

Abstract
CRISPR (clustered regularly interspaced short palindromic repeats) is a microbial immune system against foreign DNA. Recognition sequences (spacers) encoded within the CRISPR array mediate the immune reaction in a sequence-specific manner. The known mechanisms for the evolution of CRISPR arrays include spacer acquisition from foreign DNA elements at the time of invasion and array erosion through spacer deletion. Here, we consider the contribution of genetic recombination between homologous CRISPR arrays to the evolution of spacer repertoire. Acquisition of spacers from exogenic arrays via recombination may confer the recipient with immunity against unencountered antagonists. For this purpose, we develop a novel method for the detection of recombination in CRISPR arrays by modeling the spacer order in arrays from multiple strains from the same species. Because the evolutionary signal of spacer recombination may be similar to that of pervasive spacer deletions or independent spacer acquisition, our method entails a robustness analysis of the recombination inference by a statistical comparison to resampled and perturbed data sets. We analyze CRISPR data sets from four bacterial species: two Gammaproteobacteria species harboring CRISPR type I and two Streptococcus species harboring CRISPR type II loci. We find that CRISPR array evolution in Escherichia coli and Streptococcus agalactiae can be explained solely by vertical inheritance and differential spacer deletion. In Pseudomonas aeruginosa, we find an excess of single spacers potentially incorporated into the CRISPR locus during independent acquisition events. In Streptococcus thermophilus, evidence for spacer acquisition by recombination is present in 5 out of 70 strains. Genetic recombination has been proposed to accelerate adaptation by combining beneficial mutations that arose in independent lineages. However, for most species under study, we find that CRISPR evolution is shaped mainly by spacer acquisition and loss rather than recombination. Since the evolution of spacer content is characterized by a rapid turnover, it is likely that recombination is not beneficial for improving phage resistance in the strains under study, or that it cannot be detected in the resolution of intraspecies comparisons.
Keywords: evolutionary microbiology, lateral gene transfer, bacterial genomics
Introduction
The CRISPR/Cas immune system is a microbial defense mechanism against invasive mobile genetic elements such as plasmids or bacteriophages. The system is encoded by an array of clustered regularly interspaced short palindromic repeats (CRISPR) and adjacent CRISPR associated (Cas) proteins. The CRISPR repeat sequences in each locus are identical and are typically 28–37 bp long. The repeats alternate with variable spacer sequences that are 21–72 bp long (Barrangou and Marraffini 2014). The CRISPR/Cas mechanism of action consists of three main stages (Barrangou and Marraffini 2014). In the acquisition or adaptation stage, new spacers that originate from foreign DNA are incorporated into the CRISPR array at its 5′-leader-end. In the biogenesis stage, the CRISPR locus is transcribed and subsequently processed into multiple CRISPR RNAs (crRNAs) that contain one spacer each. Finally in the targeting stage, the complementary match between the crRNA spacer and a protospacer on the target DNA or RNA molecule elicits cleavage of the target. CRISPR/Cas is thus an adaptive and heritable immune system where sequence specificity is encoded in the spacer sequences. The system is carried by the majority of archaea (84%) and about 45% of the bacteria whose genomes have been sequenced so far (CRISPRfinder database, status 2014-08-05, Grissa et al. 2007). Based on the Cas protein collection and CRISPR sequence properties, several types of CRISPR/Cas systems have been defined (Makarova et al. 2011).
The spacer repertoire encoded in the CRISPR array determines the CRISPR/Cas immunity range and may change over time due to spacer acquisition and loss dynamics. Thus, the adaptation process is the most important stage for the evolution of the system. The insertion of new spacers at the 5′-leader-end results in increasing conservation along homologous arrays where spacer variability is higher at the 5′-end (e.g., Weinberger, Sun, et al. 2012). In most CRISPR/Cas types, the acquisition of new spacers requires a short protospacer associated motif (Shah et al. 2013). In addition, hybridization of spacers that are only partially complementary to foreign DNA can guide the acquisition machinery to uptake novel spacers from nearby locations (Datsenko et al. 2012; Fineran et al. 2014). This process, called priming, results in a biased acquisition of spacers in the proximity of already existing protospacers and leads to nonrandom sampling of spacers from mobile genetic elements (Paez-Espino et al. 2013; Savitskaya et al. 2013). An additional adaptation bias results from the preferred selection of spacers at stalled replication forks and from degradation intermediates of RecBCD activity during processing of DNA double strand breaks (Levy et al. 2015).
In addition to spacer acquisition, spacer evolutionary dynamics is also affected by deletions of single or multiple adjacent spacers. Pervasive deletions in CRISPR arrays have been observed in comparisons of homologous CRISPR regions (e.g., Horvath et al. 2008; Lopez-Sanchez et al. 2012). Deletions of specific spacers were observed under controlled laboratory growth conditions (e.g., Deveau et al. 2008; Gudbergsdottir et al. 2011). Since the repeat-spacer boundaries are perfectly maintained during deletion events, it has been suggested that homologous recombination is involved in the deletion of regions between repeats (Gudbergsdottir et al. 2011). Thus, replacing recombination at the array locus can lead to spacer acquisition that is coupled with the deletion of previously existing spacers (Deveau et al. 2008). Alternatively, DNA polymerase slippage during replication can also result in spacer deletions (Yosef et al. 2012).
Genetic recombination, that is, the exchange of DNA within the population, has been shown experimentally to occur after lateral DNA transfer by conjugation and transduction (Milkman et al. 1999). Acquired DNA is usually integrated into the chromosome by homologous recombination, which involves the pairing of homologous DNA strands and the resolution of the branched DNA structures into duplex DNA molecules (e.g., Lovett et al. 2002; Spies and Kowalczykowski 2005; Persky and Lovett 2008). Thus, the frequency of genetic recombination depends on sequence similarity between the acquired DNA and the chromosome (Majewski and Cohan 1999). In addition to DNA sequence divergence, known barriers to recombination include the presence of restriction-modification systems (Budroni et al. 2011, Stucken et al. 2013), genetic isolation resulting from speciation (Retchless and Lawrence 2007) and ecological differentiation (Shapiro et al. 2012). The impact of recombination may vary between different bacterial species (Feil et al. 2001; Vos and Didelot 2009). Examples for the impact of recombination on microbial genome evolution include archael and bacterial species (e.g., Smith and Smith 1993; Matic et al. 1996; Holmes et al. 1999; Papke et al. 2004; Denef and Banfield 2012). Recombination within the population is thought to be advantageous to the lineage as it may reduce the impact of clonal interference and accelerate microbial adaptation (Vos 2009). In addition to the replacement of alternative alleles, genetic recombination can also lead to variation in gene content within microbial populations (Shapiro et al. 2012; Kong et al. 2013).
Sequence similarity between repeats of homologous CRISPR arrays can, potentially, facilitate the integration of acquired DNA by homologous recombination. Spacer recombination can occur inside the same locus, between loci on the same chromosome or between CRISPR arrays from different cells. Spacer replication as a result of recombination within the locus has been observed in CRISPR arrays sampled from numerous species, including Streptococcus thermophilus (Bolotin et al. 2005), Streptococcus mutans (van der Ploeg 2009), Erwinia amylovora (Rezzonico et al. 2011), Streptococcus agalactiae (Lopez-Sanchez et al. 2012), Salmonella (Fabre et al. 2012), Synechocystis (Scholz et al. 2013), and Methanosarcina mazei (Nickel et al. 2013). Such within-locus rearrangements may increase the spacer effectiveness whereas the immunity range remains unchanged. Recombination between different CRISPR loci encoded on the same chromosome can result in shared spacers (e.g., Lillestøl et al. 2009). This type of recombination may have an effect on the immunity range if the CRISPR loci are differentially regulated.
CRISPR loci are frequently observed on mobile elements. Examples are a prophage of Clostridium difficile (Sebaihia et al. 2006) and plasmids sampled from Sulfolobus solfataricus (Lillestøl et al. 2009), Lactococcus lactis (Millen et al. 2012), and Synechocystis (Scholz et al. 2013). The presence of CRISPR arrays in mobile elements led to the suggestion that the CRISPR/Cas long-term evolution is affected by lateral transfer of whole CRISPR/Cas loci (e.g., Godde and Bickerton 2006; Horvath et al. 2009). Recombination with exogenic arrays—such as those encoded on mobile genetic elements—may introduce immunity against as yet unencountered antagonists (i.e., a “transferred immunity”). Acquisition of exogenic spacers potentially provides a large immune benefit by transferring immunity from other cells into an existing CRISPR array. In Sulfolobus islandicus, alleles from three different CRISPR loci occur in different combinations, indicating that CRISPR alleles in the population have been reassorted by genetic recombination (Held et al. 2013). In Escherichia coli, the incongruence between strain typing based on multilocus sequencing and strain classification based on CRISPR spacer information is interpreted as evidence for recombination of the CRISPR locus among E. coli strains (Almendros et al. 2014).
Here, we study the lateral component of CRISPR spacer evolution and estimate the frequency of recombination-mediated spacer acquisition into preexisting CRISPR loci. Lateral spacer transfer leads to changes in spacer order that can be recognized by a comparative analysis. To detect recombination events, we compare the spacer order in CRISPR arrays from multiple strains of a single species. In the absence of recombination, we expect the ordering to be conserved on the 3′ (leader-distal) end of the CRISPR array and diversified on the 5′ (leader-proximal) end. Lateral spacer transfer can introduce an additional pattern of spacer content similarity, which we term order divergence events (ODEs). These are composed of shared segments followed toward the 3′-end by diverse spacers that are termed here different segments (fig. 1). Here, we present a novel algorithm to infer ODEs in CRISPR arrays. To assess the power of our inference algorithm, we apply it to perturbations of the original data sets where additional recombination events were introduced and test its performance (fig. 2).
Fig. 1.—
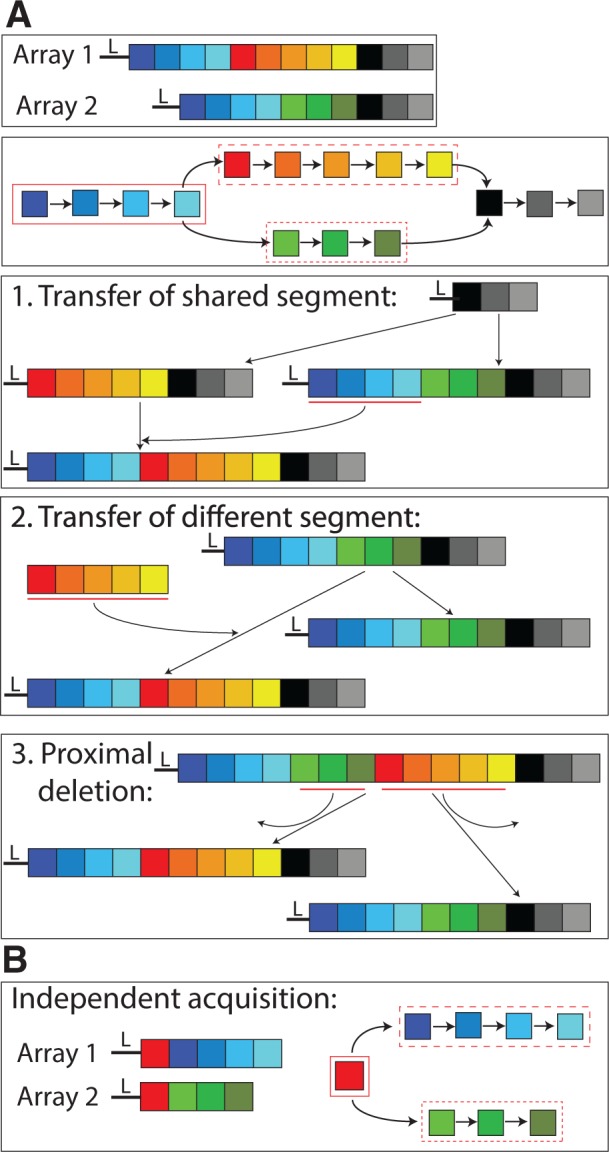
An example of ODE detection. (A) Two arrays (repeats omitted) and their corresponding spacer graph are shown at the top. Leader-proximal end (5′-end) is displayed on the left (marked by L). The spacer graph shows an ODE consisting of a shared segment (blue spacers in a solid box) followed by two different segments (red and green spacers, dashed boxes) and shared distal spacers (black boxes). Potential evolutionary scenarios that could explain the observed spacer order include: 1) lateral transfer of the blue spacers, 2) lateral transfer of the red spacers (or analogously the green spacers), and 3) proximal deletions in both arrays, omitting the green spacers in array 1 and the red spacers in array 2. (B) An independent acquisition of the red spacer that leads to an ODE of a single shared spacer.
Fig. 2.—

Overview of the analysis pipeline.
We note, however, that ODEs can be generated by two additional scenarios. Multiple independent acquisitions of the same spacer sequence due to biased sampling of protospacers from invasive genomes can lead to ODEs with shared segments of a single spacer (fig. 1B). Additionally, pervasive deletions in CRISPR arrays can create proximal deletions resulting in ODEs (fig. 1A, scenario 3). To study the impact of deletions, we apply our inference algorithm to perturbations of the original data sets, where simulated deletions were introduced. The inferred ODEs from the perturbed replicates represent the expected number of ODEs resulting from spacer deletions only. Here, we infer spacer recombination events in four bacterial species using our novel approach. This includes two Gammaproteobacteria species harboring CRISPR type I and two Streptococcus species harboring CRISPR type II.
Materials and Methods
Data
Fully sequenced genomes were downloaded from NCBI genomes (http://www.ncbi.nlm.nih.gov/genome/, last accessed August 2014) and contig-state genomes from the trace archive (http://www.ncbi.nlm.nih.gov/Traces/wgs/ , last accessed August 2014). Only CRISPR types with a considerable number of strains that encode the system were used. Additional CRISPR types that have been described for the species studied here were not included due to the limited number of strains available. For example, CRISPR type I-E that is encoded by Pseudomonas aeruginosa was detected in 20 strains only and is not included in the analysis due to an insufficient sample size.
Previously described CRISPR repeat sequences (table 1) were located in the genome and contig sequences using the EMBOSS program matcher (Rice et al. 2000). Hits of the repeat sequence on contig-state genomes are only considered if the distance of the first repeat from the beginning of the contig is longer than the length of the repeat plus the length of a typical spacer. An analogous condition was used for the distance between the last repeat and the end of the contig. In addition, scaffold state genomes containing insufficient spacer information due to stretches of unresolved nucleotides were excluded. Escherichia coli, CRISPR1 and CRISPR2 are composed of two loci that could be distinguished by the known locus structure (Díez-Villaseñor et al. 2010). For P. aeruginosa, multiple arrays were joined into a single data set regardless of multiple loci.
Table 1.
Data Sets Used as the Basis for the Analyses
| Data Set | No. of Available Genomes in NCBI (fully sequenced, draft) | No. of Genomes in NCBI with Detectable CRISPR | No. of Additional CRISPR Arrays (strains)a | Repeat Sequence | Typical Spacer Length (% of unique spacers of the typical length) | CRISPR/CAS Type |
|---|---|---|---|---|---|---|
| E. coli CRISPR1 | 77, 2106 | 55, 1247 | — | GTGTTCCCCGCGCCAGCGGGGATAAACCG | 32 (95) | I-E (Díez-Villaseñor et al. 2010) |
| E. coli CRISPR2 | 77, 2106 | 9, 133 | — | GTTCACTGCCGTACAGGCAGCTTAGAAA | 32 (91) | I-F (Díez-Villaseñor et al. 2010) |
| P. aeruginosa | 33, 254 | 15, 79 | 85 | GTTCACTGCCGTATAGGCAGCTAAGAAA | 32 (95) | I-F (Cady et al. 2011) |
| S. agalactiae | 12, 291 | 8, 235 | — | GTTTTAGAGCTGTGCTGTTTCGAATGGTTCCAAAAC | 30 (94) | II-A (Lopez-Sanchez et al. 2012) |
| S. thermophilus | 9, 8 | 8, 5 | 69 | GTTTTTGTACTCTCAAGATTTAAGTAACTGTACAAC | 30 (87) | II-A (Horvath et al. 2008) |
aData for P. aeruginosa was obtained from Cady et al. (2011). Additional data for S. thermophilus were obtained from Horvath et al. (2008).
Unique spacers were extracted from sequences flanked by repeat sequences. Pairs of spacers were aligned using the EMBOSS program needle (Rice et al. 2000). Similar spacers at syntenic positions can be the result of point mutations or sequence errors. Consequently, spacers were joined into the same unique spacer if their sequences are greater than or equal to 90% identical and at least one neighboring pair (right or left) is also 90% identical. Only few spacers were joined due to sequence similarity and synteny, including 38 spacers in E. coli CRISPR1, 3 spacers in E. coli CRISPR2, 28 spacers in P. aeruginosa, 16 spacers in S. agalactiae, and 6 spacers in S. thermophilus.
In E. coli CRISPR1, 21 spacers were found in common between the two CRISPR loci as singletons. Long spacers (at least 100 nt) occur in the data sets of E. coli CRISPR1, P. aeruginosa, and S. thermophilus. The majority of long spacers show similarities to transposons (supplementary table S4, Supplementary Material online). For the subsequent analysis, they were treated like spacers.
Inference of Events
Order Inversions
We detect two kinds of order inversions that are masked for the subsequent algorithmic steps. First, spacers that are replicated in the same array are masked from the data set. Such spacers are also masked from strains where the spacer is not replicated. Second, there may still be loops in the spacer graph and spacers causing these loops are also assigned as spacers involved in order inversions. Here, the parsimonious decision of always taking the shortest order inversion is taken (see supplementary material, Supplementary Material online, for details). If there are two paths of equal length that would resolve the loop, both are assigned to order inversions.
Deletion Events
Deletions are detected in the spacer graph after the omission of order inversions. An array contains a deletion between two successive spacers (s1,s2) if there is a further path between s1 and s2 that traverses at least one other spacer. The length of the deletion is the shortest of all nondirect paths.
Order Divergence Events
An ODE contains a shared segment and at least two different segments. We can write it as (S, D1, … , Dd) where d = 2 in most cases and S, Di are sets of spacers. An ODE is characterized by the pattern that a shared segment (S) is occurring in multiple strains. In a subset of the strains the shared segment is followed by one different segment (Di) and in another subset of strains it is followed by another different segment (fig. 1). ODEs with |S| = 1, that is, with only one shared spacer, are called single-spacer ODEs, in contrast to multiple-spacer ODEs. The details of the algorithm are described in the supplementary Materials and Methods, Supplementary Material online.
Power Analysis
For a given data set of unique arrays, the potential to detect a recombination event is assessed by perturbing the original data sets. One simulated recombination event is introduced into a recipient strain from the data set using one of several settings (supplementary fig. S1, Supplementary Material online). The lengths of introduced and replaced segments are Poisson distributed with rate λ, where λ is chosen to be about one-third of the median array length for each data set.
Estimation of Deletion Effects
The original data sets were also perturbed by introducing deletions. A deletion length is drawn randomly from the inferred deletion lengths (supplementary table S1, Supplementary Material online) and is introduced into a random array from the data set. The deletions are added sequentially so that the a data set with n simulated deletions is based on a data set with n−1 previously simulated deletions. The number of artificial deletion events, on, is the mean number of deletions detected in the perturbed replicates subtracted by the number of deletions in the original data set. The ratio of artificial to simulated deletions is calculated by on/n. ODEs in perturbed replicates (S, D1, … , Dd) are classified as artificial if there is no corresponding event in the original data set (S′,D′1, … ,D′d) with and for a permutation p. In words, the shared segments need to contain common elements and each pair of corresponding different segments needs to contain common elements. Each event in the original data set where no corresponding event is present in the perturbed replicate constitutes an absent event.
Results
Data Structure
To infer order divergence and deletion events in a set of CRISPR arrays, we begin by constructing a spacer graph from the arrays. A similar data structure was used before for inferring CRISPR arrays from metagenomic data (Skennerton et al. 2013). Nodes in the spacer graph designate unique spacers within the data set. Directed edges connect spacers that are consecutive in the CRISPR array in the 5′- to 3′-direction. Inverted spacer order, termed here order inversions, preclude a common order of all spacers and introduce loops in the spacer graph. Spacers causing order inversions are masked prior to subsequent algorithmic steps. A spacer deletion is inferred as a pair of spacers adjacent in one array that are also connected via a longer path. Genetic recombination at the CRISPR locus may lead to an ODE, which is defined as a shared segment followed by at least two different segments toward the leader-distal end (i.e., the 3′-end). In addition, we require that the different segments for one ODE are nonoverlapping and that each of them is only present in some of the strains containing the shared segment. Single-spacer ODEs contain only one shared spacer. Proximal deletions and independent spacer acquisitions can result in ODEs as well (fig. 1).
Data
For the analysis, we selected four bacterial species with known CRISPR loci and a considerable number of available genomic sequences from different strains. These include E. coli, P. aeruginosa, S. agalactiae, and S. thermophilus (table 1). For E. coli, two different CRISPR types with two loci each were analyzed separately, resulting in seven data sets in total. The number of spacers that are shared between two unique arrays is low in all data sets, ranging from an average of 0.29 shared spacers in P. aeruginosa to an average of 1.7 shared spacers in E. coli CRISPR2.2 (table 2). In all data sets, the spacer graph is composed of multiple connected components. The largest number of unique arrays connected by shared spacers varies among the data sets and ranges between 37% in S. thermophilus and 99% in E. coli CRISPR1.1.
Table 2.
CRISPR Array Statistics
| Component with Largest Number of Arrays |
||||||
|---|---|---|---|---|---|---|
| Data Set | No. Unique Arrays | Mean No. Spacers | Median No. Spacers | Avg. Pairwise Shared Spacers | No. Arrays | Avg. Pairwise Shared Spacers |
| E. coli CRISPR1.1 | ||||||
| Unique | 356 | 11.145 | 10 | 1.16 | 353 | 1.193 |
| No order inversions, unique | 299 | 9.666 | 8 | 0.9308 | 294 | 0.962 |
| E. coli CRISPR1.2 | ||||||
| Unique | 348 | 10.112 | 9 | 0.358 | 161 | 0.6825 |
| No order inversions, unique | 324 | 8.852 | 8 | 0.2513 | 156 | 0.5875 |
| E. coli CRISPR2.1 | ||||||
| Unique | 41 | 9.073 | 9 | 1.045 | 27 | 1.997 |
| No order inversions, unique | 38 | 8.184 | 8 | 0.667 | 15 | 2.4 |
| E. coli CRISPR2.2 | ||||||
| Unique | 44 | 11.318 | 11 | 1.739 | 32 | 3.008 |
| No order inversions, unique | 34 | 8.588 | 9 | 1.021 | 23 | 1.925 |
| P. aeruginosa | ||||||
| Unique | 198 | 13.87 | 13 | 0.2936 | 148 | 0.4875 |
| No order inversions, unique | 191 | 13.41 | 12 | 0.272 | 141 | 0.4578 |
| S. agalactiae | ||||||
| Unique | 210 | 11.69 | 10.5 | 0.6394 | 139 | 1.143 |
| No order inversions, unique | 204 | 8.7 | 8 | 0.3067 | 131 | 0.5948 |
| S. thermophilus | ||||||
| Unique | 70 | 23.07 | 23 | 0.7627 | 26 | 1.523 |
| No order inversions, unique | 70 | 21.23 | 20 | 0.7288 | 26 | 1.523 |
The proportion of duplicated spacers within the same array that are recognized as order inversion events ranges between 1% in P. aeruginosa and 11% in E. coli CRISPR2.2 (table 3). The maximum proportion of spacers that are involved in order inversions without being duplicated reaches 2% in S. agalactiae (table 3). The masking of spacers involved in order inversions results in decreased pairwise overlap between arrays in most data sets. After masking, the array length decreases by 1–2 spacers on average, with the exception of P. aeruginosa, where array length decreases only marginally (table 2).
Table 3.
Events Detected in the Data Sets
|
E. coli |
|||||||
|---|---|---|---|---|---|---|---|
| CRISPR1.1 | CRISPR1.2 | CRISPR2.1 | CRISPR2.2 | P. aeruginosa | S. agalactiae | S. thermophilus | |
| Arrays | 1,302 | 1,302 | 142 | 142 | 289 | 243 | 84 |
| Unique arrays | 356 | 345 | 41 | 44 | 198 | 210 | 70 |
| Median length | 10 | 9 | 9 | 11 | 13 | 10.5 | 23 |
| Unique spacers | 746 | 766 | 146 | 140 | 1,315 | 750 | 896 |
| Long spacers (≥100 nt) | 6 | 1 | 0 | 0 | 5 | 0 | 2 |
| Order inversions | |||||||
| Replicated spacers | 35 (4.7%) | 35 (4.6%) | 3 (2.1%) | 15 (11%) | 19 (1.4%) | 30 (4%) | 35 (3.9%) |
| Others | 7 (0.94%) | 2 (0.26%) | 0 | 0 | 0 | 15 (2%) | 0 |
| Deletions | |||||||
| Deletion events | 257 | 316 | 25 | 28 | 134 | 159 | 34 |
| Unique deleted spacers | 278 (37%) | 314 (41%) | 31 (21%) | 43 (31%) | 280 (21%) | 158 (21%) | 120 (13%) |
| ODEs | 40 | 46 | 0 | 4 | 40 | 30 | 15 |
| Multiple-spacer ODEs | 26 | 20 | 0 | 3 | 17 | 19 | 9 |
| Min. number of spacers in events | 45 (6.1%) | 58.5 (7.6%) | 0 | 7 (0.5%) | 45 (3.4%) | 35 (4.7%) | 27 (3.0%) |
| Max. number of spacers in events | 170.2 (23%) | 220 (29%) | 0 | 19 (14%) | 309 (23%) | 128 (17%) | 120 (13%) |
| Max. number of spacers in multiple-spacer events | 129.5 (17%) | 109 (14%) | 0 | 17 (12%) | 112 (8.5%) | 80 (11%) | 71 (7.9%) |
| Ratio ODEs to deletion events | 0.1556 | 0.1456 | 0 | 0.1429 | 0.2985 | 0.1887 | 0.4412 |
| Ratio multiple-spacer ODEs to deletion events | 0.1012 | 0.0633 | 0 | 0.1071 | 0.1269 | 0.1195 | 0.2647 |
| Estimation of deletion effects | |||||||
| Artificial to simulated deletion events | 0.2574 | 0.2192 | 0.1997 | 0.1165 | 0.2909 | 0.121 | 0.2989 |
| Proximal deletions to deletion events | 0.2084 | 0.2389 | 0.1169 | 0.3991 | 0.1947 | 0.3997 | 0.1234 |
| Multiple-spacer proximal deletions to deletion events | 0.156 | 0.1497 | 0.0944 | 0.2275 | 0.1258 | 0.2083 | 0.1105 |
Note.—Estimation of deletion effects: Ratios are given using the median for 1–30 deletions, except for E. coli CRISPR2 and S. thermophilus. There only up to 15 deletions are used, because the ratio of inferred to simulated deletions is decreasing with high numbers of deletions for these data sets (data not shown).
Inference of Events
After the masking of order inversions, the spacer graphs contain a considerable number of deletion events per data set ranging from 25 in E. coli CRISPR2.1 to 316 in E. coli CRISPR1.2 (table 3). Single-spacer deletions are the most frequent in all data sets (supplementary table S1, Supplementary Material online). The fraction of unique spacers that are affected by at least one deletion ranges from 13% in S. thermophilus to 41% in E. coli CRISPR1.2. The number of ODEs per data set is highly variable ranging from no events in E. coli CRISPR2.1 to a maximum of 46 events in E. coli CRISPR1.2 (table 3). About half of the ODEs involve a single shared spacer. The maximum number of multiple-spacer ODEs is observed in E. coli CRISPR1.2 as well. To estimate the number of different spacers participating in an ODE, we count the spacers in the smallest and largest segments (table 3). Estimating by the smallest segment, at most 7.6% (E. coli CRISPR1.2) of the spacers are part of an ODE (E. coli CRISPR1.2). According to the largest segment approach, up to 29% (E. coli CRISPR1.2) of spacers may be included in an ODE and up to 17% (E. coli CRISPR1.1) of the spacers participate in a multiple-spacer ODE (table 3). Assuming that recombination causes all the inferred ODEs, these estimates yield a lower and upper bound, respectively, for the proportion of detectable laterally transferred spacers in the analyzed strains.
Power Analysis
For evaluating the power of our approach to detect array recombination as segments of ODEs, we created perturbations of the original data sets and fed them into the inference algorithm. In the power analysis, perturbed data sets contain one additional simulated recombination event. Here, we model several recombination scenarios that include replacing a segment by a new segment that is not in the data set (supplementary fig. S1A, Supplementary Material online), inserting a segment that exists in the data set (supplementary fig. S1B, Supplementary Material online), and replacing a segment by a segment from the data set (supplementary fig. S1C, Supplementary Material online). The resulting spacer graphs were analyzed including the donor and original recipient arrays and then, again, excluding those arrays. A recombination is considered as successfully detected if it was found as a shared or different segment of an ODE.
The recovery rate of simulated recombination events varies among the data sets depending on the recombination scenario and the inclusion of the original donor or recipient in the perturbed data set (fig. 3; supplementary table S2, Supplementary Material online). On average, 52% of the recombination events are detected in the perturbed E. coli data sets, with a slightly higher average detection rate of 63% in the other data sets. The highest accuracy is achieved when an existing segment is replacing a segment from another strain, with the donor and original recipient both included in the data set (supplementary fig. S1C, Supplementary Material online). The detection rate in this scenario ranges between 45% (E. coli CRISPR1.1) and 91% (S. thermophilus). Excluding the donor array from the spacer graph decreases the accuracy more than omitting the original recipient. Notably, between 2.4% (P. aeruginosa) and 26% (E. coli CRISPR2.2) of the simulated recombination events cannot be detected due to the preliminary masking of spacers that induce order inversions.
Fig. 3.—

Recovery rates for the power analysis (in %, for 1,000 replications). Recombination scenarios: (A) Replacing a segment by a new segment that is not in the data set, (B) inserting a segment that exists in the data set, and (C) replacing a segment by a segment from the data set (supplementary fig. S1, Supplementary Material online). “Donor” and “Recipient” mark the presence of donor and original recipient in the data set, respectively. Both: The spacers were detected both as a shared segment and as a different segment. Inversion: All the spacers are involved in order inversions and cannot be detected as part of an ODE.
Robustness Analysis
To test the impact of sample size on event detection rate, we employed a resampling technique, where an increasing proportion of the strains is included in the data set. To this end, the unique arrays in each data set were randomly resampled including between 10% and 90% of the strains in the data set to create a total of 100 spacer graph replicates for each sample size. The resulting detection rates reveal a strong positive correlation between the sample size and detection rate. When fewer strains are included in the analysis, fewer deletion and ODEs can be detected (fig. 4; supplementary fig. S2, Supplementary Material online).
Fig. 4.—
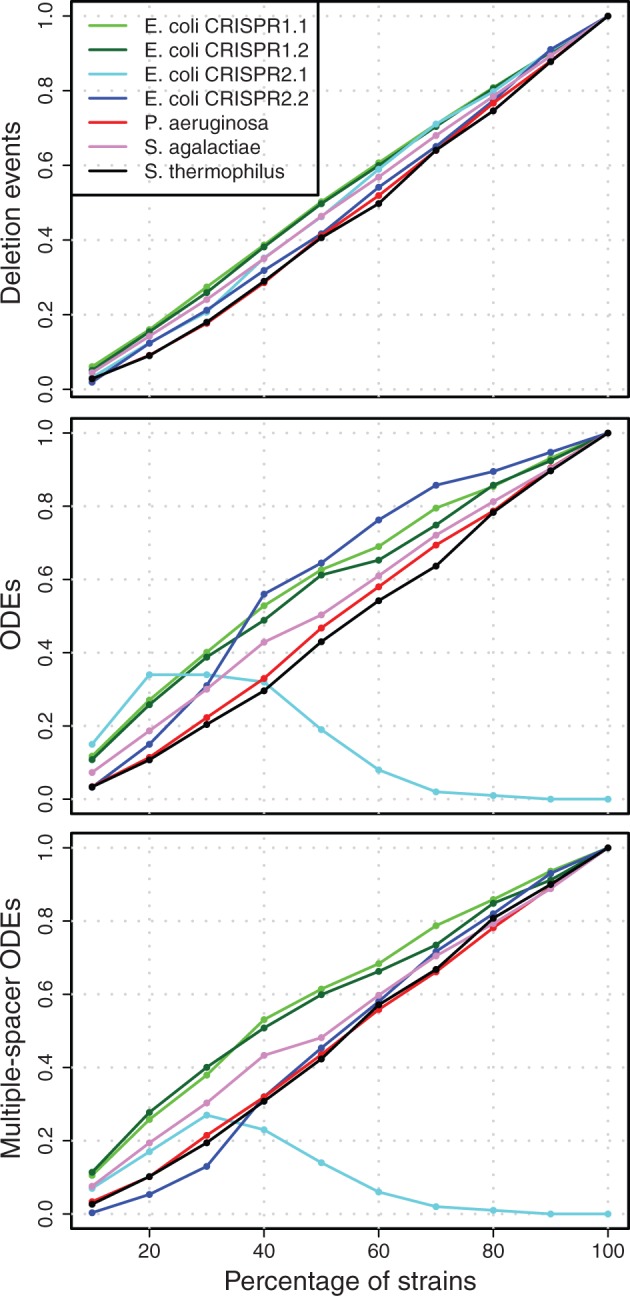
Fraction of events for the robustness analysis. The mean fraction of events is calculated as the mean number of events in the resampled data sets (100 replications) divided by the number of events in the complete data set. In data sets where no events have been observed, a denominator of 1 was used. Variation of the data can be found in supplementary figure S2, Supplementary Material online.
The impact of multiple deletions of adjacent spacers on the inference of recombination events was evaluated by inspecting the ratio of ODEs to deletion events in the resampled spacer graphs. The highest ratio is observed in S. thermophilus with a ratio of 0.44 for all ODEs and 0.26 for multiple-spacer ODEs (fig. 5 and table 3). For all data sets, we observe that the ratio of inferred ODEs to deletion events is highest when the sample size is small, but approaches a stable asymptotic ratio with larger sample sizes (fig. 5 and table 3).
Fig. 5.—
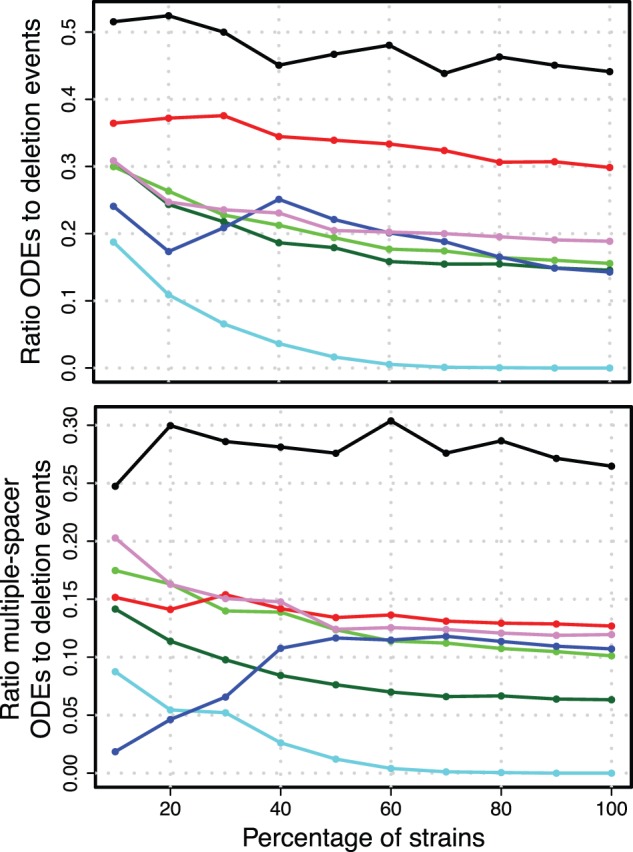
Ratios for the robustness analysis. Ratios of mean number of ODEs to mean number of deletion events. For color legend, see figure 4.
Estimation of Deletion Effects
The observed stable ratios can be the result of either a constant rate of deletions and recombination events or a constant rate of deletions and the ensuing proximal deletions. To distinguish between these two alternatives, we performed additional perturbations of the original data sets. Simulated deletions are introduced into the data sets where the number of successive spacers being deleted is randomly chosen from the observed distribution of deletion length in the respective data set (supplementary table S1, Supplementary Material online). We define artificial deletions as the deletions that are inferred in the data sets following the perturbation. This analysis reveals that increasing the number of simulated deletions in the data set results in an increased number of artificial deletions (fig. 6). However, only a minority of the simulated deletions is detectable, with a ratio of artificial to simulated deletions ranging from 0.12 (S. agalactiae) to 0.3 (S. thermophilus) (fig. 6 and table 3; supplementary fig. S3, Supplementary Material online).
Fig. 6.—

Estimation of deletion effects from 1,000 perturbed replicates. Left: Distribution of artificial deletions observed after introducing simulated deletions. The number of inferred deletions (left y axis) is calculated as the number of deletions inferred minus the number of deletions inferred for the original data set. The boxplot whisker range includes the outlier points. Middle: Artificial ODEs are present in the perturbed data set and not present in the original data set (see Materials and Methods for details). Absent events are present in the original data set and missing in the perturbed data set. Right: Ratio of mean number of artificial ODEs to deletion events. The median of each line is given as a thicker horizontal line. For comparison, the ratio from the original data set is shown. For the remaining E. coli data sets, see supplementary figure S3, Supplementary Material online.
In addition, we infer ODEs in each of the perturbation replicates. Those events are divided into observed events, where there is a corresponding event in the original data set, and artificial events, which are introduced due to the perturbation. We define absent events as events that are present in the original data set but not in the perturbed replicate. The ratio of artificial ODEs to artificial deletion events can be used as an estimate for the extent of ODEs that is expected if all events are created by proximal deletions (i.e., deletions of proximal spacers) rather than recombination. Thus, this ratio is called proximal deletion to deletion events. The comparison of this ratio with the ratio of ODEs to deletion events inferred from the original data sets reveals considerable differences among the four data sets.
In E. coli, the number of artificial ODEs increases faster than the number of ODEs for all CRISPR types except for CRISPR1.2 (supplementary fig. S3, Supplementary Material online). In these data sets, the number of ODEs generated by proximal deletions is higher than the number of events that are not detected due to deletion of the shared or different segments (that are necessary for the ODE detection). The ratio of proximal deletions to deletion events clearly exceeds the ratio of ODEs to deletion events in the original data for all four E. coli data sets, for single-spacer and multiple-spacer events alike.
In P. aeruginosa, the ratio of proximal deletions to deletion events (0.19) is lower than the ratio of ODEs to deletion events in the original data set (0.30). However, these two ratios are approximately equal when comparing multiple-spacer events (0.13). Thus, multiple-spacer events in this data set can be well explained by proximal deletions. In contrast, for single-spacer ODEs independent acquisitions or recombination should be considered. Because spacer acquisition typically occurs one spacer at a time, whereas no known restriction on the number of spacers exists for recombination events, independent acquisitions is the more likely explanation for the excess of single-spacer ODEs in P. aeruginosa.
In S. agalactiae, we observe a stable ratio of 0.40 proximal deletions to deletion events. This ratio is considerably higher than the ratio of ODEs to deletion events in the original data set (0.19). This observation holds for both single- and multiple-spacer ODEs.
In S. thermophilus, the ratio of proximal deletions to deletion events is 0.12, which is lower than the ratio inferred from the original data set (0.44). A similar trend was observed for multiple-spacer events where the ratio of proximal deletions to deletion events (0.11) is lower than the ratio of ODEs to deletion events (0.26). Thus, spacer deletions alone cannot explain the extent of ODEs observed in this data set. In agreement with this observation, S. thermophilus also has the highest ratio of ODEs to deletion events among the data sets analyzed in our study. Five strains were found to contain two ODEs with multiple shared spacers (fig. 7; supplementary fig. S4, Supplementary Material online), hence they may be particularly prone to recombination.
Fig. 7.—

Connected component of the S. thermophilus data set showing most ODEs in this data set. Only strains with multiple-spacer ODEs are shown. Leader-end is displayed on the left. Spacers are coded by colors. Unique spacers are shaded in gray. Multiple-spacer ODEs are color coded by marking the shared segment. In the legend, the number of strains and the number of spacers in the shared segment are given. The complete data set can be found in supplementary figure S4, Supplementary Material online.
Data Set Characteristics
The analyzed data sets differ in several important characteristics that are potentially related to the results of our recombination inference. First, the arrays in S. thermophilus are substantially longer than those encoded in the other species. Longer arrays have a higher potential to detect similarities in spacer order and thus also to detect ODEs. To test for a possible bias in our detection approach that is related to the number of spacers, we split the S. thermophilus arrays into two data sets. The Head data set contains the first half of all arrays and the Tail data set contains the second half of all arrays. The middle spacers in arrays of uneven length are assigned randomly to one of the two data sets. This results in Head and Tail data sets of median length 11 and 12 spacers, respectively (supplementary table S3, Supplementary Material online). Applying our inference approach to the data sets yields a total of six ODEs in the Head data set, whereas eight ODEs are inferred in the Tail data set. Furthermore, the estimation of deletion effects shows that the ratio of ODEs to deletion events exceeds the ratio of proximal deletions to deletion events in both data sets (supplementary fig. S5 and table S4, Supplementary Material online). These results demonstrate that the shortened data sets also show signatures of recombination. Consequently, the difference between S. thermophilus and the other species cannot be explained by the long arrays encoded in that species.
Additional characteristics differ between S. thermophilus and the other species. The frequency of unique spacers per strain is 12.8 for S. thermophilus whereas it is 6.6 (P. aeruginosa) or less for the other species. In the S. thermophilus data set, the proportion of pairs of unique arrays having overlapping spacers is the lowest (9%), yet, the average frequency of shared spacers between pairs with overlap is the largest observed among the data sets. Furthermore, in S. thermophilus, only 13% of the unique spacers are affected by deletions in comparison to more than 20% for the other data sets. In summary, the characteristics of S. thermophilus CRISPR arrays are clearly exceptional in comparison to the other species analyzed here and they may serve as predictors for a successful detection of ODEs created by genetic recombination.
Discussion
Methods for horizontal gene transfer inference are commonly based on the detection of conflicting phylogenetic signals between a reference species phylogeny and the gene in question (see Zhaxybayeva 2009 for a review). Conflicting phylogenies are also utilized for detecting recombination events in homologous genomic sequences (e.g., McGuire and Wright 2000; Ané 2011). However, the use of such methods strongly depends on the inferred reference species tree and the sequence alignment quality (Roettger et al. 2009). Here, we present a novel recombination inference algorithm that does not rely on a reference phylogeny but instead searches for spacer ODEs. Such patterns can however be created by three different evolutionary scenarios including genetic recombination at the array locus, independent spacer acquisition, and proximal deletions. To test the performance of our inference approach, we analyzed perturbed CRISPR arrays where simulated recombination and deletion events have been introduced.
Perturbing the data sets by introducing simulated deletion events reveals a strong bias in the detection of ODEs due to proximal deletions. In the analysis of perturbed E. coli and S. agalactiae data sets, the ratio of proximal deletions to deletion events clearly exceeds the ratio of ODEs to deletion events observed in the original data sets. This indicates that ODEs in these CRISPR arrays are better explained by proximal deletion rather than genetic recombination. In the analysis of P. aeruginosa those indicator ratios are similar only when multiple-spacer events are considered. However, the ratio of all ODEs to deletion events exceeds the ratio of proximal deletions to deletion events in the original data set. This indicates that the ODEs are probably not the result of genetic recombination but of proximal deletions and independent acquisitions. Since independent acquisitions result in single-spacer ODEs, they can better explain the excess of such events in the P. aeruginosa data set. In the arrays sampled from S. thermophilus, the ratio of ODEs to deletion events cannot be explained by proximal deletions and independent acquisitions alone, indicating that genetic recombination is contributing to the evolution of the CRISPR locus in this species.
Notably, recombination at other loci has been observed for all the species under study (Lefébure and Stanhope 2007; Rasmussen et al. 2008; Luo et al. 2011; Dettman et al. 2014). However, recombination seems only to affect the CRISPR locus in one of the four species analyzed. Although we cannot rule out the possibility that recombination at the CRISPR locus occurred in the other species under study, we can conclude that their spacer order does not include a detectable recombination pattern given the present sample. Indeed, recombination at the E. coli CRISPR1 locus has been reported (Almendros et al. 2014). However, we find no evidence for recombination events in that CRISPR locus but rather that proximal deletions are more likely to explain the observed ODEs. Notably, the evolution of E. coli CRISPR loci has been described to involve rare and radical turnovers instead of gradual change (Touchon et al. 2011). This would result in a low number of shared spacers and a spacer graph with many small connected components. We observe that the size of the connected components is largest for E. coli CRISPR1 and the average number of pairwise shared spacers is higher for E. coli than for the other species analyzed here. Thus, the characteristics described by Touchon et al. (2011) are not specific to E. coli but a similar or even more extreme pattern is exhibited by other species.
The frequency of recombination presented here may be an underestimation because we do not consider order inversions as a signal of recombination. A common order is expected when a CRISPR array evolves exclusively by insertions of unique spacers and deletions. In the presence of independent acquisitions, spacer replication or recombination, a common order might be disrupted and order inversions are observed. Among the spacers involved in order inversions, replicated spacers are the most frequently observed pattern, suggesting that spacer duplication is the most common mechanism for order inversions. However, we cannot rule out a contribution of recombination to some of the observed order inversions.
Another possible factor that can result in underestimating recombination is the assembly quality of the CRISPR arrays in the data set. Many bacterial genome sequences are deposited only in contig-state where long CRISPR arrays may not be assembled correctly onto a single contig. Here, only genomes where the locus is present on one contig and not close to the border of that contig are considered. This step may filter strains with potential recombination events. Unfortunately, this property also precludes the exploitation of CRISPR information from metagenomes. There, CRISPR loci that show diversity in the sequenced population are problematic for assembly (e.g., Rho et al. 2012; Skennerton et al. 2013).
The four model species analyzed here belong to two different bacterial phyla, Proteobacteria and Firmicutes, and harbor CRISPR arrays of type I or type II. Thus our results may not extend to other taxonomic groups or other CRISPR types. Archaea frequently show very long CRISPR arrays (e.g., Vestergaard et al. 2014) and are thus promising candidates for detecting recombination in CRISPR arrays. With the sequencing of large strain data sets for additional species, the prevalence of recombination within CRISPR arrays for different taxa and CRISPR systems can be assessed using the methods presented here.
CRISPR loci evolve much faster than other genetic elements encoded in the same genome as their content is under a strong selection pressure induced by phage predation (Stern and Sorek 2011). Thus, spacer content can be used to discriminate among microbial lineages. It is used for strain typing where other markers are lacking the necessary resolution (reviewed in Shariat and Dudley 2014). This would result in erroneous classifications, if recombination was indeed a major factor in the evolution of CRISPR arrays. Our results suggest that strain typing based on spacer content is not expected to be biased by recombination. However, because spacer deletion events can rapidly eradicate spacer information, whole array information should be preferred for strain typing.
Existing models for population dynamics of bacteria and their phages or plasmids in the presence of CRISPR immunity include only spacer acquisition and deletion events (reviewed in Koonin and Wolf 2015). Similarly, current estimates for the evolutionary rate of spacer composition are based on insertions and deletions only (Kupczok and Bollback 2013). Conditions for the maintenance of CRISPR/Cas systems were studied by including the transfer of whole CRISPR/Cas systems in the model (Weinberger, Wolf, et al. 2012). Furthermore, a model including genetic recombination of phage genomes (but not of CRISPR loci) shows that recombination may allow phages to escape CRISPR recognition more effectively than does point mutation alone (Han et al. 2013). Our results indicate that, for most of the species analyzed, recombination does not play a major role in the evolution of CRISPR arrays. This supports the predictions made by models that include spacer gain and loss only. Our findings are also consistent with the model and data analysis by Weinberger, Sun, et al. (2012) that suggests the presence of persistent spacers at the leader-distal end. These persistent spacers are a signal for vertical evolution of the CRISPR system.
Recombination has been proposed to accelerate the process of adaptation by combining beneficial mutations that arose in independent lineages (Fisher 1930; Muller 1932). Notably, recombination is of cardinal importance for the vertebrate immune system. Antibody diversity created by somatic recombination is a prerequisite for the recognition of a wide range of antigens (Gellert 2002). Indeed, lateral gene transfer is an important component of CRISPR/Cas evolution where whole cassettes are frequently transferred. However, here we find that the evolution of CRISPR arrays is shaped mainly by spacer acquisition and pervasive loss rather than recombination. Immunity to lateral gene transfer has been exemplified in several systems and is currently thought to be related to dose effect of the acquired gene (Sorek et al. 2007; Wellner and Gophna 2008). For the CRISPR/Cas system, dose effect of laterally acquired spacers is unlikely. Since the evolution of spacer content is characterized by a rapid turnover, it is likely that either recombination is not beneficial for an improved phage resistance, or that the resolution at which it occurs cannot be detected in intraspecies comparisons.
Supplementary Material
Supplementary Material and Methods, tables S1–S4, and figures S1–S5 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
The authors thank David Bogumil and Fernando Tria for their helpful comments on the manuscript and acknowledge support from the Excellence Cluster The Future Ocean and the European Research Council (Grant No. 281357).
Literature Cited
- Almendros C, Mojica FJM, Díez-Villaseñor C, Guzmán NM, García-Martínez J. 2014. CRISPR-Cas functional module exchange in Escherichia coli. mBio 5:e00767–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ané C. 2011. Detecting phylogenetic breakpoints and discordance from genome-wide alignments for species tree reconstruction. Genome Biol Evol. 3:246–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrangou R, Marraffini LA. 2014. CRISPR-Cas systems: prokaryotes upgrade to adaptive immunity. Mol Cell. 54:234–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolotin A, Quinquis B, Sorokin A, Ehrlich SD. 2005. Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology 151:2551–2561. [DOI] [PubMed] [Google Scholar]
- Budroni S, et al. 2011. Neisseria meningitidis is structured in clades associated with restriction modification systems that modulate homologous recombination. Proc Natl Acad Sci U S A. 108:4494–4499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cady KC, et al. 2011. Prevalence, conservation and functional analysis of Yersinia and Escherichia CRISPR regions in clinical Pseudomonas aeruginosa isolates. Microbiology 157:430–437. [DOI] [PubMed] [Google Scholar]
- Datsenko KA, et al. 2012. Molecular memory of prior infections activates the CRISPR/Cas adaptive bacterial immunity system. Nat Commun. 3:945. [DOI] [PubMed] [Google Scholar]
- Denef VJ, Banfield JF. 2012. In situ evolutionary rate measurements show ecological success of recently emerged bacterial hybrids. Science 336:462–466. [DOI] [PubMed] [Google Scholar]
- Dettman JR, Rodrigue N, Kassen R. 2014. Genome-wide patterns of recombination in the opportunistic human pathogen Pseudomonas aeruginosa. Genome Biol Evol. 7:18–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deveau H, et al. 2008. Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. J Bacteriol. 190:1390–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díez-Villaseñor C, Almendros C, García-Martínez J, Mojica FJM. 2010. Diversity of CRISPR loci in Escherichia coli . Microbiology 156:1351–1361. [DOI] [PubMed] [Google Scholar]
- Fabre L, et al. 2012. CRISPR typing and subtyping for improved laboratory surveillance of Salmonella infections. PLoS One 7:e36995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feil EJ, et al. 2001. Recombination within natural populations of pathogenic bacteria: short-term empirical estimates and long-term phylogenetic consequences. Proc Natl Acad Sci U S A. 98:182–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fineran PC, et al. 2014. Degenerate target sites mediate rapid primed CRISPR adaptation. Proc Natl Acad Sci U S A. 111:E1629–E1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher RA. 1930. The genetical theory of natural selection. Oxford: Clarendon Press. [Google Scholar]
- Gellert M. 2002. V(D)J recombination: RAG proteins, repair factors, and regulation. Annu Rev Biochem. 71:101–132. [DOI] [PubMed] [Google Scholar]
- Godde JS, Bickerton A. 2006. The repetitive DNA elements called CRISPRs and their associated genes: evidence of horizontal transfer among prokaryotes. J Mol Evol. 62:718–729. [DOI] [PubMed] [Google Scholar]
- Grissa I, Vergnaud G, Pourcel C. 2007. CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 35:W52–W57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudbergsdottir S, et al. 2011. Dynamic properties of the Sulfolobus CRISPR/Cas and CRISPR/Cmr systems when challenged with vector-borne viral and plasmid genes and protospacers. Mol Microbiol. 79:35–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han P, Niestemski LR, Barrick JE, Deem MW. 2013. Physical model of the immune response of bacteria against bacteriophage through the adaptive CRISPR-Cas immune system. Phys Biol. 10:25004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Held NL, Herrera A, Whitaker RJ. 2013. Reassortment of CRISPR repeat-spacer loci in Sulfolobus islandicus . Environ Microbiol. 15:3065–3076. [DOI] [PubMed] [Google Scholar]
- Holmes E, Urwin R, Maiden M. 1999. The influence of recombination on the population structure and evolution of the human pathogen Neisseria meningitidis. Mol Biol Evol. 16:741–749. [DOI] [PubMed] [Google Scholar]
- Horvath P, et al. 2008. Diversity, activity, and evolution of CRISPR loci in Streptococcus thermophilus. J Bacteriol. 190:1401–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath P, et al. 2009. Comparative analysis of CRISPR loci in lactic acid bacteria genomes. Int J Food Microbiol. 131:62–70. [DOI] [PubMed] [Google Scholar]
- Kong Y, et al. 2013. Homologous recombination drives both sequence diversity and gene content variation in Neisseria meningitidis. Genome Biol Evol. 5:1611–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin EV, Wolf YI. 2015. Evolution of the CRISPR-Cas adaptive immunity systems in prokaryotes: models and observations on virus-host coevolution. Mol Biosyst. 11:20–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupczok A, Bollback JP. 2013. Probabilistic models for CRISPR spacer content evolution. BMC Evol Biol. 13:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefébure T, Stanhope MJ. 2007. Evolution of the core and pan-genome of Streptococcus: positive selection, recombination, and genome composition. Genome Biol. 8:R71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy A, et al. 2015. CRISPR adaptation biases explain preference for acquisition of foreign DNA. Nature 520:410–505. [Google Scholar]
- Lillestøl RK, et al. 2009. CRISPR families of the crenarchaeal genus Sulfolobus: bidirectional transcription and dynamic properties. Mol Microbiol. 72:259–272. [DOI] [PubMed] [Google Scholar]
- Lopez-Sanchez MJ, et al. 2012. The highly dynamic CRISPR1 system of Streptococcus agalactiae controls the diversity of its mobilome. Mol Microbiol. 85:1057–1071. [DOI] [PubMed] [Google Scholar]
- Lovett ST, Hurley RL, Sutera VA, Aubuchon RH, Lebedeva MA. 2002. Crossing over between regions of limited homology in Escherichia coli. RecA-dependent and RecA-independent pathways. Genetics 160:851–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo C, et al. 2011. Genome sequencing of environmental Escherichia coli expands understanding of the ecology and speciation of the model bacterial species. Proc Natl Acad Sci U S A. 108:7200–7205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majewski J, Cohan F. 1999. DNA sequence similarity requirements for interspecific recombination in Bacillus. Genetics 1533:1525–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova KS, et al. 2011. Evolution and classification of the CRISPR-Cas systems. Nat Rev Microbiol. 9:467–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matic I, Taddei F, Radman M. 1996. Genetic barriers among bacteria. Trends Microbiol. 4:69–72. [DOI] [PubMed] [Google Scholar]
- McGuire G, Wright F. 2000. TOPAL 2.0: improved detection of mosaic sequences within multiple alignments. Bioinformatics 16:130–134. [DOI] [PubMed] [Google Scholar]
- Milkman R, et al. 1999. Molecular evolution of the Escherichia coli chromosome. V. Recombination patterns among strains of diverse origin. Genetics 153:539–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millen AM, Horvath P, Boyaval P, Romero DA. 2012. Mobile CRISPR/Cas-mediated bacteriophage resistance in Lactococcus lactis. PLoS One 7:e51663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller HJ. 1932. Some genetic aspect of sex. Am Nat. 77:118–138. [Google Scholar]
- Nickel L, et al. 2013. Two CRISPR-Cas systems in Methanosarcina mazei strain Gö1 display common processing features despite belonging to different types I and III. RNA Biol. 10:779–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paez-Espino D, et al. 2013. Strong bias in the bacterial CRISPR elements that confer immunity to phage. Nat Commun. 4:1430. [DOI] [PubMed] [Google Scholar]
- Papke RT, Koenig JE, Rodríguez-Valera F, Doolittle WF. 2004. Frequent recombination in a Saltern population of Halorubrum. Science 306:1928–1929. [DOI] [PubMed] [Google Scholar]
- Persky NS, Lovett ST. 2008. Mechanisms of recombination: lessons from E. coli. Crit Rev Biochem Mol Biol. 43:347–370. [DOI] [PubMed] [Google Scholar]
- Rasmussen TB, et al. 2008. Streptococcus thermophilus core genome: comparative genome hybridization study of 47 strains. Appl Environ Microbiol. 74:4703-4710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Retchless AC, Lawrence JG. 2007. Temporal fragmentation of speciation in bacteria. Science 317:1093–1096. [DOI] [PubMed] [Google Scholar]
- Rezzonico F, Smits THM, Duffy B. 2011. Diversity, evolution, and functionality of clustered regularly interspaced short palindromic repeat (CRISPR) regions in the fire blight pathogen Erwinia amylovora. Appl Environ Microbiol. 77:3819–3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rho M, Wu YW, Tang H, Doak TG, Ye Y. 2012. Diverse CRISPRs evolving in human microbiomes. PLoS Genet. 8:e1002441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice P, Longden I, Bleasby A. 2000. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 16:276–277. [DOI] [PubMed] [Google Scholar]
- Roettger M, Martin W, Dagan T. 2009. A machine-learning approach reveals that alignment properties alone can accurately predict inference of lateral gene transfer from discordant phylogenies. Mol Biol Evol. 26:1931–1939. [DOI] [PubMed] [Google Scholar]
- Savitskaya E, Semenova E, Dedkov V, Metlitskaya A, Severinov K. 2013. High-throughput analysis of type I-E CRISPR/Cas spacer acquisition in E. coli . RNA Biol. 10:716–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz I, Lange SJ, Hein S, Hess WR, Backofen R. 2013. CRISPR-Cas systems in the cyanobacterium Synechocystis sp. PCC6803 exhibit distinct processing pathways involving at least two Cas6 and a Cmr2 protein. PLoS One 8:e56470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebaihia M, et al. 2006. The multidrug-resistant human pathogen Clostridium difficile has a highly mobile, mosaic genome. Nat Genet. 38:779–786. [DOI] [PubMed] [Google Scholar]
- Shah SA, Erdmann S, Mojica FJM, Garrett RA. 2013. Protospacer recognition motifs: mixed identities and functional diversity. RNA Biol. 10:891–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro BJ, et al. 2012. Population genomics of early events in the ecological differentiation of bacteria. Science 336:48–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shariat N, Dudley EG. 2014. CRISPRs: molecular signatures used for pathogen subtyping. Appl Environ Microbiol. 80:430–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skennerton CT, Imelfort M, Tyson GW. 2013. Crass: identification and reconstruction of CRISPR from unassembled metagenomic data. Nucleic Acids Res. 41:e105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J, Smith N. 1993. How clonal are bacteria. Proc Natl Acad Sci U S A. 90:4384–4388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorek R, et al. 2007. Genome-wide experimental determination of barriers to horizontal gene transfer. Science 318:1449–1452. [DOI] [PubMed] [Google Scholar]
- Spies M, Kowalczykowski SC. 2005. Homologous recombination by RecBCD and RecF Pathways. In Higgins NP, editor. The bacterial chromosome. Washington (DC): ASM Press. p. 389–403. [Google Scholar]
- Stern A, Sorek R. 2011. The phage-host arms race: shaping the evolution of microbes. Bioessays 33:43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stucken K, Koch R, Dagan T. 2013. Cyanobacterial defense mechanisms against foreign DNA transfer and their impact on genetic engineering. Biol Res. 46:373–382. [DOI] [PubMed] [Google Scholar]
- Touchon M, et al. 2011. CRISPR distribution within the Escherichia coli species is not suggestive of immune-associated diversifying selection. J Bacteriol. 193:2460–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Ploeg JR. 2009. Analysis of CRISPR in Streptococcus mutans suggests frequent occurrence of acquired immunity against infection by M102-like bacteriophages. Microbiology 155:1966–1976. [DOI] [PubMed] [Google Scholar]
- Vestergaard G, Garrett RA, Shah SA. 2014. CRISPR adaptive immune systems of archaea. RNA Biol. 11:156–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos M. 2009. Why do bacteria engage in homologous recombination? Trends Microbiol. 17:226–232. [DOI] [PubMed] [Google Scholar]
- Vos M, Didelot X. 2009. A comparison of homologous recombination rates in bacteria and archaea. ISME J. 3:199–208. [DOI] [PubMed] [Google Scholar]
- Weinberger AD, Sun CL, et al. 2012. Persisting viral sequences shape microbial CRISPR-based Immunity. PLoS Comput Biol. 8:e1002475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberger AD, Wolf YI, Lobkovsky AE, Gilmore MS, Koonin EV. 2012. Viral diversity threshold for adaptive immunity in prokaryotes. mBio 3:e00456–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellner A, Gophna U. 2008. Neutrality of foreign complex subunits in an experimental model of lateral gene transfer. Mol Biol Evol. 25:1835–1840. [DOI] [PubMed] [Google Scholar]
- Yosef I, Goren MG, Qimron U. 2012. Proteins and DNA elements essential for the CRISPR adaptation process in Escherichia coli. Nucleic Acids Res. 40:5569–5576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhaxybayeva O. 2009. Detection and quantitaive assessment of horizontal gene transfer. In: Gogarten MB, Gogarten JP, Olendzenski LC, editors. Horizontal gene transfer: genomes in Flux, Vol. 532 of Methods in Molecular Biology. Humana Press. chap. 11, p. 195 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.