

Abstract
Background and Purpose
To investigate whether a narrow spectrum kinase inhibitor RV1088, which simultaneously targets specific MAPKs, Src and spleen tyrosine kinase (Syk), is more effective at inhibiting inflammatory signalling in rheumatoid arthritis (RA) than single kinase inhibitors (SKIs).
Experimental Approach
elisas were used to determine the efficacy of RV1088, clinically relevant SKIs and the pharmaceutical Humira on pro-inflammatory cytokine production by activated RA synovial fibroblasts, primary human monocytes and macrophages, as well as spontaneous cytokine synthesis by synovial membrane cells from RA patients. In human macrophages, RNAi knockdown of individual kinases was used to reveal the effect of inhibition of kinase expression on cytokine synthesis.
Key Results
RV1088 reduced TNF-α, IL-6 and IL-8 production in all individual activated cell types with low, nM, IC50s. SKIs, and combinations of SKIs, were significantly less effective than RV1088. RNAi of specific kinases in macrophages also caused only modest inhibition of pro-inflammatory cytokine production. RV1088 was also significantly more effective at inhibiting IL-6 and IL-8 production by monocytes and RA synovial fibroblasts compared with Humira. Finally, RV1088 was the only inhibitor that was effective in reducing TNF-α, IL-6 and IL-8 synthesis in RA synovial membrane cells with low nM IC50s.
Conclusions and Implications
This study demonstrates potent anti-inflammatory effect of RV1088, highlighting that distinct signalling pathways drive TNF-α, IL-6 and IL-8 production in the different cell types found in RA joints. As such, targeting numerous signalling pathways simultaneously using RV1088 could offer a more powerful method of reducing inflammation in RA than targeting individual kinases.
Tables of Links
| TARGETS | ||
|---|---|---|
| Catalytic receptorsa | Enzymesb | |
| TLR4 | HCK | MK2 |
| JAK1 | MK3 | |
| JAK3 | MSK | |
| MAPK12 (p38γ) | Syk | |
| MAPK14 (p38α) | Src |
| LIGANDS | ||
|---|---|---|
| BIRB 796 | IL-1ra | LPS |
| Dasatinib | IL-6 | M-CSF |
| Fostamatinib (R788) | IL-8 | R406 |
| Humira | IL-10 | TNF-α |
| IFNβ | IL-17 | Tofacitinib |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,bAlexander et al., 2013a,b).
Introduction
Monocytes, macrophages and synovial fibroblasts are abundant in the hyperplastic rheumatoid arthritis (RA) synovium and are key contributors to synovitis and joint destruction (Huber et al., 2006; Kinne et al., 2007; Muller-Ladner et al., 2007; McInnes and Schett, 2011). Pro-inflammatory cytokine expression, including TNF-α and IL-6, by effector cells induces persistent cell activation and expression of additional cytokines and chemokines, including IL-8, maintaining a state of unresolved inflammation in the joint (McInnes and Schett, 2007; Brennan and McInnes, 2008; Lee et al., 2013). Despite significant progress in the application of anti-inflammatory drugs that target cytokines or their receptors to treat RA, the lack or loss of response by significant numbers of patients to currently available therapies highlights a need for improved or complementary approaches (Genovese et al., 2005; van Schouwenburg et al., 2013).
A number of kinases are highly expressed in the RA synovium (Suzuki et al., 2000; Huber et al., 2006; Hammaker and Firestein, 2010; Lindstrom and Robinson, 2010; Page et al., 2010). Activation of specific isoforms of the p38 MAPK, p38α and p38γ (Hammaker and Firestein, 2010), spleen tyrosine kinase (Syk) (Weinblatt et al., 2010), JAKs, particularly JAK1 and JAK3 (Lindstrom and Robinson, 2010), and the family of Src protein tyrosine kinases (Shahrara et al., 2007) has been shown to drive pro-inflammatory signalling cascades inducing cytokine expression in RA synovial fibroblasts and activated monocytes and macrophages.
Small molecule inhibitors, which target specific, individual kinases, have been developed for the treatment of autoimmune diseases (Lindstrom and Robinson, 2010; O’Shea et al., 2013). Many of these, however, demonstrated significant toxicity and poor efficacy in the clinic (Cohen et al., 2009; O’Shea et al., 2013). Kinase inhibitors targeting both JAK (tofacitinib) and Syk (fostamatinib; R788) have been developed and clinically tested for the treatment of RA (Weinblatt et al., 2010; 2013,; Tanaka et al., 2011; Burmester et al., 2013), but these also suffered from suboptimal efficacy (release Ap, 2013; release Pp, 2013).
Ideally, kinase inhibitors designed for use in treating RA should target several kinases simultaneously to overcome low efficacy caused by kinase redundancy. They should also target with high specificity to reduce off-target effects caused by blockade of non-pathogenic kinases. One approach to address this issue has emerged with the development of narrow spectrum kinase inhibitor (NSKIs). Unlike single kinase inhibitors (SKIs), NSKIs are designed to target multiple signalling kinases with high specificity in order to hit a number of distinct inflammatory pathways simultaneously. These have proved extremely efficacious in treating patients with corticosteroid-insensitive pulmonary inflammatory disease (Charron et al., 2011; Knobloch et al., 2011). Similar kinase cascades are activated in inflammatory lung disease as in RA; therefore, we hypothesized that NSKIs may offer a valuable therapeutic approach to dampening joint inflammation. Here, we examine the ability of RV1088, a NSKI, to reduce pro-inflammatory cytokine synthesis in RA synovial membrane cells and in isolated immune cells compared with SKIs that target p38, c-Src, Syk or JAK individually.
Methods
Reagents
RPMI 1640 with 25 mM L-glutamine with or without phenol red, 100× penicillin-streptomycin and Trypsin-EDTA were purchased from PAA Laboratories (GE Healthcare, Fairfield, CT, USA). DMEM was purchased from Lonza BioWhittaker (Walkersville, MD, USA). All growth media were supplemented with 1× penicillin-streptomycin (100 U·mL−1 penicillin and 100 μg·mL−1 streptomycin) unless stated. Heat-inactivated FCS and Opti-MEM® were from Gibco (Thermo Fisher, Rockford, IL, USA). Recombinant human macrophage colony stimulating factor (M-CSF) was from Peprotech (Rocky Hill, NJ, USA). Cell dissociation solution (non-enzymatic, 1× Bioreagent) was from Sigma-Aldrich (St. Louis, MO, USA). LPS was purchased from Enzo Life Sciences (Farmingdale, NY, USA). DMSO was from Merck & Co. (Kenilworth, NJ, USA). Kinase inhibitors were obtained from RespiVert Ltd (London, UK).
Cells and cell culture
RA synovial membrane cells were isolated from human RA joints removed for joint replacement therapy as previously described (Brennan et al., 1989; Sacre et al., 2007). RA synovial membrane cells were plated at a density of 1 × 106 mL−1 in 96-well dishes in RPMI 1640 medium supplemented with 10% heat-inactivated FBS. Cells were treated immediately with the NSKI RV1088 (N-[4-[[4-[[[[3-(1,1-dimethylethyl)-1-(4-methylphenyl)-1H-pyrazol-5-yl]amino]carbonyl]amino]-1-naphthalenyl]oxy]-2-pyridinyl]-2-methoxyacetamide), SKIs or Humira at concentrations stated or with an equivalent volume of DMSO and then cultured for 24 h. Synovial fibroblasts were isolated from RA synovial tissue by collagenase digestion as described previously (Foxwell et al., 1998; Miller et al., 2009). RA synovial fibroblasts were used after three to four passages in DMEM supplemented with 10% FBS before being plated at 5 × 104 mL−1 in 96-well dishes. Cells were rested for 24 h before stimulation with 10 ng·mL−1 LPS and treatment with NSKI RV1088, SKIs or Humira at concentrations stated or with an equivalent volume of DMSO and cultured for a further 24 h. Human monocytes were isolated from peripheral blood mononuclear cells of individual healthy human blood donors as described previously (Foxwell et al., 1998; Sacre et al., 2007). Cells were plated at a density of 1 × 106 mL−1 in 96-well dishes in RPMI 1640 medium supplemented with 5% FBS. Cells were treated immediately with 1 ng·mL−1 LPS and the NSKI, SKIs or Humira at the concentrations stated or with an equivalent volume of DMSO and then cultured for 24 h. When required, monocytes were differentiated into macrophages by culturing with 100 ng·mL−1 recombinant human M-CSF for 5 d in RPMI 1640 medium supplemented with 5% FBS. A total of 5 × 105 mL−1 macrophages were plated in 96-well dishes and rested for 24 h before activation with 1 ng·mL−1 LPS and treatment with NSKI, SKIs or Humira at the concentrations stated or with an equivalent volume of DMSO before culturing for a further 90 min, 4 h or 24 h. For siRNA knockdown studies, cells were plated at 4.8 × 105 mL−1 in 48-well dishes and rested for 24 h before siRNA knockdown and cell stimulation. All cell cultures were incubated at 37°C in 5% CO2 in a humidified incubator. Cell viability was confirmed using the 3-(4,5-dimethythiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay (Sigma).
elisa
Sandwich elisas were used to determine TNF-α, IL-6 and IL-8 concentrations [BD Pharmingen (BD Biosciences), Oxford, Oxfordshire, UK] in conditioned media of cultured cells following the manufacturer’s protocol as described previously (Ruhmann et al., 2012). elisa plates were read at 450 nm on a spectrophotometric microplate reader (FLUOstar Omega; BMG Labtech, Ortenberg, Germany).
Determination of IC50s
Cytokine concentrations were determined and normalized to the DMSO control (set at 100%). The normalized data for each independent donor was then plotted as an inhibition dose-response curve using the GraphPad Prism software (GraphPad Software, Inc. La Jolla, CA, USA) and the IC50 for each inhibitor was calculated from this.
RNA interference
The protocol is described previously (Peirce et al., 2010; Smallie et al., 2010). In brief, commercially available Silencer® Select siRNAs targeting STAT-3 (6793), SRC (s13414), MAPK12 (s12468), MAPK14 (s3586), SYK (s13679), HCK (s6479), JAK1 (s7646), JAK3 (s7652) or a scrambled Silencer Select negative control #1 (#4390843) (Applied Biosciences, Life Technologies, Paisley, UK) were pre-incubated with Dharmafect-1 (Dharmacon/GE Healthcare, Fairfield, CT, USA) and serum-free Opti-MEM. This mix was added to macrophages in phenol red-free serum-free RPMI 1640 medium. Macrophages were incubated for 2 h to allow the siRNA transfection complexes to work. Transfection complexes were then removed and replaced with RPMI 1640 medium supplemented with 5% FBS and incubated for a further 3 days before cell stimulation with 1 ng·mL−1 LPS.
Real-time quantitative qPCR
To confirm successful gene silencing, total RNA was extracted from macrophages using the RNeasy Mini Kit (Qiagen, Venlo, Limburg, Netherlands). 200 ng RNA was then reverse transcribed to generate cDNA templates as described previously (Piccinini and Midwood, 2012). Expression of mRNA was determined using TaqMan® Universal PCR Master Mix and primer/probe sets for: STAT-3 (Hs00374280_m1), SRC (Hs01082246_m1), MAPK14 (Hs00176247_m1), MAPK12 (Hs00268060_m1), SYK (Hs00895377_m1), HCK (Hs00176654_m1), JAK1 (Hs01026983_m1), JAK3 (Hs00169663_m1), PI3KCD (Hs00192399_m1), PI3KCG (Hs00277090_m1) (Applied Biosystems). Reactions were performed in a Rotor-Gene 6000 instrument (Corbett Life Science, Qiagen, Venlo, Limburg, Netherlands) and relative gene expression was determined by ΔΔCt algorithm (Ruhmann et al., 2012).
Statistics
Data are all expressed as mean ± SEM unless stated otherwise. Data were analysed by one-way anova with Tukey’s post hoc test or two-way anova with Bonferroni’s post test.
Results
RV1088 inhibits cytokine production by primary human macrophages, monocytes and synovial fibroblasts
Synovial membranes from the joints of RA patients are composed of a mixture of cell types including macrophages, monocytes and synovial fibroblasts (Huber et al., 2006; McInnes and Schett, 2011; Cooper et al., 2012). We first examined the ability of the NSKI RV1088 to inhibit cytokine production by these distinct cell types. RV1088 inhibits the activity of p38-α, p38-γ, Src family members c-Src and Hck, and Syk. Therefore, SKIs targeting p38 (BIRB 796; all p38 isoforms), c-Src (dasatinib) and Syk (R406), as well as a JAK inhibitor (tofacitinib; JAK-1, -2 and -3) (Supporting Information Table S1) were included for comparison. LPS was used to activate primary human monocytes and macrophages isolated from healthy blood donors and fibroblasts isolated from the synovia of RA patients. The effect of increasing concentrations of kinase inhibitors on TNF-α, IL-6 and IL-8 levels were determined for each cell type. Cell viability was confirmed using the MTT assay and cell morphology did not change in response to these drugs (data from RA synovial fibroblasts and macrophages treated with DMSO, RV1088, BIRB and Humira shown in Supporting Information Fig. S1A–D). The anti-TNF-α antibody Humira was also included as a clinical standard for comparison. Inhibitory dose-response curves were plotted and the IC50 values were calculated for each inhibitor (Table 2012).
Table 1.
RV1088 is the most effective inhibitor of pro-inflammatory cytokine production by distinct RA cell types
| Macrophages (IC50 nM) | Monocytes (IC50 nM) | RA synovial fibroblasts (IC50 nM) | ||||||
|---|---|---|---|---|---|---|---|---|
| TNF-α | IL-6 | IL-8 | TNF-α | IL-6 | IL-8 | IL-6 | IL-8 | |
| BIRB 796 | 4.97 | 505.68 | ND | ND | ND | no effect | 8 225.38 | no effect |
| RV1088 | 0.63 | 1.66 | ND | 0.97 | 1.10 | 1.32 | 5.45 | 1.61 |
| Dasatinib | 7.81 | 37.05 | 3284.84 | 12.58 | no effect | no effect | 217.21 | 136.84 |
| R406 | 1.06 | 738.09 | no effect | 1181.70 | 2030.21 | 898.72 | 670.64 | no effect |
| Tofacitinib | no effect | no effect | no effect | no effect | no effect | no effect | 127 147.44 | no effect |
| Humira | 4.60 × 10−3 | 1.72 × 10−3 | 5.34 × 10−3 | 0.06 | ND | ND | no effect | no effect |
IC50s were calculated from concentration-dependent inhibitory dose-response curves of percentage inhibition normalized to DMSO controls. Data are the average from at least three independent donors. ND; not determined as the inhibition did not reach 50% within the concentration range that did not affect cell toxicity.
In LPS-activated macrophages, all inhibitors except tofacitinib, dose-dependently reduced TNF-α levels. RV1088 was the most efficacious of the kinase inhibitors (IC50 = 0.63 nM) while the strongest inhibitor was Humira (IC50 = 0.0046 nM). RV1088 and Humira also reduced IL-6 production effectively (IC50 = 1.66 nM and IC50 = 0.00172 nM respectively). A weaker effect on IL-6 was observed for BIRB 796, dasatinib and R406 with only greater than 50% inhibition observable at higher concentrations. Humira showed very potent, dose-dependent inhibition of IL-8 levels (IC50 = 0.00534 nM), whereas BIRB 796 and RV1088 did not reach 50% inhibition at the concentrations tested where cell viability was unaffected. Dasatinib only inhibited IL-8 at very high concentrations and R406 had no effect. In contrast, tofacitinib had no effect on macrophage TNF-α, IL-6 or IL-8 production (Figure 1, Table 2012). Cytokine induction by macrophages in response to LPS was assessed over time showing detectable TNF-α and IL-8 by 90 min, but negligible synthesis of IL-6 at this time point. Synthesis of all three cytokines was observed at 4 and 24 h (Supporting Information Fig. S1E). Analysis of RV1088 activity over time revealed that 0.002 μg·mL−1 achieved maximal inhibition of TNF-α and IL-6 at 4 h and of IL-8 by 24 h. Comparison of the activity of BIRB and RV1088 at the equivalent dose of 0.001 μg·mL−1 showed RV1088 to reduce levels of all three cytokines earlier than BIRB. However, high concentrations of BIRB could more effectively inhibit both TNF-α and IL-8 at 90 min (Supporting Information Fig. S1F–H).
Figure 1.
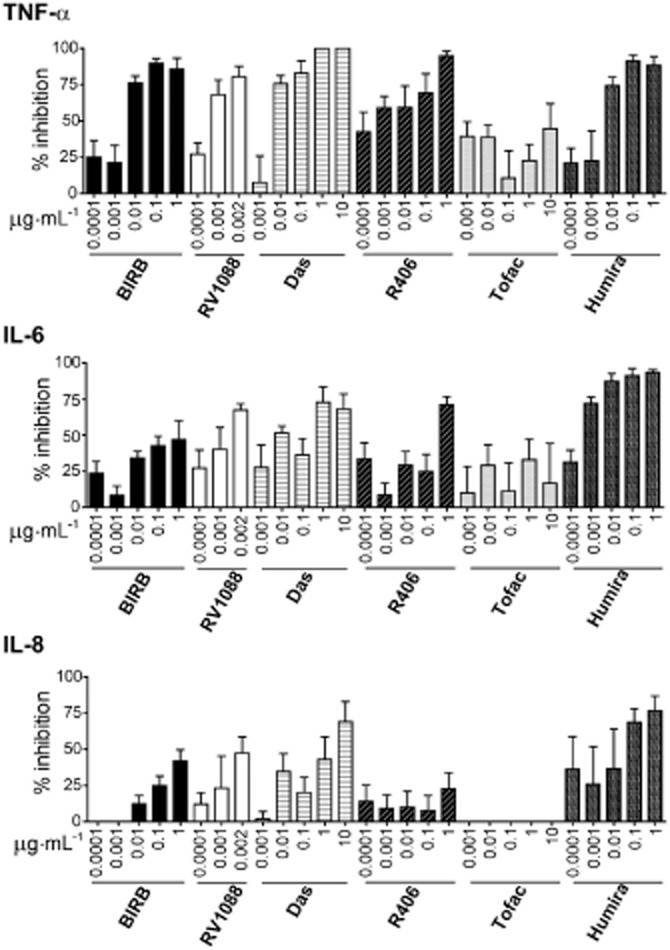
Specific effects of RV1088, SKIs and Humira on pro-inflammatory cytokine production by stimulated macrophages. TNF-α, IL-6 and IL-8 levels in conditioned media, from LPS-stimulated monocyte-derived macrophages treated with increasing concentrations of all inhibitors (BIRB, BIRB 796; Das, dasatinib; Tofac, tofacitinib) were determined by elisa. Data are shown as percentage inhibition normalized to DMSO controls and are the average from at least four donors ± SEM. Results were analysed by two-way anova with Bonferroni’s post test comparing all inhibitors to BIRB 796; no significant differences were detected.
In LPS-activated monocytes, as for macrophages, all inhibitors except tofacitinib reduced TNF-α levels. Humira was the most effective overall (IC50 = 0.06 nM). RV1088 showed significantly increased inhibition of TNF-α compared with BIRB 796 (P < 0.001 at 0.001 and 0.003 μg·mL−1) and exhibited the lowest IC50 of the kinase inhibitors (0.97 nM). However, in contrast to macrophages, RV1088 was the only compound that showed any efficacy for inhibiting IL-6 or IL-8 production in monocytes (IC50 = 1.10 and 1.32 nM respectively) and was also a significantly better inhibitor compared with BIRB 796 at concentrations above 0.001 μg·mL−1. Dasatinib did show significant inhibition of IL-8 production compared with BIRB 796 at 0.005 and 0.01 μg·mL−1 (P < 0.05), but the IC50 could not be determined as inhibition did not reach 50%. Humira did not significantly affect IL-6 or IL-8 levels (Supporting Information Fig. S2, Table 2012).
RA synovial fibroblasts do not produce TNF-α upon LPS stimulation (Ryzhakov et al., 2011; Wahamaa et al., 2011). Dose-dependent inhibition of IL-6 and IL-8 production by LPS-stimulated RA synovial fibroblasts was observed with kinase inhibitors RV1088, dasatinib and R406. RV1088 and dasatinib demonstrated significantly increased inhibition at 0.01–1 and 1 μg·mL−1 respectively compared with BIRB 796. RV1088 was the most effective inhibitor tested (IL-6, IC50 = 5.45 nM; IL-8, IC50 = 1.61 nM). Dasatinib only showed significant inhibitions at the highest concentration tested (IC50s: IL-6 = 217.21 nM; IL-8 = 136.84 nM). Neither Humira nor tofacitinib had any effect on IL-6 or IL-8 synthesis by RA synovial fibroblasts (Supporting Information Fig. S3, Table 2012).
In summary, RV1088 was consistently the most effective inhibitor, reducing TNF-α, IL-6 and IL-8 levels in each of the three cell types tested, whereas tofacitinib consistently had no effect on any cytokine in each cell type. Interestingly, the SKIs, as well as Humira, exhibited a cell type-specific effect, demonstrating good inhibition of some, but not all, cytokines in distinct cell types.
Knockdown of kinase expression modestly inhibits the synthesis of specific cytokines by macrophages
To further assess the effect of kinase inhibition on cytokine production, the expression of individual kinases in primary human macrophages was knocked down by siRNA. Knockdown of gene expression was assessed by RT-PCR compared with scrambled control siRNAs. STAT3 was included as a validated positive control (Peirce et al., 2010). An average of 74% to 56.7% reduction in gene expression was achieved (Figure 2A). siRNA knockdown of MAPK14 and MAPK12, which encode p38-α and -γ respectively, inhibited TNF-α production by LPS-stimulated macrophages (MAPK14, 31.80 ± 19.52 SEM %; MAPK12, 21.78 ± 15.64 SEM %) as did knockdown of SYK (22.50 ± 16.20 SEM %), but inhibition never exceeded 50%. No effect was observed on IL-6 or IL-8 levels. Knockdown of SRC, HCK and JAK3 had minimal effects on TNF-α, IL-6 or IL-8 levels. In contrast, knockdown of JAK1 expression resulted in a high increase of TNF-α, IL-6 and IL-8 in LPS-stimulated macrophages as did STAT3, consistent with previous reports (Takeda et al., 1999; Agbanoma et al., 2012; Pattison et al., 2012; Figure 2B). These data suggest that the reduction in expression of individual kinases has a modest impact on cytokine production upon LPS stimulation of macrophages. It also highlights the potential problems with globally inhibiting kinase families whose members exhibit opposing inflammatory properties, such as JAK1 and JAK3.
Figure 2.
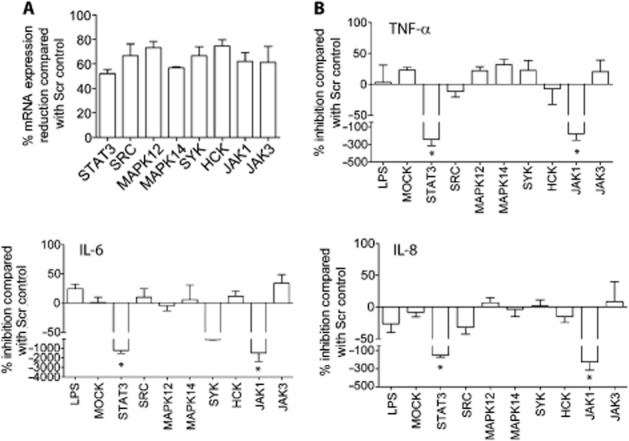
The effect of siRNA-mediated knockdown of SKIs on pro-inflammatory cytokine production. (A) Gene expression levels in macrophages for specific siRNAs targeting individual kinases: SRC, MAPK12, MAPK14, SYK, HCK, JAK1 and JAK3 were determined by RT-PCR and levels shown as percentage inhibition compared with the scrambled siRNA control. STAT3 siRNA was included as a positive control. Data are averaged from at least three donors ± SEM. Macrophages treated with specific siRNAs to knockdown expression of individual kinases were then stimulated with LPS and (B) TNF-α, IL-6 and IL-8 levels in conditioned media were determined by elisa. ‘LPS’ denotes cells that were treated only with LPS. ‘MOCK’ denotes cells that were treated with Dharmafect-1 reagent and serum-free Opti-MEM only without addition of siRNA. Data are shown as percentage inhibition normalized to scrambled siRNA controls and are the average from at least three donors ± SEM. Results were analysed by one-way anova. Only data that are statistically significant are labelled *P < 0.05.
RV1088 is a better inhibitor of cytokine synthesis than combinations of SKIs
The NSKI RV1088 targets multiple kinases. Therefore, we wanted to determine whether combining multiple SKIs: BIRB 796, dasatinib and R406, would result in additive or synergistic effects on cytokine inhibition to recapitulate the effects of RV1088. From the data in Figure 1, the lowest effective concentration of each SKI was determined (inhibition < 50%) and double or triple combinations of each SKI were compared with each inhibitor alone. A dose response of RV1088 was included for comparison. Cell viability was confirmed using the MTT assay (data not shown).
In LPS-stimulated macrophages, a combination of BIRB 796, dasatinib and R406 (69.6 ± 5.80%, SEM) showed significantly better inhibition of TNF-α levels than each inhibitor alone and reduced TNF-α to a similar level as the highest concentrations of RV1088 (0.005 μg·mL−1, 80.4 ± 0.731%, SEM; 0.001 μg·mL−1, 66.73 ± 6.93% SEM) (Figure 3A). In contrast, this combination of SKIs did not inhibit IL-6 (18.73 ± 10.60% SEM) or IL-8 (40.93 ± 17.91% SEM) to a level comparable with RV1088 [0.005 μg·mL−1: 97.3 ± 2.7% SEM (IL-6); 87.43 ± 7.58% SEM (IL-8)] (Supporting Information Fig. S4). Similarly, combinations of BIRB 796, dasatinib and R406 were not as effective as RV1088 at inhibiting TNF-α, IL-6 or IL-8 levels in LPS-stimulated monocytes nor IL-6 and IL-8 levels in synovial fibroblasts (Figure 3B and C, Supporting Information Fig. S4). These data indicate that combining SKIs can only inhibit TNF-α levels in macrophages to a similar level as RV1088, but otherwise are not as effective.
Figure 3.
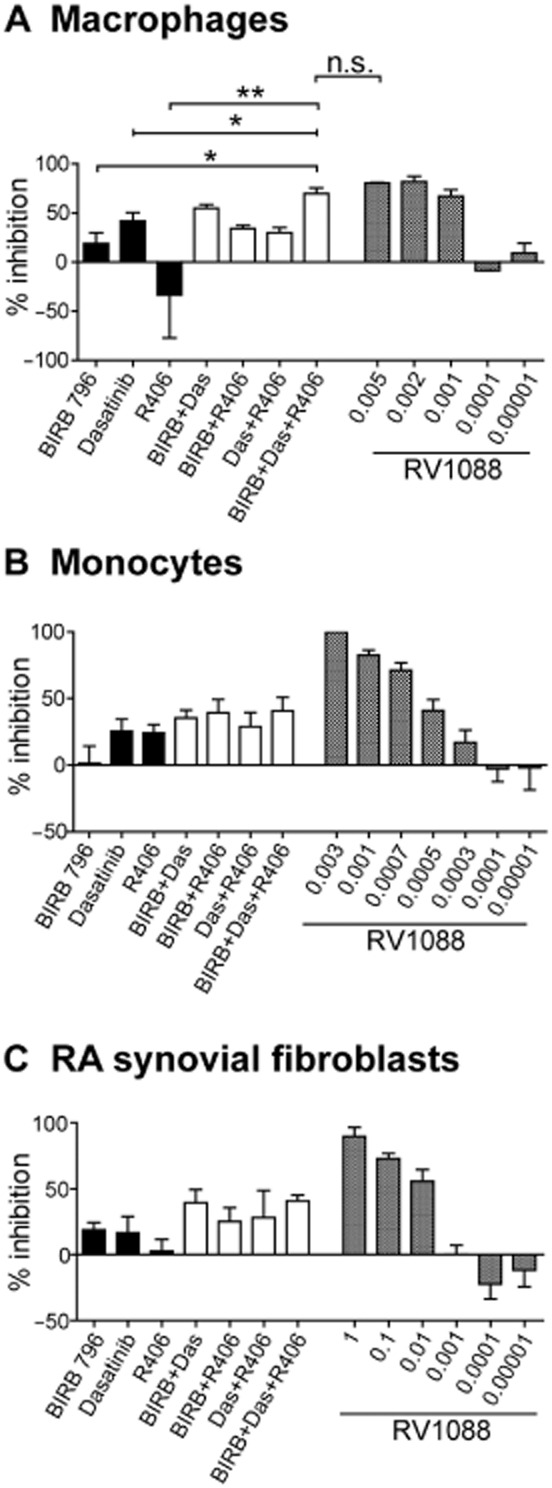
Comparison of NSKI RV1088 and combinations of SKIs. (A and B) TNF-α and (C) IL-6 were determined by elisa in conditioned media from LPS-stimulated (A) macrophages, (B) monocytes or (C) RA synovial fibroblasts after treatment with inhibitors targeting specific kinases: BIRB 796 (BIRB), dasatinib (Das) and R406, either individually or in double or multiple combinations. The weakest effective concentrations (where inhibition was observed at less than 50% of the DMSO control) were tested: (A) BIRB 796 and dasatinib at 0.001 μg·mL−1, R406 at 0.1 μg·mL−1; (B) BIRB 796 and dasatinib at 0.005 μg·mL−1, R406 at 0.0001 μg·mL−1; (C) BIRB 796 at 0.1 μg·mL−1, dasatinib and R406 at 0.01 μg·mL−1. The range of inhibition by the NSKI RV1088, which targets multiple kinases, is included for comparison. Data are shown as percentage inhibition normalized to DMSO controls and are the average from at least four donors ± SEM. Results were analysed by one-way anova comparing each inhibitor alone to combinations of inhibitors, (A) *P < 0.05; **P < 0.01; n.s., not significant (B,C): no significance was detected.
RV1088 is a better inhibitor of RA synovial membrane cytokine synthesis than SKIs
RV1088 is not currently formulated for systemic administration in vivo. However, mixed populations of synovial cells isolated from RA patients are routinely used as an ex vivo model of RA and can be a good indicator for potential in vivo drug efficacy (Brennan et al., 1989; Sacre et al., 2007). These cells spontaneously produce cytokines without the need for exogenous stimulation. Increasing concentrations of RV1088, SKIs and Humira were assessed for their effect on endogenous TNF-α, IL-6 and IL-8 production by RA synovial membrane cells. Cell viability was confirmed using the MTT assay (data not shown). Figure 4 shows the normalized inhibition dose-response curves plotted using data from at least three RA patients. Supporting Information Fig. S5 shows the effect of each kinase inhibitor on cytokine synthesis in synovial cells from one representative RA patient. The IC50 for each inhibitor was subsequently calculated (Table 2013a).
Figure 4.
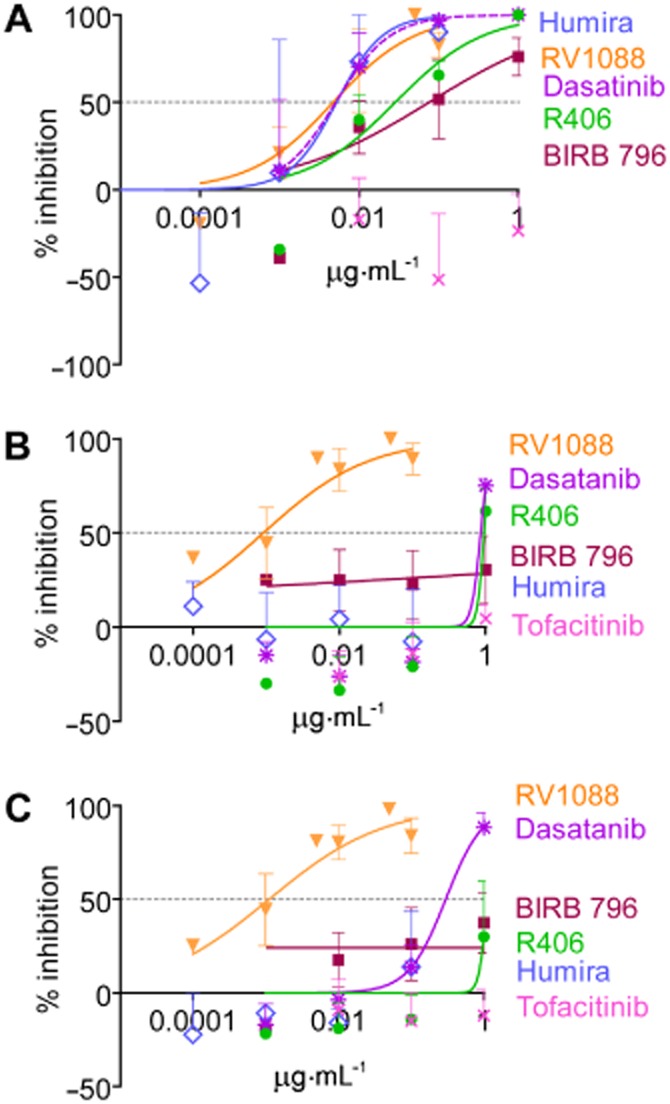
Inhibitory dose-response curves to determine the IC50 for each inhibitor on cytokine production by RA synovial membrane cells. The effects of BIRB 796, RV1088, dasatinib, R406, tofacitinib and anti-TNF-α (Humira) on (A) TNF-α, (B) IL-6 and (C) IL-8 levels in conditioned media of RA synovial membrane cells were determined by elisa. The concentration-dependent inhibitory dose-curve data were plotted as percentage inhibition normalized to DMSO controls with applied curve fits calculated using GraphPad Prism. Results are shown as the mean of at least three independent donors ± SEM. Results were analysed by two-way anova with Bonferroni’s post test comparing all inhibitors to RV1088. RV1088 was significantly more effective at inhibiting TNF-α synthesis than R406*, BIRB796* and tofacitinib** (A), and significantly more effective at inhibiting IL-6 (B) and IL-8 (C) than all inhibitors tested**. (*P < 0.05; **P < 0.01).
Table 2.
RV1088 is more effective than SKIs at inhibiting synovial membrane cell pro-inflammatory cytokine production
| TNF-α (IC50, nM) | IL-6 (IC50 nM) | IL-8 (IC50, nM) | |
|---|---|---|---|
| BIRB 796 | 144.53 | no effect | no effect |
| RV1088 | 5.72 | 3.47 | 1.94 |
| Dasatinib | 10.48 | 1753.69 | 613.93 |
| R406 | 60.91 | 2026.81 | 2363.83 |
| Tofacitinib | no effect | no effect | no effect |
| Humira | 0.04 | no effect | no effect |
IC50s were determined by plotting concentration-dependent inhibitory dose-response curves of percentage inhibition normalized to DMSO controls and subsequent analysis using GraphPad Prism software. Data are the average from at least three independent donors ± SEM.
Humira was very effective at inhibiting TNF-α synthesis (IC50 = 0.04 nM), but had no effect on IL-6 or IL-8 production by RA synovial membrane cells. Of the kinase inhibitors tested, RV1088 was the most effective, inhibiting all three cytokines with high efficacy [IC50 = 5.72 nM (TNF-α), 3.47 nM (IL-6) and 1.94 nM (IL-8)]. Dasatinib and R406 were less effective at reducing TNF-α, IL-6 and IL-8 levels compared with RV1088. BIRB 796 only showed inhibition of TNF-α levels (IC50 = 144.53 nM). Tofacitinib showed no effect (Figure 4 and Table 2013a).
These data reveal that RV1088 is the also most efficacious kinase inhibitor in RA synovial membrane cultures and further supports the theory that targeting multiple signalling kinases may be more effective than targeting p38, Src, Syk or JAK alone in RA.
Discussion
Despite significant progress in the application of anti-inflammatory drugs to treat RA, there is still an unmet need for effective treatments in large subsets of patients. The development of kinase inhibitors to target inflammation has shown much promise and offers many advantages including oral administration, high kinase isoform specificity and cost-effective production (Lindstrom and Robinson, 2010; O’Shea et al., 2013). In this study, we found that the NSKI RV1088 was more efficacious than SKIs targeting p38, c-Src, Syk or JAKs in reducing TNF-α, IL-6 and IL-8 production by LPS-stimulated primary human macrophages and monocytes and IL-6 and IL-8 by RA synovial fibroblasts. SKIs, in contrast, exhibited different cytokine- and cell type-specific effects.
These data reveal interesting insights into the signalling pathways used by LPS to stimulate cytokine production in primary human cells. TNF-α production in macrophages was effectively inhibited by individual SKIs: BIRB 796 (p38; IC50, 4.97 nM), dasatinib (c-Src; IC50, 7.81 nM) and R406 (Syk; IC50, 1.06 nM), although not as effectively as RV1088 (IC50, 0.63 nM). This suggests that all three kinases contribute to LPS-mediated TNF-α production in macrophages. Similarly, only low levels of inhibition on LPS-stimulated TNF-α production could be observed after knockdown of MAPK14, MAPK12 (encoding p38-α and p38-γ respectively) or SYK. This suggests that targeting of single kinases is insufficient to impede TNF-α production. Furthermore, the additive inhibition of TNF-α observed with LPS-stimulated macrophages cultured with suboptimal concentrations of a combination of SKIs (BIRB 796, dasatinib and R406) indicates that low levels of blockade of all three kinases, p38, c-Src and Syk, are sufficient to effectively prevent TNF-α synthesis in this cell type.
In contrast, the fact that IL-6 production by LPS-stimulated macrophages was only inhibited by dasatinib alone, but not by BIRB 796 or R406, suggests that c-Src signalling may be dominant in driving IL-6 production in this cell type. However, RV1088 was more effective than dasatinib (IC50s of 1.66 nM and 37.05 nM respectively), implying either that p38 or Syk signalling pathways may compensate in the absence of Src signalling and/or that RV1088 is a better inhibitor of c-Src than dasatinib. None of the SKIs were very effective in blocking IL-8 production at concentrations that did not impact cell viability. These data suggest signalling pathways other than p38, Src and Syk drive IL-8 expression downstream of LPS activation in this cell type.
In LPS-activated monocytes, RV1088 demonstrated effective inhibition of all cytokines with IC50s in the low nM range. Only inhibition of c-Src with dasatinib had comparably low IC50 for TNF-α (12.58 nM). Together, these data suggest that c-Src may be the major driver of LPS-induced TNF-α synthesis in monocytes and that c-Src inhibition alone may be sufficient for blockade of this cytokine. In contrast, inhibition of p38, Syk or c-Src alone appears to be insufficient to prevent IL-6 and IL-8 synthesis in this cell type. Likewise, in LPS-activated RA synovial fibroblasts, only RV1088 showed effective, dose-dependent inhibition of IL-6 and IL-8; other SKIs inhibited only at higher concentrations. These data imply that knocking out all three pathways is also required for effective inhibition of IL-6 and IL-8 in fibroblasts. The fact that combinations of p38, Syk and c-Src inhibitors at low concentrations did not improve efficacy in these cells types indicate that higher levels of inhibition of each cytokine is required.
LPS signals to cells through a Toll-like receptor 4 (TLR4)-MD-2 receptor complex (reveiwed in Piccinini and Midwood, 2010). Our data show distinct responses whereby different kinases drive cell-specific cytokine expression downstream of LPS stimulation. It will be interesting to further dissect at which point in TLR signalling these pathways diverge. However, it is still unclear which stimuli activate these cells types during RA pathogenesis (Imboden, 2009; McInnes and Schett, 2011). Chronic joint inflammation is more likely triggered or sustained by endogenous activators of inflammation rather than pathogenic stimuli. Inflammatory cytokines including TNF-α, IL-1 and IL-17 have a clearly documented role in mediating joint inflammation in RA. Moreover, evidence is emerging that highlights a role for endogenous TLR ligands in the pathogenesis of RA, potentially acting upstream of the synthesis of inflammatory mediators (Midwood et al., 2009; Piccinini and Midwood, 2010; Wahamaa et al., 2011; Ruhmann et al., 2012). It will be interesting to compare the effect of RV1088 on cytokine production induced both by cytokines, such as TNF-α and IL-1, as well as TLR stimuli including high-mobility group box protein-1, myeloid-related proteins, tenascin-C and others, either alone or in combination, to mimic the complex pro-inflammatory environment of the RA synovium.
The RA synovium comprises a mixture of cells. Consistent with the effects on the distinct RA cell types, RV1088 also effectively reduced TNF-α, IL-6 and IL-8 production by RA membrane synovial cells. In this model system, the IC50 for TNF-α inhibition by effective SKIs (dasatinib, R406 and less effectively by BIRB 796) was around the same order of magnitude to that of RV1088. This indicates signalling by c-Src, Syk and, to a lesser extent, p38 are important for TNF-α production in the RA synovium. This inhibition pattern is similar to that observed with LPS-activated macrophages, confirming reports that this cell type constitutes the major source of TNF-α in the RA joint (Cooper et al., 2012). Conversely, synovial fibroblasts are one of the major sources of IL-6 in the RA joint (Okamoto et al., 1997; Miyazawa et al., 1998; Lee et al., 2013). Accordingly, RV1088 exhibited IC50s several orders of magnitude higher than any SKI for IL-6 and IL-8 inhibition and could effectively prevent IL-6 synthesis in synovial membrane cells, as observed in LPS-activated fibroblasts.
Humira is a fully humanized recombinant monoclonal IgG1 antibody, which binds to TNF-α with high affinity to prevent its interaction with TNF-α receptors to reduce TNF-TNF receptor-dependent cell signalling and cytokine production (Mease, 2007). In our experiments, Humira showed strong inhibition of TNF-α, but not IL-6 or IL-8, in RA synovial membrane cells, whereas Humira was very effective at inhibiting all three of these cytokines in macrophages. These data may suggest that TNF-α signals to macrophages to promote IL-6 and IL-8 secretion. Humira has indeed been shown to reduce serum concentrations of IL-6 and IL-8 (Mease, 2007). In LPS-stimulated monocytes, however, only TNF-α was inhibited by Humira suggesting TNF-α-independent signalling pathways drive LPS-induced production of IL-6 and IL-8. Meusch et al. (2009) have also shown that the anti-TNF-α antibody, infliximab, also had no effect on IL-6 or IL-8 production by monocytes from either healthy or RA donors (Meusch et al., 2009). Humira had no effect on cytokine production by LPS-stimulated RA synovial fibroblasts, which is not surprising as these do not produce endogenous TNF-α. As such, targeting intracellular signalling kinases is likely to be a more powerful way to inhibit cytokine production across a range of cell types.
In our study, the JAK inhibitor tofacitinib had no effect on cytokine production by any of the cell types tested including synovial membrane cells. This might suggest that JAK signalling is not a major downstream pathway in these cells. Consistent with these data, previous studies show that even high concentrations of tofacitinib do not directly affect LPS-driven production of IL-6 or IL-8 by monocytes or RA synovial fibroblasts (Maeshima et al., 2012). Rather, tofacitinib has been shown to act on CD4+ T-cell activity with subsequent downstream inhibition of IL-8 and IL-6 by monocytes and RA synovial fibroblasts (Maeshima et al., 2012). T-cells are important players in RA pathogenesis and a prominent cell type in arthritic joints and synovial membrane cultures (Brennan et al., 1989; Imboden, 2009; McInnes and Schett, 2011). It would therefore be interesting to examine both the effect of tofacitinib and RV1088 in T-cell activity and in T-cell-related outputs in synovial membrane cultures. Alternatively, the lack of efficacy of tofacitinib inhibition may derive from simultaneous inhibition of both pro- and anti-inflammatory JAK-mediated signalling pathways. For example, in LPS-activated myeloid cells, pro-inflammatory JAK signalling is accompanied by initiation of homeostatic feedback loops including IFNβ/STAT1-mediated synthesis of IL-10. Blockade of both these arms of JAK activity by tofacitinib would therefore be predicted, at best, to result in no net change in the inflammatory status of the cell, and at worse, to enhance pro-inflammatory signalling. These effects are also likely to be cell-type specific, given the inability of RA synovial fibroblasts to produce and respond to IL-10 (reviewed in Chakravarty et al., 2013).
Similarly, Syk-mediated inflammatory signalling in macrophages is counterbalanced by the synthesis of a number of immune-suppressing molecules, including inhibitors such as ABIN3 and SOCS3, and transcriptional repressors, such as STAT3, as well as degradation of key intracellular components of the TLR signalling machinery. Likewise, p38 activation in macrophages results in parallel pro-inflammatory (MK2/3-TTP/TNF-α) and anti-inflammatory (MSK1/2-CREB-IL-10/DUSP1/IL-1ra) events, where the inflammatory axis is predominantly determined by the activity of the regulatory, homeostatic pathways (reviewed in Chakravarty et al., 2013). These data may explain why p38 inhibitors including BIRB 796 have shown poor efficacy for RA and high hepatotoxicity (Cohen et al., 2009; Hammaker and Firestein, 2010; Iwano et al., 2011). The ability of p38 to regulate the synthesis of both anti-inflammatory as well as pro-inflammatory cytokines (Suzuki et al., 2000; Kim et al., 2008; Page et al., 2010) might imply that targeting of p38-α and -γ isoforms as well as multiple pro-inflammatory signalling cascades by RV1088 may circumvent these issues and the high efficacy of RV1088 means that lower doses could be used effectively to limit toxicity. Going forward, such potential issues must be assessed in response to RV1088. These in vitro experiments also do not predict how well these effects will translate in disease in vivo. Further work is therefore required to confirm the efficacy of NSKIs in joint disease in vivo. RV1088 was initially developed to treat pulmonary inflammation, via inhalation, and has been designed to exhibit low systemic availability outside of the lung. This format precludes our testing these compounds in vivo in rodent models of arthritis. However, data presented here indicate that re-engineering of RV1088 for use in this type of experiment would be useful to determine if these drugs can be repurposed for treating RA.
Together, our data suggest that RV1088 may be a very potent inhibitor for use in suppressing cytokine production in RA. RV1088 demonstrates high efficacy at inhibiting TNF-α, IL-6 and IL-8 production in a wide range of cell types as well as in RA synovial membrane cell cultures. Here, we show that it is also more effective than SKIs and Humira at targeting the production of multiple pro-inflammatory cytokines. Thus, RV1088 warrants further development as a potential candidate for RA treatment.
Acknowledgments
This work was funded by RespiVert Ltd (to W. S. T. and K. S. M). The authors would like to thank: Dr. Tim Smallie for advice about siRNA; Dr. Lynn Williams for guidance on STAT3 siRNA, provision of RA synovial fibroblasts and RA synovial membrane cells; Khaja Syed for procurement of RA synovial membrane cells; Iwona Majkowska for provision of RA synovial fibroblasts; Patricia Green for provision of macrophages and Dr. Anna Piccinini for technical assistance.
Glossary
- M-CSF
macrophage colony stimulating factor
- NSKI
narrow spectrum kinase inhibitor
- RA
rheumatoid arthritis
- SKI
single kinase inhibitors
- Syk
spleen tyrosine kinase
- TLR4
toll-like receptor 4
Author contributions
W. S. T., A. J. C. and S. R. A. acquired the data presented in this study. K. I. and K. S. M. conceived and designed the study. All authors contributed to analysis of the data and the drafting and revising of the manuscript. All authors have approved the final version of the work and are accountable for all aspects of the work.
Conflict of interest
All kinase inhibitors used in this study were provided by RespiVert. K. I. was an employee of RespiVert up to 2013.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Figure S1 Comparative effects of RV1088, BIRB and Humira on cell viability and morphology and of RV1088 and BIRB on cytokine synthesis over time. (A) Images of RA synovial fibroblasts stimulated with LPS and treated with 1.0 μg·mL−1 RV1088, BIRB or Humira or with DMSO for 24 h. (B) MTT assay of LPS-activated RA synovial fibroblasts treated with 1.0 or 0.001 μg·mL−1 inhibitors compared with vehicle control (c) for 24 h. (C) Images of monocyte-derived macrophages stimulated with LPS and treated with 0.002 μg·mL−1 RV1088, 1.0 μg·mL−1 BIRB or 1.0 μg·mL−1 Humira or with DMSO for 24 h. (D) MTT assay of LPS-activated macrophages treated with 0.002 or 0.001 μg·mL−1 RV1088 or with 1.0 or 0.001 μg·mL−1 BIRB or Humira compared with vehicle control (c) for 24 h. (E) TNF-α, IL-6 and IL-8 levels in conditioned media, from monocyte-derived macrophages stimulated with LPS over time were determined by elisa. (F–H) TNF-α (F), IL-6 (G) and IL-8 (H) levels in conditioned media from monocyte-derived macrophages stimulated with LPS over time and treated with increasing concentrations (μg·mL−1) of RV1088 or BIRB. Data are shown as percentage inhibition normalized to vehicle controls and are the average from three donors ± SEM.
Figure S2 Effects of RV1088, SKIs and Humira on pro-inflammatory cytokine production by monocytes. TNF-α, IL-6 and IL-8 levels in conditioned media from LPS-stimulated monocytes treated with increasing concentrations of all inhibitors (BIRB, BIRB 796; Das, Dasatinib; Tofac, Tofacitinib) were determined by elisa. Data are shown as percentage inhibition normalized to DMSO controls and are the average from at least four donors ± SEM. Results were analysed by two-way anova with Bonferroni’s post test comparing all inhibitors to BIRB 796. Only data that are statistically significant are labelled: *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Figure S3 Effects of RV1088, SKIs and Humira on pro-inflammatory cytokine production by RA synovial fibroblasts. TNF-α, IL-6 and IL-8 levels in conditioned media from LPS-stimulated RA synovial fibroblasts treated with increasing concentrations of all inhibitors (BIRB, BIRB 796; Das, Dasatinib; Tofac, Tofacitinib) were determined by elisa. Data are shown as percentage inhibition normalized to DMSO controls and are the average from at least four donors ± SEM. Results were analysed by two-way anova with Bonferroni’s post test comparing all inhibitors to BIRB 796. Only data that are statistically significant are labelled: *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Figure S4 Comparison of NSKIs and combinations of SKIs. IL-6 and IL-8 in conditioned media from LPS-stimulated (A) macrophages, (B) monocytes or IL-8 levels from LPS-stimulated (C) RA synovial fibroblasts after treatment with inhibitors targeting specific kinases: BIRB 796 (BIRB), dasatinib (Das) and R406, either individually or in double or multiple combinations were determined by elisa. The weakest effective concentrations (where inhibition is observed at less than 50% of the DMSO control) were tested: (A) BIRB 796 and dasatinib at 0.001 μg·mL−1, R406 at 0.1 μg·mL−1; (B) BIRB 796 and dasatinib at 0.005 μg·mL−1, R406 at 0.0001 μg·mL−1; (C) BIRB 796 at 0.1 μg·mL−1, dasatinib and R406 at 0.01 μg·mL−1. The range of inhibition by the NSKI RV1088, which targets multiple kinases, is included for comparison. Data are shown as percentage inhibition normalized to DMSO controls and are the average from at least four donors ± SEM. Results were analysed by one-way anova, comparing each inhibitor alone to combinations of inhibitors. Only data that are statistically significant are labelled, *P < 0.05.
Figure S5 RV1088 is the most effective inhibitor of pro-inflammatory cytokine production by RA synovial membrane cells. TNF-α, IL-6 and IL-8 levels in conditioned media from synovial membrane cell cultures treated with increasing concentrations of NSKI RV1088 or SKIs were determined by elisa. Results from a single representative donor are shown as the mean of triplicate values ± SD.
Table S1 List of inhibitors. Includes brand name synonyms and the major kinase or cytokine targets (in bold) and off-target or alternative kinase targets (not bold) for each inhibitor are denoted.
References
- Agbanoma G, Li C, Ennis D, Palfreeman AC, Williams LM, Brennan FM. Production of TNF-α in macrophages activated by T cells, compared with lipopolysaccharide, uses distinct IL-10-dependent regulatory mechanism. J Immunol. 2012;188:1307–1317. doi: 10.4049/jimmunol.1100625. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: catalytic receptors. Br J Pharmacol. 2013a;170:1676–1705. doi: 10.1111/bph.12449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: enzymes. Br J Pharmacol. 2013b;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan FM, McInnes IB. Evidence that cytokines play a role in rheumatoid arthritis. J Clin Invest. 2008;118:3537–3545. doi: 10.1172/JCI36389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan FM, Chantry D, Jackson AM, Maini RN, Feldmann M. Cytokine production in culture by cells isolated from the synovial membrane. J Autoimmun. 1989;2:177–186. doi: 10.1016/0896-8411(89)90129-7. [DOI] [PubMed] [Google Scholar]
- Burmester GR, Blanco R, Charles-Schoeman C, Wollenhaupt J, Zerbini C, Benda B, et al. Tofacitinib (CP-690,550) in combination with methotrexate in patients with active rheumatoid arthritis with an inadequate response to tumour necrosis factor inhibitors: a randomised phase 3 trial. Lancet. 2013;381:451–460. doi: 10.1016/S0140-6736(12)61424-X. [DOI] [PubMed] [Google Scholar]
- Chakravarty SD, Poulikos PI, Ivashkiv LB, Salmon J, Kalliolias G. Kinase inhibitors: a new tool for the treatment of rheumatoid arthritis. Clin Immunol. 2013;148:66–78. doi: 10.1016/j.clim.2013.04.007. [DOI] [PubMed] [Google Scholar]
- Charron C, Coates M, Ito K. 2011. Effects of Rv1088, a Novel Narrow Spectrum Kinase Inhibitor, on Pro-Inflammatory Cytokine Production In Epithelial Cells Isolated From Asthma and COPD. American Thoracic Society International Conference 2011; Denver, Colorado. American Journal of Respiratory and Critical Care Medicine, A2817.
- Cohen SB, Cheng TT, Chindalore V, Damjanov N, Burgos-Vargas R, Delora P, et al. Evaluation of the efficacy and safety of pamapimod, a p38 MAP kinase inhibitor, in a double-blind, methotrexate-controlled study of patients with active rheumatoid arthritis. Arthritis Rheum. 2009;60:335–344. doi: 10.1002/art.24266. [DOI] [PubMed] [Google Scholar]
- Cooper DL, Martin SG, Robinson JI, Mackie SL, Charles CJ, Nam J, et al. FcγRIIIa expression on monocytes in rheumatoid arthritis: role in immune-complex stimulated TNF production and non-response to methotrexate therapy. PLoS ONE. 2012;7:e28918. doi: 10.1371/journal.pone.0028918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foxwell B, Browne K, Bondeson J, Clarke C, de Martin R, Brennan F, et al. Efficient adenoviral infection with IkappaB alpha reveals that macrophage tumor necrosis factor alpha production in rheumatoid arthritis is NF-kappaB dependent. Proc Natl Acad Sci U S A. 1998;95:8211–8215. doi: 10.1073/pnas.95.14.8211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genovese MC, Becker JC, Schiff M, Luggen M, Sherrer Y, Kremer J, et al. Abatacept for rheumatoid arthritis refractory to tumor necrosis factor alpha inhibition. N Engl J Med. 2005;353:1114–1123. doi: 10.1056/NEJMoa050524. [DOI] [PubMed] [Google Scholar]
- Hammaker D, Firestein GS. ‘Go upstream, young man’: lessons learned from the p38 saga. Ann Rheum Dis. 2010;69:i77–i82. doi: 10.1136/ard.2009.119479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber LC, Distler O, Tarner I, Gay RE, Gay S, Pap T. Synovial fibroblasts: key players in rheumatoid arthritis. Rheumatology. 2006;45:669–675. doi: 10.1093/rheumatology/kel065. [DOI] [PubMed] [Google Scholar]
- Imboden JB. The immunopathogenesis of rheumatoid arthritis. Annu Rev Pathol. 2009;4:417–434. doi: 10.1146/annurev.pathol.4.110807.092254. [DOI] [PubMed] [Google Scholar]
- Iwano S, Asaoka Y, Akiyama H, Takizawa S, Nobumasa H, Hashimoto H, et al. A possible mechanism for hepatotoxicity induced by BIRB-796, an orally active p38 mitogen-activated protein kinase inhibitor. J Appl Toxicol. 2011;31:671–677. doi: 10.1002/jat.1622. [DOI] [PubMed] [Google Scholar]
- Kim C, Sano Y, Todorova K, Carlson BA, Arpa L, Celada A, et al. The kinase p38 alpha serves cell type-specific inflammatory functions in skin injury and coordinates pro- and anti-inflammatory gene expression. Nat Immunol. 2008;9:1019–1027. doi: 10.1038/ni.1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinne RW, Stuhlmuller B, Burmester GR. Cells of the synovium in rheumatoid arthritis. Macrophages. Arthritis Res Ther. 2007;9:224. doi: 10.1186/ar2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knobloch J, Urban K, Jungck D, Stoelben E, Ito K, Koch A. 2011. Effects of Rv1088, a Narrow Spectrum Kinase Inhibitor, on Pro-Inflammatory Cytokine Production in Smooth Muscle Cells Obtained from COPD Patients. American Thoracic Society International Conference. American Journal of Respiratory and Critical Care Medicine, A3616.
- Lee A, Qiao Y, Grigoriev G, Chen J, Park-Min KH, Park SH, et al. Tumor necrosis factor alpha induces sustained signaling and a prolonged and unremitting inflammatory response in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 2013;65:928–938. doi: 10.1002/art.37853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindstrom TM, Robinson WH. A multitude of kinases – which are the best targets in treating rheumatoid arthritis? Rheum Dis Clin North Am. 2010;36:367–383. doi: 10.1016/j.rdc.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeshima K, Yamaoka K, Kubo S, Nakano K, Iwata S, Saito K, et al. The JAK inhibitor tofacitinib regulates synovitis through inhibition of interferon-gamma and interleukin-17 production by human CD4+ T cells. Arthritis Rheum. 2012;64:1790–1798. doi: 10.1002/art.34329. [DOI] [PubMed] [Google Scholar]
- McInnes IB, Schett G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat Rev Immunol. 2007;7:429–442. doi: 10.1038/nri2094. [DOI] [PubMed] [Google Scholar]
- McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. 2011;365:2205–2219. doi: 10.1056/NEJMra1004965. [DOI] [PubMed] [Google Scholar]
- Mease PJ. Adalimumab in the treatment of arthritis. Ther Clin Risk Manag. 2007;3:133–148. doi: 10.2147/tcrm.2007.3.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meusch U, Rossol M, Baerwald C, Hauschildt S, Wagner U. Outside-to-inside signaling through transmembrane tumor necrosis factor reverses pathologic interleukin-1beta production and deficient apoptosis of rheumatoid arthritis monocytes. Arthritis Rheum. 2009;60:2612–2621. doi: 10.1002/art.24778. [DOI] [PubMed] [Google Scholar]
- Midwood K, Sacre S, Piccinini AM, Inglis J, Trebaul A, Chan E, et al. Tenascin-C is an endogenous activator of Toll-like receptor 4 that is essential for maintaining inflammation in arthritic joint disease. Nat Med. 2009;15:774–780. doi: 10.1038/nm.1987. [DOI] [PubMed] [Google Scholar]
- Miller MC, Manning HB, Jain A, Troeberg L, Dudhia J, Essex D, et al. Membrane type 1 matrix metalloproteinase is a crucial promoter of synovial invasion in human rheumatoid arthritis. Arthritis Rheum. 2009;60:686–697. doi: 10.1002/art.24331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazawa K, Mori A, Yamamoto K, Okudaira H. Constitutive transcription of the human interleukin-6 gene by rheumatoid synoviocytes: spontaneous activation of NF-kappaB and CBF1. Am J Pathol. 1998;152:793–803. [PMC free article] [PubMed] [Google Scholar]
- Muller-Ladner U, Ospelt C, Gay S, Distler O, Pap T. Cells of the synovium in rheumatoid arthritis. Synovial fibroblasts. Arthritis Res Ther. 2007;9:223. doi: 10.1186/ar2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto H, Yamamura M, Morita Y, Harada S, Makino H, Ota Z. The synovial expression and serum levels of interleukin-6, interleukin-11, leukemia inhibitory factor, and oncostatin M in rheumatoid arthritis. Arthritis Rheum. 1997;40:1096–1105. doi: 10.1002/art.1780400614. [DOI] [PubMed] [Google Scholar]
- O’Shea JJ, Laurence A, McInnes IB. Back to the future: oral targeted therapy for RA and other autoimmune diseases. Nat Rev Rheumatol. 2013;9:173–182. doi: 10.1038/nrrheum.2013.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page TH, Brown A, Timms EM, Foxwell BM, Ray KP. Inhibitors of p38 suppress cytokine production in rheumatoid arthritis synovial membranes: does variable inhibition of interleukin-6 production limit effectiveness in vivo. Arthritis Rheum. 2010;62:3221–3231. doi: 10.1002/art.27631. [DOI] [PubMed] [Google Scholar]
- Pattison MJ, Mackenzie KF, Arthur JS. Inhibition of JAKs in macrophages increases lipopolysaccharide-induced cytokine production by blocking IL-10-mediated feedback. J Immunol. 2012;189:2784–2792. doi: 10.4049/jimmunol.1200310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucl Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peirce MJ, Brook M, Morrice N, Snelgrove R, Begum S, Lanfrancotti A, et al. Themis2/ICB1 is a signaling scaffold that selectively regulates macrophage Toll-like receptor signaling and cytokine production. PLoS ONE. 2010;5:e11465. doi: 10.1371/journal.pone.0011465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccinini AM, Midwood KS. DAMPening inflammation by modulating TLR signalling. Mediators Inflamm. 2010;2010:672395. doi: 10.1155/2010/672395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccinini AM, Midwood KS. Endogenous control of immunity against infection: tenascin-C regulates TLR4-mediated inflammation via microRNA-155. Cell Rep. 2012;2:914–926. doi: 10.1016/j.celrep.2012.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- release Ap. 2013. AstraZeneca announces top-line results of OSKIRA-4 Phase IIb study of fostamatinib as a monotherapy for rheumatoid arthritis.
- release Pp. 2013. Pfizer receives CHMP negative opinion regarding marketing authorization in Europe for rheumatoid arthritis treatment XELJANZ® (tofacitinib citrate)
- Ruhmann M, Piccinini AM, Kong PL, Midwood KS. Endogenous activation of adaptive immunity: tenascin-C drives interleukin-17 synthesis in murine arthritic joint disease. Arthritis Rheum. 2012;64:2179–2190. doi: 10.1002/art.34401. [DOI] [PubMed] [Google Scholar]
- Ryzhakov G, Lai CC, Blazek K, To KW, Hussell T, Udalova I. IL-17 boosts proinflammatory outcome of antiviral response in human cells. J Immunol. 2011;187:5357–5362. doi: 10.4049/jimmunol.1100917. [DOI] [PubMed] [Google Scholar]
- Sacre SM, Andreakos E, Kiriakidis S, Amjadi P, Lundberg A, Giddins G, et al. The Toll-like receptor adaptor proteins MyD88 and Mal/TIRAP contribute to the inflammatory and destructive processes in a human model of rheumatoid arthritis. Am J Pathol. 2007;170:518–525. doi: 10.2353/ajpath.2007.060657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Schouwenburg PA, Rispens T, Wolbink GJ. Immunogenicity of anti-TNF biologic therapies for rheumatoid arthritis. Nat Rev Rheumatol. 2013;9:164–172. doi: 10.1038/nrrheum.2013.4. [DOI] [PubMed] [Google Scholar]
- Shahrara S, Castro-Rueda HP, Haines GK, Koch AE. Differential expression of the FAK family kinases in rheumatoid arthritis and osteoarthritis synovial tissues. Arthritis Res Ther. 2007;9:R112. doi: 10.1186/ar2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smallie T, Ricchetti G, Horwood NJ, Feldmann M, Clark AR, Williams LM. IL-10 inhibits transcription elongation of the human TNF gene in primary macrophages. J Exp Med. 2010;207:2081–2088. doi: 10.1084/jem.20100414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki M, Tetsuka T, Yoshida S, Watanabe N, Kobayashi M, Matsui N, et al. The role of p38 mitogen-activated protein kinase in IL-6 and IL-8 production from the TNF-alpha- or IL-1beta-stimulated rheumatoid synovial fibroblasts. FEBS Lett. 2000;465:23–27. doi: 10.1016/s0014-5793(99)01717-2. [DOI] [PubMed] [Google Scholar]
- Takeda K, Clausen BE, Kaisho T, Tsujimura T, Terada N, Forster I, et al. Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of Stat3 in macrophages and neutrophils. Immunity. 1999;10:39–49. doi: 10.1016/s1074-7613(00)80005-9. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Suzuki M, Nakamura H, Toyoizumi S, Zwillich SH. Phase II study of tofacitinib (CP-690,550) combined with methotrexate in patients with rheumatoid arthritis and an inadequate response to methotrexate. Arthritis Care Res (Hoboken) 2011;63:1150–1158. doi: 10.1002/acr.20494. [DOI] [PubMed] [Google Scholar]
- Wahamaa H, Schierbeck H, Hreggvidsdottir HS, Palmblad K, Aveberger AC, Andersson U, et al. High mobility group box protein 1 in complex with lipopolysaccharide or IL-1 promotes an increased inflammatory phenotype in synovial fibroblasts. Arthritis Res Ther. 2011;13:R136. doi: 10.1186/ar3450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinblatt ME, Kavanaugh A, Genovese MC, Musser TK, Grossbard EB, Magilavy DB. An oral spleen tyrosine kinase (Syk) inhibitor for rheumatoid arthritis. N Engl J Med. 2010;363:1303–1312. doi: 10.1056/NEJMoa1000500. [DOI] [PubMed] [Google Scholar]
- Weinblatt ME, Kavanaugh A, Genovese MC, Jones DA, Musser TK, Grossbard EB, et al. Effects of fostamatinib (R788), an oral spleen tyrosine kinase inhibitor, on health-related quality of life in patients with active rheumatoid arthritis: analyses of patient-reported outcomes from a randomized, double-blind, placebo-controlled trial. J Rheumatol. 2013;40:369–378. doi: 10.3899/jrheum.120923. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Comparative effects of RV1088, BIRB and Humira on cell viability and morphology and of RV1088 and BIRB on cytokine synthesis over time. (A) Images of RA synovial fibroblasts stimulated with LPS and treated with 1.0 μg·mL−1 RV1088, BIRB or Humira or with DMSO for 24 h. (B) MTT assay of LPS-activated RA synovial fibroblasts treated with 1.0 or 0.001 μg·mL−1 inhibitors compared with vehicle control (c) for 24 h. (C) Images of monocyte-derived macrophages stimulated with LPS and treated with 0.002 μg·mL−1 RV1088, 1.0 μg·mL−1 BIRB or 1.0 μg·mL−1 Humira or with DMSO for 24 h. (D) MTT assay of LPS-activated macrophages treated with 0.002 or 0.001 μg·mL−1 RV1088 or with 1.0 or 0.001 μg·mL−1 BIRB or Humira compared with vehicle control (c) for 24 h. (E) TNF-α, IL-6 and IL-8 levels in conditioned media, from monocyte-derived macrophages stimulated with LPS over time were determined by elisa. (F–H) TNF-α (F), IL-6 (G) and IL-8 (H) levels in conditioned media from monocyte-derived macrophages stimulated with LPS over time and treated with increasing concentrations (μg·mL−1) of RV1088 or BIRB. Data are shown as percentage inhibition normalized to vehicle controls and are the average from three donors ± SEM.
Figure S2 Effects of RV1088, SKIs and Humira on pro-inflammatory cytokine production by monocytes. TNF-α, IL-6 and IL-8 levels in conditioned media from LPS-stimulated monocytes treated with increasing concentrations of all inhibitors (BIRB, BIRB 796; Das, Dasatinib; Tofac, Tofacitinib) were determined by elisa. Data are shown as percentage inhibition normalized to DMSO controls and are the average from at least four donors ± SEM. Results were analysed by two-way anova with Bonferroni’s post test comparing all inhibitors to BIRB 796. Only data that are statistically significant are labelled: *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Figure S3 Effects of RV1088, SKIs and Humira on pro-inflammatory cytokine production by RA synovial fibroblasts. TNF-α, IL-6 and IL-8 levels in conditioned media from LPS-stimulated RA synovial fibroblasts treated with increasing concentrations of all inhibitors (BIRB, BIRB 796; Das, Dasatinib; Tofac, Tofacitinib) were determined by elisa. Data are shown as percentage inhibition normalized to DMSO controls and are the average from at least four donors ± SEM. Results were analysed by two-way anova with Bonferroni’s post test comparing all inhibitors to BIRB 796. Only data that are statistically significant are labelled: *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Figure S4 Comparison of NSKIs and combinations of SKIs. IL-6 and IL-8 in conditioned media from LPS-stimulated (A) macrophages, (B) monocytes or IL-8 levels from LPS-stimulated (C) RA synovial fibroblasts after treatment with inhibitors targeting specific kinases: BIRB 796 (BIRB), dasatinib (Das) and R406, either individually or in double or multiple combinations were determined by elisa. The weakest effective concentrations (where inhibition is observed at less than 50% of the DMSO control) were tested: (A) BIRB 796 and dasatinib at 0.001 μg·mL−1, R406 at 0.1 μg·mL−1; (B) BIRB 796 and dasatinib at 0.005 μg·mL−1, R406 at 0.0001 μg·mL−1; (C) BIRB 796 at 0.1 μg·mL−1, dasatinib and R406 at 0.01 μg·mL−1. The range of inhibition by the NSKI RV1088, which targets multiple kinases, is included for comparison. Data are shown as percentage inhibition normalized to DMSO controls and are the average from at least four donors ± SEM. Results were analysed by one-way anova, comparing each inhibitor alone to combinations of inhibitors. Only data that are statistically significant are labelled, *P < 0.05.
Figure S5 RV1088 is the most effective inhibitor of pro-inflammatory cytokine production by RA synovial membrane cells. TNF-α, IL-6 and IL-8 levels in conditioned media from synovial membrane cell cultures treated with increasing concentrations of NSKI RV1088 or SKIs were determined by elisa. Results from a single representative donor are shown as the mean of triplicate values ± SD.
Table S1 List of inhibitors. Includes brand name synonyms and the major kinase or cytokine targets (in bold) and off-target or alternative kinase targets (not bold) for each inhibitor are denoted.