

ABSTRACT
In our study, we characterized the effect of monensin, an ionophore that is known to raise the intracellular pH, on the hepatitis C virus (HCV) life cycle. We showed that monensin inhibits HCV entry in a pangenotypic and dose-dependent manner. Monensin induces an alkalization of intracellular organelles, leading to an inhibition of the fusion step between viral and cellular membranes. Interestingly, we demonstrated that HCV cell-to-cell transmission is dependent on the vesicular pH. Using the selective pressure of monensin, we selected a monensin-resistant virus which has evolved to use a new entry route that is partially pH and clathrin independent. Characterization of this mutant led to the identification of two mutations in envelope proteins, the Y297H mutation in E1 and the I399T mutation in hypervariable region 1 (HVR1) of E2, which confer resistance to monensin and thus allow HCV to use a pH-independent entry route. Interestingly, the I399T mutation introduces an N-glycosylation site within HVR1 and increases the density of virions and their sensitivity to neutralization with anti-apolipoprotein E (anti-ApoE) antibodies, suggesting that this mutation likely induces conformational changes in HVR1 that in turn modulate the association with ApoE. Strikingly, the I399T mutation dramatically reduces HCV cell-to-cell spread. In summary, we identified a mutation in HVR1 that overcomes the vesicular pH dependence, modifies the biophysical properties of particles, and drastically reduces cell-to-cell transmission, indicating that the regulation by HVR1 of particle association with ApoE might control the pH dependence of cell-free and cell-to-cell transmission. Thus, HVR1 and ApoE are critical regulators of HCV propagation.
IMPORTANCE Although several cell surface proteins have been identified as entry factors for hepatitis C virus (HCV), the precise mechanisms regulating its transmission to hepatic cells are still unclear. In our study, we used monensin A, an ionophore that is known to raise the intracellular pH, and demonstrated that cell-free and cell-to-cell transmission pathways are both pH-dependent processes. We generated monensin-resistant viruses that displayed different entry routes and biophysical properties. Thanks to these mutants, we highlighted the importance of hypervariable region 1 (HVR1) of the E2 envelope protein for the association of particles with apolipoprotein E, which in turn might control the pH dependency of cell-free and cell-to-cell transmission.
INTRODUCTION
Hepatitis C virus (HCV) infection is a global public health problem affecting over 130 million individuals worldwide. Chronic HCV infection can result in liver cirrhosis and hepatocellular carcinoma (1). While previous interferon (IFN)-based therapies have been limited by drug resistance and marked toxicity (2), the recently clinically licensed direct-acting antivirals are expected to cure the large majority of infected patients without major adverse effects (3). Nevertheless, several challenges remain: high costs limit access to therapy even in high-resource settings, and certain subgroups of difficult-to-treat patients may need adjunctive therapeutic approaches (4). Furthermore, a vaccine is not available, and vaccine development is hampered by viral evasion of host immune responses (5).
HCV is a small, enveloped, single-stranded RNA virus that belongs to the Hepacivirus genus in the Flaviviridae family (6). This virus, which circulates in the bloodstream of infected patients as lipoviral particles, mainly targets hepatocytes. Infection begins with the attachment of viral particles to the cell surface of the hepatocytes through attachment factors and then proceeds to a complex multistep process involving a series of specific cellular entry factors (reviewed in reference 7). These molecules include scavenger receptor class B type I (SRB1) (8), the tetraspanin CD81 (9), the tight junction proteins claudin-1 (CLDN1) (10) and occludin (OCLN) (11), and the receptor tyrosine kinases epidermal growth factor receptor (EGFR) and ephrin receptor A2 (EphA2) (12). More recently, the Niemann-Pick C1-like 1 (NPC1L1) cholesterol absorption receptor and the iron uptake receptor transferrin receptor 1 (TfR1) were also shown to play a role in HCV entry (13, 14). The interaction of HCV particles through their associated apolipoproteins and envelope proteins (E1 and E2) with the different entry factors leads to the internalization of particles via a clathrin-mediated endocytosis (15, 16) followed by fusion at low pH with the membrane of an early endosome (17, 18). Although in the last few years the use of pseudotyped viruses (HCVpp) (18, 19) and infectious cell culture-produced particles (HCVcc) (20–22) has greatly advanced the knowledge of the HCV life cycle, the exact sequence of events leading from HCV interaction with host factors at the plasma membrane to internalization and viral fusion still remains elusive. In particular, cellular and viral actors involved in the fusion of cellular and viral membranes remain to be identified.
Besides transmission by circulating particles, referred to as cell-free infection, HCV particles can be transmitted directly into neighboring cells through so-called cell-to-cell transmission. This route of transmission was first suggested when infected cell foci were seen in infected human livers by RNA imaging analysis (23) and was recently confirmed using a similar approach (24). Later, it was shown that HCV can be transmitted to neighboring cells in the presence of monoclonal antibodies (MAbs) or patient-derived antibodies that are able to neutralize cell-free infectivity (25–27). Recently, it was proposed that exosomes from HCV-infected cells are capable of transmitting infection to naive human hepatoma cells (28, 29). Although several entry factors have been implicated in this process, the viral determinants, entry factor requirements, and molecular mechanisms involved in the cell-to-cell transmission route still need to be characterized further.
In our study, we used monensin A to evaluate its antiviral potential against HCV and to gain new insights into the understanding of HCV transmission. Monensin is a well-known polyether antibiotic whose structure has been described (30) and which was isolated from Streptomyces cinnamonensis (31). It specifically exchanges Na+ for H+, resulting in an elevated cytosolic Ca2+ concentration and pH (32). Monensin is used extensively in the beef and dairy industries to prevent coccidiosis, enhance growth, or prevent bloat (33–36). Monensin also has antiviral effects on different viruses, such as human immunodeficiency virus (HIV) (37, 38), simian virus 40 (SV40) (39), and herpes simplex virus (HSV) (40).
We showed that monensin efficiently inhibits cell-free and cell-to-cell transmissions of HCV. Monensin induced an alkalization of intracellular organelles, probably leading to an inhibition of the fusion between viral and cellular membranes, indicating that both cell-free and cell-to-cell transmission pathways require a fusion step in acidic compartments. Interestingly, an HCV strain (FL-8) resistant to monensin but also to chloroquine (CQ) and chlorpromazine was selected by sequential passages in the presence of monensin. Characterization of the FL-8 mutant showed that resistance to monensin was conferred by two mutations in envelope proteins, i.e., the Y297H mutation in E1 and the I399T mutation in hypervariable region 1 (HVR1) of E2, which allow HCV to use an entry route that is less dependent on the acidic vesicular pH. We further characterized these two mutations and showed that they likely induce conformational changes in E2, increase the density of particles, and increase virus sensitivity to neutralization by anti-apolipoprotein E (anti-ApoE) antibodies. Most importantly, the I399T mutation dramatically reduces cell-to-cell transmission, indicating that we describe for the first time a mutant specifically defective in this transmission route.
MATERIALS AND METHODS
Chemicals and cell culture.
Dulbecco's modified Eagle's medium (DMEM), Opti-MEM, phosphate-buffered saline (PBS), Glutamax-I, nonessential amino acids (NEAA), goat serum, and fetal bovine serum (FBS) were purchased from Invitrogen. 4′6-Diamidino-2-phenylindole (DAPI) and 5-chloromethylfluorescein diacetate (CMFDA) were purchased from Molecular Probes (Invitrogen). Monensin and Mowiol 3-88 were purchased from Calbiochem. Monensin was resuspended in ethanol at 100 mM. Boceprevir was kindly provided by Philippe Halfon (Hôpital Ambroise Paré, Marseille, France). Chlorpromazine was purchased from Alexis. Dimethyl sulfoxide (DMSO), bafilomycin A1, brefeldin A, and chloroquine were obtained from Sigma.
Huh-7 cells were described previously (41). Huh-7-Lunet-CD81-FLuc-BLR cells (42) were kindly provided by T. Pietschmann (Twincore, Hannover, Germany). HEp-2 cells were obtained from BioWhittaker. 786-O cells were kindly provided by Mariana Varna (Hôpital St. Louis, Paris, France).
Antibodies.
Mouse anti-HCV E1 (A4) (43) and anti-yellow fever virus (YFV) envelope (2D12; ATCC CRL-1689) (44) monoclonal antibodies (MAbs) were produced in vitro by using a MiniPerm apparatus (Heraeus) as recommended by the manufacturer. The 3/11 (anti-HCV E2) (45) hybridoma was kindly provided by J. McKeating (University of Birmingham, United Kingdom). An anti-CD81 MAb (5A6) (46) was kindly provided by S. Levy (Stanford University). An anti-CD151 MAb (TS151) (47) was kindly provided by E. Rubinstein (Inserm, Villejuif, France). The anti-CLDN1 (OM 8A9-A3) MAb has been described previously (48). The AR3A and AR5A MAbs (anti-E2 and anti-E1E2, respectively) were kindly provided by M. Law (The Scripps Research Institute, La Jolla, CA) (49). Anti-SRB1 (ClaI) and anti-CD81 (JS81) MAbs were obtained from BD Biosciences Pharmingen. Anti-EGFR (cocktail R19/48), anti-OCLN (ZMD481), and anti-beta-tubulin were purchased from Invitrogen. The polyclonal goat anti-ApoE (AB947) antibody was obtained from Millipore. The anti-NS5 (clone 2F6/G11) MAb was obtained from Austral Biologicals. Secondary antibodies were obtained from Jackson ImmunoResearch (West Grove, PA).
HCVcc and infection assays.
To produce HCVcc, we used a modified version of a plasmid carrying the JFH1 genome (genotype 2a; GenBank accession number AB237837), kindly provided by T. Wakita (National Institute of Infectious Diseases, Tokyo, Japan) (20). Huh-7 cells were electroporated with in vitro-transcribed RNA of JFH1, containing (HCVcc-Luc) or not containing (HCVcc or JFH1) the Gaussia luciferase reporter gene, or with the JFH1-ΔHVR1 construct (50) and engineered to express the A4 epitope (51) and titer-enhancing mutations (52). JFH1 stocks were produced by further amplification in Huh-7 cells. The intergenotypic HCVcc chimeras GT3a(S52)/JFH1 and GT5a(SA13)/JFH1, kindly provided by J. Bukh (University of Copenhagen, Denmark) (53), were also used in this work.
For infection assays, HCVcc were added to Huh-7 cells (multiplicity of infection [MOI] = 1) seeded the day before in 24-well or 96-well plates and incubated for 2 h at 37°C. The supernatants were then removed, and the cells were incubated in DMEM with 10% FBS at 37°C. At 24, 30, or 48 h postinfection, Renilla/Gaussia luciferase assays were performed as indicated by the manufacturer (Promega), or infection levels were detected by indirect immunofluorescence.
HCVpp and infection assays.
HCVpp were produced as described previously (19, 54), using plasmids kindly provided by B. Bartosch and F. L. Cosset (Lyon, France). Plasmids encoding HCV envelope glycoproteins of genotypes 1b (UKN1B-5.23), 2b (UKN2B-1.1), and 4a (UKN4-11.1) were kindly provided by J. Ball (Nottingham University, United Kingdom) (55). The genotype 1a plasmid (strain H) has been described previously (19), and the genotype 2a plasmid (strain JFH1) was kindly provided by R. Bartenschlager (University of Heidelberg, Germany). A plasmid encoding the feline endogenous virus RD114 glycoprotein (56) was used for the production of RD114pp. Pseudotyped particles were inoculated onto Huh-7 cells for 2 h at 37°C. At 24, 48, or 72 h postinfection, firefly luciferase (FLuc) assays were performed as indicated by the manufacturer (Promega).
Other viruses.
We used the YFV strain 17D and a Sindbis virus expressing firefly luciferase (SinV; Toto1101/Luc [57]), kindly provided by M. MacDonald, Rockefeller University, NY. A recombinant adenovirus expressing green fluorescent protein (AdV) was produced as described previously (51). These viruses were inoculated onto Huh-7 cells for 2 h at 37°C and cultured for 24 h at 37°C. YFV infections were scored by indirect immunofluorescence, and SinV and AdV infections were scored by luminometry and flow cytometry, respectively.
The diabetogenic coxsackievirus B4 (CVB4) E2 strain (provided by J.-W. Yoon, J. McFarlane Diabetes Research Center, Calgary, Alberta, Canada) was produced in HEp-2 cells as described previously (58). CVB4 infections were scored by evaluating the cytopathic effect at 48 h postinfection. It has to be noted that no cytotoxicity of HEp-2 cells was induced by monensin at 0.1 μM or 1 μM.
Indirect immunofluorescence microscopy.
Cells grown on glass coverslips or in 96-well plates were infected and processed for immunofluorescence detection of the E1 envelope protein with the A4 MAb, as previously described (59). Cells were observed with a Zeiss Axiophot microscope equipped with a 10×, 0.5-numerical-aperture objective. Fluorescence signals were collected with a Coolsnap ES camera (Photometrix), using specific fluorescence excitation and emission filters. For quantification, images of randomly picked areas from each coverslip or well were recorded and processed using ImageJ software. Cells labeled with anti-E1 MAb A4 were counted as infected cells. The total number of cells was obtained by counting DAPI-labeled nuclei. The infections were scored by determining the ratio of infected cells to total cells.
Viability assay.
Cells were grown in 96-well plates and treated with increasing concentrations of monensin diluted in culture medium. The day after, an MTS [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2H-tetrazolium]-based viability assay was done as recommended by the manufacturer (CellTiter 96 aqueous nonradioactive cell proliferation assay; Promega).
Entry assay.
Cells seeded in 24-well plates were infected with HCVcc-Luc for 1 h at 4°C (attachment/binding period). Virus was removed, and the cells were washed with serum-free medium and incubated again for 1 h at 4°C (postattachment/binding period). The cells were then washed and incubated for 1 h at 37°C (endocytosis/fusion period). Finally, the cells were washed and incubated in 10% FBS-containing culture medium for 21 h. Infection levels were monitored by measuring luciferase activities.
Replication assay.
To specifically assay HCV RNA replication efficiency, Huh-7-Lunet-CD81-FLuc cells were electroporated with a JFH1-Rluc RNA deleted for the HCV envelope glycoprotein sequence (JFH1-ΔE1E2-RLuc RNA) in Opti-MEM, using a Gene Pulser apparatus (Bio-Rad). Cells were next seeded in complete medium for 5 h at 37°C. Monensin (0.1 μM or 1 μM) or boceprevir (0.5 μM) was added to the cells. Cells grown in the presence of ethanol and DMSO were used as controls for monensin and boceprevir, respectively. At 24, 48, and 72 h postelectroporation, cells were lysed and tested for replication by using a luciferase-based assay (dual-luciferase assay; Promega). Firefly luciferase activities were quantified to assess cell density and viability, whereas Renilla luciferase activities were quantified to assess HCV replication.
Assembly assay.
To test the ability of cells to assemble and produce infectious HCV particles, Huh-7 cells seeded onto 6-well plates were infected with HCVcc for 2 h at 37°C. The virus was then removed and cells cultured with monensin (0.1 μM or 1 μM). In parallel, HCV-infected Huh-7 cells were treated with brefeldin A (BFA; 0.1 μM or 1 μM) for 8 h before harvest. Thirty hours later, supernatants were collected, and infectivity titers were determined by indirect immunofluorescence assay.
Flow cytometry.
For CD81, SRB1, CLDN1, CD151, and EGFR staining, cells were cultured with or without monensin and labeled with TS81, ClaI, 8A9, TS151, and anti-EGFR antibodies, respectively, for 1 h at 4°C. Cells were washed and incubated for 45 min at 4°C with phycoerythrin (PE)-labeled secondary antibodies, rinsed again, and fixed with formalin solution. Labeled cells were analyzed using a BD FACSCalibur cytometer.
Detection of OCLN.
Huh-7 and 786-O cells were cultured with or without monensin and lysed in PBS containing 1% Triton X-100 and protease inhibitors (Complete; Roche Applied Science). Twenty-four hours later, nonreducing Laemmli buffer was added, and lysates were resolved by SDS-PAGE, transferred to a nitrocellulose membrane (GE Healthcare), and immunoblotted with anti-OCLN antibody followed by peroxidase-conjugated secondary antibody.
Colocalization of CD81 and CLDN1.
Colocalization analyses were carried out using CoLocalizer Pro software. Briefly, images representing a single optical z-section of cells were converted to tiff files and imported into CoLocalizer Pro software. Background corrections were applied, and the total number of pixels overlapping the two different channels was calculated. The number of colocalized pixels was divided by the total number of pixels for a chosen channel (as indicated in the figure legends), yielding the percent colocalization. Colocalization values for at least 10 cells were averaged to give the mean percent colocalization.
Monensin/interferon assay.
Huh-7 cells seeded into 24-well plates were incubated for 2 h at 37°C in medium containing monensin (0.04 μM), interferon (1 U/ml), or both. Cells were then infected for 2 h at 37°C with HCVcc-Luc in the presence of the drugs. After infection, cells were incubated again in complete medium supplemented with monensin, interferon, or both drugs. At 24 h postinfection, cells were lysed and luciferase activities were quantified.
AO staining.
Huh-7 cells seeded in Lab-Tek wells were either left untreated or treated for 2 h or 24 h with medium containing monensin (0.1 or 1 μM) or bafilomycin A1 (25 nM). Cells were then incubated for 20 min at 37°C with complete medium supplemented with 5 μg/ml acridine orange (AO), and stains were analyzed immediately by confocal microscopy. Quantification were performed with ImageJ software. The threshold of each field was adjusted to the same level. To calculate the arbitrary units of AO staining, the number of particles of >0.1 μm2 was divided by the number of nuclei in each field.
Cell-to-cell transmission assay.
Huh-7 cells were seeded on coverslips and infected with HCVcc (JFH1 or mutants) for 2 h at 37°C. Cells were then washed and cultured for 72 h at 37°C in culture medium containing or not containing the 3/11 MAb (50 μg/ml) in the presence or absence of monensin (0.1 μM) or chloroquine (1.75 μM). Cells were then fixed with formalin solution (4% formaldehyde; Sigma), and foci were detected using an indirect immunofluorescence assay.
In another approach, Huh-7 cells were infected with HCVcc (JFH1 or mutants) 24 h to 72 h prior to their use in the assay. Infected cells were then labeled for 30 min at 37°C with 10 μM CMFDA in medium without FBS. Cells were trypsinized, washed, and incubated for 15 min with the 3/11 MAb (50 μg/ml) to neutralize any remaining viral particles at the cell surface. After washing, CMFDA-labeled donor cells were mixed with naive Huh-7 cells (at ratios of 1:4 and 1:6) and seeded in 24-well plates in the presence (cell-to-cell transmission assay) or absence (cell-free and cell-to-cell transmission assays) of neutralizing MAb (3/11; 50 μg/ml). Cocultures were then incubated for either 24 h or 44 h at 37°C (as indicated in the figure legends). For 44-h incubations, the 3/11 MAb (50 μg/ml) was again added to the cell-to-cell transmission wells 24 h after mixing cells. De novo transmission events were determined by staining for HCV NS5 and were quantified by flow cytometry. Cell-to-cell transmission levels were defined by determining the numbers of newly infected cells in the presence of neutralizing MAb.
As controls, we harvested the supernatants of cell-to-cell transmission wells (containing the 3/11 MAb) and used them to infect naive Huh-7 cells. In contrast to supernatants of total transmission wells (wells with no antibody), which led to productive infection, the supernatants of cell-to-cell transmission wells were not infectious, demonstrating that our antibody conditions were sufficient to fully block cell-free transmission. Cocultures and reinfections were also performed with AR3A (1 μg/ml) as a neutralizing antibody, and these showed the same results (data not shown).
Selection of monensin-resistant viruses.
Supernatants of JFH1-infected cells were serially passaged with increasing concentrations of monensin (from 15 nM to 600 nM). The structural region of the HCV genome was amplified by reverse transcription-PCR (RT-PCR) and reintroduced into the JFH1 plasmid by using the AgeI/KpnI restriction enzymes. Several DNA clones were selected, amplified, and sequenced. Amino acid changes that arose during inhibitor selection were identified by analysis of the DNA sequence and compared to the initial and control passages in the presence of ethanol. Plasmids were in vitro transcribed before electroporation into Huh-7 cells. Stocks of mutated viruses were generated as described above.
Mutants with resistance to monensin.
The Y297H mutant has been described previously (60). Mutant viruses carrying the I399T or Q409R mutation or a combination of both were generated by directed mutagenesis of the JFH1-encoding plasmid, using fusion PCR. PCR-amplified segments carrying these mutations were introduced into the plasmid encoding the JFH1 virus by using the KpnI/BsiwI restriction enzymes. Plasmids were in vitro transcribed before electroporation into Huh-7 cells. Viral stocks were generated as described above.
Viral fitness.
Huh-7 cells were electroporated with JFH1, FL-8, I399T mutant, or Y297H mutant in vitro-transcribed RNAs and then seeded into 6-well plates. Supernatants were collected 24, 48, 72, and 96 h after electroporation. At each time point, supernatants were used to quantify viral RNAs or to determine the viral titer. HCV RNAs were extracted with a QIAamp viral RNA minikit (Qiagen) used according to the manufacturer's instructions. HCV RNAs were quantified by a quantitative real-time PCR assay as described previously (61). Specific infection was calculated as the relationship between the number of focus-forming infectious HCV particles and the amount of HCV RNA per milliliter of supernatant.
Neutralization assays.
For neutralization assays with the AR3A, AR5A, 3/11, and JS81 antibodies, viruses were mixed with antibodies and immediately used for inoculation (MOI = 0.05) of Huh-7 cells seeded the day before in 96-well plates. At 2 h postinoculation, complete medium was added to the cells. At 30 h postinfection, cells were fixed with methanol and processed for immunofluorescence assay to measure infectivity. For neutralization assay with anti-ApoE antibodies, viruses were incubated with the antibodies for 1 h at 37°C before inoculation onto Huh-7 cells and then were treated as described above.
Silencing experiments.
Huh-7 cells were transfected with small interfering RNA (siRNA) pools (Dharmacon) targeting CD81 or SRB1 or with a nontargeting siRNA control by using RNAiMax (Invitrogen) according to the manufacturer's instructions. The knockdown effects were determined 72 h after transfection by Western blotting (WB) at the time of virus inoculation. The effects of receptor silencing on virus infection were determined 30 h later, by indirect immunofluorescence assay.
Density gradients.
One milliliter of supernatant containing virus was layered on top of a 10 to 50% continuous iodixanol gradient (Optiprep; Proteogenix). Gradients were ultracentrifuged for 16 h at 160,000 × g at 4°C in an SW41 rotor. Twelve fractions of 1 ml each were collected. Densities and infectious titers were determined for each fraction.
RESULTS
Monensin inhibits HCV infection.
To investigate a potential inhibitory effect of monensin on HCV infectivity, we first studied the capacity of JFH1-based Gaussia luciferase (GLuc) reporter HCVcc (HCVcc-Luc) to infect Huh-7 cells in the presence of increasing amounts of monensin. As shown in Fig. 1A, infection levels decreased in a dose-dependent manner in the presence of monensin, indicating that monensin inhibits HCVcc infection. Although some toxicity began to be observed at 10 μM, the concentrations of 0.1 and 1 μM had no toxic effect as measured by an MTS assay (Fig. 1B). We therefore used the 0.1 and 1 μM concentrations of monensin in our next experiments. The inhibition of HCV infection in monensin-treated cells was confirmed with an untagged JFH1 virus (data not shown). However, due to greater technical ease, HCVcc-Luc was used in most of the other experiments. Importantly, monensin was active at nanomolar concentrations, since the half-maximal inhibitory concentration (IC50) was estimated to be 25 to 45 nM (Fig. 1C). In addition, the combination of monensin and IFN, a known HCV inhibitor, led to an additive effect of inhibition of HCV infection (Fig. 1D).
FIG 1.

Monensin inhibits HCV infection. (A) Huh-7 cells were infected with HCVcc-Luc in the presence of 0.001% ethanol (EtOH) or increasing concentrations of monensin added to the medium 2 h before infection, 2 h during infection, and full time after infection. After 24 h of infection, cells were lysed and luciferase activity quantified. (B) Monensin toxicity was assayed on Huh-7 cells. Cells were cultured with increasing concentrations of monensin for 2 or 24 h, and viability was tested using an MTS-based viability assay. (C) To assess the IC50 of monensin on HCV infection, Huh-7 cells were pretreated with monensin 2 h before, 2 h during, and full time after infection with HCVcc-Luc. Cell lysates were analyzed for luciferase activity at 24 h postinfection. (D) Cells were incubated for 2 h at 37°C in medium containing monensin (0.04 μM), interferon (1 U/ml), or both. Cells were then infected for 2 h at 37°C with HCVcc-Luc in the presence of the drug(s). After infection, cells were incubated again in complete medium supplemented with monensin, interferon, or both together. At 24 h postinfection, cells were lysed and luciferase activity was quantified. Results are presented as means ± standard deviations (SD) for at least three independent experiments. **, P < 0.01; ***, P < 0.001.
Monensin inhibits HCV entry.
To define which step of the HCV life cycle is inhibited by monensin, the drug was added for 2 h at different time points before, during, and after inoculation of Huh-7 cells with HCVcc-Luc (Fig. 2A, lanes b to d), or it was added full time after inoculation (Fig. 2A, lane e), as previously described (51, 62). Cells treated with ethanol (CTL) and cells treated with monensin before, during, and after infection (lane a) were used as controls. The results clearly showed that monensin significantly inhibited HCVcc infection when the drug was present during virus infection (Fig. 2A, lane c). There was no significant effect of the drug if it was added as a pretreatment of cells (Fig. 2A, lane b) or postinfection (Fig. 2A, lanes d and e). These results indicate that monensin likely inhibits the entry step of HCV. To confirm this hypothesis, we used the same experimental conditions with retroviral particles pseudotyped with HCV envelope glycoproteins (HCVpp2a) or with the envelope protein of the retrovirus RD114 (RD114pp), as a control (Fig. 2B). HCVpp2a infectivity was reduced 80% when monensin was present during infection (Fig. 2B, lane c). A slight decrease in HCVpp2a infection was observed under condition b, likely due to some residual monensin acting on the entry step (Fig. 2B, lane b). Similarly, the slight decrease observed under condition d was likely related to monensin acting on the entry of residual virions that had not yet completed the final steps of the entry process (Fig. 2B, lane d). When cells were incubated full time with monensin (Fig. 2B, lane a) or full time after inoculation of viruses (Fig. 2B, lane e), a significant decrease was measured for both pseudotypes, indicating that a postentry step of pseudoparticle infection was blocked by monensin. It has to be noted that HCVpp2a infection was more strongly inhibited than RD114pp infection when monensin was present full time during infection (Fig. 2B, lane a). Moreover, no inhibition was found when monensin was added after inoculation of HCVcc (Fig. 2A, lanes d and e). Altogether, our results indicate that monensin inhibits the entry step of HCV. We next investigated whether monensin has an antiviral activity on HCVpp bearing envelope glycoproteins from other genotypes. As shown in Fig. 2C, monensin significantly inhibited infection by HCVpp harboring envelope proteins from genotypes 1a, 1b, 2a, 2b, 4a, and 6a. Due to technical issues in producing HCVpp bearing envelope proteins from genotypes 3a and 5a, we next generated intergenotypic HCVcc chimeras, bearing the envelope glycoproteins from these two genotypes (Fig. 2D), that also showed great sensitivity to monensin. Together, our data indicate that monensin inhibits HCV entry in a genotype-independent manner.
FIG 2.

Monensin impairs HCV entry. Huh-7 cells were infected with HCVcc-Luc (A) or with RD114pp or HCVpp2a (B) for 2 h at 37°C and treated at different time points with 1 μM monensin. Monensin was added full time during the experiment (a), for 2 h before infection (b), for 2 h during infection (c), for 2 h after infection (d), or full time after infection (e). (C) Huh-7 cells were infected with HCVpp harboring envelope glycoproteins of the indicated genotypes or with RD114pp, in the presence or absence of 1 μM monensin. To avoid the nonspecific effect of monensin on the postentry step of pseudoparticles, monensin was added only for 2 h during inoculation of pseudoparticles or chimeras, as described for panel B, lane c. Cells were then cultured for 72 h at 37°C. (D) Huh-7 cells were infected with intergenotypic chimeras harboring envelope glycoproteins of genotypes 3a or 5a, in the presence (as described for panel B, lane a) or absence of 1 μM monensin. Cells were then cultured for 30 h at 37°C. For panels A to C, infection levels were quantified by luminometry, whereas for panel D, infection levels were quantified by flow cytometry. Results are presented as means and SD for three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Monensin does not inhibit HCV replication and egress.
Although the above data indicate that monensin strongly affects HCV entry, we could not exclude additional effects on other steps of the HCV life cycle. To analyze the effect of monensin on HCV replication, we used Lunet-CD81 cells, a Huh-7-derived cell clone expressing the firefly luciferase enzyme (FLuc), thus permitting assessments of cell density and viability, which are proportional to the level of FLuc expressed in the cells (42). Lunet-CD81-FLuc cells were electroporated with in vitro-transcribed assembly-defective JFH1-ΔE1E2-Rluc RNA to bypass the entry step and avoid any interference with late steps of the HCV life cycle. Boceprevir was used in parallel as a control of inhibition of viral replication. As shown in Fig. 3A, monensin had no effect on HCV replication, even after a long period of treatment (at 72 h postelectroporation).
FIG 3.

Monensin does not affect HCV replication and secretion. (A) Huh-7-Lunet-CD81-FLuc cells were electroporated with JFH1-ΔE1E2-RLuc RNA, cultured for 5 h at 37°C in complete culture medium, and then treated with monensin (0.1 μM or 1 μM) or boceprevir (0.5 μM). Cells treated with ethanol (solvent of monensin) or DMSO (solvent of boceprevir) were used as controls (CTL). Cells were lysed at 24, 48, and 72 h postelectroporation. Firefly luciferase activities were quantified to assess cell density and viability, and Renilla luciferase activities were quantified to assess HCV replication. Renilla luciferase activities were normalized to firefly luciferase activities. (B) Huh-7 cells were infected with HCVcc. Two hours later, virus was removed and replaced by culture medium containing monensin at the indicated concentrations. In parallel, HCV-infected Huh-7 cells were treated with brefeldin A (BFA) for 8 h before harvest. Supernatants were collected at 36 h postinfection and used to infect naive Huh-7 cells. Titers were determined 40 h later by indirect immunofluorescence assay. ffu, focus-forming units. Results are presented as means and SD for three independent experiments. *, P < 0.05; **, P < 0.01.
To investigate whether monensin impairs HCV assembly or secretion, Huh-7 cells were infected with HCVcc for 2 h and then cultured with 0.1 or 1 μM monensin. Brefeldin A (BFA) was used in parallel as a control of inhibition of HCV release. Thirty hours later, supernatants were collected and infectivity titers determined by indirect immunofluorescence assay. As shown in Fig. 3B, monensin had no effect on HCVcc infectious titers, indicating that this molecule does not affect egress of HCV.
Monensin does not act on viral particles.
Since we showed that monensin blocks the entry step of HCV, we next wondered whether monensin would act directly on viral particles. For this purpose, HCVcc were preincubated with 1 μM monensin before contact with target cells and then diluted to 0.1 μM monensin during viral inoculation. Under these experimental conditions, if monensin acts directly on virions, it should have a stronger inhibitory effect on infection, or at least the same as that observed with the same concentration (1 μM) during the inoculation, without preincubation, as shown in Fig. 2A. Otherwise, no inhibitory effect should be observed, because the presence of 0.1 μM monensin during viral inoculation does not significantly inhibit HCV infection (Fig. 4A, 0.1 CTL lane, and data not shown). As shown in Fig. 4A, preincubation of virus with 1 μM monensin did not significantly affect HCV infection, demonstrating that monensin does not act on HCV particles.
FIG 4.

Monensin affects neither viral particles nor HCV receptor expression levels. (A) Huh-7 cells were infected with HCVcc-Luc (diluted 10 times) in the presence of 0.05 or 0.1 μM monensin or with HCVcc-Luc pretreated for 1 h at 37°C with 0.5 or 1 μM monensin and then diluted 10 times, to reach final concentrations during infection of 0.05 or 0.1 μM, respectively. (B) Huh-7 cells were treated with 0 (CTL), 0.1, or 1 μM monensin and stained for SRB1, CD81, CLDN1, EGFR, and CD151, using corresponding MAbs. Expression levels were analyzed by flow cytometry, and results are presented as percentages of the CTL condition for each protein. OCLN levels in treated Huh-7 cells were analyzed by Western blotting (OCLN) and compared to the level in 786-O cells, which have been described to express low levels of OCLN. The expression of tubulin was also analyzed to control the protein amounts. The results shown in the histograms in panels A and B are presented as means and SD for three independent experiments. (C) Huh-7 cells were cultured with the indicated concentrations of monensin and fixed after 24 h. Cells were labeled with the TS81 anti-CD81 and 8A9 anti-CLDN1 MAbs followed by secondary antibodies conjugated with Alexa 488 (green staining) and Alexa 555 (red staining), respectively. Colocalization was calculated as the number of yellow pixels (green and red pixels) over the number of yellow and red pixels, using CoLocalizer Pro software.
Monensin does not modulate HCV entry factor expression and localization.
HCV entry is a multistep process involving several cellular attachment and entry factors. Therefore, we next determined whether monensin inhibits HCV entry by modulating the expression levels of the aforementioned cellular factors. Huh-7 cells were treated with 0.1 μM or 1 μM monensin for 24 h, and entry factors were analyzed by flow cytometry or Western blotting (WB). Cell surface expression levels of SRB1, CD81, CLDN1, and EGFR, for which antibodies against extracellular domains are available, were analyzed by flow cytometry. In contrast, OCLN, for which no such antibody is available, was analyzed by WB. CD151, another tetraspanin not involved in HCV entry, was used as a control. As shown in Fig. 4B, no significant effect of monensin on the expression levels of HCV entry factors was found, indicating that monensin does not block HCV entry by affecting HCV entry factor expression. Since it has been shown that CD81 and CLDN1 colocalize and interact in areas of the plasma membrane to form a complex that is essential to HCV entry (63, 64), we also performed colocalization studies as previously described (65). As shown in Fig. 4C, treatment of Huh-7 cells with different concentrations of monensin did not affect the colocalization of CD81 with CLDN1.
Monensin inhibits a late step of HCV entry.
After attachment to the cell surface and binding to entry factors, HCV virions are internalized by clathrin-mediated endocytosis (15, 16). Following internalization, HCV is transported to early endosomes along actin stress fibers, where fusion seems to take place (16, 66). To determine at which step HCV entry is impaired by monensin, we administered monensin at different intervals during the early phase of infection. Virus attachment and binding were performed at 4°C (Fig. 5A, steps 1 and 2), and cells were then shifted to 37°C to allow endocytosis and fusion (Fig. 5A, step 3). Strikingly, the addition of monensin during the third step led to a strong inhibition of HCV infection, which was almost as strong as that observed when monensin was present during all three steps. In contrast, no inhibition was observed when monensin was added during the attachment/binding steps. Together, these results indicate that monensin blocks either the endocytosis or fusion step of HCV entry.
FIG 5.

Monensin inhibits HCV entry at a late step and also inhibits enveloped virus infection. (A) Huh-7 cells were infected with HCVcc-Luc for 1 h at 4°C (step 1; attachment/binding), and then the virus was removed and cells were incubated again at 4°C for 1 h (step 2; postattachment/binding). Finally, cells were shifted to 37°C for 1 h (step 3; endocytosis/fusion) and left at 37°C for 21 h. Monensin was added to 1 μM during either step 1, step 2, step 3, or steps 1 to 3. (B) Huh-7 or HEp-2 cells (for CVB4) were infected with the indicated viruses in the presence of 0 (CTL), 0.1, or 1 μM monensin added to the medium 2 h before infection, during infection, and full time after infection. HCV and SinV infections were scored by luminometry. YFV and AdV infections were scored by indirect immunofluorescence assay and flow cytometry, respectively. CVB4 infections were scored by evaluating the cytopathic effect.
Next, in order to define its activity spectrum and better understand the mechanism of action of monensin, we analyzed its antiviral activity against other viruses. We used yellow fever virus (YFV), another member of the Flaviviridae family, and Sindbis virus (SinV), which is unrelated to HCV but is an enveloped virus believed to exploit the same entry route as that used by HCV. In addition, we used human adenovirus 5 (AdV), a nonenveloped, pH-independent virus (67) that enters cells via clathrin-mediated endocytosis, and coxsackievirus B4 (CVB4), a nonenveloped virus whose entry mechanism is not well defined but which is believed to be pH independent, partly involving clathrin-mediated endocytosis. For all infections, monensin at 0.1 μM or 1 μM was added 2 h before infection and kept during and after viral inoculation (Fig. 5B). Interestingly, monensin led to a strong inhibition of YFV infection at both concentrations that was even stronger than that observed on HCV (Fig. 5B). Moreover, monensin at 0.1 μM barely reduced SinV infection, whereas monensin at 1 μM abrogated SinV infection. On the other hand, no reduction of AdV and CVB4 infections was observed (Fig. 5B).
Collectively, these results indicate that (i) monensin can affect other members of the Flaviviridae family and other enveloped viruses, (ii) monensin does not interfere with nonenveloped virus infection, (iii) monensin does not substantially affect clathrin-mediated viral endocytosis, and (iv) the antiviral activity of monensin is likely related to its effect on membrane fusion by acting on endosome acidification.
To confirm the last claim, we next analyzed the effect of monensin on organelle acidification in Huh-7 cells. Acridine orange (AO) was used to test for acidic organelles. AO is a weakly basic fluorescent probe that emits green fluorescence at low concentrations and gives a red fluorescence at high concentrations. AO accumulates in acidic compartments, where it oligomerizes and fluoresces. In contrast, alkalization of the endocytic structure with bafilomycin A1 is accompanied by a change in AO fluorescence (68, 69). Indeed, AO produced a highly punctate red staining in Huh-7 cells (Fig. 6A) that we quantified by using arbitrary units grossly corresponding to acidic compartments stained with AO (Fig. 6B). The addition of bafilomycin A1 for 2 h or 24 h led to a drastic decrease of red AO fluorescence (Fig. 6A and B). Interestingly, this effect was also observed in Huh-7 cells treated with monensin at 1 μM for 2 h or 24 h, indicating that the treatment of Huh-7 cells with monensin leads to an alkalization of intracellular organelles. Therefore, the antiviral activity of monensin is likely related to its effect on organelle acidification.
FIG 6.
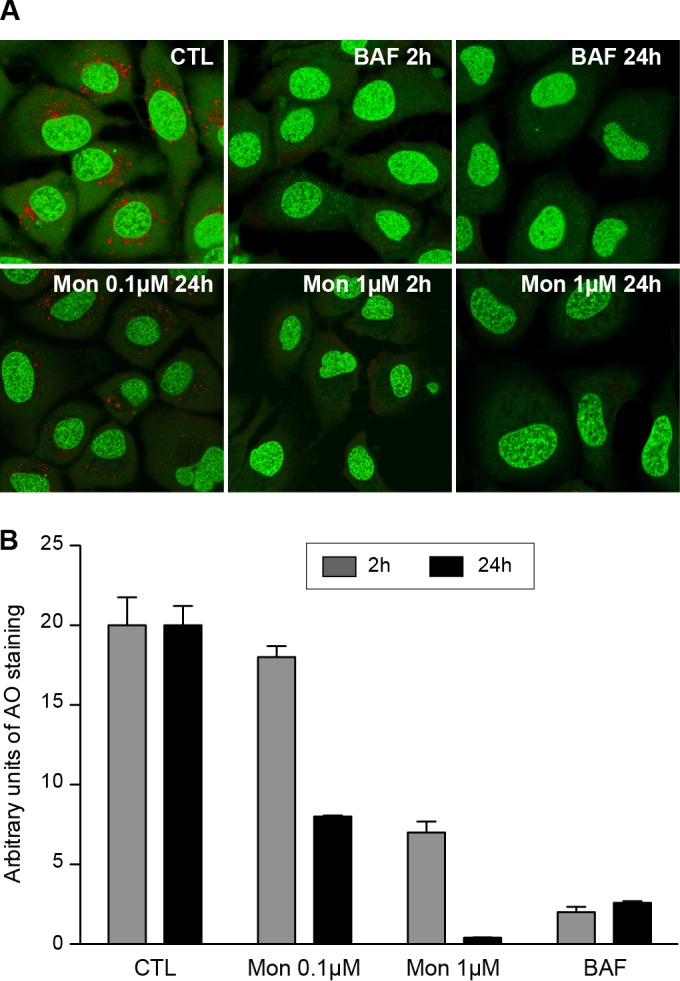
Monensin leads to an alkalization of intracellular organelles. (A) Huh-7 cells were treated for 2 h or 24 h with monensin (Mon; 0.1 or 1 μM) or bafilomycin A1 (BAF; 25 nM) or were left untreated (CTL). After incubation with AO, cells were observed by confocal microscopy. (B) Quantification was performed with ImageJ software. The threshold of each field was adjusted to the same level. To calculate the arbitrary units of AO staining, the number of particles of >0.1 μm2 was divided by the number of nuclei in each field. Results are presented as means and SD for two independent experiments.
HCV cell-to-cell transmission is a pH-dependent process.
As described previously, in addition to cell-free infection, HCV can also propagate by cell-to-cell transmission. Since cell-to-cell transmission has been suggested to be an important route of transmission for HCV (25), we next analyzed the effect of monensin on this process. For this purpose, Huh-7 cells were infected with HCVcc for 2 h and then cultured with neutralizing anti-E2 antibody (3/11) in the presence or absence of monensin (0.1 μM and 1 μM). At 3 days postinfection, foci were visualized by immunofluorescence (Fig. 7A), and sizes of foci were measured by counting the number of cells per focus. Our results showed that monensin at 0.1 μM led to a strong reduction of the number of cells per focus (Fig. 7B), whereas monensin at 1 μM totally inhibited HCV transmission (data not shown). We next used another approach to analyze cell-to-cell transmission, as described previously (65). Briefly, HCVcc-infected Huh-7 cells were labeled with 5-chloromethylfluorescein diacetate (CMFDA) and then mixed with naive Huh-7 cells and cultured in the presence of neutralizing MAb (3/11; 50 μg/ml) and monensin (0.1 μM and 1 μM). Cocultures were incubated for 24 h, and cell-to-cell transmission levels were quantified by flow cytometry. It has to be noted that to avoid false-positive results in such an assay, it is beneficial to use short incubation times (24 to 48 h). As shown in Fig. 7C, monensin at 0.1 μM and 1 μM significantly inhibited HCV cell-to-cell transmission, indicating that this process is likely dependent on the vesicular pH. To strengthen our hypothesis, we next treated cells with chloroquine (CQ), a lysosomotropic weak base that inhibits acidification of lysosomes and endosomes. CQ is a widely used molecule for the treatment of malaria and exerts inhibitory effects against several RNA viruses, including HCV (15, 70, 71). As shown in Fig. 7A and B, chloroquine also led to a large decrease of the number of cells per focus, indicating that it abrogates HCV cell-to-cell transmission. Together, these results indicate that HCV cell-to-cell transmission is a pH-dependent process.
FIG 7.

Monensin blocks cell-to-cell transmission. (A) Huh-7 cells were seeded on coverslips and infected with HCVcc for 2 h at 37°C. Cells were then washed and cultured for 72 h at 37°C in culture medium containing the 3/11 neutralizing MAb (50 μg/ml), in the presence or absence of monensin (Mon; 0.1 μM) or chloroquine (CQ; 1.75 μM), as indicated. Foci of infected cells were detected using an A4 anti-E1 indirect immunofluorescence assay. (B) The number of infected cells per focus was determined in the presence of no drug (CTL), 0.1 μM monensin, or 1.75 μM chloroquine. (C) HCVcc-infected donor cells were labeled with CMFDA, mixed with naive Huh-7 cells (ratios of 1:4 and 1:6), and cultured in the presence of a saturating concentration of the 3/11 MAb (50 μg/ml) and with monensin at the indicated concentrations. Cocultures were incubated for 24 h at 37°C and then stained for HCV NS5. Cell-to-cell transmission levels were quantified by flow cytometry. ***, P < 0.001.
Selection of a monensin-resistant virus.
To further investigate the mechanism of action of monensin, we next aimed to select monensin-resistant viruses by propagating HCVcc for several passages in the presence of increasing concentrations of monensin (JFH1-Mon). Passages were performed in parallel with virus cultured in the presence of corresponding amounts of ethanol (JFH1-EtOH). After 25 passages, both viral stocks were used to infect naive Huh-7 cells in the presence of monensin during viral inoculation. Interestingly, viruses amplified in the presence of monensin (JFH1-Mon) were 2.5-fold more resistant to increasing concentrations of monensin than control viruses (JFH1-EtOH) (Fig. 8A). Total RNAs were extracted from cells infected with JFH1-Mon or JFH1-EtOH, and viral genomes were amplified by RT-PCR and sequenced. We identified several mutations in the E1 and E2 JFH1-Mon sequences which were not found in the JFH1-EtOH or JFH1wt sequences. We next replaced the JFH1 DNA fragment corresponding to the structural HCV proteins with that of JFH1-Mon. Several JFH1-Mon DNA clones carrying different combinations of mutations in E1 and E2 were selected, in vitro transcribed, and electroporated into Huh-7 cells to generate viral clone stocks carrying different combinations of mutations. The analysis of their resistance toward monensin showed that one clone, named FL-8, was resistant to monensin compared to JFH1 (Fig. 8B). Dose-response experiments (Fig. 8E) showed that the IC50s of monensin for the JFH1 and FL-8 viruses were 0.23 μM and 0.43 μM, respectively (Table 1). Thus, our results suggested that we had generated HCV particles that were resistant to monensin and therefore less dependent on acidic pH for their entry. To confirm this assumption, we next assayed the capacity of the JFH1-Mon and FL-8 viruses to infect Huh-7 cells in the presence of CQ. As shown in Fig. 8C and D, the JFH1-Mon and FL-8 viruses were able to infect Huh-7 cells in the presence of increasing concentrations of CQ, whereas infection by control viruses (JFH1-EtOH and JFH1) was dramatically reduced. Dose-response experiments (Fig. 8E) showed that the IC50s of CQ for the JFH1 and FL-8 viruses were 1.9 μM and 6 μM, respectively (Table 1). Together, these results indicate that we managed to select viruses that use an entry route that is less dependent on the acidic endosomal pH.
FIG 8.

Generation of mutant viruses using an entry route that is less dependent on endosomal pH and clathrin. (A) Huh-7 cells were pretreated for 2 h with monensin and then infected with JFH1-EtOH or JFH1-Mon for 2 h in the presence of monensin or EtOH (CTL). Cells were then incubated for 30 h in complete medium supplemented with monensin at the indicated concentrations. (B) Huh-7 cells were infected with JFH1 or FL-8 as described for panel A. (C) Huh-7 cells were pretreated for 2 h with chloroquine at the indicated concentrations and then infected with JFH1-EtOH or JFH1-Mon viral populations for 2 h in the presence of chloroquine. Cells were then incubated for 30 h in complete medium supplemented with chloroquine or H2O (CTL). (D) Huh-7 cells were infected with JFH1 or FL-8 as described for panel C. (E) Dose-response curves for the JFH1 and FL-8 viruses and monensin or chloroquine. Infections and treatments were performed as described for the previous panels. (F) Huh-7 cells were pretreated with chlorpromazine (5 μg/ml) for 30 min and then infected with JFH1 or FL-8 for 2 h in the presence of chlorpromazine or H2O (CTL). Cells were then incubated for 30 h in complete medium supplemented with chlorpromazine. Results are presented as means ± SD for three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
TABLE 1.
E1 and E2 amino acid changes that emerged under the selective pressure of monensin
| Virus | Amino acid and position |
Titer (FFU/ml) | Monensin IC50 (μM) | Chloroquine IC50 (μM) | ||||
|---|---|---|---|---|---|---|---|---|
| E1 | E2 (HVR1) | |||||||
| JFH1 | Y297 | V343c | I399d | S404e | Q409f | 1.7 × 105 | 0.23 | 1.9 |
| FL-8 | H297b | A343 | T399d | G404 | R409 | 1.1 × 105 | 0.43 | 6 |
| Y297H mutant | H297 | 8.3 × 104 | 0.44 | ND | ||||
| I399T mutant | T399 | 1.7 × 105 | 0.44 | ND | ||||
| Q409R mutant | R409 | 0 | NDa | ND | ||||
| T+R mutant | T399 | R409 | 0 | ND | ND | |||
ND, not determined.
Mutation previously described for viruses cultured in the presence of ferroquine (60).
Nonconserved amino acid (A in other genotypes).
Highly conserved amino acid (the I399T mutation introduces an N-glycosylation site into HVR1).
Conserved amino acid.
Amino acid that is totally conserved among genotypes.
HCV enters target cells through clathrin-mediated endocytosis followed by fusion at low pH with the membrane of an early endosome (15–18). Since we generated HCV particles that were less pH dependent for their entry, we next studied whether their endocytosis was still dependent on clathrin-coated pits. Huh-7 cells were pretreated for 30 min with chlorpromazine (5 μg/ml), an inhibitor of clathrin assembly at the plasma membrane, and then infected with the FL-8 or JFH1 virus (Fig. 8F). Chlorpromazine dramatically reduced JFH1 infection levels, as previously described (15). Surprisingly, the FL-8 virus was partially inhibited by chlorpromazine, indicating that a fraction of this virus can potentially enter cells independently of clathrin-mediated endocytosis.
Collectively, our results indicate that under the selective pressure of monensin, we selected HCV particles that had evolved to use a new entry route that appears to be less dependent on clathrin and endosomal pH.
Characterization of the Y297H and I399T mutations.
The FL-8 clone displayed several mutations in envelope glycoproteins: two mutations in E1 and three mutations in HVR1 of E2 (Table 1). The E1 Y297H mutation has previously been described for viruses cultivated in the presence of ferroquine but did not confer any resistance to this drug (60). The E1 V343A mutation was on a nonconserved amino acid frequently found as an A in other genotypes. The E2 I399T mutation potentially introduced an N-glycosylation site in HVR1, the E2 S404G mutation was on a residue that is conserved among genotypes, and the E2 Q409R mutation was on a glutamine residue that is totally conserved among genotypes (Table 1). We made the hypothesis that an adaptive mutation(s) involved in the partial pH independence of the FL-8 virus was likely to occur at a highly or totally conserved residue(s). Therefore, the Y297H mutation in E1 and the I399T and Q409R mutations in E2 were introduced into strain JFH1 by reverse genetics (Table 1). However, introduction of the Q409R mutation alone or in combination with the I399T mutation led to the production of noninfectious viruses (Table 1). Additional mutations are likely necessary to compensate for the effect of the Q409R mutation and to generate infectious particles. In contrast, Y297H and I399T mutants were infectious and showed a significant amount of resistance to monensin (Fig. 9A) compared to that of the JFH1 virus. Indeed, the Y297H and I399T mutations were able to confer the same level of resistance to monensin as that of the FL-8 virus, which carries several adaptive mutations (Table 1). Together, these results indicate that resistance to monensin maps to the E1 and E2 envelope proteins, at amino acids Y297 and I399, respectively. It is worth noting that FL-8 and Y297H and I399T mutant virus resistance to monensin was not due to differences in viral fitness and/or titers of mutants (Fig. 9B and Table 1).
FIG 9.

Generation of monensin-resistant viruses. (A) Huh-7 cells were pretreated for 2 h with monensin and then infected with mutant viruses for 2 h in the presence of monensin or EtOH (CTL). Cells were then incubated for 30 h in complete medium supplemented with monensin at the indicated concentrations. (B) Huh-7 cells were electroporated with JFH1, FL-8, or I399T or Y297H mutant RNAs. Supernatants were collected 24, 48, 72, and 96 h after electroporation, and virus titers as well as viral RNA amounts were determined. Specific infections were calculated by dividing the virus titer by the corresponding number of viral RNA copies quantified in the same volume of supernatant. Results are presented as means ± SD for four independent experiments. (C) (Top) Amino acid sequences of HVR1 of the JFH1 and I399T viruses. (Bottom) Analysis of infected cell lysates by 8% SDS-PAGE followed by 3/11 Western blotting. The dashed line allowed us to evaluate the shift in migration profile, which was due to the presence of an additional N-glycosylation site in E2 HVR1 of the FL-8 and I399T viruses. The sizes (in kilodaltons) of protein molecular mass markers are indicated on the left.
As described previously, the I399T mutation generates an N-V-T sequon in HVR1 that might be used for the addition of an N-glycan on N397 (Fig. 9C). Interestingly, the presence of this additional glycosylation site was confirmed by the change in the migration profile of E2 bearing the I399T mutation (Fig. 9C, FL-8 and I399T lanes) compared to that of wild-type E2 (Fig. 9C, JFH1 lane).
Since mutations in envelope glycoproteins can affect the sensitivity of HCV to antibody neutralization and modulate the use of entry factors (72), we next tested the mutants in antibody neutralization assays. We first used AR3A and AR5A, two well-characterized human MAbs (73), and the rat 3/11 MAb. AR3A is an anti-E2 conformation-sensitive human antibody recognizing a discontinuous epitope located within the CD81 binding site, whereas the AR5A MAb recognizes a discontinuous epitope on E1E2 which does not interfere with CD81 binding (49). 3/11 is an antibody recognizing a linear epitope located within the CD81 binding region of E2 (18). As shown in Fig. 10A, the I399T mutation reduced the sensitivity of the virus to neutralization with low concentrations of the AR3A and AR5A MAbs compared to that of JFH1, whereas the Y297H mutation had no effect. The FL-8 mutant virus was also less neutralized by the AR3A and AR5A MAbs, likely due to the presence of the I399T mutation in this virus. In contrast, the FL-8 and I399T viruses were more sensitive than JFH1 to neutralization with the 3/11 MAb, whereas the Y297H mutation had no significant effect.
FIG 10.

Neutralization and densities of monensin-resistant viruses. (A) Viruses were incubated on Huh-7 cells with the AR3A, AR5A, 3/11, and JS81 antibodies at the indicated concentrations for 2 h in complete medium. Cells were then incubated for 30 h in complete medium. Infection levels were quantified by indirect immunofluorescence assay. Results are presented as means ± SD for three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001. (B) SRB1 and CD81 expression is downregulated by siRNA. A nontargeting siRNA was used as a control. The knockdown efficiency in Huh-7 cells was determined by Western blotting with anti-SRB1 and anti-CD81 antibodies 72 h after transfection. An anti-β-tubulin antibody was used as a loading control. Cells were infected 72 h after transfection for 2 h in serum-free medium. Cells were then washed and incubated for 30 h in complete medium. Virus infectivity was measured by immunofluorescence assay. Results are presented as means and SD for three independent experiments. (C) Viruses were preincubated with a polyclonal anti-ApoE antibody at the indicated concentrations for 1 h at 37°C before inoculation of cells in the presence of the antibody. Cells were then incubated for 30 h in complete medium. Infection levels were quantified by indirect immunofluorescence assay. Results are presented as means ± SD for three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001. (D) One-milliliter supernatant samples containing viruses were layered on an iodixanol gradient and ultracentrifuged. Twelve fractions were collected, and their densities were measured. The JFH1-ΔHVR1 virus was used as a positive control for particles with an increased density. Fractions were titrated, and the result for each fraction is expressed as the percentage of the total virus infectivity in the gradient. Results are presented as means for three independent experiments. SD were omitted for clarity.
These results indicate that the I399T mutation likely induces some conformational changes in the E2 structure, possibly in the CD81 binding site. The use of an anti-CD81 MAb in the neutralization assay (Fig. 10A, JS81 graph) showed that the Y297H mutation likely also leads to conformational changes in the E2 structure, since neutralization of the Y297H virus was significantly reduced compared to that of the JFH1 virus. Unexpectedly, the FL-8 mutant carrying both mutations did not show any significant difference in neutralization with the JS81 MAb. Together, our results suggest that the interaction between E2 and CD81 might be different in the context of the I399T and Y297H mutations. However, CD81 remains an essential entry factor for viruses carrying these mutations, since the silencing of CD81 totally abrogated their infection (Fig. 10B).
HVR1 is a region of E2 involved in viral immune escape and the use of SRB1 and CD81 (8, 74). It also plays a role in virus infectivity by modulating the association of viral particles with apolipoproteins (50, 74–76). In this context, we sought to determine if the I399T and Y297H mutations, which likely induce conformational changes in the E2 structure, would affect the usage of SRB1 and the association with apolipoproteins. We used a panel of anti-SRB1 antibodies in neutralization assays (data not shown), but we did not see any significant neutralization of infection. We next analyzed the effect of silencing SRB1 expression on the infectivity of mutated viruses. As shown in Fig. 10B, the infectivity of mutant viruses was reduced to the same level as that for JFH1, indicating that the I399T and Y297H mutations did not affect SRB1 usage. We next used an antibody directed against apolipoprotein E (ApoE) in the neutralization assay (Fig. 10C). We used a polyclonal anti-ApoE antibody because previous studies on viruses lacking HVR1 (ΔHVR1) have shown that polyclonal anti-ApoE antibodies differently neutralize ΔHVR1 viruses, while monoclonal anti-ApoE antibodies do not (50, 76, 77). As shown in Fig. 10C, we found that neutralization of the I399T and FL-8 viruses was significantly increased compared to that of the JFH1 virus. These results indicate that, as described for ΔHVR1 viruses (50, 76), the I399T mutation might modulate the association of viral particles with ApoE. In agreement with this, the density of infectious viral particles bearing the I399T mutation, as analyzed by density gradient ultracentrifugation, was increased (Fig. 10D), with a peak of infectivity at a density of 1.12 g/ml, compared to that of the JFH1 virus, which produces infectious particles with a density of 1.08 g/ml. Interestingly, the densities of infectious particles produced from the I399T, FL-8, and ΔHVR1 viruses were strictly identical (1.12 g/ml), whereas the density of Y297H particles was slightly lower (1.11 g/ml). Together, these results indicate that the I399T mutation in HVR1 leads to the production of viral particles with a higher density.
The I399T mutation in HVR1 drastically reduces HCV cell-to-cell transmission.
We showed above that cell-to-cell transmission of HCV is likely a pH-dependent process. Therefore, we next sought to determine the levels of cell-to-cell transmission for the monensin-resistant viruses that are less dependent on acidic pH for their entry. First, Huh-7 cells were infected with the JFH1, FL-8, and I399T and Y297H mutant viruses for 2 h and then cultured in the presence of the anti-E2 neutralizing antibody 3/11. It has to be noted that even though we previously showed that the FL-8 and I399T viruses were more sensitive to neutralization with 3/11, the concentration of 3/11 (50 μg/ml) used in our cell-to-cell assays totally abolished cell-free infection (Fig. 10A). At 3 days postinfection, foci were visualized by immunofluorescence (Fig. 11A), and sizes of foci were measured by counting the number of cells per focus. Surprisingly, we found that cell-to-cell transmission of viruses bearing the I399T mutation was dramatically reduced (Fig. 11B) and that the transmission of the Y297H mutant was also reduced, but to a lesser extent. To confirm these results, we next made cocultures of differentially labeled infected donor and naive acceptor cells and evaluated cell-to-cell transmission in the presence of the neutralizing 3/11 MAb by flow cytometry (Fig. 11C). As observed previously, the cell-to-cell transmission of the FL-8 and I399T viruses was highly reduced, indicating that the I399T mutation affects this transmission route. In contrast, we did not observe any effect of the Y297H mutation with this approach of cell-to-cell transmission analysis. It has to be noted that the use of the AR3A MAb as the neutralizing antibody led to the same results (data not shown), indicating that the effect of the I399T mutation on cell-to-cell propagation was not related to a differential susceptibility of viral particles to antibody neutralization.
FIG 11.

The I399T mutation in HVR1 inhibits cell-to-cell transmission. Huh-7 cells were seeded on coverslips and infected with JFH1 or mutated viruses for 2 h at 37°C. Cells were then washed and cultured for 72 h at 37°C in culture medium containing the 3/11 neutralizing MAb (50 μg/ml). (A) Foci of infected cells were detected using an A4 indirect immunofluorescence assay. (B) The number of infected cells per focus was determined for each virus. (C) Huh-7 cells infected with JFH1 or a mutant virus were labeled with CMFDA, mixed with naive Huh-7 cells, and cultured in the presence of a saturating concentration of the 3/11 MAb (50 μg/ml). Cocultures were incubated for 44 h at 37°C and then stained for HCV NS5. Cell-to-cell transmission levels were quantified by flow cytometry. ***, P < 0.001.
Together, our data show that the I399T mutation, which likely introduces an N-glycan in HVR1, drastically reduces viral cell-to-cell spread, indicating that HVR1 plays a major role in this process.
DISCUSSION
The aim of this study was to characterize the effects of monensin A on the HCV life cycle and to gain new insights into the understanding of HCV transmission. We showed that monensin inhibits HCV infection in a dose-dependent manner by blocking the entry step. Indeed, monensin leads to an alkalization of intracellular organelles in Huh-7 cells that affects a late step of entry, probably the fusion step between viral and cellular membranes, which is known to be pH dependent. In addition to the inhibition of cell-free transmission, monensin also inhibits cell-to-cell transmission, demonstrating for the first time that this process is dependent on the vesicular pH. Interestingly, by long-term culture of HCV in the presence of increasing concentrations of monensin, we selected viruses that were resistant to monensin but also to chloroquine and chlorpromazine. These adapted viruses evolved to use a new entry route that is probably less dependent on clathrin and the endosomal pH. We identified two mutations, Y297H in E1 and I399T in E2 HVR1, that enable HCV to use an entry route that is less dependent on the acidic endosomal pH. The characterization of monensin-resistant mutants showed that the Y297H and I399T mutations likely induce conformational changes in the E2 structure and modify the association of viral particles with lipoproteins. Most strikingly, we found that the I399T mutation in HVR1 dramatically reduces the cell-to-cell transmission pathway.
Due to monensin toxicity in humans (78, 79), it will be difficult to use monensin as a new therapeutic treatment against HCV. However, as shown in this study, monensin is a valuable tool for analyzing the late steps of HCV entry, i.e., membrane fusion. In addition, due to a broad spectrum of biological activities, monensin derivatives are a major focus of drug development aiming at reducing the toxicity and obtaining new compounds with improved efficacy. In particular, certain derivatives of monensin, such as urethanes, seem promising (80–82). Thus, it will be of interest to evaluate the anti-HCV activity of monensin derivatives in the future.
Enveloped viruses enter cells either by direct fusion at the plasma membrane or by receptor-mediated endocytosis. For a large number of retroviruses, penetration into the cytoplasm of the target cell occurs directly at the plasma membrane by a fusion mechanism triggered through the interaction of viral envelope proteins with their cognate receptors. In contrast, for numerous viruses, such as influenza virus and SinV, following attachment to receptors on the cell surface, the virus-receptor complex is internalized via clathrin-coated pits and delivered to intracellular vesicles of the endosomal compartment. In these vesicles, the acidic environment induces a conformational change in the viral fusogenic envelope glycoproteins, which is believed to expose a hydrophobic domain that interacts with the endosomal membrane. This interaction leads to the fusion between cellular and viral membranes, delivering the nucleocapsid into the cytosol. Such a mechanism is likely used by HCV. Indeed, it has been demonstrated that virions enter target cells by clathrin-mediated endocytosis (15) and that fusion occurs in the early endosomes (16). The use of vacuolar acidification inhibitors (15–18, 83–85; this study) showed that HCVcc and HCVpp entry is pH dependent, indicating that the acidic pH of endosomes triggers the fusion machinery, likely by inducing conformational changes in E1E2 envelope glycoproteins. However, exposure of cell surface-bound HCV to an acidic pH followed by a return to neutral pH does not affect viral infectivity (16, 17). In addition, Tscherne et al. have shown that low-pH-triggered entry of HCVcc requires incubation at 37°C, indicating that productive HCV entry needs interactions/processes that do not occur at 4°C (17). These data indicate that HCV envelope proteins require a priming event, which occurs at 37°C, to become sensitive to low pH. Concurring with this, it has been shown that HCV pretreatment with the large extracellular loop (LEL) of CD81 enhances infectivity, induces conformational changes in E1 and E2, allows E2 binding to liposomes, irreversibly inactivates particles at low pH in the absence of a target membrane, and leads to the fusion with the plasma membrane of permissive cells at acidic pH, indicating that CD81 plays a central role in HCV entry by priming HCV for low-pH-dependent conformational changes (83). In accordance with the studies described above, our results show that HCV entry is inhibited by monensin, a Na+/H+-exchanging ionophore that raises the cytosolic Ca2+ concentration and the pH. Importantly, we demonstrated that monensin blocks HCV infection with maximal potency when added during the first hour of the shift to 37°C, after the 4°C postattachment/binding step. Almost complete insensitivity to monensin was achieved about 2 h after infection, indicating that almost all particles have undergone fusion and reached the cytosol at this time. The use of monensin on infections with viruses from different families indicated that monensin does not block internalization but rather acts on the fusion machinery, likely by inducing an alkalization of intracellular organelles, as demonstrated by AO staining. Importantly, we demonstrated that both monensin and chloroquine block the cell-to-cell transmission route of HCV, for which precise mechanisms need to be defined. Our data indicate that cell-free and cell-to-cell transmission of HCV may share common mechanisms involving a pH-dependent fusion step between cellular and viral membranes.
Until now, the mechanism mediating HCV fusion has not been elucidated, and the precise role in membrane fusion played by E1E2 envelope proteins needs to be defined. It has been suggested that the fusion mechanism occurring for other viruses of the Flaviviridae may apply to HCV. The use of vacuolar acidification inhibitors (15–18, 83–85; this study) showed that HCVcc and HCVpp entry is pH dependent, thus allowing HCV RNA delivery into the cytoplasm, supporting the hypothesis of similar fusion mechanisms between HCV and other Flaviviridae viruses. Indeed, the HCV envelope glycoproteins have long been assumed to belong to class II fusion proteins, based on the fact that HCV is a member of the Flaviviridae family. In addition, some amino acids in E1 (residues 262 to 290) and E2 (residues 416 to 430, 504 to 522, and 604 to 624) were identified as being important for fusion (86, 87). However, the structure of the E2 core protein (excluding the HVR1) was recently solved and showed that the E2 core has an architecture made of a central β sandwich flanked by front and back layers consisting of loops, helices, and β sheets. E2 has a compact globular shape that does not fit the highly extended three-domain class II fusion fold (88, 89). These new data strongly suggest that E1 should be the fusion protein, or at least a fusion partner of an E1E2 fusion complex formed upon conformational rearrangements (87, 90). The results of our study concur with this hypothesis. With the FL-8 mutant, we identified two residues, one in E1 and one in E2 HVR1, that, when mutated, led to a less pH-dependent entry route. The Y297H mutation in E1 did confer resistance to monensin, indicating that this residue by itself is able to modify the HCV entry process, or at least the pH requirements. Interestingly, this mutation also appeared following the use of ferroquine, an analog of chloroquine with weak base properties (91) that modifies the fusogenic properties of E1E2 (60). Together, these observations suggest that the Y297 residue in E1 might be involved in the fusion process, possibly by acting in concert with other residues in E1 and/or E2. In concurrence with this, we identified an additional mutation in E2 HVR1 that also confers resistance to monensin. This mutation (I399T) generates an N-V-T sequon that is used for the addition of an N-glycan on N397 in HVR1. This additional posttranslational modification likely leads to conformational changes in HVR1 and/or other regions in E2. Thus, residues in E1 and in HVR1 of E2 likely play a role in the fusion process. HVR1 might therefore contribute to the E2 folding rearrangement that likely occurs during the fusion step. The Y297H and I399T mutations might induce conformational changes in an envelope protein(s) similar to those that occur in acidic compartments and thus allow entry in a less pH-dependent manner.
Interestingly, we found that the FL-8 mutant was also able to infect cells in the presence of chlorpromazine, indicating that this mutant likely uses an alternative entry route that is less dependent on vesicular pH and clathrin-coated pits. It has to be noted that a clathrin-independent but still pH-dependent entry route was described very recently (92). Thus, under the selective pressure of monensin, we generated viral particles that adapted to the drug and managed to use a new entry route.
Beside its involvement in immune escape, HVR1 is involved in the use of SRB1 and conceals the viral CD81 binding site (8, 74). In addition, HVR1 plays a role in virus infectivity (74, 75, 93) by modulating the association of viral particles with apolipoproteins, notably ApoE (76). In our study, we showed that the Y297H mutation and, more significantly, the I399T mutation increase neutralization with anti-ApoE antibodies. These results indicate that the I399T mutation might reduce the incorporation levels of ApoE into particles; further studies would be necessary to test this hypothesis. Another possibility is that ApoE may display a different conformation and/or epitope exposure in the presence of the I399T mutation and therefore have a different sensitivity to antibodies, as described recently for Jc1 particles lacking HVR1 (76), but incorporating ApoE at similar levels in JFH1 and mutated viruses. In addition, we observed that the I399T mutation dramatically reduces cell-to-cell spread, a transmission pathway for which it has been shown that ApoE incorporated into particles plays a major role (94). Combined with our observation that the I399T mutation reduces the pH dependence for viral entry, our results suggest that the association levels of lipids and ApoE with viral particles might regulate the pH-dependent fusion process during HCV entry. Further studies would be necessary to test this hypothesis.
In conclusion, the use of monensin allowed us to demonstrate that cell-free and cell-to-cell transmission pathways are both pH dependent, likely by sharing common mechanisms. We identified two monensin resistance mutations, Y297H in E1 and I399T in E2 HVR1, the latter of which leads to the addition of an N-glycan in HVR1. These mutations enable viruses to enter their target cells by an entry route that is less dependent on the acidic endosomal pH, likely through changes in the conformation of the E1E2 envelope proteins. Our results suggest that the I399T mutation in HVR1 likely decreases the association of particles with ApoE. Importantly, this mutation drastically reduces cell-to-cell transmission. Taken together, our results indicate that the regulation by HVR1 of particle association with lipids, notably ApoE, might control the pH dependence of cell-free and cell-to-cell transmission. Thus, HVR1 and ApoE incorporated into viral particles are critical regulators of HCV transmission.
ACKNOWLEDGMENTS
We thank S. Ung and A. Pillez for their technical assistance. We are grateful to S. Levy, J. McKeating, E. Rubinstein, T. Pietschmann, J. Bukh, T. Wakita, J. Ball, M. MacDonald, J.-W. Yoon, and J. McFarlane for providing us with reagents. We thank the BioImaging Center Lille-Nord de France for access to instruments.
This work was supported by the Agence Nationale de Recherches sur le Sida et les Hépatites Virales (ANRS). L.F., N.C., and T.V. were supported by a fellowship from ANRS. L.F., J.P., N.C., and T.V. were supported by a fellowship from the French Ministry of Research. S.B. was supported by a Marie Curie International Reintegration Grant (grant PIRG-GA-2009-256300).
REFERENCES
- 1.Lemon SM, Walker C, Alter MJ, Yi M. 2007. Hepatitis C virus, p 1253–1304. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Tai AW, Chung RT. 2009. Treatment failure in hepatitis C: mechanisms of non-response. J Hepatol 50:412–420. doi: 10.1016/j.jhep.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pawlotsky J-M. 2014. New hepatitis C therapies: the toolbox, strategies, and challenges. Gastroenterology 146:1176–1192. doi: 10.1053/j.gastro.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 4.Chung RT, Baumert T-F. 2014. Curing chronic hepatitis C—the arc of a medical triumph. N Engl J Med 370:1576–1578. doi: 10.1056/NEJMp1400986. [DOI] [PubMed] [Google Scholar]
- 5.Baumert T-F, Fauvelle C, Chen DY, Lauer GM. 2014. A prophylactic hepatitis C virus vaccine: a distant peak still worth climbing. J Hepatol 61:S34–S44. doi: 10.1016/j.jhep.2014.09.009. [DOI] [PubMed] [Google Scholar]
- 6.Lindenbach BD, Thiel H-J, Rice CM. 2007. Flaviviridae: the viruses and their replication, p 1101–1152. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 7.Fénéant L, Levy S, Cocquerel L. 2014. CD81 and hepatitis C virus (HCV) infection. Viruses 6:535–572. doi: 10.3390/v6020535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Scarselli E, Ansuini H, Cerino R, Roccasecca RM, Acali S, Filocamo G, Traboni C, Nicosia A, Cortese R, Vitelli A. 2002. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J 21:5017–5025. doi: 10.1093/emboj/cdf529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pileri P, Uematsu Y, Campagnoli S, Galli G, Falugi F, Petracca R, Weiner AJ, Houghton M, Rosa D, Grandi G, Abrignani S. 1998. Binding of hepatitis C virus to CD81. Science 282:938–941. doi: 10.1126/science.282.5390.938. [DOI] [PubMed] [Google Scholar]
- 10.Evans MJ, von Hahn T, Tscherne DM, Syder AJ, Panis M, Wölk B, Hatziioannou T, McKeating JA, Bieniasz PD, Rice CM. 2007. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 446:801–805. doi: 10.1038/nature05654. [DOI] [PubMed] [Google Scholar]
- 11.Ploss A, Evans MJ, Gaysinskaya VA, Panis M, You H, De Jong YP, Rice CM. 2009. Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature 457:882–886. doi: 10.1038/nature07684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lupberger J, Zeisel M-B, Xiao F, Thumann C, Fofana I, Zona L, Davis C, Mee CJ, Turek M, Gorke S, Royer C, Fischer B, Zahid MN, Lavillette D, Fresquet J, Cosset F-L, Rothenberg SM, Pietschmann T, Patel AH, Pessaux P, Doffoel M, Raffelsberger W, Poch O, McKeating JA, Brino L, Baumert T-F. 2011. EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat Med 17:589–595. doi: 10.1038/nm.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sainz B, Barretto N, Martin DN, Hiraga N, Imamura M, Hussain S, Marsh KA, Yu X, Chayama K, Alrefai WA, Uprichard SL. 2012. Identification of the Niemann-Pick C1-like 1 cholesterol absorption receptor as a new hepatitis C virus entry factor. Nat Med 18:281–285. doi: 10.1038/nm.2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martin DN, Uprichard SL. 2013. Identification of transferrin receptor 1 as a hepatitis C virus entry factor. Proc Natl Acad Sci U S A 110:10777–10782. doi: 10.1073/pnas.1301764110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blanchard E, Belouzard S, Goueslain L, Wakita T, Dubuisson J, Wychowski C, Rouille Y. 2006. Hepatitis C virus entry depends on clathrin-mediated endocytosis. J Virol 80:6964–6972. doi: 10.1128/JVI.00024-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meertens L, Bertaux C, Dragic T. 2006. Hepatitis C virus entry requires a critical postinternalization step and delivery to early endosomes via clathrin-coated vesicles. J Virol 80:11571–11578. doi: 10.1128/JVI.01717-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tscherne DM, Jones CT, Evans MJ, Lindenbach BD, McKeating JA, Rice CM. 2006. Time- and temperature-dependent activation of hepatitis C virus for low-pH-triggered entry. J Virol 80:1734–1741. doi: 10.1128/JVI.80.4.1734-1741.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hsu M, Zhang J, Flint M, Logvinoff C, Cheng-Mayer C, Rice CM, McKeating JA. 2003. Hepatitis C virus glycoproteins mediate pH-dependent cell entry of pseudotyped retroviral particles. Proc Natl Acad Sci U S A 100:7271–7276. doi: 10.1073/pnas.0832180100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bartosch B, Dubuisson J, Cosset F-L. 2003. Infectious hepatitis C virus pseudo-particles containing functional E1-E2 envelope protein complexes. J Exp Med 197:633–642. doi: 10.1084/jem.20021756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Kräusslich H-G, Mizokami M, Bartenschlager R, Liang TJ. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med 11:791–796. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhong J, Gastaminza P, Cheng G, Kapadia S, Kato T, Burton DR, Wieland SF, Uprichard SL, Wakita T, Chisari FV. 2005. Robust hepatitis C virus infection in vitro. Proc Natl Acad Sci U S A 102:9294–9299. doi: 10.1073/pnas.0503596102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lindenbach BD, Evans MJ, Syder AJ, Wölk B, Tellinghuisen TL, Liu CC, Maruyama T, Hynes RO, Burton DR, McKeating JA, Rice CM. 2005. Complete replication of hepatitis C virus in cell culture. Science 309:623–626. doi: 10.1126/science.1114016. [DOI] [PubMed] [Google Scholar]
- 23.Chang M, Williams O, Mittler J, Quintanilla A, Carithers RL, Perkins J, Corey L, Gretch DR. 2003. Dynamics of hepatitis C virus replication in human liver. Am J Pathol 163:433–444. doi: 10.1016/S0002-9440(10)63673-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wieland S, Makowska Z, Campana B, Calabrese D, Dill MT, Chung J, Chisari FV, Heim MH. 2014. Simultaneous detection of hepatitis C virus and interferon stimulated gene expression in infected human liver. Hepatology 59:2121–2130. doi: 10.1002/hep.26770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brimacombe CL, Grove J, Meredith LW, Hu K, Syder AJ, Flores MV, Timpe JM, Krieger SE, Baumert T-F, Tellinghuisen TL, Wong-Staal F, Balfe P, McKeating JA. 2011. Neutralizing antibody-resistant hepatitis C virus cell-to-cell transmission. J Virol 85:596–605. doi: 10.1128/JVI.01592-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Timpe JM, Stamataki Z, Jennings A, Hu K, Farquhar MJ, Harris HJ, Schwarz A, Desombere I, Roels GL, Balfe P, McKeating JA. 2008. Hepatitis C virus cell-cell transmission in hepatoma cells in the presence of neutralizing antibodies. Hepatology 47:17–24. [DOI] [PubMed] [Google Scholar]
- 27.Catanese MT, Loureiro J, Jones CT, Dorner M, von Hahn T, Rice CM. 2013. Different requirements for scavenger receptor class B type I in hepatitis C virus cell-free versus cell-to-cell transmission. J Virol 87:8282–8293. doi: 10.1128/JVI.01102-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ramakrishnaiah V, Thumann C, Fofana I, Habersetzer F, Pan Q, de Ruiter PE, Willemsen R, Demmers JAA, Stalin Raj V, Jenster G, Kwekkeboom J, Tilanus HW, Haagmans BL, Baumert T-F, van der Laan LJW. 2013. Exosome-mediated transmission of hepatitis C virus between human hepatoma Huh7.5 cells. Proc Natl Acad Sci U S A 110:13109–13113. doi: 10.1073/pnas.1221899110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bukong TN, Momen-Heravi F, Kodys K, Bala S, Szabo G. 2014. Exosomes from hepatitis C infected patients transmit HCV infection and contain replication competent viral RNA in complex with Ago2-miR122-HSP90. PLoS Pathog 10:e1004424. doi: 10.1371/journal.ppat.1004424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Agtarap A, Chamberlin JW, Pinkerton M, Steinrauf L. 1967. The structure of monensic acid, a new biologically active compound. J Am Chem Soc 89:5737–5739. doi: 10.1021/ja00998a062. [DOI] [PubMed] [Google Scholar]
- 31.Haney ME, Hoehn MM. 1967. Monensin, a new biologically active compound. I. Discovery and isolation. Antimicrob Agents Chemother 7:349–352. [DOI] [PubMed] [Google Scholar]
- 32.Mollenhauer HH, Morré DJ, Rowe LD. 1990. Alteration of intracellular traffic by monensin; mechanism, specificity and relationship to toxicity. Biochim Biophys Acta 1031:225–246. doi: 10.1016/0304-4157(90)90008-Z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meinert RA, Yang CM, Heinrichs AJ, Varga GA. 1992. Effect of monensin on growth, reproductive performance, and estimated body composition in Holstein heifers. J Dairy Sci 75:257–261. doi: 10.3168/jds.S0022-0302(92)77760-1. [DOI] [PubMed] [Google Scholar]
- 34.Zinn RA, Plascencia A, Barajas R. 1994. Interaction of forage level and monensin in diets for feedlot cattle on growth performance and digestive function. J Anim Sci 72:2209–2215. [DOI] [PubMed] [Google Scholar]
- 35.Goodrich RD, Garrett JE, Gast DR, Kirick MA, Larson DA, Meiske JC. 1984. Influence of monensin on the performance of cattle. J Anim Sci 58:1484–1498. [DOI] [PubMed] [Google Scholar]
- 36.Haberkorn A. 1996. Chemotherapy of human and animal coccidioses: state and perspectives. Parasitol Res 82:193–199. doi: 10.1007/s004360050094. [DOI] [PubMed] [Google Scholar]
- 37.Dewar RL, Vasudevachari MB, Natarajan V, Salzman NP. 1989. Biosynthesis and processing of human immunodeficiency virus type 1 envelope glycoproteins: effects of monensin on glycosylation and transport. J Virol 63:2452–2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pal R, Gallo RC, Sarngadharan MG. 1988. Processing of the structural proteins of human immunodeficiency virus type 1 in the presence of monensin and cerulenin. Proc Natl Acad Sci U S A 85:9283–9286. doi: 10.1073/pnas.85.23.9283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Clayson ET, Brando LV, Compans RW. 1989. Release of simian virus 40 virions from epithelial cells is polarized and occurs without cell lysis. J Virol 63:2278–2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ghosh-Choudhury N, Graham A, Ghosh HP. 1987. Herpes simplex virus type 2 glycoprotein biogenesis: effect of monensin on glycoprotein maturation, intracellular transport and virus infectivity. J Gen Virol 68:1939–1949. doi: 10.1099/0022-1317-68-7-1939. [DOI] [PubMed] [Google Scholar]
- 41.Nakabayashi H, Taketa K, Miyano K, Yamane T, Sato J. 1982. Growth of human hepatoma cells lines with differentiated functions in chemically defined medium. Cancer Res 42:3858–3863. [PubMed] [Google Scholar]
- 42.Gentzsch J, Hinkelmann B, Kaderali L, Irschik H, Jansen R, Sasse F, Frank R, Pietschmann T. 2011. Hepatitis C virus complete life cycle screen for identification of small molecules with pro- or antiviral activity. Antiviral Res 89:136–148. doi: 10.1016/j.antiviral.2010.12.005. [DOI] [PubMed] [Google Scholar]
- 43.Dubuisson J, Hsu HH, Cheung RC, Greenberg HB, Russell DG, Rice CM. 1994. Formation and intracellular localization of hepatitis C virus envelope glycoprotein complexes expressed by recombinant vaccinia and Sindbis viruses. J Virol 68:6147–6160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schlesinger JJ, Brandriss MW, Monath TP. 1983. Monoclonal antibodies distinguish between wild and vaccine strains of yellow fever virus by neutralization, hemagglutination inhibition, and immune precipitation of the virus envelope protein. Virology 125:8–17. doi: 10.1016/0042-6822(83)90059-4. [DOI] [PubMed] [Google Scholar]
- 45.Cocquerel L, Meunier JC, Pillez A, Wychowski C, Dubuisson J. 1998. A retention signal necessary and sufficient for endoplasmic reticulum localization maps to the transmembrane domain of hepatitis C virus glycoprotein E2. J Virol 72:2183–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oren R, Takahashi S, Doss C, Levy R, Levy S. 1990. TAPA-1, the target of an antiproliferative antibody, defines a new family of transmembrane proteins. Mol Cell Biol 10:4007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Charrin S, Le Naour F, Oualid M, Billard M, Faure G, Hanash SM, Boucheix C, Rubinstein E. 2001. The major CD9 and CD81 molecular partner. Identification and characterization of the complexes. J Biol Chem 276:14329–14337. [DOI] [PubMed] [Google Scholar]
- 48.Fofana I, Krieger SE, Grunert F, Glauben S, Xiao F, Fafi-Kremer S, Soulier E, Royer C, Thumann C, Mee CJ, McKeating JA, Dragic T, Pessaux P, Stoll-Keller F, Schuster C, Thompson J, Baumert T-F. 2010. Monoclonal anti-claudin 1 antibodies prevent hepatitis C virus infection of primary human hepatocytes. Gastroenterology 139:953–964, 964.e1–964.e4. doi: 10.1053/j.gastro.2010.05.073. [DOI] [PubMed] [Google Scholar]
- 49.Giang E, Dorner M, Prentoe JC, Dreux M, Evans MJ, Bukh J, Rice CM, Ploss A, Burton DR, Law M. 2012. Human broadly neutralizing antibodies to the envelope glycoprotein complex of hepatitis C virus. Proc Natl Acad Sci U S A 109:6205–6210. doi: 10.1073/pnas.1114927109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xu Y, Martinez P, Séron K, Luo G, Allain F, Dubuisson J, Belouzard S. 2015. Characterization of hepatitis C virus interaction with heparan sulfate proteoglycans. J Virol 89:3846–3858. doi: 10.1128/JVI.03647-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Goueslain L, Alsaleh K, Horellou P, Roingeard P, Descamps V, Duverlie G, Ciczora Y, Wychowski C, Dubuisson J, Rouille Y. 2010. Identification of GBF1 as a cellular factor required for hepatitis C virus RNA replication. J Virol 84:773–787. doi: 10.1128/JVI.01190-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Delgrange D, Pillez A, Castelain S, Cocquerel L, Rouille Y, Dubuisson J, Wakita T, Duverlie G, Wychowski C. 2007. Robust production of infectious viral particles in Huh-7 cells by introducing mutations in hepatitis C virus structural proteins. J Gen Virol 88:2495–2503. doi: 10.1099/vir.0.82872-0. [DOI] [PubMed] [Google Scholar]
- 53.Gottwein JM, Scheel TKH, Jensen TB, Lademann JB, Prentoe JC, Knudsen ML, Hoegh AM, Bukh J. 2009. Development and characterization of hepatitis C virus genotype 1-7 cell culture systems: role of CD81 and scavenger receptor class B type I and effect of antiviral drugs. Hepatology 49:364–377. doi: 10.1002/hep.22673. [DOI] [PubMed] [Google Scholar]
- 54.Op De Beeck A, Voisset C, Bartosch B, Ciczora Y, Cocquerel L, Keck Z, Foung S, Cosset F-L, Dubuisson J. 2004. Characterization of functional hepatitis C virus envelope glycoproteins. J Virol 78:2994–3002. doi: 10.1128/JVI.78.6.2994-3002.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lavillette D, Tarr AW, Voisset C, Donot P, Bartosch B, Bain C, Patel AH, Dubuisson J, Ball JK, Cosset F-L. 2005. Characterization of host-range and cell entry properties of the major genotypes and subtypes of hepatitis C virus. Hepatology 41:265–274. doi: 10.1002/hep.20542. [DOI] [PubMed] [Google Scholar]
- 56.Sandrin V, Boson B, Salmon P, Gay W, Nègre D, Le Grand R, Trono D, Cosset F-L. 2002. Lentiviral vectors pseudotyped with a modified RD114 envelope glycoprotein show increased stability in sera and augmented transduction of primary lymphocytes and CD34+ cells derived from human and nonhuman primates. Blood 100:823–832. doi: 10.1182/blood-2001-11-0042. [DOI] [PubMed] [Google Scholar]
- 57.Bick MJ, Carroll J-WN, Gao G, Goff SP, Rice CM, MacDonald MR. 2003. Expression of the zinc-finger antiviral protein inhibits alphavirus replication. J Virol 77:11555–11562. doi: 10.1128/JVI.77.21.11555-11562.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chehadeh W, Kerr-Conte J, Pattou F, Alm G, Lefebvre J, Wattré P, Hober D. 2000. Persistent infection of human pancreatic islets by coxsackievirus B is associated with alpha interferon synthesis in beta cells. J Virol 74:10153–10164. doi: 10.1128/JVI.74.21.10153-10164.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rocha-Perugini V, Montpellier C, Delgrange D, Wychowski C, Helle F, Pillez A, Drobecq H, Le Naour F, Charrin S, Levy S, Rubinstein E, Dubuisson J, Cocquerel L. 2008. The CD81 partner EWI-2wint inhibits hepatitis C virus entry. PLoS One 3:e1866. doi: 10.1371/journal.pone.0001866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vausselin T, Calland N, Belouzard S, Descamps V, Douam F, Helle F, François C, Lavillette D, Duverlie G, Wahid A, Fénéant L, Cocquerel L, Guérardel Y, Wychowski C, Biot C, Dubuisson J. 2013. The antimalarial ferroquine is an inhibitor of hepatitis C virus. Hepatology 58:86–97. doi: 10.1002/hep.26273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Castelain S, Descamps V, Thibault V, François C, Bonte D, Morel V, Izopet J, Capron D, Zawadzki P, Duverlie G. 2004. TaqMan amplification system with an internal positive control for HCV RNA quantitation. J Clin Virol 31:227–234. doi: 10.1016/j.jcv.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 62.Calland N, Albecka A, Belouzard S, Wychowski C, Duverlie G, Descamps V, Hober D, Dubuisson J, Rouille Y, Séron K. 2012. (−)-Epigallocatechin-3-gallate is a new inhibitor of hepatitis C virus entry. Hepatology 55:720–729. doi: 10.1002/hep.24803. [DOI] [PubMed] [Google Scholar]
- 63.Harris HJ, Farquhar MJ, Mee CJ, Davis C, Reynolds GM, Jennings A, Hu K, Yuan F, Deng H, Hubscher SG, Han JH, Balfe P, McKeating JA. 2008. CD81 and claudin 1 coreceptor association: role in hepatitis C virus entry. J Virol 82:5007–5020. doi: 10.1128/JVI.02286-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Harris HJ, Davis C, Mullins JGL, Hu K, Goodall M, Farquhar MJ, Mee CJ, McCaffrey K, Young S, Drummer H, Balfe P, McKeating JA. 2010. Claudin association with CD81 defines hepatitis C virus entry. J Biol Chem 285:21092–21102. doi: 10.1074/jbc.M110.104836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Potel J, Rassam P, Montpellier C, Kaestner L, Werkmeister E, Tews BA, Couturier C, Popescu C-I, Baumert T-F, Rubinstein E, Dubuisson J, Milhiet P-E, Cocquerel L. 2013. EWI-2wint promotes CD81 clustering that abrogates hepatitis C virus entry. Cell Microbiol 15:1234–1252. doi: 10.1111/cmi.12112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Coller KE, Berger KL, Heaton NS, Cooper JD, Yoon R, Randall G. 2009. RNA interference and single particle tracking analysis of hepatitis C virus endocytosis. PLoS Pathog 5:e1000702. doi: 10.1371/journal.ppat.1000702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Suomalainen M, Luisoni S, Boucke K, Bianchi S, Engel DA, Greber UF. 2013. A direct and versatile assay measuring membrane penetration of adenovirus in single cells. J Virol 87:12367–12379. doi: 10.1128/JVI.01833-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yoshimori T, Yamamoto A, Moriyama Y, Futai M, Tashiro Y. 1991. Bafilomycin A1, a specific inhibitor of vacuolar-type H(+)-ATPase, inhibits acidification and protein degradation in lysosomes of cultured cells. J Biol Chem 266:17707–17712. [PubMed] [Google Scholar]
- 69.Harada M, Shakado S, Sakisaka S, Tamaki S, Ohishi M, Sasatomi K, Koga H, Sata M, Tanikawa K. 1997. Bafilomycin A1, a specific inhibitor of V-type H+-ATPases, inhibits the acidification of endocytic structures and inhibits horseradish peroxidase uptake in isolated rat sinusoidal endothelial cells. Liver 17:244–250. [DOI] [PubMed] [Google Scholar]
- 70.Ashfaq UA, Javed T, Rehman S, Nawaz Z, Riazuddin S. 2011. Lysosomotropic agents as HCV entry inhibitors. Virol J 8:163. doi: 10.1186/1743-422X-8-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mizui T, Yamashina S, Tanida I, Takei Y, Ueno T, Sakamoto N, Ikejima K, Kitamura T, Enomoto N, Sakai T, Kominami E, Watanabe S. 2010. Inhibition of hepatitis C virus replication by chloroquine targeting virus-associated autophagy. J Gastroenterol 45:195–203. doi: 10.1007/s00535-009-0132-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Grove J, Nielsen S, Zhong J, Bassendine MF, Drummer HE, Balfe P, McKeating JA. 2008. Identification of a residue in hepatitis C virus E2 glycoprotein that determines scavenger receptor BI and CD81 receptor dependency and sensitivity to neutralizing antibodies. J Virol 82:12020–12029. doi: 10.1128/JVI.01569-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Law M, Maruyama T, Lewis J, Giang E, Tarr AW, Stamataki Z, Gastaminza P, Chisari FV, Jones IM, Fox RI, Ball JK, McKeating JA, Kneteman NM, Burton DR. 2008. Broadly neutralizing antibodies protect against hepatitis C virus quasispecies challenge. Nat Med 14:25–27. doi: 10.1038/nm1698. [DOI] [PubMed] [Google Scholar]
- 74.Bankwitz D, Steinmann E, Bitzegeio J, Ciesek S, Friesland M, Herrmann E, Zeisel M-B, Baumert T-F, Keck Z-Y, Foung SKH, Pécheur E-I, Pietschmann T. 2010. Hepatitis C virus hypervariable region 1 modulates receptor interactions, conceals the CD81 binding site, and protects conserved neutralizing epitopes. J Virol 84:5751–5763. doi: 10.1128/JVI.02200-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dao Thi VL, Granier C, Zeisel MB, Guerin M, Mancip J, Granio O, Penin F, Lavillette D, Bartenschlager R, Baumert TF, Cosset FL, Dreux M. 2012. Characterization of hepatitis C virus particle subpopulations reveals multiple usage of the scavenger receptor BI for entry steps. J Biol Chem 287:31242–31257. doi: 10.1074/jbc.M112.365924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bankwitz D, Vieyres G, Hueging K, Bitzegeio J, Doepke M, Chhatwal P, Haid S, Catanese MT, Zeisel MB, Nicosia A, Baumert TF, Kaderali L, Pietschmann T. 2014. Role of hypervariable region 1 for the interplay of hepatitis C virus with entry factors and lipoproteins. J Virol 88:12644–12655. doi: 10.1128/JVI.01145-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Prentoe J, Serre SBN, Ramirez S, Nicosia A, Gottwein JM, Bukh J. 2014. Hypervariable region 1 deletion and required adaptive envelope mutations confer decreased dependency on scavenger receptor class B type I and low-density lipoprotein receptor for hepatitis C virus. J Virol 88:1725–1739. doi: 10.1128/JVI.02017-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kouyoumdjian JA, Morita MP, Sato AK, Pissolatti AF. 2001. Fatal rhabdomyolysis after acute sodium monensin (Rumensin) toxicity: case report. Arq Neuropsiquiatr 59:596–598. doi: 10.1590/S0004-282X2001000400022. [DOI] [PubMed] [Google Scholar]
- 79.Caldeira C, Neves WS, Cury PM, Serrano P, Baptista MA, Burdmann EA. 2001. Rhabdomyolysis, acute renal failure, and death after monensin ingestion. Am J Kidney Dis 38:1108–1112. doi: 10.1053/ajkd.2001.28618. [DOI] [PubMed] [Google Scholar]
- 80.Aowicki D, Huczyński A. 2013. Structure and antimicrobial properties of monensin A and its derivatives: summary of the achievements. Biomed Res Int 2013:742149. doi: 10.1155/2013/742149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Westley JW, Liu CM, Evans RH, Sello LH, Troupe N, Hermann T. 1983. Preparation, properties and biological activity of natural and semisynthetic urethanes of monensin. J Antibiot 36:1195–1200. doi: 10.7164/antibiotics.36.1195. [DOI] [PubMed] [Google Scholar]
- 82.Tanaka R, Nagatsu A, Mizukami H, Ogihara Y, Sakakibara J. 2001. Studies on chemical modification of monensin IX. Synthesis of 26-substituted monensins and their Na+ ion transport activity. Chem Pharm Bull 49:711–715. [DOI] [PubMed] [Google Scholar]
- 83.Sharma NR, Mateu G, Dreux M, Grakoui A, Cosset F-L, Melikyan GB. 2011. Hepatitis C virus is primed by CD81 protein for low pH-dependent fusion. J Biol Chem 286:30361–30376. doi: 10.1074/jbc.M111.263350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bartosch B, Vitelli A, Granier C, Goujon C, Dubuisson J, Pascale S, Scarselli E, Cortese R, Nicosia A, Cosset F-L. 2003. Cell entry of hepatitis C virus requires a set of co-receptors that include the CD81 tetraspanin and the SR-B1 scavenger receptor. J Biol Chem 278:41624–41630. doi: 10.1074/jbc.M305289200. [DOI] [PubMed] [Google Scholar]
- 85.Koutsoudakis G, Kaul A, Steinmann E, Kallis S, Lohmann V, Pietschmann T, Bartenschlager R. 2006. Characterization of the early steps of hepatitis C virus infection by using luciferase reporter viruses. J Virol 80:5308–5320. doi: 10.1128/JVI.02460-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Krey T, d'Alayer J, Kikuti CM, Saulnier A, Damier-Piolle L, Petitpas I, Johansson DX, Tawar RG, Baron B, Robert B, England P, Persson MAA, Martin A, Rey FA. 2010. The disulfide bonds in glycoprotein E2 of hepatitis C virus reveal the tertiary organization of the molecule. PLoS Pathog 6:e1000762. doi: 10.1371/journal.ppat.1000762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lavillette D, Pécheur E-I, Donot P, Fresquet J, Molle J, Corbau R, Dreux M, Penin F, Cosset F-L. 2007. Characterization of fusion determinants points to the involvement of three discrete regions of both E1 and E2 glycoproteins in the membrane fusion process of hepatitis C virus. J Virol 81:8752–8765. doi: 10.1128/JVI.02642-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Khan AG, Whidby J, Miller MT, Scarborough H, Zatorski AV, Cygan A, Price AA, Yost SA, Bohannon CD, Jacob J, Grakoui A, Marcotrigiano J. 2014. Structure of the core ectodomain of the hepatitis C virus envelope glycoprotein 2. Nature 509:381–384. doi: 10.1038/nature13117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kong L, Giang E, Nieusma T, Kadam RU, Cogburn KE, Hua Y, Dai X, Stanfield RL, Burton DR, Ward AB, Wilson IA, Law M. 2013. Hepatitis C virus E2 envelope glycoprotein core structure. Science 342:1090–1094. doi: 10.1126/science.1243876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Douam F, Thi VLD, Maurin G, Fresquet J, Mompelat D, Zeisel M-B, Baumert T-F, Cosset F-L, Lavillette D. 2014. A critical interaction between E1 and E2 glycoproteins determines binding and fusion properties of hepatitis C virus during cell entry. Hepatology 59:776–788. doi: 10.1002/hep.26733. [DOI] [PubMed] [Google Scholar]
- 91.Biot C, Taramelli D, Forfar-Bares I, Maciejewski LA, Boyce M, Nowogrocki G, Brocard JS, Basilico N, Olliaro P, Egan TJ. 2005. Insights into the mechanism of action of ferroquine. Relationship between physicochemical properties and antiplasmodial activity. Mol Pharm 2:185–193. [DOI] [PubMed] [Google Scholar]
- 92.Matsuda M, Suzuki R, Kataoka C, Watashi K, Aizaki H, Kato N, Matsuura Y, Suzuki T, Wakita T. 2014. Alternative endocytosis pathway for productive entry of hepatitis C virus. J Gen Virol 95:2658–2667. doi: 10.1099/vir.0.068528-0. [DOI] [PubMed] [Google Scholar]
- 93.Prentoe J, Jensen TB, Meuleman P, Serre SBN, Scheel TKH, Leroux-Roels G, Gottwein JM, Bukh J. 2011. Hypervariable region 1 differentially impacts viability of hepatitis C virus strains of genotypes 1 to 6 and impairs virus neutralization. J Virol 85:2224–2234. doi: 10.1128/JVI.01594-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hueging K, Doepke M, Vieyres G, Bankwitz D, Frentzen A, Doerrbecker J, Gumz F, Haid S, Wölk B, Kaderali L, Pietschmann T. 2014. Apolipoprotein E codetermines tissue tropism of hepatitis C virus and is crucial for viral cell-to-cell transmission by contributing to a postenvelopment step of assembly. J Virol 88:1433–1446. doi: 10.1128/JVI.01815-13. [DOI] [PMC free article] [PubMed] [Google Scholar]