

Abstract
Repetitive elements have lately emerged as key components of genome, performing varieties of roles. It has now become necessary to have an account of repeats for every genome to understand its dynamics and state. Recently, genomes of two major Solanaceae species, Solanum tuberosum and Solanum lycopersicum, were sequenced. These species are important crops having high commercial significance as well as value as model species. However, there is a reasonable gap in information about repetitive elements and their possible roles in genome regulation for these species. The present study was aimed at detailed identification and characterization of complex repetitive elements in these genomes, along with study of their possible functional associations as well as to assess possible transcriptionally active repetitive elements. In this study, it was found that ~50–60% of genomes of S. tuberosum and S. lycopersicum were composed of repetitive elements. It was also found that complex repetitive elements were associated with >95% of genes in both species. These two genomes are mostly composed of LTR retrotransposons. Two novel repeat families very similar to LTR/ERV1 and LINE/RTE-BovB have been reported for the first time. Active existence of complex repeats was estimated by measuring their transcriptional abundance using Next Generation Sequencing read data and Microarray platforms. A reasonable amount of regulatory components like transcription factor binding sites and miRNAs appear to be under the influence of these complex repetitive elements in these species, while several genes appeared to possess exonized repeats.
Introduction
Very recently two important Solanaceae species, S. tuberosum and S. lycopersicum, genomes have been sequenced, reporting 810.6 Mb and 781.6 Mb of genome size for S. tuberosum and S. lycopersicum, respectively [1,2]. These genomes have been annotated for various genomic elements including repetitive elements, giving a total of 404,861 repetitive elements for S. tuberosum and 719,453 repetitive elements for S. lycopersicum. Initial studies with repetitive elements focused upon understanding their structure and functional aspects [3,4]. In one of the first attempts to study repeat content of Solanaceae genomes, Ganal et al. [5] had identified four different repeat families accounting for 0.15% of the genome of S. lycopersicum. Osborne et al. [4] studied the transposition behavior of Ac elements in S. lycopersicum by employing cloning and IPCR method to evaluate the transposition site preferences of these elements. Later, the importance of these elements in transposon tagging was discussed by Belzile & Yoder [6], who also studied the transposition behavior of maize Ac elements introduced in recombinant lines. Similar kind of study was performed by Stadler and colleagues [7] where they demonstrated the utility of repetitive elements in somatic hybridization technique and also identified novel repetitive elements having variable species specificity in different Solanum genomes. These studies were shown to be important for selection of different agronomically important traits in hybrid genomes.
A novel in-silico approach to identify the repetitive elements in different Solanaceae genomes was attempted by Oosumi et al. [8] where the authors searched the GenBank nucleic acid database for the presence of inverted repeats. The authors made observations that several genes possessed repetitive elements either in their 5' or 3' flanking regions or in introns, proposing functional aspects of repetitive elements in plants and their associations with gene-coding regions of the genome. Later studies on Solanaceae were focused on different satellite repetitive elements which could be developed as markers. Many genetic markers were developed for distinguishing different cultivars of S. tuberosum, S. lycopersicum and other Solanaceae species [9–12]. Tandem repeated DNA sequences were studied comprehensively and many new tandem repetitive elements were discovered in Solanaceae [13–18]. A novel method for estimation of repeat-content of a genome was proposed by Zhu et al. [19] where they estimated the genomic repeat-content by studying ~10% of a genome sequence. The authors studied 89.9 Mb of the genomic sequence of S. tuberosum in the form of BAC sequences and identified that repetitive content of the S. tuberosum BAC sequences was ~34% while for S. lycopersicum BAC sequences repetitive content was ~46% on the basis of homology. In both species, majority of the identified repetitive elements were not characterized and the authors observed prevalence of LTR retrotransposons, specifically LTR/Gypsy [19]. Thus, the authors provided a way to estimate the repetitive content of a genome even before the availability of the complete genome sequence. Studying the distribution of repetitive elements provides a view of the genomic localization as well as the abundance of different elements in a genome. Such a study was undertaken in the S. lycopersicum genome, FISH and fibre-FISH technique were employed to study the distribution of microsatellites as well as complex repeats [20]. Functional influence of a repetitive element on protein-coding genes was studied in S. lycopersicum, where the fruit shape gene was observed to be under the influence of a retrotransposon named Rider [21]. Kuang et al. [22] identified 22 MITE families in Solanaceae out of which fifteen were reported as novel repeat families. The authors also studied the functional roles of these MITE families and identified different exonized genes in their study [22] as well as active families and associated them with the biogenesis of different siRNA sequences. Active MITE elements were also reported in S. tuberosum genome, where the active nature of these elements produced phenotypic changes in S. tuberosum plants [23]. tRNA derived SINE elements were identified in different plant families including Solanaceae by Wenke et al. [24] and the chromosomal distribution of SINE elements was later assessed by using FISH. The authors also developed a tool to identify these elements in-silico and reported many novel SINE elements [24]. Another in-silico analysis was performed for the identification and study of MULE elements in different plant genomes including S. tuberosum and S. lycopersicum [25]. Identification of different LTR elements was also performed by Yadav and Singh [26] by using the tool LTR Finder on the EST sequences which were further validated by matching their prediction with the RepeatMasker output. Another most widely studied type of repetitive elements were the elements residing in the telomeric and centromeric regions. Centromeres are important structural components of a chromosome which aid the correct segregation of chromosomes during cell division. These repeats and the evolution of centromeres has been studied a lot. It was reported that centromeres are composed of specific histone proteins and long arrays of satellite repeats and retrotransposons [27]. These repeats were proposed to evolve rapidly and show divergence even in closely related species, thus the utility of these elements to identify different molecular characteristics of transgenic plants was studied [28–30]. Tang et al. [31] also studied the repeat-content of S. tuberosum using FISH and identified three repeat families. Thus as observed, most of the studies performed on the repetitive elements of Solanaceae genomes focused on derivation of agronomically important markers while limited studies were performed on the complex repetitive fraction of plant genomes.
From commercial point of view, many major crop species like potato (Solanum tuberosum), tomato (Solanum lycopersicum), pepper (Capsicum annum) and other Capsicum species, many ornamental plants, and biological model systems like Nicotiana spp and Datura spp belong to this family. S. tuberosum and S. lycopersicum, are two closely related species of Solanaceae family [32,33] which diverged very recently (~8 Mya) [34]. To reveal the genomes and provide a single stop molecular information about different Solanaceae species Sol Genomics Network (SGN) was established as a clade-oriented database [35,36]. Initially, SGN was developed to store the data like EST sequences and data of genetic mapping. The main focus of SGN was the identification of protein coding genes important for the development of different plant species and to understand the genetic basis of plant diversity [37]. SGN provides free access to all the information about the different Solanaceae families from a single web portal and has emerged as an important comprehensive resource for Solanaceae and other closely related families. It houses information about genomic (BAC sequences and genome sequences), transcriptomic (EST, unigene sequences, high throughput sequencing data and microarray data), proteomic, genetic and phenotypic (physical maps and markers), taxonomic and functional annotation of the different Solanaceae genomes [36,37]. SGN has been sequencing several species of Solanaceae genomes simultaneously. Currently in SGN, the complete as well as draft genome sequences are available for fourteen Solanaceae species. SGN initiated the tomato genome sequencing in 2004 using BAC-by-BAC sequencing methodology and later added the whole genome shotgun sequencing approach to the sequencing methodology [36], while the complete genome sequence of S. lycopersicum, was reported in 2012 [2]. The genome sequence of the S. tuberosum was released in 2011 [1] which is still being updated [38]. Although, SGN provided annotations and information for different repetitive elements of S. tuberosum and S. lycopersicum, there appears an enormous scope to carry out dedicated study with respect to the detailing of the repetitive elements in these species mainly but due to limited characterization of de-novo and species-specific repetitive elements where homology based methods have been applied predominantly. Also as mentioned above, most of the initial studies on Solanaceae repetitive elements were focused on tandem repetitive elements like satellite repeats while studies of complex repetitive element were mostly performed on isolated groups of repetitive elements. The functional impacts of these elements on the genome dynamics in Solanaceae genomes were seldom studied in detail. This all has been the motivation to carry out the present study on a genome-wide scale and identify the potential impacts of these elements on the genome dynamics of these two species in detailed comparative manner. In this study, an attempt has been made to identify and characterize the known as well as novel complex repetitive elements in the two most commercially valuable Solanaceae species of S. tuberosum and S. lycopersicum whose genomes have been reported recently [1,2]. The genomes of these two species appear to hold more complex repetitive elements than previously appreciated. Several of these repetitive elements appeared transcriptionally active while several were found associated with potential to carry out some regulatory impact. It was discovered that the repetitive elements had a huge influence over the host protein coding genes as >95% of the genes in the two Solanaceae genomes overlapped with repetitive elements, suggesting a major role being played by repetitive elements in gene formation and transcriptionally active elements. The impacts of repetitive elements with respect to regulatory elements was also studied. The present study is mainly focused around the complex repeats and has excluded analysis over simple and tandem repeats.
Materials and Methods
Sequence information
The genome sequences, co-ordinates of various genomic elements, protein and transcript sequences of S. tuberosum (S. tuberosum group phureja doubled monoploid clone) and S. lycopersicum (S. lycopersicum cv. Heinz 1706) were downloaded from Ensembl plants (http://plants.ensembl.org/index.html). Co-ordinates of introns and upstream regions with respect to gene start sites were extracted using an in-house PERL script. Syntenic regions between S. tuberosum and S. lycopersicum genomes were identified using Symap (version 42) [39]. The gene co-ordinates along-with sequences were submitted as an input in the form of GTF file to Symap for both species. BedTools [40] was used to merge the overlapping genomic co-ordinates of various genomic elements. Pre-miRNA sequences for S. tuberosum and S. lycopersicum were downloaded from miRBase (version 20) [41]. The non-coding RNA sequences were downloaded from Rfam database version 11 [42]. Orthologous genes in S. tuberosum and S. lycopersicum were identified by matching the respective protein sequences using BLASTP [43].
Repetitive element identification
RepeatModeler (http://www.repeatmasker.org/RepeatModeler.html) is a de-novo repeat identification tool, which also provides annotation to identified sequences utilizing three different repeat identification algorithms namely RECON [44], RepeatScout [45] and TRF [46]. For identification of known (based on homology) as well as novel repetitive sequences, a database of the genome sequences of S. tuberosum and S. lycopersicum was generated using BuildDatabase command, on which RepeatModeler was executed. RepeatModeler (http://www.repeatmasker.org/RepeatModeler.html) generated the consensus sequences of identified repeat families, which was used by RepeatMasker (http://www.repeatmasker.org/RMDownload.html) to annotate the repeats in the genomes of S. tuberosum and S. lycopersicum. Repeat family sequences identified using RepeatModeler were matched to library sequences of RepeatMasker/Repbase to identify the already known repeats among the novel repeats.
To verify the annotation provided by RepeatModeler for the identified repetitive elements, two different approaches were followed. Firstly, the repetitive library sequences were provided as an input to RepeatMasker which identifies repeats based on homology search against Repbase annotations. Secondly, RepeatProteinMasker (http://www.repeatmasker.org/cgi-bin/RepeatProteinMaskRequest), a tool which annotates repeats based on their amino acids domains over their translated frames, was executed on the repetitive sequence library to identify the conserved protein domains in the library sequences. Annotation of library sequences was done by mapping the annotations provided by the mentioned approaches. If a library sequence had same annotation in all methods, the sequence was annotated with highest confidence. However, if the annotations provided by the mentioned approaches were not found converging, annotation provided by the RepeatProteinMasker was assigned. If RepeatProteinMasker could not identify any protein domain in any given sequence, then, the annotation of the sequence was determined based on RepeatMasker and RepeatModeler. Besides this, sequences were also subjected to manual characterization processes based on the defining features like sequence, domain, internal elements, and target site duplications properties of various families.
All those repetitive families which could not be assigned any annotation were specified as “Unknown”. Unknown families of S. tuberosum and S. lycopersicum were matched against each other to identify similar families in both species. For further annotation of the “Unknown” repeat families identified by RepeatModeler, the consensus repeat family sequences were matched to non-coding RNA sequences downloaded from Rfam database (version 11) [42]. Unknown repeat family sequences were matched to the non-coding RNA sequences using BLASTN and best hits were used to characterize the unknown families. The remaining unknown repeat family sequences were scanned against the NCBI NT database using TBLASTX to identify any multi-copy genes/pseudo-genes. Remaining “Unknown” repetitive element families were characterized by searching for repeat family specific properties mentioned above. The co-ordinates of the repetitive elements identified in this study were also matched with the co-ordinates of the repetitive elements provided in the PGSC_DM_v4.03 (http://solanaceae.plantbiology.msu.edu/pgsc_download.shtml) for S. tuberosum and the ITAG v2.3 [2] for S. lycopersicum. This was performed in order to identify the commonly reported repeats by various approaches, as well as the novel repeats reported in this study.
Multiple sequence alignments of some families was performed using ClustalW [47], while phylogenetic trees were created using the Neighbor Joining method with a bootstrap value of 1000.
Genomic Analysis of Repetitive elements
To identify the distribution pattern of repetitive elements across the genomes of S. tuberosum and S. lycopersicum, different analyses were performed. Percentage of a chromosome's length occupied by repetitive elements was calculated as the total number of base-pairs of repetitive element coverage for a given chromosome with respect to the total length of the respective chromosome. For this analysis, the co-ordinates of repetitive elements provided by RepeatMasker using consensus library sequences were analyzed while merging the overlapping regions using BedTools [40]. The count of base-pairs of repetitive elements was then used to calculate the percentage of chromosome occupied by repeats. To assess the most common repeat families for a particular chromosome, percentage coverage of every repeat family for each chromosome was also calculated.
To identify the overall distribution of repetitive sequences in terms of proximity to gene rich regions, the percentage of repetitive sequences within a gene or in its 5kb upstream region was calculated. For this study, repeat sequences having overlap with the coding sequence and 5kb upstream region were identified. Repetitive sequences found overlapping with the aforementioned regions were considered as those preferring gene rich regions. Percentage of repeat family sequences found in either upstream region of the gene or in the coding regions were also calculated using the above mentioned relation. All other repetitive sequences which were not found in the vicinity of genes (coding region + 5kb upstream), were considered as repetitive sequences preferring intergenic regions. To test whether there is any significant enrichment of genes around repetitive regions, binomial test was applied on the count of genes found to overlapping with repetitive elements. The null hypothesis stated was: “There is no significant enrichment of genes near repetitive elements”. The test was implemented in R.
A protein coding gene usually consists of exons, introns and UTRs. It was previously studied that the repetitive elements which were found residing within a coding region also showed differential preferences with respect to exonic or intronic regions [48]. Therefore, in order to identify such differential preference, repetitive elements found overlapping exclusively with either exons or introns were identified. For this analysis, the repetitive sequences within the co-ordinates of exons or introns were considered. UTRs were not considered in this study as UTRs have not yet been identified in both species.
To further assess the relationship between genomic location of repetitive elements and coding regions (exons) of the genome, Pearson Correlation Coefficient (PCC) was calculated between percentage coverage of repetitive elements and percentage coverage of coding region (exons) for every chromosome. The correlation between gene coverage and repeat coverage was then statistically validated by calculating the p-value of PCC via implementing t-test. This analysis was performed to identify whether gene density has any association with the accumulation of repetitive elements. Post ENCODE the scenario has changed a lot in terms of functionality of genomes and perceptions about so called junk elements. Many previous works have showcased how these elements have emerged as regulatory engines of genes, and their general influences over genes have been increasingly revealed in many recent reports [49–54]. This provided motivation to understand the distribution of genes with respect to repetitive elements in a genome.
Analysis of Exonization
Exonization is the process of insertion of a non-protein coding region in the coding region of a gene, where this region starts functioning as a part of exon [48,53,55,56]. This occurs due to the presence of pseudosplice or splice donor sites within repetitive elements which lead to generation of a new gene sequence [55]. Along-with protein coding genes, certain long non-coding RNAs were also reported to be generated in a similar manner [57]. However, the fraction of such exonized transcripts in the transcriptome is low [58,59]. If any exonized gene provides improved functionality or a novel function, then such events may become fixed in the genome. To study the presence of such exonized transcripts in the two genomes, the sequence and structure of orthologous genes were analyzed. The orthologous gene-pairs were subjected to global alignment using EMBOSS Stretcher program to measure the sequence differences. The identified indels and substitutions were mapped against the identified repetitive elements.
The positions of indels and substitutions were mapped to the corresponding amino acid sequence to identify its impact over the protein structure. This was performed by comparing the six frames translated transcript sequences for respective amino acid sequence for both species. Amino acid sequences of S. tuberosum and S. lycopersicum were also subjected to global alignment using EMBOSS Stretcher. The indels and substitutions in the protein sequences were transformed to the nucleotide sequences. The changes in the amino acid sequences which were found within the repeat-overlapping region in the nucleotide sequences were recorded. To identify effects of these changes on protein structures, the secondary structure of protein sequences was studied using PsiPred [60]. Orthologous proteins showing changes in the secondary structure were then subjected to three dimensional (3D) structure prediction via threading using RaptorX [61]. Due to unavailability of suitable template (having sequence identity ≥ 30%) for these proteins in Protein Data Bank, homology protein models could not be built. Therefore, threading based structure prediction was performed [62,63]. This method realizes upon unique protein folds present in several resolved protein 3D structures [64]. Thus, in the absence of suitable protein structure template it was the best way to search for a protein fold present in target protein sequence against the fold library of resolved 3D structures. The 3D structures obtained were validated using web-server of RAMPAGE (http://mordred.bioc.cam.ac.uk/~rapper/rampage.php). Structures formed represented only part of the complete sequence due to threading based tertiary structure modeling. Therefore, residues showing changes in orthologous proteins were matched with the structures. 3D structures were then used as input in LigPlot+ [65] to visualize residues targeted hydrogen bonding and hydrophobic contacts showing changes in orthologous proteins due to repetitive elements.
Transcription Factor Analysis
Many transcription factor binding sites (TFBS) were previously reported to be exapted from repetitive elements and regulate the downstream genes [51,66,67]. Thus, the TFBS within repeat overlapping regions in 2kb upstream sequences were identified and significant gain/loss of TFBS in the repeat overlapping region of every pair of orthologous genes was studied.
To analyze the possible TFBS hosted by repetitive elements, TFBS on the 2kb upstream region with respect to the start co-ordinate of the genes were identified using pPromotif [50,68]. pPromotif is a tool to identify TFBS on plant genomic sequences based on probabilistic models for various TFBS derived using large amount of experimental and high throughput data. So far, pPromotif has matrices modeled for 57 transcription factor families. pPromotif was executed using the default parameters. Count of every TFBS on 2kb upstream region of every orthologous gene pair was calculated and difference between the total number of sites was calculated. Gain/loss of TFBS for orthologous genes was also estimated. Binomial test was applied to statistically test the significance of the observed gain/loss of TFBS.
Role of Repetitive elements in miRNA evolution
To analyze the influence of repetitive regions over miRNAs, miRNAs reported so far from both species were identified for their overlap with repeats. Pre-miRNA sequences were downloaded from miRBase (version 20) [41] and mapped to both the genomes. Using the identified co-ordinates, overlaps between miRNAs and repetitive sequences were calculated. Orthologous miRNAs were identified in S. tuberosum and S. lycopersicum by comparing the pre-miRNAs with each other, while considering their annotations. The repetitive content across and around them was analyzed to find out any potential association of repetitive elements with the evolution of miRNAs in the two species. Binomial test was also applied to identify any possible influence of repetitive elements on the accumulation of miRNAs with the null hypothesis that repetitive elements are not associated with miRNAs and thus would not be enriched in miRNA sequences. The orthologous miRNAs were also studied for the presence of conserved motifs and their positional arrangements in 2kb upstream and downstream regions around the pre-miRNA sequence, considering any possibility of detecting the eroded repetitive regions hosting the miRNAs. Using WATER tool from the EMBOSS package [69], local alignment was performed between these sequences and conserved motifs between these orthologous miRNAs were extracted. Motifs at least 5 bp in length and having overlap with a repetitive element in at least one of the orthologous miRNAs were considered. Moreover, multiple motifs present in the same arrangement for both orthologous miRNAs were considered as strong candidates for being footprints of some repetitive elements which might have been eroded during evolution [70,71].
Abundance analysis of repetitive elements
Transcriptional activity and abundance of repetitive elements identified in this study was calculated using data from two different platforms namely, Next Generation Sequencing and Microarrays. For S. tuberosum, RNA-Seq data was taken from NCBI SRA (SRP005965) [1] having a total of 40 different experimental conditions, while for S. lycopersicum RNA-seq data was taken from three different experiments namely SRP019504 [72], SRP007969 [73] and SRP026374 [74] having a total of 55 experimental conditions. Normalized microarray data and the probe sequences were downloaded from Array Express with the accessions E-MTAB-629 and E-MTAB-634 for S. tuberosum [75] for 42 conditions. For S. lycopersicum, microarray data was downloaded from GEO with the accession ID GSE22300 [76], making a total of 10 conditions.
For abundance calculation of repetitive elements and genes of both species, RPKM was calculated using SeqMap and R-seq with default commands. From the RPKM value, the average expression of every repeat family was then calculated by using the following equation:
For calculating expression from microarray platform, methodology as proposed by Reichmann et al. [77] was used. Probe sequences were searched against the genomes using BLAST. From BLAST result, the best match for every probe was identified and the co-ordinates of the probes were recorded. Co-ordinates of the probes were matched with the co-ordinates of the genes. If a probe overlapped with a gene with more than 90% of its length, expression values of the probe was assigned to the respective gene. The probes which could not be matched to genes were further matched with co-ordinates of repeats and expression of the probe whose length coverage was more than 90% with repeats was assigned to the corresponding repeat. Average expression of repeat family was then calculated using the above mentioned equation.
Although RPKM and microarray based abundance estimations were done for repetitive elements, small RNA sequencing data was also used, considering their reported association with small RNA biogenesis. For S. tuberosum, sRNA read data was downloaded from NCBI SRA under accession ID SRP033230 [78]. For S. lycopersicum, sRNA read data was downloaded from GEO (GSE18110) [79]. These sRNA reads were processed by removing adapter sequences and only those sRNA reads were selected which were at least 17 bp long. These processed sRNA reads were first mapped to ncRNA sequences downloaded from Rfam (version 11) and to the transcript sequences to remove any read sequence which could be a degradation product. The remaining reads were mapped to the repetitive elements using Bowtie [80] with maximum of one mismatch.
All metadata associated with this work has been made freely available at: http://scbb.ihbt.res.in/SCBB_dept/solanum_metadata.php and https://github.com/mrigayamehrajha/Solanum-Repeats-Metadata.
Results and Discussion
The total genome size of S. tuberosum is 810.6 MB, ~85% of the genome is sequenced and total number of yet to be sequenced regions contributes towards 15.78% (127,958,425 bp) of the genome. For S. lycopersicum, the total genome size is 781.6 MB with ~5% (44,030,063 bp) of the genome as yet not sequenced. Thus, the sequences of these two genomes were sufficiently complete for this study. The repetitive elements have been reported to occupy a significant percentage of different genomes ranging up to ~80% of the genome sequence in many plant species like wheat and Capsicum spp [81,82]. S. tuberosum and S. lycopersicum, both have moderately large genomes with significant portion of their genome being represented as complex repetitive elements. Detailed identification and characterization of repetitive elements in these genomes provided a more complete map of repetitive elements of these two genomes and their impact over the associated genomes, several of which were not reported earlier. It was identified that repetitive elements occupy ~49% and ~60% of the genomic sequence of S. tuberosum and S. lycopersicum, respectively, with chromosome 12 as the most repeat rich chromosome in both S. tuberosum and S. lycopersicum (Fig 1, Table 1). The repeat families identified in both species included DNA transposons and retrotransposons, some of which could not be assigned to any particular DNA transposon or retrotransposon order. Repetitive elements are multifaceted which are increasingly being associated with several key functional aspects of the genome. Repetitive elements identified in this study were investigated with respect to their distribution across the genomes and it was found that these elements were remarkably abundant in the gene-coding regions. Since the presence of repetitive elements around genes could have a plethora of outcomes, the functional relevance of these elements was studied by analyzing their impacts on the associated genes via cis-regulation, exonization and miRNA evolution. It was found that ~4% (41,966) of repetitive elements in S. tuberosum and ~0.13% (1,358) repetitive elements in S. lycopersicum overlap with regions of the genome which are still not sequenced. Therefore, it is unlikely to see any major change in the findings made here even after sequencing of these regions.
Fig 1. Distribution of repetitive elements, genes, pseudo-genes and positions of orthologous miRNAs having footprints of old repetitive elements in (A) Solanum tuberosum (B) Solanum lycopersicum.
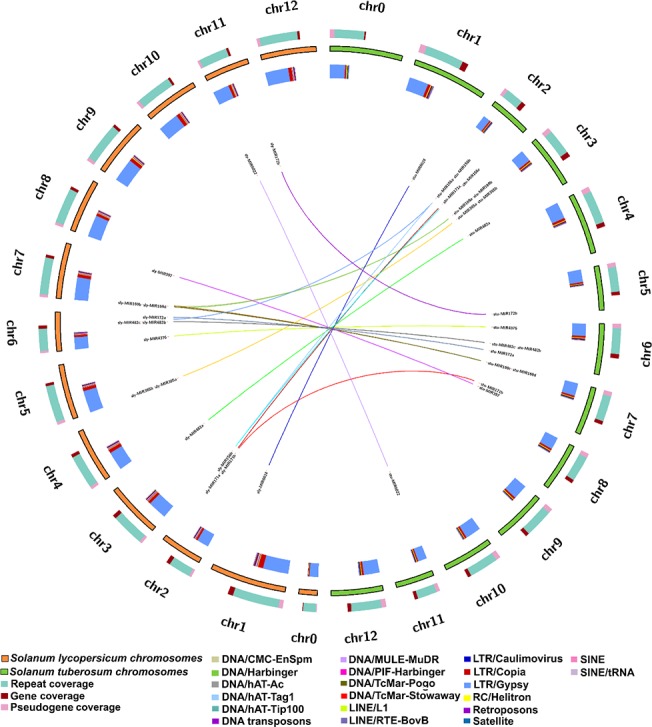
Table 1. Chromosome wise coverage of repetitive elements.
Coverage of different repeat super-families was calculated as the percentage of nucleotides represented by repetitive elements out of total nucleotides of every chromosome.
| Solanum tuberosum | |||
| Chromosome | Total number of base pairs occupied by repeats | Total number of base pairs in the chromosome | Percentage of chromosome occupied by repeats |
| chr0 | 33,415,365 | 85,736,662 | 38.97 |
| chr1 | 43,197,010 | 88,663,952 | 48.72 |
| chr2 | 19,955,974 | 48,614,681 | 41.05 |
| chr3 | 28,514,945 | 62,190,286 | 45.85 |
| chr4 | 36,487,020 | 72,208,621 | 50.53 |
| chr5 | 27,570,804 | 52,070,158 | 52.95 |
| chr6 | 28,789,650 | 59,532,096 | 48.36 |
| chr7 | 28,734,258 | 56,760,843 | 50.62 |
| chr8 | 28,479,417 | 56,938,457 | 50.02 |
| chr9 | 32,022,384 | 61,540,751 | 52.03 |
| chr10 | 32,804,959 | 59,756,223 | 54.90 |
| chr11 | 21,872,185 | 45,475,667 | 48.10 |
| chr12 | 33,669,946 | 61,165,649 | 55.05 |
| Total | 395,513,917 | 810,654,046 | 48.79 |
| Solanum lycopersicum | |||
| Chromosome | Total number of base pairs occupied by repeats | Total number of base pairs in the chromosome | Percentage of chromosome occupied by repeats |
| chr0 | 14,457,253 | 21,805,821 | 66.30 |
| chr1 | 52,060,945 | 90,304,244 | 57.65 |
| chr2 | 24,994,446 | 49,918,294 | 50.07 |
| chr3 | 36,771,573 | 64,840,714 | 56.71 |
| chr4 | 37,594,132 | 64,064,312 | 58.68 |
| chr5 | 41,130,811 | 65,021,438 | 63.26 |
| chr6 | 24,862,196 | 46,041,636 | 54.00 |
| chr7 | 41,046,411 | 65,268,621 | 62.89 |
| chr8 | 39,637,992 | 63,032,657 | 62.88 |
| chr9 | 43,430,035 | 67,662,091 | 64.19 |
| chr10 | 40,148,283 | 64,834,305 | 61.92 |
| chr11 | 31,877,585 | 53,386,025 | 59.71 |
| chr12 | 42,301,100 | 65,486,253 | 64.60 |
| Total | 470,312,762 | 781,666,411 | 60.17 |
Repetitive elements identified in S. tuberosum and S. lycopersicum
The initial identification of repetitive elements in S. tuberosum genome was performed on 66,245 super-scaffolds [1] using similarity based approaches like RepeatMasker and RepeatProteinMask. 62.2% of the genome of S. tuberosum was reported to be represented by repetitive elements where ~25% of these repeats were not characterized. The super-scaffolds were distributed into twelve chromosomes and those which could not be assembled and assigned to any chromosome were merged to create a thirteenth chromosome, Chromosome 0. Therefore, sections of many previously identified repetitive elements were divided into different chromosomes, while some were merged together. This might lead to the identification of many new repetitive elements as well as removal of some previously identified repetitive elements across the super-scaffolds. Similarly, in S. lycopersicum, though the LTRs were found using LTR_STRUC program, most of the repetitive elements were identified using RepeatMasker [2]. Initially, the total repetitive content of these two genomes were reported as 57.6% and 62.2% in S. lycopersicum and S. tuberosum (at scaffold level), respectively. However, the observed repetitive content has reduced once the contigs and scaffolds were merged and distributed across the chromosomes with values lower than expected repetitive content (19.49% for S. tuberosum). As already mentioned, the previous repeat annotations for both the genomes had considered similarity based approaches predominantly to report the repeats, leaving ample scope for novel elements' discovery using combination of similarity and de novo based approaches. The similarity based approaches usually miss out sparsely similar and divergent members as well as species specific repeats. The present work applied a combination of similarity based, de novo based and manual analysis to identify the repetitive elements in the two species.
RepeatModeler identified 1,921 and 1,438 consensus repeat family sequences in S. tuberosum and S. lycopersicum respectively (Table 2, S1 Table). Of these identified repeat families, 892 repeat family consensus sequences in S. tuberosum and 474 repeat family consensus sequences in S. lycopersicum were labeled as “Unknown” as no significant similarity with known repeat families could be detected for these families. For verifying the annotation of all repeat consensus families and to provide annotations to the non-characterized repeat consensus family sequences, using RepeatProteinMasker and RepeatMasker, the annotations were searched against the the annotated library sequences as mentioned in the methods section. Thus, annotation was verified and provided for 1,233 out of 1,921 and 1,075 out of 1,438 consensus repeat families for S. tuberosum and S. lycopersicum, respectively. A total of 204 “Unknown” repeat families in S. tuberosum and 111 “Unknown” repeat family consensus sequences in S. lycopersicum were annotated (Fig 2). A total of 1,061,377 and 793,890 repetitive sequences were identified in S. tuberosum and S. lycopersicum genomes, respectively. After removing the sequences annotated as rRNA, snRNA, tRNA, simple repeats, low complexity and unannotated elements, 629,713 and 589,561 repetitive elements belonging to different complex repeat families were obtained for S. tuberosum and S. lycopersicum, respectively (Table 2). DNA transposons identified in this study included hAT elements like hAT-Ac, hAT-Tag1, hAT-Tip100, Harbinger/PIF-Harbinger, CMC-EnSpm, MULE-MuDR, TcMar-Pogo/TcMar-Stowaway and Helitrons. Retrotransposons identified included non-LTR elements like LINE L1 and RTE-BovB, SINE elements, LTR elements including Gypsy, Copia, Caulimovirus and ERVs (Table 2).
Table 2. Distribution of identified repeat families in S. tuberosum and S. lycopersicum.
Total number of families, super-families and elements identified in both species are presented.
| Repeat Family | Repeat Super-family | Total number of families | Total number of elements |
|---|---|---|---|
| Solanum tuberosum | |||
| DNA transposons | DNA/CMC-EnSpm | 43 | 17,198 |
| DNA/CMC-EnSpm? | 4 | 2,801 | |
| DNA/Harbinger | 18 | 14,341 | |
| DNA/hAT-Ac | 22 | 10,951 | |
| DNA/hAT-Tag1 | 3 | 1,987 | |
| DNA/hAT-Tag1? | 2 | 1,201 | |
| DNA/hAT-Tip100 | 21 | 12,689 | |
| DNA/MULE-MuDR | 16 | 8,318 | |
| DNA/PIF-Harbinger | 9 | 9,469 | |
| DNA transposons | 35 | 29,354 | |
| DNA/TcMar-Pogo | 6 | 1,967 | |
| DNA/TcMar-Stowaway | 13 | 27,720 | |
| RC/Helitron | 5 | 1,957 | |
| Retrotransposons | LINE/L1 | 63 | 23,521 |
| LINE/RTE-BovB | 17 | 27,091 | |
| LTR/Caulimovirus | 23 | 5,858 | |
| LTR/Copia | 172 | 72,026 | |
| LTR/Gypsy | 558 | 334,474 | |
| Retroposon | 11 | 9,926 | |
| SINE | 2 | 3,853 | |
| SINE/tRNA | 6 | 10,567 | |
| Uncategorized | Unknown | 688 | 329,727 |
| snRNA | 2 | 487 | |
| rRNA | 5 | 1,845 | |
| Satellite | 4 | 2,444 | |
| Solanum lycopersicum | |||
| DNA transposons | DNA/CMC-EnSpm | 28 | 21,007 |
| DNA/CMC-EnSpm? | 2 | 2,442 | |
| DNA/Harbinger | 22 | 13,390 | |
| DNA/hAT-Ac | 30 | 15,164 | |
| DNA/hAT-Tag1 | 8 | 3,655 | |
| DNA/hAT-Tag1? | 2 | 1,021 | |
| DNA/hAT-Tip100 | 16 | 11,757 | |
| DNA/MULE-MuDR | 34 | 16,344 | |
| DNA/PIF-Harbinger | 9 | 6,118 | |
| DNA transposons | 38 | 34,059 | |
| DNA/TcMar-Pogo | 4 | 5,132 | |
| DNA/TcMar-Stowaway | 14 | 22,343 | |
| RC/Helitron | 2 | 2,398 | |
| Retrotransposons | LINE/L1 | 28 | 13,653 |
| LINE/RTE-BovB | 14 | 21,154 | |
| LTR/Caulimovirus | 4 | 1,223 | |
| LTR/Copia | 165 | 71,179 | |
| LTR/ERV1 | 1 | 56 | |
| LTR/Gypsy | 525 | 306,511 | |
| Retroposon | 11 | 9,062 | |
| SINE | 4 | 3,772 | |
| SINE/tRNA | 6 | 7,614 | |
| Uncategorized | Unknown | 363 | 145,776 |
| snRNA | 1 | 157 | |
| rRNA | 1 | 272 | |
| tRNA | 3 | 838 | |
| Satellite | 1 | 507 | |
Fig 2. Venn diagram showing annotation provided to the non-characterized repeat families using different approaches (A) Solanum tuberosum (B) Solanum lycopersicum.
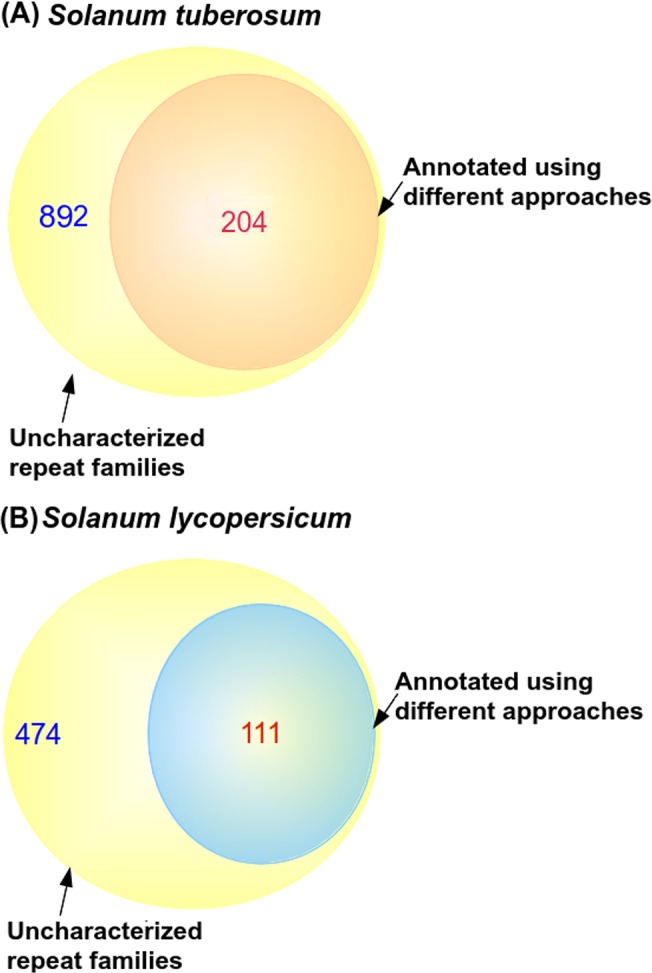
The inner circle represents the repeat consensus sequences assigned annotations using the different approaches. 204 (out of 892) non-characterized repeat families were characterized in S. tuberosum and 111 (out of 474) non-characterized repeat families were annotated in S. lycopersicum.
The remaining non-characterized consensus repeat families amounted to a total of 688 repetitive element families in S. tuberosum and 363 repetitive element families in S. lycopersicum (Table 2). The non-characterized families might account for some novel repeat families or pseudo-genes for which further characterization of these unknown repeat families was performed. These sequences were searched against the non-coding RNA sequences downloaded from the Rfam database. Of the 688 unknown repeat family consensus sequences in S. tuberosum, only nine repeat family consensus sequences matched the annotated non-coding RNA sequences which included introns, miRNA genes, 5S rRNA and SRP while the remaining 679 repeat family sequences did not match any non-coding RNAs (S2 Table). Similarly, out of 363 unknown repeat family sequences of S. lycopersicum only two repeat family consensus sequences matched the non-coding RNA sequences in Rfam (S2 Table) which included introns and miRNA genes. The remaining 679 repeat family consensus sequences of S. tuberosum and 361 repeat family consensus sequences of S. lycopersicum were searched against the NCBI nucleotide (NT) database using TBLASTX (S2 Table). In this analysis, 574 repeat family consensus sequences of S. tuberosum and 326 repeat family consensus sequences of S. lycopersicum matched with known nucleotide sequences as pseudo-genes (S2 Table). The remaining 105 repeat family consensus sequences of S. tuberosum and 35 repeat family consensus sequences of S. lycopersicum were then searched for conserved known motifs which would enable their classification. Motifs for internal Pol III promoters, A-Box and B-box, which are usually found within the SINE elements were searched in the remaining consensus sequences. Same was done for 5' and 3' conserved motifs for B. oleraceae SINE elements [83,84]. 105 repeat family consensus sequences in S. tuberosum and 34 repeat family consensus sequences in S. lycopersicum showed the presence of at least one of these motifs (S2 Table), suggesting their possible association with SINE elements. These non-characterized repetitive families of S. tuberosum and S. lycopersicum were also compared with each other to identify the common non-characterized families. In this analysis, it was found that 41 out of 688 non-characterized families in S. tuberosum matched with 44 out of 363 non-characterized families of S. lycopersicum, where some repeat family consensus sequences of S. lycopersicum exhibited multi-homologs for same repeat families in S. tuberosum.
The repeat families identified here were compared with the repeat families identified in the 4.03 version of the S. tuberosum genome released by the Potato genome sequencing consortium (PGSC_DM_v4.03) (http://solanaceae.plantbiology.msu.edu/pgsc_download.shtml) and the initially identified repetitive elements in S. lycopersicum by Tomato genome consortium (ITAG 2.3). 681 (35.45%) families of S. tuberosum identified in this study belonging to 7 repeat super-families matched with 7 repeat super-families (DNA transposons, DNA/Harbinger, DNA/hAT, LTR/Copia, LTR/Gypsy, RC/Helitron and SINE) identified by the potato genome sequencing consortium (PGSC_DM_v4.03), while 732 (50.90%) families of S. lycopersicum identified in this study belonging to 5 repeat super-families matched with 5 repeat super-families (DNA transposons, DNA/hAT, LTR/Copia, LTR/Gypsy and SINE) identified by the ITAG2.3 in S. lycopersicum genome. Although, a total of 699,160 (88.07%) elements identified in this study matched with 673,145 (93.56%) elements identified by the ITAG2.3 in S. lycopersicum genome and a total of 346,179 (32.62%) elements identified in this study matched with 331,009 (81.76%) elements identified by the PGSC_DM_v4.03 in S. tuberosum. Some annotations, however, differed from the previously done annotations at family level. Details are provided in S3 Table.
Distribution of repetitive elements across the genomes
It has been observed that in mammalian species, non-LTR elements (LINEs and SINEs) are more abundant while in plants LTR elements are more prevalent [85]. Such differential accumulation of repetitive elements has been proposed to be either due to some species-specific amplifications or deletions of specific elements [85]. Even within a species the distribution of repetitive elements is highly dependent upon the family of complex repeats [85]. Some repeats have been found enriched in the regulatory regions upstream the protein coding genes, while some are found within introns where some get exonized and domesticated [86]. Thus, to understand the genomic hot spots for association of repetitive elements and their overall spread, the distribution patterns of these elements in the two genomes were studied.
The repeat families most prevalent in the genomes of S. tuberosum and S. lycopersicum were analyzed in two ways: 1) the total number of copies of every repeat family, and 2) genome-wide distribution of repeat families were calculated. The super-family having maximum copies in both species was LTR/Gypsy (Table 2). The total number of repetitive elements belonging to LTR Gypsy were 334,474 and 306,511 in S. tuberosum and S. lycopersicum, respectively. Similarly, coverage of each repeat super-family on every chromosome was also calculated which revealed LTR Gypsy occupying the largest number of bases on every chromosome in both the genomes (Fig 1, S4 Table). Two other repeat super-families which occupied a significant percentage of the genome sequence were LTR Copia and LINE elements L1 (S4 Table). This trend is in sync with earlier observations which report LTR Gypsy as the most prevalent element in plant genomes and presence of LTR elements higher than any other complex repetitive element in overall. Pearson Correlation Coefficient (PCC) was calculated between the gene coverage and coverage of repeat super-family for every chromosome (Table 3) to identify the association between gene coverage and accumulation of repetitive elements. It was found that coverages of DNA transposons and TcMar-Stowaway were significantly correlated with the gene coverage, supported by significant p-values (Table 3). When the correlation of gene coverage and coverage of LTR/Gypsy elements was calculated while considering chromosome 0, it was significant only in S. lycopersicum. When correlation was estimated excluding chromosome 0, correlation coefficient was significant in both species. Chromosome 0 symbolizes the yet incomplete and unassigned parts of the genome. This chromosome is ~2.5 times larger in S. tuberosum than in S. lycopersicum. Similar density of repetitive elements on the chromosomes of S. tuberosum and S. lycopersicum thus encouraged the identification of syntenic regions in the two genomes. It was found that all the twelve chromosomes of S. tuberosum and S. lycopersicum were highly syntenic to each other (Fig 3), sharing high similarity for genes and repeats distribution in overall.
Table 3. Correlation between coverage of repeat family and gene coverage on every chromosome.
| Solanum tuberosum | |||
| Repeat Type | PCC | T-statistic | P-value |
| DNA/TcMar-Stowaway | 0.9620917122 | 11.7000058689 | 1.51E-007 |
| DNA | 0.9596712079 | 11.3219072983 | 2.11E-007 |
| DNA/Harbinger | 0.9347881483 | 8.7282979872 | 2.83E-006 |
| DNA/PIF-Harbinger | 0.9334391543 | 8.6299382074 | 3.16E-006 |
| DNA/TcMar-Pogo | 0.8493600038 | 5.3371237214 | 2.38E-004 |
| DNA/MULE-MuDR | 0.8462555467 | 5.2680784776 | 2.65E-004 |
| DNA/hAT-Tag1 | 0.7845382608 | 4.1962606691 | 1.49E-003 |
| DNA/hAT-Tip100 | 0.7474276283 | 3.7314099879 | 3.32E-003 |
| SINE/tRNA | 0.7375728291 | 3.622641389 | 4.01E-003 |
| DNA/hAT-Ac | 0.6500795482 | 2.8374338255 | 1.62E-002 |
| SINE | 0.6215187549 | 2.6312820978 | 2.34E-002 |
| LINE/RTE-BovB | 0.6028804797 | 2.5062037609 | 2.92E-002 |
| LTR/Caulimovirus | -0.5729613412 | 2.3186171403 | 4.07E-002 |
| LTR/Copia | 0.4799071858 | 1.8142449766 | 9.70E-002 |
| Satellite | -0.4216051498 | 1.542057425 | 1.51E-001 |
| Retroposon | 0.3105955013 | 1.0837275421 | 3.02E-001 |
| RC/Helitron | -0.2474331189 | 0.8469796697 | 4.15E-001 |
| LTR/Gypsy | -0.2390211119 | 0.8164074281 | 4.32E-001 |
| LINE/L1 | 0.1577892178 | 0.5299666206 | 6.07E-001 |
| DNA/CMC-EnSpm | -0.0200565794 | 0.0665335318 | 9.48E-001 |
| Solanum lycopersicum | |||
| Repeat Type | PCC | T-statistic | P-value |
| DNA/Harbinger | 0.9544544177 | 10.6100060709 | 4.08E-007 |
| DNA/TcMar-Stowaway | 0.9455427665 | 9.6345035426 | 1.07E-006 |
| LTR/Gypsy | -0.9252673099 | 8.0902647726 | 5.87E-006 |
| DNA | 0.8976427794 | 6.7551253297 | 3.14E-005 |
| DNA/PIF-Harbinger | 0.8687875924 | 5.818921915 | 1.16E-004 |
| DNA/hAT-Tip100 | 0.7270065462 | 3.511652681 | 4.87E-003 |
| DNA/TcMar-Pogo | 0.7137034022 | 3.379389004 | 6.15E-003 |
| DNA/MULE-MuDR | 0.612600281 | 2.5705764119 | 2.60E-002 |
| SINE/tRNA | 0.6050936111 | 2.5207028186 | 2.84E-002 |
| DNA/hAT-Ac | 0.6041348528 | 2.5144103905 | 2.88E-002 |
| DNA/hAT-Tag1 | 0.5995442809 | 2.4845184335 | 3.03E-002 |
| SINE | 0.5610626717 | 2.2479999158 | 4.61E-002 |
| LINE/RTE-BovB | 0.3807148849 | 1.3655225463 | 1.99E-001 |
| LINE/L1 | -0.3745541236 | 1.3397850768 | 2.07E-001 |
| LTR/ERV1 | 0.3642191812 | 1.2970698665 | 2.21E-001 |
| LTR/Copia | -0.3101460642 | 1.0819923379 | 3.02E-001 |
| RC/Helitron | 0.3012442711 | 1.0477870658 | 3.17E-001 |
| DNA/CMC-EnSpm | 0.2752760876 | 0.9496781064 | 3.63E-001 |
| LTR/Caulimovirus | -0.2157779953 | 0.7329204594 | 4.79E-001 |
| Retroposon | -0.0845703336 | 0.2814965229 | 7.84E-001 |
| Satellite | 0.0297194853 | 0.0986119407 | 9.23E-001 |
Fig 3. Syntenic regions across S. tuberosum and S. lycopersicum.
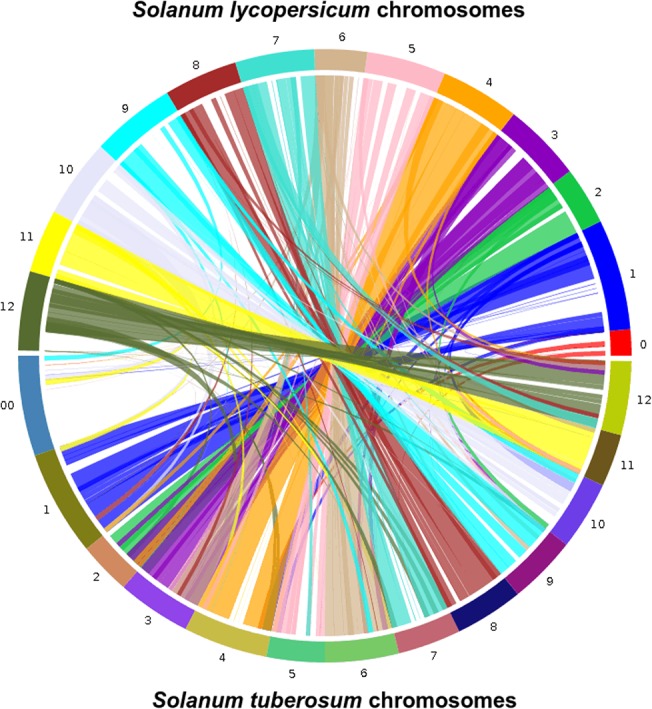
All the 12 chromosomes in S. tuberosum and S. lycopersicum were observed to show high syntenic relationship, showing similar distribution of genes in both species.
The co-ordinates of the repetitive elements and genic regions (5kb upstream + coding region) were studied to find any possible overlaps between the genic and repetitive regions. It was found that in S. tuberosum, 99.29% (38,740 genes out of 39,021 genes) genes had repeats overlapping either with their coding sequence or with the 5kb upstream region, whereas in S. lycopersicum 98.92% (34,303 genes out of 34,675 genes) genes had repeats overlapping with their genic regions and/or with the 5kb upstream region. This suggests that majority of protein coding genes in both species might be under the influence of repetitive sequences. This was also supported by a binomial test performed in both species suggesting a significant association between coding genes and repetitive elements (p-value <2.2E-16). When similar analysis was performed for the total repetitive elements identified and their genomic preferences, it was found that in S. tuberosum 33.72% of repetitive sequences (357,893 out of 1,061,377 repetitive sequences) were found overlapping with the genic regions while in S. lycopersicum, 30.37% of the repetitive sequences (283,295 out of 932,559 repetitive sequences) were found overlapping with the genic regions, suggesting the repetitive elements were more enriched in the intergenic regions rather than the genic regions. This distribution pattern was also studied for every repeat family on the basis of the count of the repetitive elements found in the genic or intergenic regions. It was identified that most of the repeat families had more number of elements in the intergenic regions than the genic regions. This pattern is quite understandable as compared to the intergenic regions the amount of genic region is very small. When the relative distribution of repeats was compared for the genic regions with the same for the intergenic regions, the association with intergenic region was found significantly higher. A t-test was also performed to validate significant enrichment of repetitive elements in the intergenic regions which gave a highly significant p-value for the enrichment of repetitive elements in the intergenic regions (p-value = 0.0001957 in S. tuberosum and p-value = 6.018e-11 in S. lycopersicum).
When analyzed for different repeat families, for LINE elements RTE-BovB, SINE elements and DNA transposon Stowaway, the larger proportions of the repetitive elements were found within the genic regions (Fig 4) in S. tuberosum. In S. lycopersicum, the DNA transposons hAT-Tag1, hAT-Tip100, PIF-Harbinger, RC Helitron and SINEs showed preferential abundance in the genic regions while the genic region preference of LTR/ERV1 repeat family was found very pronounced. The other repeat super-families were found within the intergenic regions are shown in Fig 4. The presence of repetitive elements within the coding regions points towards possible exonization event, while the presence of repetitive elements within the upstream region of gene might provide evidence for the possible exaptation of cis-regulatory elements. To study the repetitive elements' contribution towards such events, an analysis was performed and abundance of repetitive elements within the boundaries of gene coding regions or upstream regions was analyzed. It was found that most of the repeat families were more prevalent in the upstream regions of the genes (Fig 5(A)). Complex repetitive elements like LTR elements possess promoter elements which may provide regulatory elements for the downstream gene and in turn influence the gene expression in a tissue or stage-specific manner. A large number of previous studies highlighted the importance of accumulation of repetitive elements near genes where these elements served as sources of variation [87,88]. Presence of a repetitive elements in introns has been shown to influence the spatio-temporal expression of genes, creation of cryptic splices sites and other effects, whereas insertion of repetitive elements has been considered to be more devastating and associated with many disease conditions [89–94]. Therefore, identification of the different insertion spots of repetitive elements would provide insights into the possible mechanisms through which repetitive elements might be influencing genes and their products. To identify the preferential insertion of these elements in exonic or intronic regions, percentage count of repetitive elements overlapping with the exonic or intronic regions was calculated. It was found that in S. tuberosum, the distribution appeared uniform for both the regions. However, for S. lycopersicum, DNA transposon MULE/MuDR and LTR/ERV1 showed preferential accumulation within the exonic regions, while DNA transposons, TcMar-Stowaway, LINE elements RTE-BovB and SINE elements displayed a preferential association with the intronic regions (Fig 5(B)).
Fig 4. Percentage of repetitive elements found in the genic and intergenic regions in (A) Solanum tuberosum and (B) Solanum lycopersicum.
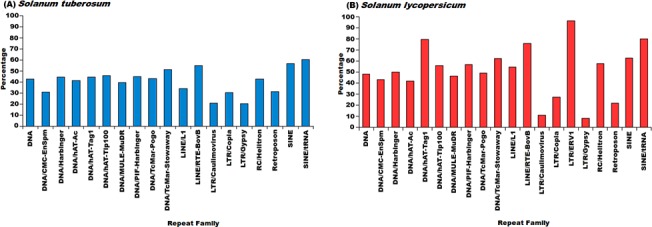
Percentage calculated from the total elements identified for every repeat super-family. Most of the repeat super-families in both species prefer intergenic regions as their insertion sites.
Fig 5. (a): Percentage of repetitive elements found in the upstream and genic regions in (A) Solanum tuberosum and (B) Solanum lycopersicum. In comparison to genic regions, repeat super-families prefer to be inserted within the upstream regions harboring regulatory elements. (b): Percentage of repetitive elements found in the exonic and intronic regions in (A) Solanum tuberosum and (B) Solanum lycopersicum.
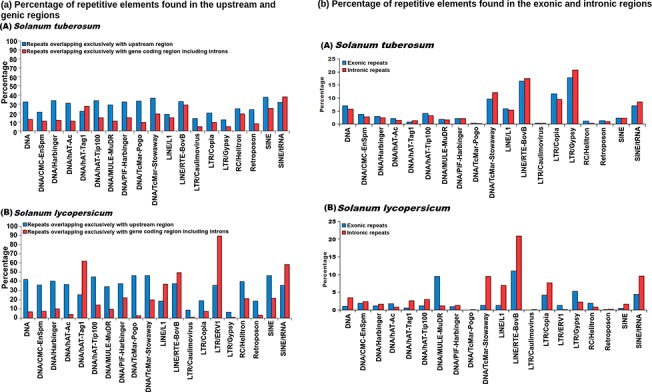
Distribution of repeat super-families did not show any preferential enrichment in exonic and intronic region in S. tuberosum, while in S. lycopersicum, different repeat super-families show differential enrichment in intronic and exonic region.
Impact of exonized repeats on protein coding gene's structure
As mentioned by Jacob [95], “to create is to recombine”, thus there is a high probability that applying various permutations and combinations to existing genomic materials, evolution shapes a genome. In this context, it is presumable that exonization is a favored mechanism of evolution as creating new combinations by incorporating segments of repetitive elements seems much easier than de novo generation of functional elements. Exonization occurs due to the presence of splice-sites within the repetitive elements which are found overlapping with genes. There are many possible outcomes of exonization, most of which lead towards beneficial inclusion and fixation [96,97]. Exonization of repetitive elements and their impact on shaping the transcriptome of various species has been studied widely [48,55,98–100]. SINE elements, specifically Alu elements, have been associated with many exonized transcripts having consequential effects [48,49,101,102]. Although, the protein coding potential of exonized elements has been under speculation, the contributions of repetitive elements to provide exons to transcript sequences is undeniable [56,59]. In A. thaliana, ~2000 loci have been reported to be derived from the segments of repetitive elements [93]. In rice, exonization of Ds elements has been studied for epsps gene [96]. The increasing examples being uncovered with regard to exonization of different repetitive elements might be viewed as a widely opted mechanism by genome to create new genes. However, this process can also have negative impacts on the fitness of the genome [101]. The exonized genes might become causative agents for different diseases and subject to selective screening [101]. However, to minimize the possibility of negative effects of exonization, most of these events occur in the duplicated genes where the possibility of interference with the gene function is lowered as well as ways to evolve new genic isoforms are opened. To study any possible impact of exonization of repetitive elements over the structures of the genes and possible variations, the orthologous sequences were studied and their secondary and tertiary structures were compared with respect to exonized repeats and possible variations. A comparative analysis for homologous amino acids sequences of these two species revealed that there were 27,923 orthologous gene pairs (S5 Table). Global alignment was performed for each orthologous pair to identify the changes in the sequences in the form of indels and substitutions. However, only 2,968 gene-pairs had indels associated with repeat-inhabited genic regions. Similar analysis for substitution positions was performed and it was found that 27,923 genes had substitutions in their alignments, whereas only 4,723 gene-pairs had substitutions which were found within the repeat-overlapping region of the genes. These changes (indels and substitutions) in nucleotide sequences were translated at the level of amino acids and it was identified that only 120 gene-pairs had changes in amino acid sequences corresponding to the changes in repeat-inhabited regions of the genes. It was further identified that, 61 gene pairs had changes in their secondary structure for the corresponding amino acid change (indels and substitution) positions. The amino acid sequences of these 61 gene-pairs were then subjected to 3D structure modeling using threading as the homology between these sequences and known protein structures in PDB was extremely poor (identity < 30%). After the prediction of the 3D structures, residue positions which were identified to have undergone changes in the secondary structures due to the presence of repetitive elements were mapped to 3D structures of their respective proteins. This was performed in order to look for the variations in orthologous proteins due to these changes. It was identified that many of these residues were at locations for which 3D structures were not available. Therefore, after removing such residues only 23 orthologous genes remained available for analysis. These remaining residues were studied using LigPlot+ and changes in the local environments of these residues (S6 Table) were observed. For the sake of clarity, these changes were categorized into three parts: substitutions, insertions in S. tuberosum genes and insertion in S. lycopersicum genes. Overall protein stability and conformation is determined by hydrogen bonds, van der Waals' forces and hydrophobic contacts formed among amino acid residues. As given in Table 4, barring a few cases, most of the residues which were substituted or inserted either in S. tuberosum or S. lycopersicum, had a comparatively more hydrophobic local environment. These changes might have consequential effects on the functioning of proteins and their binding specificity. Due to substitution and insertions at corresponding positions, the local environments having hydrophobic contacts and hydrogen bonding patterns varied leading to changed conformation of orthologous proteins. Hydrogen bonds contribute little to overall protein stability, but they align molecular groups in a specific orientation giving proteins a defined structure. When different non polar residues come closer, the extent of solvation decreases due to availability of less surface to water resulting in increase in entropy and thereby providing more stability to the protein structure. Thus, changes in the hydrophobic or H-bonding patterns may lead to alterations in the activity of orthologous proteins. The current analysis underlined the role of repetitive elements in bringing structural changes in proteins through exonization, however, for a limited number of genes. Most of these changes did not influence the protein structure and the critical regions significantly, an observation very similar to previous findings [51]. A comparison of the exonized repetitive regions of genes with the non repetitive coding regions of the genes for partition of changes between these two regions also suggested no significant difference, corroborating that repeats had no significant impact over the structure of the genes and their protein products. All these findings are in concordance with the previous studies which reported least structural changes by the repeats and maintenance of neutral evolution [51,59].
Table 4. Local environment of inserted or substituted residues in orthologous proteins.
The count of total H-bonds and hydrophobic contacts as represented in LigPlot+ results for orthologous genes of S. tuberosum and S. lycopersicum.
| Types of changes | Orthologous Proteins | Hydrophobic contacts | Hydrogen bonds |
|---|---|---|---|
| Substitutions | PGSC0003DMT400026140/Solyc02g050240.2.1 | 7/3 | 3/2 |
| PGSC0003DMT400034983/ Solyc03g095680.1.1 | 3/0 | 0/0 | |
| PGSC0003DMT400049939/ Solyc06g010200.2.1 | 2/2 | 2/4 | |
| PGSC0003DMT400053786/ Solyc01g057550.1.1 | 6/6 | 2/0 | |
| PGSC0003DMT400070945/ Solyc06g006040.1.1 | 4/6 | 2/2 | |
| Insertion in S. tuberosum | PGSC0003DMT400000956 | 10 | 0 |
| PGSC0003DMT400021686 | 5 | 2 | |
| PGSC0003DMT400021824 | 2 | 0 | |
| PGSC0003DMT400039198 | 2 | 1 | |
| PGSC0003DMT400039552 | 2 | 1 | |
| PGSC0003DMT400049812 | 3 | 2 | |
| PGSC0003DMT400053083 | 6 | 1 | |
| PGSC0003DMT400053786 | 6 | 0 | |
| PGSC0003DMT400056487 | 3 | 0 | |
| PGSC0003DMT400064637 | 2 | 1 | |
| PGSC0003DMT400074273 | 3 | 1 | |
| PGSC0003DMT400074709 | 2 | 3 | |
| PGSC0003DMT400078131 | 3 | 0 | |
| PGSC0003DMT400078833 | 0 | 1 | |
| PGSC0003DMT400077130 | 1 | 0 | |
| Insertion in S. lycopersicum | Solyc00g025650.1.1 | 5 | 1 |
| Solyc09g074850.2.1 | 2 | 2 | |
| Solyc09g074850.2.1 | 3 | 0 |
Repetitive regions influence the distribution of regulatory spots across the genome
Compared to the influence of repetitive elements over structural variations in genes, some previous studies have given enough reasons for speciation through regulatory variability caused by the repetitive elements. The contribution of repetitive elements has been acknowledged widely for gene regulations by exaptation of various cis-regulatory elements, enhancers and silencers [103,104]. Evolution of Brassica species has been associated with regulatory evolution carried out through TEs like MITE elements [105]. Similarly, the evolution of sunflower has also been attributed to transposable elements like LTR elements [103]. In P. abies, the large genome size has been attributed to slow accumulation transposable elements [106], while in olive genome, accumulation of tandem repeats has influenced its genome size [107]. In mammalian and primate genomes, several studies have reported about some major roles being played by the repetitive elements in the distribution and evolution of regulatory sites [50,51,108,109]. Therefore, it becomes imperative to assess the possible regulatory impacts of the repetitive elements over the Solanum genomes, especially when it is found that >95% genes of these two Solanum species are associated with the repetitive elements. The TFBS gained/lost in the 2kb upstream regions of orthologous genes and present within the repetitive elements were identified. Probability of gain/loss of TFBS for every TF in every orthologous gene pair was elucidated using binomial test. In this analysis, null hypotheses assumed was that there was no significant gain/loss of TFBS in the 2kb upstream region of orthologous genes due to repetitive element. From this analysis, only those TFBS which showed significant p-value (≤ 0.05) for gain/loss of TFBS while being present within repeat sequences were retained, rejecting the null hypothesis. In S. tuberosum, it was found that of the total binding sites of I-box gained/lost in the 2kb upstream region of the genes, ~36% were found overlapping with repetitive elements (Fig 6). The I-box promoter motif has been found to be present in the upstream region of genes involved in light based responses. I-box has been found associated with tomato genes and classified as a member of Myb-group of transcription factors. Similarly, another transcription factor (TF), SORLIP2 (Sequences Over-Represented in Light-Induced Promoters (SORLIPs)), was found to have a significant gain of their TFBS in the orthologous genes of S. tuberosum with ~23% of the gained sites occurring within the repetitive regions (Fig 6). This transcription factor has been associated with light-induced genes in cotyledon and roots of plants including A. thaliana. Another TF G-Box, which has been found involved in the regulation of expression of genes in response to light, anaerobic stress, abscissic acid and other metabolites. It was identified that 13% of the gained sites of G-box were within the repeat overlapping regions. Also, MADS family of transcription factors had ~14% of the TFBS gained in S. tuberosum. MADS TF possesses the MADS domain and these transcription factors have been associated critically with all sorts of development processes in plants, including flower development and gametophyte, embryo and seed development.
Fig 6. Gain/loss of Transcription factor binding sites (TFBS) in the upstream regions of orthologous genes in S. tuberosum and S. lycopersicum.
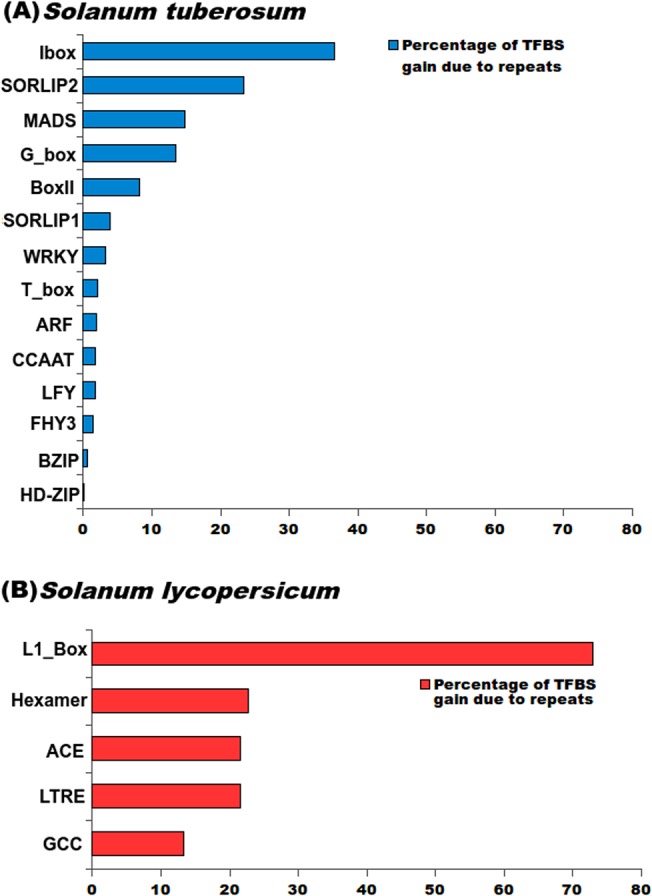
Plot showing the percentage of TFBS gained in orthologous genes contributed by repetitive elements.
In S. lycopersicum, gain of sites for five transcription factor families (L1-box, LTRE, Hexamer, GCC box and ACE) occurred in the repeat overlapping regions (Fig 6). GCC box binding factors have been shown to play significant roles in response to different secondary metabolites like jasmonate, which is involved in the activation of several pathogen responsive genes. GCC box also acts as an ethylene responsive element, which regulates some defense responsive genes. GCC box has also been found to be the binding site for PTi transcription factor which regulates the expression of defense related genes. In this study, ~12% of the gained sites of GCC in S. lycopersicum have been found within repeat-overlapping regions, showing that many defense responses are under indirect regulations by repetitive elements. Another transcription factor, whose binding sites were found overlapping with repetitive element was ACE (ACGT containing element). It was shown to have about 21% of the gained sites within the repeat-overlapping regions. ACE motifs are also associated with the light-responsive genes and anthocyanin biosynthetic genes. This shows another important plant specific functions being indirectly regulated by repetitive elements. LTRE is a low temperature response element found specifically in genes responsive to low temperature in plant species including A. thaliana and Barley, and it also had a significant number of gained sites overlapping with repetitive elements (~21%). Hexamer promoter element also seems to have been distributed by repetitive elements as ~22% of the gained hexamer sites were found within the repeat-overlapping regions. Hexamer promoter elements are reported to regulate histone H3 and H4 in different plant species including A. thaliana and Maize, where they were found regulating the expression of genes in meristems. The hexamer motif was seen to function in alliance with nonamer motifs. The case of L1 box response element requires exclusive mentioning, as the majority of gain of these sites could be attributed to the repetitive elements (~70%) (Fig 6). L1 box promoters are involved in L1-layer specific expression of genes, and it contains a L1-binding homeodomain and Myb binding motif. L1 layer corresponds to the outermost layer in a shoot apical meristem and is responsible for its growth. L1 box was identified to be 8 bp long cis-regulatory element essential for the expression of L1-layer specific genes. Other transcription factors whose TFBS have been gained in S. tuberosum and S. lycopersicum are described in Table 5 and S7 Table. All the transcription factors have been discussed elsewhere in good details [110].
Table 5. Gain of transcription factor binding sites due to repetitive elements in the promoter regions of orthologous genes.
| Solanum tuberosum | |||
| TF name | Gain in repeat overlapping region | Gain in Total 2kb region | Percentage of TFBS gain due to repeats |
| ARF | 11 | 574 | 1.92 |
| BoxII | 102 | 1,245 | 8.19 |
| BZIP | 1,369 | 265,990 | 0.51 |
| CCAAT | 437,839 | 25,186,369 | 1.74 |
| FHY3 | 45,132 | 3,328,200 | 1.36 |
| G_box | 12 | 89 | 13.48 |
| HD-ZIP | 1,519 | 3,453,104 | 0.04 |
| Ibox | 49 | 134 | 36.57 |
| LFY | 386 | 22,590 | 1.71 |
| MADS | 2,952 | 19,959 | 14.79 |
| SORLIP1 | 6 | 154 | 3.90 |
| SORLIP2 | 580 | 2,489 | 23.30 |
| T_box | 153 | 7,237 | 2.11 |
| WRKY | 458 | 14,375 | 3.19 |
| Solanum lycopersicum | |||
| TF name | Gain in Repeat overlapping region | Gain in Total 2kb region | Percentage of TFBS gain due to repeats |
| ACE | 45 | 217 | 20.74 |
| GCC | 25 | 195 | 12.82 |
| Hexamer | 484 | 2,221 | 21.79 |
| L1_box | 35,998 | 51,380 | 70.06 |
| LTRE | 194 | 936 | 20.73 |
All transcription factors mentioned above were found involved in major metabolic pathways, defense response, regulating specifically the genes showing response to light-induced stimuli and normal plant growth and development. This displays the extent to which many repetitive elements have been domesticated by the plants for their own survival purpose, contradicting the tags like “genomic parasites” or “junks” given to the repetitive elements initially.
Repetitive elements in miRNA genesis
miRNAs are a class of small non-coding RNAs with ~21–25 nucleotides in length [111–113]. These small RNA species have received enormous attention due to the regulatory roles played by them through post transcriptional gene silencing as well as RNA directed DNA methylation (RdDM) [68,114–118]. miRNAs have been shown to regulate ~60–70% of genes in an organism while displaying broad range of target interactions as well the modes of their biogenesis [112,114,119,120]. Due to such important implications posed by miRNAs in different biological processes and disease conditions, miRNAs have gained considerable importance. Many new miRNAs and their expressions have been studied with regard to many different diseases, where their roles in regulating these processes have been strengthened. miRNA sequences identified in different organisms have grown exponentially over the years [121]. However, the process of miRNA evolution and biogenesis is still intriguing and suggestive of multiple sources. In recent times, some dedicated studies by certain groups has helped a lot to identify the repetitive elements origin of miRNAs, more so with plants [122–125]. The biogenesis of miRNAs from transposable elements was first proposed by Smallheiser and Torvik in 2005 [123], but this did not get due attention until it was also observed by many other authors [124]. Since then multiple hypotheses have been proposed for the evolution of miRNAs from transposable elements [124]. Approximately half of the human genome and ~80% of several plant genomes are composed of transposable elements, making the origin of miRNAs from such elements more likely. Although many models for the origin of miRNAs from repetitive elements have been proposed, the one proposed by Smalheiser and Torvik remains the most highlighted one [126]. Plant miRNAs have been reported to be derived from different families of transposable elements. miRNAs like TamiR1123 was shown to be derived from MITE elements in wheat [70], which regulates the expression of a vernalization gene by influencing its promoter element. Similarly, many miRNAs in O. sativa and A. thaliana were also reported to be derived from different transposable elements [122]. The most common transposable elements have been associated with many conserved miRNAs include MITE (Miniature Inverted Transposable Elements) [70,122], LINE elements [123] and SINEs [125]. In the present study also, a close association between miRNAs and repetitive elements was observed. Thus, for identification of such transposable element-derived miRNAs, overlap between miRNAs and identified complex repetitive elements was assessed for the entire genome of both species. In S. tuberosum, 224 miRNA sequences were found across its genome. A total of 30 pre-miRNA sequences were found originating from multiple loci in the genome of S. tuberosum (S8 Table), displaying repetitiveness and suggesting a repetitive origin associated with them. All the multiple loci of these miRNAs were studied for the presence of repetitive elements and footprints in the 2kb upstream and downstream regions. Most of these multiple loci were observed to be overlapping with different repetitive elements. In S. lycopersicum, 77 pre-miRNA were found and all of them were found originating from single locus. The identified co-ordinates of pre-miRNA sequences were used to extract the 2kb upstream and downstream sequences in both species. For this range of 2kb upstream and downstream regions around the pre-miRNA sequences (~4kb), overlapping repeats were identified in both species. Considering all the multiple loci of miRNAs, 242 loci of miRNAs in S. tuberosum were identified to be overlapping with repetitive elements. Similarly, in S. lycopersicum 77 loci of miRNAs were identified as overlapping with repetitive elements. It was also identified that LTR/Gypsy was the most prevalent repeat family in S. tuberosum miRNAs while in S. lycopersicum DNA transposons were more prevalent (Fig 7). It was further found that same members of the multi-loci miRNAs were overlapping with different repeat families. This analysis led support to the previous reports that transposable elements might serve as precursors to enrich the miRNA repertoire in many plant species [122–126]. The probability of enrichment of miRNAs around repetitive elements was elucidated using binomial test. Significant p-values were obtained for S. tuberosum (4.136e-10) and S. lycopersicum (1.819e-12), suggesting that miRNAs were enriched around repetitive elements.
Fig 7. Repeat families most prevalent in miRNAs in S. tuberosum and S. lycopersicum.
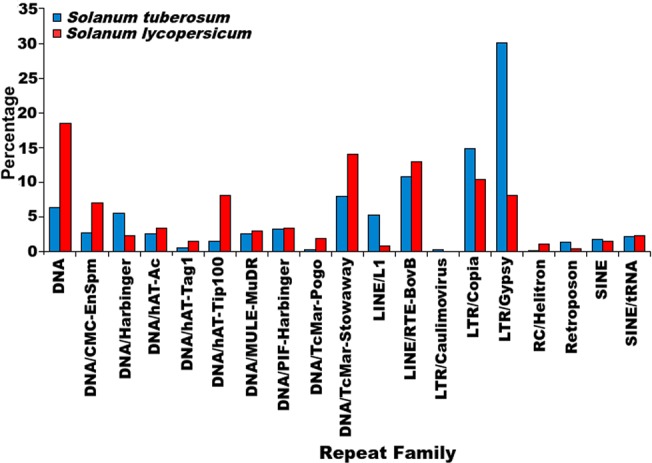
DNA transposons, LTR elements and LINE elements were observed as the most common in the miRNAs of both species.
During evolutionary course of an organism, repetitive elements may become unrecognizable as repeats due to different mutational events, sometimes leaving behind their footprints [70,71,127]. The above mentioned findings indicate that miRNA sequences might have taken birth from repetitive elements. Therefore, there could be a fare possibility that several miRNAs could have been contributed by complex repeats which gradually eroded with time and became unrecognizable into the genome. An attempt to discover such phenomenon through hunt for some sequential signatures in the flanking regions could support such view to some extent. The 2kb upstream and downstream regions including pre-miRNA sequences in both species were extracted from the genome. Orthologous pre-miRNAs were identified and local alignment between orthologous pre-miRNA pairs was performed. Common motifs in the orthologous sequences present in the same orientation and with same arrangement was found for some miRNAs, suggesting a repetitive origin for them despite of no clear presence of any full length or substantially long repetitive element around it. From the alignment, it was identified that in 14 orthologous miRNA pairs the selected motifs were present within repeat overlapping regions in both species. While in seven orthologous pairs, the motifs were found within the repeat overlapping region in S. tuberosum only, and in two orthologous miRNA pairs the motifs were found within the repeat overlapping region in S. lycopersicum only. Thus, these seven orthologous miRNAs in S. lycopersicum and two orthologous miRNAs in S. tuberosum might be have been generated through some transposable elements (Fig 8). In this study, it was also identified that the motifs which were found in the resulting orthologous genes, were originating from complex repetitive elements including LTR elements Copia and Gypsy, SINE/tRNA, DNA transposons TcMar-Stowaway, RTE-BovB, hAT-AC and CMC/EnSpm. However, the contribution of DNA transposons was observed to be more than retrotransposons.
Fig 8. Evolution of miRNAs from repetitive elements.
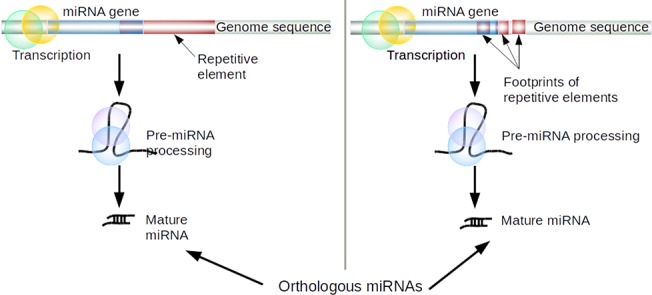
To search for candidate miRNAs which evolved from repetitive elements, footprints of repetitive elements around miRNA genes were identified.
Transcriptional activity of repetitive elements
Repetitive elements are generally under high constraints and characterized by high DNA methylation making them silent components of the genome. Although transcriptional activity of repetitive elements has been observed under stress conditions, pathogen attack and tissue culture conditions [128,129]. Also a low level of activity for different repetitive elements has been reported in normal conditions which is one of the reasons for their amplifications in a genome. Many plant species, specially flowering plants, have been shown to possess active repetitive elements belonging to both classes of transposable elements [130,131]. Active nature of repetitive elements has been associated with the generation of small non-coding RNA (siRNAs) which through post transcriptional gene silencing mechanisms (PTGS) create a feed-back loop silencing the repetitive elements themselves [132]. Other than providing control to repetitive elements, transcriptional activity of repetitive elements may also provide tissue specific expression of certain genes [23]. These elements upon transcription can also alter the expression of certain genes by RNA interference or through antisense RNA as well as through different epigenetic modifications. Therefore, identification of active repetitive elements in these two genomes would help in defining the functional boundaries generated by these elements with regard to their host genes' expression patterns. Abundance of transcript sequences and repetitive elements was calculated using digital expression data from two different platforms. Using sequence read count based RPKM abundance measure, it was found that RC/Helitron was transcriptionally most active repeat super-family in S. tuberosum (Fig 9). Helitrons were initially discovered in A. thaliana, C. elegans and O. sativa using different in-silico methods [133,134] and since then, they have been identified in numerous eukaryotes. Helitrons transpose by rolling circle transposition rather than by traditional “cut and paste” mechanism as is followed by other DNA transposons [134,135]. Although, helitrons make only a small portion of genomes of eukaryotes, they have been known to contribute significantly to the evolution of genes by capturing exons as has been demonstrated in maize [135]. The other transcriptionally active repeat super-families in S. tuberosum on the basis of RPKM abundances include DNA transposon Harbinger and TcMar-Stowaway (Fig 9). TcMar-Stowaway was also transcriptionally most active repeat super-families identified using microarray data. DNA transposon Harbinger was the first super-family of DNA transposons to be identified in A. thaliana using in-silico analysis which was identified as the most transcriptionally active repetitive element on the basis of RPKM and microarray expression data (S1 Fig) [136]. A few of Harbinger elements have been reported to be active members of their respective genomes [137]. DNA transposons TcMar-Stowaway were first discovered in S. bicolor as the elements inserted within Tourist elements [89]. Stowaway elements can form a hairpin shaped structure [89] and have shown to be able to generate miRNAs [138]. DNA transposon TcMar-Pogo was another repeat super-family which was identified as highly transcriptionally active in S. tuberosum on the basis of microarray data (S1 Fig). Pogo super-family of repeats was first identified in Drosophila and since then has been identified in many other species [139,140]. Pogo elements have been associated with exaptation of the CENP-B gene in mammals [141] and of some MITE elements in A. thaliana [142]. SINE elements activity was also evident through S. tuberosum microarray data. However, in S. lycopersicum, a repeat super-family very similar to LTR/ERV1 repeat super-family was found as the transcriptionally most active repeat family in the RPKM based expression estimates (Fig 9). The other transcriptionally active repeat super-families according to NGS expression measures observed in S. lycopersicum belonged to SINEs and LINE element RTE-BovB, which was also observed to be transcriptionally active in microarray data (Fig 9). Other highly transcriptionally active repeat super-families in the NGS expression measures included DNA transposons CMC-EnSpm. Information regarding the annotation of repeat super-families and the method of annotation is provided in S9 Table.
Fig 9. Transcriptionally most active repeat families on the basis of average RPKM expression.
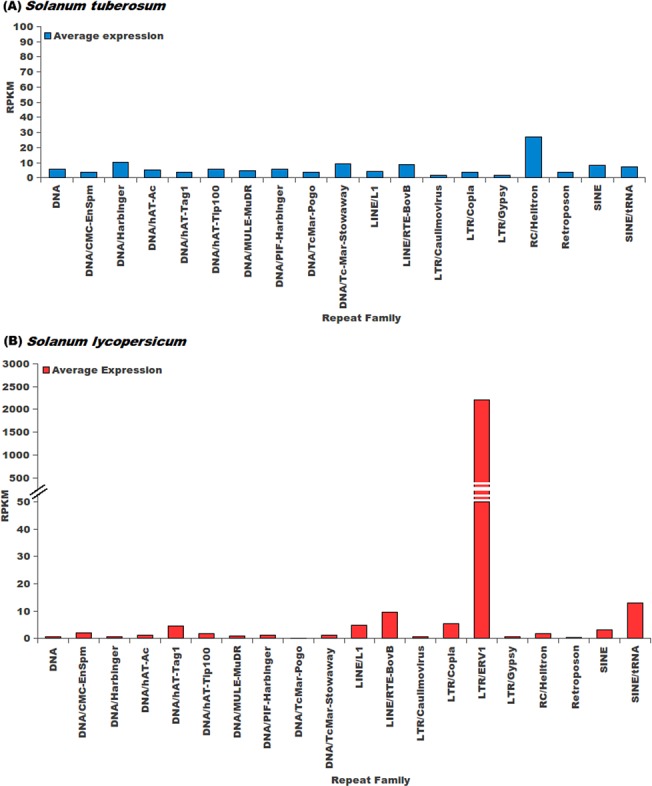
RC/Helitron and LTR/ERV1 were the transcriptionally most active repeat super-families in S. tuberosum and S. lycopersicum, respectively.
Further, expression of repetitive elements was also compared with the expression of housekeeping genes to obtain the view about their relative abundance. It was observed that though several repetitive elements were active in the system and expressing themselves, their relative abundance with respect to the housekeeping genes was found much lower, with exception of LTR/ERV1 (Fig 10). Being an exonic part of transcribing genes makes such repeats to be detected easily as an expressing element. Abundance of repetitive element transcripts found within exons and introns was also calculated. This analysis showed that the repetitive elements found within exonic regions were having higher abundance in both the species (S2 Fig).
Fig 10. Comparison of average RPKM expression of repeat families and housekeeping genes.
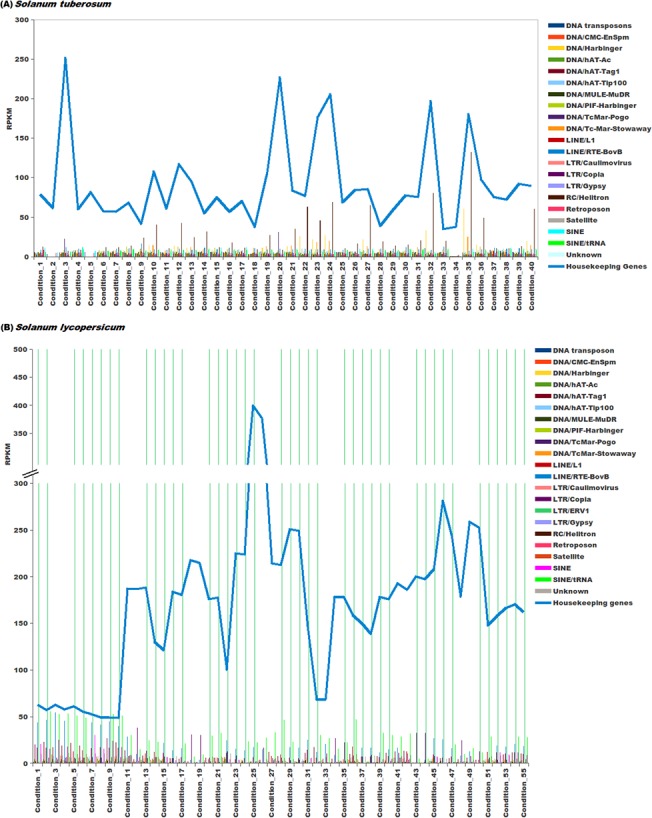
The details about experimental conditions is given in S11 Table.
An interesting finding of this study has been observation for two novel repeat super-families, LTR/ERV1 and LINE/RTE-BovB, reported first time for any plant species, and therefore, deserve special mention. RTE-BovB was first discovered in reptiles and it was shown to be horizontally transmitted from reptiles to ruminants and marsupials [143,144]. Super-families very similar to RTE-BovB were identified in S. tuberosum and S. lycopersicum, although RTE-BovB has not been reported in plants so far. Interestingly, ERV1 was also not reported in any plant species, previously. LTR/ERV1 are the endogenous retroviral elements and their active nature has been observed previously in mouse [77,145] and they have also been found enriched in human linc RNAs [146]. The members for this repeat family were identified here primarily on the basis of the identification of conserved protein domains, sparse sequence similarity and conservation of certain signature spots. Most of the LINE/RTE-BovB elements exhibited >90% length coverage with the consensus given in Repbase. The information regarding the species with known LTR/ERV1 and LINE/RTE-BovB families is provided in S10 Table. Further, multiple sequence alignment (MSA) of the consensus sequences of identified LINE/RTE-BovB elements and LTR/ERV1 elements was performed with known LINE/RTE-BovB and LTR/ERV1 elements for various species. Phylogenetic trees were drawn using Neighbor Joining method with a bootstrap value of 1000. It was observed that in both the species, the identified families of LINE/RTE-BovB emerged as an outgroup compared to the rest of species, an expected result (S3 Fig LINE/RTE-BovB in S. tuberosum and S. lycopersicum and S4 Fig for LTR/ERV1 in S. lycopersicum). In the MSA, the central regions in the alignments of LINE/RTE-BovB and LTR/ERV1 sequences were observed as characteristically the most conserved ones. These regions are known to harbor the important genes specially the ORFs encoding endonuclease and reverse transcriptase genes in LINE/RTE-BovB elements, while gag, pol and env for LTR/ERV1 elements. It would be difficult to annotate such elements just based on traditional sequence similarity search against Repbase alone, as the overall similarity varied a lot (46% to 85% for LINE/RTE-BovB and 6% to 62% for LTR/ERV1 when considering indels while when considering only substitutions, similarity ranged from ~50% to 92% for LINE/RTE-BovB and ~33% to 90% for LTR/ERV1). For the LTR/ERV1 family identified in this study, the similarity was observed within range for known species (~21% when considering indels, and 71.97% when only substitutions were considered). The LINE/RTE-BovB showed similarity ranges upto from 40–98% (~82–98% similarity when substitution was considered only) for S. tuberosum while 71–95% (71–94% approximately, when substitution was considered only) similarity range for S. lycopersicum when compared with the consensus. However, as apparent from the MSA and structural domains study, certain spots and regions of this family exhibited high conservation across all families in a very characteristic manner (S5 Fig for LINE/RTE-BovB elements in S. tuberosum and S. lycopersicum and S6 Fig for LTR/ERV1 in S. lycopersicum).
A large number of expressing repetitive elements have been reported to be involved in small RNAs/siRNA biogenesis [147,148]. Several of such sRNAs regulate the genes in either cis or trans manner. While some sRNAs are involved in post transcriptional gene silencing [148], a good number of such small RNAs are involved in de novo DNA methylation in plant genome [147,149–151]. Measuring the abundance of such small RNAs could also mirror the expression of repetitive elements which could have regulatory roles in the system. The small RNA sequencing reads were mapped to the repetitive elements as mentioned in the Methods section. Out of 35,992,757 unique small RNA reads in S. tuberosum and 9,620,265 unique reads in S. lycopersicum, ~0.23% (85,013) and ~33.41% (3,214,301) unique reads mapped to different repetitive elements, respectively. It was also found that most of the sRNA reads mapped to LTR/Gypsy elements in both species (Fig 11). To further verify the nature of these small RNAs, length distribution plot of the small RNA reads was made and it was identified that a high percentage of sRNA reads in S. tuberosum and S. lycopersicum were around 24bp, a length most prevalent with small regulatory RNAs like endogenous siRNAs and miRNAs (Fig 12). The overall comparison between the coverage of repeat family, the average abundance of repeat families and the percentage of sRNA reads mapped has been illustrated in Fig 13. It appears that expression of a repeat family is not correlated to its genomic prevalence.
Fig 11. Percentage of sRNA reads mapping to different repeat families.
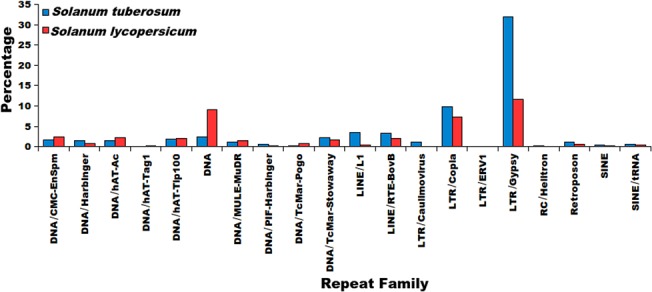
It was observed that most of the sRNAs were originating from LTR elements Gypsy and Copia in both species.
Fig 12. Length distribution plot of the sRNA reads mapping to repetitive elements.
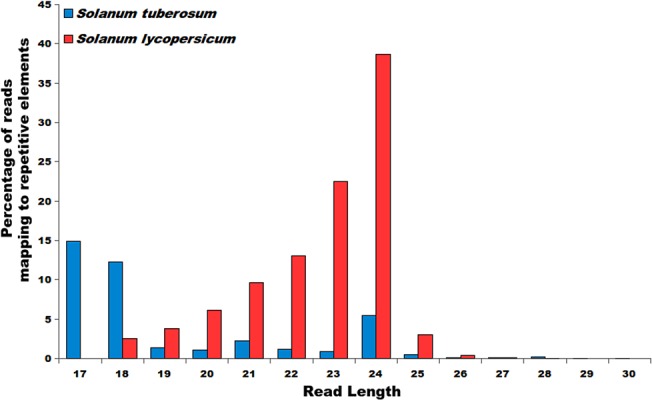
sRNA reads of length 24 bp were observed to be most enriched in S. lycopersicum, while in S. tuberosum, 17 bp long sRNAs were more prevalent.
Fig 13. Comparative plots showing coverage of repeat families, average expression of repeat families and percentage of small RNA reads mapping to repeat families in S. tuberosum and S. lycopersicum.
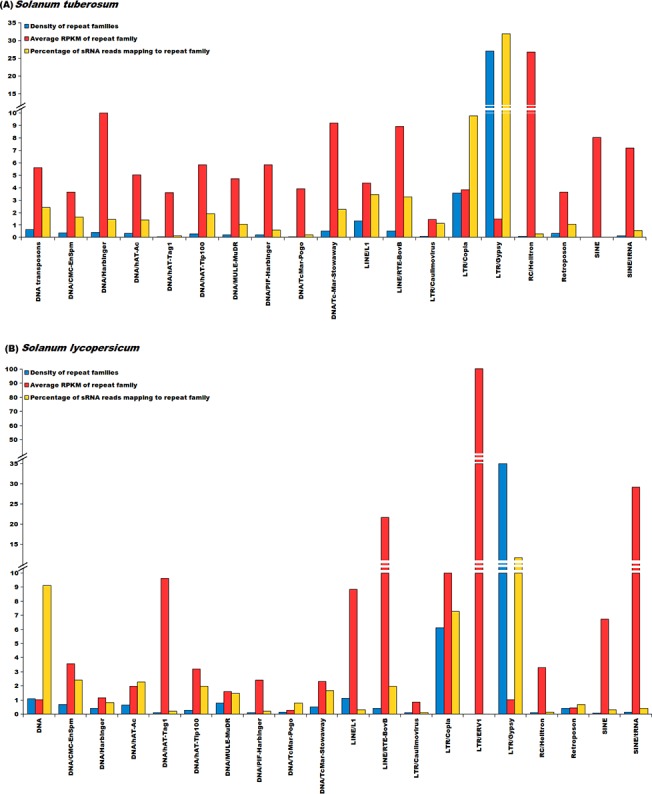
The plot shows that density of repetitive elements is not determining the abundance of repeat super-families and the generation of sRNAs from repeat super-families.
The sRNAs were found closely associated with repetitive elements of these species, concordant with some recent reports [152–154] that there is a big stake of repetitive elements in sRNA biogenesis, which in turn are now considered core members of post transcriptional as well as RdDM based transcriptional gene regulatory processes. It opens a door for further studies with repetitive elements and their impact over Solanum gene regulatory system.
Supporting Information
(TIFF)
(TIFF)
The LINE/RTE-BovB super-families identified in S. tuberosum are mentioned with the prefix “Stu”, while those identified in S. lycopersicum are mentioned with the prefix “Sly”.
(TIFF)
The consensus sequence of LTR/ERV1 identified in this study was matched with known consensus sequences of LTR/ERV1 and phylogenetic tree was created using Neighbor joining method with a bootstrap value of 1000. The LTR/ERV1 family identified in this study was named as “rnd-6_family-7426-LTR-ERV1”.
(TIFF)
The LINE/RTE-BovB super-families identified in S. tuberosum are mentioned with the prefix “Stu”, while those identified in S. lycopersicum are mentioned with the prefix “Sly”.
(PDF)
The consensus sequence of LTR/ERV1 identified in this study was matched with known consensus sequences of LTR/ERV1 and all the sequences matching with LTR/ERV1 were used for MSA.
(PDF)
(DOC)
Characterization of uncharacterized elements in ncRNA, pseudo-genes and SINE elements identified by matching A-box motif, B-box motif, 5' and 3' conserved motifs of B. oleraceae SINE elements.
(XLS)
(XLS)
Coverage of different repeat super-families was calculated as the percentage of nucleotides represented by repetitive elements out of total nucleotides for the given chromosome.
(DOC)
The best matches in BLAST result for every protein were considered as orthologs.
(XLS)
(XLS)
(XLS)
(XLS)
(XLS)
The list of species was prepared using RepBase.
(XLS)
(XLS)
Acknowledgments
MM and IG are thankful to Council of Scientific and Industrial Research for their Senior Research Fellowship. We are thankful to open source communities like Linux and R, and freely available experimental data and publications which helped us shape this work. We would like to dedicate this work in the loving memories of one of the biggest authorities in the area of transposable elements research, Dr Jerzy Jurka, and our beloved colleague, Dr RD Singh, who always helped us in becoming better in every sense. Institute of Himalayan Bioresource Technology communication number for this manuscript is 3876. All the metadata has been made freely available at http://scbb.ihbt.res.in/SCBB_dept/solanum_metadata.php and https://github.com/mrigayamehrajha/Solanum-Repeats-Metadata.
Data Availability
Data are freely available from http://scbb.ihbt.res.in/SCBB_dept/solanum_metadata.php and https://github.com/mrigayamehrajha/Solanum-Repeats-Metadata.
Funding Statement
This study was funded by SERB (Science and Engineering Research Board) young scientist fast track grant award (SR/FT/LS-97/2010) to Ravi Shankar and BSC-118. MM and IG are thankful to CSIR for their SRFship. IHBT communication number for this MS is 3876. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Potato Genome Sequencing Consortium, Xu X, Pan S, Cheng S, Zhang B, Mu D et al. Genome sequence and analysis of the tuber crop potato. Nature. 2011; 475(7355): 189–195. 10.1038/nature10158 [DOI] [PubMed] [Google Scholar]
- 2. Tomato Genome Consortium, Sato S, Tabata S, Hirakawa H, Asamizu E, Shirasawa K et al. The tomato genome sequence provides insights into fleshy fruit evolution. Nature. 2012; 485(7400): 635–641. 10.1038/nature11119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. McClintock B. The Origin and Behavior of Mutable Loci in Maize. Proc Natl Acad Sci USA. 1950; 36: 344–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Osborne BI, Corr CA, Prince JP, Hehl R, Tanksley SD, McCormick S et al. Ac transposition from a T-DNA can generate linked and unlinked clusters of insertions in the tomato genome. Genetics. 1991; 129(3): 833–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ganal MW, Lapitan NLV, Tanksley SD. A molecular and cytogenetic survey of major repeated DNA sequences in tomato (Lycopersicon esculentum). MGG. 1988; 213(2–3): 262–268. [Google Scholar]
- 6. Belzile F, Yoder JI. Pattern of somatic transposition in a high copy Ac tomato line. Plant J. 1992; 2(2): 173–179. [PubMed] [Google Scholar]
- 7. Stadler M, Stelzer T, Borisjuk N, Zanke C, Schilde-Rentschler L, Hemleben V. Distribution of novel and known repeated elements of Solanum and application for the identification of somatic hybrids among Solanum species. Theor Appl Genet. 1995; 91: 1271–1278. 10.1007/BF00220940 [DOI] [PubMed] [Google Scholar]
- 8. Oosumi T, Garlick B, Belknap WR. Identification and characterization of putative transposable DNA elements in solanaceous plants and Caenorhabditis elegans . Proc. Natl. Acad. Sci. USA. 1995; 92: 8886–8890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zanke C, Hemleben V. A new Solanum satellite DNA containing species-specific sequences which can be used for identification of genome parts in somatic hybrids of potato. Plant Sci. 1997; 126(2): 185–191. [Google Scholar]
- 10. Provan J, Powell W, Waugh R. Microsatellite analysis of relationships within cultivated potato (Solanum tuberosum). Theor Appl Genet. 1996; 92(8): 1078–1084. 10.1007/BF00224052 [DOI] [PubMed] [Google Scholar]
- 11. McGregor CE, Greyling MM, Warnich L. The use of Simple Sequence Repeats (SSRs) to identify commercially important potato (Solanum tuberosum L.) cultivars in South Africa. S. Afr. J. Plant Soil. 2000; 17(4). [Google Scholar]
- 12. Tam SM, Mhiri C, Vogelaar A, Kerkveld M, Pearce SR, Grandbastien MA. Comparative analyses of genetic diversities within tomato and pepper collections detected by retrotransposon-based SSAP, AFLP and SSR. Theor Appl Genet. 2005; 110(5):819–831. Epub 2005 Feb 8. [DOI] [PubMed] [Google Scholar]
- 13. Stupar RM, Song J, Tek AL, Cheng Z, Dong F, Jiang J. Highly condensed potato pericentromeric heterochromatin contains rDNA-related tandem repeats. Genetics. (2002) 162(3): 1435–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tek AL, Jiang J. The centromeric regions of potato chromosomes contain megabase-sized tandem arrays of telomere-similar sequence. Chromosoma. 2004; 113(2): 77–83. Epub 2004 Jul 16. [DOI] [PubMed] [Google Scholar]
- 15. Jo SH, Koo DH, Kim JF, Hur CG, Lee S, Yang TJ, et al. Evolution of ribosomal DNA-derived satellite repeat in tomato genome. BMC Plant Biol. 2009; 9: 42 10.1186/1471-2229-9-42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kwon YS, Parkland SG, Yi SI. Assessment of Genetic Variation among Commercial Tomato (Solanum Iycopersicum L) Varieties Using SSR Mariters and Morphological Characteristics. GENES and GENOMICS 2009; 3 1(I):1–10. [Google Scholar]
- 17. Torres GA, Gong Z, Iovene M, Hirsch CD, Buell CR, Bryan GJ, et al. Organization and evolution of subtelomeric satellite repeats in the potato genome. G3 (Bethesda). 2011; 1(2): 85–92. 10.1534/g3.111.000125 Epub 2011 Jul 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Adeniji OT, Kusolwa P, Reuben SOWM, Deo P. Molecular diversity among seven Solanum (eggplant and relatives) species assessed by simple sequence repeats (SSRs) markers. African Journal of Biotechnology. 2012; 11(90): 15643–15653. [Google Scholar]
- 19. Zhu W, Ouyang S, Iovene M, O'Brien K, Vuong H, Jiang J et al. Analysis of 90 Mb of the potato genome reveals conservation of gene structures and order with tomato but divergence in repetitive sequence composition. BMC Genomics. 2008; 9: 286 10.1186/1471-2164-9-286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chang SB, Yang TJ, Datema E, van Vugt J, Vosman B, Kuipers A, et al. (2008) FISH mapping and molecular organization of the major repetitive sequences of tomato. Chromosome Res. 16(7): 919–33. 10.1007/s10577-008-1249-z Epub 2008 Aug 13. [DOI] [PubMed] [Google Scholar]
- 21. Jiang N, Gao D, Xiao H, van der Knaap E. Genome organization of the tomato sun locus and characterization of the unusual retrotransposon Rider. Plant J. 2009; 60(1): 181–193. 10.1111/j.1365-313X.2009.03946.x [DOI] [PubMed] [Google Scholar]
- 22. Kuang H, Padmanabhan C, Li F, Kamei A, Bhaskar PB, Ouyang S, et al. Identification of miniature inverted-repeat transposable elements (MITEs) and biogenesis of their siRNAs in the Solanaceae: new functional implications for MITEs. Genome Res. 2009; 19(1): 42–56. 10.1101/gr.078196.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Momose M, Abe Y, Ozeki Y. Miniature Inverted-Repeat Transposable Elements of Stowaway Are Active in Potato. Genetics. 2010; 186: 59–66. 10.1534/genetics.110.117606 Epub 2010 Jul 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wenke T, Döbel T, Sörensen TR, Junghans H, Weisshaar B, Schmidt T. Targeted identification of short interspersed nuclear element families shows their widespread existence andextreme heterogeneity in plant genomes. Plant Cell. 2011; 23(9):3117–3128. 10.1105/tpc.111.088682 Epub 2011 Sep 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ferguson AA, Jiang N. Mutator-like elements with multiple long terminal inverted repeats in plants. Comp Funct Genomics. 2012; 2012: 695827 10.1155/2012/695827. Epub 2012 Mar 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yadav CB, Singh HN . In-Silico Identification of LTR type Retrotransposons and Their Transcriptional Activities in Solanum Tuberosum. IJSCE. 2013; 3(1). [Google Scholar]
- 27. Henikoff S, Ahmad K, Malik HS. The centromere paradox: stable inheritance with rapidly evolving DNA. Science. 2001; 293: 1098–1102. [DOI] [PubMed] [Google Scholar]
- 28. Zhang H, Koblížková A, Wang K, Gong Z, Oliveira L, Torres GA, et al. Boom-Bust Turnovers of Megabase-Sized Centromeric DNA in Solanum Species: Rapid Evolution of DNA Sequences Associated with Centromeres. Plant Cell. 2014; 26(4):1436–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Melters DP, Bradnam KR, Young HA, Telis N, May MR, Ruby JG, et al. Comparative analysis of tandem repeats from hundreds of species reveals unique insights into centromere evolution. Genome Biol. 2013; 14(1):R10 10.1186/gb-2013-14-1-r10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gong Z, Wu Y, Koblízková A, Torres GA, Wang K, Iovene M, et al. Repeatless and repeat-based centromeres in potato: implications for centromere evolution. Plant Cell. 2012; 24(9):3559–3574. 10.1105/tpc.112.100511 Epub 2012 Sep 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tang X, Datema E, Guzman MO, de Boer JM, van Eck HJ, Bachem CW et al. Chromosomal organizations of major repeat families on potato (Solanum tuberosum) and further exploring in its sequenced genome. Mol Genet Genomics. 2014; 289(6):1307–1319. 10.1007/s00438-014-0891-8 Epub 2014 Aug 9. [DOI] [PubMed] [Google Scholar]
- 32. Melotto-Passarin DM, Berger IJ, Dressano K, De Martin VDF, Oliveira GCX, Bock R, et al. Phylogenetic relationships in Solanaceae and related species based on cpDNA sequence from plastid trn E-trn T region. Crop Breeding and Applied Biotechnology. 2008; 8: 85–95. [Google Scholar]
- 33. Borisjuk N, Borisjuk L, Petjuch G, Hemleben V. Comparison of nuclear ribosomal RNA genes among Solanum species and other Solanaceae . Genome. 1994; 37: 271–279. [DOI] [PubMed] [Google Scholar]
- 34. Särkinen T, Bohs L, Olmstead RG, Knapp S. A phylogenetic framework for evolutionary study of the nightshades (Solanaceae): a dated 1000-tip tree. BMC Evol Biol. 2013; 13:214 10.1186/1471-2148-13-214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mueller LA, Mills AA, Skwarecki B, Buels RM, Menda N, Tanksley SD. The SGN comparative map viewer. Bioinformatics. 2008; 24(3): 422–423. 10.1093/bioinformatics/btm597 Epub 2008 Jan 17. [DOI] [PubMed] [Google Scholar]
- 36. Bombarely A, Menda N, Tecle IY, Buels RM, Strickler S, Fischer-York T, et al. The Sol Genomics Network (solgenomics.net): growing tomatoes using Perl. Nucleic Acids Res. 2011; 39(Database issue): D1149–1155. 10.1093/nar/gkq866 Epub 2010 Oct 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mueller LA, Solow TH, Taylor N, Skwarecki B, Buels R, Binns J, et al. The SOL Genomics Network: a comparative resource for Solanaceae biology and beyond. Plant Physiol. 2005; 138(3): 1310–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sharma SK, Bolser D, de Boer J, Sønderkær M, Amoros W, Carboni MF, et al. Construction of Reference Chromosome-Scale Pseudomolecules for Potato: Integrating the Potato Genome with Genetic and Physical Maps. G3 (Bethesda). 2013; 3: 2031–2047. 10.1534/g3.113.007153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Soderlund C, Nelson W, Shoemaker A, Paterson A. SyMAP: A system for discovering and viewing syntenic regions of FPC maps. Genome Res. 2006; 16: 1159–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26(6): 841–842. 10.1093/bioinformatics/btq033 Epub 2010 Jan 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kozomara A, Griffiths-Jones S. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011; 39(Database issue): D152–D157. 10.1093/nar/gkq1027 Epub 2010 Oct 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Griffiths-Jones S, Bateman A, Marshall M, Khanna A, Eddy SR. Rfam: an RNA family database. Nucleic Acids Res. 2003; 31(1): 439–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J. Mol. Biol. 1990; 215: 403–410. [DOI] [PubMed] [Google Scholar]
- 44. Bao Z, Eddy SR. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res. 2002; 12(8): 1269–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Price AL, Jones NC, Pevzner PA. De novo identification of repeat families in large genomes. Bioinformatics. 2005; 21 Suppl 1: i351–i358. [DOI] [PubMed] [Google Scholar]
- 46. Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999; 27(2): 573–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994; 22(22):4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nekrutenko A, Li WH. Transposable elements are found in a large number of human protein-coding genes. Trends Genet. 2001; 17(11): 619–621. [DOI] [PubMed] [Google Scholar]
- 49. Hoen D, Bureau T. Transposable Element Exaptation in Plants In: Grandbastien Marie-Angèle, Casacuberta Josep M., editors. Transposable Elements: Topics in Current Genetics. Springer-Verlag Berlin and Heidelberg GmbH & Co; 2012. pp. 219–251. [Google Scholar]
- 50. Shankar R, Grover D, Brahmachari SK, Mukerji M. Evolution and distribution of RNA polymerase II regulatory sites from RNA polymerase III dependant mobile Alu elements. BMC Evol Biol. 2004; 4: 37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Shankar R, Chaurasia A, Ghosh B, Chekmenev D, Cheremushkin E, Kel A, et al. Non-random genomic divergence in repetitive sequences of human and chimpanzee in genes of different functional categories. Mol Genet Genomics. 2007; 277(4): 441–455. Epub 2007 Mar 9. [DOI] [PubMed] [Google Scholar]
- 52. Feschotte C, Pritham EJ. DNA transposons and the evolution of eukaryotic genomes. Annu Rev Genet. 2007; 41:331–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Britten RJ. DNA sequence insertion and evolutionary variation in gene regulation. Proc. Natl. Acad. Sci. USA. 1996; 93: 9374–9377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Volff JN. Turning junk into gold: domestication of transposable elements and the creation of new genes in eukaryotes. BioEssays. 2006; 28: 913–922. [DOI] [PubMed] [Google Scholar]
- 55. Huang KC, Yang HC, Li KT, Liu LY, Charng YC. Ds transposon is biased towards providing splice donor sites for exonization in transgenic tobacco. Plant Mol Biol. 2012; 79(4–5): 509–519. 10.1007/s11103-012-9927-9 Epub 2012 May 27. [DOI] [PubMed] [Google Scholar]
- 56. Piskurek O, Austin CC, Okada N. Sauria SINEs: Novel short interspersed retroposable elements that are widespread in reptile genomes. J Mol Evol. 2006; 62(5): 630–644. Epub 2006 Apr 11. [DOI] [PubMed] [Google Scholar]
- 57. Kapusta A, Kronenberg Z, Lynch VJ, Zhuo X, Ramsay L, Bourque G, et al. Transposable Elements Are Major Contributors to the Origin, Diversification, and Regulation of Vertebrate Long non-coding RNAs. PLoS Genet. 2013; 9(4): e1003470 10.1371/journal.pgen.1003470 Epub 2013 Apr 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sela N, Mersch B, Hotz-Wagenblatt A, Ast G. Characteristics of Transposable Element Exonization within Human and Mouse. PLoS ONE. 2010; 5(6): e10907 10.1371/journal.pone.0010907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Piriyapongsa J, Rutledge MT, Patel S, Borodovsky M, Jordan IK (2007) Evaluating the protein coding potential of exonized transposable element sequences. Biol Direct. 2: 31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Jones DT. Protein Secondary Structure Prediction Based on Position-specific Scoring Matrices. J. Mol. Biol. 1999; 292: 195–202. [DOI] [PubMed] [Google Scholar]
- 61. Peng J, Xu J. RaptorX: exploiting structure information for protein alignment by statistical inference. Proteins. 2011; 79 Suppl 10: 161–171. 10.1002/prot.23175 Epub 2011 Oct 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Brenner SE. Target selection for structural genomics. Nat. Struct. Biol. 2000; 7(Suppl): 967–969. [DOI] [PubMed] [Google Scholar]
- 63. Vitkup D, Melamud E, Moult J, Sander C. Completeness in structural genomics. Nat. Struct. Biol. 2001; 8: 559–566. [DOI] [PubMed] [Google Scholar]
- 64. Bowie J, Luthy R, Eisenberg D. A method to identify protein sequences that fold into a known three-dimensional structure. Science. 1991; 253: 164–170. [DOI] [PubMed] [Google Scholar]
- 65. Laskowski RA, Swindells MB. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011; 51: 2778–2786. 10.1021/ci200227u Epub 2011 Oct 5. [DOI] [PubMed] [Google Scholar]
- 66. Jordan IK, Rogozin IB, Glazko GV, Koonin EV. Origin of a substantial fraction of human regulatory sequences from transposable elements. Trends Genet. 2003; 19(2): 68–72. [DOI] [PubMed] [Google Scholar]
- 67. Labrador M, Farre M,Utzet F, Fontdevila A. Interspecific Hybridization Increases Transposition Rates of Osvaldo. Mol. Biol. Evol. 1999; 16(7): 931–937. [DOI] [PubMed] [Google Scholar]
- 68. Jha A, Shankar R. MiRNAting control of DNA methylation. J Biosci. 2014; 39(3): 365–380. [DOI] [PubMed] [Google Scholar]
- 69. Rice P, Longden I, Bleasby A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000; 16(6): 276–277. [DOI] [PubMed] [Google Scholar]
- 70. Yu M, Carver BF, Yan L. TamiR1123 originated from a family of miniature inverted-repeat transposable elements (MITE) including one inserted in the Vrn-A1a promoter in wheat. Plant Sci. 2014; 215–216: 117–123. 10.1016/j.plantsci.2013.11.007 Epub 2013 Nov 16. [DOI] [PubMed] [Google Scholar]
- 71. Gogvadze E, Buzdin A. Retroelements and their impact on genome evolution and functioning. Cell Mol Life Sci. 2009; 66(23): 3727–3742. 10.1007/s00018-009-0107-2 Epub 2009 Aug 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Koenig D, Jiménez-Gómez JM, Kimura S, Fulop D, Chitwood DH, Headland LR, et al. Comparative transcriptomics reveals patterns of selection in domesticated and wild tomato. PNAS. 2013; 110(28): E2655–E2662. 10.1073/pnas.1309606110 Epub 2013 Jun 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Hamilton JP, Sim SC, Stoffel K, Deynze AV, Buell CR, Francis DM. Single nucleotide polymorphism discovery in cultivated tomato via sequencing by synthesis. Plant Genome. 2012; 5(1): 17–29. [Google Scholar]
- 74. Wang Y, Tao X, Tang XM, Xiao L, Sun JL, Yan XF, et al. Comparative transcriptome analysis of tomato (Solanum lycopersicum) in response to exogenous abscisic acid. BMC Genomics. 2013; 14: 841 10.1186/1471-2164-14-841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Hammond JP, Broadley MR, Bowen HC, Spracklen WP, Hayden RM, White PJ. Gene Expression Changes in Phosphorus Deficient Potato (Solanum tuberosum L.) Leaves and the Potential for Diagnostic Gene Expression Markers. PLoS ONE. 2011; 6(9): e24606 10.1371/journal.pone.0024606 Epub 2011 Sep 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Lopez-Gomollon S, Mohorianu I, Szittya G, Moulton V, Dalmay T. Diverse correlation patterns between microRNAs and their targets during tomato fruit development indicates different modes of microRNA actions. Planta. 2012; 236(6): 1875–1887. 10.1007/s00425-012-1734-7 Epub 2012 Aug 26. [DOI] [PubMed] [Google Scholar]
- 77. Reichmann J, Crichton JH, Madej MJ, Taggart M, Gautier P, Garcia-Perez JL, et al. Microarray analysis of LTR retrotransposon silencing identifies Hdac1 as a regulator of retrotransposon expression in mouse embryonic stem cells. PLoS Comput Biol. 2012; 8(4): e1002486 10.1371/journal.pcbi.1002486 Epub 2012 Apr 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Lakhotia N, Joshi G, Bhardwaj AR, Agarwal SK, Agarwal M, Jagannath A, et al. Identification and characterization of miRNAome in root, stem, leaf and tuber developmental stages of potato (Solanum tuberosum L.) by high-throughput sequencing. BMC Plant Biol. (2014) 14: 6 10.1186/1471-2229-14-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Mohorianu I, Schwach F, Jing R, Lopez-Gomollon S, Moxon S, Szittya G, et al. Profiling of short RNAs during fleshy fruit development reveals stage-specific sRNAome expression patterns. Plant J. 2011; 67(2): 232–246. 10.1111/j.1365-313X.2011.04586.x Epub 2011 May 9. [DOI] [PubMed] [Google Scholar]
- 80. Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009; 10: R25 10.1186/gb-2009-10-3-r25 Epub 2009 Mar 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Qin C, Yu C, Shen Y, Fang X, Chen L, Min J, et al. Whole-genome sequencing of cultivated and wild peppers provides insights into Capsicum domestication and specialization. PNAS. 2014; 111(14): 5135–5140. 10.1073/pnas.1400975111 Epub 2014 Mar 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Brenchley R, Spannag M, Pfeifer M, Barker GLA, D’Amore R, Allen AM, et al. Analysis of the bread wheat genome using whole-genome shotgun sequencing. Nature. 2012; 491: 705–709. 10.1038/nature11650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Wenke T, Döbel T, Sörensen TR, Junghans H, Weisshaar B, Schmidt T. Targeted identification of short interspersed nuclear element families shows their widespread existence and extreme heterogeneity in plant genomes. Plant Cell. 2011; 23(9): 3117–3128. 10.1105/tpc.111.088682 Epub 2011 Sep 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Zhang X, Wessler SR. BoS: a large and diverse family of short interspersed elements (SINEs) in Brassica oleracea . J Mol Evol. 2005; 60(5): 677–687. [DOI] [PubMed] [Google Scholar]
- 85. Böhne A, Brunet F, Galiana-Arnoux D, Schultheis C, Volff JN. Transposable elements as drivers of genomic and biological diversity in vertebrates. Chromosome Res. 2008; 16: 203–215. 10.1007/s10577-007-1202-6 [DOI] [PubMed] [Google Scholar]
- 86. van de Lagemaat LN, Landry JR, Mager DL, Medstrand P. Transposable elements in mammals promote regulatory variation and diversification of genes with specialized functions. Trends Genet. 2003; 19(10): 530–536. [DOI] [PubMed] [Google Scholar]
- 87. Muotri AR, Marchetto MC, Coufal NG, Gage FH. The necessary junk: new functions for transposable elements. Hum Mol Genet. 2007; 2: R159–R167. [DOI] [PubMed] [Google Scholar]
- 88. Makałowski W. Genomic scrap yard: how genomes utilize all that junk. Gene. 2000; 259(1–2): 61–67. [DOI] [PubMed] [Google Scholar]
- 89. Bureau TE, Wessler SR. Stowaway: A New Family of lnverted Repeat Elements Associated with the Genes of Both Monocotyledonous and Dicotyledonous Plants. Plant Cell. 1994; 6: 907–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Bureau TE, Wessler SR. Tourist: a large family of small inverted repeat elements frequently associated with maize genes. Plant Cell. 1992; 4: 1283–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Bureau TE, Wessler SR. Mobile inverted-repeat elements of the Tourist family are associated with the genes of many cereal grasses. Proc Natl Acad Sci USA. 1994; 91: 1411–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Sampath P, Lee SC, Lee J, Izzah NK, Choi BS, Jin M, et al. Characterization of a new high copy Stowaway family MITE, BRAMI-1 in Brassica genome. BMC Plant Biol. 2013; 13: 56 10.1186/1471-2229-13-56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Lockton S, Gaut BS. The contribution of transposable elements to expressed coding sequence in Arabidopsis thaliana . J Mol Evol. 2009; 68: 80–89. 10.1007/s00239-008-9190-5 Epub 2009 Jan 3. [DOI] [PubMed] [Google Scholar]
- 94. Feschotte C. The contribution of transposable elements to the evolution of regulatory networks. Nat Rev Genet. 2008; 9(5): 397–405. 10.1038/nrg2337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Jacob F. Evolution and Tinkering. Science. 1977; 196: 1163. [DOI] [PubMed] [Google Scholar]
- 96. Liu LY, Charng YC. Genome-wide survey of ds exonization to enrich transcriptomes and proteomes in plants. Evol Bioinform Online. 2012; 8: 575–587. 10.4137/EBO.S10324 Epub 2012 Oct 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Chien TY, Liu LD, Charng YC. Analysis of New Functional Profiles of Protein Isoforms Yielded by Ds exonization in Rice. Evol Bioinform Online. 2013; 9: 417–427. 10.4137/EBO.S12757 ECollection 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Mason JM, Frydrychova RC, Biessmann H. Drosophila telomeres: an exception providing new insights. BioEssays. 2007; 30: 25–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Beauregard A, Curcio MJ, Belfort M. The take and give between retrotransposable elements and their hosts. Annu Rev Genet. 2008; 42:587–617. 10.1146/annurev.genet.42.110807.091549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Schmitz J, Brosius J. Exonization of transposed elements: A challenge and opportunity for evolution. Biochimie. 2011; 93(11):1928–1934. 10.1016/j.biochi.2011.07.014 Epub 2011 Jul 26. [DOI] [PubMed] [Google Scholar]
- 101. Sorek R. The birth of new exons: Mechanisms and evolutionary consequences. RNA. 2007; 13: 1603–1608. Epub 2007 Aug 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Lev-Maor G, Sorek R, Shomron N, Ast G. The birth of an alternatively spliced exon: 3' splice-site selection in Alu exons. Science. 2003; 300: 1288–1291. [DOI] [PubMed] [Google Scholar]
- 103. Staton SE, Bakken BH, Blackman BK, Chapman MA, Kane NC, Tang S, et al. The sunflower (Helianthus annuus L.) genome reflects a recent history of biased accumulation of transposable elements. Plant J. 2012; 72(1): 142–153. 10.1111/j.1365-313X.2012.05072.x Epub 2012 Jul 30. [DOI] [PubMed] [Google Scholar]
- 104. Houck CM, Rinehart FP, Schmid CW. A ubiquitous family of repeated DNA sequences in the human genome. J Mol Biol. 1979; 132(3): 289–306. [DOI] [PubMed] [Google Scholar]
- 105. Sampath P, Murukarthick J, Izzah NK, Lee J, Choi HI, Shirasawa K, et al. Genome-wide comparative analysis of 20 miniature inverted-repeat transposable element families in Brassica rapa and B. oleracea . PLoS One. 2014; 9(4): e94499 10.1371/journal.pone.0094499 ECollection 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Nystedt B, Street NR, Wetterbom A, Zuccolo A, Lin YC, Scofield DG, et al. The Norway spruce genome sequence and conifer genome evolution. Nature. 2013; 497(7451): 579–584. 10.1038/nature12211 Epub 2013 May 22. [DOI] [PubMed] [Google Scholar]
- 107. Barghini E, Natali L, Cossu RM, Giordani T, Pindo M, Cattonaro F, et al. The peculiar landscape of repetitive sequences in the olive (Olea europaea L.) genome. Genome Biol Evol. 2014; 6(4): 776–791. 10.1093/gbe/evu058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Khaitovich P, Hellmann I, Enard W, Nowick K, Leinweber M, Franz H, et al. Parallel patterns of evolution in the genomes and transcriptomes of humans and chimpanzees. Science. 2005; 309(5742): 1850–1854. Epub 2005 Sep 1. [DOI] [PubMed] [Google Scholar]
- 109. Tomilin NV, Bozhkov VM. Human nuclear protein interacting with a conservative sequence motif of Alu-family DNA repeats. FEBS Lett. 1989; 251(1–2): 79–83. [DOI] [PubMed] [Google Scholar]
- 110. Pérez-Rodríguez P, Riaño-Pachón DM, Corrêa LGG, Rensing RA, Kersten B, Mueller-Roeber B. PlnTFDB: updated content and new features of the plant transcription factor database. Nucleic Acids Res. 2010; 38(Database issue): D822–D827. 10.1093/nar/gkp805 Epub 2009 Oct 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993; 75: 843–854. [DOI] [PubMed] [Google Scholar]
- 112. Bartel DP. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell. 2004; 116: 281–297. [DOI] [PubMed] [Google Scholar]
- 113. Winter J, Jung S, Keller S, Gregory RI, Diederichs S. Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat Cell Biol. 2009; 11(3): 228–234. 10.1038/ncb0309-228 [DOI] [PubMed] [Google Scholar]
- 114. Bonnet E, Wuyts J, Rouzé P, Van de Peer Y. Detection of 91 potential conserved plant microRNAs in Arabidopsis thaliana and Oryza sativa identifies important target genes. Proc Natl Acad Sci U S A. 2004; 101(31): 11511–11516. Epub 2004 Jul 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Reinhart BJ, Weinstein EG, Rhoades MW, Bartel B, Bartel DP. MicroRNAs in plants. Genes Dev. 2002; 16(13): 1616–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Bushati N, Cohen SM. miRNA functions. Annu. Rev. Cell Dev. Biol. 2007; 23: 175–205. [DOI] [PubMed] [Google Scholar]
- 117. Hu W, Wang T, Xu J, Li H. MicroRNA mediates DNA methylation of target genes. Biochem Biophys Res Commun. 2014; 444(4): 676–681. 10.1016/j.bbrc.2014.01.171 Epub 2014 Feb 4. [DOI] [PubMed] [Google Scholar]
- 118. Wu L, Zhou H, Zhang Q, Zhang J, Ni F, Liu C, et al. DNA methylation mediated by a microRNA pathway. Mol Cell. 2010; 38(3): 465–475. 10.1016/j.molcel.2010.03.008 Epub 2010 Apr 8. [DOI] [PubMed] [Google Scholar]
- 119. Jha A, Mehra M, Shankar R. The regulatory epicenter of miRNAs. J Biosci. 2011; 36(4): 621–638. [DOI] [PubMed] [Google Scholar]
- 120. Axtell MJ, Westholm JO, Lai EC. Vive la différence: biogenesis and evolution of microRNAs in plants and animals. Genome Biol. 2011; 12:221 10.1186/gb-2011-12-4-221 Epub 2011 Apr 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Jha A, Chauhan R, Mehra M, Singh HR, Shankar R. miR-BAG: bagging based identification of microRNA precursors. PLoS One. 2012; 7(9): e45782 10.1371/journal.pone.0045782 Epub 2012 Sep 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Piriyapongsa J, Jordan IK. Dual coding of siRNAs and miRNAs by plant transposable elements. RNA. 2008; 14(5): 814–821. 10.1261/rna.916708 Epub 2008 Mar 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Smalheiser NR, Torvik VI. Mammalian microRNAs derived from genomic repeats. Trends Genet. 2005; 21: 322–326. [DOI] [PubMed] [Google Scholar]
- 124. Roberts JT, Cardin SE, Borchert GM. Burgeoning evidence indicates that microRNAs were initially formed from transposable element sequences. Mob Genet Elements. 2014; 4: e29255 eCollection 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Smalheiser NR, Torvik VI. Alu elements within human mRNAs are probable microRNA targets. Trends Genet. 2006; 22: 532–536. [DOI] [PubMed] [Google Scholar]
- 126. Roberts JT, Cooper EA, Favreau CJ, Howell JS, Lane LG, Mills JE, et al. Continuing analysis of microRNA origins: Formation from transposable element insertions and noncoding RNA mutations. Mob Genet Elements. 2013; 3(6):e27755 Epub 2014 Jan 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Kidwell MG. The evolutionary history of the P family of transposable elements. J Hered. 1994; 85:339–346 [DOI] [PubMed] [Google Scholar]
- 128. Grandbastien MA. Activation of plant retrotransposons under stress conditions. Trends Plant Sci. 1998; 3: 181–187. [Google Scholar]
- 129. Takeda S, Sugimoto K, Otsuki H, Hirochika H. Transcriptional activation of the tobacco retrotransposon Tto1 by wounding and methyl jasmonate. Plant Mol. Biol. 1998; 36: 365–376. [DOI] [PubMed] [Google Scholar]
- 130. Jiang N, Bao Z, Zhang X, Hirochika H, Eddy SR, McCouch SR, et al. An active DNA transposon family in rice. Nature. 2003; 421(6919): 163–167. [DOI] [PubMed] [Google Scholar]
- 131. Rudenko GN, Walbot V. Expression and Post-Transcriptional Regulation of Maize Transposable Element MuDR and Its Derivatives. Plant Cell. 2001; 13: 553–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Okamoto H, Hirochika H. Silencing of transposable elements in plants. Trends Plant Sci. 2001; 6: 527–534. [DOI] [PubMed] [Google Scholar]
- 133. Kapitonov VV, Jurka J. Rolling-circle transposons in eukaryotes. PNAS. 2001; 98(15): 8714–8719. Epub 2001 Jul 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Kapitonov VV, Jurka J. Helitrons on a roll: eukaryotic rolling-circle transposons. Trends Genet. 2007; 23(10): 521–529. Epub 2007 Sep 11. [DOI] [PubMed] [Google Scholar]
- 135. Gupta S, Gallavotti A, Stryker GA, Schmidt RJ, Lal SK. A novel class of Helitron-related transposable elements in maize contain portions of multiple pseudo-genes. Plant Mol Biol. 2005; 57(1): 115–127. [DOI] [PubMed] [Google Scholar]
- 136. Kapitonov VV, Jurka J. Molecular paleontology of transposable elements from Arabidopsis thaliana . Genetica. 1999; 107(1–3):27–37. [PubMed] [Google Scholar]
- 137. Grzebelus D, Lasota S, Gambin T, Kucherov G, Gambin A. Diversity and structure of PIF/Harbinger-like elements in the genome of Medicago truncatula . BMC Genomics. 2007; 8: 409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Ou-Yang F, Luo QJ, Zhang Y, Richardson CR, Jiang Y, Rock CD. Transposable element-associated microRNA hairpins produce 21-nt sRNAs integrated into typical microRNA pathways in rice. Funct Integr Genomics. 2013; 13(2): 207–216. 10.1007/s10142-013-0313-8 Epub 2013 Feb 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Alzohairy AM, Gyulai G, Jansen RK, Bahieldin A. Transposable elements domesticated and neofunctionalized by eukaryotic genomes. Plasmid. 2013; 69: 1–15. 10.1016/j.plasmid.2012.08.001 Epub 2012 Aug 30. [DOI] [PubMed] [Google Scholar]
- 140. Guermonprez H, Loot C, Casacuberta JM. Different Strategies to Persist: The pogo-Like Lemi1 Transposon Produces Miniature Inverted- Repeat Transposable Elements or Typical Defective Elements in Different Plant Genomes. Genetics. 2008; 180: 83–92. 10.1534/genetics.108.089615 Epub 2008 Aug 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Casola C, Hucks D, Feschotte C. Convergent domestication of pogo-like transposases into centromere-binding proteins in fission yeast and mammals. Mol Biol Evol. 2008; 25(1): 29–41. Epub 2007 Oct 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Feschotte C, Mouchès C. Evidence that a family of miniature inverted-repeat transposable elements (MITEs) from the Arabidopsis thaliana genome has arisen from a pogo-like DNA transposon. Mol Biol Evol. 2000; 17(5): 730–737. [DOI] [PubMed] [Google Scholar]
- 143. Kordis D, Gubensek F. Unusual horizontal transfer of a long interspersed nuclear element between distant vertebrate classes. Proc. Natl. Acad. Sci. USA. 1998; 95: 10704–10709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Adelson DL, Raison JM, Edgar RC. Characterization and distribution of retrotransposons and simple sequence repeats in the bovine genome. PNAS. 2009; 106(31): 12855–12860. 10.1073/pnas.0901282106 Epub 2009 Jul 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Webster KE, O’Bryan MK, Fletcher S, Crewther PE, Aapola U, Craig J, et al. Meiotic and epigenetic defects in Dnmt3L-knockout mouse spermatogenesis. PNAS. 2005; 102(11): 4068–4073. Epub 2005 Mar 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Kelley D, Rinn J. Transposable elements reveal a stem cell-specific class of long non-coding RNAs. Genome Biol. 2012; 13: R107 10.1186/gb-2012-13-11-r107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Matzke MA, Mosher RA. RNA-directed DNA methylation: an epigenetic pathway of increasing complexity. Nat Rev Genet. 2014; 15(6):394–408. 10.1038/nrg3683 Epub 2014 May 8 [DOI] [PubMed] [Google Scholar]
- 148. Malone CD, Hannon GJ. Small RNAs as guardians of the genome. Cell. 2009; 136(4): 656–668. 10.1016/j.cell.2009.01.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Slotkin RK, Vaughn M, Borges F, Tanurdzić M, Becker JD, Feijó JA, et al. Epigenetic reprogramming and small RNA silencing of transposable elements in pollen. Cell. 2009; 136(3): 461–472. 10.1016/j.cell.2008.12.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Zeh DW, Zeh JA, Ishida Y. Transposable elements and an epigenetic basis for punctuated equilibria. BioEssays. 2009; 31: 715–726. 10.1002/bies.200900026 [DOI] [PubMed] [Google Scholar]
- 151. Elgin SC, Grewal SI. Heterochromatin: silence is golden. Curr Biol. 2003; 13(23): R895–R898. [DOI] [PubMed] [Google Scholar]
- 152. Menon DU, Coarfa C, Xiao W, Gunaratne PH, Meller VH. siRNAs from an X-linked satellite repeat promote X-chromosome recognition in Drosophila melanogaster . Proc Natl Acad Sci U S A. 2014; 111(46): 16460–16465. 10.1073/pnas.1410534111 Epub 2014 Nov 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Matzke M, Kanno T, Huettel B, Daxinger L, Matzke AJ. RNA-directed DNA methylation and Pol IVb in Arabidopsis . Cold Spring Harb Symp Quant Biol. 2006; 71:449–459. [DOI] [PubMed] [Google Scholar]
- 154. Vagin VV, Sigova A, Li C, Seitz H, Gvozdev V, Zamore PD. A distinct small RNA pathway silences selfish genetic elements in the germline. Science. 2006; 313(5785): 320–324. Epub 2006 Jun 29. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(TIFF)
(TIFF)
The LINE/RTE-BovB super-families identified in S. tuberosum are mentioned with the prefix “Stu”, while those identified in S. lycopersicum are mentioned with the prefix “Sly”.
(TIFF)
The consensus sequence of LTR/ERV1 identified in this study was matched with known consensus sequences of LTR/ERV1 and phylogenetic tree was created using Neighbor joining method with a bootstrap value of 1000. The LTR/ERV1 family identified in this study was named as “rnd-6_family-7426-LTR-ERV1”.
(TIFF)
The LINE/RTE-BovB super-families identified in S. tuberosum are mentioned with the prefix “Stu”, while those identified in S. lycopersicum are mentioned with the prefix “Sly”.
(PDF)
The consensus sequence of LTR/ERV1 identified in this study was matched with known consensus sequences of LTR/ERV1 and all the sequences matching with LTR/ERV1 were used for MSA.
(PDF)
(DOC)
Characterization of uncharacterized elements in ncRNA, pseudo-genes and SINE elements identified by matching A-box motif, B-box motif, 5' and 3' conserved motifs of B. oleraceae SINE elements.
(XLS)
(XLS)
Coverage of different repeat super-families was calculated as the percentage of nucleotides represented by repetitive elements out of total nucleotides for the given chromosome.
(DOC)
The best matches in BLAST result for every protein were considered as orthologs.
(XLS)
(XLS)
(XLS)
(XLS)
(XLS)
The list of species was prepared using RepBase.
(XLS)
(XLS)
Data Availability Statement
Data are freely available from http://scbb.ihbt.res.in/SCBB_dept/solanum_metadata.php and https://github.com/mrigayamehrajha/Solanum-Repeats-Metadata.