

Abstract
Colorectal cancer remains the most prevalent malignancy in humans. The impact of epigenetic alterations on the development of this complex disease is now being recognized. The dynamic and reversible nature of epigenetic modifications makes them a promising target in colorectal cancer chemoprevention and treatment. Curcumin (CUR), the major component in Curcuma longa, has been shown as a potent chemopreventive phytochemical that modulates various signaling pathways. Deleted in lung and esophageal cancer 1 (DLEC1) is a tumor suppressor gene with reduced transcriptional activity and promoter hypermethylation in various cancers, including colorectal cancer. In the present study, we aimed to investigate the inhibitory role of DLEC1 in anchorage-independent growth of the human colorectal adenocarcinoma HT29 cells and epigenetic regulation by CUR. Specifically, we found that CUR treatment inhibited colony formation of HT29 cells, whereas stable knockdown of DLEC1 using lentiviral short hairpin RNA vector increased cell proliferation and colony formation. Knockdown of DLEC1 in HT29 cells attenuated the ability of CUR to inhibit anchorage-independent growth. Methylation-specific polymerase chain reaction (MSP), bisulfite genomic sequencing, and methylated DNA immunoprecipitation revealed that CUR decreased CpG methylation of the DLEC1 promoter in HT29 cells after 5 days of treatment, corresponding to increased mRNA expression of DLEC1. Furthermore, CUR decreased the protein expression of DNA methyltransferases and subtypes of histone deacetylases (HDAC4, 5, 6, and 8). Taken together, our results suggest that the inhibitory effect of CUR on anchorage-independent growth of HT29 cells could, at least in part, involve the epigenetic demethylation and up-regulation of DLEC1.
Keywords: Colorectal cancer, curcumin, DLEC1, anchorage-independent growth, epigenetics
Introduction
Colorectal cancer is one of the leading causes of cancer-related morbidity and mortality worldwide. As one of the most well-studied malignancies, colorectal cancer is now considered a complex disease that results from the accumulation of genetic and epigenetic alterations [1]. Extensive studies of colorectal cancer have identified several significant genetic mutations implicated in proliferation, differentiation, adhesion, apoptosis, cell cycle, and DNA repair [2]. In recent years, emerging evidence has suggested that the aberrant epigenetic landscape (heritable alterations in gene expression without changes in DNA sequence) may add an additional layer of complexity to the initiation and progression of colorectal cancer. The reversible and dynamic nature of these epigenetic alterations has enabled their development as potential biomarkers for diagnostic, prognostic, and therapeutic targets in colorectal cancer [3]. Among epigenetic mechanisms, DNA methylation is perhaps the most extensively studied epigenetic alteration in colorectal cancer. Importantly, inactivation of tumor suppressor genes by promoter CpG island hypermethylation has been recognized as one of the hallmarks of cancer [4] and is frequently detected even in the early stages in colorectal cancer patients [5, 6]. Multiple genes, including MLH1, p16, RASSF1A, and APC, are frequently silenced in colorectal cancer by promoter hypermethylation [7]. Enhanced understanding of the aberrant methylation patterns in colorectal cancer has shed light on the development of agents that target enzymes responsible for reactivating epigenetically silenced genes. For example, several epigenetic therapeutics have been approved for cancer treatment by the U.S. Food and Drug Administration (FDA), such as DNA methyltransferase (DNMT) inhibitors and histone deacetylase (HDAC) inhibitors [8]. However, adverse effects after chronic exposure have hindered their use in chemoprevention [9]. By contrast, multiple lines of evidence have suggested that dietary and environmental factors may be important contributors to cancer development by dynamically modifying the epigenetic landscape [10]. Therefore, great effort has been applied to evaluate the capacity of chemopreventive nutritional phytochemicals to alter the profile of adverse epigenetic marks in cancer cells to attenuate tumor growth.
Curcumin (CUR) is the major active component in the golden spice Curcuma longa (also known as turmeric). Turmeric has been used as a common food spice for millennia. According to epidemiological reports, the consumption of Curcuma longa is associated with lower cancer incidence [11]. Accumulating evidence has indicated that CUR may be a potent chemopreventive agent by targeting various molecular signaling pathways involved in carcinogenesis [12]. Despite the high safety and tolerability of oral CUR as evidenced in phase I studies, CUR was also found to have low systemic bioavailability because of rapid metabolism [13]. However, it has been suggested that favorable effects of CUR can be achieved through accumulation of CUR and its metabolites in tissues by long-term exposure. Studies of CUR have focused on colorectal diseases (most notably colorectal cancer) because of the preferential distribution of orally administered CUR in the colon mucosa compared with that in other organs [14]. Garcea et al. reported that CUR concentration in human colorectal mucosa after oral consumption of up to 3600 mg may be sufficient to obtain pharmacological effects [15]. It has been suggested that epigenetic modifications, which are achievable at lower concentrations, may be involved in the mechanism of chemoprevention by CUR. For example, CUR has been reported to regulate the activity of histone acetyltransferase (HAT), HDAC, and, more recently, DNMT in different model systems [16]. Recent studies in our laboratory have demonstrated that CUR decreases the CpG methylation of Nrf2 and Neurog 1 in murine tramp C1 prostate cancer cells and human LnCap prostate cancer cells, respectively [17, 18]. However, few studies have demonstrated the effect of CUR in modulating the CpG hypermethylation of specific tumor suppressor genes related to colorectal cancer. We believe the development of CUR as an epigenetic agent warrants further studies to explore its diversity and efficacy in preventing colorectal cancer.
Deleted in lung and esophageal cancer 1 (DLEC1) was initially discovered in 1999 as a candidate tumor suppressor gene in lung, esophageal, and renal cancers [19]. DLEC1 is located at chromosome 3p22-p21.3, a region recognized as a hot spot likely to contain tumor suppressor genes with frequent genetic abnormalities during carcinogenesis, including colorectal cancer [20]. Tumor suppressor genes in this region such as RASSF1 and BLU have been found to be frequently silenced by promoter CpG methylation [21, 22]. Similarly, inactivation of DLEC1 by promoter CpG hypermethylation has been reported in a wide spectrum of cancers, such as lung [23], hepatocellular [24], ovarian [25], renal [26], nasopharyngeal [27], and breast cancers [28]. Additionally, these studies have also provided evidence that overexpression of DLEC1 significantly suppresses the clonogenicity of tumor cells. Recently, Ying et al. demonstrated for the first time that expression of DLEC1 was decreased and underwent promoter hypermethylation in various colorectal cancer cell lines and primary tumor samples but not in DKO (HCT116 DNMT1-/- DNMT3B-/-) cells, CCD-841 (normal colon epithelial cells), and paired normal tissues [29]. To the best of our knowledge, potential epigenetic interventions targeting DLEC1 using phytochemicals have not been evaluated. Hence, the present study was undertaken to investigate the involvement of DLEC1 in the chemopreventive effects of CUR in suppressing anchorage-independent growth of HT29 cells. Furthermore, the potential of CUR to restore DLEC1 expression in HT29 cells through epigenetic mechanisms was evaluated.
Materials and Methods
Materials
CUR, azadeoxycytidine (5AZA), trichostatin A (TSA), bacteriological agar, puromycin, ethidium bromide, and Basal Medium Eagle (BME) were purchased from Sigma-Aldrich (St. Louis, MO, USA). All the enzymes used in this study were obtained from New England Biolabs Inc. (Ipswich, MA, USA). The Cell Titer 96 Aqueous One Solution Cell Proliferation Assay Kit, the luciferase reporter vector pGL4.15, the pSV-β-Galactosidase control vector, the luciferase assay system, and the β-Galactosidase enzyme assay system were purchased from Promega (Madison, WI, USA).
Cell culture, cell viability assay, and lentiviral transduction
The human colorectal adenocarcinoma HT29 and SW48 cell line, human colorectal carcinoma HCT116 cell line, and human embryonic kidney HEK293 cell line were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA). HT29 cells, HCT116 cells, and HEK293 cells were routinely maintained in Dulbecco's modified Eagle medium (DMEM; Gibco, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS; Gibco). SW48 cells were cultured in RPMI-1640 medium (Gibco) with 10% FBS. All the cells were grown at 37°C in a humidified 5% CO2 atmosphere.
HT29 cells were seeded in 96-well plates at an initial density of 1,000 cells/well for 24 h. The cells were then treated with CUR (1-25 μM) for 5 days. The medium was changed every other day. On day 5, a MTS assay was performed using the Cell Titer 96 Aqueous One Solution Cell Proliferation Assay Kit as described previously [30].
Lentivirus mediated short hairpin RNAs were used to establish stable mock (scramble control, sh-Mock) and DLEC1 knockdown (sh-DLEC1) HT29 cells. The shRNA clone sets were obtained from Genecopoeia (Rockville, MD, USA), and lentiviral transduction was performed according to the manufacturer's manual. After selection in DMEM medium supplemented with 10% FBS and 2 μg/mL puromycin for 3 weeks, the sh-Mock and sh-DLEC1 cells were further used to evaluate the functional role of DLEC1. To examine the proliferation rate of sh-Mock and sh-DLEC1 HT29 cells, the cells were seeded in 60-mm tissue culture plates at an initial density of 10,000 cells. The cell number was counted and recorded after 24, 48, and 72 h of incubation using a TC20 automated cell counter (Bio-rad, Hercules, CA, USA).
DNA methylation analysis
HT29 cells were plated in 10-cm plates for 24 h and then treated with 0.1% DMSO (control), 2.5 μM 5AZA and 100 nM TSA, or CUR at 2.5 and 5 μM for 5 days. The medium was changed every other day. For the 5AZA and TSA combined treatment, 100 nM TSA was added 20 h before harvest. On day 5, the cells were harvested for further analyses. Genomic DNA was isolated from the treated cells using the QIAamp DNA Mini Kit (Qiagen, Valencia, CA, USA). Next, 750 ng of genomic DNA was subjected to bisulfite conversion using EZ DNA Methylation Gold Kits (Zymo Research Corp., Orange, CA, USA) following the manufacturer's instructions. To obtain products for sequencing, the converted DNA was amplified by PCR using Platinum PCR Taq DNA polymerase (Invitrogen, Carlsbad, CA, USA) using the forward and reverse primers: 5′- CGA AGA TAT AAA TGT TTA TAA TGA TT-3′ and 5′-CAA CTA CAA CCC CAA ATC CTA A-3′. The PCR products were cloned into a pCR4 TOPO vector using the TOPO TA Cloning Kit (Invitrogen), as previously described [17, 18, 31]. For each sample, at least 10 clones were randomly selected and sequenced (Genewiz, Piscataway, NJ, USA). The percentage of methylated CpG was calculated as the number of methylated CpG sites over the total number of CpG sites examined.
Methylation-specific PCR (MSP) was performed on bisulfite-converted genomic DNA. The primer sequences for the methylated reactions were 5′-GAT TAT AGC GAT GAC GGG ATT C-3′ (forward) and 5′- ACC CGA CTA ATA ACG AAA TTA ACG-3′ (reverse), and the primer sequences for the unmethylated reactions were 5′- TGA TTA TAG TGA TGA TGG GAT TTG A3′ (forward) and 5′-CCC AAC TAA TAA CAA AAT TAA CAC C-3′ (reverse). The amplification products were separated by agarose gel electrophoresis and visualized by ethidium bromide staining using a Gel Documentation 2000 system (Bio-Rad, Hercules, CA, USA). The bands were semi-quantitated by densitometry using ImageJ (Version 1.48d; NIH, Bethesda, Maryland, USA).
To verify the DNA methylation changes, methylated DNA was captured and quantified using methylated DNA immunoprecipitation coupled with quantitative real-time polymerase chain reaction analysis (MeDIP-qPCR) as described previously [32, 33]. Briefly, extracted DNA from treated HT29 cells was sheared in ice-cold water using a Bioruptor sonicator (Diagenode Inc., Sparta, NJ, USA) to approximately 200-1000 base pair. The fragmented DNA was further denatured at 95 °C for 2 min. Methylated DNA was isolated by immunoprecipitation with anti-5′-methylcytosine antibody using Methylamp Methylated DNA capture Kit (Epigentek, Farmingdale, NY, USA) according to the manufacturer's manual. After final purification and elution, the methylation status was quantified by qPCR amplification of MeDIP-enriched DNA using the primer set 5′- AAA CGC GGA GGT CTT TAG C-3′ (forward) and 5′- GCA GAC GAA GCA GCT GAG -3′ (reverse). The enrichment of methylated DNA in each treatment was calculated according to the standard curve of the serial dilution of input DNA. The relative methylated DNA ratios were then calculated with the basis of the control as 100% of DNA methylation.
RNA isolation and qPCR
Total RNA was extracted from the treated HT29 cells using the RNeasy Mini Kit (Qiagen, Valencia, CA, USA) and reverse-transcribed to cDNA using the SuperScript III First-Strand Synthesis System (Invitrogen, Carlsbad, CA, USA). Relative DLEC1 mRNA expression was determined by qPCR using cDNA as the template and the Power SYBR Green PCR Master Mix (Applied Biosystems, Carlsbad, CA, USA) in an ABI7900HT system (Applied Biosystems). The forward and reverse primers for DLEC1 amplification were 5′- CGA ACC CTT CGC CTG AAT AA-3′ and 5′- GGG AAA GGT GGC CCA TAA A-3′, respectively. Primers for GAPDH (internal control) were 5′- GGT GTG AAC CAT GAG AAG TAT GA-3′ (forward) and 5′-GAG TCC TTC CAC GAT ACC AAA G-3′ (reverse).
Protein lysate preparation and western blotting
Protein lysates were prepared using radioimmunoprecipitation assay (RIPA) buffer (Sigma-Aldrich St. Louis, MO, USA) supplemented with protein inhibitor cocktail (Sigma-Aldrich). The detailed procedure for western blotting was previously described [34]. Briefly, 20 μg of total protein as determined using the bicinchoninic acid (BCA) method (Pierce, Rockford, IL, USA) was separated by 4-15% SDS polyacrylamide gel electrophoresis (Bio-Rad, Hercules, CA, USA) and electro-transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, Bedford, MA, USA). After blocking with 5% BSA (Fisher Scientific, Pittsburgh, PA, USA) in Tris-buffered saline-0.1% Tween 20 (TBST) buffer (Boston Bioproducts, Ashland, MA, USA), the membranes were sequentially incubated with specific primary antibodies and horseradish peroxidase-conjugated secondary antibodies. The blots were visualized using Supersignal West Femto chemiluminescent substrate (Pierce, Rockford, IL, USA) and documented using a Gel Documentation 2000 system (Bio-Rad, Hercules, CA, USA). Densitometry of the bands was analyzed using ImageJ (Version 1.48d; NIH). The primary antibodies were obtained from different sources: anti-β-ACTIN from Santa Cruz Biotechnology (Santa Cruz, CA, USA); anti-DNMT1, 3A, and 3B from IMGENEX (San Diego, CA, USA); anti-HDAC1-7 from Cell Signaling Technology (Boston, MA, USA); and anti-HDAC8 from Proteintech (Chicago, IL, USA). The secondary antibodies were purchased from Santa Cruz Biotechnology.
Plasmids, transfection, and luciferase reporter assay
Human DLEC1 promoter was amplified from genomic DNA isolated from HT29 cells using the following primers: 5′- GAC ACA AAT GTT TAC AAT GAC C-3′ (forward) and 5′- TTT CTC AAC TGC AGC CCC AGA T-3′ (reverse). The PCR products were cloned into pCR4 TOPO vector using a TOPO TA Cloning kit (Invitrogen, Carlsbad, CA, USA), digested with KpnI and XhoI enzymes, and inserted into pGL4.15 luc2P/Hygro vector using T4 ligase as previously described [31]. All the recombinant plasmids were verified by sequencing (Genewiz, Piscataway, NJ, USA). To further generate the methylated luciferase reporter, the constructs were treated with methyl-trasferase M. SssI. Briefly, 5 μg reporter constructs were incubated with 5 unites of M. SssI in NEBuffer 2 (New England Biolabs Inc. Ipswich, MA, USA) supplemented with 160 μM S-adenosylmethionine (SAM, New England Biolabs Inc) at 37°C for 1 h. After the reaction, the methylated luciferase reporter plasmids were purified by gel extraction using the QIAquick gel extraction kit (Qiagen, Valencia, CA, USA). The methylation-dependent HhaI and HpaII restriction endonucleases were used to confirm the efficiency of the methylation reaction.
The transfection efficiency using HT29 cells were not optimal, human colon cancer cell lines HCT116, SW48, and human embryonic kidney HEK293 cells with higher transfection efficiency were used. The cells were seeded in 12-well plates for 24 h, then transfected with 500 ng of the methylated or unmethylated reporter plasmids using Lipofectamine 3000 transfection reagent (Invitrogen, Carlsbad, CA, USA) according to manufacturer's instructions. 500 ng of the pSV-β-Galactosidase control vector was co-transfected as internal control. 24 h after the transfection, the cells were lysed in 1X Reporter Lysis Buffer (Promega, Madison, WI, USA). 10 μL aliquiots of the cell lysate were assayed using the luciferase assay system with a Sirius luminometer (Berthold Technologies, Pforzheim, Germany). 30 μL aliquiots were assayed using the β-Galactosidase enzyme assay system and the absorbance was read at 420 nm by Infinite 200 Pro microplate reader (Tecan, Mannedorf, Switzerland). The transcriptional activities of the methylated or unmethylated constructs were calculated by normalizing the luciferase activities with the corresponding β-Galactosidase activities, and were reported as the folds of induction compared with the activity of the empty pGL 4.15 vector.
Colony formation assay
The colony-formation assay was performed as described previously with some modifications [34, 35]. The HT29, sh-Mock, and sh-DLEC1 cells (8 × 103 / well) were transferred to 1 mL of BME containing 0.33% agar over 3 mL of BME containing 0.5% agar with 10% FBS in 6-well plates. The cells were maintained with 0.1% DMSO, 2.5 μM and 5 μM CUR at 37°C in a humidified 5% CO2 atmosphere for 14 days.
In another set of experiment, the HT29 cells were first treated with control (0.1% DMSO), or CUR at 2.5 and 5 μM for 5 days similar to that described for the DNA methylation assays. On day 5, the pretreated HT29 cells (8 × 103 / well) were transferred to 1 mL of BME containing 0.33% agar over 3 mL of BME containing 0.5% agar with 10% FBS in 6-well plates. The cells were maintained in soft agar without the presence of CUR at 37°C in a humidified 5% CO2 atmosphere for additional 14 days.
The colonies were photographed using a computerized microscope system with the Nikon ACT-1 program (Version 2.20) and counted using ImageJ (Version 1.48d; NIH).
Statistical Analysis
The data are presented as the mean ± SEM (standard error of the mean). The statistical analyses were performed using Student's t-test. P values less than 0.05 were considered statistically significant and are indicated with *; P values less than 0.01 are indicated with **.
Results
CUR suppressed anchorage-independent growth of HT29 cells
To investigate the effect of CUR on the anchorage-independent growth of HT29 cells, the soft agar assay was employed. Firstly, HT29 cells were grown in soft agar containing CUR for 14 days. As illustrated in Figure 1A, colony formation of HT29 cells was significantly reduced by CUR at 2.5 μM and 5 μM by 32.2% and 37.8%, respectively. The cell viability of HT29 cells was not affected by CUR treatment at concentrations of 2.5 μM and 5 μM after 5 days when examined by the MTS assay (Figure 1C). However, a continuous cell counting with trypan blue staining for 14 days revealed that the number of viable cells was significantly reduced by CUR at 5 μM after 12 days (data not shown). To further confirm that the inhibition of colony formation by CUR is not a result from cell death, HT29 cells were pretreated with CUR (2.5 μM and 5 μM) for 5 days before transferred to agar. The pretreated cells were grown in agar for additional 14 days without the presence of CUR. As shown in Figure 1B, although the suppression of colony size by CUR pretreatment is not as pronounced as when CUR is present in agar medium, HT29 cells pretreated with CUR resulted in a significant reduced colony number in a similar trend. These results indicated that CUR inhibits the anchorage-independent growth of HT29 cells in soft agar.
Figure 1. CUR inhibited anchorage-independent growth of HT29 cells.

(A) HT29 cells (8,000 cells/well) were plated in soft agar containing 0.1% DMSO (Control) and CUR (2.5 μM or 5 μM) in 6-well plates for 14 days. The colonies were counted under a microscope and analyzed using ImageJ software. The colony number percentage was calculated by dividing the number of colonies formed in the CUR treatment groups by the number of colonies formed in the control group. Representative images of each group under a microscope are shown in the left panel. Graphical data are presented as the mean ± SEM of triplicate results from three independent experiments. * P<0.05 versus the control group and ** P<0.01 versus the control group. (B) HT29 cells were firstly treated with 0.1% DMSO (Control) and CUR (2.5 μM or 5 μM) for 5 days. On day 5, pretreated cells (8,000 cells/well) were transferred and grown in agar for additional 14 days without presence of CUR. The colonies were counted under a microscope and analyzed using ImageJ software. The colony number percentage was calculated by dividing the number of colonies formed with pretreated cells by the number of colonies formed in the control group. Representative images of each group under a microscope are shown in the left panel. Graphical data are presented as the mean ± SEM of triplicate results from three independent experiments. * P<0.05 versus the control group and ** P<0.01 versus the control group. (C) HT29 Cells were plated in 96-well plates at an initial density of 1,000 cells/ well for 24 h. The cells were then incubated in fresh medium with the presence of CUR (1-25 μM) for 5 days. Cell viability was determined by MTS assay. The data are presented as mean ± SEM of three independent experiments. * P<0.05 versus control group, ** P<0.01 versus control group.
Knockdown of DLEC1 reduced the inhibitory effect of CUR against colony formation in HT29 cells
DLEC1 is a candidate tumor suppressor whose overexpression is associated with repression of colony formation in many cancer cell lines [24-27, 29]. To investigate whether DLEC1 plays a critical role in the inhibitory effect of CUR in the anchorage-independent growth of HT29 cells, sh-Mock and sh-DLEC1 cells were established using lentivirus shRNAs vectors. Deficient mRNA expression of DLEC1 was confirmed in sh-DLEC1 cells by qPCR (Figure 2A). Significantly higher cell proliferation in sh-DLEC1 HT29 cells than in sh-Mock HT29 cells was observed from 24 h to 72 h (Figure 2B). This result was in agreement with previous reports that cells overexpressing DLEC1 grew at a reduced rate [24, 27]. Importantly, knockdown of DLEC1 significantly increased the anchorage-independent growth of HT29-shDLEC1 cells in soft agar by approximately 1.5-fold compared with sh-Mock cells (Figure 2C). Similar to the inhibitory effect in HT29 cells, CUR at the concentrations of 2.5 μM and 5 μM significantly suppressed colony formation of sh-Mock cells by 41% and 45.4%, respectively (Figure 1A and 2C). By contrast, the CUR-mediated inhibition of colony formation was remarkably reduced in sh-DLEC1 cells (Figure 2C and 2D). The inhibition of colony formation by CUR in sh-DLEC1 cells was only approximately 12.8% to 16.4%. These results suggest that DLEC1 played an important role in the CUR-mediated suppression of anchorage-independent growth of HT29 cells.
Figure 2. DLEC1 knockdown increased proliferation and attenuated the inhibitory effects of CUR on the anchorage-independent growth of HT29 cells.
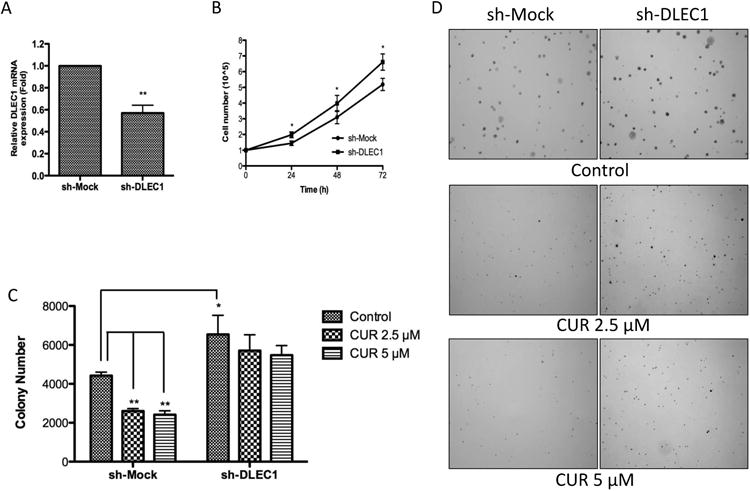
Stable mock (scramble-sequence control, sh-Mock) and DLEC1 knockdown (sh-DLEC1) HT29 cells were established using lenti-virus mediated short hairpin RNAs and were selected with puromycin for 3 weeks. (A) Reduced mRNA expression of DLEC1 in knockdown cells was confirmed by qPCR. (B) The growth of HT29 sh-DLEC1 cells was compared with that of sh-Mock cells over a period of 72 h. (C) Anchorage-independent growth of sh-Mock and sh-DLEC1 with or without the presence of CUR in soft agar for 14 days. (D) Representative images of each group under a microscope. All the data are presented as the mean ± SEM of triplicate results from at least three independent experiments. * P<0.05 versus the control group and ** P<0.01 versus the control group.
CUR decreased the methylation of the DLEC1 promoter in HT29 cells
Considering the role that DLEC1 played in CUR-mediated inhibition of colony formation in HT29 cells (Figure 2), we further investigated the effect of CUR treatment in regulating DLEC1 activity. DLEC1 has been found to be down-regulated in many human colorectal cancer cell lines and colorectal tumors with aberrant hypermethylated promoter regions [29]. To test whether CUR treatment could reverse the methylation of the DLEC1 gene promoter, MSP, bisulfite genomic sequencing, and MeDIP-qPCR were performed. In agreement with previous reports, we found that the methylated MSP gel bands are with higher density than the unmethylated MSP gel bands, indicating that the CpG sites in the promoter of DLEC1 gene was hypermethylated in HT29 cells (Figure 3A). Sequencing results showed an average of 95.8% methylation in the CpG island (-66 to +516 with translation initiation site designated as +1) in the control sample (data not shown). However, when the cells were treated with 5 μM CUR or a combination of 2.5 μM 5AZA and 100 nM TSA (serving as a positive control as previously described [17, 18, 31, 34]) for 5 days, the density of unmethylated MSP gel bands was significantly increased by approximately 50% (Figure 3A). To further confirm the demethylation effect of CUR observed in MSP, the methylation status of individual CpG site was examined using bisulfite genomic sequencing. The percentage of methylated CpG sites in the CpG island (-66 to +516) was slightly decreased (data not shown). Within this CpG island, the reduction was most significant at CpG sites 10-30 (+15 to +269) after 5 days of treatment with CUR 2.5 μM (p= 0.04) and CUR 5 μM (p=0.005) or a combination of 2.5 μM 5AZA and 100 nM TSA (p=0.004) (Figure 3B). To further quantify the methylation changes by CUR treatment, MeDIP-qPCR analysis was performed. Unlike methods based on bisulfite conversion, MeDIP experiment directly isolates methylated DNA fragments by immunoprecipitation with 5′-methylcytosine-specific antibody. qPCR analysis was then followed to quantitatively measure the enrichment of methylated DNA in the DLEC1 promoter region. As shown in Figure 3C, 5 μM CUR and the combination of 5AZA/TSA treatment significantly reduced the relative amount of methylated DNA containing DLEC1 promoter in HT29 cells. Together with the results obtained from MSP and bisulfite genomic sequencing, we showed that CpG methylation of DLEC1 promoter was decreased by CUR treatment.
Figure 3. Effects of CUR on CpG methylation in the DLEC1 promoter region.
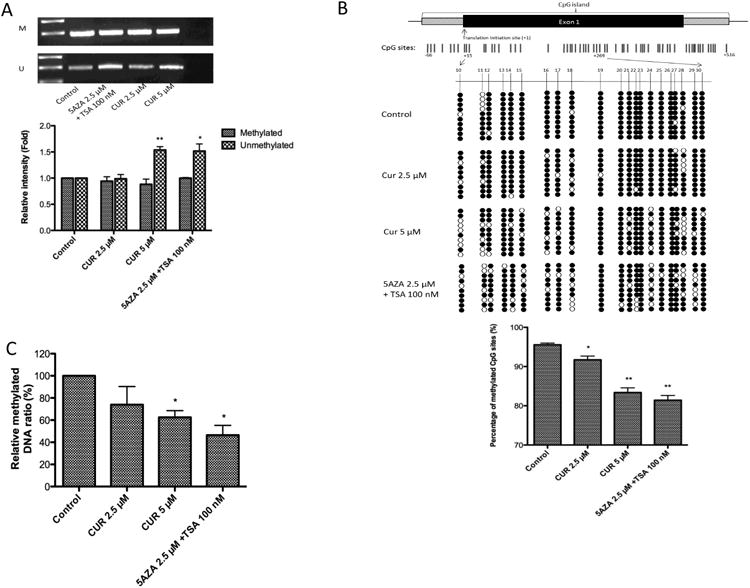
HT29 cells (3 × 104 /10-cm dish) were incubated with CUR (2.5 μM and 5 μM) for 5 days. The control group was treated with 0.1% DMSO, and the positive control group was treated with 2.5 μM 5AZA and 100 nM TSA (TSA was added 20 h before harvesting). (A) DLEC1 methylation as measured by methylation-specific PCR (MSP) in HT29 cells after 5 days of treatment. Genomic DNA was extracted, and bisulfite conversion was performed. M: methylated, U: unmethylated. Representative images are presented in the top panel. The relative intensity of the methylated and unmethylated band was measured by ImageJ and presented in the bottom panel. (B) The detailed methylation patterns of 10-30 CpGs (+15 to +269) in the promoter regions of the DLEC1 gene in HT29 cells were confirmed by bisulfite genomic sequencing. Filled circles indicate methylated CpGs, and empty circles indicate unmethylated CpGs. Ten clones were selected to represent the three independent experiments. The percentage of methylated CpG sites is shown in the bottom panel. The methylation percentage was calculated from three independent experiments as the number of methylated CpG sites over the total number of CpG sites examined. (C) The enrichment of the methylated DNA fragments captured by MeDIP was determined by qPCR according to the standard curve from a serial dilution of the inputs. Relative methylatied ratio was calculated by normalizing with control group (defined as 100% methylated DNA). All of the data are presented as the mean ± SEM. * P<0.05 versus the control group, ** P<0.01 versus the control group.
Figure 5. CUR reduced the protein expression of DNMTs in HT29 cells.
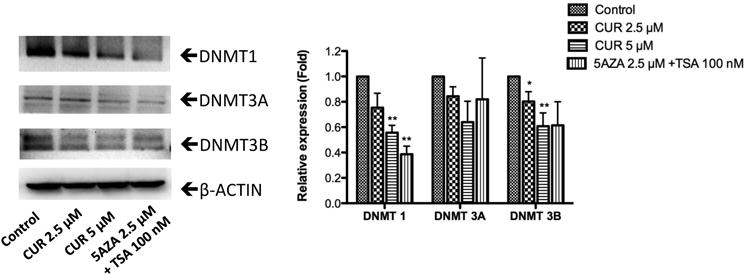
Proteins were extracted and examined by western blotting. The fold relative expression was calculated by dividing the intensity of each sample by that of the control sample and then normalizing to the intensity of β-actin using ImageJ. Representative bands are shown in the left panel. The bar chart in the right panel presents the mean ± SEM of three independent experiments. * P<0.05 versus the control group, ** P<0.01 versus the control group.
CUR increased the transcription of DLEC1 in HT29 cells
It has been reported that down-regulation of DLEC1 expression is correlated with hypermethylation of the DLEC1 promoter in various cancer cells and tissues [23-26, 29, 36]. In the present study, we constructed a luciferase reporter driven by the DLEC1 promoter (-66 to +516) to confirm the repression of gene transcription by CpG methylation. In vitro CpG methylation of the plasmid by M.sssI CpG methytransferase resulted in a significant decrease in DLEC1 transcriptional activity by 81.3%, 60.1%, and 51.5% in HCT116, SW48, and HEK293 cell lines, respectively (Figure 4A). Since CUR decreased the methylation of the DLEC1 promoter in HT29 cells, we hypothesized that the transcriptional activity of the DLEC1 gene could be enhanced by CUR treatment. qPCR analysis revealed that the mRNA expression of DLEC1 was significantly induced in HT29 cells after 5 days of treatment with 5 μM CUR or a combination of 2.5 μM 5AZA and 100 nM TSA (Figure 4B).
Figure 4. CUR increased the mRNA expression of DLEC1.
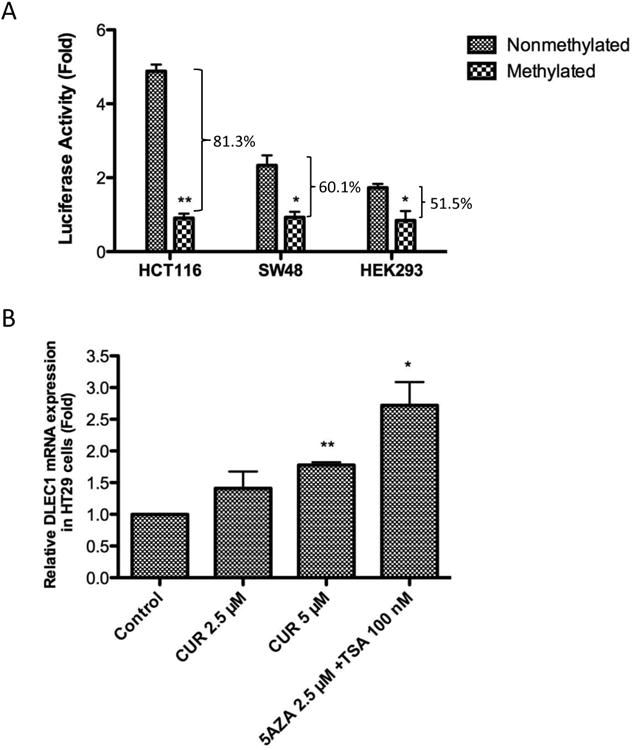
(A) Methylation of the CpGs inhibited the transcriptional activity of DLEC1. The DLEC1 CpG island (-66 to +516) was amplified from genomic DNA and inserted into pGL4.15 vector. The luciferase reporter construct, either methylated in vitro by CpG methyltransferase or not, were co-transfected with β-Galactosidase control vector into several cell lines. And the luciferase activities were measured 24 h post transfection. The luciferase activities were calculated by normalizing the firefly luciferase activities with corresponding β-Galactosidase activities, and are represented as fold change compared with the activity of empty pGL4.15 vector. The data are presented as the mean ± SEM of three independent experiments. * P<0.05 versus nonmethylated construct, ** P<0.01 versus nonmethylated construct. (B) Effect of CUR on the DLEC1 mRNA expression in HT29 cells. Total mRNA was isolated and analyzed using quantitative real-time PCR. The data are presented as the mean ± SEM of four independent experiments. * P<0.05 versus the control group, ** P<0.01 versus the control group.
CUR altered the expression of epigenetic modifying enzymes in HT29 cells
We next examined the effect of CUR on the expression of epigenetic modifying enzymes to explore the epigenetic mechanism by which CUR demethylated the DLEC1 promoter and increased DLEC1 transcription. DNA methylation at the 5-position of cytosine through the addition of a methyl group is catalyzed by DNA methyltransferases (DNMTs), including DNMT1, DNMT3A, and DNMT3B [37]. As shown in Figure 5, CUR reduced the protein expression of DNMT1, DNMT 3A, and DNMT 3B in a concentration-dependent manner in HT29 cells after 5 days of treatment. In addition, HDAC inhibition activity of CUR was previously reported in a molecular docking study [38]. Therefore, western blotting was also performed to evaluate the effect of CUR in modifying the protein expression of HDAC1-8. We found that the protein levels of HDAC4, HDAC5, HDAC6, and HDAC8 were significantly reduced in a concentration-dependent manner after treatment with CUR for 5 days in HT29 cells, whereas no considerable changes in the protein expression of HDAC1, HDAC2, HDAC3, and HDAC7 were detected (Figure 6). These results suggest that down-regulation of DNMTs and subtypes of HDACs by CUR may result in reduced methylation of DLEC1 promoter and activation of DLEC1 transcription in HT29 cells.
Figure 6. CUR altered the protein expression of HDACs in HT29 cells.
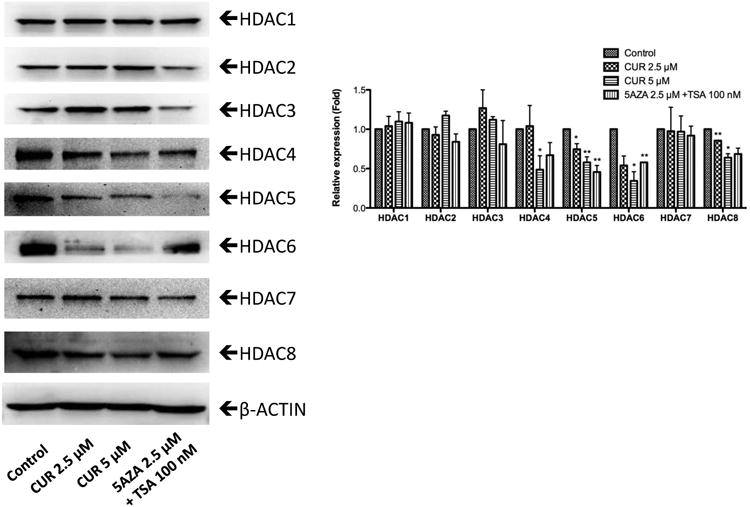
Protein expression of HDAC1-8 was measured by western blotting. The fold relative expression was calculated by dividing the intensity of each sample by that of the control sample and then normalizing to the intensity of β-actin using ImageJ. Representative bands are shown in the left panel. The bar chart in the right panel presents the mean ± SEM of three independent experiments. * P<0.05 versus the control group, ** P<0.01 versus the control group.
Discussion
The tumor-suppressing properties of DLEC1 have been supported by the observation of tumor-specific reduced transcription and decreased colony formation, in addition to reduced growth rate, in tumor cells with DLEC1 exogenous expression [23-26, 29, 36]. Moreover, Kwong et al. reported that no tumors formed in nude mice injected with the nasopharyngeal carcinoma cell line HONE1 transfected with DLEC1 cDNA, whereas tumors with an average size over 200 mm3 formed in control mice after 55 days [27]. However, growth suppression could result from overexpression of any gene; thus, experiments with DLEC1 knockdown cells are necessary. To further confirm the tumor suppressor role of DLEC1, we generated stable DLEC1 knockdown HT29 cells via shRNA expression through lentiviral transduction. We observed a significantly higher proliferation rate (Figure 2B) and enhanced anchorage-independent growth in HT29-shDLEC1 cells compared to HT29-shMock cells (Figure 2C). These observations, together with those of previous reports, demonstrate the tumor suppressor roles of DLEC1 in colorectal cancer.
Anchorage-independent growth (colony-forming capacity in semisolid medium) is an in vitro characteristic of tumorigenic cells and has served as a marker to distinguish transformed cells from normal cells [39]. Programmed cell death occurs when non-transformed cells are deprived of attachment for an extended period of time, whereas malignant cells proliferate without attachment to a solid substrate during tumor progression. The anchorage-independent growth of tumor cells has been correlated to their tumorigenic and metastatic potential in vivo [40]. The potential of suppressing the anchorage-independent growth of cancer cells by numerous phytochemicals, including CUR, has been investigated. Our results (Figure 1A and 1B) are in good agreement with published data supporting that CUR inhibits anchorage-independent growth of colorectal cancer cells [41, 42]. Multiple genetic changes such as those related to the Myc, Notch, β-catenin, and PI3K/Akt signaling pathways have been shown to be required for anchorage-independent growth and may be involved in the inhibition of colony formation by chemopreventive agents [43-45]. However, the molecular basis for these effects remains complex and poorly understood. Chen et al. reported that CUR inhibited the colony formation of HCT116 cells through down-regulation of the transcription of Sp-1, a genetic factor associated with the suppression of anchorage-independent growth in fibrocarcinoma cells [41, 46]. In our study, inhibition of anchorage-independent growth by CUR was considerably attenuated by the ablation of DLEC1 expression in HT29 cells (Figure 2C and 2D). Hence, we show for the first time that CUR inhibits the anchorage-independent growth of HT29 cells, at least partially through the modulation of DLEC1 expression. Since DLEC1 expression was reported to be associated with cell growth rate and cell cycle [23-26], it is possible that the inhibitory effect of CUR against colony formation of HT29 cells involves cell growth inhibition via DLEC1. The involvement of DLEC1 in the protective role of CUR against colorectal cancer requires further investigation in in vivo models.
DLEC1 encodes a 1755-amino-acid protein with no significant homology to any known protein or domains, but contains 27 potential casein kinase II (CK2) phosphorylation sites [19]. CK2 regulates the phosphorylation of more than 300 important substrates, and one-third of them are implicated in cell division and the cell cycle. Recently, CK2α (subunit α) was shown to be associated with the malignant transformation of several tissues, including colorectal cancer [47]. Based on the finding that ectopic expression of DLEC1 induced G1 arrest of the hepatocellular carcinoma cell cycle, Qiu et al. proposed that phosphorylation of DLEC1 by CK2 facilitates its nuclear localization and causes G1 arrest [24]. Notably, adhesion of integrin receptors to the extracellular matrix is required for attachment-dependent cells to transit through the G1 phase [44]. Thus, the potential involvement of CK2, DLEC1, and G1 cell cycle arrest in the inhibitory effect of CUR on the anchorage-independent growth of HT29 cells should be examined in the future.
CUR is a multi-targeting chemopreventive phytochemical that has been studied extensively in colorectal cancer. Recent studies have recognized the effect of CUR in modifying epigenetic mechanisms. For example, using DNA promoter methylation microarrays and gene expression arrays, Link et al. assessed global methylation and the gene expression profiles in colorectal cancer cells upon CUR treatment [42]. Interestingly, the results indicated that CUR modulates gene-specific DNA methylation, whereas the global hypomethylation induced by 5AZA is nonspecific. However, data regarding the ability of CUR to regulate the methylation levels of specific genes in colorectal cancer are relatively scarce. Our present study provides evidence that CUR decreases the CpG methylation of the DLEC1 promoter (Figure 3), which regulates a tumor suppressor gene potentially involved in the anchorage-independent growth of HT29 cells. Consequently, we revealed elevated mRNA expression of DLEC1 after CUR treatment (Figure 4B), which may be mediated by reduced CpG methylation in the DLEC1 promoter (Figure 3). Moreover, our results suggest that this demethylation effect may be associated with CUR-mediated inhibition of the protein expression of all subtypes of DNMTs in HT29 cells (Figure 5). Similarly, the importance of DNMT expression in regulating the methylation level and transcriptional activity of DLEC1 was demonstrated by Ying et al., who showed that the hypermethylated DLEC1 promoter and down-regulated DLEC1 transcription were demethylated and reactivated, respectively, only in HCT116 DKO (deficient in DNMT1 and DNMT3B) cells and not in DNMT1KO or DNMT3BKO cells [29]. This finding reflected that the combination of DNMT1 and DNMT3B, but not a specific DNMT, may be crucial for regulating DLEC1 promoter methylation and transcription. However, the effects of CUR in modulating the expression of DNMTs remain controversial. For example, Liu et al. used a molecular docking approach to suggest that CUR inhibits DNMT1 through covalent binding to the catalytic thiolate of C1226 in DNMT1 [48], whereas little or no alteration of the expression of DNMTs upon CUR treatment was observed in colorectal cancer cells (the specific cell type was not provided) [42] and LnCap cells [17]. Here, we clearly showed that CUR reduced the protein expression of DNMT1, DNMT3A, and DNMT3B (the inhibitory effect of DNMT3A was not statistically significant) in a concentration-dependent manner in HT29 cells after 5 days of treatment (Figure 5).
In addition to DNA methylation, deacetylation of histone H3 and H4 at the DLEC1 promoter may be involved in the epigenetic regulation of DLEC1 in human ovarian cancer and nasopharyngeal cancer cells [25, 27]. Thus, histone modification may also contribute to the regulation of transcriptional activity of DLEC1. Molecular docking studies predicted that CUR is a potential HDAC inhibitor [38]. Lee et al. reported that total HDAC activity was blocked by CUR treatment in medulloblastoma cells, although HDAC4 was the only HDAC subtype with decreased protein expression [49]. Similarly, the effect of CUR in inhibiting HDAC activity in myeloproliferative neoplasm cells was documented as a result of the reduced protein level of HDAC8 [50]. In the present study, we found that CUR significantly reduced the protein expression of HDAC4, HDAC5, HDAC6, and HDAC8 in HT29 cells (Figure 6), possibly resulting in impaired HDAC activity after CUR treatment. Although the exact mechanism of how CUR activates the transcription of DLEC1 requires further investigation, our results, together with previously reported evidence, suggest that CUR may epigenetically regulate the transcriptional activity of DLEC1 through alterations of DNMTs and HDACs.
In conclusion, our present study confirmed the tumor suppressor role of DLEC1 and suggested the involvement of DLEC1 in the suppression of anchorage-independent growth of HT29 cells by CUR treatment. Furthermore, we demonstrated that CUR could epigenetically up-regulate DLEC1 and reducing CpG methylation in HT29 cells, an activity that may be associated with lower protein expression of DNMTs and HDACs. Collectively, we propose a new mechanism underlying the chemopreventive effect of CUR in attenuating the clonogenicity of HT29 cells: the epigenetic regulation of DLEC1 expression and modification of the protein expression of DNMTs and HDACs. These findings provide valuable information for the future development of CUR and other phytochemicals as epigenetic modulators for preventing colorectal cancer.
Acknowledgments
The authors thank all the members in Dr. Kong's laboratory for their helpful discussion and preparation of this manuscript.
Grant Support: This work was supported in part by institutional funds and by R01AT007065 from the National Center for Complementary and Alternative Medicines (NCCAM) and the Office of Dietary Supplements (ODS).
Footnotes
Disclosure of Potential Conflicts of Interest: No potential conflicts of interest were disclosed.
References
- 1.Grady WM, Carethers JM. Genomic and epigenetic instability in colorectal cancer pathogenesis. Gastroenterology. 2008;135:1079–99. doi: 10.1053/j.gastro.2008.07.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, Leary RJ, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–13. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- 3.Lao VV, Grady WM. Epigenetics and colorectal cancer. Nature reviews Gastroenterology & hepatology. 2011;8:686–700. doi: 10.1038/nrgastro.2011.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. The New England journal of medicine. 2003;349:2042–54. doi: 10.1056/NEJMra023075. [DOI] [PubMed] [Google Scholar]
- 5.Glockner SC, Dhir M, Yi JM, McGarvey KE, Van Neste L, Louwagie J, et al. Methylation of TFPI2 in stool DNA: a potential novel biomarker for the detection of colorectal cancer. Cancer research. 2009;69:4691–9. doi: 10.1158/0008-5472.CAN-08-0142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yi JM, Dhir M, Guzzetta AA, Iacobuzio-Donahue CA, Heo K, Yang KM, et al. DNA methylation biomarker candidates for early detection of colon cancer. Tumour biology : the journal of the International Society for Oncodevelopmental Biology and Medicine. 2012;33:363–72. doi: 10.1007/s13277-011-0302-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van Engeland M, Derks S, Smits KM, Meijer GA, Herman JG. Colorectal cancer epigenetics: complex simplicity. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2011;29:1382–91. doi: 10.1200/JCO.2010.28.2319. [DOI] [PubMed] [Google Scholar]
- 8.Dhanak D, Jackson P. Development and classes of epigenetic drugs for cancer. Biochemical and biophysical research communications. 2014 doi: 10.1016/j.bbrc.2014.07.006. [DOI] [PubMed] [Google Scholar]
- 9.Miyamoto K, Ushijima T. Diagnostic and therapeutic applications of epigenetics. Japanese journal of clinical oncology. 2005;35:293–301. doi: 10.1093/jjco/hyi088. [DOI] [PubMed] [Google Scholar]
- 10.Dolinoy DC, Weidman JR, Jirtle RL. Epigenetic gene regulation: linking early developmental environment to adult disease. Reproductive toxicology. 2007;23:297–307. doi: 10.1016/j.reprotox.2006.08.012. [DOI] [PubMed] [Google Scholar]
- 11.Wargovich MJ. Experimental evidence for cancer preventive elements in foods. Cancer letters. 1997;114:11–7. doi: 10.1016/s0304-3835(97)04616-8. [DOI] [PubMed] [Google Scholar]
- 12.Shehzad A, Wahid F, Lee YS. Curcumin in cancer chemoprevention: molecular targets, pharmacokinetics, bioavailability, and clinical trials. Archiv der Pharmazie. 2010;343:489–99. doi: 10.1002/ardp.200900319. [DOI] [PubMed] [Google Scholar]
- 13.Hsu CH, Cheng AL. Clinical studies with curcumin. Advances in experimental medicine and biology. 2007;595:471–80. doi: 10.1007/978-0-387-46401-5_21. [DOI] [PubMed] [Google Scholar]
- 14.Garcea G, Jones DJ, Singh R, Dennison AR, Farmer PB, Sharma RA, et al. Detection of curcumin and its metabolites in hepatic tissue and portal blood of patients following oral administration. British journal of cancer. 2004;90:1011–5. doi: 10.1038/sj.bjc.6601623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garcea G, Berry DP, Jones DJ, Singh R, Dennison AR, Farmer PB, et al. Consumption of the putative chemopreventive agent curcumin by cancer patients: assessment of curcumin levels in the colorectum and their pharmacodynamic consequences. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2005;14:120–5. [PubMed] [Google Scholar]
- 16.Reuter S, Gupta SC, Park B, Goel A, Aggarwal BB. Epigenetic changes induced by curcumin and other natural compounds. Genes & nutrition. 2011;6:93–108. doi: 10.1007/s12263-011-0222-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shu L, Khor TO, Lee JH, Boyanapalli SS, Huang Y, Wu TY, et al. Epigenetic CpG demethylation of the promoter and reactivation of the expression of Neurog1 by curcumin in prostate LNCaP cells. The AAPS journal. 2011;13:606–14. doi: 10.1208/s12248-011-9300-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khor TO, Huang Y, Wu TY, Shu L, Lee J, Kong AN. Pharmacodynamics of curcumin as DNA hypomethylation agent in restoring the expression of Nrf2 via promoter CpGs demethylation. Biochemical pharmacology. 2011;82:1073–8. doi: 10.1016/j.bcp.2011.07.065. [DOI] [PubMed] [Google Scholar]
- 19.Daigo Y, Nishiwaki T, Kawasoe T, Tamari M, Tsuchiya E, Nakamura Y. Molecular cloning of a candidate tumor suppressor gene, DLC1, from chromosome 3p21.3. Cancer research. 1999;59:1966–72. [PubMed] [Google Scholar]
- 20.Imreh S, Klein G, Zabarovsky ER. Search for unknown tumor-antagonizing genes. Genes, chromosomes & cancer. 2003;38:307–21. doi: 10.1002/gcc.10271. [DOI] [PubMed] [Google Scholar]
- 21.Agathanggelou A, Dallol A, Zochbauer-Muller S, Morrissey C, Honorio S, Hesson L, et al. Epigenetic inactivation of the candidate 3p21.3 suppressor gene BLU in human cancers. Oncogene. 2003;22:1580–8. doi: 10.1038/sj.onc.1206243. [DOI] [PubMed] [Google Scholar]
- 22.Hesson L, Bieche I, Krex D, Criniere E, Hoang-Xuan K, Maher ER, et al. Frequent epigenetic inactivation of RASSF1A and BLU genes located within the critical 3p21.3 region in gliomas. Oncogene. 2004;23:2408–19. doi: 10.1038/sj.onc.1207407. [DOI] [PubMed] [Google Scholar]
- 23.Seng TJ, Currey N, Cooper WA, Lee CS, Chan C, Horvath L, et al. DLEC1 and MLH1 promoter methylation are associated with poor prognosis in non-small cell lung carcinoma. British journal of cancer. 2008;99:375–82. doi: 10.1038/sj.bjc.6604452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qiu GH, Salto-Tellez M, Ross JA, Yeo W, Cui Y, Wheelhouse N, et al. The tumor suppressor gene DLEC1 is frequently silenced by DNA methylation in hepatocellular carcinoma and induces G1 arrest in cell cycle. Journal of hepatology. 2008;48:433–41. doi: 10.1016/j.jhep.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 25.Kwong J, Lee JY, Wong KK, Zhou X, Wong DT, Lo KW, et al. Candidate tumor-suppressor gene DLEC1 is frequently downregulated by promoter hypermethylation and histone hypoacetylation in human epithelial ovarian cancer. Neoplasia. 2006;8:268–78. doi: 10.1593/neo.05502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Q, Ying J, Li J, Fan Y, Poon FF, Ng KM, et al. Aberrant promoter methylation of DLEC1, a critical 3p22 tumor suppressor for renal cell carcinoma, is associated with more advanced tumor stage. The Journal of urology. 2010;184:731–7. doi: 10.1016/j.juro.2010.03.108. [DOI] [PubMed] [Google Scholar]
- 27.Kwong J, Chow LS, Wong AY, Hung WK, Chung GT, To KF, et al. Epigenetic inactivation of the deleted in lung and esophageal cancer 1 gene in nasopharyngeal carcinoma. Genes, chromosomes & cancer. 2007;46:171–80. doi: 10.1002/gcc.20398. [DOI] [PubMed] [Google Scholar]
- 28.Park SY, Kwon HJ, Lee HE, Ryu HS, Kim SW, Kim JH, et al. Promoter CpG island hypermethylation during breast cancer progression. Virchows Archiv : an international journal of pathology. 2011;458:73–84. doi: 10.1007/s00428-010-1013-6. [DOI] [PubMed] [Google Scholar]
- 29.Ying J, Poon FF, Yu J, Geng H, Wong AH, Qiu GH, et al. DLEC1 is a functional 3p22.3 tumour suppressor silenced by promoter CpG methylation in colon and gastric cancers. British journal of cancer. 2009;100:663–9. doi: 10.1038/sj.bjc.6604888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saw CL, Guo Y, Yang AY, Paredes-Gonzalez X, Ramirez C, Pung D, et al. The berry constituents quercetin, kaempferol, and pterostilbene synergistically attenuate reactive oxygen species: Involvement of the Nrf2-ARE signaling pathway. Food and chemical toxicology : an international journal published for the British Industrial Biological Research Association. 2014;72:303–11. doi: 10.1016/j.fct.2014.07.038. [DOI] [PubMed] [Google Scholar]
- 31.Yu S, Khor TO, Cheung KL, Li W, Wu TY, Huang Y, et al. Nrf2 expression is regulated by epigenetic mechanisms in prostate cancer of TRAMP mice. PloS one. 2010;5:e8579. doi: 10.1371/journal.pone.0008579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Su ZY, Khor TO, Shu L, Lee JH, Saw CL, Wu TY, et al. Epigenetic reactivation of Nrf2 in murine prostate cancer TRAMP C1 cells by natural phytochemicals Z-ligustilide and Radix angelica sinensis via promoter CpG demethylation. Chemical research in toxicology. 2013;26:477–85. doi: 10.1021/tx300524p. [DOI] [PubMed] [Google Scholar]
- 33.Zhang C, Su ZY, Khor TO, Shu L, Kong AN. Sulforaphane enhances Nrf2 expression in prostate cancer TRAMP C1 cells through epigenetic regulation. Biochemical pharmacology. 2013;85:1398–404. doi: 10.1016/j.bcp.2013.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Su ZY, Zhang C, Lee JH, Shu L, Wu TY, Khor TO, et al. Requirement and epigenetics reprogramming of Nrf2 in suppression of tumor promoter TPA-induced mouse skin cell transformation by sulforaphane. Cancer prevention research. 2014;7:319–29. doi: 10.1158/1940-6207.CAPR-13-0313-T. [DOI] [PubMed] [Google Scholar]
- 35.Wang L, Zhang C, Guo Y, Su ZY, Yang Y, Shu L, et al. Blocking of JB6 Cell Transformation by Tanshinone IIA: Epigenetic Reactivation of Nrf2 Antioxidative Stress Pathway. The AAPS journal. 2014 doi: 10.1208/s12248-014-9666-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang Z, Li L, Su X, Gao Z, Srivastava G, Murray PG, et al. Epigenetic silencing of the 3p22 tumor suppressor DLEC1 by promoter CpG methylation in non-Hodgkin and Hodgkin lymphomas. Journal of translational medicine. 2012;10:209. doi: 10.1186/1479-5876-10-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Subramaniam D, Thombre R, Dhar A, Anant S. DNA methyltransferases: a novel target for prevention and therapy. Frontiers in oncology. 2014;4:80. doi: 10.3389/fonc.2014.00080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bora-Tatar G, Dayangac-Erden D, Demir AS, Dalkara S, Yelekci K, Erdem-Yurter H. Molecular modifications on carboxylic acid derivatives as potent histone deacetylase inhibitors: Activity and docking studies. Bioorganic & medicinal chemistry. 2009;17:5219–28. doi: 10.1016/j.bmc.2009.05.042. [DOI] [PubMed] [Google Scholar]
- 39.Cifone MA, Fidler IJ. Correlation of patterns of anchorage-independent growth with in vivo behavior of cells from a murine fibrosarcoma. Proceedings of the National Academy of Sciences of the United States of America. 1980;77:1039–43. doi: 10.1073/pnas.77.2.1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mori S, Chang JT, Andrechek ER, Matsumura N, Baba T, Yao G, et al. Anchorage-independent cell growth signature identifies tumors with metastatic potential. Oncogene. 2009;28:2796–805. doi: 10.1038/onc.2009.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen CC, Sureshbabul M, Chen HW, Lin YS, Lee JY, Hong QS, et al. Curcumin Suppresses Metastasis via Sp-1, FAK Inhibition, and E-Cadherin Upregulation in Colorectal Cancer. Evidence-based complementary and alternative medicine : eCAM. 2013;2013:541695. doi: 10.1155/2013/541695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Link A, Balaguer F, Shen Y, Lozano JJ, Leung HC, Boland CR, et al. Curcumin modulates DNA methylation in colorectal cancer cells. PloS one. 2013;8:e57709. doi: 10.1371/journal.pone.0057709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sjolund J, Johansson M, Manna S, Norin C, Pietras A, Beckman S, et al. Suppression of renal cell carcinoma growth by inhibition of Notch signaling in vitro and in vivo. The Journal of clinical investigation. 2008;118:217–28. doi: 10.1172/JCI32086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Orford K, Orford CC, Byers SW. Exogenous expression of beta-catenin regulates contact inhibition, anchorage-independent growth, anoikis, and radiation-induced cell cycle arrest. The Journal of cell biology. 1999;146:855–68. doi: 10.1083/jcb.146.4.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dang CV, O'Donnell KA, Zeller KI, Nguyen T, Osthus RC, Li F. The c-Myc target gene network. Seminars in cancer biology. 2006;16:253–64. doi: 10.1016/j.semcancer.2006.07.014. [DOI] [PubMed] [Google Scholar]
- 46.Lou Z, O'Reilly S, Liang H, Maher VM, Sleight SD, McCormick JJ. Down-regulation of overexpressed sp1 protein in human fibrosarcoma cell lines inhibits tumor formation. Cancer research. 2005;65:1007–17. [PubMed] [Google Scholar]
- 47.Zou J, Luo H, Zeng Q, Dong Z, Wu D, Liu L. Protein kinase CK2alpha is overexpressed in colorectal cancer and modulates cell proliferation and invasion via regulating EMT-related genes. Journal of translational medicine. 2011;9:97. doi: 10.1186/1479-5876-9-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu Z, Xie Z, Jones W, Pavlovicz RE, Liu S, Yu J, et al. Curcumin is a potent DNA hypomethylation agent. Bioorganic & medicinal chemistry letters. 2009;19:706–9. doi: 10.1016/j.bmcl.2008.12.041. [DOI] [PubMed] [Google Scholar]
- 49.Lee SJ, Krauthauser C, Maduskuie V, Fawcett PT, Olson JM, Rajasekaran SA. Curcumin-induced HDAC inhibition and attenuation of medulloblastoma growth in vitro and in vivo. BMC cancer. 2011;11:144. doi: 10.1186/1471-2407-11-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen CQ, Yu K, Yan QX, Xing CY, Chen Y, Yan Z, et al. Pure curcumin increases the expression of SOCS1 and SOCS3 in myeloproliferative neoplasms through suppressing class I histone deacetylases. Carcinogenesis. 2013;34:1442–9. doi: 10.1093/carcin/bgt070. [DOI] [PubMed] [Google Scholar]