 min at 12,000 g at 4°C. The supernatants were aspirated and mixed with 1 M Na-acetate buffer, pH 3 at a ratio 5:1 (volume/volume) and filtered through a 0.22 μm centrifugal filter for 4 min at 12,000 g at 4°C. The filtrates were stored at −80°C before HPLC analysis.
Concentrations of NA, DA, and 5-HT in the brain tissue samples were determined by HPLC with electrochemical detection as described elsewhere.58 Briefly, the HPLC system consisted of a HTEC500 unit (Eicom, Kyoto, Japan), and a CMA/200 Refrigerated Microsampler (CMA Microdialysis, Stockholm, Sweden) equipped with a 20 μL loop and operating at 4°C. The potential of the glassy carbon working electrode was +450 mV versus the Ag/AgCl reference electrode. The separation was achieved on a 200×2.0 mm Eicompak CAX column (Eicom). The mobile phase was a mixture of methanol and 0.1 M phosphate buffer (pH 6.0; 30:70, volume/volume) containing 40 mM potassium chloride and 0.13 mM EDTA-2Na. The chromatograms were recorded and integrated by use of a computerized data acquisition system Clarity (DataApex, Prague, Czech Republic). The detection limit (signal-to-noise ratio=3) for NA, DA, and 5-HT was 0.05 nM (i.e., 0.75 fmoL in 15 μL injected onto the column).
Concentrations of DOPAC, HVA, and 5-HIAA were determined by a separate HPLC system with electrochemical detection (HTEC500). The potential of the glassy carbon working electrode was +750 mV versus the Ag/AgCl reference electrode. The separation was achieved on a 150×3.0 mm Eicompak SC-5ODS column (Eicom). The mobile phase was a mixture of methanol and 0.1 M citrate/0.1 M sodium acetate buffer solution (pH 3.5; 16:84, volume/volume) and contained 210 mg/L octanesulphonic acid sodium salt and 5 mg/L EDTA-2Na. The detection limit (signal-to-noise ratio=3) for DOPAC, HVA, and 5-HIAA was 2 nM (i.e., 10 fmoL in 5 μL injected onto the column). The chromatograms were recorded and integrated by use of the computerized data acquisition system Clarity (DataApex).
Statistical analysis
All statistical analyses were performed using GraphPad Prism version 5 (GraphPad Software, CA).
For the FST differences in the total time spent climbing and immobile during each test session (baseline and 1 d, 14 d, and 35 d post-exposure) were independently evaluated across all three groups (exposed, n=8; sham, n=7; control, n=6) using analysis of variance (ANOVA), repeated measures ANOVA, and Dunnett's t-test.
For ISH, the MGDs were determined for each of the ROIs (DRN, MRN, and LC) for each transcript (TH and TPH2). The MGDs of individual ROIs from each specific time-point (2 h, n=5; 1 d, n=5, 3 d, n=4; and 7 d, n=8; and a total of 11 shams) and transcript were separately compared with sham MGDs of corresponding ROIs and transcript, and analyzed using ANOVA and followed up by the Tukey-Kramer multiple comparison test, which enables comparisons across all five experimental groups. No difference between shams of the different time-points was found as evaluated by ANOVA, so the groups were collapsed. The MGD of each ROI was normalized to its corresponding sham level to clearly show increases and/or decreases in transcript level on sham levels.
For HPLC, each specific monoamine/ metabolite (NA, DA, 5-HT and DOPAC, HVA, 5-HIAA) was evaluated exclusively from one brain region against its corresponding sham region. The two terminal time-points post-exposure were evaluated separately, using an unpaired t-test, 1 d=6, 6 and 7 d=6, 6, exposed and sham, respectively. Since none of the brain regions showed a significant difference between the left and right, these groups were collapsed in the analysis.
All data are presented as the mean±standard error of the mean (SEM). The level of significance is as follows: *p<0.05, **p<0.01, and ***p<0.001.
Results
Anxiety-/depression-like behavior
Anxiety-/depression-like behavior was assessed in the second day of the FST, during a 5-min test session ran at 1 d pre-exposure (baseline) and 1 d, 14 d, and 35 d post-exposure (immobility, Fig. 1A; climbing, Fig. 1B). A one-way ANOVA comparing the behavioral differences across the groups revealed no differences in the three groups pre-exposure (baseline), as expected. However, after 1 d, the exposed group exhibited decreased immobility relative to the control and sham groups, whereas the values for the climbing behavior were not statistically significant (between the groups). In the remaining test sessions, even after 35 d, similar climbing and immobility behaviors were observed across the groups.
FIG. 1.
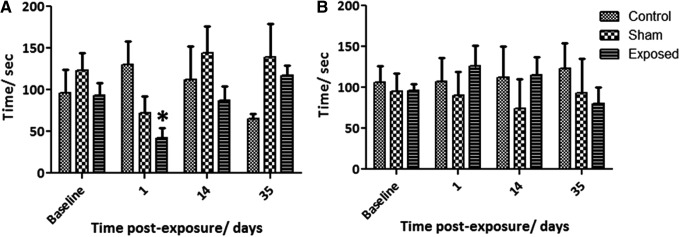
Time of immobility (A) and climbing (B) in the forced swim test measured at 1 d pre-exposure (baseline), and 1 d, 14 d, and 35 d post-exposure. Immobility behavior decreased in the exposed group 1 d post-exposure relative to sham and controls (a). Data are presented as means±standard error of the mean in the three groups; control (n=6), sham (n=7), and exposed (n=8). *p<0.05.
Further analysis of the data using a repeated measures ANOVA, which looks at the changes in the behavior of the individuals in each group across the different time-points, revealed a significant change in the amount of time spent climbing in the exposed group (p=0.0472). Thus, 1 d post-exposure climbing behavior increased versus baseline in the exposed group, but returned to baseline levels 35 d post-exposure. No change in climbing behavior was seen in the control and sham group (p=0.9691 and 0.7075, respectively). The same statistical parameters were used to look at the immobility behavior, again no difference was seen in the control group (p=0.3404). Both the sham and exposed group exhibited increased immobility behavior as time progressed, this trend towards an increase in immobility was far more apparent in the exposed group as evident in the p values (0.0298 and 0.0003 for sham and exposed, respectively).
Transcript of TPH2 and TH as measured by ISH
ISH analysis of the transcript levels of the two key biosynthetic enzymes, TPH2 and TH, showed an early and significant upregulation. TH was upregulated bilaterally in the LC and TPH2 increased in the mid/caudal, but not rostral, DRN as early as 2 h post-exposure (Fig. 2 and Fig. 3). The results indicate TH transcript was distinctly sensitive to the blast exposure, remaining significantly elevated at 1 and 3 d post-exposure, and returning to sham levels after 7 d. The increase in TPH2 transcript was modest, being limited to the mid/caudal region of the DRN and returned to sham levels 3 d post-exposure. TPH2 levels in the median raphe were also measured but no changes were observed at any of the time-points.
FIG. 2.

Representative dark field in situ hybridization photomicrographs of emulsion-dipped sections showing the distribution of tryptophan hydroxylase 2 (TPH2; A-D) and tyrosine hydroxylase (TH; E–H), messenger ribonucleic acid (mRNA) in the DRN and LC, post-exposure to a mild blast-induced traumatic brain injury in the rat. TPH2 mRNA is found upregulated 2 h post-exposure in the mid/caudal DRN (A) but not in the rostral DRN (B), relative to shams, mid/caudal DRN (C), and rostral (D). TH mRNA is upregulated 2 h post-exposure in both the right (E) and left (F) LC, relative to sham right (G) and left (H) LC. Aq, aqueduct; PAG, periaqueductal gray; DRN, dorsal raphe nucleus; MRN, median raphe nucleus; LC, locus coeruleus.
FIG. 3.

Quantification of messenger ribonucleic acid (mRNA) levels of the biosynthetic enzymes tryptophan hydroxylase 2 (TPH2) in the dorsal raphe nucleus (DRN; A-B) and of tyrosine hydroxylase (TH) in the locus coeruleus (LC; C-D), at 2 h, 1 d, 3d, and 7 d post-exposure in the rat. The expression levels of the mRNA at each time-point were normalized to sham levels (shams of the different time-points showed no statistical difference and were pooled). There is an initial increase in TPH2 mRNA levels immediately post-exposure, which lasts up to 1 d, and is statistically significant in the mid/caudal part of the DRN (A). An increase in TH mRNA in both the right LC (C) and left (D) is detected as early as 2 h post-exposure continuing to rise up to 3d and returning to sham levels at 7d. Data are presented as mean±standard error of the mean. *p<0.05, **p<0.01, ***p<0.001.
Nerve cell death, blood vessel leakage and APP accumulation assessed by IHC
Sections from the LC, DRN and both dHiFo and vHiFo were tested for nerve cell death, blood vessel leakage and APP accumulation by IHC. No differences were seen between shams and exposed rats terminated at 1 and 7 d post-exposure. Exposed rats did not appear to have leakages of any major blood vessels in the brain stem or forebrain regions post-exposure, nor was cell death seen in any of these areas. Evaluation of the white matter tracts by staining for APP accumulation, revealed no difference to the shams (data not shown).
Monoamine levels as measured by HPLC
The HPLC analysis, performed at 1 and 7 d post-exposure, showed regional and varying changes in the levels of the neurotransmitters NA, DA, and 5-HT (Table 1A), along with their metabolites DOPAC, HVA, and 5-HIAA (Table 1B), in a number of forebrain regions (i.e., projection areas of the LC NA and DRN/MRN 5-HT neurones). No statistically significant differences were observed between left versus right sides; therefore, these groups were collapsed. The major findings were significant increases in NA levels 1 d post-exposure in CX, ERC, and dHiFo. The changes did not reach significance in the vHiFo. A significant increase in DA levels was observed in both the dHiFo and vHiFo on 1 d post-exposure. No significant differences were seen in 5-HT levels. With regard to the metabolites, a significant decrease in DOPAC was measured in the CX, and HVA levels were significantly decreased in the ERC and dHiFo. These changes occurred at 7 d post-exposure. Finally, a significant reduction in 5-HIAA levels was seen in the PFC at 1 d post-exposure. Taken together, the NA system appears most sensitive to the blast but only in the early phase (i.e., 1 d post-exposure).
Table 1.
HPLC Raw Data Values for Monoamines (a), and Their Metabolites (b), at 1 d and 7 d Post-Exposure
| (A) | Monoamines [pg/mg] | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Group | NA | DA | 5-HT | |||||||||
| Mean±SEM | Sham 1 d | Exposed 1 d | Sham 7 d | Exposed 7 d | Sham 1 d | Exposed 1 d | Sham 7 d | Exposed 7 d | Sham 1 d | Exposed 1 d | Sham 7 d | Exposed 7 d |
| Hypothalamus | 2176±153.4 | 2516±282.7 | 1468±176.7 | 1236±93.1 | 241.4±58.0 | 368.0±31.01 | 199.5±28.2 | 208.2±43.4 | 261.5±47.1 | 334.9±34.3 | 263.2±40.2 | 288.8±31.3 |
| Prefrontal cortex | 322.5±12.8 | 320.3±13.4 | 291.8±23.5 | 288.1±30.8 | 78.1±7.3 | 87.6±8.4 | 154.8±47.1 | 120.0±24.4 | 237.9±24.9 | 242.1±11.9 | 186.5±15.5 | 180.3±16.7 |
| Occipital cortex | 238.3±10.5 | 293.3±8.3*** | 196.2±13.2 | 194.1±18.8 | 21.8±3.5 | 25.1±3.7 | 5.8±0.39 | 6.1±0.7 | 159.2±13.5 | 190.2±8.2 | 72.7±5.3 | 84.1±9.2 |
| Entorhinal cortex | 348.7±22.8 | 510.0±59.4* | 325.9±21.3 | 327.7±28.4 | 193.5±38.7 | 335.5±82.1 | 54.7±9.9 | 46.1±9.1 | 519.7±46.5 | 526.6±59.6 | 183.6±22.0 | 204.8±22.9 |
| Dorsal hippocampus | 295.7±15.5 | 399.2±25.0* | 232.2±21.9 | 234.3±19.8 | 17.1±1.3 | 26.4±3.9* | 5.9±0.8 | 8.2±1.0 | 251.5±20.4 | 291.4±20.6 | 112.4±11.5 | 142.9±10.7 |
| Ventral hippocampus | 473.4±17.1 | 533.9±38.5 | 266.5±15.9 | 307.5±15.3 | 17.7±1.5 | 26.5±3.0* | 7.0±0.8 | 7.4±0.6 | 390.5±31.5 | 435.2±39.3 | 125.5±9.6 | 130.2±9.7 |
| (B) | Metabolites [pg/mg] | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Group | DOPAC | HVA | 5-HIAA | |||||||||
| Mean±SEM | Sham 1 d | Exposed 1 d | Sham 7 d | Exposed 7 d | Sham 1 d | Exposed 1 d | Sham 7 d | Exposed 7 d | Sham 1 d | Exposed 1 d | Sham 7 d | Exposed 7 d |
| Hypothalamus | 115.7±11.1 | 88.9±9.3 | 88.7±5.8 | 83.3±9.7 | 45.7±3.9 | 36.1±6.9 | 54.3±7.9 | 49.5±6.4 | 760.2±31.9 | 665.3±42.2 | 427.7±21.5 | 418.7±26.4 |
| Prefrontal cortex | 43.7±3.5 | 35.8±2.6 | 151.0±37.1 | 125.3±10.2 | 94.9±9.7 | 87.8±12.3 | 114.0±10.7 | 144.9±21.7 | 305.1±10.9 | 257.9±8.0** | 336.6±26.0 | 371.5±25.3 |
| Occipital cortex | 5.2±0.2 | 5.7±0.7 | 8.2±0.5 | 6.0±0.5** | 6.3±0.3 | 7.2±0.9 | 7.5±0.7 | 7.2±0.6 | 172.3±5.2 | 179.0±6.3 | 198.3±8.4 | 184.9±6.5 |
| Entorhinal cortex | 49.9±10.7 | 47.0±14.0 | 31.1±4.3 | 22.1±4.3 | 40.9±8.9 | 44.0±11.2 | 33.2±2.3 | 18.1±2.5*** | 319.0±23.4 | 344.8±38.9 | 327.7±13.4 | 318.9±24.7 |
| Dorsal hippocampus | 6.3±0.5 | 8.8±1.5 | 4.0±0.8 | 3.7±0.4 | 9.5±0.9 | 9.0±1.0 | 9.9±1.1 | 5.9±0.5** | 388.2±15.7 | 384.6±11.4 | 324.2±25.8 | 359.4±15.6 |
| Ventral hippocampus | 7.0±1.1 | 4.9±0.3 | 4.1±0.4 | 3.8±0.5 | 6.3±0.4 | 6.2±0.5 | 10.4±1.4 | 11.4±2.0 | 382.2±16.9 | 376.6±16.8 | 336.7±19.0 | 329.3±13.9 |
Data are presented as mean±S.E.M.
p<0.05; ** p<0.01; ***p<0.001.
HPLC, high-performance liquid chromatography; NA, noradrenaline; DA, dopamine; 5-HT, 5-hydroxytryptamine; DOPAC, 3,4-dihydroxyphenylacetic acid; HVA, homovanillic acid; 5-HIAA, 5-hydroxyindoleacetic acid; SEM, standard error of the mean.
Discussion
Here, we show that exposure to a single blast wave with short duration and relatively high peak pressure results in an apparent mTBI. The blast caused decreased immobility in the FST only during the first day and in parallel, an early and only transient increase in extracellular levels of NA 1 d post-exposure. The changes in TH and TPH2 transcripts occurred as early as 2 h post-exposure and remained elevated for few days (TPH2 only for one day; TH for 3 but not 7 d). Moreover, we could not detect white matter injury, blood–brain barrier disruption, or cell death. All of these findings support the involvement of the noradrenergic system originating in the LC in the mild form of blast TBI.
The interpretation of the behavioral state of our rats at this time-point is complex. Estanislau and colleagues59 evaluated FST behavior in the elevated plus-maze60 and found that the most anxious animals have a higher latency to immobility versus a stronger immobility behavior in less anxious animals. According to these results, our rats exhibit anxiety-like behavior, a term that we use in this discussion. It should be noted that the behavior associated with increased NA levels has often been termed hyperarousal.61
Except for 1 d post-exposure, the rats did not show any changes in the FST, compared with control and shams (i.e., either at 14 or 35 d post-exposure). Thus, no signs of increased immobility and decreased climbing were recorded, at the later time points, i.e., there was no sign of behavioral despair (failure of continuing an escape-directed behavior), nor of development of learned helplessness (passive behavior).47,62 Failure to cope with stressful events is considered a feature of the depression syndrome and has been associated with dysfunction of monoamine systems, in particular NA, and 5-HT neurones.63
TBI and monoamine turnover
Our HPLC analysis shows that mild blast increases NA levels in several forebrain regions, in concert with the noradrenergic LC system being activated by stress.9,21–25 Also, a second catecholamine, DA, showed a significant increase 1 d post-exposure but only in the hippocampal formation (HiFi). No changes were seen in 5-HT levels; however, a decrease in the serotonin metabolite 5-HIAA was seen in the PFC 1 d post-exposure.
The increase in monoamine levels could indicate a substantial release of neurotransmitters followed by compensatory re-synthesis. This also would explain the increase in the transcript levels for the synthesizing enzymes, and the decrease in metabolite levels, as the cell tries to replenish its neurotransmitter stores. The surge in catecholamine levels in the brain also may have a relation to inflammation.64 It has been shown by Cernak65 that there is a systemic inflammatory response to blast and that this includes the brain. Interestingly, no effects on monoamine transmitter levels were seen in the PFC, in spite of the fact that PFC is a key region in cognition and emotions.63,66–68
Monoamine turnover has previously been monitored after various types of TBI in animal experiments. Six days after unilateral, ventrolateral cortical lesions, 5-HT and NA were decreased ipsilaterally, whereas 5-HIAA was increased.69 Unilateral, focal cortical freezing or heat lesion both decreased 5-HT levels bilaterally after 1 d, whereas 5-HIAA increased and remained elevated up to 10 ld.70 Subsequently, this group could show an ipsilateral increase in 5-HT synthesis in the cortex, hippocampus, and DRN using the same model.71 Finally, the monoamine system also has been monitored shortly after concussion/head injury. Thus, Tanaka and colleagues72 found a strong, very rapid decrease in cortical NA level at 25 and 210 sec using a cortical weight drop model but no effects on DA or 5-HT, again pointing to the high reactivity and sensitivity of the noradrenergic LC system.
Brain monoamines and PTSD have also been analyzed in models not involving physical lesions (e.g., various types of stress). Using sequential restraint stress, swim stress, and halothane exposure, NA, 5-HT, and DA concentrations were elevated in the hippocampus after 7 d but not after 1 h.73 Wilson and colleagues74 exposed rats to a predator (cat) plus daily cage cohort changes. Using HPLC, they observed increased levels of NA and DOPAC in the hippocampus and PFC in the stress group, whereas 5-HT was lower in the PFC.
Taken together, these studies suggest that the monoamine systems react in response to various TBIs and psychosocial stress and that the increased transmitter release represents a generalized reaction. The NA neurons appear to be particularly sensitive and rapidly activated, in agreement with our findings in the blast model.
The employed blast tube with the rigid montage of the animals represents a generic model for primary blast only, in which any rotational acceleration injury or focal impacts has been limited. No accumulation of APP was observed in white matter. Such accumulations have been reported in blast and are generally attributed to rotational acceleration rather than to primary blast.46,75 However, effects by smoke emission and heating (quaternary blast) cannot be excluded. Gene expression in the hippocampus has been shown to be very different in this model for primary blast, compared with rotational injury.46 It should therefore be of interest to analyze catecholamines and serotonin also in models for rotational acceleration.
Clinical trauma materials are generally very heterogeneous in terms of age and type of trauma. One interesting exception is the Vietnam head injury study, in which most patients were young men with a penetrating TBI.76 In addition, the patients were tested before they were injured (i.e., at the time they entered the armed forces), and outcome data have been sampled for decades. This material has been used to analyze the relation between the TBI and PTSD77 or other disorders. A new model for experimental penetrating TBI has been used to analyze changes in brain-derived neurotrophic factor (BDNF) and neurotrophin receptors,78 since it was shown that BDNF polymorphism has a strong effect on outcome in the Vietnam head injury study.79 Thus, an analysis of catecholamines and serotonin in penetrating TBI could be of interest.
TBI and monoamine synthesizing enzymes
In agreement with the HPLC results, we observed bilaterally increased transcript levels for TH in the LC and, less pronounced, for TPH2 in the DRN. There are several reports on elevation of both TH and TPH2 following exposure to various other types of stressors.80–84 Supporting our observations, Tümer and colleagues85 also have reported significant increases in TH protein levels in the adrenal medulla (20%) and an even higher increase in nucleus tractus solitarii of the brain stem (49%), and showed that this increase is associated with elevated plasma norepinephrine levels (23%) 6 h post-exposure to blast. The authors have linked this upregulation of the sympathetic system to elevated oxidative stress in the hypothalamus. However, our HPLC analysis of the hypothalamus did not reveal any changes in the monoamines or their metabolites.
George and colleagues86 found differences in both the firing rate of the LC neurones and elevations of TH mRNA in a validated rat PTSD model. Associations between TPH2 genetic variants and “risk ” of development of PTSD have been reported.10 However, Wilson and colleagues74 showed elevated TH protein levels in stressed hippocampus and PFC but reduced TPH2 levels in both regions.
Mild TBI and other trauma/stress models
Milman and colleagues87 have analyzed concussive head trauma in mice and shown increased immobility in the FST at 7 and 90 d post-injury. Another recent study reported increased immobility in the FST of rats 3 d post-exposure in a moderate fluid percussion model.88 Neither of these two studies examined short term consequences, as in our study, and we could not reproduce their demonstration of depression-like behavior at later time-points. These variances may in part be explained by the different injury models, species and severity of injury, such as weight drop versus blast, with the former causing a focal injury and carried out on mice;87 while the latter resulting in a diffuse injury and on rats.88 Even if both these injuries are considered mild, our blast model appears even less severe. In contrast, the fluid percussion model is considered moderate in severity.
PTSD in humans
Dysfunction of the monoamine system has been found in patients suffering from PTSD. NA has a prominent role in arousal, autonomic stress responses and encoding of emotional memories in humans, thus being of central importance in PTSD.89 Studies using yohimbine, an α2 receptor antagonist, prompting increased NA release, induced flashbacks, and increased stress responses in PTSD patients.90 Indirect evidence implicates that 5-HT plays a role in stress and PTSD89,91 due to its extensive role in a number of mood and anxiety disorders. Importantly, 5-HT interacts with NA in the LC exerting inhibition,89 hence possibly modulating the “fight-or-flight” response. Interestingly, in a recent genetic association analysis a risk haplotype in SLC18A2 (also known as vesicular monoamine transporter 2, VMAT2) for PTSD was identified,92 thus reinforcing the role of monoamines in PTSD.
Limitations
The limitations of this study include modeling highly complex physical events, such as explosive blast. The short duration of the blast wave in our model is not representative for all possible types of explosive blasts occurring in various environmental situations. An under-belly detonation of a vehicle by a road-bomb is likely to result in a much more complex waveform with reflections and longer duration. Also, the timetable of pathological changes is likely different in rats, compared with in humans. A larger number of animals and more time-points may reveal even more detailed information about the temporal pattern of changes that occur following exposure. Rat strains also may differ in their neuroendocrine responses,93 and even the same rat strain obtained from different suppliers can have different behaviors in the FST.50 We have not determined changes in NA metabolites in this experiment, and we only used young male rats.
Conclusions
Our present findings indicate that exposure to a single mild blast triggers rapid and transient changes in both NA levels in several forebrain brain regions, and of an NA-synthesising enzyme in the LC. Effects on the serotonin system were essentially confined to changes in the 5-HT synthesising enzyme TPH2, but they were of small magnitude with a short duration, compared with the LC system. Taken together, these findings provide novel information for the further characterization of the pathobiology of mbTBI. A further step could be to analyze biomarkers related to metabolites of NA in animal models for blast and in comparable human populations, such as breachers, which is a situation with a good control of physical parameters and possibilities for sampling of biomarkers.94
Acknowledgments
This study was supported by the Swedish Armed Forces R&D (MR), the Swedish Research Council (04X-2887) (TH) and Karolinska Institutet Funds (TH). The valuable contribution of Maria Angeria is gratefully acknowledged. We thank Ms. Alaa Kamnaksh for her advice on the statistical analysis and for read proofing the paper.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Kessler R.C., Sonnega A., Bromet E., Hughes M., and Nelson C.B. (1995). Posttraumatic stress disorder in the National Comorbidity Survey. Arch. Gen. Psychiatry 52, 1048–1060 [DOI] [PubMed] [Google Scholar]
- 2.Bryant R.A., O'Donnell M.L., Creamer M., McFarlane A.C., Clark C.R., and Silove D. (2010). The psychiatric sequelae of traumatic injury. Am. J. Psychiatry 167, 312–320 [DOI] [PubMed] [Google Scholar]
- 3.Fann J.R., Burington B., Leonetti A., Jaffe K., Katon W.J., and Thompson R.S. (2004). Psychiatric illness following traumatic brain injury in an adult health maintenance organization population. Arch. Gen. Psychiatry 61, 53–61 [DOI] [PubMed] [Google Scholar]
- 4.Chapman J.C. and Diaz-Arrastia R. (2014). Military traumatic brain injury: a review. Alzheimer's Dement. 10, S97–S104 [DOI] [PubMed] [Google Scholar]
- 5.Rosenfeld J.V., McFarlane A.C., Bragge P., Armonda R.A., Grimes J.B., and Ling G.S. (2013). Blast-related traumatic brain injury. Lancet Neurol. 12, 882–893 [DOI] [PubMed] [Google Scholar]
- 6.Agoston D.V., and Elsayed M. (2012). Serum-based protein biomarkers in blast-induced traumatic brain injury spectrum disorder. Front. Neurol. 3, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sherin J.E. and Nemeroff C.B. (2011). Post-traumatic stress disorder: the neurobiological impact of psychological trauma. Dialogues Clin. Neurosci. 13, 263–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pitman R.K., Rasmusson A.M., Koenen K.C., Shin L.M., Orr S.P., Gilbertson M.W., Milad M.R., and Liberzon I. (2012). Biological studies of post-traumatic stress disorder. Nat. Rev. Neurosci. 13, 769–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heim C. and Nemeroff C.B. (2009). Neurobiology of posttraumatic stress disorder. CNS Spectr. 14, 13–24 [PubMed] [Google Scholar]
- 10.Goenjian A.K., Bailey J.N., Walling D.P., Steinberg A.M., Schmidt D., Dandekar U., and Noble E.P. (2012). Association of TPH1, TPH2, and 5HTTLPR with PTSD and depressive symptoms. J. Affect. Disord. 140, 244–252 [DOI] [PubMed] [Google Scholar]
- 11.Kobeissy F., Mondello S., Tümer N., Toklu H.Z., Whidden M.A., Kirichenko N., Zhang Z., Prima V., Yassin W., Anagli J., Chandra N., Svetlov S., and Wang K.K.W. (2013). Assessing neuro-systemic & behavioral components in the pathophysiology of blast-related brain injury. Front. Neurol. 4, 186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yehuda R., McFarlane A.C., and Shalev A.Y. (1998). Predicting the development of posttraumatic stress disorder from the acute response to a traumatic event. Biol. Psychiatry 44, 1305–1313 [DOI] [PubMed] [Google Scholar]
- 13.Goldstein L.E., Fisher A.M., Tagge C. a, Zhang X.-L., Velisek L., Sullivan J. a, Upreti C., Kracht J.M., Ericsson M., Wojnarowicz M.W., Goletiani C.J., Maglakelidze G.M., Casey N., Moncaster J. a, Minaeva O., Moir R.D., Nowinski C.J., Stern R. a, Cantu R.C., Geiling J., Blusztajn J.K., Wolozin B.L., Ikezu T., Stein T.D., Budson A.E., Kowall N.W., Chargin D., Sharon A., Saman S., Hall G.F., Moss W.C., Cleveland R.O., Tanzi R.E., Stanton P.K., and McKee A.C. (2012). Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci. Transl. Med. 4, 134ra60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Drake A.I., Meyer K.S., Cessante L.M., Cheung C.R., Cullen M.A., McDonald E.C., and Holland M.C. (2010). Routine TBI screening following combat deployments. NeuroRehabilitation 26, 183–189 [DOI] [PubMed] [Google Scholar]
- 15.Korf J., Aghajanian G.K., and Roth R.H. (1973). Increased turnover of norepinephrine in the rat cerebral cortex during stress: role of the locus coeruleus. Neuropharmacology 12, 933–938 [DOI] [PubMed] [Google Scholar]
- 16.Abercrombie E.D., Keller R.W., and Zigmond M.J. (1988). Characterization of hippocampal norepinephrine release as measured by microdialysis perfusion: pharmacological and behavioral studies. Neuroscience 27, 897–904 [DOI] [PubMed] [Google Scholar]
- 17.Valentino R.J., Foote S.L., and Page M.E. (1993). The locus coeruleus as a site for integrating corticotropin-releasing factor and noradrenergic mediation of stress responses. Ann. N. Y. Acad. Sci. 697, 173–188 [DOI] [PubMed] [Google Scholar]
- 18.Foote S. and Aston-Jones G. (1995). Pharmacology and physiology of central Noradrenergic Systems. Raven Press: New York [Google Scholar]
- 19.Schatzberg A.F. and Schildkraut J. (1995). Recent studies on norepinephrine systems im mood disorders. In: Psychopharmacology: The Fourth Generation of Progress. Bloom F. and Kupfer D. (eds). Raven Press: New York [Google Scholar]
- 20.Robbins T. and Everitt B. (1995). Central Norepinephrine Neurons and Behavior. Raven Press: New York [Google Scholar]
- 21.Bremner J.D., Krystal J.H., Southwick S.M., and Charney D.S. (1996). Noradrenergic mechanisms in stress and anxiety: I. Preclinical studies. Synapse 23, 28–38 [DOI] [PubMed] [Google Scholar]
- 22.Harro J. and Oreland L. (2001). Depression as a spreading adjustment disorder of monoaminergic neurons: a case for primary implication of the locus coeruleus. Brain Res. Brain Res. Rev. 38, 79–128 [DOI] [PubMed] [Google Scholar]
- 23.Aston-Jones G. and Cohen J.D. (2005). An integrative theory of locus coeruleus-norepinephrine function: adaptive gain and optimal performance. Annu. Rev. Neurosci. 28, 403–450 [DOI] [PubMed] [Google Scholar]
- 24.Berridge C.W. (2008). Noradrenergic modulation of arousal. Brain Res. Rev. 58, 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goddard A.W., Ball S.G., Martinez J., Robinson M.J., Yang C.R., Russell J.M., and Shekhar A. (2010). Current perspectives of the roles of the central norepinephrine system in anxiety and depression. Depress. Anxiety 27, 339–350 [DOI] [PubMed] [Google Scholar]
- 26.Mathew S.J., Manji H.K., and Charney D.S. (2008). Novel drugs and therapeutic targets for severe mood disorders. Neuropsychopharmacology 33, 2080–2092 [DOI] [PubMed] [Google Scholar]
- 27.Millan M.J. (2006). Multi-target strategies for the improved treatment of depressive states: Conceptual foundations and neuronal substrates, drug discovery and therapeutic application. Pharmacol. Ther. 110, 135–370 [DOI] [PubMed] [Google Scholar]
- 28.Maes M. and Meltzer H. (1995). The serotonin hypothesis of major depression. In: Psychopharmacology: The Fourth Generation of Progress. Bloom F. and Kupfer D. (eds). Raven Press: New York [Google Scholar]
- 29.Mann J.J. (1999). Role of the serotonergic system in the pathogenesis of major depression and suicidal behavior. Neuropsychopharmacology 21, 99S–105S [DOI] [PubMed] [Google Scholar]
- 30.Deakin J.F., Pennell I., Upadhyaya A.J., and Lofthouse R. (1990). A neuroendocrine study of 5HT function in depression: evidence for biological mechanisms of endogenous and psychosocial causation. Psychopharmacology (Berl). 101, 85–92 [DOI] [PubMed] [Google Scholar]
- 31.Asberg M., Träskman L., and Thorén P. (1976). 5-HIAA in the cerebrospinal fluid. A biochemical suicide predictor? Arch. Gen. Psychiatry 33, 1193–1197 [DOI] [PubMed] [Google Scholar]
- 32.Canli T. and Lesch K.-P. (2007). Long story short: the serotonin transporter in emotion regulation and social cognition. Nat. Neurosci. 10, 1103–9 [DOI] [PubMed] [Google Scholar]
- 33.Graeff F.G., Guimarães F.S., De Andrade T.G., and Deakin J.F. (1996). Role of 5-HT in stress, anxiety, and depression. Pharmacol. Biochem. Behav. 54, 129–41 [DOI] [PubMed] [Google Scholar]
- 34.Blanchard E.B., Kolb L.C., Prins A., Gates S., and McCoy G.C. (1991). Changes in plasma norepinephrine to combat-related stimuli among Vietnam veterans with posttraumatic stress disorder. J. Nerv. Ment. Dis. 179, 371–373 [DOI] [PubMed] [Google Scholar]
- 35.Dahlstrom A. and Fuxe K. (1964). Evidence for the existence of monoamine-containing neurons in the central nervous system. I. Demonstration of monoamines in the cell bodies of the brain stem neurons. Acta Physiol. Scand. Suppl. SUPPL 232, 1–55 [PubMed] [Google Scholar]
- 36.Nagatsu T., Levitt M., and Udenfriend S. (1964). Tyrosine hydroxylase. The initial step in norepinephrine biosynthesis. J. Biol. Chem. 239, 2910–2917 [PubMed] [Google Scholar]
- 37.Grima B., Lamouroux A., Boni C., Julien J.F., Javoy-Agid F., and Mallet J. (1987). A single human gene encoding multiple tyrosine hydroxylases with different predicted functional characteristics. Nature 326, 707–711 [DOI] [PubMed] [Google Scholar]
- 38.Moore R.Y. and Bloom F.E. (1979). Central catecholamine neuron systems: anatomy and physiology of the norepinephrine and epinephrine systems. Annu. Rev. Neurosci. 2, 113–68 [DOI] [PubMed] [Google Scholar]
- 39.Ungerstedt U. (1971). Stereotaxic mapping of the monoamine pathways in the rat brain. Acta Physiol. Scand. Suppl. 367, 1–48 [DOI] [PubMed] [Google Scholar]
- 40.Bjorklund A. and Lindvall O. (1986). Catecholaminergic regulatory systems. In: Handbook of Physiology. Section 1: The Nervous System. Intrinsic Regulatory Systems of the Brain. Bloom F.E. (ed). American Physiology Society: Washington, DC [Google Scholar]
- 41.Steinbusch H.W. (1981). Distribution of serotonin-immunoreactivity in the central nervous system of the rat-cell bodies and terminals. Neuroscience 6, 557–618 [DOI] [PubMed] [Google Scholar]
- 42.Jacobs B.L. and Azmitia E.C. (1992). Structure and function of the brain serotonin system. Physiol. Rev. 72, 165–229 [DOI] [PubMed] [Google Scholar]
- 43.Walther D.J., Peter J.-U., Bashammakh S., Hörtnagl H., Voits M., Fink H., and Bader M. (2003). Synthesis of serotonin by a second tryptophan hydroxylase isoform. Science 299, 76. [DOI] [PubMed] [Google Scholar]
- 44.Xiong Y., Mahmood A., and Chopp M. (2013). Animal models of traumatic brain injury. Nat. Rev. Neurosci. 14, 128–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cernak I. (2005). Animal models of head trauma. NeuroRx 2, 410–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Risling M., Plantman S., Angeria M., Rostami E., Bellander B.-M., Kirkegaard M., Arborelius U., and Davidsson J. (2011). Mechanisms of blast induced brain injuries, experimental studies in rats. Neuroimage 54 Suppl 1, S89–S97 [DOI] [PubMed] [Google Scholar]
- 47.Porsolt R.D., Anton G., Blavet N., and Jalfre M. (1978). Behavioral despair in rats: a new model sensitive to antidepressant treatments. Eur. J. Pharmacol. 47, 379–391 [DOI] [PubMed] [Google Scholar]
- 48.Detke M.J. and Lucki I. (1996). Detection of serotonergic and noradrenergic antidepressants in the rat forced swimming test: the effects of water depth. Behav. Brain Res. 73, 43–46 [DOI] [PubMed] [Google Scholar]
- 49.Detke M.J., Rickels M., and Lucki I. (1995). Active behaviors in the rat forced swimming test differentially produced by serotonergic and noradrenergic antidepressants. Psychopharmacology (Berl). 121, 66–72 [DOI] [PubMed] [Google Scholar]
- 50.Bogdanova O. V, Kanekar S., D'Anci K.E., and Renshaw P.F. (2013). Factors influencing behavior in the forced swim test. Physiol. Behav. 118, 227–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Clemedson C.J. (1956). Shock wave transmission to the central nervous system. Acta Physiol. Scand. 37, 204–214 [DOI] [PubMed] [Google Scholar]
- 52.Clemedson C.J., Jonsson A., and Pettersson H. (1956). Propagation of an air-transmitted shock wave in muscular tissue. Nature 177, 380–381 [DOI] [PubMed] [Google Scholar]
- 53.Säljö A., Bao F., Haglid K.G., and Hansson H.A. (2000). Blast exposure causes redistribution of phosphorylated neurofilament subunits in neurons of the adult rat brain. J. Neurotrauma 17, 719–726 [DOI] [PubMed] [Google Scholar]
- 54.Clemedson C.J. and Criborn C.O. (1955). A detonation chamber for physiological blast research. J. Aviat. Med. 26, 373–381 [PubMed] [Google Scholar]
- 55.Paxinos G. and Watson C. (2007). The Rat Brain in Streotaxic Coordinates, 6th ed. Elsevier: Amsterdam [Google Scholar]
- 56.Dagerlind A., Friberg K., Bean A.J., and Hökfelt T. (1992). Sensitive mRNA detection using unfixed tissue: combined radioactive and non-radioactive in situ hybridization histochemistry. Histochemistry 98, 39–49 [DOI] [PubMed] [Google Scholar]
- 57.Rostami E., Davidsson J., Ng K.C., Lu J., Gyorgy A., Walker J., Wingo D., Plantman S., Bellander B.-M., Agoston D. V, and Risling M. (2012). A Model for Mild Traumatic Brain Injury that Induces Limited Transient Memory Impairment and Increased Levels of Axon Related Serum Biomarkers. Front. Neurol. 3, 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kehr J. and Yoshitake T. (2006). Monitoring brain chemical signals by microdialysis. In: Encyclopedia of Sensors, Volume 6 Grimes C.A., Dickey E.C., and Pishko M.V., (eds). American Scientific Publishers, USA [Google Scholar]
- 59.Estanislau C., Ramos A.C., Ferraresi P.D., Costa N.F., de Carvalho H.M.C.P., and Batistela S. (2011). Individual differences in the elevated plus-maze and the forced swim test. Behav. Processes 86, 46–51 [DOI] [PubMed] [Google Scholar]
- 60.Pellow S., Chopin P., File S.E., and Briley M. (1985). Validation of open:closed arm entries in an elevated plus-maze as a measure of anxiety in the rat. J. Neurosci. Methods 14, 149–167 [DOI] [PubMed] [Google Scholar]
- 61.Shinba T., Ozawa N., Yoshii M., and Yamamoto K. (2010). Delayed increase of brain noradrenaline after acute footshock stress in rats. Neurochem. Res. 35, 412–417 [DOI] [PubMed] [Google Scholar]
- 62.Cryan J.F., Markou A., and Lucki I. (2002). Assessing antidepressant activity in rodents: recent developments and future needs. Trends Pharmacol. Sci. 23, 238–245 [DOI] [PubMed] [Google Scholar]
- 63.Robbins T.W., and Arnsten A.F.T. (2009). The neuropsychopharmacology of fronto-executive function: monoaminergic modulation. Annu. Rev. Neurosci. 32, 267–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Flierl M.A., Rittirsch D., Huber-Lang M., Sarma J.V., and Ward P.A. (2008). Catecholamines-crafty weapons in the inflammatory arsenal of immune/inflammatory cells or opening pandora's box? Mol. Med. 14, 195–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cernak I. (2010). The importance of systemic response in the pathobiology of blast-induced neurotrauma. Front. Neurol. 1, 151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Arnsten A.F.T. (2009). Stress signalling pathways that impair prefrontal cortex structure and function. Nat. Rev. Neurosci. 10, 410–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.McEwen B.S. and Morrison J.H. (2013). The brain on stress: vulnerability and plasticity of the prefrontal cortex over the life course. Neuron 79, 16–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lucassen P.J., Pruessner J., Sousa N., Almeida O.F.X., Van Dam A.M., Rajkowska G., Swaab D.F., and Czéh B. (2014). Neuropathology of stress. Acta Neuropathol. 127, 109–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Finklestein S., Campbell A., Stoll A.L., Baldessarini R.J., Stinus L., Paskevitch P.A., and Domesick V.B. (1983). Changes in cortical and subcortical levels of monoamines and their metabolites following unilateral ventrolateral cortical lesions in the rat. Brain Res. 271, 279–288 [DOI] [PubMed] [Google Scholar]
- 70.Pappius H.M. and Dadoun R. (1987). Effects of injury on the indoleamines in cerebral cortex. J. Neurochem. 49, 321–325 [DOI] [PubMed] [Google Scholar]
- 71.Tsuiki K., Takada A., Nagahiro S., Grdisa M., Diksic M., and Pappius H.M. (1995). Synthesis of serotonin in traumatized rat brain. J. Neurochem. 64, 1319–1325 [DOI] [PubMed] [Google Scholar]
- 72.Tanaka K., Ogawa N., Asanuma M., and Kondo Y. (1997). Thyrotropin releasing hormone prevents abnormalities of cortical acetylcholine and monoamines in mice following head injury. Regul. Pept. 70, 173–178 [DOI] [PubMed] [Google Scholar]
- 73.Harvey B.H., Brand L., Jeeva Z., and Stein D.J. (2006). Cortical/hippocampal monoamines, HPA-axis changes and aversive behavior following stress and restress in an animal model of post-traumatic stress disorder. Physiol. Behav. 87, 881–890 [DOI] [PubMed] [Google Scholar]
- 74.Wilson C.B., Ebenezer P.J., McLaughlin L.D., and Francis J. (2014). Predator exposure/psychosocial stress animal model of post-traumatic stress disorder modulates neurotransmitters in the rat hippocampus and prefrontal cortex. PLoS One 9, e89104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Risling M. and Davidsson J. (2012). Experimental animal models for studies on the mechanisms of blast-induced neurotrauma. Front. Neurol. 3, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Raymont V., Salazar A.M., Krueger F., and Grafman J. (2011). “Studying injured minds”—the Vietnam head injury study and 40 years of brain injury research. Front. Neurol. 2, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Koenigs M., Huey E.D., Raymont V., Cheon B., Solomon J., Wassermann E.M., and Grafman J. (2008). Focal brain damage protects against post-traumatic stress disorder in combat veterans. Nat. Neurosci. 11, 232–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rostami E., Krueger F., Plantman S., Davidsson J., Agoston D., Grafman J., and Risling M. (2014). Alteration in BDNF and its receptors, full-length and truncated TrkB and p75(NTR) following penetrating traumatic brain injury. Brain Res. 1542, 195–205 [DOI] [PubMed] [Google Scholar]
- 79.Rostami E., Krueger F., Zoubak S., Dal Monte O., Raymont V., Pardini M., Hodgkinson C.A., Goldman D., Risling M., and Grafman J. (2011). BDNF polymorphism predicts general intelligence after penetrating traumatic brain injury. PLoS One 6, e27389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ong L.K., Guan L., Damanhuri H., Goodchild A.K., Bobrovskaya L., Dickson P.W., and Dunkley P.R. (2014). Neurobiological consequences of acute footshock stress: effects on tyrosine hydroxylase phosphorylation and activation in the rat brain and adrenal medulla. J. Neurochem. 128, 547–560 [DOI] [PubMed] [Google Scholar]
- 81.McDevitt R.A., Szot P., Baratta M. V, Bland S.T., White S.S., Maier S.F., and Neumaier J.F. (2009). Stress-induced activity in the locus coeruleus is not sensitive to stressor controllability. Brain Res. 1285, 109–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chang M.S., Sved A.F., Zigmond M.J., and Austin M.C. (2000). Increased transcription of the tyrosine hydroxylase gene in individual locus coeruleus neurons following footshock stress. Neuroscience 101, 131–139 [DOI] [PubMed] [Google Scholar]
- 83.Evans A.K., Heerkens J.L.T., and Lowry C.A. (2009). Acoustic stimulation in vivo and corticotropin-releasing factor in vitro increase tryptophan hydroxylase activity in the rat caudal dorsal raphe nucleus. Neurosci. Lett. 455, 36–41 [DOI] [PubMed] [Google Scholar]
- 84.Chamas F.M., Underwood M.D., Arango V., Serova L., Kassir S.A., Mann J.J., and Sabban E.L. (2004). Immobilization stress elevates tryptophan hydroxylase mRNA and protein in the rat raphe nuclei. Biol. Psychiatry 55, 278–283 [DOI] [PubMed] [Google Scholar]
- 85.Tümer N., Svetlov S., Whidden M., Kirichenko N., Prima V., Erdos B., Sherman A., Kobeissy F., Yezierski R., Scarpace P.J., Vierck C., and Wang K.K.W. (2013). Overpressure blast-wave induced brain injury elevates oxidative stress in the hypothalamus and catecholamine biosynthesis in the rat adrenal medulla. Neurosci. Lett. 544, 62–67 [DOI] [PubMed] [Google Scholar]
- 86.George S.A., Knox D., Curtis A.L., Aldridge J.W., Valentino R.J., and Liberzon I. (2013). Altered locus coeruleus-norepinephrine function following single prolonged stress. Eur. J. Neurosci. 37, 901–909 [DOI] [PubMed] [Google Scholar]
- 87.Milman A., Rosenberg A., Weizman R., and Pick C.G. (2005). Mild traumatic brain injury induces persistent cognitive deficits and behavioral disturbances in mice. J. Neurotrauma 22, 1003–10 [DOI] [PubMed] [Google Scholar]
- 88.Kuo J.-R., Cheng Y.-H., Chen Y.-S., Chio C.-C., and Gean P.-W. (2013). Involvement of extracellular signal regulated kinases in traumatic brain injury-induced depression in rodents. J. Neurotrauma 30, 1223–1231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Blier P. (2001). Crosstalk between the norepinephrine and serotonin systems and its role in the antidepressant response. J. Psychiatry Neurosci. 26 Suppl, S3–S10 [PMC free article] [PubMed] [Google Scholar]
- 90.Bremner J.D., Innis R.B., Ng C.K., Staib L.H., Salomon R.M., Bronen R.A., Duncan J., Southwick S.M., Krystal J.H., Rich D., Zubal G., Dey H., Soufer R., and Charney D.S. (1997). Positron emission tomography measurement of cerebral metabolic correlates of yohimbine administration in combat-related posttraumatic stress disorder. Arch. Gen. Psychiatry 54, 246–254 [DOI] [PubMed] [Google Scholar]
- 91.Davis L.L., Suris A., Lambert M.T., Heimberg C., and Petty F. (1997). Post-traumatic stress disorder and serotonin: new directions for research and treatment. J. Psychiatry Neurosci. 22, 318–326 [PMC free article] [PubMed] [Google Scholar]
- 92.Solovieff N., Roberts A.L., Ratanatharathorn A., Haloosim M., De Vivo I., King A.P., Liberzon I., Aiello A., Uddin M., Wildman D.E., Galea S., Smoller J.W., Purcell S.M., and Koenen K.C. (2014). Genetic Association Analysis of 300 Genes Identifies a Risk Haplotype in SLC18A2 for Post-traumatic Stress Disorder in Two Independent Samples. Neuropsychopharmacology 39, 1872–1879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pardon M.-C., Gould G.G., Garcia A., Phillips L., Cook M.C., Miller S.A., Mason P.A., and Morilak D.A. (2002). Stress reactivity of the brain noradrenergic system in three rat strains differing in their neuroendocrine and behavioral responses to stress: implications for susceptibility to stress-related neuropsychiatric disorders. Neuroscience 115, 229–242 [DOI] [PubMed] [Google Scholar]
- 94.Tate C.M., Wang K.K.W., Eonta S., Zhang Y., Carr W., Tortella F.C., Hayes R.L., and Kamimori G.H. (2013). Serum brain biomarker level, neurocognitive performance, and self-reported symptom changes in soldiers repeatedly exposed to low-level blast: a breacher pilot study. J. Neurotrauma 30, 1620–1630 [DOI] [PubMed] [Google Scholar]