

Abstract
Phosphatidylethanolamine (PE) is the most abundant inner membrane phospholipid. PE synthesis from ethanolamine and diacylglycerol is regulated primarily by CTP:phosphoethanolamine cytidylyltransferase (Pcyt2). Pcyt2+/− mice have reduced PE synthesis and, as a consequence, perturbed glucose and fatty acid metabolism, which gradually leads to the development of hyperlipidemia, obesity, and insulin resistance. Glucose and fatty acid uptake and the corresponding transporters Glut4 and Cd36 are similarly impaired in male and female Pcyt2+/− hearts. These mice also have similarly reduced phosphatidylinositol 3-kinase (PI3K)/Akt1 signaling and increased reactive oxygen species (ROS) production in the heart. However, only Pcyt2+/− males develop hypertension and cardiac hypertrophy. Pcyt2+/− males have upregulated heart AceI expression, heart phospholipids enriched in arachidonic acid and other n-6 polyunsaturated fatty acids, and dramatically increased ROS production in the aorta. In contrast, Pcyt2+/− females have unmodified heart phospholipids but have reduced heart triglyceride levels and altered expression of the structural genes Acta (low) and Myh7 (high). These changes together protect Pcyt2+/− females from cardiac dysfunction under conditions of reduced glucose and fatty acid uptake and heart insulin resistance. Our data identify Pcyt2 and membrane PE biogenesis as important determinants of gender-specific differences in cardiac lipids and heart function.
INTRODUCTION
The mammalian heart requires an enormous amount of energy to perform (1). The heart can use various sources of energy (2), but in a normal adult heart, lipids are the main source, and they account for ∼70 to 80% of the total energy requirements (3). Phosphatidylethanolamine (PE) is the most important phospholipid and is located in the inner leaflet of cellular membranes. PE has many biological functions, including in membrane integrity, cell division, cytokinesis, autophagy, apoptosis, and blood coagulation, as reviewed by Bakovic et al. (4). The de novo synthesis of PE occurs through the ethanolamine Kennedy pathway. In this pathway, ethanolamine is phosphorylated by ethanolamine kinase to produce phosphoethanolamine. The CTP:phosphoethanolamine cytidylyltransferase (Pcyt2) then converts phosphoethanolamine to CDP-ethanolamine. Finally, CDP-ethanolamine:1,2-diacylglycerol phosphotransferase catalyzes the production of PE from CDP-ethanolamine and diacylglycerol (DAG). CDP-ethanolamine is also alternatively coupled with alkylacylglycerols derived in peroxisomes, to produce plasmanyl-PE, which is further modified in mitochondria to the final vinyl-ether (plasmalogen) PE. Various isoforms of kinases and phosphotransferases share substrates for the choline and ethanolamine branches of the Kennedy pathway; however, Pcyt2 is a product of a single gene (5) and therefore is highly specific for the ethanolamine branch of the pathway. The Pcyt2 gene has been cloned and characterized in human (5), Saccharomyces cerevisiae (6), rat (7), and mouse (5, 8).
The total deletion of Pcyt2 in mouse (9) or Arabidopsis (10) causes embryonic lethality. Heterozygous (Pcyt2+/−) mice, despite the reduced flux through the Kennedy pathway, are able to maintain normal PE levels by reducing PE turnover (11). Decreased PE synthesis in Pcyt2+/− mice increases the availability of unused DAG, which in turn esterifies with free fatty acids (FAs) into triacylglyceride (TAG), causing the development of obesity, hypertriglyceridemia, and insulin resistance. A liver-specific Pcyt2 knockout (12) further affirms the DAG hypothesis for TAG accumulation and disease development, when PE synthesis by the Kennedy pathway becomes reduced. Important to this study, there is a strong relationship between increased TAG levels and cardiomyopathy (13); however, there are no in vivo mammalian models available that relate membrane phospholipid deregulation with cardiac dysfunction. Several studies indicate that this is crucial, as reduced PE synthesis can impair cardiac function in mutant fruit flies (Drosophila melanogaster) when three separate genes in the PE Kennedy pathway are affected (14, 15). A Drosophila ethanolamine kinase (eas) mutant had reduced PE synthesis, accumulated heart lipids, and developed multiple heart pathologies, including tachycardia, diastolic dysfunction, atrial fibrillation, and cardiac arrest under stress (14). Similarly to Pcyt2-deficient mice (11, 12), the eas mutants made TAG from excess DAG not used in PE synthesis and FAs produced by increased lipogenesis. The silencing of two other Drosophila genes, the Pcyt2 (pect) and choline/ethanolamine phosphotransferase (cept) genes, produces similar heart pathologies, strongly affirming the importance of PE and the Kennedy pathway for heart function (15).
In this study, we investigate cardiac function and pathophysiology in Pcyt2-deficient (Pcyt2+/−) mice. These animals develop a metabolic syndrome phenotype with abdominal obesity, hypertriglyceridemia, and insulin resistance (11). Notably, although both male and female Pcyt2+/− mice had impaired cardiac insulin signaling, only Pcyt2+/− males had elevated levels of membrane arachidonic acid and n-6 long-chain polyunsaturated fatty acid (LCPUFA) and, as a consequence, developed systemic hypertension and cardiac hypertrophy. Our data demonstrate for the first time that impaired PE synthesis in Pcyt2+/− mice leads to gender-related differences in cardiac membrane phospholipid metabolism and gene expression that appear particularly detrimental to the heart function of males.
MATERIALS AND METHODS
Animals.
Pcyt2+/− mice were generated as described previously (9). Pcyt2+/− mice were cross-bred, and the heterozygous colony was maintained at the University of Guelph, according to procedures approved by the University Animal Care Committee and in agreement with guidelines of the Canadian Council on Animal Care. Pcyt2+/− and Pcyt2+/+ genotypes were identified from genomic DNA as described previously (9, 11). Mice were housed with a 12-h light/12-h dark cycle, had free access to water, and were fed a standardized chow diet (catalog number S-2335; Harlan Teklad). Four groups were used for these studies: (i) 3-month-old Pcyt2+/− and Pcyt2+/+ males, (ii) 8-month-old Pcyt2+/+ and Pcyt2+/− males, (iii) 3-month-old Pcyt2+/− and Pcyt2+/+ females, and (iv) 8-month-old Pcyt2+/− and Pcyt2+/+ females. All animals were sacrificed 3 to 4 h after lights were turned on to control for circadian time-of-day effects. Samples taken for biochemical assays (heart, liver, and pancreas) were snap-frozen in liquid nitrogen and stored at −80°C for later use. For histopathology, hearts were washed in phosphate-buffered saline (PBS), weighed, and fixed in 10% neutral buffered formalin.
Transmission electron microscopy.
Liver tissues were prepared as recommended by the Electron Microscopy Facility of the University of Guelph (16). Briefly, liver samples were incubated at 4°C overnight in fixing buffer (2.5% glutaraldehyde and 1.0% paraformaldehyde in PBS), washed in 0.1 M HEPES, and suspended in 1.0% osmium tetroxide for 4 h. Tissues were washed 3 times in 100 mM HEPES, suspended in 2% uranyl acetate for 3 h, washed 3 times in 0.1% HEPES, and dehydrated by incubation in a graded ethanol series (i.e., 25% to 100% ethanol). Tissue was infiltrated with resin by suspending the tissue in 50% ethanol–50% resin (LR White; London Resin Company) for 4 h and then in pure resin for 4 h, using a rotating mixer. Tissue was embedded in pure resin overnight at 60°C to polymerize the resin. Sections (100 nm) were laid onto 200-mesh Formvar-carbon copper grids and stained with 2% uranyl acetate and Reynolds lead citrate. A minimum of three sections were placed onto each grid. Images were obtained from each sample in a randomized systematic order. Samples were viewed on a Philips CM 10 transmission electron microscope at 80 kV, and images were obtained with an Olympus/SIS Morada charge-coupled-device (CCD) camera using Olympus/SIS iTEM software.
Tissue sectioning and staining.
For liver histopathology, tissues were fixed in 10% neutral buffered formalin, processed, and stained with either hematoxylin and eosin (H&E) for steatosis or Picrosirius red for fibrosis. Pancreatic islet cells reactive to anti-somatostatin 14 and anti-pancreatic polypeptide were characterized by using antiperoxidase immunohistochemistry (17). A minimum of 3 to 5 different images were analyzed for each mouse group. Images were taken, using Q-Capture software, at a magnification of ×100 to ×200.
Echocardiography and hemodynamics.
Cardiovascular pathophysiology in mice at 3 and 8 months of age was investigated by using our standard approach (18). First, transthoracic echocardiography was performed in a blind manner, using a GE Vivid 7 ultrasound machine equipped with an i13L 14-MHz linear array transducer, on mice anesthetized with 1% isoflurane (maintenance dose). Standard B-mode and M-mode short-axis-view images were taken at the level of the papillary muscles to determine the left ventricle (LV) internal diameter at diastole (LVIDd), LV external diameter at systole (LVEDs), LV anterior wall thickness (AWT), LV posterior wall thickness (PWT), percent fractional shortening (FS) [calculated as (LVIDd − LVIDs)/LVIDd × 100], percent ejection fraction (EF), and heart rate (HR). At least 3 different images were analyzed for each mouse heart, with 6 mice per group. Body temperature was monitored and maintained at 37°C throughout the experiment.
Values for in vivo hemodynamic parameters were also obtained in a blind manner. Animals (n = 6 mice/group) were anesthetized with and maintained at 2% isoflurane (mixed with 99% oxygen), intubated (22-gauge 1.00-in. catheter), and ventilated (model 687; Harvard Apparatus, St. Laurent, QC, Canada), and body temperature was maintained at 37°C throughout the experiment. The right common carotid artery was exposed and cannulated with a 1F (0.33-mm) microtip catheter (Millar, Houston, TX), which was advanced through the ascending aorta into the LV for measurements. Data were acquired by using Powerlab and Lab Chart 7 (AD Instruments, Colorado Springs, CO) and analyzed for systolic blood pressure (SBP), diastolic blood pressure (DBP), mean arterial blood pressure (MAP), LV end systolic pressure (LVESP), LV end diastolic pressure (LVEDP), LV developed pressure (LVDP), and the maximum and minimum first derivatives of LV pressure (dP/dt max and dP/dt min, respectively).
Cardiomyocyte cross-sectional area and fibrosis.
Hearts were arrested in diastole with 1 M KCl, fixed in 10% neutral buffered formalin for 24 h, processed, and paraffin embedded. Next, 5-μm sections were stained with H&E. Images were taken by using a Q-Capture instrument (QImaging, Surrey, BC) at a ×200 magnification and analyzed by using ImageJ software. Total field size was measured, and the cardiomyocyte cross-sectional area was determined for at least 50 myocytes per heart.
Gene expression analyses.
RNA extraction and first-strand cDNA synthesis were performed as described previously (11). Genes of interest were analyzed in the exponential phase of PCR amplification, using the optimal amount of cDNA and the reaction cycle number. Each gene level is expressed relative to the internal Gapdh control. The genes tested include cardiac tissue-specific genes (Myh6, Myh7, Acta, and Nppa), lipolysis genes (Atgl, Hsl, and Lpl), genes for fatty acid transport and oxidation (Cd36 and Pparα), the mitochondrial biogenesis gene Pgc1α, and the lipogenic stearoyl coenzyme A (stearoyl-CoA) desaturase gene Scd1. Also included were the gene for the membrane G protein (estrogen) receptor (Gpr30), the mitochondrial lactate/glutamate transporter gene (Mct1), and the angiotensin-converting enzyme gene (Ace). The experiments were performed by using heart samples collected from young (3-month-old) and adult (8-month-old) Pcyt2+/− and Pcyt2+/+ male and female mice (n = 6 mice/group). ImageJ 1.46 software was used to quantify band density, and gene levels are expressed as fold changes relative to the controls. The primers used are listed in Table S4 in the supplemental material.
Quantitative real-time PCR.
RNA was isolated from the heart, kidney, and liver tissues of male and female adult (8-month-old) Pcyt2+/− and wild-type mice (n = 3/group) with TRIzol (Invitrogen) and assessed for high quality (RNA/protein absorbance ratio [260 nm/280 nm] > 2.0) by using a NanoDrop ND1000 instrument (Thermo-Scientific). The angiotensin-converting enzyme 1, angiotensin-converting enzyme 2, renin, and angiotensinogen genes were amplified from 200 ng of RNA, using primers (30 μM), the Power SYBR green RNA-to-CT 1-Step kit (Applied Biosystems), and RNA/DNA-free H2O (Invitrogen) on a Viia7 real-time PCR (RT-PCR) machine (Applied Biosystems). The temperature cycles were as follows: reverse transcription for 30 min at 48°C followed by 10 min at 95°C and then an amplification step (15 s at 95°C and 1 min at 60°C) for 40 cycles. Data were normalized to histone gene values and analyzed by using the ΔΔCT method (19). The primers used are listed in Table S4 in the supplemental material.
Immunoblotting.
Frozen heart samples (n = 6/group) were homogenized in cold lysis buffer (10 mM HEPES, 10 mM KCl, 1.5 mM MgCl2 containing protease [1/10] and phosphatase [1/100] inhibitor cocktails [Sigma], and NP-40). The lysate was centrifuged at 2,000 × g for 20 min at 4°C to remove cell debris. Proteins (25 μg) were separated by 10% SDS-PAGE and transferred onto polyvinylidene difluoride membranes. Ponceau S stain was used to ensure proper protein transfer and loading. Membranes were blocked with 5% milk in a solution containing 20 mM Tris-HCl (pH 7.5), 500 mM NaCl, and 0.05% Tween 20 (TBS-T) for 1 h at room temperature, followed by a brief wash in TBS-T. Membranes were incubated with primary antibody to AMPKα (Cell Signaling), phosphorylated AMPK (pAMPKα) (Cell Signaling), Akt1/2 (Upstate), pS473-Akt, pT308-Akt (Upstate), or phosphatidylinositol 3-kinase (PI3K) (Cell Signaling) at a 1:1,000 dilution in TBS-T at 4°C overnight. Membranes were washed 4 times for 15 min in TBS-T, incubated with the secondary horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG antibody (1:10,000) for 1 h at room temperature, and then washed four times for 15 min each in TBS-T. Bands were visualized by using enhanced chemiluminescence (Amersham). The β-tubulin control was detected with anti-β-tubulin antibody (Biovision Inc.) at a dilution of 1:10,000 (1% bovine serum albumin [BSA] in TBS-T).
For Glut4, Fat/Cd36, Fabp4, and caveolin 1 (Cav1), whole-heart homogenates (5 μg) were separated by SDS-PAGE, transferred onto a polyvinylidene difluoride membrane, and incubated in a 7.5% BSA blocking solution. Thereafter, primary antibodies for Fat/Cd36 and Fabp4 (Santa Cruz), Cav1 (BD Biosciences), Glut4 (Chemicon), and α-tubulin (Abcam) and the corresponding secondary IgG antibody, as specified by the supplier, were used. Membrane proteins were detected by enhanced chemiluminescence (ChemiGenius2 bioimaging system; SynGene, Cambridge, United Kingdom), and Ponceau S staining was used to verify equal loading. Relative band densities were used for quantification of proteins in Pcyt2+/− versus Pcyt2+/+ hearts (n = 4 per group).
Quantification of heart lipids.
Lipid analysis of Pcyt2+/− and Pcyt2+/+ male and female hearts was performed two times on 4 mice per group by “shotgun” lipidomics, as described previously (20). The lipids were extracted according to the method of Bligh and Dyer (21), and analysis was performed by direct infusion of the lipid extract into an ABSciex Qtrap5500 instrument in both the positive and negative electrospray ionization (ESI) modes. Samples were infused at a constant rate of 5 μl/min and added into the solvent at a flow rate of 100 μl per min. Data were acquired by precursor-ion and neutral-loss scans. Data analyses were performed with LipdView software (ABSciex). Results are based on the peak area of each lipid species and expressed as the percentage of each species in the total lipid species identified. The total cardiac TAG content was measured with a Wako L-type TAG assay kit, in accordance with the manufacturer's specifications.
2-Bromopalmitate and 2-deoxyglucose uptake by cardiac tissue.
After 12 h of fasting, mice (n = 6 per group) were injected retroorbitally with 5 μCi 2-deoxy[14C]glucose and 1.5 μCi BSA-complexed 2-bromo-[3H]palmitate (22). Blood was drawn after 5 min of injection, 10 μl of serum was isolated, and the radioactivity was determined by liquid scintillation counting immediately. One hour after injection, the hearts were harvested and homogenized in 250 μl of PBS. The radioactivity of total heart homogenates was determined and normalized to the radioactivity present in 5 μl of serum collected 5 min after injection.
Lipid peroxidation.
Hearts and aortas were isolated after 12 h of fasting (n = 6), and the TBARS assay kit (Caymen) was used to measure malondialdehyde (MDA) derived from polyunsaturated fatty acid (PUFA) peroxidation. Samples were processed according to the instruction manual provided with the kit, and the amount of MDA was quantified colorimetrically at 532 nm.
Hormone analyses.
Eight-month-old mice (n = 12) of both genders were sacrificed 4 h into the light period, and blood samples were collected by cardiac puncture into 2-ml microcentrifuge tubes. Serum was separated by centrifugation for 10 min at 5,000 × g and snap-frozen in liquid nitrogen. Samples were sent to the University of Tennessee for steroid hormone quantification using radioimmunoassays (RIAs). Cortisol and testosterone levels were determined with a Coat-a-Count kit (Siemens Medical Solution Diagnostics, Los Angeles, CA), and androstenedione, estradiol, and 17-OH-progesterone levels were determined with an ImmunChem Double Antibody kit (MP Biomedicals, Solon, OH).
Blood biochemistry.
Blood was collected after 12 h of fasting (n = 8). Serum was separated immediately and sent to the Animal Health Laboratory (University of Guelph) for biochemical analyses of kidney and liver function.
Microarrays and bioinformatics analysis.
Total liver RNA was isolated from 3-month-old Pcyt2+/− males (n = 6) and wild-type littermates (n = 6) and analyzed on Affymetrix Mouse Gene 1.1 ST arrays (whole transcriptome). RNA quality and integrity were confirmed by using the Agilent BioAnalyzer. Principal component analysis (PCA) and variance component analysis (VCA) were performed for 6 array data sets. A volcano plot was used to estimate global variations in gene expression at a P value of <0.05. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis was performed by using FunNet.
Statistical analyses.
Data are expressed as means ± standard errors of the means (SEM). Statistical significance (Pcyt2+/− mice relative to Pcyt2+/+ littermate controls) was determined by one- and two-tailed Student t tests using GraphPad Prism 4 software. For echocardiography and hemodynamic measurements, analysis of variance (ANOVA) followed by Tukey's post hoc test was performed. Densitometry analyses were done by using ImageJ 1.46 software. Data from at least 6 independent measurements for each group were used for statistical analysis.
Microarray data accession number.
The microarray data were submitted to the GEO database under accession number GSE55617.
RESULTS
Multiple-organ dysfunction in adult Pcyt2+/− mice.
Reduced PE synthesis in Pcyt2+/− mice causes hyperlipidemia, insulin resistance, and, as a result, TAG accumulation in the liver and adipocytes, as anticipated (11). Figure 1A shows that compared to wild-type mice, 8-month-old Pcyt2+/− mice had an increased amount of lipid droplets and a reduced number of mitochondria in the liver. The reduced number of mitochondria was accompanied by increased autophagy, as evidenced by the increased population of autophagosomes (specific two-membrane organelles) in Pcyt2+/− liver. Liver steatosis in Pcyt2+/− mice was also visible by H&E staining (Fig. 1B), and it was accompanied by collagen accumulation (liver fibrosis) (Fig. 1C), which are well-known characteristics of nonalcoholic fatty liver disease. Staining of pancreatic islets for somatostatin and pancreatic polypeptide further demonstrated islet hypertrophy in Pcyt2+/− mice in comparison to littermate controls (Fig. 1D), in agreement with previously established hyperinsulinemia in older Pcyt2+/− mice (11). Surprisingly, only older male Pcyt2+/− mice had an enlarged heart compared to those of the controls (Fig. 1E), but the Pcyt2+/− females of the same age had unchanged heart size compared to those of the littermate controls. The heart size was further confirmed by a thorough measurement of the heart weight and heart weight relative to body weight (HW/BW), and a significantly (P < 0.0001) increased weight was observed only for the older Pcyt2+/− males but not the older Pcyt2+/− females (Table 1). Increased heart size was not observed for young Pcyt2+/− mice of any gender.
FIG 1.

Multiorgan abnormalities in the context of Pcyt2+/− metabolic syndrome. (A) Electron microscopy reveals an increased amount of liver lipid droplets (LD), reduced and damaged mitochondria (m) (black arrow), and mitophagy (m with two membrane layers) (white arrows) in 8-month-old Pcyt2+/− liver. (B and C) Liver sections stained with H&E (B) and Picrosirius red (C) reveal severe liver steatosis and fibrosis in Pcyt2+/− mice. Phenotypes in panels A to C are present in both male and female Pcyt2+/− mice. (D) Staining for somatostatin 14 and pancreatic polypeptide demonstrates pancreatic islet hypertrophy in Pcyt2+/− mice. (E) Representative images illustrating that 8-month-old Pcyt2+/− male hearts are larger than the hearts of control male littermates.
TABLE 1.
Morphometry, echocardiography, and hemodynamic analyses of 8-month-old male and female Pcyt2+/− and wild-type control micea
| Parameter | Mean value ± SEM for: |
P value |
||||
|---|---|---|---|---|---|---|
| Pcyt2+/− males | WT males | Pcyt2+/− females | WT females | ANOVA | t test | |
| BW (g) | 45.01 ± 1.49 | 44.48 ± 1.53 | 43.30 ± 2.22 | 37.13 ± 4.98 | NS | NS |
| HW (mg) | 231.50 ± 8.15** | 197.33 ± 3.15 | 143.00 ± 4.19 | 139.50 ± 8.83 | <0.01 | <0.001 |
| HW/BW | 5.09 ± 0.04** | 4.46 ± 0.13 | 3.39 ± 0.20 | 3.98 ± 0.39 | <0.01 | <0.0001 |
| HW/TL | 12.50 ± 0.46* | 10.56 ± 0.11 | 7.98 ± 0.24 | 7.67 ± 0.51 | <0.05 | 0.002 |
| HR (bpm) | 498.42 ± 17.57 | 490.74 ± 4.45 | 511.90 ± 10.46 | 503.20 ± 18.34 | NS | NS |
| LVIDd (mm) | 4.68 ± 0.08** | 4.14 ± 0.06 | 4.08 ± 0.07 | 3.94 ± 0.06 | <0.01 | <0.001 |
| LVIDs (mm) | 3.18 ± 0.11** | 2.41 ± 0.05 | 2.48 ± 0.09 | 2.34 ± 0.04 | <0.01 | <0.0001 |
| FS (%) | 32.13 ± 1.19** | 41.39 ± 0.79 | 39.34 ± 1.42 | 40.41 ± 0.83 | <0.01 | <0.0001 |
| EF (%) | 66.77 ± 1.73** | 78.44 ± 0.87 | 76.09 ± 1.63 | 77.38 ± 0.91 | <0.01 | <0.0001 |
| LVESP (mm Hg) | 148.73 ± 4.27§ | 102.48 ± 0.61 | 101.64 ± 0.66 | 100.35 ± 1.05 | <0.0001 | <0.00001 |
| LVEDP (mm Hg) | 1.57 ± 1.61 | −1.20 ± 0.68 | −0.90 ± 0.81 | 0.00 ± 0.76 | NS | NS |
| LDP (mm Hg) | 147.16 ± 3.92§ | 103.68 ± 1.19 | 102.54 ± 1.36 | 100.35 ± 1.29 | <0.0001 | <0.00001 |
| SBP (mm Hg) | 137.50 ± 2.29§ | 100.82 ± 0.53 | 99.50 ± 0.68 | 99.83 ± 0.93 | <0.0001 | 0.0000001 |
| DBP (mm Hg) | 83.34 ± 1.51§ | 64.04 ± 0.62 | 57.79 ± 1.62 | 59.96 ± 2.00 | <0.0001 | 0.000008 |
| MAP (mm Hg) | 101.39 ± 0.96§ | 76.22 ± 0.48 | 72.50 ± 1.00 | 73.25 ± 1.40 | <0.0001 | <0.000001 |
| dP/dt max (mm Hg/s) | 10,756.43 ± 876 | 9,591.04 ± 667 | 9,699.95 ± 752 | 7,991.47 ± 479 | NS | NS |
| dP/dt min (mm Hg/s) | 10,116.59 ± 1,163 | 9,348.37 ± 314 | 9,519.56 ± 860 | 8,136.27 ± 395 | NS | NS |
*, P < 0.05; **, P < 0.01; §, P < 0.0001 (for 8-month-old Pcyt2+/− mice versus wild-type [WT] mice [n = 6], determined by t test for male Pcyt2+/− versus male wild-type mice [n = 6] or by two-way ANOVA and a post hoc Tukey test for all other groups). BW, body weight; HW, heart weight; HW/BW, ratio of heart weight to body weight; TL, tibia length; HR, heart rate; LVIDd, left ventricle internal diameter at diastole; LVIDs, left ventricle internal diameter at systole; FS, fractional shortening; EF, ejection fraction; LVESP, LV end systolic pressure; LVEDP, LV end diastolic pressure; LDP, LV developed pressure; SBP, systolic blood pressure; DBP, diastolic blood pressure; MAP, mean arterial blood pressure; dP/dt, first derivative of LV pressure. NS, not significant.
Pcyt2+/− males exclusively develop cardiac hypertrophy and hypertension.
We first documented significant cardiac dysfunction in 8-month-old Pcyt2+/− males versus littermate controls by echocardiography. Table 1 shows that male Pcyt2+/− mice exhibited cardiac hypertrophy with increased (P < 0.001) left ventricle (LV) internal systolic diameter (LVIDs) (3.18 ± 0.11 mm versus 2.41 ± 0.05 mm) and increased (P < 0.0001) LV internal diastolic diameter (LVIDd) (4.68 ± 0.08 mm versus 4.14 ± 0.06 mm), accompanied by decreased (P < 0.0001) fractional shortening (FS) (32.13% ± 1.19% versus 41.39% ± 0.79%) and a decreased (P < 0.0001) ejection fraction (EF) (66.77% ± 1.73% versus 78.44% ± 0.87%). These male Pcyt2+/− mice also exhibited profound hypertension compared to male littermate controls, as determined by in vivo catheterization and hemodynamics (Table 1). The parameters included increased (P < 0.0001) (i) LV end systolic pressure (LVESP) (148.73 ± 4.27 mm Hg versus 102.48 ± 0.61 mm Hg), (ii) LV developed pressure (LVDP) (147.16 ± 3.92 mm Hg versus 103.68 ± 1.19 mm Hg), (iii) systolic blood pressure (SBP) (137.50 ± 2.29 mm Hg versus 100.82 ± 0.53 mm Hg), (iv) diastolic blood pressure (DBP) (83.34 ± 1.51 mm Hg versus 64.04 ± 0.62 mm Hg), and (v) mean arterial pressure (MAP) (101.39 ± 0.96 mm Hg versus 76.22 ± 0.48 mm Hg). Thus, these data show severe myocardial hypertrophy and hypertension in 8-month-old males.
We then excluded the possibility that the cardiac phenotype in aging male Pcyt2+/− mice was congenital in nature rather than being due to reduced PE synthesis and consequential changes in membrane and FA metabolism. Young Pcyt2+/− mice at 3 months of age appeared healthy, with all cardiovascular parameters, including echocardiography and hemodynamic parameters, being similar to those of the control littermates (see Table S1 in the supplemental material). Although this does not exclude congenital cardiac conditions, these conditions were clearly not obvious at a young age. Moreover, this suggests that the hypertension and hypertrophy that develop in male Pcyt2+/− mice do so over an extended period of deregulation. Taken together, abnormal PE synthesis appears to act as a pathophysiological stimulus, and heart disease develops over time.
We next demonstrated that the cardiovascular phenotype in aging male Pcyt2+/− mice is gender specific. Consistent with this notion, pathophysiology was restricted to male mice, as age-matched female Pcyt2+/− mice did not develop cardiac hypertrophy or hypertension. As might be expected from the functional data, 8-month-old female Pcyt2+/− mice did not exhibit significantly different (P > 0.05) values for echocardiography (LVIDd, LVIDs, FS, and EF) or hemodynamic (LVESP, LVEDP, LDP, SBP, DBP, and MAP) parameters compared to female littermate control mice (Table 1). Furthermore, young female Pcyt2+/− mice also showed no evidence of cardiovascular dysfunction (see Table S1 in the supplemental material). Thus, these data establish the presence of profound hypertension with compensatory cardiac hypertrophy that appears to specifically develop in older male Pcyt2+/− mice.
Increased cardiomyocyte size in Pcyt2+/− male hearts.
The functional pathologies in older male Pcyt2+/− hearts were associated with increased cardiomyocyte size, as shown in Fig. 2A and B. Cardiomyocyte hypertrophy (P < 0.05) was detected in 8-month-old Pcyt2+/− males compared to the littermate controls (284.13 ± 12.08 μm2 versus 228.57 ± 9.37 μm2). As expected, there was no myocyte hypertrophy in young Pcyt2+/− male hearts compared to the littermate controls (178.73 ± 8.36 μm2 versus 169 ± 11.75 μm2) (Fig. 2C and D). There was also no difference in cardiomyocyte sizes in young and old Pcyt2+/− female mice.
FIG 2.

Pcyt2+/− males have cardiomyocyte hypertrophy with age. (A and B) Representative H&E-stained images of littermate control hearts and hearts of 8-month-old Pcyt2+/− males (A), with Pcyt2+/− hearts exhibiting significantly increased myocyte cross-sectional area (MSCA) (n = 5 hearts/group) (B). (C and D) In contrast, representative images from a littermate control heart and a 3-month-old Pcyt2+/− male heart (C) show no difference in cardiomyocyte size (n = 5 hearts/group) (D). These findings are consistent with the notion that changes in cardiac pathophysiology in 8-month-old Pcyt2+/− mice are associated with aging and are not congenital. *, P < 0.05.
Gender-specific regulation of gene expression in Pcyt2+/− heart.
We next examined the gender-related variations in cardiac gene expression. Differences established for 8-month-old male and female mice are summarized in Fig. 3 and 4. As shown in Fig. 3A, the genes responsible for FA supply from circulating lipoproteins, the FA transporter gene Cd36 and the lipoprotein lipase gene Lpl, are specifically upregulated in male Pcyt2+/− heart. In contrast, genes encoding the TAG- and FA-degrading hormone-sensitive lipase (Hsl), adipose triglyceride lipase (Atgl), and peroxisome proliferator-activated receptor alpha (Pparα) were not significantly modified, and as expected, the peripheral G protein (estrogen) receptor gene Gpr30 was not detected in hearts of any group of males (Fig. 3A). The expression of genes encoding the structural proteins α-actin (Acta), myosin heavy chain 6 (Myh6), and myosin heavy chain 7 (Myh7) was also unaltered in Pcyt2+/− male heart. The level of the natriuretic peptide type A gene Nppa was significantly reduced in the Pcyt2+/− male heart (Fig. 3A). Altogether, Lpl and Cd36 levels were 27% to 30% higher (P < 0.05) (Fig. 3C and D), while the Nppa level was 30% lower (P < 0.05) (Fig. 3E), in Pcyt2+/− male hearts than in Pcyt2+/+ male hearts.
FIG 3.

Differential expression of heart biomarkers and lipid genes in male and female Pcyt2+/− hearts. (A and B) Representative expression of heart failure genes and genes involved in the heart fatty acid transport and metabolism in 8-month-old male and female hearts. Each lane (n = 3) represents separate Pcyt2+/− and Pcyt2+/+ animals. (C to E) Quantification (n = 6 for each group) reveals significant upregulation of the fatty acid transporter gene Cd36 (C) and the lipoprotein lipase gene Lpl (D) and significant downregulation of Nppa (E) in Pcyt2+/− male hearts. (F to H) Conversely, there is significant downregulation of Cd36 (G) and α-actin (Acta) (F) and upregulation of Myh7 (H) in Pcyt2+/− female hearts. The surface G-protein (estrogen) receptor Gpr30 is detected only in Pcyt2+/− and Pcyt2+/+ female hearts. The values are means ± SEM. *, P < 0.05 by a two-tailed or one-tailed Student t test.
FIG 4.

Gender-dependent expression of metabolic and hypertension genes in Pcyt2+/− heart. (A) Genes identified as differently expressed in 8-month-old male and female hearts. (B) The monocarboxylate (lactate) transporter gene Mct1 is significantly upregulated in both Pcyt2+/− male and female hearts; however, the Mct1 expression level is significantly higher in Pcyt2+/− female hearts than in Pcyt2+/− male hearts. (C) The angiotensin-converting enzyme 1 gene AceI is significantly upregulated in Pcyt2+/− male heart versus littermate controls. Control female hearts have low AceI expression levels, and the expression level remained relatively low in Pcyt2+/− female hearts in comparison to Pcyt2+/− males. (D) The mitochondrial regulator Pgc1α is significantly upregulated in control female hearts relative to control male hearts. Pgc1α expression was also significantly upregulated in Pcyt2+/− female hearts relative to Pcyt2+/− male hearts. (E) Expression of the stearoyl-CoA desaturase gene Scd1 is significantly upregulated in Pcyt2+/− female versus Pcyt2+/− male hearts. Scd1 expression is significantly upregulated in Pcyt2+/− female hearts versus littermate controls. Values are means ± SEM (n = 6 in each group). *, P < 0.05; **, P < 0.01; ***, P < 0.001 (by two-tailed Student t test).
In Pcyt2+/− female hearts, the levels of Cd36, Acta, and Myh7 mRNAs were significantly altered (Fig. 3B). Cd36 and Acta levels were reduced 60% and 55%, respectively (P < 0.05) (Fig. 3F and G), while the Myh7 level was 70% higher (P < 0.05) (Fig. 3H), in Pcyt2+/− female hearts than in Pcyt2+/+ female hearts. The expression levels of Nppa, Atgl, Hsl, Lpl, Paraα, and Grp30 were unchanged in Pcyt2+/− female hearts. Taken together, the data in Fig. 3 demonstrate significantly altered expression levels of genes for FA supply and heart remodeling in 8-month-old Pcyt2+/− mice. Gene expression patterns are gender specific, underlying the cardiac dysfunction that develops only in Pcyt2+/− males and the cardiac protection in Pcyt2+/− females, even though both genders develop chronic obesity, hyperlipidemia, and insulin resistance (11).
Next, we examined the expression of genes that can be directly linked to Pcyt2+/− energy metabolism. We investigated the genes for tricarboxylic acid (TCA) cycle activity and mitochondrial biogenesis (monocarboxylate [lactate/glutamate] transporter 1 [Mct1] and peroxisome proliferator-activated receptor gamma coactivator 1 alpha [Pgc1α]) and FA synthesis (stearoyl-CoA desaturase 1 [Scd1]) (Fig. 4). As shown in Fig. 4A and B, Mct1 was 56% upregulated in Pcyt2+/− male hearts and 20% upregulated in female Pcyt2+/− hearts relative to the levels in the male and female controls. When Pcyt2+/− male and female hearts were compared, female Pcyt2+/− hearts still had 33% higher Mct1 expression levels (P < 0.05). Interestingly, the basal Ace expression level was 73% (P < 0.05) higher in male hearts than in female hearts (Fig. 4A and C). Pcyt2+/− male hearts also had an additional 11% upregulation of Ace in comparison to the basal levels. At the basal level, Pgc1α was 19% upregulated in female hearts in comparison to male hearts, and a similar difference was maintained in Pcyt2+/− female and male hearts (16% increase; P < 0.05) (Fig. 4A and D). The modest but significant upregulation of Pgc1α is consistent with the notion that mitochondrial biogenesis is unaltered in Pcyt2-deficient hearts of both genders. Scd1 expression was unaltered in Pcyt2+/− male hearts and was strongly upregulated (88%) in Pcyt2+/− female hearts (Fig. 4A and E). Altogether, the data in Fig. 4 suggest gender-specific alterations of substrate supply (lactate/glutamate transporter [Mct1] and monounsaturated fatty acid synthesis [Scd1]) and renin-angiotensin system (RAAS) (AceI) genes in Pcyt2+/− hearts.
The cardiac insulin signaling pathway is impaired in Pcyt2+/− mice of both genders.
We next tested if insulin signaling contributes to cardiac dysfunction in 8-month-old Pcyt2+/− males. There was a dramatic decrease in the PI3K protein level in both male and female Pcyt2+/− hearts relative to Pcyt2+/+ control hearts. In addition, Akt1 activation by phosphorylation (pAkt1/Akt1 ratio) was reduced 38 to 40% (P < 0.05) (Fig. 5A), and the amount of the glucose transporter Glut4 was similarly decreased by 30% (P < 0.05) in both male and female Pcyt2+/− hearts (Fig. 5B). Therefore, in agreement with previously established systemic insulin resistance (11), heart insulin signaling and glucose transport are impaired in Pcyt2+/− mice of both genders. Reduced heart PI3K and pAkt1/Akt levels were also observed in 3-month-old Pcyt2+/− mice of both genders (see Fig. S1 in the supplemental material). Similarly to Glut4, the Fat/Cd36 protein level was reduced by 36 to 41% in Pcyt2+/− male and female hearts (Fig. 5B). The levels of Fabpm, another protein important for cardiac FA uptake, and the membrane-specific protein caveolin 1 (Cav1), were not significantly different between Pcyt2+/− and Pcyt2+/+ hearts (Fig. 5B).
FIG 5.

Gender-independent reduction in insulin signaling and substrate uptake in Pcyt2+/− heart. (A and B) The insulin signaling pathway is impaired in 8-month-old male and female Pcyt2+/− hearts. Levels of PI3K and pT308Akt/Akt (A) and of Fat/Cd36 and Glut4 (B) are reduced in hearts of Pcyt2+/− mice of both genders. The heart Fabpm, caveolin 1, and beta-tubulin control levels are not affected by Pcyt2 deficiency. (C) Glucose and fatty acid uptake was similarly reduced in hearts of Pcyt2+/− mice of both genders. [14C]deoxyglucose and [3H]bromopalmitate were injected, and heart incorporation (disintegrations per minute/milligram/hour) in Pcyt2+/− and Pcyt2+/+ mice of both sexes was determined. (D) Pcyt2 gene expression in Pcyt2+/− mice did not vary between different tissue types, showing no link with male-specific cardiac dysfunction. The values are means ± SEM (n = 6 per group). *, P < 0.05; **, P < 0.01.
To relate the levels of Glut4 and Fat/Cd36 transport proteins with the heart's capacity for substrate uptake, we measured the uptake of the respective nondegradable substrates [3H]deoxyglucose and [14C]bromopalmitate in male and female Pcyt2+/− and Pcyt2+/+ hearts (Fig. 5C). In accordance with the reduced Glut4 levels, glucose uptake was reduced 47% (P < 0.01) in Pcyt2+/− male hearts and 32% (P < 0.001) in Pcyt2+/− female hearts (Fig. 5C). In accordance with the reduced Fat/Cd36 levels, the [14C]bromopalmitate uptake in Pcyt2+/− male hearts decreased 44.7% (P < 0.01), and that in Pcyt2+/− female hearts decreased 29.7% (P < 0.01) (Fig. 5C). Altogether, these data establish that hearts of Pcyt2+/− mice of both genders have impaired insulin signaling and a significantly reduced capacity for exogenous substrate (glucose and fatty acid) uptake, mediated mostly by Glut4 and Fat/Cd36. However, since the impairments are present in Pcyt2+/− mice of both genders, they are unrelated to male-specific cardiac dysfunction.
The composition of Pcyt2+/− heart lipids is gender dependent.
To identify the underlying mechanism(s) responsible for the male-specific cardiac dysfunction, we also tested the gender-related differences in Pcyt2 expression in various tissues. As shown in Fig. 5D, Pcyt2 mRNA levels in the hearts, kidneys, and livers of male and female Pcyt2+/− mice did not significantly differ, and therefore, the Pcyt2 expression level cannot be related to the male-specific phenotype. Since deletion of the Pcyt2 gene directly affects membrane phospholipids, we performed a detailed analysis of the heart lipids by shotgun lipidomics. The content and fatty acid composition of the heart phospholipids and triglycerides are shown in Fig. 6A to C. Figure 6A shows variations in individual phospholipids (PE, phosphatidylcholine [PC], phosphatidylserine [PS], phosphatidylinositol [PI], and cardiolipin [CL]) in Pcyt2+/− and Pcyt2+/+ male and female hearts. While PS and CL levels did not differ among groups, PE, PC, and PI levels showed a gender-dependent difference. The total PE content was 11.9% (P < 0.05) higher in Pcyt2+/− male hearts and 8.6% (P < 0.05) lower in Pcyt2+/− female hearts (Fig. 6B). The total PC content was reduced (6.2%; P < 0.05) in Pcyt2+/− male hearts and was unchanged in Pcyt2+/− female hearts. The PI content was dramatically increased (47%; P < 0.05) only in Pcyt2+/− male hearts (Fig. 6A). These changes altered the membrane PC/PE ratio only in Pcyt2+/− male hearts, where the ratio was 17.6% (P < 0.01) reduced relative to that of Pcyt2+/+ male hearts. Pcyt2+/− and Pcyt2+/+ female hearts had similar membrane PC/PE ratios (Fig. 6B), showing that Pcyt2 heterozygosity alters the membranes of only the male heart.
FIG 6.

Modified phospholipid ratio and fatty acid composition in Pcyt2+/− male heart. (A) Contribution of individual phospholipids (percent) to total heart phospholipids. PE, phosphatidylethanolamine; PC, phosphatidylcholine; PI, phosphatidylinositol; PS, phosphatidylserine; CL, cardiolipin; SM, sphingomyelin. (B) Specific contribution of PE, PC, and the PC/PE ratio in Pcyt2-deficient and wild-type male and female hearts (8-month-old Pcyt2+/− and Pcyt2+/+ mice). The PE level is increased in Pcyt2+/− male hearts and reduced in Pcyt2+/− female hearts, while the PC/PE ratio is lower only in Pcyt2+/− male hearts. The PI level is specifically higher in Pcyt2+/− male hearts, and CL and PS levels remained unchanged. (C and D) Fatty acid profiles of cardiac phospholipids (C) and triglycerides (D) showing sexual dimorphism and male-specific changes in Pcyt2-deficient heart. Values are means ± SEM for Pcyt2+/− and Pcyt2+/+ male and female hearts (2× n = 4 per group). *, P < 0.05; **, P < 0.01.
Detailed analyses of the phospholipid fatty acid composition further reveal that wild-type male hearts have a fatty acid composition different from that of wild-type female hearts and that the fatty acid composition is altered by Pcyt2 deficiency only in males (Fig. 6B). Wild-type Pcyt2+/+ male hearts had the lowest content of palmitic acid (18:0), otherwise the most abundant saturated fatty acid in other groups, and the highest content of linoleic acid (18:2n-6), the essential fatty acid and the precursor of arachidonic acid (20:4n-6) and other n-6 long-chain PUFAs. Wild-type male heart TAG has a reduced content of oleic acid (18:1) and an increased content of stearic acid (16:0), which is also different from all other groups (Fig. 6C). These data for the first time clearly establish a sexual dimorphism in the phospholipid and TAG fatty acid compositions between male and female hearts that is specifically perturbed in the Pcyt2+/− male heart.
Arachidonic acid and n-6 LCPUFAs are enriched in Pcyt2+/− male heart phospholipids.
Since the altered fatty acid composition is the most critical alteration caused by Pcyt2 deficiency, we also performed a detailed lipidomic analysis of the formation of arachidonic acid by desaturation/elongation of linoleic acid (18:2n-6). As shown in Fig. 7A and B, there is a clear sexual dimorphism in the heart n-6 elongation/desaturation products in wild-type males versus wild-type female (white bars). The wild-type male heart is enriched in 18:2n-6 and contains ∼37% (P < 0.01) more 18:2n-6 than does the wild-type female heart. The wild-type female heart, however, contains a higher proportion of the elongation/desaturation products, including arachidonic acid (20:4n-6) and other n-6 PUFAs (20:3n-6, 22:3n-6, and 22:4n-6).
FIG 7.

Accumulation of arachidonic acid and long-chain n-6 PUFAs in Pcyt2+/− male heart. (A) Desaturation and elongation of the essential 18:2n-6 PUFA linoleic acid are sexually dimorphic and more active in wild-type female heart than in wild-type male heart (white bars). The production of arachidonic acid (20:4n-6) and longer-chain n-6 PUFAs from linoleic acid (18:2n-6) is upregulated only in Pcyt2-deficient male hearts (black versus white bars). (B) Cardiac levels of very-long-chain n-6 and n-3 PUFAs and total triglycerides also show sexual dimorphism and are elevated in wild-type female hearts (white bars). The level of docosahexaenoic acid (22:6n-3) additionally is increased only in Pcyt2+/− female heart. (C) ROS production is increased in both male and female Pcyt2+/− hearts, while in the aorta, ROS production is specifically increased in Pcyt2+/− males, showing male-specific vascular oxidative stress in Pcyt2 deficiency. (D and E) Pcyt2+/− male-specific overexpression of the AceI gene in the heart, with no differences in the rest of the renin-angiotensin system, i.e., Ace2 (heart and kidney), angiotensinogen (Agt) (liver), renin (Ren kidney), and angiotensin receptor (Agtr1) (heart). Values are means ± SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Deletion of Pcyt2 causes remodeling of the membrane n-6 PUFAs that is more specific in deficient males (Fig. 7A and B, black bars). While the 18:2n-6 content in Pcyt2+/− males is reduced 55% (P < 0.001), the levels of most of the elongation/desaturation products (20:3n-6, 20:4n-6, 22:3n-6, and 22:4n-6) are increased and in similar increments of 38 to 56%. On the other hand, the n-6 PUFA content in the female heart is not significantly affected by Pcyt2 deficiency. The Pcyt2+/− female heart, however, is enriched in docosahexaenoic acid (DHA) (22:6n-3). The DHA level is elevated in wild-type female hearts compared to wild-type male hearts (20%; P < 0.01) and is further elevated in Pcyt2-deficient female hearts. The hearts of Pcyt2+/− females have 30% (P < 0.001) more DHA than do the hearts of Pcyt2+/− males (Fig. 7B). Together, these data firmly establish that increased linoleic acid (18:2n-6) desaturation/elongation leading to the accumulation of arachidonic acid and other n-6 PUFAs is specific to Pcyt2-deficient male hearts (which develop cardiomyopathy), while the unmodified n-6 PUFA phospholipids and accumulation of n-3 DHA are characteristics of Pcyt2-deficient female hearts (which are protected from cardiomyopathy).
Finally, in addition to the heart TAG fatty acid composition (Fig. 6C), total heart TAGs (Fig. 7B) are also sexually dimorphic, with wild-type females having 44% more TAG than wild-type males. However, the total heart TAG level was decreased 15.6% in Pcyt2+/− females relative to that in wild-type females, while in Pcyt2+/− males, the TAG level was increased 16% relative to that in wild-type males, additionally showing a gender-dependent variation in TAG turnover and utilization. While females accumulated more TAG, they also used more TAG in Pcyt2 deficiency. On the other hand, male hearts contained less TAG than female hearts and used less TAG in Pcyt2 deficiency.
Lipid peroxidation is increased in the heart and aorta of Pcyt2+/− males.
Since Pcyt2+/− male hearts have increased contents of arachidonic acid and long-chain n-6 PUFAs, we also analyzed malondialdehyde levels (a measure of PUFA peroxidation [reactive oxygen species {ROS}]) in cardiac tissue and aorta (Fig. 7C). The cardiac tissues of Pcyt2+/− mice of both genders had increased ROS production, but cardiac ROS production increased 62% (P < 0.05) in Pcyt2+/− males and 32.8% (P < 0.01) in Pcyt2+/− females in comparison to the same-gender controls. ROS production was dramatically elevated and 2.6-fold higher in the aortas of Pcyt2+/− males than in the aortas of wild-type males. Aortas of Pcyt2+/− females, on the other hand, had reduced ROS content relative to that of the wild-type females. Therefore, it seems likely that the Pcyt2 deficiency causes a higher level of desaturation of the membrane phospholipids and higher n-6 PUFA levels, which increase oxidative stress in the male heart and vascular system and predispose Pcyt2-deficient males to hypertension and cardiac dysfunction.
AceI is specifically upregulated in Pcyt2+/− male hearts.
Blood biochemical analyses show that male-specific hypertension cannot be caused by hepatic or renal dysfunction, since Pcyt2+/− mice of both genders had normal test results for liver and kidney function (see Table S2 in the supplemental material). To examine if the RAAS (Fig. 7D and E) has a causative role in male-specific hypertension, we tested the heart, liver, and kidney arms of the RAAS. We established that the level of AceI but not that of Ace2 was specifically elevated (2.6-fold increase) in Pcyt2+/− male hearts and that liver angiotensinogen (Agt), kidney renin (Ren), and cardiac angiotensin receptor (Agtr1) levels were not significantly different for both genders and genotypes. In agreement with the well-known role of AceI in male hypertension (23, 24), we conclude that the upregulation of AceI is most critical for male-specific RAAS activity and the development of hypertension.
Pcyt2+/− males have a modified hormonal status.
We also analyzed the steroid hormone levels in male and female mice in a gender-specific manner (Fig. 8). The levels of the steroid hormone precursor 17-OH-progesterone and aldosterone were unaltered by Pcyt2 deficiency versus wild-type controls (Fig. 8A and B). The estrogen levels were modestly elevated in Pcyt2+/− females versus control females (Fig. 8C). Pcyt2+/− males, however, had dramatically reduced circulating testosterone and cortisol levels (Fig. 8D and E). The testosterone level was reduced 3.6-fold (P < 0.05) (Fig. 8D) and the cortisol level was reduced 2.2-fold (P < 0.05) (Fig. 8E) in Pcyt2+/− males in comparison to control males. No significant changes in serum cortisol levels were detected in female Pcyt2+/− mice, and as expected, a low level of testosterone (basal level) was present in all female mice. These data strongly correlate Pcyt2 deficiency and steroid hormone homeostasis with specific cardiac dysfunction in male Pcyt2+/− mice.
FIG 8.

Pcyt2+/− males have reduced amounts of serum testosterone and cortisol. (A and B) Serum levels of the 17-OH-progesterone (A) and aldosterone (B) precursors do not show any differences between groups. (C) The serum estradiol level is upregulated in female Pcyt2+/− mice versus control littermates. (D and E) Serum testosterone (D) and cortisol (E) levels are significantly reduced in male Pcyt2+/− mice versus control littermates. Data are for 8-month-old mice. Data are expressed as means ± SEM (n = 6 per group), and the analysis was performed by using two-tailed Student t test. *, P < 0.05; **, P < 0.01.
Young Pcyt2+/− males have modified fatty acid metabolism and cholesterol homeostasis.
The liver is a very important regulator of cholesterol and steroid hormone homeostasis. To capture early changes that might have occurred in sterol metabolism and to further investigate Pcyt2 deficiency in steroid hormone homeostasis, we performed a genome-wide microarray analysis on liver samples from young, asymptomatic male Pcyt2+/− and control mice. The microarray data are shown in Fig. 9. Quality analysis established a clear separation between groups, indicating that the gene expression profiles for the Pcyt2+/− and control groups are distinct (Fig. 9A). A mixed-model ANOVA with a relaxed P value (α = 0.05) was used to prepare a volcano plot and further identify differentially expressed genes (Fig. 9B). The number of differentially expressed transcripts identified (P < 0.05) was 1,458 out of 28,996 transcripts, with 536 genes being upregulated and 714 genes being downregulated in Pcyt2+/− mice relative to littermate controls. Functional KEGG pathway analyses revealed that 67.9% of the downregulated genes are metabolic pathway genes, while 44.4% of the upregulated genes belong to the steroid biosynthesis pathways (Fig. 9C).
FIG 9.

Differences in regulation of metabolic pathways and steroid hormone biosynthesis. (A) Quality analysis establishes a clear separation between groups. (B) Volcano plot with a mixed-model ANOVA and a relaxed P value (α = 0.05) identifies differentially expressed genes. (C) Functional KEGG pathway analysis showing that 67.9% of the downregulated genes are metabolic pathway genes, while 44.4% of the upregulated genes are steroid biosynthesis genes. (D) Multiple mitochondrial FA oxidation and Pparα-regulated genes are significantly downregulated in Pcyt2+/− liver versus controls. (E) While the steroid hormone genes Cyp2c70, Comt1, and Hsd3b4 are upregulated, the cholesterol biosynthesis genes Sqle, Ebp, Pgrmc1, Hsd17b1, Tm7sf2, Cyp3a25, Hmgcs2, Cyp7a1, and Cyp2a5 are downregulated in 3-month-old Pcyt2+/− male liver. All units are arbitrary (P < 0.05).
Among the downregulated metabolic pathway genes, the Pparα signaling and the fatty acid oxidation genes contributed 14.5% and 12.2%, respectively, and in most cases, they were shared genes (Fig. 9C). This group includes the aldehyde dehydrogenase 3a2 gene Aldh3a2, the carnitine palmitoyl transferase gene Cpt1a, the acyl-CoA dehydrogenase member 11 gene Acad11, the acyl-CoA dehydrogenase C4-C12 straight-chain gene Acadm, the acyl-CoA synthase gene Acsl1, the cytochrome 450 gene Cyp11A1, the fatty acid desaturase 1 gene Fads1, the fatty acid desaturase 2 gene Fads2, and the acyl-CoA oxidase 1 gene Acox1 (Fig. 9D). Interestingly, based on the lipidomic data in Fig. 7A, 18:2 desaturation steps catalyzed by Fads1 and Fads2 activities are activated in Pcyt2-deficient male hearts, suggesting that the reduced gene expression is most likely compensatory.
The most upregulated genes are the genes for sex steroid hormone metabolism; however, many cholesterol metabolic genes are downregulated (Fig. 9E). The upregulated group includes the key regulatory gene in steroid hormone biosynthesis, the 3-beta hydroxyl steroid dehydrogenase gene Hsd3b4, the male-specific testosterone-degrading gene Cyp2c70, and the estrogen-degrading catechol-O-methyltransferase gene Comt1. In fact, these genes are the top upregulated genes in the Pcyt2+/− male liver (Fig. 9E). Conversely, the most downregulated steroidogenic genes are as follows: the hydroxysteroid (17-beta) dehydrogenase 13 gene Hsd17b13; the progesterone receptor membrane component 1 gene Pgrmc1; the C14 sterol reductase gene that regulates the conversion of lanosterol to cholesterol, Tm7sf2; the testosterone 6-beta hydroxylase gene Cyp3a25; the 3-hydroxy 3-methyl glutaryl coenzyme A synthase cholesterol biosynthesis gene Hmgcs2; the major bile acid synthesis gene encoding cholesterol 7-alpha hydroxylase, Cyp7a1; the testosterone- and xenobiotic-oxidizing gene Cyp2a5; the sterol isomerase/emopamil binding protein gene Ebp; and the squalene epoxidase gene Sqle (Fig. 9E). Taken together, the results of the steroidogenic gene expression analyses agree with the plasma hormonal data for Pcyt2+/− males (Fig. 8), at least in the case of low testosterone levels, predicting that increased steroid metabolism (not reduced steroidogenesis) contributes to the reduced blood testosterone and, perhaps, cortisol contents.
DISCUSSION
In this study, we used a heterozygous murine model for Pcyt2 that has reduced membrane PE synthesis by the CDP-ethanolamine Kennedy pathway. This is the first study to directly link deregulated membrane phospholipid homeostasis with mammalian cardiac dysfunction. This study emphasizes different consequences of reduced membrane PE synthesis on fatty acid metabolism and steroidogenesis, leading to cardiac dysfunction in Pcyt2-deficient males and providing protection in Pcyt2-deficient females.
Our findings are consistent with reduced PE synthesis in the Drosophila Kennedy pathway, in which mutants accumulate TAG and experience cardiac dysfunction (15). Pcyt2+/− male mice have elevated TAG levels and significantly increased left ventricular systolic and diastolic diameters along with decreased fractional shortening (FS) and ejection fraction (EF). These alterations occur along with profound hypertension, including significantly increased systemic and left ventricular blood pressure. Interestingly, the cardiovascular pathology was detected only in Pcyt2+/− males and not females. Despite reduced PE synthesis, Pcyt2+/− females did not have modified membrane phospholipids, had unimpaired EF and FS, and were protected from cardiac dysfunction.
Reduced substrate supply to the insulin-resistant Pcyt2+/− heart is gender independent.
Normal cardiac function depends on FA metabolism and, to a lesser degree, on glucose metabolism as an energy source (3, 25, 26). In contrast, the hypertrophied heart requires alternative energy sources and tends to rely more on glucose metabolism (25, 27–29). Both male and female Pcyt2+/− hearts have impaired PI3K/pAkt1/Glut4 signaling and glucose uptake; however, cardiac hypertrophy is present only in males. Myocardial FA uptake is mediated predominantly by the Cd36 membrane transporter (30). Cd36-null mice develop cardiac conduction abnormalities, altered membrane phospholipids, and impairments in calcium homeostasis (31). Moreover, CD36 deficiency in humans is linked to hypertrophic cardiomyopathy (32). However, both Pcyt2+/− males and females have reduced cardiac FA uptake and Cd36 protein levels that cannot be responsible for the male-specific hypertrophy. Both Pcyt2+/− male and female hearts have elevated levels of the lactate/glutamate transporter gene Mct1, probably to compensate for the reduced glucose uptake. As a compensation for the reduced FA uptake, the Scd1 expression level in female but not male Pcyt2+/− mice was highly elevated to increase the intracellular amount of fatty acids. Altogether, we conclude that under the underlying conditions of heart insulin resistance and similarly reduced glucose and fatty acid uptake in Pcyt2+/− mice of both genders, a different mechanism is responsible for male-specific cardiac dysfunction.
Specific remodeling of Pcyt2+/− male heart phospholipids.
The ratio and composition of the bilayer phospholipids are vital for membrane integrity and function, and a number of animal and human studies have established that the membrane phospholipid compositions in males and females differ (33–37). Because deletion of the Pcyt2 gene specifically modifies membrane PE (9, 11), we expected that membrane phospholipid remodeling could be the main initiator of male-specific cardiac dysfunction. Indeed, the lipidomics analysis established a strong sexual dimorphism in cardiac n-6 PUFA and a decreased PC/PE ratio in the Pcyt2+/− male heart, reflecting differential phospholipid remodeling in male and female Pcyt2-deficient hearts. Arachidonic acid (20:4n-6) and longer n-6 PUFAs are produced from linoleic acid (18:2n-6) by sequential desaturation and elongation reactions. Pcyt2+/− female heart phospholipids have unmodified n-6 PUFA and elevated n-3 PUFA DHA (22:6n-3) levels, known to be cardioprotective (34, 36, 37). The wild-type male heart is enriched in 18:2n-6 and has low 20:4n-6 levels. However, the Pcyt2+/− male heart became deficient in 18:2n-6 and enriched in 20:4n-6, showing that Pcyt2 deficiency stimulates n-6 PUFA elongation and desaturation in these animals.
It is well established that estrogen stimulates, whereas testosterone inhibits, the conversion of PUFAs into their longer-chain metabolites (36). Since Pcyt2+/− males have reduced testosterone levels, this may be an additional contributing factor in the release of the inhibition of 18:2n-6 conversion to 20:4n-6 and accumulation of n-6 PUFA in the Pcyt2+/− male heart. Androgen deficiency has been widely accepted to have a negative effect on metabolic diseases and promotes heart disese (38). Benefits of testosterone supplementation were evident in heart failure patients, in patients with metabolic syndrome, and with performance enhancement in athletes (39). Testosterone deficiency is a marker for early death in men (40, 41), and as shown in this study, it could be an important factor in male-specific cardiovascular disease.
Gender-dependent phospholipid remodeling is critical for the Pcyt2+/− heart phenotype.
Our proposed model for the male-specific heart phenotype in insulin-resistant Pcyt2+/− mice is shown in Fig. 10. Pcyt2 gene deletion primarily alters cardiac phospholipid homeostasis and remodeling. We establish a strong sexual dimorphism in heart phospholipid and fatty acid contents. Pcyt2 deficiency specifically alters heart phospholipids by reducing the PC/PE ratio and increasing n-6 PUFA levels in the male heart. The increased levels of arachidonic acid and other n-6 PUFAs instigate oxidative stress in the Pcyt2+/− male heart and aorta, leading to the development of male-specific cardiac pathology. Dysregulated steroid homeostasis and low testosterone levels in Pcyt2+/− males contribute to phospholipid remodeling and cardiac dysfunction. Conversely, unmodified membrane n-6 PUFA and increased n-3 PUFA levels protect the female Pcyt2+/− heart and vasculature from oxidative stress, hypertension, and cardiac remodeling.
FIG 10.
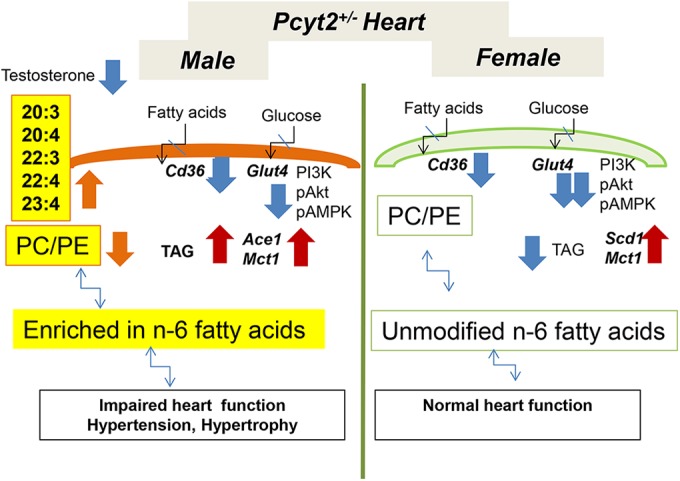
Proposed mechanism for the male-specific heart phenotype in insulin-resistant Pcyt2+/− mice. Both male and female Pcyt2+/− hearts exhibit similarly reduced fatty acid and glucose uptake and insulin resistance. The Pcyt2 gene deficiency directly alters membrane phospholipid homeostasis and side-chain fatty acid remodeling. There is a strong sexual dimorphism in the heart n-6 PUFA elongation/desaturation pathway. Pcyt2 deficiency specifically increases the conversion of linoleic acid (18:2n-6) to arachidonic acid (20:4n-6) in Pcyt2+/− male heart. This instigates oxidative stress in the heart and vasculature and elevates the AceI expression level in the Pcyt2+/− male heart, leading to the development of hypertension and cardiac hypertrophy. Low testosterone levels in Pcyt2+/− males are the result of deregulated steroidogenesis and possibly add to cardiovascular dysfunction. On the contrary, Pcyt2+/− females are protected from heart disease. Unmodified membrane phospholipids and n-6 PUFA and elevated n-3 PUFA levels protect female Pcyt2+/− heart and vasculature from oxidative stress, cardiac dysfunction, and hypertension. Although females experience reduced substrate uptake as much as the male Pcyt2+/− heart, the female Pcyt2+/− heart has more TAG, utilizes TAG more efficiently, and stimulates the expression of Scd1 for oleic acid synthesis and Mct1 for lactic and glutamic acid transport to compensate for the lack of exogenous substrates.
Male and female Pcyt2+/− hearts are insulin resistant and exhibit similarly reduced fatty acid and glucose transport, and these defects cannot be considered a source of male-specific hypertrophy. Interestingly, Mct1 expression levels are increased in both male and female Pcyt2+/− hearts, probably to increase the transport of alternative substrates (lactic acid and glutamine) to compensate for a reduced glucose supply. Although females experience reduced substrate uptake as much as males, the female Pcyt2+/− heart has more TAG available and utilizes TAG more efficiently. The female Pcyt2+/− heart also has elevated expression levels of Scd1 to contribute additional oleic acid (18:1) for TAG and β-oxidation, rendering the female Pcyt2+/− heart less sensitive to insulin resistance than the male Pcyt2+/− heart (Fig. 10).
Finally, due to the lack of previous research on Pcyt2, we examined gene expression profiles in the GEO database (http://www.ncbi.nlm.nih.gov/geoprofiles/) to search for any relationship between heart function and Pcyt2. We found that in most heart-related diseases in humans and in animal models, Pcyt2 expression is downregulated. Examples include human heart failure (GEO database accession number GDS651), human dilated cardiomyopathy (accession number GDS2205), murine heart failure (accession number GDS3386), rat diabetic heart (accession number GPL341), rat heart failure (accession number GDS1959), rat cardiac aging (accession number GPL85), human isoproterenol- or exercise-induced cardiac hypertrophy (accession number GDS3596), murine hypertension (accession number GPL9734), and rat hypertension (accession number GPL85). We conclude that Pcyt2 is a new and important regulator of heart phospholipid homeostasis, and if functionally impaired, it may have serious consequences, particularly for male heart function under conditions of insulin resistance and obesity. Pcyt2-deficient mice are the first mammalian model to directly establish a firm relationship between gender-specific remodeling of phospholipid essential fatty acids and the development of heart disease, also providing an explanation for why females are protected with their blood pressure and cardiac function preserved in a similar diabetic state.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by an Ontario Trillium Scholarship (P.B.), CIHR grant MOP86448 (M.B.), and the Canadian Institutes of Health Research (T.A.M.).
We declare that we have no conflicts of interest.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00380-15.
REFERENCES
- 1.Kolwicz SC Jr, Purohit S, Tian R. 2013. Cardiac metabolism and its interactions with contraction, growth, and survival of cardiomyocytes. Circ Res 113:603–616. doi: 10.1161/CIRCRESAHA.113.302095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Neely JR, Morgan HE. 1974. Relationship between carbohydrate and lipid metabolism and the energy balance of heart muscle. Annu Rev Physiol 36:413–459. doi: 10.1146/annurev.ph.36.030174.002213. [DOI] [PubMed] [Google Scholar]
- 3.Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC. 2010. Myocardial fatty acid metabolism in health and disease. Physiol Rev 90:207–258. doi: 10.1152/physrev.00015.2009. [DOI] [PubMed] [Google Scholar]
- 4.Bakovic M, Fullerton MD, Michel V. 2007. Metabolic and molecular aspects of ethanolamine phospholipid biosynthesis: the role of CTP:phosphoethanolamine cytidylyltransferase (Pcyt2). Biochem Cell Biol 85:283–300. doi: 10.1139/O07-006. [DOI] [PubMed] [Google Scholar]
- 5.Poloumienko A, Cote A, Quee AT, Zhu L, Bakovic M. 2004. Genomic organization and differential splicing of the mouse and human Pcyt2 genes. Gene 325:145–155. doi: 10.1016/j.gene.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 6.Min-Seok R, Kawamata Y, Nakamura H, Ohta A, Takagi M. 1996. Isolation and characterization of ECT1 gene encoding CTP:phosphoethanolamine cytidylyltransferase of Saccharomyces cerevisiae. J Biochem 120:1040–1047. doi: 10.1093/oxfordjournals.jbchem.a021497. [DOI] [PubMed] [Google Scholar]
- 7.Bladergroen BA, Houweling M, Geelen MJ, van Golde LM. 1999. Cloning and expression of CTP:phosphoethanolamine cytidylyltransferase cDNA from rat liver. Biochem J 343(Part 1):107–114. [PMC free article] [PubMed] [Google Scholar]
- 8.Tie A, Bakovic M. 2007. Alternative splicing of CTP:phosphoethanolamine cytidylyltransferase produces two isoforms that differ in catalytic properties. J Lipid Res 48:2172–2181. doi: 10.1194/jlr.M600536-JLR200. [DOI] [PubMed] [Google Scholar]
- 9.Fullerton MD, Hakimuddin F, Bakovic M. 2007. Developmental and metabolic effects of disruption of the mouse CTP:phosphoethanolamine cytidylyltransferase gene (Pcyt2). Mol Cell Biol 27:3327–3336. doi: 10.1128/MCB.01527-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mizoi J, Nakamura M, Nishida I. 2006. Defects in CTP:phosphoryl ethanolamine affect embryonic and postembryonic development in Arabidopsis. Plant Cell 18:3370–3385. doi: 10.1105/tpc.106.040840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fullerton MD, Hakimuddin F, Bonen A, Bakovic M. 2009. The development of a metabolic disease phenotype in CTP:phosphoethanolamine cytidylyltransferase-deficient mice. J Biol Chem 284:25704–25713. doi: 10.1074/jbc.M109.023846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leonardi R, Frank MW, Jackson PD, Rock CO, Jackowski S. 2009. Elimination of the CDP-ethanolamine pathway disrupts hepatic lipid homeostasis. J Biol Chem 284:27077–27089. doi: 10.1074/jbc.M109.031336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Khan RS, Drosatos K, Goldberg IJ. 2010. Creating and curing fatty hearts. Curr Opin Clin Nutr Metab Care 13:145–149. doi: 10.1097/MCO.0b013e3283357272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lim HY, Bodmer R. 2011. Phospholipid homeostasis and lipotoxic cardiomyopathy: a matter of balance. Fly (Austin) 5:234–236. doi: 10.4161/fly.5.3.15708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lim HY, Wang W, Wessells RJ, Ocorr K, Bodmer R. 2011. Phospholipid homeostasis regulates lipid metabolism and cardiac function through SREBP signaling in Drosophila. Genes Dev 25:189–200. doi: 10.1101/gad.1992411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Holloway GP, Snook LA, Harris RJ, Glatz JF, Luiken JJ, Bonen A. 2011. In obese Zucker rats, lipids accumulate in the heart despite normal mitochondrial content, morphology and long-chain fatty acid oxidation. J Physiol 589:169–180. doi: 10.1113/jphysiol.2010.198663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ku SK, Lee HS, Lee JH. 2002. An immunohistochemical study of pancreatic endocrine cells in SKH-1 hairless mice. Eur J Histochem 46:229–236. [DOI] [PubMed] [Google Scholar]
- 18.Alibhai FJ, Tsimakouridze EV, Chinnappareddy N, Wright DC, Billia F, O'Sullivan ML, Pyle WG, Sole MJ, Martino TA. 2014. Short-term disruption of diurnal rhythms after murine myocardial infarction adversely affects long-term myocardial structure and function. Circ Res 114:1713–1722. doi: 10.1161/CIRCRESAHA.114.302995. [DOI] [PubMed] [Google Scholar]
- 19.Tsimakouridze EV, Straume M, Podobed PS, Chin H, LaMarre J, Johnson R, Antenos M, Kirby GM, Mackay A, Huether P, Simpson JA, Sole M, Gadal G, Martino TA. 2012. Chronomics of pressure overload-induced cardiac hypertrophy in mice reveals altered day/night gene expression and biomarkers of heart disease. Chronobiol Int 29:810–821. doi: 10.3109/07420528.2012.691145. [DOI] [PubMed] [Google Scholar]
- 20.Schenkel LC, Singh RK, Michel V, Zeisel SH, da Costa KA, Johnson AR, Mudd HS, Bakovic M. 2 December 2014. Mechanism of choline deficiency and membrane alteration in postural orthostatic tachycardia syndrome primary skin fibroblasts. FASEB J doi: 10.1096/fj.14-258566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bligh EG, Dyer WJ. 1959. A rapid method of total lipid extraction and purification. Can J Biochem Physiol 37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 22.Paul S, Grevengoed TJ, Pascual F, Ellis JM, Willis MS, Coleman RA. 2014. Deficiency of cardiac acylCoA synthetase 1 induces diastolic dysfunction, but pathologic hypertrophy is reversed by rapamycin. Biochim Biophys Acta 1841:880–887. doi: 10.1016/j.bbalip.2014.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mosca L. 2004. Cardiology patient page. Heart disease prevention in women. American Heart Association. Circulation 109:e158–e160. doi: 10.1161/01.CIR.0000124449.48800.C3. [DOI] [PubMed] [Google Scholar]
- 24.Frohlich ED, Apstein C, Chobanian AV, Devereux RB, Dustan HP, Dzau V, Fauad-Tarazi F, Horan MJ, Marcus M, Massie B, Pfeffer MA, Re RN, Roccella EJ, Savage D, Shub C. 1992. The heart in hypertension. N Engl J Med 327:998–1008. doi: 10.1056/NEJM199210013271406. [DOI] [PubMed] [Google Scholar]
- 25.Sambandam N, Lopaschuk GD, Brownsey RW, Allard MF. 2002. Energy metabolism in the hypertrophied heart. Heart Fail Rev 7:161–173. doi: 10.1023/A:1015380609464. [DOI] [PubMed] [Google Scholar]
- 26.Tappia PS, Singal T. 2008. Phospholipid-mediated signaling and heart disease. Subcell Biochem 49:299–324. doi: 10.1007/978-1-4020-8831-5_11. [DOI] [PubMed] [Google Scholar]
- 27.Abel ED, O'Shea KM, Ramasamy R. 2012. Insulin resistance: metabolic mechanisms and consequences in the heart. Arterioscler Thromb Vasc Biol 32:2068–2076. doi: 10.1161/ATVBAHA.111.241984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sharma S, Adrogue JV, Golfman L, Uray I, Lemm J, Youker K, Noon GP, Frazier OH, Taegtmeyer H. 2004. Intramyocardial lipid accumulation in the failing human heart resembles the lipotoxic rat heart. FASEB J 18:1692–1700. doi: 10.1096/fj.04-2263com. [DOI] [PubMed] [Google Scholar]
- 29.Ruberg FL. 2007. Myocardial lipid accumulation in the diabetic heart. Circulation 116:1110–1112. doi: 10.1161/CIRCULATIONAHA.107.721860. [DOI] [PubMed] [Google Scholar]
- 30.Kienesberger PC, Pulinilkunnil T, Nagendran J, Dyck JR. 2013. Myocardial triacylglycerol metabolism. J Mol Cell Cardiol 55:101–110. doi: 10.1016/j.yjmcc.2012.06.018. [DOI] [PubMed] [Google Scholar]
- 31.Febbraio M, Abumrad NA, Hajjar DP, Sharma K, Cheng W, Pearce SF, Silverstein RL. 1999. A null mutation in murine CD36 reveals an important role in fatty acid and lipoprotein metabolism. J Biol Chem 274:19055–19062. doi: 10.1074/jbc.274.27.19055. [DOI] [PubMed] [Google Scholar]
- 32.Pietka TA, Sulkin MS, Kuda O, Wang W, Zhou D, Yamada KA, Yang K, Su X, Gross RW, Nerbonne JM, Efimov IR, Abumrad NA. 2012. CD36 protein influences myocardial Ca2+ homeostasis and phospholipid metabolism: conduction anomalies in CD36-deficient mice during fasting. J Biol Chem 287:38901–38912. doi: 10.1074/jbc.M112.413609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tanaka T, Sohmiya K, Kawamura K. 1997. Is CD36 deficiency an etiology of hereditary hypertrophic cardiomyopathy? J Mol Cell Cardiol 29:121–127. doi: 10.1006/jmcc.1996.0257. [DOI] [PubMed] [Google Scholar]
- 34.Slater-Jefferies JL, Hoile SP, Lillycrop KA, Townsend PA, Hanson MA, Burdge GC. 2010. Effect of sex and dietary fat intake on the fatty acid composition of phospholipids and triacylglycerol in rat heart. Prostaglandins Leukot Essent Fatty Acids 83(4–6):219–223. doi: 10.1016/j.plefa.2010.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Crowe FL, Skeaff CM, Green TJ, Gray AR. 2008. Serum n-3 long-chain PUFA differ by sex and age in a population-based survey of New Zealand adolescents and adults. Br J Nutr 99:168–174. doi: 10.1017/S000711450779387X. [DOI] [PubMed] [Google Scholar]
- 36.Burdge GC, Slater-Jefferies JL, Grant RA, Chung WS, West AL, Lillycrop KA. 2008. Sex, but not maternal protein or folic acid intake, determines the fatty acid composition of hepatic phospholipids, but not of triacylglycerol, in adult rats. Prostaglandins Leukot Essent Fatty Acids 78:73–79. doi: 10.1016/j.plefa.2007.10.028. [DOI] [PubMed] [Google Scholar]
- 37.Decsi T, Kennedy K. 2011. Sex-specific differences in essential fatty acid metabolism. Am J Clin Nutr 94(6 Suppl):1914S–1919S. doi: 10.3945/ajcn.110.000893. [DOI] [PubMed] [Google Scholar]
- 38.Burdge GC, Calder PC. 2006. Dietary alpha-linolenic acid and health-related outcomes: a metabolic perspective. Nutr Res Rev 19:26–52. doi: 10.1079/NRR2005113. [DOI] [PubMed] [Google Scholar]
- 39.Jones TH. 2010. Testosterone deficiency: a risk factor for cardiovascular disease? Trends Endocrinol Metab 21:496–503. doi: 10.1016/j.tem.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 40.Kaushik M, Sontineni SP, Hunter C. 2010. Cardiovascular disease and androgens: a review. Int J Cardiol 142:8–14. doi: 10.1016/j.ijcard.2009.10.033. [DOI] [PubMed] [Google Scholar]
- 41.Traish AM, Kypreos KE. 2011. Testosterone and cardiovascular disease: an old idea with modern clinical implications. Atherosclerosis 214:244–248. doi: 10.1016/j.atherosclerosis.2010.08.078. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.