

Abstract
A unified strategy for enantioselective total synthesis of all stereoisomers of the 2+2 family of quadrigemine alkaloids is reported. In this approach, two enantioselective intramolecular Heck reactions are carried out at the same time on precursors fashioned in four steps from either meso- or (+)-chimonanthine to form the two critical quaternary carbons of the peripheral cyclotryptamine rings of these products. Useful levels of catalyst control are realized in either desymmetrizing a meso precursor or controlling diastereoselectivity in elaborating C2-symmetic intermediates. None of the synthetic quadrigemines are identical with alkaloids isolated previously and referred to as quadrigemines A and E. In addition, we report improvements in our previous total syntheses of (+)- or (−)-quadrigemine C that shortened the synthetic sequence to 10 steps and provided these products in 2.2% overall yield from tryptamine.
Keywords: Stereocontrolled total synthesis, alkaloid, enantioselective catalysis, intramolecular Heck reaction
1. Introduction
Alkaloids composed of multiple 1,2,3,3a,8,8a-hexahydropyrrolo[2,3-b]indole (cyclotryptamine or pyrrolidinoindoline) units have been isolated from a variety of natural sources, including bacteria, fungi, plants and amphibians (Figure 1).1 In the predominant family of these alkaloids, cyclotryptamine units are joined at their benzylic C3a carbon to generate dimers, trimers and higher-order oligomers. In this linkage, two types of quaternary stereogenic centers are produced: (a) vicinal quaternary carbons joining benzylic (C3a) quaternary stereocenters of two cyclotryptamine units, and (b) aryl-substituted quaternary carbons linking the peri (C7) carbon of one pyrrolidinoindoline unit and the benzylic quaternary stereocenter of another. Both types of quaternary stereocenters present formidable challenges for stereocontrolled synthesis. As a result, when our efforts in this area began in 1995, no stereocontrolled methods were available for linking cyclotryptamine fragments at C3a.2 In the intervening years, this challenging problem in total synthesis has been addressed by a number of researchers and many imaginative methods are currently available.2,3 Nonetheless, stereocontrolled total synthesis of the more complex members of this group remains largely an unmet challenge.4
Figure 1.
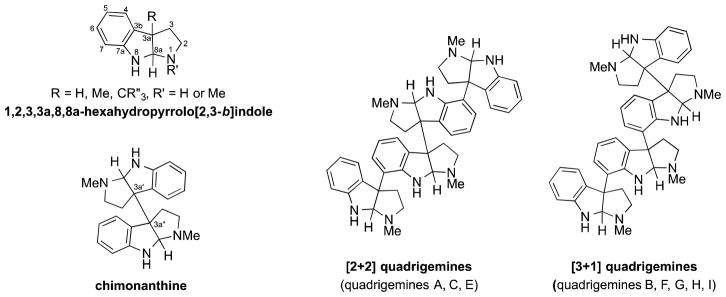
A pyrrolidinoindoline fragment and the connectivity of the chimonanthine and quadrigemine alkaloids.
The first cyclotryptamine alkaloids containing four tricyclic units, quadrigemines A and B, were reported by Perry and Smith in 1978 from the leaves of Hodgkinsonia frutesecens.5,6 Analysis of quadrigemine A revealed a base peak of m/z 344 corresponding to half the molecular weight of quadrigemine A along with ions m/z 345 and 690 in the EI mass spectrum. In comparison, quadrigemine B showed a parent ion of m/z 516, along with ions m/z 690, 517, and 172. The difference in fragmentation patterns corresponds to the location of the labile σ-bond connecting the 3a′–3a″-vicinal quaternary carbon centers of the chimonanthine subunit. 7 This fragmentation pattern has been used to designate the two groups of constitutional isomers: the [2+2] and [3+1] quadrigemine alkaloids (Figure 1).8
Quadrigemines A,5 C9, and E10 are the three reported members of the [2+2] quadrigemine family. The complex NMR spectra and amorphous nature of these higher-order polypyrrolidinoindoline alkaloids has made determination of their three-dimensional structures particularly difficult. The relative and absolute configuration of only quadrigemine C (1) is known with certainty (Figure 2). Sévenet, who initially isolated quadrigemine C from an extract of Psychotria oleoides found in New Caledonia,9a provided evidence for the R absolute configuration of the two outer quaternary carbons of 1 by chemical correlation with hodgkinsine, whose absolute and relative configuration had been determined by single-crystal X-ray analysis.9b,11 However rigorous proof of the absolute configuration of the central chimonanthine unit was not secured until the enantioselective total synthesis of (−)-quadrigemine C was reported by our group in 2002.4 The optical rotations reported for quadrigemines A, C, and E in alcoholic solvents ([α]D A, +32 (EtOH);5 C, +40 (MeOH);9d E, +33 (EtOH)10 are similar; suggesting that these alkaloids, which were isolated by different laboratories, could be identical.12 Resolving this issue from the published NMR characterization data is impossible, because of differences in the reported NMR solvent and spectrometer field strength. Even if directly comparable data were available for evaluation, the presence of several interconverting low-energy conformations of these alkaloids results in broad peaks on the NMR timescale at 298K. Attempts to coalesce these signals at elevated temperatures are typically compromised by the lability of σ-bond connecting the vicinal quaternary carbons.7 Cooling the NMR sample can result in enhanced resolution of the major conformers, as is the case for quadrigemine C;4,9c however, fully analyzing these complex spectra is challenging. Quadrigemines A and E, if different from quadrigemine C, could be one of six C2-symmetric stereoisomers (5, 6, 7 and their enantiomers, Figure 2). Overall there are ten possible [2+2] quadrigemine stereoisomers: 4 enantiomeric pairs and two meso compounds (Figure 2).
Figure 2.

Structure of quadrigemine C and six stereoisomers.
In an attempt to clarify the structures of quadrigemines A and E, we initiated stereocontrolled total syntheses of the remaining chiral members of the [2+2] quadrigemine alkaloid family. Moreover such an investigation would allow us to investigate the degree of catalyst control achieved in enantioselective Heck cyclizations carried out with C2-symmetric intermediates; in our original total synthesis of quadrigemine C (1), the pivotal enantioselective cyclizations were realized with a meso precursor. During these studies, several improvements to our original synthesis of quadrigemine C were attained, allowing the synthetic route to be shortened and the overall yield improved. Finally, with access to an expanded group of [2+2] quadrigemines, the effect of relative and absolute configuration on their antitumor activity was evaluated.
2. Results and discussion
2.1 Inside-out approach to the [2+2] quadrigemines
In the approach we developed to synthesize members of the [2+2] family of quadrigemines, the outer two pyrrolidinoindoline fragments are elaborated simultaneously on a functionalized chimonanthine core (Scheme 1). The formation of decacyclic dioxindole A by double enantioselective intramolecular Heck cyclization of dibutenanilide B is the pivotal step in this sequence.13 The Heck cyclization precursor B is formed by a double Stille coupling of a chimonanthine diiodide C with two equiv of stannane 8, an intermediate that contains all the heavy atoms of a cyclotryptamine fragment. The natural products meso-chimonanthine and its enantiopure C2-symmetric stereoisomers, which are now readily available by stereocontrolled chemical synthesis,14,15 serve as the starting point of the synthetic sequence.
Scheme 1.

Retrosynthetic analysis: an inside-out synthetic strategy.
2.2 Enantioselective synthesis of [2+2] quadrigemines having a meso core
In our initial synthesis of quadrigemine C,4 meso-chimonanthine (9) was prepared from oxindole and isatin in 13 steps and ~30% overall yield by a stereocontrolled sequence that we had defined previously.14b To expedite access to larger quantities of meso-chimonanthine (9), we have since adopted the non-stereocontrolled oxidative dimerization method reported by Takayama and co-workers.16 This method involves the oxidative dimerization of Nb-carbomethoxytryptamine with phenyliodine(III) bis(trifluoroacetate) and a subsequent reduction with Red-Al® to generate meso-chimonanthine (9) in two steps and 20–30% yield. Willis and coworkers recently improved this procedure by enhancing overall scalability and purification of product 9.17 Utilizing our previously published procedure, meso-chimonanthine diiodide 10 was prepared from meso-chimonanthine in three steps and 66% overall yield by di-Boc protection, di-ortho-iodination, and removal of the Boc groups (Scheme 2).4
Scheme 2.

Optimized double-Stille cross coupling.
In our first generation synthesis, the Stille cross coupling of meso-diiodide 10 with 3 equiv of stannane aryl triflate 8 to provide the meso-dibutenanilide 11 was achieved using a catalyst system of Pd2dba3·CHCl3, tri-2-furylphosphine and CuI in 1-methyl-2-pyrrolindoline (NMP).4,18 This Stille coupling was sluggish, requiring long reaction times (>36 h) at room temperature, and provided variable yields of 11 (55–71%). In the hope of both accelerating the rate of the reaction and improving the overall yield, several additional conditions for accomplishing this double cross coupling were investigated. Attempts to accelerate the reaction rate by increasing the reaction temperature were thwarted by slow decomposition of the aryl triflate functionality of stannane triflate 8. The use of fluoride ion (CsF) to accelerate the reaction did not have a substantial effect on improving the overall conversion. 19 The use of copper(I) thiophene-2-carboxylate (CuTC), 20 or CuTC in addition to [Ph2PO2][NBu4]21 did accelerate the reaction rate, but did not improve the yield. After further experimentation, a Stille coupling procedure reported by Corey and co-workers using Pd(PPh3)4/CuCl/LiCl was found to be optimal.22 Thus, reaction of meso-diiodide 10 with 3 equiv of stannane aryl triflate 8, 0.5 equiv of Pd(PPh3)4, 5 equiv of CuCl, and 6 equiv of LiCl in DMSO at room temperature for 20 h provided meso-dibutenanilide 11 in 96% yield.
As the outer pyrrolidinoindolines of quadrigemine C (1) have the same absolute configuration, our strategy was to utilize an enantioselective intramolecular Heck reaction to access the desired C1-symmetric dioxindole stereoisomer using a two-directional synthesis strategy.23 In accordance with the Horeau principle, a double enantioselective transformation has the potential to amplify the enantiopurity of the product over that provided by a single enantioselective transformation.24 Although the overall yield of the desired product is diminished, the product’s enantiomeric purity should by equal to the square of the enantioselectivity of a single reaction.
Several conditions were examined to optimize the double enantioselective Heck cyclization of meso-dibutenanilide 11 (Scheme 3, Table 1). Of the diphosphine ligands screened, (R)-tol-BINAP in MeCN provided superior diastereo- and enantioselection, providing pentacyclic dioxindole 12 and its two meso stereoisomers in a ratio of 9.3:2.0:1.0. The C1-symmetric product 12 (62%, 90% ee) and the meso products 13 and 14 (14% and 7% respectively) 25 were separated by preparative HPLC. The related enantioselective Heck cyclization employed in the enantioselective synthesis of the nonacyclic cyclotryptamine alkaloids hodgkinsines A and B from meso-chimonanthine gave a 1:1 ratio of these products in high yield and 79 and 83% ee.26,27 As the observed enantiomeric purity of the C1-symmetric product 12 (90% ee) is somewhat less than the predicted (98% ee) from the results in the hodgkinsine series, it can be inferred that the two enantioselective intramolecular Heck reactions are not completely independent. It is important to note that representing these molecules in two dimensions is deceiving. This decrease in enantioselectivity from the predicted model most likely results from stereoinduction across the meso-chimonanthine backbone during the second Heck cyclization. A sense that such interactions would be possible can be gleaned from a three-dimensional model of quadrigemine C (Figure 3). Carrying out the cyclization of ditriflate 11 under identical conditions using (S)- rather than (R)-BINAP gave dioxindole ent-12 in 80% yield; the higher selectivity in forming the C1-symmetric product in this double enantioselective Heck reaction suggests that catalyst and substrate control are matched with the catalyst formed from (S)-BINAP.
Scheme 3.

Double enantioselective Heck cyclization of meso-dibutenanilide 11 (one of the meso stereoisomers has the S,S,R,R absolute configuration and the other the R,S,R,S absolute configuration).
Table 1.
Optimization of the double enantioselective Heck cyclization of meso-dibutenanilide 11.
| Entry | Conditionsa | Solvent | ds ratio (12:13:14)b | % ee 12b |
|---|---|---|---|---|
|
| ||||
| 1 | (R)-BINAP | THF | 2.8:1.7:1.0 | 65 |
| 2 | (R)-BINAP | DMA | 2.5:1.0:1.1 | 64 |
| 3 | (R)-BINAP | MeCN | 3.5:1.7:1.0 | 80 |
| 4 | (R)-BINAP | PhMe | 3.1:2.7:1.0 | 54 |
| 5c | (S,S)-BDPP | THF | 2.4:1.0:1.5 | 35 |
| 6 | (R)-tol-BINAP | THF | 7.2:2.0:1.0 | 85 |
| 7 | (R)-tol-BINAP | DMA | 6.7:1.2:1.0 | 81 |
| 8 | (R)-tol-BINAP | NMP | 5.6:1.2:1.0 | 80 |
| 9 | (R)-tol-BINAP | NMP | 5.1:1.7:1.0 | 79 |
| 10 | (R)-tol-BINAP | MeCN | 9.3:2.0:1.0 | 90 |
50 mol% Pd(OAc)2, 100 mol% phosphine ligand, 4 equiv 1,2,2,6,6-pentamethylpiperidine (PMP), 80 °C;
ratio determined by HPLC;
ent-12 was formed preferentially.
Figure 3.
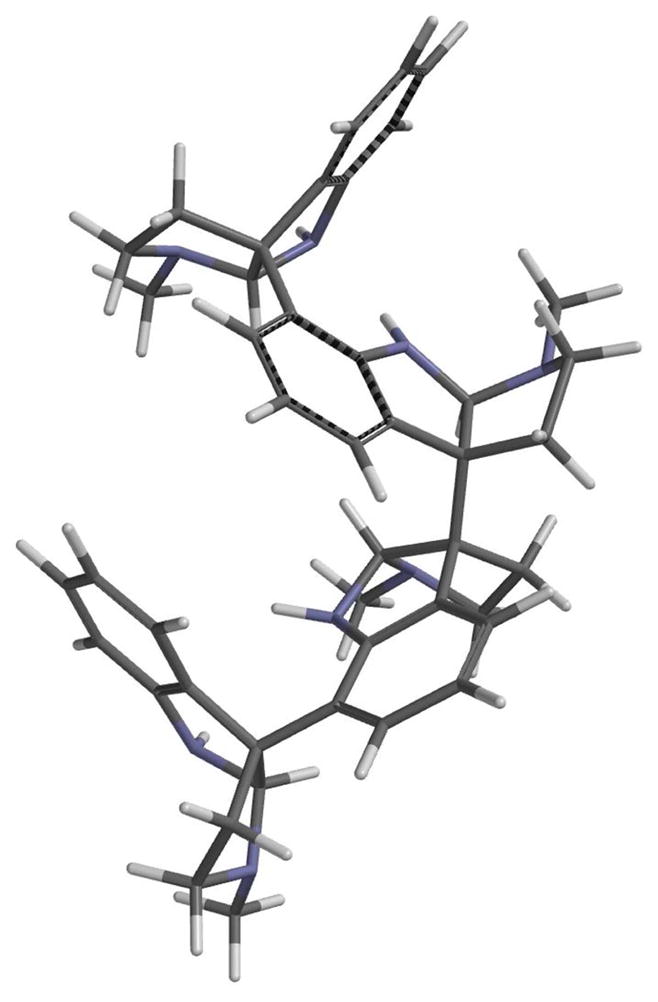
Model of a low-energy conformation of quadrigemine C.28
In our original synthesis of quadrigemine C, diene disulfonamide 12 was hydrogenated at 100 psi using Pd(OH)2/C in 10:1 EtOH-MeOH at 80 °C to saturate the two double bonds.4 These conditions often resulted in yields of the tetrahydro product 15 that varied depending upon the batch of substrate and catalyst.29 Since our initial report, we discovered that including 8 equiv of K2CO3 resulted in enhanced reproducibility for this transformation, which we attribute to preventing any acid-catalyzed decomposition30,31 Additionally, increasing the hydrogen pressure to 1000 psi in EtOH at 80 °C helped to ensure full conversion. Using this procedure, 12 reproducibly was transformed to its tetrahydro congener in yields of 90–95% (Scheme 4). Exposure of this unpurified intermediate to a large excess of Na (50 equiv) in THF/NH3 containing 4 equiv of tert-butanol for 20 min at −78 °C, followed by quenching with NH4Cl and HPLC purification afforded (−)-quadrigemine C (1). Modification of our previous conditions by the incorporation of tert-butanol32led to significant improvements in reproducibility, reliably yielding (−)-quadrigemine C in 22–24% for the two steps
Scheme 4.

Completion of an optimized total synthesis of quadrigemine C.
The improvements made in this second generation synthesis of (−)-quadrigemine C shortened the synthetic sequence to 10 steps and provided (−)-quadrigemine C (1), [α]23D −67 (c = 0.2, CHCl3) and [α]23D −30 (c = 0.2, EtOH), in 2.2% overall yield from tryptamine. Synthetic (−)-quadrigemine C showed physical properties (1H NMR, 13C NMR, HRMS) consistent with those of the natural product,9a–d and was indistinguishable from an authentic sample by HPLC and CD analysis.33,34 Moreover, heating a sample of (−)-quadrigemine C at 100 °C in the presence of dilute aqueous acetic acid gave synthetic (−)-psycholeine (16) in 38% yield, which showed 1H NMR and 13C NMR spectra identical to those reported for the natural product.9a–c Using the optimized procedures described herein, ent-(+)-quadrigemine C (2), [α]23D +70 (c = 0.12, CHCl3) and [α]23D +19 (c = 0.15, EtOH), as well as the two meso-quadrigemine congeners 3 and 4 were prepared in an identical fashion (see summary in Table 3).
Table 3.
Yield of the final two steps in the synthesis of [2+2] quadrigemines and their optical rotations.
| Starting Material | Product | Yield (over 2 steps) | [α]Da |
|---|---|---|---|
|
| |||
| 12 | 1 (R,S,R,R)-quadrigemine C | 22–24% | −30 (−67)b |
| ent-12 | 2 (S,R,S,S)-ent-quadrigemine C | 24% | +20 (+70)b |
| 13 | major meso-quadrigeminec,e | 40% | – |
| 14 | minor meso-quadrigemined,e | 16% | – |
| 20 | 7 (R,R,R,R) | 23% | +277 |
| 21 | 6 (S,R,R,R) | 29% | +140 |
| 22 | 5 (S,R,R,S) | 29% | +231 |
[α]D taken in EtOH.
[α]D taken in CHCl3.
The dodecacyclic product resulting from transformation of the major meso Heck product 13;
The dodecacyclic product resulting from transformation of the minor meso Heck product 14;
The relative configuration could not be confirmed, the product is either meso quadrigemine 3 or 4.
2.3 Enantioselective total synthesis of [2+2] quadrigemines having a C2-symmetric chimonanthine core
The starting material for our studies in this series was (+)-chimonanthine (17), which we prepared in gram quantities from α-methoxycarbonyl-L-tryptophan methyl ester using the 8-step biomimetic sequence developed by Movassaghi and co-workers.14c The necessary C2-symmetric diiodide 18 was synthesized, as previously optimized in the meso-chimmonanthine series, by di-Boc protection of the aniline nitrogens, di-ortho-iodination, and removal of the Boc groups to provide C2-symmetric diiodide 18 in 72% yield. The di-Stille cross coupling of diiodide 18 with stannane 8 using the recently optimized conditions provided C2-symmetric dibutenanilide 19 in 98% yield.
We turned to examine the double enantioselective Heck cyclization in the C2-symmetric series, wherein the chiral substrate would influence diastereoselectivity.31,35 To examine the inherent substrate bias, the double enantioselective Heck cyclization of ditriflate 19 was carried out initially using rac-tol-BINAP, which resulted in modest substrate control to give the C2- and C1-symmetric dioxindole products 20 and 21 in a 1.0:2.3 ratio. Utilizing the catalyst generated from 0.5 equiv Pd(OAc)2, 1.0 equiv of (R)-tol-BINAP and 4 equiv of 1,2,2,6,6-pentamethylpiperidine (PMP) at 80 °C, various solvents were evaluated. A decrease in solvent polarity from N-methylpyrrolidinone (NMP) to THF resulted in a decrease in diastereoselectivity (entries 2–4, Table 2). To some surprise, diastereoselection was inverted in toluene, giving the C1-symmetric stereoisomer 21 as the major product. The combined yield of the decacyclic dioxindole products 20 and 21 produced in NMP was lower (58%) than we had anticipated. Analysis of the crude product mixture by mass spectrometry identified the formation of byproducts in which one of the pyrrolidinoindoline nitrogens had been acetylated. This product is a likely culprit of the decreased selectivity and undoubtedly arose from the formation of acetic anhydride during the reduction of Pd(OAc)2 by the phosphine ligand.36 Inclusion of 10 equiv of N-methyl-p-anisidine as a scavenger for Ac2O diminished formation of the acetylated byproducts, providing the C2-symmetric dioxindole 20 and its C1-symmetric stereoisomer 21 in 4.3:1 ratio and 79% combined yield. Identical Heck cyclization of 19 using Pd-(S)-tol-BINAP afforded C2-symmetric (S,R,R,S) dioxindole 22 and dioxindole 21 in a 2:1 ratio and 81% yield. The diastereomeric dioxindole products products were separated by silica gel chromatography and subsequently processed independently to their respective [2+2] quadrigemines.
Table 2.
Diastereoselective double Heck cyclization in the C2-symmetric series.
| Entry | Conditionsa | Solvent | de ratio (20:21; C2:C1)b |
|---|---|---|---|
|
| |||
| 1 | rac-tol-BINAP | NMP | 1.0:2.3 |
| 2 | (R)-tol-BINAP | NMP | 4.3:1.0 |
| 3 | (R)-tol-BINAP | MeCN | 3.0:1.0 |
| 4 | (R)-tol-BINAP | THF | 2.5:1.0 |
| 5 | (R)-tol-BINAP | PhMe | 1.0:2.3 |
50 mol% Pd(OAc)2, 100 mol% phosphine ligand, 4.0 equiv 1,2,2,6,6-pentamethylpiperidine (PMP), 80 °C;
ratio determined by HPLC
Utilizing the procedures developed during our optimized total synthesis of quadrigemine C (1), pentacyclic dioxindole products 20–22 were hydrogenated (1000 psi) with Pd(OH)2/C in EtOH at 80 °C. Subsequent exposure of the resulting tetrahydro products to a large excess of sodium (50 equiv) in THF/t-BuOH/NH3 at −78 °C for 20 min, followed by quenching with NH4Cl provided the respective [2+2] quadrigemine products 5–7, in 23–29% yield for the final two steps. The yields of the final steps in the total syntheses of the quadrigemine stereoisomers prepared in this study and the optical rotations at the sodium D line of the quadrigemine products are summarized in Table 3. The CD spectra of synthetic quadrigemines 1, 2, 5–7 are shown in Fig. 4.
Figure 4.

Circular dichroism of [2+2] quadrigeminesa
aall data taken ~2.9 × 10−4 M in EtOH
The optical rotation data and 13C NMR spectra for the synthetic quadrigemines 5–7 confirm that these C2-symmetic [2+2] quadrigemines are not identical to natural quadrigemines A and E. 37 Moreover, HPLC comparison of these products with a crude isolate of Psychotria muscosa showed that these synthetic quadrigemines were not identical to the structurally uncharacterized higher-order polypyrrolidindoline alkaloids identified in this sample by Verotta and coworkers. 38,39 These findings suggest that quadrigemines A and E could be identical to quadrigemine C.
2.4 Antitumor evaluation
A diversity of biological activities–antiviral, antibacterial, antifungal, and anticandidal–are described for quadrigemine alkaloids.1c In addition, selected quadrigemines are reported to be analgesics,31,38,40 antagonists of the somatostain receptor (SRIF),9b inhibitors of human platelet aggregation,41 and to display cytotoxic activity against solid and blood tumors.42 To gain some insight into the relationship between the three-dimensional structures of higher-order polypyrrolidinoindoline alkaloids and their antitumor activity, the in vitro cytotoxicities of the 2+2 quadrigemines prepared in this study, and several additional polypyrrolindoline alkaloids prepared in our laboratories (depicted in Figure 5), were determined against two invasive cancer cell lines: DU145 (human prostate cancer) and A2058 (human melanoma). Several trends emerge from the cytotoxicity data summarized in Table 4: (a) In general, cytotoxicity increases as a function of molecular weight (increasing number of pyrrolidinoindoline units); only one dodecacyclic alkaloid (the minor meso-quadrigemine 4, entry 3) was less active than the nonacyclic alkaloids (entries 7–11). (b) Relative configuration of higher-order polypyrrolidinoindolines makes only a minor contribution to cytotoxicity. For example, (−)-quadrigemine C is only 2–4 fold more active than the other [2+2] quadrigemine alkaloids. (c) (−)-Psycholeine (16), the isomer of (−)-quadrigemine C having a calycanthine core, showed little cytotoxicity (entry 7). The first two of these trends are in accord with cytotoxicity data reported previously for a few cyclotryptamine alkaloids.42
Figure 5.
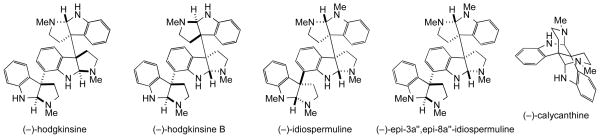
Additional pyrrolidinoinoline alkaloids.
Table 4.
Cytotoxic activity against prostate cancer (DU145) and melanoma (A2508) cell lines (IC50).a
| Entry | Compound | DU145 | A2058 |
|---|---|---|---|
|
| |||
| 1 | (−)-quadrigemine C (1) | 2.2 μM | 1.7 μM |
| 2 | meso-quadrigeminea,c | 4 μM | 5 μM |
| 3 | meso-quadrigemineb,c | >10 μM | >10 μM |
| 4 | (S,R,R,S)-quadrigemine (5) | 8.8 μM | 4.1 μM |
| 5 | (S,R,R,R)-quadrigemine (6) | 4.7 μM | 3.5 μM |
| 6 | (R,R,R,R)-quadrigemine (7) | 5.0 μM | 4.1 μM |
| 7 | (−)-psycholeine (16) | >10 μM | >10 μM |
| 8 | (−)-hodgkinsine | 7.2 μM | 7.2 μM |
| 9 | (−)-hodgkinsine B | 8.0 μM | 8.1 μM |
| 10 | (−)idiospermuline | >10 μM | >10 μM |
| 11 | (−)-epi-idiospermuline | >10 μM | >10 μM |
| 12 | meso-chimonanthine (9) | >10 μM | >10 μM |
| 13 | (−)-chimonanthine (ent-17) | >10 μM | >10 μM |
| 14 | (−)-calycanthine | >10 μM | >10 μM |
The dodecacyclic product resulting from transformation of the major meso Heck product 13;
The dodecacyclic product resulting from transformation of the minor meso Heck product 14;
The relative configuration could not be confirmed, the product is either meso quadrigemine 3 or 4.
3. Conclusion
The inside-out strategy (Scheme 1) that we first introduced in 2002 to synthesize (−)- (1) and (+)-quadrigemine C (2) is utilized in this investigation to prepare all potential stereoisomers of dodecacyclic quadrigemines having chimonanthine cores (Figure 2). As a prelude to these studies, two steps—the double Stille cross coupling to introduce the heavy atoms of the peripheral cyclotryptamine rings and the subsequent catalytic hydrogenation–were optimized to improve the yields and make these transformations more robust. The second-generation total synthesis of natural (−)-quadrigemine C (1) reported herein was accomplished in 10 steps and 2.2% overall yield from tryptamine. This short enantioselective total synthesis, and the other concise constructions of quadrigemine stereoisomers we report, are testaments to the power of two-directional synthesis strategies23 and the utility of catalytic enantioselective transformations–in this case intramolecular Heck reactions13–to stereoselectively generate structurally intricate polycyclic molecules. The total syntheses reported herein are rare examples of using two-directional strategies to stereoselectively elaborate cyclic molecules.43
This total synthesis study showed that the previously reported alkaloids referred to as quadrigemines A5 and E10 are not identical to any of the synthetic quadrigemine stereoisomers, and most likely are the same as quadrigemine C. In addition, investigations of the in vitro antitumor activities of products prepared in this study showed that the relative configuration of the 2+2 family of quadrigemines influences cytotoxicity in only a minor way.
4. Experimental section
Experimental procedures and characterization data for the preparation of compounds 1, 8, 11–15 have been reported previously.4
4.1 Enantioselective total synthesis of quadrigemine C (1) and ent-quadrigemine C (ent-1)
meso-Dibutenanilide 11
Diiodide 10 (262 mg, 0.44 mmol) and stannane 8 (1.14 g, 1.31 mmol) were combined in a round bottom flask and azeotroped to dryness with dry THF (3 × 6 mL). The mixture was placed under vacuum (1.0 mmHg) for 30 min and then pumped into an inert atmosphere (N2) drybox. A stirbar followed by Pd(PPh3)444 (252 mg, 0.219 mmol), LiCl (111 mg, 2.60 mmol), and CuCl (216 mg, 2.19 mmol) were added to the flask and the mixture was suspended in dry DMSO (15 mL). The flask was capped and stirred at room temperature in the drybox for 20 h. After removing the flask from the drybox, the black solution was partitioned between 5% v/v aqueous solution of NH4OH (20 mL) and EtOAc (30 mL) and the aqueous phase was extracted with EtOAc (3 × 15 mL). The combined organic layers were washed consecutively with water and brine. The organic layer was dried over MgSO4, filtered, and concentrated under reduced pressure. The dark residue was purified by flash column chromatography (10 %KF/SiO245 100% CH2Cl2 → 10:1 CH2Cl2/MeOH; product 11 (635 mg, 96%) elutes with 3–5% MeOH in CH2Cl2 as a brown foam, which showed 1H and 13C NMR spectra identical to those reported.4
Double enantioselective Heck cyclization of meso-dibutenanalide 11
A sample of ditriflate 11 (150 mg, 0.100 mmol) was azeotroped to dryness in benzene (3 × 2 mL) in a sealable Schlenk tube and placed under vacuum (1.0 mm) for 1 h. To the Schlenk flask was added a stirbar, Pd(OAc)2 (23 mg, 0.10 mmol), and (R)-tol-BINAP (0.140 g, 0.200 mmol). The flask was evacuated and backfilled with N2 (3 ×). Dry MeCN (2 mL) and 1,2,2,6,6-pentamethylpiperidine50 (73 μL, 0.40 mmol) were added and the reaction mixture was degassed using the freeze–pump–thaw technique (3 cycles, liquid N2 cooling bath, 0.1 mmHg, backfill with N2). The heterogeneous brown-red mixture was then heated to 80 °C for 16 h. After cooling to room temperature, the deep red solution was partitioned between a 20% w/w aqueous solution of NaCN (10 mL) and EtOAc (10 mL). The aqueous layer was extracted with EtOAc (3 × 20 mL). The combined organic layers were washed with water and brine. The organic layer was dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by column chromatography (SiO2: 100:0 CH2Cl2 → 97:3 CH2Cl2/MeOH → 94:5:1 CH2Cl2/MeOH/NH4OH → 89:10:1 CH2Cl2/MeOH/NH4OH) to provide a mixture of 12, 13, and 14 in a 9:2:1 ratio (ratio determined by reverse-phase HPLC analysis). The three diastereomers could be further purified by preparative reverse-phase HPLC (Phenomenex Gemini-NX, 250 × 21.2 mm), 80:20 MeCN-H2O (1% NH4OH), 25 mL/min, UV detection at 254 nm; to afford 74 mg (62%) of 12 (rt = 30.9 min), 17 mg (14%) of 13 (rt = 37.6 min), and 8 mg (7%) of 14 (rt = 23.7 min) as colorless foams.
(R,R,S,R)-Isomer 12.4
[α]28D −60, [α]28577 −62, [α]28546 −72, [α]28435 −153, [α]28405 −207 (c = 0.25, CH2Cl2).
Major meso-isomer 13
1H NMR (500 MHz, (CD3)2SO, 376K) δ 7.49 (d, J = 7.5 Hz, 4H), 7.35 (d, J = 7.8 Hz, 6H), 7.34–7.31 (m, 4H), 7.27–7.23 (m, 8H), 7.06–7.01 (m, 4H), 6.96 (d, J = 7.2 Hz, 2H), 6.79 (d, J = 7.8 Hz, 2H), 6.65 (d, J = 14.3 Hz, 2H), 6.41 (br s, 2H), 5.32 (d, J = 14.2 Hz, 2H), 4.97 (d, J = 15.4 Hz, 2H), 4.90 (d, J = 15.5 Hz, 2H), 4.25 (br s, 2H), 4.09 (s, 2H), 2.85 (s, 6H), 2.38 (s, 6H), 2.30–2.24 (m, 4H), 1.98–1.94 (m, 2H), 1.78–1.73 (m, 2H), 1.71 (s, 6H); 13C NMR (125 MHz, (CD3)2SO, 376 K) δ 175.9 (C), 148.9 (C), 143.3 (C), 141.3 (C), 135.7 (C), 133.8 (C), 133.7 (C), 130.6 (C), 129.7 (CH), 129.3 (CH), 128.0 (CH), 127.6 (CH), 126.92 (CH), 126.88 (CH), 126.2 (CH), 126.0 (CH), 124.0 (CH), 122.7 (C), 122.0 (CH), 118.6 (C), 116.8 (CH), 110.0 (CH), 108.9 (CH), 82.3 (CH), 61.7 (C), 55.6 (C), 50.8 (CH2), 42.8 (CH2), 36.0 (CH2), 33.8 (CH3), 31.9 (CH3), 20.2 (CH3); IR (thin film) 3358, 2934, 1706, 1610, 1467, 1355, 1162, 745 cm−1; LRMS-ESI (m/z) [M + H]+ calcd for C72H70N8O6S2H 1207.5; found, 1207.5.
Minor meso-isomer 14
1H NMR (500 MHz, (CD3)2SO, 376K) δ 7.42 (d, J = 7.8 Hz, 4H), 7.32–7.27 (m, 6H), 7.22 (br s, 14 H), 6.97 (br s, 4H), 6.94 (d, J = 7.6 Hz, 2H), 6.70 (br s, 2H), 6.58 (d, J = 14.3 Hz, 2H), 5.37 (d, J = 14.2 Hz, 2H), 4.98 (d, J = 15.8 Hz, 2H), 4.75 (d, J = 15.8 Hz, 2H), 2.90 (br s, 2H), 2.85 (s, 6H), 2.69–2.59 (m, 2H), 2.46 (s, 6H), 2.38–2.35 (m, 2H), 2.34 (s, 6H), 2.16–2.09 (m, 2H), 2.04 (br s, 4H), 1.82–1.79 (m, 2H); 13C NMR (125 MHz, (CD3)2SO, 376 K) δ 175.9 (C), 148.9 (C), 143.3 (C), 141.3 (C), 135.7 (C), 133.7 (C), 130.6 (C), 129.7 (CH), 129.3 (CH), 129.2 (CH), 128.0 (CH), 127.6 (CH), 126.9 (CH), 126.5 (CH), 126.2 (CH), 125.9 (CH), 124.0 (CH), 122.7 (CH), 122.1 (CH), 118.6 (C), 116.7 (C), 110.1 (CH), 108.9 (CH), 82.3 (CH), 61.7 (C), 55.6 (C), 50.8 (CH2), 42.8 (CH2), 36.0 (CH3), 33.8 (CH2), 31.8 (CH3), 20.3 (CH3); IR (thin film) 3405, 2934, 1710, 1610, 1455, 1359, 1162, 748 cm−1; LRMS-ESI (m/z) [M + H]+ calcd for C72H70N8O6S2H 1207.5; found, 1207.5.
(S,R,S,S)-Isomer (ent-12)
Carrying out the double Heck cyclization in similar fashion using (S)-tol-BINAP and 77 mg (0.051 mmol) of meso-dibutenanalide 11 gave as the major product dioxindole ent-12 (51 mg, 83%): 1H NMR (500 MHz, (CD3)2SO, 376K)46 δ 7.49–7.45 (m, 4H), 7.36–7.30 (m, 8H), 7.27–7.19 (m, 10H), 7.11 (br d, J = 6.4 Hz, 1H), 7.06–6.97 (m, 5H), 6.90 (br d, J = 7.7 Hz, 1H), 6.74 (br s, 2H), 6.65 (d, J = 15.0 Hz, 1H), 6.61 (d, J = 15.8 Hz, 1H), 6.47 (br s, 1H), 6.34 (br s, 1H), 5.42 (d, J = 14.3 Hz, 1H), 5.29 (d, J = 14.3 Hz, 1H), 5.05 (d, J = 15.8 Hz, 1H), 4.98 (d, J = 15.5 Hz, 1H), 4.85 (d, J = 15.6 Hz, 1H), 4.78 (d, J = 15.8 Hz, 1H), 4.40–4.10 (br s, 3H), 2.88 (s, 3H), 2.83 (s, 3H), 2.66 (br t, J = 6.7 Hz, 1H), 2.52–2.48 (m, 2H), 2.38 (s, 3H), 2.36 (s, 3H), 2.35–2.30 (m, 1H), 2.29–2.21 (m, 1H), 2.14 (s, 3H), 2.07–1.89 (m, 1H), 1.86–1.82 (m, 1H), 1.78–1.71 (m, 1H), 1.72 (s, 3H); 13C NMR (125 MHz, (CD3)2SO, 376 K) δ 176.2 (C), 176.0 (C), 150.0 (C), 149.0 (C), 143.4 (C), 143.3 (C), 141.3 (C), 141.2 (C), 135.66 (C), 135.64 (C), 134.5 (C), 133.9 (C), 133.7 (C), 133.4 (br, C), 130.5 (CH), 130.4 (C), 129.8 (CH), 129.5 (CH), 129.24 (2 peaks, CH), 127.93 (CH), 127.92 (CH), 127.68 (CH), 127.65 (CH), 127.58 (CH), 127.0 (CH), 126.8 (CH), 126.7 (CH), 126.4 (CH), 126.1 (CH), 126.0 (CH), 125.95 (CH), 124.5 (CH), 124.2 (CH), 122.8 (CH), 122.6 (CH), 122.1 (C), 121.9 (C), 118.24 (br, 2 peaks, C), 117.3 (CH), 116.6 (CH), 110.0 (CH), 109.8 (CH), 108.99 (CH), 108.95 (CH), 82.2 (CH), 62.6 (C), 61.8 (C), 55.7 (C), 50.98 (CH2), 50.97 (CH2), 42.8 (CH2), 42.7 (CH2), 35.8 (br, 2 peaks, CH2), 34.3 (CH3), 33.8 (CH3), 31.79 (CH3), 31.75 (CH3), 20.25 (CH3), 20.23 (CH3); IR (thin film) 3354, 2930, 1710, 1610, 1359, 1162, 745 cm−1; LRMS-ESI (m/z) [M + H]+ calcd for C72H70N8O6S2H 1207.5; found, 1207.5; [α]23D +68, [α]23577 +69, [α]23546 +79, [α]23435 +157 (c = 0.33, CH2Cl2).
(−)-Quadrigemine C (1)
The procedure for hydrogenation of the enesulfonamide side chains and reductive cyclization for the ultimate conversion of tetrahydro intermediate to (−)-quadrigemine C that is reported herein (see the optimized general procedures described for the preparation of (R,R,R,R)-quadrigemine 7) has been found to be more reproducible than the procedure described previously.4 Using the optimized general procedure for hydrogenation of the enesulfonamide side chains, tetrahydro derivative 15 was prepared in >90% yield: [α]28D −144, [α]28577 −154, [α]28546 −177, [α]28435 −369, [α]28405 −492 (c = 0.10, CH2Cl2). Reduction of this intermediate with Na/NH3/t-BuOH gave (−)-quadrigemine (C) (8.3 mg, 24% overall yield from 12), [α]23D −67 (c = 0.2, CHCl3), HRMS-ESI (m/z) [M + H]+ calcd for C44H50N8H 691.4236; found, 691.4232; NMR and IR and optical rotation data were identical to those reported previously.4
ent-(+)-Quadrigimine C (2)
Following the optimized general procedure for hydrogenation of the enesulfonamide side chains, 104 mg of ent-12 was converted to tetrahydro derivative ent-15. This product was then directly subjected to the optimized reductive cyclization conditions to afford ent-(+)-quadrigemine C (2) (14 mg, 24%); IR (thin film) 3271, 3054, 2929, 2854, 2791, 1604, 1481, 1452 cm−1; HRMS-ESI (m/z) [M + H]+ calcd for C44H50N8H 691.4236; found, 691.4226; [α]23D +20, [α]23577 +17, [α]23546 +18, [α]23435 +48 (c = 0.15, EtOH); [α]23D +70, [α]23577 +67, [α]23546 +60, [α]23435 +110 (c = 0.12, CHCl3).
4.2 Synthesis of meso-[2+2] quadrigemines
Tetrahydro derivative of the major meso-dioxindole isomer 13
Following the optimized general procedure for hydrogenation of the enesulfonamide side chains, tetrahydro-13 (28.4 mg, 0.024 mmol, 98% yield) was obtained as a colorless solid from 29.6 mg of 13. 1H NMR (500 MHz, (CD3)2SO, 396K) δ 7.50 (d, J = 8.1 Hz, 4H), 7.33 (d, J = 7.6 Hz, 8H), 7.29–7.20 (m, 10H), 7.13 (d, J = 6.9 Hz, 2H), 7.09 (t, J = 7.1 Hz, 2H), 7.00 (d, J = 7.7 Hz, 2H), 6.85 (d, J = 7.7 Hz, 2H), 6.50 (br s, 2H), 6.43 (br s, 2H), 4.96 (d, J = 15.5 Hz, 2H), 4.85 (d, J = 15.5 Hz, 2H), 4.78 (s, 2H), 4.29 (s, 2H), 2.96 (dt, J = 5.0, 12.6 Hz, 2H), 2.79 (br s, 2H), 2.70–2.66 (m, 2H), 2.61 (s, 6H), 2.53–2.51 (m, 2H), 2.44–2.40 (m, 2H), 2.38 (s, 6H), 2.35–2.25 (m, 2H), 2.10–2.07 (m, 2H), 1.90 (s, 6H), 1.80–1.77 (m, 2H); 13C NMR (125 MHz, (CD3)2SO, 396K) δ 176.8 (C), 149.3 (C), 142.3 (C), 141.8 (C), 135.5 (C), 134.8 (C), 130.0 (C), 128.8 (CH), 127.8 (CH), 127.5 (CH), 127.1 (C), 126.9 (CH), 126.7 (CH), 126.1 (CH), 125.2 (CH), 123.8 (CH), 122.6 (CH), 121.9 (CH), 117.7 (C), 116.9 (CH), 108.7 (CH), 82.6 (CH), 61.7 (C), 53.2 (CH2), 50.9 (C), 45.1 (CH2), 42.9 (CH2), 35.9 (CH2), 34.1 (CH3), 33.8 (CH3), 31.6 (CH2), 20.0 (CH3); IR (thin film) 3336, 3060, 2928, 2865, 2791, 1697, 1610, 1487, 1454, 1342 cm−1; LRMS-ESI (m/z) [M + H]+ calcd for C72H74N8O6S2H 1211.5; found, 1211.5.
Tetrahydro derivative of the minor meso-dioxindole isomer 14
Following the optimized general procedure for hydrogenation of the enesulfonamide side chains, tetrahydro-14 (28.2 mg, 0.024 mmol, 94% yield) was obtained as a colorless solid from 31.2 mg of dioxindole 14. 1H NMR (500 MHz, (CD3)2SO, 396K) δ 7.45 (d, J = 8.2 Hz, 4H), 7.31 (d, J = 8.4 Hz, 4H), 7.27–7.21 (m, 8H), 7.18 (d, J = 7.6 Hz, 2H), 7.07 (t, J = 7.4 Hz, 2H), 6.97(d, J = 7.8 Hz, 2H), 6.71 (br d, J = 7.2 Hz, 2H), 4.89 (d, J = 15.7 Hz, 2H), 4.78 (d, J = 15.7 Hz, 2H), 4.27 (br s, 2H), 2.95–2.83 (m, 4H), 2.80 (s, 10H), 2.70–2.67 (m, 4H), 2.66–2.63 (m, 2H), 2.61 (s, 6H), 2.41–2.33 (m, 10H), 2.27–2.23 (m, 2H), 2.21 (s, 4H), 1.86 (dd, J = 4.7 & 11.4 Hz, 2H); 13C NMR (125 MHz, (CD3)2SO, 396K) δ 177.0 (C), 142.3 (C), 141.9(C), 135.6 (C), 134.6 (C), 129.5 (C), 129.8 (CH), 127.7 (CH), 127.2 (CH), 126.9 (C), 126.6 (CH), 126.5 (CH), 126.1 (CH), 125.2 (C), 124.3 (CH), 122.5 (CH), 121.7 (CH), 117.43 (C), 108.8 (CH), 82.0 (CH), 62.1 (C), 53.6 (CH2), 50.9 (C), 45.4 (CH2), 42.8 (CH2), 35.3 (CH2), 34.3 (CH3), 33.9 (CH3), 30.6 (CH2), 20.0 (CH3); IR (thin film) 3328, 3060, 2926, 2851, 2789, 1695, 1611, 1488, 1466, 1343 cm−1; LRMS-ESI (m/z) [M + H]+ calcd for C72H74N8O6S2H 1211.5; found, 1211.5.
Major meso-quadrigemine isomer
Following the optimized general reductive cyclization procedure, 21 mg (0.017 mmol) of tetrahydro-13 was transformed to quadrigemine derived from the major meso-dioxindole Heck product 13. The solid was purified by preparative reverse-phase HPLC (Zorbax Extend C18, 100 × 21.2 mm, 80:20 MeOH-H2O (1% NH4OH), 16 mL/min, UV detection at 254 nm) to afford 4.8 mg (40%) of the major meso-quadrigemine isomer (NOTE: relative configuration was not established; this product is either meso quadrigemine 3 or 4.) (rt = 9.7 min) as a colorless solid. 1H NMR (500 MHz, (CD3)2SO, 376K) δ 7.43–7.22 (m, 7H), 6.92 (d, J = 7.5 Hz, 3H), 6.54 (d, J – 16.7 Hz, 2H), 6.17 (s, 2H), 5.65 (s, 3H), 5.13 (s, 1H), 4.96 (br s, 1H), 4.27 (d, J = 6.6 Hz, 2H), 3.22 (s, 2H), 3.76 (br s, 7H), 2.57 (d, J = 6.9 Hz, 2H), 2.50–2.11 (m, 11H), 1.61 (s, 3H), 1.32–1.28 (m, 4H); 13C NMR (125 MHz, (CD3)2SO, 376K) δ 147.0 (C), 146.7 (C), 140.9 (CH), 134.3 (C), 129.1 (CH), 128.2 (CH), 127.0 (CH), 120.5 (CH), 120.1 (C), 118.7 (CH), 107.4 (CH), 107.1 (CH), 100.5 (C), 69.0 (CH), 60.6 (C), 58.0 (C), 54.0 (CH2), 48.0 (CH2), 37.7 (CH2), 37.2 (CH2), 28.2 (CH3), 22.9 (CH3); IR (thin film) 3365, 2923, 2851 1658, 1605, 1451, 1247, 1158, 1035, 744 cm−1; HRMS-ESI (m/z) [M + H]+ calcd for C44H50N8H 691.4236; found, 691.4244.
Minor meso-Quadrigemine isomer
Following the optimized general reductive cyclization procedure, 45 mg (0.037 mmol) of tetrahydro-14 was converted the quadrigemine derived from the minor meso-dioxindole Heck product 14. The resulting solid was purified by preparative reverse-phase HPLC (Zorbax Extend C18, 100 × 21.2 mm, 80:20 MeOH-H2O (1% NH4OH), 10 mL/min, UV detection at 254 nm) to afford 4.0 mg (16%) of minor meso-quadrigemine isomer (NOTE: relative configuration was not established; this product is either meso quadrigemine 3 or 4.) (rt = 70.7 min) as a colorless solid. 1H NMR (500 MHz, (CD3)2SO, 376K) δ 6.98–6.93 (m, 4H), 6.60 (app t, J = 7.1 Hz, 2H), 6.54 (d, J = 7.8 Hz, 2H), 6.35 (br s, 2H), 5.88 (br s, 2H), 5.71 (br s, 2H), 4.81 (s, 2H), 4.55 (br s, 2H), 3.55 (s, 2H), 2.96 (6H, br s), 2.66–2.58 (m, 4H), 2.50 (s, 4H), 2.08 (br s, 10H), 1.91–1.89 (m, 2H), 1.82–1.81 (m, 2H), 1.27 (s, 2H); 13C NMR (125 MHz, (CD3)2SO, 376K) δ 150.9 (C), 149.3 (C), 133.0 (C) 131.8 (C), 126.8 (CH), 124.4 (CH), 123.9 (CH), 122.6 (C), 121.5 (CH), 116.7 (CH), 115.1 (CH), 107.4 (CH), 85.1 (CH), 82.3 (CH), 61.8 (C), 59.6 (C), 51.1 (CH2), 37.8 (CH2), 36.3 (CH2), 34.8 (CH3), 34.3 (CH3); HRMS-ESI (m/z) [M + H]+ calcd for C44H50N8H 691.4236; found, 691.4248; IR (thin film) 3271, 2926, 2854, 2791, 1674, 1604, 1486, 1448, 1253, 1246, 1154, 1032, 743 cm−1; HRMS-ESI (m/z) [M + H]+ calcd for C44H50N8H 691.4236; found, 691.4237.
4.3 Enantioselective total synthesis of [2+2] quadrigemines having a C2-symmetric core
(R,R)-1,1′-Dimethyl-1,2,3,8,8a,1′,2′,3′,8,8a′-octahydro-1H,1H′-[3a,3a′]bi[pyrrolo[2,3-b]indolyl]-8,8′-dicarboxylic acid di-tert-butyl ester (E1)
Following the procedure employed in the meso series,4 a THF solution of sodium bis(trimethylsilyl)amide (15.3 mL, 30.6 mmol, 2 M) was added dropwise via syringe pump over 3 h to a solution of (+)-chimonanthine (2.95 g, 8.51 mmol),14c di-tert-butyldicarbonate (Boc2O) (8.16 g, 37.4 mmol), and THF (130 mL) at −78 °C. Upon completion of the addition the solution was maintained for 30 min at −78 °C and then partitioned between saturated aqueous NH4Cl (50 mL) and EtOAc (50 mL). The layers were separated and the aqueous phase was extracted with EtOAc (3 × 50 mL). The combined organic layers were washed with brine (50 mL), dried over MgSO4, filtered, and concentrated under reduced pressure. The residue was purified by flash column chromatography (2:98 → 5:95 MeOH/EtOAc) to yield the di-Boc derivative E1 as a colorless foam (4.07 g, 87%): 1H NMR (600 MHz, (CD3)2SO, 373K) δ 7.29 (d, J = 7.9 Hz, 2H), 7.04 (d, J = 7.5 Hz, 2H), 6.98 (dd, J = 7.7, 0.9 Hz, 2H), 6.74 (t, J = 7.4 Hz, 2H), 2.71–2.67 (m, 2H), 2.47–2.44 (m, 2H), 2.44 (s, 6H), 2.38–2.30 (m, 2H), 2.10–2.06 (m, 2H), 1.57 (s, 18H); 13C NMR (150 MHz, (CD3)2SO, 373K) δ 152.3 (C), 143.0 (C), 135.1 (C), 128.1 (CH), 123.1 (CH), 122.4 (CH), 115.4 (CH), 85.8 (CH), 81.1 (C), 61.1 (C), 52.9 (CH2), 37.6 (CH3), 34.4 (CH2), 28.5 (CH3); IR (thin film): 2974, 2942, 2793, 1696, 1483, 1384, 1366, 1164 cm−1; HRMS-ESI (m/z) [M + H]+ calcd for C32H42O4N4H 547.3284; found, 547.3267; [α]23D +163, [α]23577 +168, [α]23546 +191, [α]23435 +355 (c = 0.66, CH2Cl2).
(R,R)-7,7′-diiodo-1,1′-dimethyl-1,2,3,8a,1′,2′,3′,8a′-octahydro-[3a,3a′]bi[pyrrolo[2,3-b]indolyl]-8,8′-dicarboxylic acid di-tert-butyl ester (E2)
Following the procedure employed in the meso series,4 a cyclohexane solution of s-BuLi (23.5 mL, 23.0 mmol, 0.98 M) was added dropwise over 1.5 h maintaining an internal temperature below −70 °C to a solution of dicarbamate E1 (2.79 g, 5.10 mmol), N,N,N′,N′-tetramethylethylenediamine 47 (TMEDA) (4.55 mL, 30.1 mmol) and Et2O (51 mL) at −78 °C. The solution was aged at −78 °C for 45 min. Then a solution of diiodoethane (14.5 g, 51.0 mmol) and Et2O (51 mL) was added dropwise by syringe. The resulting solution was maintained at −78 °C for 10 min, then warmed to 0 °C and maintained at 0 °C for 3 h. The mixture was partitioned between saturated aqueous Na2S2O4 (50 mL), saturated aqueous NaHCO3 (50 mL), and EtOAc (50 mL). The phases were separated and the aqueous layer was extracted with EtOAc (3 × 50 mL). The combined organic layers were washed with brine (100 mL) dried over MgSO4, filtered, and concentrated under reduced pressure. The residue was purified by flash column chromatography (2:1→0:100 hexane/EtOAc) to provide diiodide E2 as a beige solid (3.11 g, 84%): 1H NMR (600 MHz, (CD3)2SO, 373K) δ 7.62 (d, J = 7.9 Hz, 2H), 7.06 (d, J = 7.5 Hz, 2H), 6.74 (t, J = 7.7 Hz, 2H), 5.32 (s, 2H), 2.65–2.62 (m, 2H), 2.21–2.18 (m, 2H), 2.05–2.02 (m, 2H), 1.79–1.75 (m, 2H), 1.59 (s, 18H); 13C NMR (150 MHz, (CD3)2SO, 373K) δ 152.2 (C), 146.6 (C), 139.40 (CH), 126.3 (CH), 124.1(CH), 88.9 (CH), 87.3 (C), 85.8 (C), 82.0 (C), 61.8 (C), 51.9 (CH2), 36.4 (CH3), 35.7 (CH2), 28.4 (CH3); mp = 197–199 °C; IR (thin film): 2975, 2796, 1702, 1442, 1367, 1352, 1299, 1245, 1158 cm−1; HRMS-ESI (m/z) [M + H]+ calcd for C32H40N4O4I2H 799.1218; found, 799.1224; [α]23D +186, [α]23577 +195, [α]23546 +225, [α]23435 +399, [α]23405 442 (c = 0.63, CH2Cl2).
(R,R)-7,7′-diiodo-1,1′-dimethyl-1,2,3,8,8a,1′,2′,3′,8,8a′-octahydro-1H,1H′-[3a,3a′]bi[pyrrolo[2,3-b]indolyl] (18)
Following the procedure employed in the meso series,4 neat TMSOTf (4.44 mL, 24.5 mmol) was added dropwise to a solution of diiodide E2 (4.45 g, 5.57 mmol) and CH2Cl2 (140 mL). The flask was left open to the air so that adventitious H2O would create a small amount of triflic acid. After 3 h, the solution was partitioned between saturated aqueous NaHCO3 (50 mL) and CH2Cl2 (100 mL). The phases were separated and the aqueous phase was extracted with CH2Cl2 (3 × 150 mL). The combined organic layers were dried over MgSO4, filtered, and concentrated under reduced pressure. The residue was purified by flash column chromatography with a gradient elution (9:1:0 → 9:1:0.1 CH2Cl2/MeOH/Et3N) to yield 18 as a pale yellow foam (3.28 g, 99%): 1H NMR (500 MHz, (CD3)2SO, 376K) δ 7.32 (d, J = 7.9 Hz, 2H), 7.20 (d, J = 7.3 Hz, 2H), 6.37 (t, J = 7.6 Hz, 2H), 5.56 (s, 2H), 4.88 (br s, 2H), 3.04 (s, 1H), 2.78 (dt, J = 2.8, 7.5 Hz, 2H), 2.65 (br s, 3H), 2.61–2.55 (m, 2H), 2.48–2.45 (m, 2H), 2.01–1.97 (m, 2H); 13C NMR (125 MHz, (CD3)2SO, 376K) δ 152.3 (C), 135.1 (CH), 133.2 (C), 122.7 (CH), 118.1 (CH), 82.2 (CH), 72.8 (C), 64.3 (C), 50.7 (CH2), 35.2 (CH2), 35.1 (CH3); IR (thin film) 3402, 3062, 2845, 2789, 1596, 1470, 1246, 734 cm−1; HRMS-ESI (m/z) [M + H]+ calcd for C22H24N4I2H 599.0168; found, 599.0167; mp = 193–195 °C; [α]23D +363, [α]23577 +383, [α]23546 +445, [α]23435 +909, [α]23405 +1058 (c = 1.1, CH2Cl2).
(R,R)-Dibutenanalide 19
Diiodide 18 (0.300 g, 0.501 mmol) and stannane 8 (1.31 g, 1.50 mmol) were combined in a round bottom flask and azeotroped in dry THF (3 × 6 mL). The mixture was then placed under vacuum (1.0 mmHg) for 30 min and then pumped into an inert atmosphere (N2) drybox. A stirbar followed by Pd(PPh3)448 (0.290 g, 0.251 mmol), LiCl (0.128 g, 3.01 mmol), and CuCl (0.250 g, 2.51 mmol) were added to the flask and the mixture was suspended in dry DMSO (17 mL). The flask was capped and stirred at room temperature in the drybox for 20 h. Upon completion the flask was removed from the drybox and the black solution was partitioned between 5% v/v aqueous solution of NH4OH (40 mL) and EtOAc (50 mL) and the aqueous phase was extracted with EtOAc (3 × 30 mL). The combined organic layers were washed with water (3 × 100 mL) followed by brine (1 × 100 mL). The organic layer was dried over MgSO4, filtered, and concentrated under reduced pressure. The black residue was purified by flash column chromatography (10%KF/SiO245: 100:0 CH2Cl2 → 98:2 CH2Cl2/MeOH 94:5:1 → CH2Cl2/MeOH/NH4OH → 88:10:2 CH2Cl2/MeOH/NH4OH) to provide 19 (0.741g, 98%) as a brown foam: 1H NMR (500 MHz, CDCl3) δ complex due to the presence of multiple conformations on the NMR time-scale, see copy of spectra; 13C NMR (125 MHz, CDCl3) complex due to the presence of multiple conformations on the NMR time-scale, see copy of spectra (only major peaks listed) 134.22, 129.7, 128.8, 128.6, 128.1, 128.0, 127.6, 127.4, 117.1, 83.9, 49.3, 35.8, 29.7, 27.8, 26.8, 21.5, 17.5, 13.6; 19F NMR (376 MHz, CDCl3) −73.6; IR (thin film) 3415, 3063, 2863, 2791, 1649, 1494, 1455, 1421, 1339, 1207, 1162, 893, 736 cm−1; LRMS-ESI (m/z) [M + H]+ calcd for C74H72N8F6O12S4H 1507.4; found, 1507.4; [α]23D +158, [α]23577 +167, [α]23546 +192, [α]23435 +433, [α]23405 403 (c = 0.46, CH2Cl2).
Double enantioselective Heck cyclization of (R,R)-Dibutenanalide 19
Ditriflate 19 (0.200 g, 0.133 mmol) was azeotroped in benzene (3 × 2 mL) in a sealable Schlenk tube and placed under vacuum (1 mm) for 1 h. To the Schlenk flask was added a stirbar followed by Pd(OAc)2 (30 mg, 0.13 mmol), (R)-tol-BINAP (0.181 g, 0.266 mmol), and N-Me-p-anisidine (0.143 g, 1.33 mmol). The flask was evacuated and backfilled with N2 (3 times). The mixture was then suspended in 1-methyl-2-pyrrolidinone (4.4 mL)49 and 1,2,2,6,6-pentamethylpiperidine50 (97 μL, 0.53 mmol). The reaction mixture was degassed using the freeze–pump–thaw technique (3 cycles, liquid N2 cooling bath, 0.1 mmHg, backfill with N2). The heterogeneous brown-red mixture was heated to 80 °C for 16 h. After cooling to room temperature the deep red solution was partitioned between a 20% w/w aqueous solution of NaCN (20 mL) and EtOAc (20 mL). The aqueous layer was extracted with EtOAc (3 ×20 mL). The combined organic layers were washed with water (3 × 40 mL) followed by brine (1 × 20 mL). The organic layer was dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by column chromatography (SiO2: 100:0 CH2Cl2 → 97:3 CH2Cl2/MeOH → 94:5:1 CH2Cl2/MeOH/NH4OH → 89:10:1 CH2Cl2/MeOH/NH4OH) to provide eluting first C1-symmetric dioxindole 21 (17.0 mg, 11%) followed by C2-symmetric dioxindole 20 (74.0 mg, 46%) as tan foams: (R,R,R,R)-dioxindole 20: 1H NMR (500 MHz, (CD3)2SO, 376K) δ 7.47 (d, J = 8.3 Hz, 4H), 7.33 (d, J = 8.1 Hz, 4H), 7.31 (dd, J = 7.8, 1.1 Hz, 2H), 7.20 (dd, J = 6.0, 1.9 Hz, 4H), 7.13 (dd, J = 5.7, 1.9 Hz, 8H), 7.01 (d, J = 7.1 Hz, 2H), 6.95 (d, J = 7.8 Hz, 2H), 6.70 (br d, J = 7.2 Hz, 2H), 6.62 (d, J = 14.3 Hz, 2H), 6.41 (d, J = 7.8 Hz, 2H), 6.14 (t, J = 7.7 Hz, 2H), 5.45 (d, J = 14.3 Hz, 2H), 4.95 (d, J = 15.9 Hz, 2H), 4.90 (d, J = 15.9 Hz, 2H), 4.83 (br s, 4H), 2.90 (s, 6H), 2.63 (br m, 2H), 2.37 (s, 6H), 2.37–2.31 (m, 2H), 2.21 (br s, 6H), 1.80–1.77 (m, 2H); 13C NMR (125 MHz, (CD3)2SO, 376K) δ 176.3 (C), 149.3 (C), 143.3 (C), 141.4 (C), 135.7 (C), 135.0 (C), 133.9 (C), 130.4 (CH), 129.4 (CH), 129.2 (CH), 127.8 (CH), 126.5 (CH), 126.3 (CH), 125.9 (CH), 125.2 (CH), 124.7 (CH), 121.9 (CH), 121.7 (CH), 118.6 (C), 117.0 (CH), 109.8 (CH), 109.0 (CH), 82.8 (CH), 78.5 (C), 62.5 (C), 55.7 (C), 50.1 (CH2), 42.5 (CH2), 34.9 (CH2), 34.5 (CH3), 31.8 (CH3), 20.2 (CH3); IR (thin film) 3367, 3061, 3032, 2853, 2788, 1700, 1647, 1609, 1466, 1355, 1246, 1159, 743 cm−1; HRMS-ESI (m/z) [M + H]+ calcd for C72H70N8O6S2H 1207.4938; found, 1207.4950; [α]23D +28, [α]23577 +29, [α]23546 +34, [α]23435 +74 (c = 1.6, CH2Cl2). (S,R,R,R)-Dioxindole 21: 1H NMR (500 MHz, (CD3)2SO, 376K) δ 7.51 (d, J = 13.6 Hz, 2H), 7.48 (d, J = 13.6 Hz, 2H), 7.44–7.19 (overlapping multiplets, 16H), 7.11–7.03 (m, 6H), 6.98 (d, J = 7.9 Hz, 1H), 6.91 (d, J = 7.4 Hz, 1H), 6.74 (br d, J = 6.4 Hz, 1H), 6.63 (dd, J = 14.3, 3.7 Hz, 2H), 6.57 (br dd, J = 10.6, 7.9 Hz, 2H), 6.40 (t, J = 7.5 Hz, 1H), 6.28 (t, J = 7.1 Hz, 1H), 5.45 (d, J = 14.3 Hz, 1H), 5.34 (d, J = 14.3 Hz, 1H), 5.01 (d, J = 9.1 Hz, 1H), 4.97 (s, 2H), 4.85 (d, J = 15.8 Hz, 1H), 4.68 (br s, 1H), 4.44 (br s, 1H), 4.37 (br s, 1H), 2.92 (s, 3H), 2.90 (s, 3H), 2.65–2.58 (m, 1H), 2.48–2.41 (m, 1H), 2.40 (s, 3H), 2.39 (s, 3H), 2.27–2.26 (m, 2H), 2.22–2.19 (m, 1H), 2.18 (s, 3H), 2.02–1.98 (m, 1H), 1.82 (s, 3H), 1.77 (br dd, J = 11.9, 4.9 Hz, 1H), 1.69–1.66 (m, 1H); 13C NMR (125 MHz, (CD3)2SO, 376 K) δ 176.3 (C), 175.9 (C), 149.2 (C), 148.4 (C), 143.35 (C), 143.31 (C), 141.4 (C), 141.3 (C), 135.8 (C), 135.6 (C), 134.7 (C), 134.2 (C), 133.9 (C), 133.8 (C), 130.6 (C), 130.5 (C), 129.5 (CH), 129.4 (CH), 129.28 (CH), 129.26 (CH), 127.94 (CH), 127.91 (CH), 127.8 (CH), 127.7 (CH), 127.0 (CH), 126.9 (CH), 126.7 (CH), 126.5 (CH), 126.45 (CH), 126.0 (CH), 125.9 (CH), 125.7 (CH), 125.6 (CH), 124.5 (CH), 124.2 (CH), 122.3 (CH), 122.1 (CH), 121.9 (CH), 119.1 (C), 118.7 (C), 117.2 (CH), 116.7 (CH), 110.2 (CH), 110.0 (CH), 109.0 (CH), 108.9 (CH), 83.2 (CH), 83.0 (CH), 62.2 (C), 61.9 (C), 55.7 (C), 55.6 (C), 50.4 (CH2), 50.3 (CH2), 42.8 (CH2), 42.6 (CH2), 35.2 (CH2), 35.1 (CH2), 34.5 (CH3), 34.3 (CH3), 31.84 (CH3), 31.78 (CH3), 20.27 (CH3), 20.24 (CH3); IR (thin film) 3378, 3060, 2857, 2790, 1703, 1609, 1465, 1356, 1159, 742 cm−1; LRMS-ESI (m/z) [M + H]+ calcd for C72H70N8O6S2H 1207.5; found, 1207.5; [α]23D +116, [α]23577 +121, [α]23546 +142, [α]23435 +298, [α]23405 +276 (c = 0.80, CH2Cl2).
Following the same procedure utilizing (S)-tol-BINAP instead of (R)-tol-BINAP, dibutenanilide 19 (0.282 mg, 0.187 mmol) was converted into C2-symmetric dioxindole product 22 and C1-symmetric isomer 21. These products were purified by column chromatography (SiO2: 100:0 CHCl3 → 98:2 CHCl3/MeOH → 94:3:1 CHCl3/MeOH/NH4OH → 94:5:1 CHCl3/MeOH/NH4OH) to elute first C2-symmetric 22 (123 mg, 55%) followed by C1-symmetric 21 (87.0 mg, 39%) as tan foams. (S,R,R,S)-dioxindole 22: 1H NMR (500 MHz, (CD3)2SO, 376K) δ 7.50 (d, J = 8.2 Hz, 4H), 7.40 (t, J = 6.3 Hz, 8H), 7.32 (t, J = 6.9 Hz, 4H), 7.28 (dt, J = 1.9, 9.0 Hz, 4H), 7.07 (dd, 7.2, 14.8 Hz, 8H), 6.90 (d, J = 7.7 Hz, 2H), 6.68 (d, J = 7.9 Hz, 2H), 6.64 (d, J = 14.3 Hz, 2H), 6.44 (t, J = 7.8 Hz, 2H), 5.34 (d, J = 14.3 Hz, 2H), 4.97 (d, J = 15.8 Hz, 2H), 4.95 (d, J = 15.7 Hz, 2H), 4.39 (br d, 12.6 Hz, 2H), 2.90 (s, 6H), 2.40 (m, 8H), 2.25–2.19 (m 2H), 2.01–1.90 (m, 2H), 1.80 (s, 6H), 1.65–1.62 (m, 2H); 13C NMR (125 MHz, (CD3)2SO, 376K) δ 176.0 (C), 148.6 (C), 143.4 (C), 141.3 (C), 135.7 (C), 133.8 (C), 130.5 (CH), 129.6 (CH), 129.3 (CH), 127.9 (CH), 127.7 (CH), 127.0 (CH), 126.9 (CH), 126.5 (C), 126.99 (CH), 125.95 (CH), 124.2 (CH), 122.7 (CH), 122.1 (CH), 118.8 (C), 116.8 (CH), 110.2 (CH), 108.9 (CH), 83.3 (CH), 61.9 (C), 55.7 (C), 50.6 (CH2), 42.8 (CH2), 35.3 (CH2), 34.2 (CH3), 31.8 (CH3), 20.3 (CH3); IR (thin film) 3417, 3376, 3061, 3032, 2867, 2791, 2244, 1708, 1650, 1609, 1485, 1465, 1357, 1247, 1160, 732 cm−1; LRMS-ESI (m/z) [M + H]+ calcd for C72H70N8O6S2H 1207.5; found, 1207.5; [α]23D +147, [α]23577 +156, [α]23546 +184, [α]23435 +379, [α]23405 +201 (c = 0.81, CH2Cl2).
4.4 Optimized general Procedure for Hydrogenation of the enesulfonamide side chains of the dioxindole Heck products
Synthesis of the tetrahydro derivative of 20
A solution of 20 (140 mg, 0.116 mmol) in warm EtOH (4.0 mL) was added to a glass sleeve containing Pd(OH)2/C (0.28 g, 20 wt %), K2CO3 (130 mg, 0.928 mmol) and a stirbar. The sleeve was fitted inside a pressure reactor (Parr bottle 250 mL) and sealed. The Parr bottle was charged with hydrogen gas (1000 psi) and heated to 80 °C for 24 h. After cooling to room temperature and venting the mixture was filtered through Celite® and the filter cake was washed with CHCl3 saturated with NH3 (20 mL). The washes were concentrated to afford the tetrahydro derivative of 20 (139 mg, 99%) as a colorless foam. An analytical sample was obtained by column chromatography (SiO2: 100:0 CH2Cl2 → 97:3 CH2Cl2/MeOH → 94:5:1 CH2Cl2/MeOH/NH4OH → 89:10:1 CH2Cl2/MeOH/NH4OH) to provide 121 mg (96%) of 21 as a colorless foam: 1H NMR (500 MHz, (CD3)2SO, 396K) δ 7.54 (d, J = 8.2 Hz, 4H), 7.31 (overlapping signals d and m, d; J = 8.0 Hz, 8H), 7.18 (t, J = 7.2 Hz, 2H), 7.14 (t, J = 7.4 Hz, 2H), 7.10 (d, J = 7.2 Hz, 4H), 7.02 (t, J = 6.8 Hz, 2H), 6.96 (t, J = 7.4 Hz, 4H), 6.89 (d, J = 7.9 Hz, 2H), 6.77 (d, J = 7.5 Hz, 2H), 6.33 (d, J = 7.7 Hz, 2H), 6.04 (t, J = 7.6 Hz, 2H), 5.77 (br s, 2H), 4.97 (d, J = 15.8 Hz, 2H), 4.75 (d, J = 15.8 Hz, 2H), 3.07–3.01 (m, 2H), 2.91–2.86 (m, 4H), 2.81 (br s, 4H), 2.63 (s, 6H), 2.38 (s, 6H), 2.37 (s, 6H), 2.32–2.23 (m, 2H), 1.93–1.87 (m, 2H) 1.26 (br s, 2H); 13C NMR (125 MHz, (CD3)2SO, 396K) δ 177.2 (C), 149.6 (C), 142.3 (C), 141.9 (C), 135.3 (C), 134.6 (C), 129.4 (C), 128.8 (CH), 127.62 (CH), 127.57 (CH), 126.3 (CH), 126.2 (CH), 126.1 (CH), 124.7 (CH), 124.5 (CH), 121.7 (CH), 121.4 (CH), 118.0 (C), 117.7 (C), 117.3 (CH), 108.9 (CH), 82.8 (CH), 62.4 (C), 53.8 (C), 50.1 (CH2), 45.5 (CH2), 42.5 (CH2), 34.7 (CH2), 34.6 (CH2), 33.9 (CH3), 30.0 (CH3), 20.0 (CH3); IR (thin film) cm−1; 3334, 3060, 3031, 2857, 2788, 1695, 1611 cm−1; HRMS-ESI (m/z) [M + H]+ calcd for C72H74N8O6S2H 1211.5251; found, 1211.5251; [α]23D −40, [α]23577 −43, [α]23546 −51, [α]23435 −102, [α]23405 −121 (c = 1.5, CH2Cl2).
Tetrahydro derivative of 22
Following the optimized general procedure for the hydrogenation of the enesulfonamide side chains, 125 mg of 22 yielded 121 mg (96%) of the tetrahydro derivative of 22: 1H NMR (500 MHz, (CD3)2SO, 396K) δ 7.51 (d, J = 7.9 Hz, 4H), 7.38–7.34 (m, 8H), 7.30–7.27 (m, 8H), 7.17 (d, J = 7.0 Hz, 2H), 7.10 (t, J = 7.4 Hz, 2H), 7.03 (d, J = 7.6 Hz, 2H), 6.94 (d, J = 6.8 Hz, 2H), 6.72 (d, J = 7.8 Hz, 2H), 6.42 (t, J = 7.3 Hz, 2H), 5.23 (s, 2H), 4.95 (d, J = 16.4 Hz, 2H), 4.89 (d, J = 15.3 Hz, 2H), 4.41 (s, 2H), 2.95–2.91 (m, 2H), 2.84–2.80 (m, 4H), 2.65 (s, 6H), 2.49–2.42 (m, 2H), 2.39 (s, 6H), 2.36–2.31 (m, 2H), 2.27–2.24 (m, 2H), 2.14–1.11 (m, 2H), 2.00 (s, 6H), 1.75–1.71 (m, 2H); 13C NMR (125 MHz, (CD3)2SO, 396K) δ 177.0 (C), 148.9 (C), 142.3 (C), 141.8 (C), 135.6 (C), 134.8 (C), 129.8 (C), 128.8 (CH), 127.8 (CH), 127.6 (CH), 126.9 (CH), 126.7 (CH), 126.2 (CH), 125.1 (CH), 124.0 (CH), 122.8 (CH), 121.9 (CH), 117.8 (C), 116.8 (CH), 116.7 (C), 108.8 (CH), 83.8 (CH), 61.7 (C), 53.3 (C), 50.9 (CH2), 45.2 (CH2), 42.9 (CH2), 35.0 (CH3), 34.8 (CH3), 33.9 (CH2), 31.5 (CH2), 20.0 (CH3); IR (thin film) 3320, 3057, 3033, 2851, 2790, 1692, 1610 cm−1; LRMS-ESI (m/z) [M + H]+ calcd for C72H74N8O6S2H 1211.5; found, 1211.5; [α]23D +254, [α]23577 +269, [α]23546 +312, [α]23435 +625 (c = 1.1, CH2Cl2).
Tetrahydro derivative of 21
Following the optimized general procedure for the hydrogenation of the enesulfonamide side chains, 230 mg of 21 yielded 224 mg (97%) of tetrahydro derivative of 21: 1H NMR (500 MHz, (CD3)2SO, 396K) δ 7.50 (d, J = 8.2 Hz, 2H), 7.48 (d, J = 8.2 Hz, 2H), 7.37 (d, J = 7.1 Hz, 2H), 7.32 (d, J = 8.2 Hz, 4H), 7.30–7.23 (m, overlapping signals, 8H), 7.22 (d, J = 7.9 Hz, 2H), 7.16–7.07 (m, 4H), 7.02 (d, J = 7.8 Hz, 1H), 6.98 (d, J = 7.8 Hz, 1H), 6.95 (d, J = 7.3 Hz, 1H), 6.80 (d, J = 7.3 Hz, 1H), 6.62 (d, J = 7.7 Hz, 1H), 6.59 (d, J = 7.9 Hz, 1H), 6.36 (t, J = 7.5 Hz, 1H), 6.27 (t, J = 7.4 Hz, 1H), 5.44 (d, J = 3.9 Hz, 1H), 5.18 (s, 1H), 4.96 (d, J = 15.6 Hz, 1H), 4.90 (s, 2H), 4.89 (d, J = 15.4 Hz, 1H), 4.70 (br s, 1H), 4.43 (br s, 1H), 3.03–2.98 (m, 1H), 2.97–2.89 (m, 2H), 2.80 (br s, 3H), 2.68–2.61 (m, 1H), 2.66 (s, 3H), 2.65 (s, 3H), 2.44–2.40 (m, 2H), 2.39 (s, 3H), 2.37 (s, 3H), 2.34–2.31 (m, 2H), 2.30 (s, 3H), 2.28–2.25 (m, 2H), 2.15–2.10 (m, 1H), 2.00 (s, 3H), 1.84–1.81 (m, 1H), 1.75–1.72 (m, 1H); 13C NMR (125 MHz, (CD3)2SO, 396K) δ 177.2 (C), 176.9 (C), 149.5 (C), 148.8 (C), 142.30 (C, overlapping signal), 142.30 (C, overlapping signal), 141.89 (C), 141.86 (C), 135.63 (C), 135.57 (C), 135.2 (C), 134.8 (C), 134.7 (C), 134.5 (C), 129.8 (C), 129.6 (C), 128.85 (CH, overlapping signal), 128.85 (CH, overlapping signal), 127.76 (CH, overlapping signal), 127.67 (CH, overlapping signal), 127.71 (CH), 127.6 (CH), 126.9 (CH), 126.7 (CH), 126.6 (CH), 126.3 (CH), 126.2 (CH), 126.1 (CH), 124.9 (CH), 124.7 (CH), 124.4 (CH), 124.0 (CH), 122.3 (CH), 122.0 (CH), 121.84 (CH), 121.77 (CH), 118.5 (C), 117.7 (CH), 117.4 (C), 116.8 (CH), 108.8 (CH), 108.7 (CH), 83.6 (CH), 82.9 (CH), 62.3 (C), 61.9 (C), 53.7 (C), 53.3 (C), 50.6 (CH2), 50.3 (CH2), 45.4 (CH2), 45.2 (CH2), 42.9 (CH2), 42.8 (CH2), 35.0 (CH2), 34.9 (CH2), 34.8 (CH3), 34.7 (CH3), 33.92 (CH3), 33.87 (CH3), 31.3 (CH2), 30.6 (CH2), 20.05 (CH3), 20.03 (CH3); IR (thin film) 3334, 3058, 2852, 2789, 1693, 1610 cm−1; LRMS-ESI (m/z) [M + H]+ calcd for C72H74N8O6S2H 1211.5; found, 1211.5; [α]23D +148, [α]23577 +155, [α]23546 +180, [α]23435 +360, [α]23405 +410 (c = 0.87, CH2Cl2).
4.5 Optimized general procedure for reductive cyclization to form dodecacyclic alkaloid products
Preparation of (R,R,R,R)-quadrigemine 7
Caution!!! Ammonia gas is toxic and should only be used in a well-ventilated fume hood. Ammonia was condensed in a 2-neck flask cooled to −78 °C and fitted with a cold finger filled with dry ice and isopropanol. After condensing ~20 mL NH3(l) a small piece of sodium (~25 mg) was added to provide a deep blue color. Using a wide-bore cannula approximately 10 mL of ammonia was distilled into another 2-neck flask cooled to −78 °C and fitted with a cold finger filled with dry ice and isopropanol. The tetrahydro derivative of 20 (68.0 mg, 0.056 mmol) in dry THF (3 mL) was added to the liquid NH3. Tert-butanol (42 μL, 0.448 mmol) was added followed by slow addition of small pieces of sodium metal (65 mg, 2.8 mmol). The heterogeneous solution was stirred vigorously at −78 °C as the solution slowly changed from clear yellow, green-brown, to blue. After persistence of the blue color for 20 min the reaction was quenched at −78 °C by slowly adding solid NH4Cl (~190 mg). The blue color disappeared and a cloudy colorless precipitate formed. The flask was allowed to slowly warm to room temperature while open to the atmosphere and the ammonia permitted to evaporate. Water (20 mL) was added to the heterogeneous mixture and extracted with CHCl3 (sat with NH3; 3 × 30 mL). The combined organic layers were washed with brine (20 mL), dried over Na2SO4, filtered, concentrated under reduced pressure, and the residue was purified by reverse phase HPLC to yield (R,R,R,R)-quadrigemine 7 (8.8 mg, 23% from 20): Analytical reverse-phase HPLC (Zorbax Extend C18, 250 × 4.6 mm), 72:28–>85:15 MeOH-H2O (1% NH4OH) over 40 min, 1.0 mL/min, UV detection at 254 nm (rt = 24.8 min); 1H NMR (500 MHz, (CD3)2SO, 376K) δ 6.93 (app t, J = 7.1 Hz, 2H), 6.87 (d, J = 7.4 Hz, 2H), 6.70–6.67 (m, 3H), 6.57 (app t, J = 7.0 Hz, 2H), 6.50 (d, J = 7.7 Hz, 2H), 6.20 (br app t, J = 7.7 Hz, 1H), 5.81 (br s, 1H), 5.75 (br s, 1H), 4.77 (br s, 1H), 4.71 (br s, 2H), 3.89–2.82 (m, 7H), 2.61 (br s, 1H), 2.41 (s, 7H), 2.36 (s, 5H), 2.26–2.23 (m, 2H), 1.83–1.79 (m, 3H), 1.34–1.25 (m, 6H), 0.85 (app t, J = 7.1 Hz, 2H); 1H NMR (500 MHz, C6D6, 345K) δ 7.10 (d, J = 6.9 Hz, 2H), 7.08 (d, J = 7.5 Hz, 1H), 7.01 (t, J = 7.6 Hz, 2H), 6.96 (br s, 1H), 6.76 (t, J = 7.3 Hz, 2H), 6.48 (br s, 1H), 6.42 (d, J = 7.5 Hz, 1H), 5.92 (s, 1H), 5.05 (s, 1H), 4.86 (s, 1H), 3.46 (br s, 1H), 3.14–3.10 (m, 1H), 2.78 (t, J = 7.8 Hz, 2H), 2.70–2.68 (m, 3H), 2.60–2.56 (m, 3H), 2.55–2.51 (m, 3H), 2.49 (s, 3H), 2.40 (br s, 5H), 2.25 (s, 4H), 1.94 (t, J = 8.1 Hz, 4H), 1.85 (dd, J = 5.0 & 11.1 Hz, 1H), 1.32–1.28 (m, 3H); 13C NMR (125 MHz, C6D6, 345K) δ 151.8 (C), 150.8 (C), 133.7 (C), 126.5 (CH), 125.7 (CH), 119.6 (CH), 117.4 (CH), 109.8 (CH), 88.2 (CH), 84.8 (CH), 64.3 (C), 61.9 (C), 53.0 (CH2), 51.8 (CH2), 48.9 (CH), 39.3 (CH2), 37.4 (CH2), 36.2 (CH3), 35.9 (CH3), 30.7 (C), 29.5 (CH), 18.1 (C); IR (thin film) 3379, 3270, 3053, 2930, 2855, 2789, 1604, 1485, 1466, 1248, 1153, 1036, 908, 737 cm−1; HRMS-ESI (m/z) [M + H]+ calcd for C44H50N8H 691.4236; found, 691.4237; [α]23D +277, [α]23577 +283, [α]23546 +328, [α]23435 +656 (c = 0.20, EtOH).
(S,R,R,S)-quadrigemine (5)
Following the optimized general for the reductive cyclization, the tetrahydro derivative of 22 (71 mg, 0.059 mmol) was converted to 5 (11.9 mg, 29%): Analytical reverse-phase HPLC (Zorbax Extend C18, 250 × 4.6 mm), 72:28–>85:15 MeOH-H2O (1% NH4OH) over 40 min, 1.0 mL/min, UV detection at 254 nm: (rt = 29.4 min); 1H NMR (500 MHz, (CD3)2SO, 376K) δ 6.98–6.94 (overlapping doublets, 6H), 6.86 (d, J = 7.2 Hz, 2H), 6.61 (t, J = 7.1 Hz, 2H), 6.56 (d, J = 7.6 Hz, 2H), 6.37 (t, J = 7.5 Hz, 2H), 6.05 (br s, 2H), 5.88 (br s, 2H), 4.84 (s, 2H), 4.72 (s, 2H), 2.94–2.90 (m, 8H), 2.76–2.69 (m, 2H), 2.41 (s, 6H), 2.37–2.34 (m, 2H), 2.13 (s, 6H), 1.90–1.88 (m, 2H), 1.78–1.75 (m, 2H); 13C NMR (125 MHz, (CD3)2SO, 376K) δ 150.9 (C), 148.9 (C), 132.8 (C), 131.7 (C), 127.6 (CH), 126.8 (CH), 124.5 (CH), 123.8 (CH), 122.9 (C), 116.6 (CH), 115.0 (CH), 107.5 (CH), 85.5 (CH), 83.2 (CH), 61.9 (C), 59.6 (C), 51.2 (CH2), 50.9 (CH2), 37.9 (CH2), 35.4 (CH2), 34.8 (CH3), 34.7 (CH3); IR (thin film) cm−1; 3379, 3245, 3050, 2858, 2791, 1683, 1604, 1486 cm−1; HRMS-ESI (m/z) [M + H]+ calcd for C44H50N8H 691.4236; found, 691.4223; [α]23D +279, [α]23577 +289, [α]23546 +332, [α]23435 +690 (c = 0.27, EtOH).
(S,R,R,R)-Quadrigemine 6
Following the optimized general procedure for the reductive cyclization, the tetrahydro derivative of 21 (73 mg, 0.06 mmol) was converted to 6 (11.9 mg, 29%): Analytical reverse-phase HPLC (Zorbax Extend C18, 250 × 4.6 mm), 72:28–>85:15 MeOH-H2O (1% NH4OH) over 40 min, 1.0 mL/min, UV detection at 254 nm: (rt = 27.3 min); 1H NMR (500 MHz, (CD3)2SO, 376K) δ 6.98 (d, J = 7.1Hz, 2H), 6.95 (d, J = 3.3 Hz, 1H), 6.93 (d, J = 7.2 Hz, 3H), 6.83 (d, J = 7.2 Hz, 1H), 6.79 (t, J = 8.1 Hz, 2H), 6.62–6.57 (m, 2H), 6.54 (dd, J = 5.7, 7.1 Hz, 2H), 6.34 (t, J = 7.2 Hz, 1H), 6.28 (t, J = 7.3 Hz, 1H), 6.00 (s, 1H), 5.86 (s, 1H), 5.82 (s, 1H), 5.75 (s, 1H), 4.80 (s, 2H), 4.69 (br s, 1H), 4.56 (br s, 1H), 2.68–2.61 (m, 3H), 2.47– 2.43 (m, 5H), 2.41 (s, 3H), 2.39 (s, 3H), 2.33 (s, 3H), 2.31–2.26 (m, 2H), 2.14–2.11 (m, 1H), 2.10 (s, 3H), 1.89–1.82 (m, 3H), 1.77–1.73 (m, 1H); 13C NMR (125 MHz, (CD3)2SO, 376K) δ 151.9 (C), 149.9 (C), 127.9 (C), 125.7 (C), 117.7 (CH), 117.5 (C), 116.4 (CH), 116.0 (CH), 108.5 (CH), 87.1 (CH), 86.5 (CH), 84.3 (CH), 83.9 (CH), 70.5 (CH), 63.0 (C), 60.7 (C), 52.3 (CH2), 51.8 (CH2), 39.0 (CH2), 38.9 (CH2), 36.2 (CH3), 35.9 (CH3); IR (thin film) 3378, 3055, 2852, 2791, 1698, 1603, 1486, 1456 cm−1; HRMS-ESI (m/z) [M + H]+ calcd for C44H50N8H 691.4236; found, 691.4237; [α]23D +347, [α]23577 +367, [α]23546 +435, [α]23435 +854 (c = 0.10, EtOH).
Supplementary Material
Scheme 5.

Synthesis of the Heck cyclization precursor in the C2-symmetric series.
Scheme 6.

Diastereoselective double Heck cyclization of C2-symmetric dibutenanalide 19.
Acknowledgments
This research was supported by NIH grants (R01-HL25854 and R01-GM098601) and an NIH postdoctoral fellowship to T.L.M.-D. (F32-GM09660). NMR and mass spectra analyses were obtained at UC Irvine using instrumentation acquired with the assistance of NSF and NIH Shared Instrumentation programs. We thank Dr. Eike Hupe and Dr. Matthew Weiss for early studies in this area, and Dr. Philip Dennison and Dr. John Greaves, UC Irvine, for their assistance with NMR and mass spectrometric analyses. We are grateful Professor Françoise Guéritte-Voegelein for a sample of natural quadrigemine C and Professor Luisella Verotta for providing a sample of the crude Psychotria muscosa extract. Biological screening at the City of Hope was supported in part by NIH P30-CA22572 and the Drug Discovery and Structural Biology Core facility.
Footnotes
Supplementary Information Available: Copies of 1H and 13C NMR spectra and HPLC traces, a description of general experimental details, summary of optical rotations reported for quadrigemine C, and HPLC and CD comparisons of synthetic and natural quadrigemine C.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.(a) Anthoni U, Christophersen C, Nielsen PH. In: Alkaloids: Chemical and Biological Perspectives. Pelletier WS, editor. Vol. 13. Pergammon; New York: 1999. pp. 163–236. [Google Scholar]; (b) Hino T, Nakagawa M. In: Alkaloids: Chemistry and Pharmacology. Brossi A, editor. Vol. 34. Academic Press; New York: 1989. pp. 1–75. [Google Scholar]; (c) Sévenet T, Pusset J. In: The Alkaloids: Chemistry and Pharmacology. Cordell GA, editor. Vol. 48. Academic Press; New York: 1996. pp. 1–73. [Google Scholar]
- 2.Steven A, Overman LE. Angew Chem Int Ed. 2007;46:5488–5508. doi: 10.1002/anie.200700612. [DOI] [PubMed] [Google Scholar]
- 3.For reviews of recent synthetic studies, see: Schmidt MA, Movassaghi M. Synlett. 2008:313–324.Ruiz-Sanchis P, Savina SA, Albericio F, Álvarez M. Chem Eur J. 2011;17:1388–1408. doi: 10.1002/chem.201001451.Tadano S, Ishikawa H. Synlett. 2014:157–162. and reference 2.
- 4.To date, the only total synthesis of a dodecacyclic members of these alkaloids is our total synthesis of (−)-quadrigemine C, see: Lebsack AD, Link JT, Overman LE, Stearns BA. J Am Chem Soc. 2002;124:9008–9009. doi: 10.1021/ja0267425.
- 5.Parry KP, Smith GF. J Chem Soc, Perkin Trans. 1978;1:1671–1682. [Google Scholar]
- 6.Parry KP. PhD Thesis. University of Manchester; Manchester, UK: 1968. Alkaloids from Hodgkinsona frutescens. The Stucture of the Quadrigemines. [Google Scholar]
- 7.The 3a′–3a″ bond lengths of meso-chimonanthine and (−)-chimonanthine dihydrobromide, determined by single-crystal X-ray crystallography, are significantly elongated: 1.556 and 1.582 Å, respectively; see: Link JT, Overman LE. J Am Chem Soc. 1996;118:8166–8177.Grant IJ, Hamor TA, Robertson JM, Sim GA. J Chem Soc. 1965:5678–5696.
- 8.Clayton E, Reed RI, Wilson JM. Tetrahedron. 1962;18:1495–1501. [Google Scholar]
- 9.(a) Libot F, Miet C, Kunesch N, Poisson JE, Pusset J, Sévenet T. J Nat Prod. 1987;50:468–473. [PubMed] [Google Scholar]; (b) Guértte-Voegelein F, Sévenet T, Pusset J, Adeline MT, Gillet B, Beloeil JC, Guénard D, Potier P, Rasolonjanahary R, Kordon C. J Nat Prod. 1992;55:923–930. doi: 10.1021/np50085a012. [DOI] [PubMed] [Google Scholar]; (c) Jannic V, Guéritte F, Laprévote O, Serani L, Martin MT, Sévenet T, Potier P. J Nat Prod. 1999;62:838–843. doi: 10.1021/np9805387. [DOI] [PubMed] [Google Scholar]; (d) Verotta L, Pilati T, Tatò M, Elisabetsky E, Amador TA, Nunes DS. J Nat Prod. 1998;61:392–396. doi: 10.1021/np9701642. [DOI] [PubMed] [Google Scholar]
- 10.Roth A. Thése de Doctorat de 3éme cycle. Université Louis Pasteur; Strasbourg: 1984. [Google Scholar]
- 11.Fridrichsons J, Mackay MF, Mathieson AMcL. Tetrahedron. 1974;36:85–92. [Google Scholar]
- 12.From initial degradation studies by Parry and Smith, quadrigemine A was believed to be the R,S,R,R isomer that was potentially contaminated with a diastereomer or meso compound resulting in a lower than expected optical rotation, see ref. 5.
- 13.For a review of the use of enantioselective intramolecular Heck reactions in total synthesis, see: Dounay A, Overman LE. The Asymmetric Intramolecular Mizoroki-Heck Reaction in Natural Product Synthesis. In: Oestreich M, editor. The Mizoroki-Heck Reaction. Ch 16. Wiley–VCH; West Sussex, UK: 2009. pp. 533–568.For a review of using Heck reactions to desymmetrize precursors, see; Shibasaki M, Ohshima T. Desymmetrizing Heck Reactions. In: Oestreich M, editor. The Mizoroki-Heck Reaction. Ch 13. Wiley–VCH; West Sussex, UK: 2009. pp. 463–483.
- 14.Stereocontrolled enantioselective total syntheses of (+)- or (−)-chimonanthine or (+)- or (−)-folicanthine: Overman LE, Paone DV, Stearns BA. J Am Chem Soc. 1999;121:7702–7703.Overman LE, Larrow JF, Stearns BA, Vance JM. Angew Chem Int Ed. 2000;39:213–215. doi: 10.1002/(sici)1521-3773(20000103)39:1<213::aid-anie213>3.0.co;2-z.Movassaghi M, Schmidt MA. Angew Chem Int Ed. 2007;46:3725–3728. doi: 10.1002/anie.200700705.Guo C, Song J, Huang JZ, Chen PH, Luo SW, Gong LZ. Angew Chem Int Ed. 2012;51:1046–1050. doi: 10.1002/anie.201107079.Mitsunuma H, Shibasaki M, Kanai M, Matsunaga S. Angew Chem Int Ed. 2012;51:5217–5221. doi: 10.1002/anie.201201132.Xie W, Jiang G, Liu H, Hu J, Pan X, Zhang H, Wan X, Lai Y, Ma D. Angew Chem Int Ed. 2013;52:12924–12927. doi: 10.1002/anie.201306774.
- 15.Stereocontrolled total syntheses of meso-chimonanthine: Link JT, Overman LE. J Am Chem Soc. 1996;118:8166–8167.Lathrop SP, Movassaghi M. Chem Sci. 2014;5:333–340. doi: 10.1039/C3SC52451E.Wada M, Murata T, Oikawa H, Oguri H. Org Biomol Chem. 2014;12:298–306. doi: 10.1039/c3ob41918e. refs 14a and 14b.
- 16.Ishikawa H, Takayama H, Aimi N. Tetrahedron Lett. 2002;43:5637. [Google Scholar]
- 17.Snell RH, Woodward RL, Willis MC. Angew Chem Int Ed. 2011;50:9116–9119. doi: 10.1002/anie.201103864. [DOI] [PubMed] [Google Scholar]
- 18.For examples of Stille cross-couplings at room temperature and the effect of tri-2-furylphosphine, see: Farina V, Krishnan B. J Am Chem Soc. 1991;113:9585–9595.For the role of CuI see: Farina V, Kapadia S, Krishnan B, Wang C, Liebeskind LS. J Org Chem. 1994;59:5905–5911.Liebeskind LS, Fengl RW. J Org Chem. 1990;55:5359–5364.
- 19.(a) Mee SPH, Lee V, Baldwin JE. Angew Chem Int Ed. 2004;43:1132–1136. doi: 10.1002/anie.200352979. [DOI] [PubMed] [Google Scholar]; (b) Mee SPH, Lee V, Baldwin JE. Chem Eur J. 2005;11:3294–3308. doi: 10.1002/chem.200401162. [DOI] [PubMed] [Google Scholar]
- 20.Allred GD, Liebeskind LS. J Am Chem Soc. 1996;118:2748–2749. [Google Scholar]
- 21.Fürstner A, Funel JA, Tremblay M, Bouchez LC, Nevado C, Waser M, Ackerstaff J, Stimson CC. Chem Comm. 2008:2873–2875. doi: 10.1039/b805299a. [DOI] [PubMed] [Google Scholar]
- 22.Han X, Stoltz BM, Corey EJ. J Am Chem Soc. 1999;121:7600–7605. [Google Scholar]
- 23.For reviews of two-directional chain synthesis see: Schreiber SL. Chem Scr. 1987;27:563–566.Poss CS, Schreiber SL. Acc Chem Res. 1994;27:9–17.Magnuson SR. Tetrahedron. 1995;51:2167–2213.Willis MC. J Chem Soc Perkins 1. 1999;13:1765–1784.Mikami K, Yoshida AJ. Syn Org Chem Japan. 2002;60:732–739.Hoffmann RW. Angew Chem Int Ed. 2003;42:1096–1109. doi: 10.1002/anie.200390291.
- 24.Vigneron JP, Dhaenens M, Horeau A. Tetrahedron. 1973;29:1055–1059. [Google Scholar]
- 25.The relative configuration of the major and minor meso-dioxindole products was not possible by spectroscopic means. In addition, we were unsuccessful in crystallizing the two meso-quadrigemines that were prepared from these diooxindole intermediates. As a result, the relative configuration of these two products (i.e., which is 3 and which is 4) is unknown.
- 26.Conditions: 10 mol% Pd(OAc)2, 20 mol% (R)-tol-BINAP, 4.0 equiv PMP, MeCN at 80 °C.27
- 27.Kodanko JJ, Overman LE. Angew Chem Int Ed. 2003;42:2528–2531. doi: 10.1002/anie.200351261. [DOI] [PubMed] [Google Scholar]
- 28.Computational model of the lowest energy conformation of (−)-quadrigemine C identified by Monte Carlo conformational searching using the MMMF force field and final optimization by DFT calculation at the EDF2-631G* level. Calculations performed using Spartan 14, Wavefunction, Inc.
- 29.Hydrogenation catalysts such as Rh/Al2O3, Ru/Al2O3, Pt2O, Ir(COD)(PCy3)PyPF6, Pd/C, and Pd(OAc)2) evaluated using a H2 atmosphere or under transfer hydrogenation conditions were less effective.
- 30.Preparation of Pd(OH)2/C involves washing with dilute AcOH, see: Pearlman W. Tetrahedron Lett. 1967;17:1663–1664.
- 31.Kodanko JJ, Hiebert S, Peterson EA, Sung L, Overman LE, de Moura Linck V, Goerck GC, Amador TA, Leal MB, Elisabetsky E. J Org Chem. 2007;72:7909–7914. doi: 10.1021/jo7013643. [DOI] [PubMed] [Google Scholar]
- 32.Rao TS, Pandey PS. Synth Commun. 2004;34:3121–3127. [Google Scholar]
- 33.Comparison CD and HPLC data is available in the Supplementary Information.
- 34.The origin of the apparent discrepancy between the levorotatory optical rotation in ethanol that we observe for ent-quadrigemine C, [α]23D −30 (c = 0.2), and that reported for natural quadrigemine C in methanol, [α]D +40 (c = 0.2),9d is unclear, as CD spectra of synthetic quadrigemine C and natural quadrigemine C in methanol under the same conditions are identical.33 Moreover, we observe [α]23D +20 (c = 0.2) for ent-quadrigemine C.
- 35.(a) Overman LE, Peterson EA. Angew Chem Int Ed. 2003;42:2525–2528. doi: 10.1002/anie.200351260. [DOI] [PubMed] [Google Scholar]; (b) Overman LE, Peterson EA. Tetrahedron. 2003;59:6905–6919. [Google Scholar]
- 36.For formation of Ac2O with Pd(OAc)2 and Ph3P see, Amatore C, Jutand A, M’Barki MA. Organometallics. 1992;11:3009–3013.
- 37.Obtaining NMR spectra in d6-DMSO at 376K of the [2+2] qudrigemine products 1, 2, meso-3, meso-4, and 5–7 resulted in some simplification of the spectra by reducing the number of significant conformers present. However, these samples gradually decomposed during extended acquisition time at this temperature. NMR samples, which were clear and colorless solutions at the outset, eventually turned dark brown; partial degradation under the analysis conditions could be confirmed by HPLC analysis.
- 38.Verotta L, Peterlongo F, Elisabetsky E, Amador TA, Nunes DS. J Chromatogr A. 1999;841:165–176. [Google Scholar]
- 39.Extract was from P. muscosa collected at Benfica, Brazil in Feb 1997 by Verotta and co-workers.
- 40.(a) Amador TA, Elisabetsky E, Souza DO. Neurochem Res. 1996;21:97–102. doi: 10.1007/BF02527677. [DOI] [PubMed] [Google Scholar]; (b) Amador TA, Verotta L, Nunes DS, Elisabetsky E. Planta Med. 2000;66:770–772. doi: 10.1055/s-2000-9604. [DOI] [PubMed] [Google Scholar]; (c) Amador TA, Verotta L, Nunes DS, Elisabetsky E. Phytomedicine. 2001;8:202–6. doi: 10.1078/0944-7113-00025. [DOI] [PubMed] [Google Scholar]; (d) Verotta L, Orsini F, Sbacchi M, Scheildler MA, Amador TA, Elisabetsky E. Bioorg Med Chem. 2002;10:2133–2142. doi: 10.1016/s0968-0896(02)00078-0. [DOI] [PubMed] [Google Scholar]
- 41.Beretz A, Roth-Georger A, Corre G, Kuballa B, Anton R, Cazenave JP. Planta Med. 1985;51:300–303. doi: 10.1055/s-2007-969496. [DOI] [PubMed] [Google Scholar]
- 42.(a) Roth A, Kuballa B, Bounthanh C, Cabalion P, Sévenet T, Beck JP, Anton R. Planta Med. 1986;52:450–453. doi: 10.1055/s-2007-969251. [DOI] [PubMed] [Google Scholar]; (b) Adjibadé Y, Kuballa B, Cabalion P, Jung ML, Beck JP, Anton R. Planta Med. 1989;52:567–568. doi: 10.1055/s-2006-962098. [DOI] [PubMed] [Google Scholar]; (c) Adjibadé Y, Saad H, Kuballa B, Beck JP, Sévenet T, Cabalion P, Anton R. J Ethnopharmacol. 1990;29:127–136. doi: 10.1016/0378-8741(90)90050-4. (d) [DOI] [PubMed] [Google Scholar]
- 43.Such strategies have generally been employed to elaborate acyclic chains, see reference 23.
- 44.Prepared as described: Coulson DR. Inorg Syntheses. 1972;13:121–124.
- 45.10% KF/SiO2 was prepared as described: Harrowven DC, Guy IL. Chem Comm. 2004:1968–1969. doi: 10.1039/b406041e.
- 46.NMR spectra were nearly identical to those obtained previously for the enantiomer.4 The slight changes observed likely result from differences in the concentrations of the NMR samples.
- 47.TMEDA was distilled from CaH2 immediately prior to use.
- 48.Prepared as describe by Coulson DR. Inorg Syntheses. 1972;13:121–124.
- 49.Dried by first azeotroping with benzene, followed by distillation from 4Å MS under vacuum and stored over 4Å MS.
- 50.Dried by distillation under vacuum from CaH2.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.