

Abstract
Scaffold attachment factor A (SAF-A), also called heterogenous nuclear ribonuclear protein U (hnRNP-U), is phosphorylated on serine 59 by the DNA-dependent protein kinase (DNA-PK) in response to DNA damage. Since SAF-A, DNA-PK catalytic subunit (DNA-PKcs), and protein phosphatase 6 (PP6), which interacts with DNA-PKcs, have all been shown to have roles in mitosis, we asked whether DNA-PKcs phosphorylates SAF-A in mitosis. We show that SAF-A is phosphorylated on serine 59 in mitosis, that phosphorylation requires polo-like kinase 1 (PLK1) rather than DNA-PKcs, that SAF-A interacts with PLK1 in nocodazole-treated cells, and that serine 59 is dephosphorylated by protein phosphatase 2A (PP2A) in mitosis. Moreover, cells expressing SAF-A in which serine 59 is mutated to alanine have multiple characteristics of aberrant mitoses, including misaligned chromosomes, lagging chromosomes, polylobed nuclei, and delayed passage through mitosis. Our findings identify serine 59 of SAF-A as a new target of both PLK1 and PP2A in mitosis and reveal that both phosphorylation and dephosphorylation of SAF-A serine 59 by PLK1 and PP2A, respectively, are required for accurate and timely exit from mitosis.
INTRODUCTION
Accurate chromosome segregation during mitosis is vital for maintaining genomic stability. Critical to mitosis is the precise attachment of mitotic chromosomes to microtubule spindles. Failure of chromosome-kinetochore attachment leads to activation of the spindle assembly checkpoint (SAC), which prevents the anaphase-promoting complex/cyclosome (APC/C) from degrading cyclin B1 and securin, thus preventing progression from metaphase to anaphase and delaying exit from mitosis (1–3). Also critical to faithful mitosis are the coordinated phosphorylation and dephosphorylation of a host of mitotic proteins (3–8). Consequently, mitotic protein kinases such as polo-like kinase 1 (PLK1), Aurora A (AurA), Aurora B (AurB), and Mps1 have attracted attention as potential anticancer drug targets, and inhibitors of mitotic protein kinases are currently being evaluated in clinical trials (9, 10).
Our lab has a long-standing interest in the role of the DNA-dependent protein kinase catalytic subunit (DNA-PKcs) in DNA double-strand break repair (11, 12). DNA-PK phosphorylates multiple proteins in vitro, and in vivo, DNA-PKcs autophosphorylation is important for regulating nonhomologous end joining and DNA double-strand break repair pathway choice (reviewed in reference 11, 12, and 13). Recently, we and others showed that DNA-PKcs also plays an important role in mitosis (14–18). DNA-PKcs localizes to mitotic spindles, and autophosphorylated forms of DNA-PKcs localize to centrosomes and the mitotic spindle as well as the midbody during cytokinesis. Moreover, small interfering RNA (siRNA) depletion of DNA-PKcs or inhibition of its protein kinase activity with highly selective inhibitors such as NU7441 led to increased misaligned mitotic chromosomes and lagging chromosomes (14–17). Furthermore, DNA-PKcs interacts with PLK1 in mitosis (14, 18), and DNA-PKcs is phosphorylated on serine 3205 (S3205) by PLK1 in mitosis (14). In addition, DNA-PKcs interacts with protein phosphatase 6 (PP6) in both interphase and mitosis (14, 19), and PP6 dephosphorylates T288 of Aurora A kinase, regulating its activity in mitosis (20). These observations prompted us to search for potential DNA-PKcs substrates in mitosis.
One of the few proteins (apart from DNA-PKcs itself) known to be phosphorylated in a DNA-PK-dependent manner in response to DNA damage in vivo is scaffold attachment factor A (SAF-A), also known as heterogenous nuclear ribonucleoprotein U (hnRNP-U) (21, 22). DNA damage-induced, DNA-PK-dependent SAF-A phosphorylation occurs on serine 59 (S59) (21, 22); however, the function of SAF-A S59 phosphorylation has not been addressed. SAF-A belongs to a family of ubiquitously expressed nuclear ribonucleoproteins and is involved in multiple cellular processes, including RNA splicing, mRNA transport, and mRNA turnover as well as transcription and protein translation (23). Recently, SAF-A was shown to localize to mitotic spindles, the spindle midzone, and cytoplasmic bridges. Moreover, siRNA depletion of SAF-A induced mitotic delay and defects in chromosome alignment and spindle assembly, indicating new roles in mitosis (24). SAF-A was also shown to interact with Aurora A and TPX2 (24), and proteomics studies identified SAF-A as a component of mitotic spindles (25, 26). Moreover, high-throughput mass spectrometry screens showed that SAF-A S59 is highly phosphorylated (85% occupancy) in mitosis (5, 8). These observations prompted us to ask whether SAF-A S59 is phosphorylated by DNA-PKcs in mitosis. We generated a phospho-specific antibody to SAF-A S59 and showed that, in keeping with high-throughput phosphoproteomics studies, SAF-A S59 is highly phosphorylated in nocodazole-treated mitotic cells. By immunofluorescence, we show that SAF-A phosphorylated on S59 localizes to centrosomes during prophase and metaphase, to mitotic spindles in anaphase, and to the midbody during cytokinesis. Our results also reveal that SAF-A is phosphorylated on S59 by PLK1 and dephosphorylated by protein phosphatase 2A (PP2A) in mitosis. Importantly, ablation of SAF-A S59 phosphorylation by mutation of S59 to a nonphosphorylatable amino acid (alanine) caused delayed passage through mitosis and resulted in misalignment of metaphase chromosomes, as well as in a high percentage of polylobed daughter cells. Moreover, incubation of cells expressing nonphosphorylatable SAF-A (SAF-A S59A) with either a microtubule poison that inhibits microtubule polymerization (nocodazole) or a clinically relevant antimitotic agent that stabilizes microtubules (paclitaxel [originally named taxol]) (27, 28) resulted in enhanced levels of APC/C targets securin and cyclin B1, suggesting that PLK1-dependent phosphorylation of SAF-A on S59 is required for progression from metaphase to anaphase and, consequently, mitotic exit. Mutation of SAF-A S59 to glutamic acid to mimic constitutive phosphorylation caused abnormal alignment of mitotic chromosomes and increased lagging chromosomes, as well as a slightly shorter time to traverse mitosis. Together, our studies identify SAF-A as a new target of PLK1 and PP2A in mitosis and reveal that PLK1-dependent phosphorylation of SAF-A is required for accurate and timely passage through mitosis.
MATERIALS AND METHODS
Reagents and antibodies.
Microcystin-LR, bovine serum albumin (BSA), phenylmethylsulfonyl fluoride (PMSF), Tris base, EGTA, leupeptin, and pepstatin were purchased from Sigma-Aldrich. Inhibitors to DNA-PK (NU7441), ATM (KU55933), PLK1 (BI2536), Aurora A (Aurora A inhibitor 1), Aurora B (hesperadin), and cyclin-dependent kinse 1 (CDK1; RO3366) were from Selleck Chemicals. Antibodies to PP6c, and PP4c were purchased from Bethyl Laboratories. The antibody to Aurora A phospho-T288 was from Cell Signaling. Antibodies to PLK1, histone H3, securin, DNA-PKcs phospho-S2056, and Ku80 were from Abcam. The antibody to a fragment of recombinant DNA-PKcs (DPK1) was raised in-house and has been described previously (19). The phospho-specific antibody to serine 10 of histone H3 was from Upstate Biotechnologies, and the antibody to TPX2 was from Novus. The antibody to cyclin B1 was from Santa Cruz. The phospho-specific antibody to T210 of PLK1 and the antibody to PP2A catalytic subunit (PP2Ac) were from BD Pharmingen, and the antibody to Aurora A (mouse) was from Serotec.
For total SAF-A, either a rabbit polyclonal antibody (Abcam number 20666) raised to the C-terminal region of hnRNP-U or a mouse monoclonal antibody (Abcam number 10297, monoclonal antibody 3G6) was used. Both antibodies recognized a major band running at approximately 125 kDa on SDS-PAGE. In some experiments, in particular when larger amounts of SAF-A were loaded onto the gel (for example, in immunoprecipitation experiments), an additional smaller band that migrated at approximately 110 kDa was observed (see Fig. 3 and 4). We suspect that the smaller, ∼110-kDa band is either an alternatively spliced form of SAF-A or a proteolytic degradation product. We also generated a phospho-specific antibody to SAF-A S59 in rabbits against the peptide EPGNG(pS)LDLGGC by GL Biochem (Shanghai) Ltd. The residue in parentheses corresponds to the phosphorylated serine. The C-terminal cysteine was added for coupling. This rabbit phospho-specific antibody was used in immunoblotting experiments. For immunofluorescence experiments, a mouse monoclonal antibody to SAF-A phospho-S59 (a kind gift from Patrick Calsou and Bernard Salles, Université de Toulouse, France) was used. This antibody was manufactured by Biotem (Le Rivier d'Apprieu, France) by using phosphopeptide EPGNGS(Ph)LDLGGDC as an antigen. Representative Western blots with molecular weight markers of whole-cell extracts from mitotic cells are provided in the appendix in the supplemental material.
FIG 3.
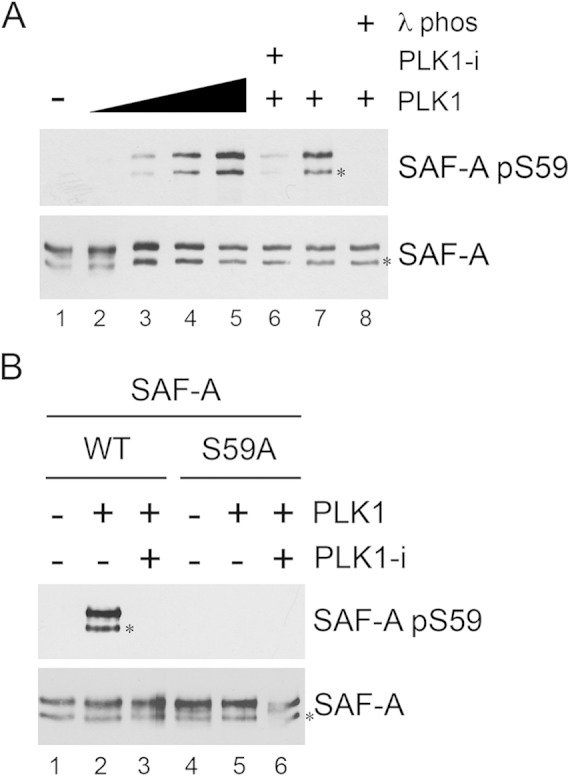
PLK1 phosphorylates SAF-A in vitro. (A) FLAG-tagged SAF-A WT was immunoprecipitated from asynchronously growing MRC5-CV cells as described in Materials and Methods and incubated under kinase assay conditions alone (lane 1) or with 0.1, 0.5, 1, or 2 μg of purified PLK1 (lanes 2 to 5, respectively). Lanes 6 to 8 contained 2 μg purified PLK1. In lane 6, the PLK1 inhibitor BI2536 was added to reaction mixtures (final concentration, 100 nM) prior to addition of PLK1. All reactions were stopped by addition of SDS sample buffer, and samples were run on SDS-PAGE and analyzed by Western blotting using the rabbit phospho-specific antibody to S59 and a rabbit polyclonal antibody to total SAF-A (Abcam number 20666). The band corresponding to full-length SAF-A runs at ∼125 kDa. The faster-migrating band, running at ∼110 kDa, indicated by the asterisk, likely represents either an alternatively spliced or a proteolytically cleaved form of SAF-A. In lanes 7 and 8, samples were incubated with lambda phosphatase buffer (lane 7) or lambda phosphatase (200 units) (lane 8) for 10 min prior to addition of SDS sample buffer and SDS-PAGE. (B) SAF-A WT or SAF-A S59A was immunoprecipitated from asynchronously growing MRC5-SV cells and incubated with purified PLK1 as described in Materials and Methods. Reactions were analyzed by SDS-PAGE followed by Western blotting and probed for SAF-A S59 phosphorylation and total SAF-A as indicated for panel A. The two bands cross-reacting with the SAF-A antibody are as in panel A.
FIG 4.
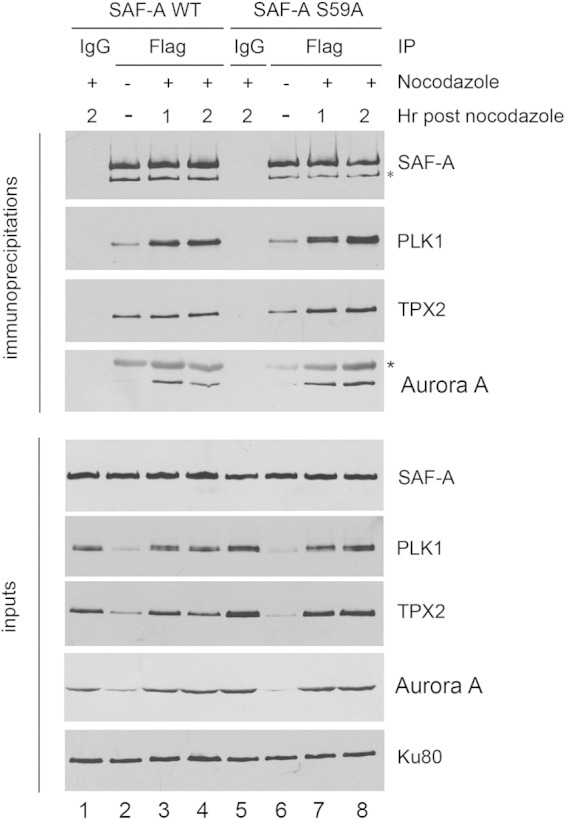
SAF-A interacts with PLK1, and the interaction does not require S59 phosphorylation. MRC5-SV cells expressing either FLAG-tagged SAF-A WT (lanes 1 to 4) or SAF-A S59A (lanes 5 to 8) were grown in the presence of nocodazole 40 ng/ml (16 h) (lanes 1, 3, 4, 5, 7, and 8) or under asynchronous conditions (lanes 2 and 6). The nocodazole was washed away, and cells were placed in nocodazole-free medium and harvested after either 1 h (lanes 3 and 7) or 2 h (lanes 1, 4, 5, and 8). SAF-A was immunoprecipitated using anti-FLAG beads, and samples were washed, run on SDS-PAGE, and analyzed by immunoblotting using the antibodies indicated. Samples in lanes 1 and 5 were from immunoprecipitates using an equivalent amount of IgG and protein G-Sepharose beads as a control. The lower panels show Western blots for 50 μg of protein for each immunoprecipitate. For immunoblotting of immunoprecipitates, a rabbit antibody to SAF-A was used. For inputs, a mouse monoclonal antibody to SAF-A was used. The two bands cross-reacting with the SAF-A antibody are as in Fig. 3A. The asterisk in the immunoblot for Aurora A indicates a cross-reacting band, likely immunoglobulin.
Cell culture.
HeLa cells were maintained at 37°C under a humidified atmosphere of 5% CO2 in Dulbecco's modified Eagle medium (DMEM) (Invitrogen) supplemented with 5% (vol/vol) fetal bovine serum (FBS) (HyClone), 50 U/ml penicillin, and 50 μg/ml streptomycin. MCF-7 cells were grown in the same medium as HeLa cells but with 10% serum. MRC5-SV cells in which endogenous SAF-A had been depleted by short hairpin RNA (shRNA) and replaced with shRNA-resistant FLAG-tagged wild type SAF-A (SAF-A WT), SAF-A with a mutation of S59 to alanine (S59A, phosphoablation), or SAF-A with a mutation of S59 to glutamate (S59E, phosphomimic) were grown in DMEM supplemented with 10% (vol/vol) FBS (HyClone), 50 U/ml penicillin, 50 μg/ml streptomycin, and 0.1 mg/ml hygromycin as described previously (21). Cells lines stably expressing FLAG-tagged SAF-A at approximately the same expression level as endogenous SAF-A were selected for further study (21) (also see Fig. S1A in the supplemental material).
Preparation of cell extracts and immunoblotting.
Where indicated, cells were incubated in the presence of 40 ng/ml nocodazole for 16 h and harvested by mitotic shake-off. Briefly, nocodazole-containing medium was removed and the cells washed with 2 ml phosphate-buffered saline (PBS) and then dislodged by shake-off. Cells were centrifuged at 2,000 × g for 5 min and then resuspended in fresh medium (without nocodazole) and returned to the CO2 incubator for 35 min or the times indicated. Asynchronously growing cells were harvested using trypsin-EDTA solution. Preparation of NETN lysates and immunoblotting were carried out as described previously (14, 19).
In some experiments, cells were incubated with nocodazole as described above for 15 h, protein kinase inhibitors were added at the concentrations indicated in the figure legends, and after 1 h, mitotic cells were collected by shake-off, as described above, then placed in nocodazole-free media containing the relevant protein kinase inhibitor and harvested after an additional incubation period of 35 min or as indicated in the figure legends.
In vitro phosphorylation of SAF-A by PLK1.
FLAG-tagged SAF-A WT and SAF-A S59A were immunoprecipitated from NETN lysates of asynchronously growing MRC5-SV cells using an anti-FLAG M2 affinity gel (Sigma). Immunoprecipitations were carried out as described previously (14, 19) but with the addition of two washes of NETN buffer without NP-40 prior to kinase assays. Immunoprecipitated SAF-A WT or SAF-A S59A from 200 μg whole-cell extract was incubated with the indicated amounts of purified, recombinant human PLK1 protein (a kind gift from James Hastie and Dario Alessi, University of Dundee) for 10 min at 30°C in kinase assay buffer (50 mM HEPES-NaOH, [pH 7.5], 75 mM KCl, 10 mM MgCl2, 0.2 mM EDTA, 1 mM dithiothreitol [DTT]) containing 0.25 mM ATP. Where indicated, the PLK1 inhibitor BI2536 was added (to 100 nM) prior to addition of ATP. Where indicated, SAF-A was dephosphorylated by incubation for 10 min at 30°C with either 200 units of lambda phosphatase (NEB) or an equivalent volume of buffer. Reactions were stopped by addition of SDS sample buffer, analyzed on SDS-polyacrylamide gels, and probed with antibodies to SAF-A phospho-S59 and total SAF-A as indicated. Because of cross-reactivity with IgG, when FLAG-tagged SAF-A was immunoprecipitated with mouse anti-FLAG antibody, rabbit antibodies to both total SAF-A (Abcam number 20666) and phospho-S59 (generated in-house) were used in Western blotting.
siRNA transfection.
SMARTpool siRNA oligonucleotides were purchased from Dharmacon (Lafayette, CO). HeLa cells were plated in antibiotic-free medium 24 h prior to transfection. Target siRNA or a scrambled siRNA control (100 nM each) was transfected using Oligofectamine (Invitrogen) for PP4 and PP6 or Dharmafect (Dharmacon) for PP2Ac (isoforms alpha and beta), according to the manufacturer's instructions. Twenty-four hours after transfection, fresh medium was added, and 80 h after transfection, cells were either left untreated or incubated with 40 ng/ml nocodazole for 16 h and harvested as described above.
Flow cytometry assays.
SAF-A WT, S59A, and S59E cells were either untreated or treated with 40 ng/ml nocodazole for 16 h, fixed, and prepared for flow cytometry analysis after propidium iodide staining (for G1, S, and G2 phases) or histone H3 phospho-S10 staining (for late G2/mitosis) as described previously (14, 19). Flow cytometry was carried out by the Cumming School of Medicine Flow Cytometry Facility as described previously (14, 19).
Immunofluorescence.
Cells were grown on poly-l-lysine-coated coverslips and analyzed by immunofluorescence using DAPI (4′,6′-diamidino-2-phenylindole), tubulin, or the indicated antibodies as described previously (14, 19). For counting misaligned chromosomes, DAPI-stained metaphase cells with abnormal tubulin staining structures were identified visually and scored from populations of at least 200 cells. The percentage of cells in which metaphase chromosomes were not correctly aligned on the metaphase plate were scored as a percentage of total mitotic cells.
Live-cell imaging.
Time-lapse imaging was carried out using a wide-field restoration deconvolution-based fluorescence microscope (DeltaVision Core; GE Healthcare) equipped with a three-dimensional motorized stage, temperature- and gas-controlled environmental chamber, and a xenon light source. Z-stacks of 5- by 1-μm images were collected using a 40×/1.35 NA Plan-Apochromat oil objective and the appropriate filter set and recorded with a CoolSNAP charge-coupled device (CCD) camera (Roper Scientific). The microscope was controlled by SoftWorX acquisition and deconvolution software (Applied Precision). DNA was stained by incubating the cells for 20 min in medium containing 0.25 μg/ml Hoechst no. 33342 (Sigma-Aldrich) and the medium replaced with phenol red-free DMEM prior to imaging.
Densitometry.
Densitometric analysis of bands was performed using ImageQuant software (GE Healthcare).
Statistical analysis.
P values were calculated using the Student t test. P values of less than 0.05 were considered statistically significant.
RESULTS
SAF-A is phosphorylated on serine 59 in mitosis.
SAF-A S59 in the sequence GNGSLDLGG (Fig. 1A and B) has been reported to be phosphorylated in a DNA-PK-dependent manner in response to DNA-damaging agents in human cells (21, 22). Serine 59 is conserved over several vertebrate species (Fig. 1B), but the functional consequences of SAF-A S59 phosphorylation are not known. To determine the function of SAF-A S59 phosphorylation, we used MRC5-SV, a simian virus 40 (SV40)-transformed human fetal lung cell line, in which endogenous SAF-A had been replaced with either FLAG-tagged SAF-A (SAF-A WT), FLAG-tagged SAF-A with a mutation of S59 to alanine (SAF-A S59A) for phosphoablation, or FLAG-tagged SAF-A in which S59 had been replaced with glutamic acid (SAF-A S59E) as a phosphomimic (21). SAF-A was expressed at approximately the same level as endogenous SAF-A in all three cell lines (21) (see Fig. S1A in the supplemental material), and the three cells lines had approximately equal growth rates under asynchronous conditions (see Fig. S1B in the supplemental material).
FIG 1.
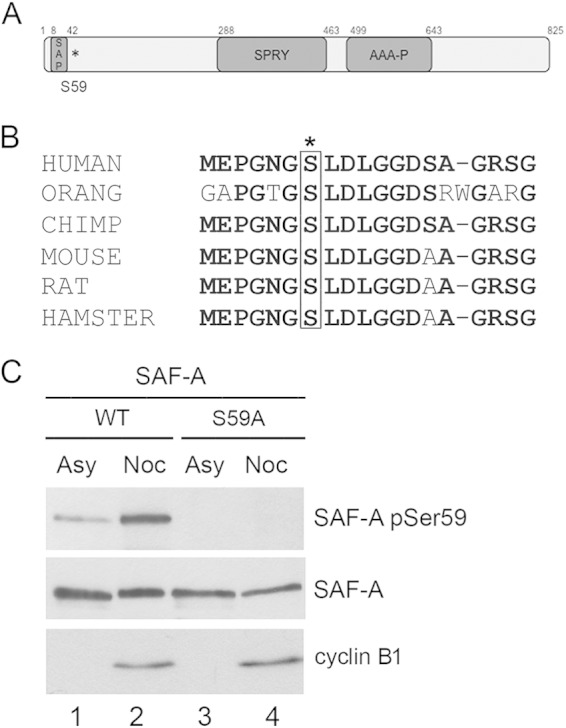
SAF-A is phosphorylated on serine 59 in mitosis. (A) Schematic showing domain structure of SAF-A and location of the S59 phosphorylation site (indicated by the asterisk). The locations of the conserved domains were obtained from the NCBI database (accession number Q00839). The SAF-A/B, Acinus, and PIAS (SAP) domain is a putative chromatin-binding domain. The function of the SPRY domain is unknown. The AAA domain contains a P-loop domain but no Walker B motif, and SAF-A has no known catalytic activity. (B) Alignment of the amino acid sequence of human SAF-A/hnRNP-U surrounding serine 59 with those of other vertebrate species. Conserved residues are in bold. Serine 59 is boxed and indicated by an asterisk. The human sequence shown starts at residue 53 and extends to residue 71. The multiple-sequence alignment was performed using Clustal-omega. The accession numbers for the sequences used were as follows: human, Q00839.6; Pongo abelii (orang), XP_009244899.1; Pan troglodytes (chimpanzee), NP_001267207; Mus musculus (mouse), Q8VEK3; Rattus norvegicus (Norwegian rat), NP_476480.2; and Mesocricetus auratus (hamster), XP_005078211. (C) Specificity of the in-house-generated rabbit phospho-specific antibody to SAF-A S59. Asynchronously growing MRC5-SV cells expressing FLAG-tagged wild-type SAF-A or SAF-A S59A were left untreated (Asy) or treated with 40 ng/ml nocodazole for 16 h (Noc). Nocodazole-treated cells were harvested by mitotic shake-off, whereas cells grown asynchronously were harvested with trypsin-EDTA. Cells were lysed using NETN lysis buffer containing 1% NP-40 with protease and protein phosphatase inhibitors. Fifty micrograms of whole-cell extract was run on an 8% acrylamide SDS gel and transferred to nitrocellulose, and blots were probed using a rabbit phospho-specific antibody to SAF-A S59 and antibodies to total SAF-A and cyclin B1 as indicated.
To confirm results from high-throughput mass spectrometry screens (5, 8), we immunoprecipitated SAF-A from nocodazole-treated cells and analyzed S59 phosphorylation by mass spectrometry. MRC5-SV cells expressing FLAG-tagged SAF-A WT (21) were incubated with nocodazole (40 ng/ml) for 16 h, harvested by mitotic shake-off, and, after incubation for 35 min in fresh medium (in the absence of nocodazole), lysed in NETN buffer as described in Materials and Methods and previously (14, 19). Asynchronously growing cells were harvested by treatment with trypsin-EDTA followed by lysis in NETN buffer as described previously (14, 19). FLAG-tagged SAF-A was immunoprecipitated from NETN lysates as described previously (14, 19), and the band corresponding to SAF-A was excised from colloidal Coomassie-stained SDS-polyacrylamide gels and analyzed by mass spectrometry as described previously (29). A peptide containing phosphorylated serine 59 (S59) was greatly enhanced in digests from nocodazole-treated SAF-A samples (see Fig. S2 in the supplemental material), confirming that S59 is phosphorylated in nocodazole-treated, mitotic MRC5-SV cells expressing SAF-A.
To further characterize the phosphorylation of SAF-A during mitosis, we generated a phospho-specific antibody in rabbits to SAF-A phosphorylated on S59 (see Materials and Methods for details). To show specificity of this antibody, MRC5-SV cells stably expressing SAF-A WT or FLAG-tagged SAF-A S59A were either untreated or treated with nocodazole as described above. Cells were either harvested by incubation with trypsin-EDTA (when grown under asynchronous conditions) or collected by mitotic shake-off and harvested 35 min after removal of nocodazole. NETN extracts were generated and run on 8% acrylamide-SDS gels and probed with the phospho-specific antibody to SAF-A S59 and antibodies to total SAF-A and cyclin B1 (as an indication of mitosis) (Fig. 1C). While some SAF-A S59 phosphorylation was detected in asynchronously growing cells (Asy), S59 phosphorylation was significantly enhanced in extracts from nocodazole-treated mitotic cells (Noc). The low level of S59 phosphorylation observed in asynchronously growing cells is likely due to the low percentage of cells in late G2 and mitosis under normal growing conditions. Importantly, no phosphorylation was detected in cells expressing FLAG-tagged SAF-A S59A, confirming specificity of the phospho-specific antibody (Fig. 1C).
To confirm mitotic phosphorylation of endogenous SAF-A (as opposed to phosphorylation of FLAG-tagged SAF-A), we also examined SAF-A S59 phosphorylation in HeLa cells (Fig. 2A, lane 2) and MCF7 cells (see Fig. S3 in the supplemental material) that had either been untreated or incubated with nocodazole for 16 h and harvested 35 min after release from nocodazole. The results show that endogenous SAF-A is phosphorylated on S59 in nocodazole-treated HeLa cells (Fig. 2A, lane 2) and MCF7 cells (see Fig. S3, lane 2, in the supplemental material).
FIG 2.

SAF-A S59 phosphorylation in nocodazole-treated cells requires PLK1 and/or CDK1. (A) Asynchronously growing HeLa cells were either left untreated (Asy) or treated with 40 ng/ml nocodazole for 16 h as for Fig. 1. One hour prior to mitotic shake-off, cells were untreated or treated with 8 μM DNA-PK inhibitor NU7441 or 5 μM ATM inhibitor KU55933. Cells were harvested by mitotic shake-off and left to recover for 35 min in fresh medium (without nocodazole) but in the presence of the protein kinase inhibitors as indicated. Cells were lysed using NETN lysis buffer containing 1% NP-40 with protease and protein phosphatase inhibitors. Fifty micrograms of whole-cell extract was run on an 8% gel and transferred to nitrocellulose, and blots were probed using the in-house-generated rabbit phospho-specific SAF-A S59 antibody and then with antibodies to SAF-A (Abcam number 10297 mouse), PLK1, PLK1-pT210, Aurora A and Aurora A-pT288, DNA-PKcs, DNA-PKcs-pS2056, Ku80 (loading control), and cyclin B1 as indicated. (B) Experiments were carried out as for panel A, but cells were incubated with 100 nM PLK1 inhibitor (BI2536), 100 nM Aurora A inhibitor 1 (AurA-i), or 100 nM Aurora B inhibitor (hesperadin) as indicated. (C) As for panels A and B, but cells were treated with the PLK1 inhibitor BI2536 at 100 nM (lane 3), the CDK1 inhibitor RO3366 at 20 μM (lane 4), or the PLK1 inhibitor BI2536 at 100 nM plus RO3366 at 20 μM (lane 5) as indicated.
SAF-A serine 59 is phosphorylated by PLK1 in mitosis.
To identify the protein kinase or kinases responsible for phosphorylating SAF-A on S59 during mitosis, HeLa cells were either untreated or treated with nocodazole as described above, but where indicated, protein kinase inhibitors were added to the medium at 15 h after addition of nocodazole, and cells were harvested by mitotic shake-off after an additional 1 h of incubation with nocodazole and protein kinase inhibitor. After shake-off, cells were allowed to recover in medium in the presence of protein kinase inhibitor but without nocodazole for 35 min, and then NETN lysates were generated and analyzed by immunoblotting as described above. Since DNA-PKcs phosphorylates SAF-A after DNA damage (21, 22) and DNA-PKcs' protein kinase activity is required for alignment of mitotic chromosomes and accurate mitoses (14, 15), we first asked whether inhibition of DNA-PK or the related protein kinase ataxia-telangiectasia mutated (ATM) reduced SAF-A phosphorylation in mitosis by incubating the cells with NU7441 (30) or KU55933 (31) to inhibit DNA-PK or ATM, respectively. Consistent with previous reports, NU7441 inhibited mitotic phosphorylation of DNA-PKcs on S2056 (14–17), but neither inhibition of DNA-PKcs nor ATM affected SAF-A S59 phosphorylation in nocodazole-treated mitotic HeLa cells (Fig. 2A, lanes 3 and 4).
We next incubated the mitotic cells with selective inhibitors of mitotic protein kinases PLK1 (BI2536), Aurora A (Aurora A inhibitor 1), or Aurora B (hesperadin). Inhibition of PLK1 reduced phosphorylation of SAF-A S59 by over 80% (82.3% ± 1.5%, n = 4), indicating that PLK1 is the major kinase responsible for phosphorylating SAF-A on S59 in mitosis (Fig. 2B, lane 3). Consistent with our previous report, BI2536 also inhibited Aurora A-T288 phosphorylation (14), consistent with PLK1 acting upstream of Aurora A (32). Inhibition of either Aurora A or Aurora B kinase activity also resulted in an approximately 50% decrease in SAF-A S59 phosphorylation; however, both inhibitors also reduced PLK1-T210 phosphorylation (Fig. 2B, lanes 4 and 5), suggesting that these protein kinases could be acting on SAF-A indirectly, by modifying PLK1 activity (Fig. 2B). Inhibition of PLK1 using BI2536 also reduced SAF-A S59 phosphorylation in MCF7 cells (see Fig. S3, lane 3, in the supplemental material), confirming a role for PLK1 in SAF-A S59 phosphorylation. As in HeLa cells, BI2536 did not completely eliminate SAF-A S59 phosphorylation in MCF7 cells, suggesting involvement of additional protein kinases (see Fig. S3 in the supplemental material).
Many PLK1 substrates require prior phosphorylation on a separate “priming” phosphorylation site by cyclin-dependent protein kinase, CDK1, prior to phosphorylation by PLK1 (33). CDK1-dependent phosphorylation of a serine or threonine that is followed by a proline in the amino acid sequence S-S/T-P, where the serine (S) or threonine (T) that is targeted for phosphorylation is shown in bold, creates a binding site for the C-terminal polo-binding domain of PLK1, targeting PLK1 to its substrate and facilitating phosphorylation of the substrate by PLK1 (33). To determine whether CDK1 was required for phosphorylation of SAF-A S59, we incubated HeLa cells with nocodazole and then added either BI2536 to inhibit PLK1 (Fig. 2C, lane 3), the CDK1 inhibitor RO3366 (34) (Fig. 2C, lane 4), or BI2536 plus RO3366 (Fig. 2C, lane 5). Extracts were processed and analyzed as described above. Incubation with either the PLK1 inhibitor BI2536 and/or the CDK1 inhibitor RO3366 significantly reduced mitotic SAF-A S59 phosphorylation (Fig. 2C), supporting a model whereby CDK1 phosphorylates SAF-A and/or PLK1 to create a polo-box-binding motif that serves to activate PLK1 and/or facilitate PLK1-dependent phosphorylation of SAF-A on S59 in vivo.
PLK1 phosphorylates SAF-A on S59 in vitro.
To determine whether PLK1 directly phosphorylates SAF-A on S59, SAF-A WT was immunoprecipitated from asynchronously growing MRC5-SV cells expressing FLAG-tagged SAF-A WT using a FLAG antibody and incubated either without (Fig. 3A, lane 1) or with increasing amounts of purified PLK1 protein (Fig. 3A, lanes 2 to 5). Samples were assayed under standard assay conditions and then run on SDS-polyacrylamide gels and immunoblotted for SAF-A phospho-S59 and total SAF-A as indicated (Fig. 3A). Where indicated, the PLK1 inhibitor BI2536 was added to reaction mixtures to inhibit PLK1 activity (Fig. 3A, lane 6). In lanes 7 and 8, after incubation with PLK1, samples were incubated with lambda phosphatase (Fig. 3A, lane 8) or buffer control (Fig. 3A, lane 7) prior to addition of SDS sample buffer to demonstrate phosphorylation specificity of the antibody. The results show that purified PLK1 phosphorylates SAF-A on S59 in vitro (Fig. 3). Given our results showing that both CDK1 and PLK1 are required for SAF-A S59 phosphorylation in vivo (Fig. 2C), we suspect that immunoprecipitated SAF-A may already be phosphorylated at the CDK1 priming site, prior to phosphorylation by PLK1. Alternatively, in vitro phosphorylation of SAF-A S59 by PLK1 may not require prior CDK1 phosphorylation. As an additional control for antibody specificity, in vitro assays were repeated but using SAF-A that had been immunoprecipitated from S59A expressing cells (Fig. 3B). No S59 phosphorylation was observed when PLK1 was incubated with SAF-A that had been immunoprecipitated from MRC5-SV cells expressing SAF-A S59A (Fig. 3B, lanes 4 to 6), again confirming specificity of the phosphoantibody.
In each experiment, immunoprecipitation of FLAG-tagged SAF-A with a mouse monoclonal antibody followed by immunoblotting with either a rabbit polyclonal antibody or a rabbit phosphospecific antibody to SAF-A S59 resulted in the detection of two bands on SDS-PAGE, an upper band migrating at approximately 125 kDa and a lower band at approximately 110 kDa (indicated by the asterisks in Fig. 3). Both bands cross-reacted with both the phospho-specific antibody to SAF-A pS59 and the rabbit polyclonal antibody to total SAF-A (Fig. 3A and B). The UniProt database (http://www.uniprot.org/uniprot/Q00839) reports that SAF-A/hnRNP-U exists in two splicing variants, a longer form of 825 amino acids and a shorter form of 806 amino acids that lacks residues 213 to 231. We suspect that both forms are immunoprecipitated by the anti-FLAG antibody. Alternatively, the smaller, ∼110-kDa band may represent a proteolytically cleaved form of SAF-A. Regardless of the nature of the two bands, our results clearly show that PLK1 phosphorylates SAF-A on S59 in vitro.
SAF-A interacts with PLK1 in mitosis.
Given that SAF-A is phosphorylated by PLK1 on S59 in mitosis (Fig. 2) and in vitro (Fig. 3), and SAF-A has been reported to interact with Aurora A and TPX2 in mitosis (24), we asked whether SAF-A interacts with PLK1 and whether this interaction is disrupted in cells in which S59 has been ablated. SAF-A WT and SAF-A S59A cells were grown under asynchronous conditions (Fig. 4, lanes 2 and 6), or treated with nocodazole (40 ng/ml for 16 h) and then placed in nocodazole-free medium and harvested after 1 or 2 h (Fig. 4, lanes 1, 3, 4, 5, 7, and 8). SAF-A was immunoprecipitated using a mouse anti-FLAG antibody, and immunoprecipitates were run on SDS-polyacrylamide gels and probed with rabbit antibodies to SAF-A, PLK1, TPX2, and Aurora A as indicated. In control samples, nocodazole-treated cells were harvested 2 h after release from nocodazole and immunoprecipitated with an equivalent amount of control IgG antibody (Fig. 4, lanes 1 and 5). Fifty micrograms of extract was also run on SDS-PAGE and probed with the antibodies as shown in the lower panels (Fig. 4, inputs). Our results show that SAF-A WT interacts with PLK1 and that this interaction is not ablated in cells expressing SAF-A S59A (Fig. 4). Our results also confirm a previous report that SAF-A interacts with TPX2 and Aurora A (24), and again this interaction was not disrupted by ablation of S59. As in Fig. 3, immunoprecipitation of FLAG-tagged SAF-A using a mouse monoclonal antibody to FLAG, followed by Western blotting with a rabbit polyclonal antibody to total SAF-A resulted in two bands of approximately 125 and 110 kDa. In contrast, straight Western blotting of samples (without immunoprecipitation by FLAG antibody) using the mouse monoclonal antibody to SAF-A resulted in a single band migrating at 125 kDa (Fig. 4, inputs).
Given that many PLK1 substrates are also phosphorylated by CDK1 to create a priming/binding site for the polo-box motif (PBM) of PLK1 (33), we also asked whether inhibition of PLK1 and/or CDK1 would affect the interaction of PLK1 with SAF-A. For these experiments, SAF-A WT cells were grown under asynchronous conditions or treated with nocodazole as described above. After 15 h, the PLK1 inhibitor BI2536 (100 nM) and/or the CDK1 inhibitor RO3366 (20 μM) was added to cells, and then the cells were harvested as described above and incubated in the absence or presence of kinase inhibitors and SAF-A was immunoprecipitated as described above (see Fig. S4 in the supplemental material). Incubation with neither the PLK1 inhibitor BI2536 alone nor the CDK1 inhibitor RO3366 alone disrupted the interaction of SAF-A with PLK1, TPX2, or Aurora A; however, addition of both PLK1 and CDK1 inhibitors caused a decrease in the amount of interacting PLK1 and Aurora A, and inhibition of CDK1 alone reduced the amount of Aurora A that interacted with SAF-A (see Fig. S4 in the supplemental material). However, we speculate that apparent loss of interactions in the presence of inhibitors of both PLK1 and CDK1 may in part reflect blocked entry into mitosis, as suggested by the low levels of total Aurora A and cyclin B1 observed under these condition (see Fig. S4, lane 6, inputs, in the supplemental material).
Phosphorylation of SAF-A S59 after DNA damage in nocodazole-treated mitotic cells is PLK1 dependent.
SAF-A S59 is phosphorylated in a DNA-PK-dependent manner after DNA damage (21, 22) (see Fig. S5A in the supplemental material). Since we show here that the same site is also phosphorylated by PLK1 in vitro and in a PLK1-dependent manner in mitosis, we asked whether DNA-PK or PLK1 would phosphorylate SAF-A if DNA damage was administered in mitosis. HeLa cells were either left untreated (see Fig. S5B, lanes 1 and 6, in the supplemental material) or treated with 40 ng/ml nocodazole for 15 h (Fig. S5B, lanes 2 to 5 and 7 to 10). One hour prior to mitotic shake-off, cells were treated with either 8 μM NU7441 (to inhibit DNA-PK) (lanes 3, 5, 8, and 10) or 100 nM BI2536 (to inhibit PLK1) (lanes 4, 5, 9, and 10). Cells were harvested by mitotic shake-off and either treated with 10 Gy irradiation (lanes 6 to 10) or not irradiated (lanes 1 to 5). All cells were left to recover in medium without nocodazole for 35 min, with inhibitors added where appropriate (lanes 3 to 5 and 8 to 10). NTEN lysates were prepared, 50 μg of lysate was run on SDS-polyacrylamide gels with various percentages of acrylamide, and immunoblots were probed with various antibodies as indicated in Fig. S5B. As expected, DNA-PKcs S2056 phosphorylation was DNA-PK dependent after nocodazole alone and also after nocodazole treatment followed by irradiation (see Fig. S5B, lanes 3, 5, 8, and 10). In contrast, SAF-A S59 phosphorylation was largely PLK1 dependent in mitosis (lane 4) and in nocodazole-treated cells that were subsequently irradiated (lane 9); however, the presence of residual phosphorylation after inhibition of both PLK1 and DNA-PKcs (see Fig. S5B, lane 10), suggests that other protein kinases may also contribute to SAF-A S59 phosphorylation after DNA damage in mitosis.
SAF-A phospho-S59 localizes to centrosomes, the midzone, and the midbody in mitosis.
To examine the localization of endogenous SAF-A S59 phosphorylation during mitosis, we performed immunostaining in asynchronously growing HeLa cells using a SAF-A S59 phospho-specific monoclonal antibody raised in mice (a kind gift from Patrick Calsou and Bernard Salles, Université de Toulouse) and costained for either TPX2, Aurora A phospho-T288, or Aurora B (Fig. 5A, B, and C, respectively). SAF-A phospho-S59 (green) localized to centrosomes in prophase and metaphase cells, to the midzone in anaphase, and to the midbody in cytokinesis (Fig. 5A, B, and C). SAF-A pS59 colocalized with TPX2 at metaphase spindles (Fig. 5A) and with Aurora A pT288 at the spindles in prophase, metaphase, and anaphase (Fig. 5B). Diffuse staining that partially overlapped with Aurora B staining at the midzone in anaphase was also observed (Fig. 5C). Incubation with a blocking peptide corresponding to the amino acid sequence surrounding S59 [EPGNG(pS)LDLGGC] blocked the signal from the SAF-A phospho-S59 antibody in immunofluorescence (see Fig. S6 in the supplemental material), and no significant staining was seen when cells expressing SAF-A S59A were probed with the SAF-A pS59 phospho-specific antibody (see Fig. S7 in the supplemental material), confirming antibody specificity. Moreover, incubation with the PLK1 inhibitor BI2536 abrogated SAF-A S59 phosphorylation, confirming the requirement of PLK1 for SAF-A S59 phosphorylation in vivo (see Fig. S8 in the supplemental material).
FIG 5.

SAF-A phosphorylated on S59 localizes to centrosomes in prophase and metaphase, to the midzone in anaphase, and to the midbody in cytokinesis. (A) HeLa cells were grown on poly-l-lysine-coated coverslips then stained with DAPI (blue), a mouse monoclonal antibody to SAF-A phospho-S59 (green), and an antibody to TPX2 (red) and processed for immunofluorescence as described in Materials and Methods. A merged view of a representative cell in interphase, prophase, metaphase, anaphase, and cytokinesis is shown in the lower panels. Size bars are shown in the lower left corner of each image and represent 10 μm. (B) As for panel A, but cells were stained for DAPI (blue), a mouse monoclonal antibody to SAF-A phospho-S59 (green), and an antibody to Aurora A phospho-T288 (red). (C) As for panel A, but cells were stained with DAPI (blue), a mouse monoclonal antibody to SAF-A phospho-S59 (green), and an antibody to Aurora B (red).
Either ablation of SAF-A Ser 59 or introduction of a phosphomimic induces defects in mitosis.
In order to investigate the functional significance of the mitotic, PLK1-dependent phosphorylation of SAF-A on S59, we compared alignment of metaphase chromosomes in MRC5-SV SAF-A WT cells with those in cells expressing SAF-A S59A (phosphoablation) or SAF-A with mutation of S59 to glutamate (S59E) (phosphomimic). In SAF-A WT cells, 76% of the cells at metaphase had properly aligned chromosomes with a properly formed bipolar spindle; however, cells expressing either SAF-A S59A or SAF-A S59E had a dramatically higher level of misaligned chromosomes with abnormal bipolar spindles (83% and 93% in SAF-A S59A and S59E, respectively), implying that either inability to phosphorylate SAF-A S59 (S59A) or constitutive phosphorylation of SAF-A (S59E) compromises accurate mitoses (Fig. 6).
FIG 6.
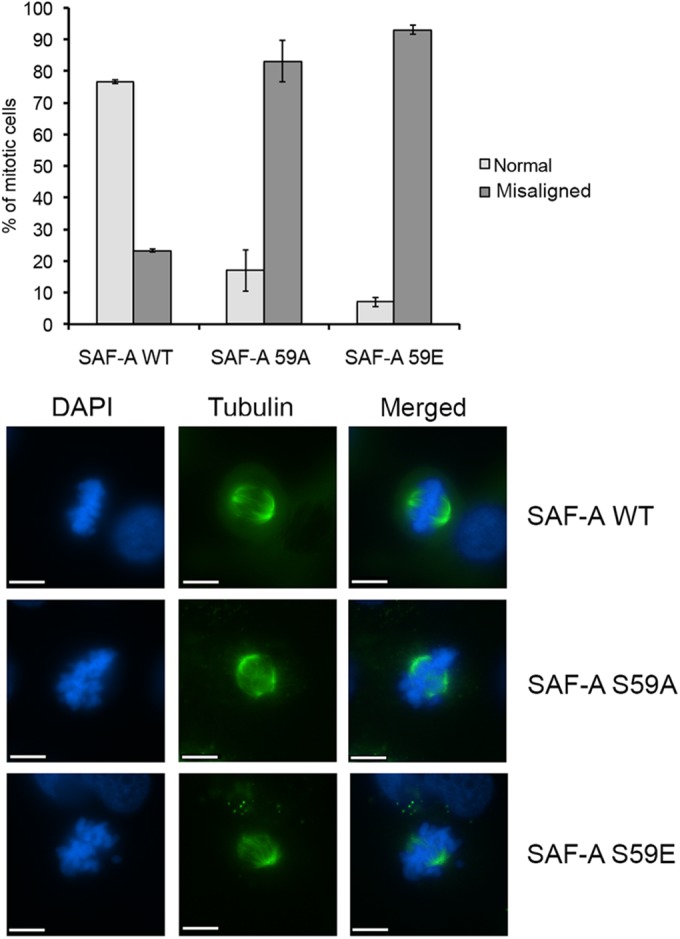
Phosphoablation or phosphomimicry at SAF-A 59A and 59E results in increased misaligned chromosomes. SAF-A WT, 59A, and 59E cells were stained with tubulin (green) and DAPI (blue) and scored for normal or misaligned chromosomes as described in Materials and Methods. The graph summarizes the average number of misaligned chromosomes in each of the three cell lines with standard deviation. Experiments were carried out 3 times, with at least 50 mitotic cells counted for each experiment. The differences in misaligned chromosomes between cells expressing SAF-A WT and SAF-A S59A and between cells expressing WT and S59E were considered highly significant, with P values of 0.0039 and 0.0002, respectively. Representative images of normal (SAF-A WT) and misaligned chromosomes (SAF-A S59A and S59E) are shown in the lower panels. Scale bars represent 10 μm.
To further explore the effects of SAF-A S59 phosphorylation on mitosis, we compared the cell cycle profile and the mitotic index (percentage of histone H3 pS10-positive cells) of SAF-A WT cells to those of sells expressing SAF-A S59A and S59E using fluorescence-activated cell sorting (FACS). In the absence of nocodazole, the percentages of cells in G1, S, and G2 were similar in the three cell lines examined (Fig. 7A). As expected, nocodazole resulted in an increase in the percentage of cells in G2 (by propidium iodide staining) in all cell lines compared to the same cell lines in the absence of nocodazole (compare Fig. 7B and A). There were no significant changes in the percentage of cells in G1 after nocodazole treatment (4.46% ± 1.95% in WT, 2.22% ± 0.79% in S59A [P = 0.112] and 4.91% ± 2.81% in S59E [P = 0.051]) (see Table S1 in the supplemental material). A small but significant increase in the proportion of cells in G2 in nocodazole-treated SAF-A S59A cells compared to SAF-A WT cells was observed (71.03% ± 5.00% in SAF-A WT cells compared to 77.51% ± 6.081% in SAF-A S59A cells; P = 0.025), and a trend toward a decreasing number of cells in G2 in SAF-A S59E cells compared to SAF-A S59A cells was observed (however, it was not statistically significant) (77.51% ± 6.081% in S59A cells compared to 65.36% ± 7.39% in SAF-A S59E cells; P = 0.059) (Fig. 7B; see Table S1 in the supplemental material). However, nocodazole-treated cells expressing the phosphomimic, SAF-A S59E, showed a significant decrease in H3 phospho-S10-positive cells (decreasing from 28.70% ± 1.83% in WT to 18.04% ± 2.90% in S59E; P = 0.0002) (Fig. 7C). We reasoned that this decrease in H3 pS10-positive/mitotic cells in SAF-A S59E cells might indicate that the cells were either stalled in early G2 or had slipped through to the subsequent G1 or S phase. However, the introduction of the SAF-A S59E mutation did not result in a detectable increase in the percentage of cells in either G1 phase (4.46% ± 1.95% in WT to 4.91% ± 2.81% in S59E; P = 0.812) or S phase (24.50% ± 3.24% in WT compared with 29.73% ± 6.87% in S59E; P = 0.103) by propidium iodide staining after nocodazole treatment (Fig. 7B). In addition, a trend toward a decrease in the percentage of cells in G2 was observed in cells expressing the S59E mutation but again was not statistically significant (71.03% ± 5.0% in WT compared to 65.36% ± 7.39% in S59E; P = 0.147) (see Table S1 in the supplemental material). We speculate that the S59E mutation may induce cell death or polyploidy or have other consequences that were not detected in our assays.
FIG 7.
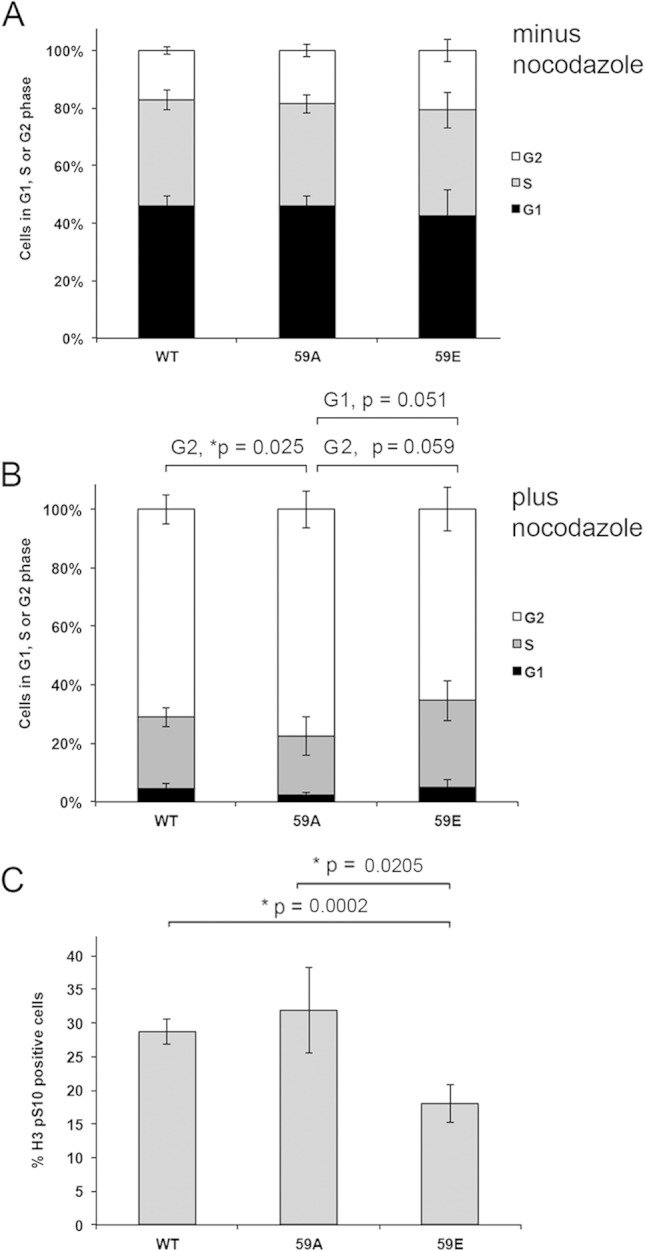
Nocodazole-treated cells expressing SAF-A S59E have a decreased mitotic index. (A) MRC5-SV cells expressing either FLAG-tagged SAF-A WT, SAF-A 59A, or SAF-A 59E were grown under asynchronous conditions, stained with propidium iodide, and analyzed for percentages of cells in G1, S, and G2 by flow cytometry. The results of five separate experiments were averaged, and the standard deviation is shown. Additional analysis of data is shown in Table S1 in the supplemental material. (B) MRC5-SV cells expressing either FLAG-tagged SAF-A WT, SAF-A 59A, or SAF-A 59E were incubated with nocodazole (16 h, 40 ng/ml), harvested using trypsin-EDTA, and then stained with propidium iodide and analyzed by FACS as for panel A. The results show the average from five biological replicates. Statistical significance was determined by Student's t test. P values of <0.05 were considered statistically significant. Additional analysis of data is shown in Table S1 in the supplemental material. (C) MRC5-SV cells expressing either FLAG-tagged SAF-A WT, SAF-A 59A, or SAF-A 59E were incubated with nocodazole and collected by trypsinization, as described for panel B. The percentage of cells in mitosis was measured by flow cytometry after histone H3 phospho-S10 staining. The percentage of mitotic cells shown represents the average from five biological replicates. Error bars represent the standard deviation. Statistical significance was determined using Student's t test. P values of less than 0.05 were considered statistically significant. Additional analysis of data is shown in Table S1 in the supplemental material.
To further investigate the functional significance of S59 phosphorylation, mitotic progression was directly visualized in MRC5-SV cells expressing SAF-A WT, SAF-A S59A, or SAF-A S59E in the absence of nocodazole. Live cells were stained with the cell-permeative DNA dye Hoechst 33342 and imaged every 10 min for 24 h, using minimal light exposure, to capture chromosome condensation and segregation. The average time from initial chromosome condensation (prophase) to mitotic exit in SAF-A WT cells was found to be 95 min (Fig. 8A and D). The majority of mitotic events that were observed were normal, with only a small fraction of cells displaying abnormal chromosome organization in the form of lagging chromosomes (Fig. 8E and F). In contrast, the period from prophase to mitotic exit in SAF-A S59A cells was significantly longer, averaging 137 min (Fig. 8B and D). This increase was primarily due to a prolonged prophase-to-anaphase transition (108 ± 6.8 min in S59A versus 60 ± 3.7 min in WT cells), which is suggestive of chromosome alignment defects. Consistent with this, the SAF-A 59A daughter cells demonstrated an increased prevalence of segregation defects (Fig. 8E and F; see Fig. S9 in the supplemental material), with lagging chromosomes observed in 50% of cells (17/34) and a large proportion (85%) containing polylobed nuclei (29/34) (Fig. 8E and F). SAF-A S59E cells showed a slightly faster mitotic progression than SAF-A WT cells (85 min in S59E versus 95 min in WT), governed primarily by a decreased prophase-to-anaphase transition (52 ± 1.7 min, versus 60 ± 3.7 min in WT cells). The SAF-A S59E daughter cells also showed defects in mitosis, with 71% containing lagging chromosomes (12/17) and 18% containing polylobed nuclei (3/17) (Fig. 8E and F). Videos of live-cell imaging results for cells expressing SAF-A WT, S59A, and S59E are provided in Fig. S9 in the supplemental material.
FIG 8.

Altered S59 phosphorylation impacts duration of mitosis and induces mitotic errors. (A to C) MRC5-SV cells stably expressing WT SAF-A (A), nonphosphorylatable mutant S59A SAF-A (B), or phosphomimic S59E SAF-A (C) were stained with Hoechst 33342 to visualize chromosomes and imaged over a 24-hour period to capture mitotic events. Elapsed time in minutes is shown in the bottom right corner of each panel. Arrows indicate lagging chromosomes. Arrowheads indicate polylobed daughter nuclei. Scale bars are 5 μm. Images are two-dimensional maximum-intensity projections of three-dimensional data sets, and intensities have been scaled so that all stages of the cell cycle are visible (hence, condensed chromosomes are saturated). (D) Graph summarizing the mitotic timing for all three stable cell lines. Data are mean ± standard error for n = 17 to 39. Asterisks indicate significant variance from the mean (P < 0.01). Statistical analyses were carried out using 2-tailed equal-variance t tests. (E) Percentage of total SAF-A WT, S59A, and S59E cells showing lagging chromosomes of polylobed nuclei. (F) Table summarizing the number of dividing cells that showed lagging chromosomes and/or polylobed nuclei, based on the total number of mitotic events captured for each cell line in 3 separate experiments. See Fig. S9 in the supplemental material for movies of live-cell imaging experiments.
Thus, the results from live-cell imaging show that preventing SAF-A S59 phosphorylation (S59A) significantly increases the time that cells require to progress through mitosis, particularly the prophase-to-anaphase transition, during which chromosome alignment is established prior to segregation. Conversely, blocking dephosphorylation of SAF-A (S59E phosphomimic mutation) slightly reduced the duration of mitosis, although this decrease was not statistically significant. In addition, the live-cell imaging results show that replacement of WT SAF-A with either S59A or S59E had significant effects on mitotic outcome, in terms of misaligned chromosomes and abnormal nuclear morphologies (Fig. 8; see Fig. S9 in the supplemental material). Together, these results suggest that regulated phosphorylation (i.e., both phosphorylation and dephosphorylation) of SAF-A on S59 is critical for accurate chromosome segregation and exit from mitosis.
Cyclin B1 and securin levels are elevated and prolonged in nocodazole- and paclitaxel-treated cells expressing SAF-A S59A.
We considered that results from in vivo imaging were consistent with ablation of S59 phosphorylation (S59A) activating the SAC to produce a more robust checkpoint (i.e., more cells in mitosis), while expression of the phosphomimic (S59E) caused a less efficient SAC (i.e., fewer cells in mitosis). If this was the case, we would expect that cells expressing SAF-A S59A would have reduced APC/C-mediated activity and thus elevated levels of cyclin B1 and securin, while cells expressing SAF-A S59E would show enhanced APC/C activity and lower levels of cyclin B1 and securin. To explore further the mechanism by which SAF-A phosphorylation regulates the timing of mitotic progression and to determine whether disruption of SAF-A phosphorylation has consequences that could be attributable to disruption of APC/C function, MRC5-SV cells expressing WT SAF-A, SAF-A S59A, or SAF-A S59E were treated with nocodazole (40 ng/ml) for 16 h to inhibit microtubule/kinetochore attachment, activate the SAC and arrest cells in mitosis. Mitotic cells were then harvested by shake-off and allowed to recover in fresh medium (without nocodazole) to allow release from mitosis. Cells were harvested either immediately (0 h) or 0.5, 1, 2, and 4 h after release from nocodazole and lysed using NTEN lysis buffer in the presence of protein phosphatase inhibitors and protease inhibitors. Fifty micrograms of each extract was run on gels of various percentages and probed for cyclin B1 and securin. Levels of cyclin B1 and securin were elevated in nocodazole-treated cells expressing SAF-A S59A compared to in cells expressing wild-type SAF-A. Conversely, cyclin B1 and securin levels were lower in cells expressing SAF-A S59E than in cells expressing WT SAF-A (Fig. 9). These results are consistent with results from live-cell imaging and suggest that blocking S59 phosphorylation (expression of S59A) prolongs mitosis by activating the SAC and/or inhibiting the APC/C, while expression of SAF-A S59E, to mimic constitutive and irreversible S59 phosphorylation, prevents activation of the SAC, allowing activation of the APC/C and degradation of cyclin B1 and securin.
FIG 9.

Inability to phosphorylate SAF-A on S59 in nocodazole-treated cells results in enhanced and prolonged expression of cyclin B1 and securin. SAF-A WT, 59A, and 59E cells were grown to 70% confluence, and extracts were prepared from either asynchronously growing cells or cells treated with nocodazole (40 ng/ml for 16 h) and harvested by mitotic shake-off either immediately (0 h) or at various times after placement in fresh, nocodazole-free medium as indicated. Fifty micrograms of total protein from NETN extracts was run on SDS-polyacrylamide gels and transferred to nitrocellulose, and blots were probed with the indicated antibodies. The antibody to SAF-A was a mouse monoclonal antibody. Bands corresponding to cyclin B1 and securin were quantitated and normalized to total Ku80 and are graphically shown below gels on the left, and right, respectively. Results are representative of 3 separate experiments. See Fig. S10 in the supplemental material for similar results with the microtubule stabilizer paclitaxel.
To confirm these results, we repeated the experiment in cells incubated with paclitaxel, which induces activation of the SAC and mitotic arrest through stabilization of microtubules, rather than inhibiting microtubule attachment (27, 35). Again, levels of cyclin B1 and securin were elevated in paclitaxel-treated cells expressing SAF-A S59A and decreased in cells expressing S59E, compared to the case in cells expressing wild-type SAF-A (see Fig. S10 in the supplemental material), adding further support for our hypothesis that SAF-A S59 phosphorylation is required for efficient activation of the SAC and/or inhibition of the APC/C.
SAF-A is dephosphorylated by PP2Ac during mitosis.
Given that both ablation of SAF-A S59 phosphorylation (S59A) and introduction of a phosphomimic (S59E), which mimics constitutive phosphorylation and/or inability to dephosphorylate SAF-A S59, had significant effects on mitosis (Fig. 7 and 8), we deemed it important to identify the protein phosphatase that dephosphorylates S59 in mitosis. SAF-A interacts with both Aurora A and TPX2 in mitosis (24) (confirmed in Fig. 4). Since PP6 dephosphorylates Aurora A on T288 in the activating T loop during mitosis (20) and PP6 interacts with DNA-PKcs (19) and dephosphorylates DNA-PKcs in mitosis (14), we first asked whether PP6 was responsible for dephosphorylating SAF-A S59 in mitosis. The PP6 catalytic subunit (PP6c) was depleted from HeLa cells using siRNA, and at 80 h posttransfection nocodazole was added to the cells and left for 16 h. Mitotic cells were isolated by shake-off, and cells were left to recover in fresh medium without nocodazole for the times indicated. Despite depletion of PP6c by approximately 90%, compared to that in cells transfected with control siRNA, the levels of SAF-A S59 phosphorylation did not change significantly after PP6 depletion, indicating that PP6 is not responsible for dephosphorylating S59 during mitosis (see Fig. S11 in the supplemental material). Of note, the level of Aurora A pT288 phosphorylation in the siRNA PP6-depleted cells was enhanced compared to that in control cells, confirming a previous report that PP6c contributes to dephosphorylation of Aurora A in mitosis (20).
We next examined the effects of depleting the PP2A-like protein phosphatases PP2Ac and PP4c on SAF-A S59 phosphorylation in mitosis. As can be seen in Fig. S12 in the supplemental material, the knockdown level of PP4c was again very efficient (approximately 90%) compared to that in control cells (lower band on blot). However, neither SAF-A S59 nor Aurora A T288 phosphorylation changed significantly in the absence of PP4c. In striking contrast, siRNA depletion of the PP2A catalytic subunit (PP2Ac) resulted in increased and sustained phosphorylation of SAF-A at S59 in nocodazole-treated cells (Fig. 10). Together our data reveal that SAF-A is phosphorylated on S59 by PLK1 and dephosphorylated by PP2A in mitosis and that phosphorylation of SAF-A on S59 is required for efficient mitotic exit in nocodazole- and paclitaxel-treated human cells (Fig. 11).
FIG 10.
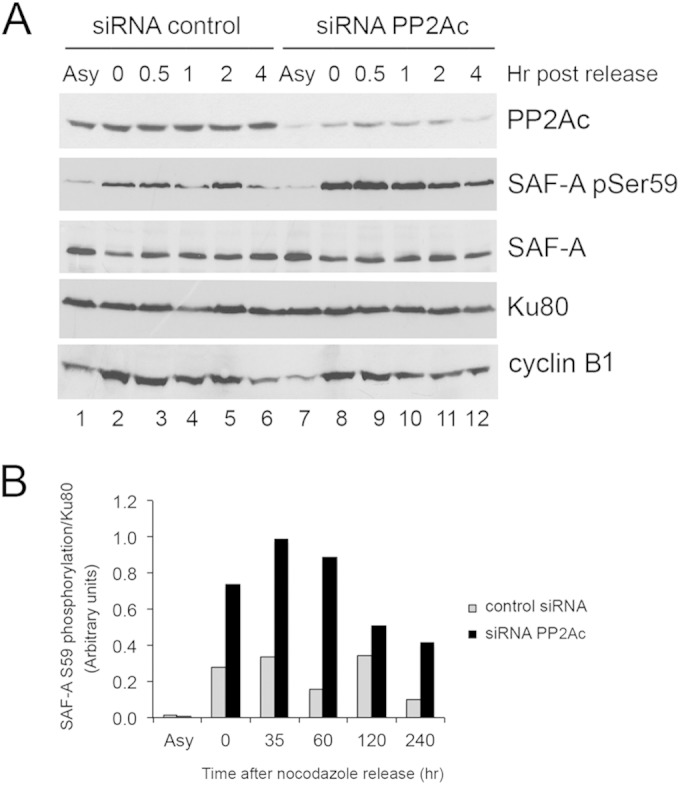
PP2A is required for dephosphorylation of SAF-A S59 in mitosis. HeLa cells were treated with 100 nM siRNA scrambled control or siRNA to PP2A catalytic subunit (PP2Ac, alpha and beta isoforms). Eighty hours after transfection, 40 ng/ml of nocodazole was added, and the cells were returned to the incubator for a further 16 h. Cells were harvested by mitotic shake-off and left to recover in fresh medium (without nocodazole) for the times indicated. (A) Fifty micrograms of extract from asynchronized cells (Asy) or from nocodazole-treated cells was run on 8% or 10% acrylamide SDS gels and probed for PP2Ac, phosphoserine 59 SAF-A, total SAF-A (mouse monoclonal, Abcam number 10297), and Ku80 as indicated. Bands corresponding to SAF-A phosphoserine 59 were quantitated and normalized to total Ku80. (B) Quantitation of the experiment. Results are representative of 3 separate experiments.
FIG 11.
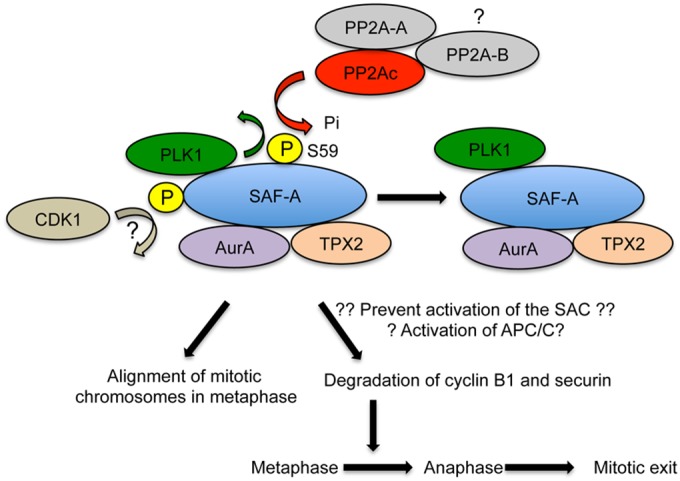
Model describing SAF-A S59 phosphorylation in mitosis SAF-A interacts with PLK1, and SAF-A S59 is phosphorylated in a PLK1-dependent manner in nocodazole-treated mitotic cells. SAF-A S59 phosphorylation also requires CDK1, possibly by phosphorylation of SAF-A on an unidentified PLK1 priming site (indicated by a question mark); however, S59 was not required for interaction of SAF-A with PLK1, Aurora A, or TPX2. SAF-A S59 is dephosphorylated by PP2A for cells to proceed from metaphase into anaphase and subsequently exit mitosis. The PP2A catalytic subunit interacts with a scaffolding A subunit and one of multiple B subunits. The PP2A-B subunit required for SAF-A S59 dephosphorylation in mitosis in not known. Inability to phosphorylate SAF-A S59 blocks degradation of cyclin B1 and securin, suggesting that SAF-A S59 phosphorylation is required for activation of the E3 ubiquitin ligase APC/C/E2 complex required for cyclin B1 and securin degradation. Alternatively, inability to phosphorylate SAF-A S59 may prevent activation of the SAC, causing misaligned mitotic chromosomes and polylobed nuclei in mitosis.
DISCUSSION
SAF-A (also called hnRNP-U) is a member of the hnRNP family of proteins and has been implicated in multiple nuclear processes. It is also phosphorylated in a DNA-PK-dependent manner after DNA damage (21, 22). Recently, a new role for SAF-A in mitosis was described (24). SAF-A was reported to form a complex with Aurora A and TPX2 in mitosis, and depletion of SAF-A resulted in a delay in exit from mitosis (24). However, neither the molecular mechanism by which SAF-A function is regulated in mitosis nor the role of SAF-A S59 phosphorylation in mitosis or after DNA damage has been addressed.
Here we show that SAF-A is dynamically phosphorylated on S59 in mitosis, confirming and extending results from quantitative high-throughput proteomics screens (5, 8). We show that PLK1 phosphorylates SAF-A on S59 in vitro and that mitotic phosphorylation of SAF-A on S59 requires PLK1 activity in vivo (Fig. 2 and 3). The canonical PLK1 consensus site has been defined as (D/E)Xp(S/T)(FLIYWVM), where X is any amino acid and p indicates the phosphorylated serine (36). At first glance, the amino acid sequence surrounding SAF-A S59 (PGNGpSLDLGGD) does not correspond to a canonical PLK1 consensus. However, a recent unbiased phosphoproteomics screen for PLK1 substrates in mitosis (8) revealed additional PLK1 consensus motifs, including NXp(S/T), where the N at position −2 was essential for PLK1 phosphorylation. Interestingly, the amino acid sequence surrounding S59 of SAF-A conforms to this novel PLK1 consensus sequence (PGNGpSLDLGGD). Together these data suggest that S59 is a direct target of PLK1 in mitosis in vivo. However, we note that inhibition of PLK1 alone did not completely abrogate SAF-A S59 phosphorylation, and additional mitotic protein kinases such as Aurora A and B may also contribute to SAF-A S59 phosphorylation in mitosis (Fig. 2B). We also show that incubation with the CDK1 inhibitor R03366 blocked SAF-A S59 phosphorylation in nocodazole-treated cells and decreased S59 phosphorylation remaining after inhibition of PLK1 with BI2536 (Fig. 2C). Many PLK1 substrates require prior phosphorylation on a priming site by CDK1 (33). PLK1 contains two C-terminal polo-box domains (PBDs) that interact with phosphorylated polo-box motifs (PBMs) either in PLK1 itself or in its interacting partners and substrates (37, 38). The optimal binding sequence for the PBM is S/T-pS-P, where pS represents a serine that is phosphorylated either by PLK1 itself (self-priming) or a proline-directed protein kinase, such as a cycle-dependent kinase (CDK) (non-self-priming) (37, 38). It is therefore interesting to note that serine 4 of SAF-A contains a putative PBM sequence (MSSSPVN) and serine 4 is highly phosphorylated (75% occupancy) in mitosis (5, 39). Thus, we speculate that phosphorylation of SAF-A by CDK1, possibly on S4, could create a PBM that could promote interaction with PLK1 and consequent phosphorylation on S59. Our finding that inhibition of CDK1 with RO3366 inhibits SAF-A S59 phosphorylation supports this hypothesis (Fig. 2C). Experiments to identify putative CDK1-dependent priming phosphorylation sites in SAF-A are in progress.
Although SAF-A S59 has been identified as a mitotic phosphorylation site in numerous phosphoproteomics studies, it was not identified as a target of PLK1, Aurora A, or Aurora B by high-throughput phosphoproteomics studies (8). As shown in Fig. 2, although PLK1 accounts for approximately 80% SAF-A S59A phosphorylation, Aurora A, Aurora B, and CDK1 also play a role in SAF-A S59 phosphorylation, and thus we speculate that the involvement of multiple mitotic protein kinases in SAF-A S59 phosphorylation in vivo may have resulted in a lower score in SILAC, placing SAF-A S59 phosphorylation below the cutoff for identification as a PLK1 or Aurora substrate in high-throughput screens.
We also show that SAF-A interacts with PLK1 in mitosis and confirm an earlier study reporting an interaction of SAF-A with TPX2 and Aurora A (24). However, none of these interactions required SAF-A S59 phosphorylation, as all three interactions were seen in cells expressing SAF-A S59A (Fig. 4). We were unable to determine whether the S59E mutation affected interaction of SAF-A with PLK1, TPX2, or Aurora because this mutant protein did not immunoprecipitate efficiently (data not shown). Experiments to determine whether inhibition of PLK1 or CDK1 (to eliminate potential PLK1- or CDK1-mediated phosphorylation sites in SAF-A) suggested that inhibition of PLK1 alone had no effect on the SAF-A/TPX2/Aurora A interaction, while inhibition of CDK1 alone reduced the amount of Aurora A but not PLK1 or TPX2 that immunoprecipitated with SAF-A. Together, our results suggest that SAF-A may be phosphorylated by multiple protein kinases in mitosis, the regulation of which may be complex. Indeed, multiple mitotic phosphorylation sites have been detected in SAF-A, in addition to S59 (5, 8); however, neither the function of these phosphorylation events nor the kinase(s) responsible for their phosphorylation is known.
SAF-A has been reported to localize to mitotic spindles, the spindle midzone, and cytoplasmic bridges (24). Using a phospho-specific antibody to SAF-A S59, we extend these studies to show that SAF-A phospho-S59 localizes at centrosomes in prophase and metaphase, at spindle microtubules in anaphase, and at the midbody in cytokinesis. How SAF-A localization in mitosis is regulated and whether phospho-SAF-A interacts with different partners at different subcellular locations to regulate mitosis remains to be determined.
Our results also show that MRC5-SV cells expressing either SAF-A S59A (phosphoablation) or S59E to mimic constitutive/irreversible phosphorylation exhibit multiple mitotic defects. MRC5-SV cells expressing either SAF-A S59A or S59E exhibited a large increase in the number of cells with misaligned mitotic chromosomes (Fig. 6), and the percentage of cells expressing histone H3 pS10, a marker of early mitosis (40), was decreased in nocodazole-treated cells expressing SAF-A S59E (Fig. 7). Also, live-cell imaging, in the absence of nocodazole, revealed that cells expressing SAF-A S59A took longer to proceed from prophase to anaphase and that cells with either phosphoablation (S59A) or phosphomimic (S59E) had increased lagging chromosomes (compared to WT), while cells expressing SAF-A S59A had a dramatic increase in polylobed nuclei (Fig. 8; see Fig. S9 in the supplemental material). These results suggest that SAF-A S59 phosphorylation is required for multiple events in mitosis.
Live-cell imaging results showing an increased time from prophase to anaphase in cells expressing S59A (Fig. 8) is in keeping with the presence of increased levels of cyclin B1 and securin in S59A-expressing cells and less expression of either cyclin B1 or securin in S59E-expressing cells that had been exposed to either nocodazole (Fig. 9) or paclitaxel (see Fig. S10 in the supplemental material) and may suggest that S59 phosphorylation of SAF-A is required for activation of the APC/C and/or to prevent activation of the SAC (Fig. 11). Experiments to determine how SAF-A S59 phosphorylation is required for progression from prophase to anaphase and degradation of cyclin B1 and securin are in progress.
Perhaps less well explained is why cells expressing S59E showed a significant decrease in histone H3 phospho-S10 staining in nocodazole-treated cells (Fig. 7C) but not a significant decrease in time taken to traverse mitosis, by live-cell imaging (Fig. 8). We speculate that the different results may be a reflection of the different methodologies used in the two assays, i.e., flow cytometry in the presence of nocodazole with histone H3 phospho-S10 staining (Fig. 7C) versus live-cell imaging in the absence of nocodazole (Fig. 8). Also, we noted that S59E cells were more prone to phototoxicity-induced death during live imaging than either WT or S59A cells; therefore, we speculate that S59E cells could be more sensitive to nocodazole, perhaps triggering cell death pathways. Regardless, both methodologies show that either ablation (S59A) or introduction of a phosphomimic (S59E) have significant effects on the percentage of cells in mitosis and/or mitotic timing.
We also show that cells expressing either SAF-A S59A or SAF-A S59E have increased levels of lagging chromosomes and that cells expressing SAF-A S59A have increased polylobed nuclei, suggesting multiple defects in chromosome segregation (Fig. 8). Together, our results point to the importance of both phosphorylation and dephosphorylation of SAF-A S59 in the regulation of mitosis. In this regard we show that SAF-A S59 is dephosphorylated by protein phosphatase PP2A in mitosis (Fig. 10). PP2A plays a critical role in regulation of mitotic exit (41–43). Our study adds SAF-A S59 as a new PP2A target in mitosis. Further work will be required to determine how dephosphorylation of SAF-A S59 by PP2A contributes to regulation of mitotic exit and to identify the appropriate PP2A-B subunit involved.
In summary, our studies show that PLK1 and CDK1 are required for phosphorylation of SAF-A on S59 in mitosis and that failure in turn to either phosphorylate or dephosphorylate SAF-A S59 results in multiple mitotic defects, including increased lagging chromosomes, misaligned chromosomes, and chromosome segregation defects resulting in daughter cells with polylobed nuclei. Thus, we speculate that dysregulation of SAF-A phosphorylation in mitosis may contribute to genomic instability and disease.
Supplementary Material
ACKNOWLEDGMENTS
This work was funded by grants from the Cancer Research Society (1022480), the Canadian Institutes of Health Research (MOP 13639), and the Engineered Air Chair in Cancer Research (to S.P.L.-M.), Cancer Research UK (to N.M.), and the Terry Fox Foundation (to L.T.-M.). Sébastien Britton is funded by a senior postdoctoral fellowship from La Ligue Nationale contre le Cancer.
We thank Edward Bartlett (University of Calgary) for helpful discussions, Aru Narendran (University of Calgary) for the generous gift of Aurora A and B inhibitors, James Hastie and Dario Alessi (University of Dundee) for purified PLK1, and Bernard Salles and Patrick Calsou (CNRS and Université de Toulouse, France) for the generous gift of cell lines (MRC5-SV SAF-A WT, S59A, and S59E) and the mouse monoclonal antibody to SAF-A phospho-S59 and their helpful comments on the manuscript.
We declare that we have no conflict of interest.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.01312-14.
REFERENCES
- 1.Lara-Gonzalez P, Westhorpe FG, Taylor SS. 2012. The spindle assembly checkpoint. Curr Biol 22:R966–R980. doi: 10.1016/j.cub.2012.10.006. [DOI] [PubMed] [Google Scholar]
- 2.Foley EA, Kapoor TM. 2013. Microtubule attachment and spindle assembly checkpoint signalling at the kinetochore. Nat Rev Mol Cell Biol 14:25–37. doi: 10.1038/nrm3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baker DJ, Dawlaty MM, Galardy P, van Deursen JM. 2007. Mitotic regulation of the anaphase-promoting complex. Cell Mol Life Sci 64:589–600. doi: 10.1007/s00018-007-6443-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bollen M, Gerlich DW, Lesage B. 2009. Mitotic phosphatases: from entry guards to exit guides. Trends Cell Biol 19:531–541. doi: 10.1016/j.tcb.2009.06.005. [DOI] [PubMed] [Google Scholar]
- 5.Olsen JV, Vermeulen M, Santamaria A, Kumar C, Miller ML, Jensen LJ, Gnad F, Cox J, Jensen TS, Nigg EA, Brunak S, Mann M. 2010. Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci Signal 3:ra3. doi: 10.1126/scisignal.2000475. [DOI] [PubMed] [Google Scholar]
- 6.Dephoure N, Zhou C, Villen J, Beausoleil SA, Bakalarski CE, Elledge SJ, Gygi SP. 2008. A quantitative atlas of mitotic phosphorylation. Proc Natl Acad Sci U S A 105:10762–10767. doi: 10.1073/pnas.0805139105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ma HT, Poon RY. 2011. How protein kinases co-ordinate mitosis in animal cells. Biochem J 435:17–31. doi: 10.1042/BJ20100284. [DOI] [PubMed] [Google Scholar]
- 8.Kettenbach AN, Schweppe DK, Faherty BK, Pechenick D, Pletnev AA, Gerber SA. 2011. Quantitative phosphoproteomics identifies substrates and functional modules of Aurora and Polo-like kinase activities in mitotic cells. Sci Signal 4:rs5. doi: 10.1126/scisignal.2001497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marzo I, Naval J. 2013. Antimitotic drugs in cancer chemotherapy: promises and pitfalls. Biochem Pharmacol 86:703–710. doi: 10.1016/j.bcp.2013.07.010. [DOI] [PubMed] [Google Scholar]
- 10.Salmela AL, Kallio MJ. 2013. Mitosis as an anti-cancer drug target. Chromosoma 122:431–449. doi: 10.1007/s00412-013-0419-8. [DOI] [PubMed] [Google Scholar]
- 11.Wang C, Lees-Miller SP. 2013. Detection and repair of ionizing radiation-induced DNA double strand breaks: new developments in nonhomologous end joining. Int J Radiat Oncol Biol Phys 86:440–449. doi: 10.1016/j.ijrobp.2013.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mahaney BL, Meek K, Lees-Miller SP. 2009. Repair of ionizing radiation-induced DNA double-strand breaks by non-homologous end-joining. Biochem J 417:639–650. doi: 10.1042/BJ20080413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shibata A, Moiani D, Arvai AS, Perry J, Harding SM, Genois MM, Maity R, van Rossum-Fikkert S, Kertokalio A, Romoli F, Ismail A, Ismalaj E, Petricci E, Neale MJ, Bristow RG, Masson JY, Wyman C, Jeggo PA, Tainer JA. 2014. DNA double-strand break repair pathway choice is directed by distinct MRE11 nuclease activities. Mol Cell 53:7–18. doi: 10.1016/j.molcel.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Douglas P, Ye R, Trinkle-Mulcahy L, Neal JA, De Wever V, Morrice NA, Meek K, Lees-Miller SP. 2014. Polo-like kinase 1 (PLK1) and protein phosphatase 6 (PP6) regulate DNA-dependent protein kinase catalytic subunit (DNA-PKcs) phosphorylation in mitosis. Biosci Rep 34:e00113. doi: 10.1042/BSR20140051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee KJ, Lin YF, Chou HY, Yajima H, Fattah KR, Lee SC, Chen BP. 2011. Involvement of DNA-dependent protein kinase in normal cell cycle progression through mitosis. J Biol Chem 286:12796–12802. doi: 10.1074/jbc.M110.212969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shang ZF, Huang B, Xu QZ, Zhang SM, Fan R, Liu XD, Wang Y, Zhou PK. 2010. Inactivation of DNA-dependent protein kinase leads to spindle disruption and mitotic catastrophe with attenuated checkpoint protein 2 phosphorylation in response to DNA damage. Cancer Res 70:3657–3666. doi: 10.1158/0008-5472.CAN-09-3362. [DOI] [PubMed] [Google Scholar]
- 17.Shang Z, Yu L, Lin YF, Matsunaga S, Shen CY, Chen BP. 2014. DNA-PKcs activates the Chk2-Brca1 pathway during mitosis to ensure chromosomal stability. Oncogenesis 3:e85. doi: 10.1038/oncsis.2013.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang B, Shang ZF, Li B, Wang Y, Liu XD, Zhang SM, Guan H, Rang WQ, Hu JA, Zhou PK. 2014. DNA-PKcs associates with PLK1 and is involved in proper chromosome segregation and cytokinesis. J Cell Biochem 115:1077–1088. doi: 10.1002/jcb.24703. [DOI] [PubMed] [Google Scholar]
- 19.Douglas P, Zhong J, Ye R, Moorhead GB, Xu X, Lees-Miller SP. 2010. Protein phosphatase 6 interacts with the DNA-dependent protein kinase catalytic subunit and dephosphorylates gamma-H2AX. Mol Cell Biol 30:1368–1381. doi: 10.1128/MCB.00741-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zeng K, Bastos RN, Barr FA, Gruneberg U. 2010. Protein phosphatase 6 regulates mitotic spindle formation by controlling the T-loop phosphorylation state of Aurora A bound to its activator TPX2. J Cell Biol 191:1315–1332. doi: 10.1083/jcb.201008106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Britton S, Froment C, Frit P, Monsarrat B, Salles B, Calsou P. 2009. Cell nonhomologous end joining capacity controls SAF-A phosphorylation by DNA-PK in response to DNA double-strand breaks inducers. Cell Cycle 8:3717–3722. doi: 10.4161/cc.8.22.10025. [DOI] [PubMed] [Google Scholar]
- 22.Berglund FM, Clarke PR. 2009. hnRNP-U is a specific DNA-dependent protein kinase substrate phosphorylated in response to DNA double-strand breaks. Biochem Biophys Res Commun 381:59–64. doi: 10.1016/j.bbrc.2009.02.019. [DOI] [PubMed] [Google Scholar]
- 23.Han SP, Tang YH, Smith R. 2010. Functional diversity of the hnRNPs: past, present and perspectives. Biochem J 430:379–392. doi: 10.1042/BJ20100396. [DOI] [PubMed] [Google Scholar]
- 24.Ma N, Matsunaga S, Morimoto A, Sakashita G, Urano T, Uchiyama S, Fukui K. 2011. The nuclear scaffold protein SAF-A is required for kinetochore-microtubule attachment and contributes to the targeting of Aurora-A to mitotic spindles. J Cell Sci 124:394–404. doi: 10.1242/jcs.063347. [DOI] [PubMed] [Google Scholar]
- 25.Nousiainen M, Sillje HH, Sauer G, Nigg EA, Korner R. 2006. Phosphoproteome analysis of the human mitotic spindle. Proc Natl Acad Sci U S A 103:5391–5396. doi: 10.1073/pnas.0507066103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sauer G, Korner R, Hanisch A, Ries A, Nigg EA, Sillje HH. 2005. Proteome analysis of the human mitotic spindle. Mol Cell Proteomics 4:35–43. doi: 10.1074/mcp.M400158-MCP200. [DOI] [PubMed] [Google Scholar]
- 27.Gascoigne KE, Taylor SS. 2008. Cancer cells display profound intra- and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell 14:111–122. doi: 10.1016/j.ccr.2008.07.002. [DOI] [PubMed] [Google Scholar]
- 28.Gascoigne KE, Taylor SS. 2009. How do anti-mitotic drugs kill cancer cells? J Cell Sci 122:2579–2585. doi: 10.1242/jcs.039719. [DOI] [PubMed] [Google Scholar]
- 29.Roget K, Ben-Addi A, Mambole-Dema A, Gantke T, Yang HT, Janzen J, Morrice N, Abbott D, Ley SC. 2012. IkappaB kinase 2 regulates TPL-2 activation of extracellular signal-regulated kinases 1 and 2 by direct phosphorylation of TPL-2 serine 400. Mol Cell Biol 32:4684–4690. doi: 10.1128/MCB.01065-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao Y, Thomas HD, Batey MA, Cowell IG, Richardson CJ, Griffin RJ, Calvert AH, Newell DR, Smith GC, Curtin NJ. 2006. Preclinical evaluation of a potent novel DNA-dependent protein kinase inhibitor NU7441. Cancer Res 66:5354–5362. doi: 10.1158/0008-5472.CAN-05-4275. [DOI] [PubMed] [Google Scholar]
- 31.Hickson I, Zhao Y, Richardson CJ, Green SJ, Martin NM, Orr AI, Reaper PM, Jackson SP, Curtin NJ, Smith GC. 2004. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res 64:9152–9159. doi: 10.1158/0008-5472.CAN-04-2727. [DOI] [PubMed] [Google Scholar]
- 32.Scutt PJ, Chu ML, Sloane DA, Cherry M, Bignell CR, Williams DH, Eyers PA. 2009. Discovery and exploitation of inhibitor-resistant aurora and polo kinase mutants for the analysis of mitotic networks. J Biol Chem 284:15880–15893. doi: 10.1074/jbc.M109.005694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lowery DM, Clauser KR, Hjerrild M, Lim D, Alexander J, Kishi K, Ong SE, Gammeltoft S, Carr SA, Yaffe MB. 2007. Proteomic screen defines the Polo-box domain interactome and identifies Rock2 as a Plk1 substrate. EMBO J 26:2262–2273. doi: 10.1038/sj.emboj.7601683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Terasawa M, Shinohara A, Shinohara M. 2014. Canonical non-homologous end joining in mitosis induces genome instability and is suppressed by M-phase-specific phosphorylation of XRCC4. PLoS Genet 10:e1004563. doi: 10.1371/journal.pgen.1004563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Musacchio A, Salmon ED. 2007. The spindle-assembly checkpoint in space and time. Nat Rev Mol Cell Biol 8:379–393. doi: 10.1038/nrm2163. [DOI] [PubMed] [Google Scholar]
- 36.Nakajima H, Toyoshima-Morimoto F, Taniguchi E, Nishida E. 2003. Identification of a consensus motif for Plk (Polo-like kinase) phosphorylation reveals Myt1 as a Plk1 substrate. J Biol Chem 278:25277–25280. doi: 10.1074/jbc.C300126200. [DOI] [PubMed] [Google Scholar]
- 37.Reinhardt HC, Yaffe MB. 2013. Phospho-Ser/Thr-binding domains: navigating the cell cycle and DNA damage response. Nat Rev Mol Cell Biol 14:563–580. doi: 10.1038/nrm3640. [DOI] [PubMed] [Google Scholar]
- 38.Zitouni S, Nabais C, Jana SC, Guerrero A, Bettencourt-Dias M. 2014. Polo-like kinases: structural variations lead to multiple functions. Nat Rev Mol Cell Biol 15:433–452. doi: 10.1038/nrm3819. [DOI] [PubMed] [Google Scholar]
- 39.Sharma K, D'Souza RC, Tyanova S, Schaab C, Wisniewski JR, Cox J, Mann M. 2014. Ultradeep human phosphoproteome reveals a distinct regulatory nature of Tyr and Ser/Thr-based signaling. Cell Rep 8:1583–1594. doi: 10.1016/j.celrep.2014.07.036. [DOI] [PubMed] [Google Scholar]
- 40.Prigent C, Dimitrov S. 2003. Phosphorylation of serine 10 in histone H3, what for? J Cell Sci 116:3677–3685. doi: 10.1242/jcs.00735. [DOI] [PubMed] [Google Scholar]
- 41.Grallert A, Boke E, Hagting A, Hodgson B, Connolly Y, Griffiths JR, Smith DL, Pines J, Hagan IM. 2015. A PP1-PP2A phosphatase relay controls mitotic progression. Nature 517:94–98. doi: 10.1038/nature14019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hunt T. 2013. On the regulation of protein phosphatase 2A and its role in controlling entry into and exit from mitosis. Adv Biol Regul 53:173–178. doi: 10.1016/j.jbior.2013.04.001. [DOI] [PubMed] [Google Scholar]
- 43.Jeong AL, Yang Y. 2013. PP2A function toward mitotic kinases and substrates during the cell cycle. BMB Rep 46:289–294. doi: 10.5483/BMBRep.2013.46.6.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.