

SUMMARY
Rad50 contains a conserved Zn2+ coordination domain (the Rad50 hook) that functions as a homodimerization interface. Hook ablation phenocopies Rad50 deficiency in all respects. Here we focused on rad50 mutations flanking the Zn2+-coordinating hook cysteines. These mutants impaired hook-mediated dimerization, but recombination between sister chromatids was largely unaffected. This may reflect that cohesin-mediated sister chromatid interactions are sufficient for double strand break repair. However, Mre11 complex functions specified by the globular domain, including Tel1 (ATM) activation, nonhomologous end-joining, and DNA double strand break end resection were affected, suggesting that dimerization exerts a broad influence on Mre11 complex function. These phenotypes were suppressed by mutations within the coiled coil and globular ATPase domain, suggesting a model in which conformational changes in the hook and globular domains are transmitted via the extended coils of Rad50. We propose that transmission of spatial information in this manner underlies the regulation of Mre11 complex functions.
Keywords: DNA repair, Rad50, Mre11 complex, hook, double strand break, coiled coil
INTRODUCTION
The Mre11 complex is an oligomeric assembly comprising dimers of Mre11, Rad50, and Xrs2 in budding yeast (or Nbs1 in fission yeast and other eukaryotes). The Mre11 complex exerts a broad influence on the DNA damage response (DDR) network, first as a sensor of DNA double strand breaks (DSBs), and subsequently as a mediator of DDR signaling and DSB repair. Mre11 complex-dependent DDR signaling primarily occurs via Tel1/ATM, the activation of which requires the Mre11 complex (Stracker and Petrini, 2011). Recent data also suggest a role for the Mre11 complex in the activation of ATR, the checkpoint kinase activated in response to DNA replication associated stress (Duursma et al., 2013; Shiotani et al., 2013; Stiff et al., 2004). The mechanistic basis of ATM (or ATR) activation by the Mre11 complex has not been established, although evidence that activation of ATM is influenced by Rad50-dependent ATP binding and hydrolysis is emerging (Al-Ahmadie et al., 2014; Deshpande et al., 2014; Hopfner, 2014; Lee et al., 2013). Underscoring its importance for ATM activation and signaling is the fact that mutations affecting the Mre11 complex in humans underlie cancer predisposition syndromes that exhibit phenotypic similarities to ataxia telangiectasia, a syndrome resulting from ATM deficiency (Stracker and Petrini, 2011; Stracker et al., 2013).
The Mre11 complex has an elongated structure characteristic of SMC (Structural Maintenance of Chromosomes) protein family members (de Jager et al., 2004; Wyman et al., 2011). It consists of a globular domain, from which the antiparallel coiled coil domains of each Rad50 extend, spanning approximately 500 amino acids in each direction (Stracker and Petrini, 2011). At the apex of the Rad50 coiled coil lies the Rad50 hook domain, which is characterized by two invariant cysteine residues separated by two hydrophobic amino acids (CXXC; Figure 1A). Two Rad50 hook domains can dimerize via tetrahedral coordination of a zinc ion by the two pairs of invariant cysteines; the hook domain is thus a Zn2+-dependent interaction interface (Hopfner et al., 2002) (Figure 1B). Based on genetic and structural data, we have proposed that the orientation of the dimerized Rad50 coiled coil domains imposed by hook domain-mediated dimerization promotes the bridging of sister chromatids to facilitate sister chromatid recombination (SCR) (Bressan et al., 1999; Hohl et al., 2011; Hopfner et al., 2002; Moreno-Herrero et al., 2005; van der Linden et al., 2009; Wiltzius et al., 2005).
Figure 1. Damage sensitivity of rad50hook mutants.
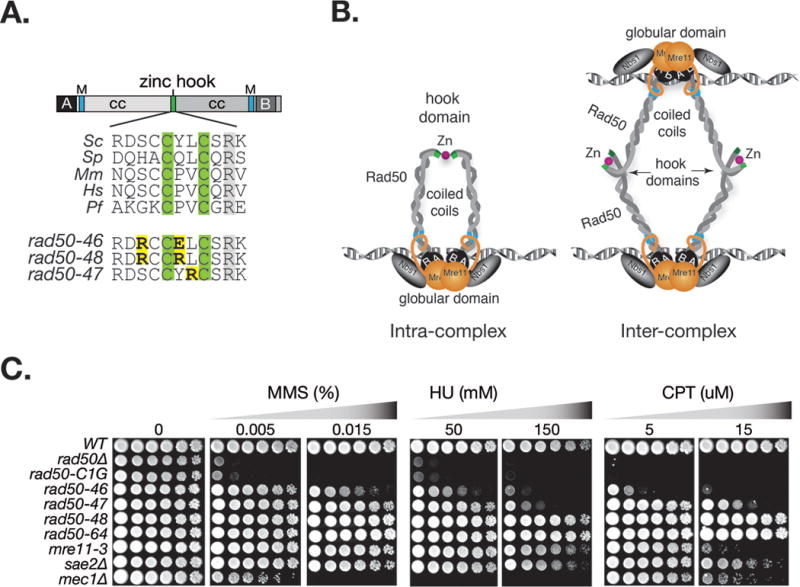
A. Rad50 primary domain structure and multiple sequence alignment of the central portion of the Rad50 hook domain. The zinc coordinating hook cysteines are highlighted in green and in yellow the S. cerevisiae rad50hook alleles, rad50-46 (S685R Y688E), rad50-4 7 (L689R) and rad50-48 (S685R Y688R). Annotations: coiled coil (cc), Walker A (A), Walker B (B), Mre11 interaction interface (M); Sc, S. cerevisiae; Sp, S. pombe; Mm, M. musculus, Hs, H. sapiens; Pf, P. furiosus. B. Schematic illustration of Rad50 hook dimerization between Rad50 proteins within a dimeric Mre11 complex assembly (intra-complex) or between Rad50 molecules in separate Mre11 complex dimeric assemblies (inter-complex). C. Sensitivities of rad50hook mutants to the indicated concentration of MMS, HU and CPT. The rad50-46 mutational residues were flipped in rad50-64 (S688E Y688R). Plates were incubated at 30°C. Outcomes at other incubation temperatures are given in Figure S1.
The influences of the hook and coiled coil domains appear to be largely structural, as the globular domain is the center of Mre11 complex enzymatic activities; the Mre11 nuclease domain and domains governing Rad50 ATP binding and hydrolysis reside within it (Stracker and Petrini, 2011; Williams et al., 2008; Williams and Tainer, 2005). DNA binding by the Mre11 complex also occurs within the globular domain; however a precise definition of DNA binding by the Mre11 complex is lacking, and it appears likely that more than one mode of DNA engagement by the globular domain is operative (Rojowska et al., 2014).
Recent structural studies revealed that Rad50 ATP binding and hydrolysis underlie large scale structural transitions of the Mre11 complex (Lammens et al., 2011; Lim et al., 2011; Williams et al., 2011). Upon ATP binding, Rad50 dimerizes and forms a closed complex, proposed to mediate DNA end binding, DNA end tethering, ATM checkpoint signaling and nonhomologous end-joining (NHEJ) (Deshpande et al., 2014; Lee et al., 2013). Upon ATP hydrolysis, the globular domain assumes an open configuration, promoting Mre11 nuclease activity, DNA end resection and DSB repair by homologous recombination (Deshpande et al., 2014; Lammens et al., 2011). Although it is several hundred angstroms distal to the globular domain, the hook domain may also influence the transition from open to closed conformation (Deshpande et al., 2014; Lee et al., 2013).
Having previously shown that mutations of the invariant hook cysteines disrupt Mre11 complex integrity (Hopfner et al., 2002), we sought to establish hypomorphic mutations that impaired, but did not abolish hook domain function. We reasoned that such alleles would reveal Mre11 complex DDR activities directly dependent on the Rad50 hook domain. We randomly mutagenized the conserved hydrophobic XX-residues situated between the invariant cysteines (CXXC), as structural and biophysical data regarding the P. furiosus hook dimer suggested that these residues constitute a hydrophobic interface stabilizing the zinc hook dimer assembly (Hopfner et al., 2002; Kochanczyk et al., 2013). We focused on three (of over 40) alleles, designated rad50-46, rad50-47, and rad50-48 (referred to collectively as rad50hook; Figure 1A).
Through genetic, biochemical, and biophysical assessments, we established evidence that the rad50hook alleles partially destabilized hook dimerization. The phenotypic outcomes were diverse, affecting myriad Mre11 complex functions, most of which appear to be specified within the globular domain. Tel1 (ATM) activation was impaired in each of the rad50hook mutants, whereas homologous recombination, which requires hook-mediated dimerization (Hohl et al., 2011; Wiltzius et al., 2005), remained largely intact.
These rad50hook phenotypes suggest that the stability of hook dimerization domain influences the disposition of the globular domain, and thereby functions mediated by domains within it. Mutations within the coiled coil and globular domains suppressed some of the rad50-46 phenotypes, supporting the interpretation that the hook and globular domains of the Mre11 complex function interdependently, and that the coiled coil domains are integral to that interaction.
RESULTS
Rad50 hook alleles with partial damage sensitivity
The Rad50 hook motif consists of two invariant cysteines separated by two hydrophobic residues (CXXC); over 80% of known hook domains conform to a C(P/Y)(L/V)C consensus (Pfam database ID: PF04423). P. furiosus Pro445 and Val446 were shown to bind in trans to a hydrophobic groove formed at the hook loop base of the distal protomer (Hopfner et al., 2002; Kochanczyk et al., 2013). In light of their conservation, it is likely that the corresponding residues in other organisms also stabilize the zinc hook dimer assembly.
In this study, we examined the biological functions influenced by the Rad50 hook domain. Having shown that mutation of the invariant cysteines phenocopied complete Rad50 inactivation (Hopfner et al., 2002), we reasoned that mutation within these hydrophobic residues of the hook would destabilize, but not destroy the Zn2+-mediated assembly, and so would confer a circumscribed effect, confined to alteration of Rad50 hook function. Random mutagenesis of the codons encoding Y688 and L689 of the S. cerevisiae RAD50 gene was carried out, and the resulting >40 mutants were screened for MMS (methyl methanesulfonate) sensitivity. In some instances, the primers used for mutagenesis also introduced a S to R change at position 685 (S685R). Most mutants obtained were essentially wild-type for MMS sensitivity, and two mutants phenocopied rad50Δ (Table S2). These two classes of mutants were not examined further. In this study we focused on the three rad50hook alleles, rad50-46, rad50-47, and rad50-48 (Figure 1A).
rad50-46 (S685R Y688E) and rad50-47 (L689R) showed partial MMS, HU (hydroxyurea) and CPT (camptothecin) sensitivity (Figure 1C). rad50-46 was slightly more sensitive than rad50-47, the later comparable to a Mre11 nuclease dead mutant (mre11-3), whereas a hook cysteine mutant (rad50-C1G) deficient in Zn2+ coordination was indistinguishable from rad50Δ. rad50-46 damage sensitivity was a composite effect of two mutations (S685R Y688E), as neither single mutant was sensitive (data not shown). rad50-48 (S685R Y688R) resulted in minimal MMS sensitivity (Figure 1C). Damage survival of rad50-46 and rad50-47, but not of rad50-48, was strongly temperature dependent, only mildly sensitive at 30°C, but highly sensitive at both 23°C and 37°C (Figure S1). As they appeared to represent hypomorphic defects in hook function, rad50-46, -47, and -48 were retained for further analysis.
Rad50 hook mutants alter zinc hook dimerization
To determine whether the partial damage sensitivity of rad50-46 and rad50-47 was a result of impaired hook function, we examined the dimerization, Zn2+ binding and accompanying conformational changes of peptides encompassing 41 or 68 amino acids. The peptides (Table S3 and S4) included the hook domain as well as the adjacent coiled coils on either side that mediate the aforementioned intermolecular hook and coiled coil interactions. As with previous studies of the archaeal hook peptide (Kochanczyk et al., 2013), fluorescence anisotropy, circular dichroism and UV spectroscopy were employed to examine the molecular outcomes of the rad50-46, -47, and -48 mutations (Figure 2A–C).
Figure 2. Rad50 hook mutations destabilize hook dimerization.
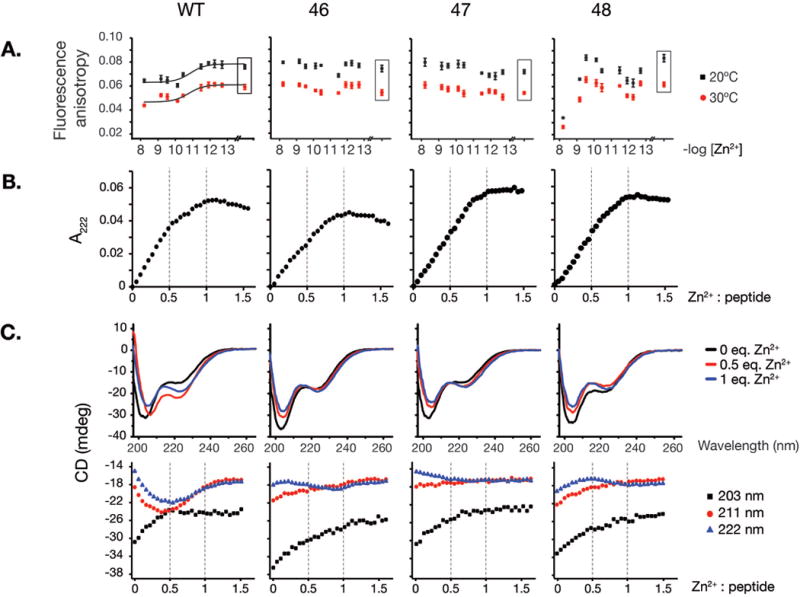
A. Homo-FRET of 5-carboxyfluorescein labeled 68 amino acid long hook peptides. Fluorescence anisotropy values for wild-type and 46, 47, 48 hook mutant peptides incubated in free Zn2+-controlled metal buffers providing various free zinc concentrations (−log[Zn2+]) are plotted. Data points shown in a box reflects buffer without Zn2+, containing chelator only (1 mM EDTA). Data were fitted to Hill’s equation with cooperativity coefficient fixed at 1.0. B. Titration of 41-mer WT and Rad50hook peptides with Zn2+ monitored by spectroscopy at 222 nm. Formation of coordinate bonds is observed as CysS-> Zn LMCT bands that can be monitored in the UV region. C. Circular dichroism (CD) spectra of wild-type and Rad50-46 hook mutant 41-mer unlabeled peptides. CD spectra of peptides with 0 (apo-peptide), 2.5 μM (0.5 equivalent of Zn2+) and 5 μM Zn2+ (1 eq. of Zn2+) at 203, 211 and 222 nm are given.
In the anisotropy studies, homotypic fluorescence energy transfer (homo-FRET) between 5-carboxyfluorescein moieties appended to the N-termini of the 68-mer peptides was monitored over a wide range of free Zn2+ concentrations. With the wild-type peptide, we observed a decrease in fluorescence anisotropy to approximately 3/4 of its initial value at subnanomolar free Zn2+(−log[Zn2+] < 10), which we interpreted as an effect of homo-FRET due to formation of Zn2+ induced hook dimers (Hamman et al., 1996). The rad50-46, -47, and -48 mutants did not show appreciable Zn2+ dependent anisotropy, suggesting either that Zn2+ binding or Zn2+-mediated dimerization is impaired (Figure 2A). An alternative explanation is that the geometry of the coiled coils is altered by the hook mutations, placing the 5-carboxyfluorescein moiety at a suboptimal distance for homotypic FRET to occur.
To address this issue, we used UV spectroscopy to monitor ligand to metal charge transfer (LMCT), which occurs upon Zn2+ coordination by cysteines (Figure 2B). In this setting LMCT is detected by UV absorbance at 222 nm. Under the conditions used, increased absorbance as a function of increasing Zn2+ concentration can be used to infer Zn2+ binding stoichiometry. In all cases we observed a linear correlation between Zn2+ concentration and UV absorbance over 0.0 to 0.5 (Figure 2B). For the wild-type peptide, but not rad50-46, -47, and -48, an inflection point was noted at the Zn2+:peptide ratio of 0.5 (Figure 2B). This is consistent with the appearance of relatively stable Zn2+ dependent species in which two peptides coordinate one Zn2+ ion. In all cases, the absorbance continues to increase over Zn2+:peptide molar ratios of 0.5 to 1.0. This indicates that presumptive higher order structures are dynamic under these conditions, and at higher Zn2+ concentrations, individual peptides bound to Zn2+ predominate. The lack of a marked inflection point in the mutants argues that dimerization is somehow aberrant, possibly due to altered secondary structure of the hook domain, as suggested by the CD spectra (Figure 2C) as well as anisotropy data (Figure 2A).
Consistent with the in vitro data, yeast 2-hybrid analysis data suggested that zinc-dependent hook homodimerization was impaired in all rad50hook mutants (Figure S2A–C).
Rad50 hook mutants destabilize the Rad50-Mre11 interaction
The Rad50 hook domain is critical for Mre11 complex-dependent DNA repair functions (Hohl et al., 2011; Hopfner et al., 2002; Wiltzius et al., 2005), and for the stability of the Mre11 complex (Hopfner et al., 2002). We assessed the effect of hook mutations on Mre11 complex stability by co-immunoprecipitation from whole cell extracts of rad50-46, -47, and -48 mutants (Figure 3A). Rad50-Mre11 interaction was strongly impaired in rad50-46, approximating the reduction seen in rad50-C2A (second invariant cysteine mutated to alanine). Rad50-Mre11 interactions were reduced to a lesser extent in rad50-47 and rad50-48, but were nonetheless clearly affected relative to wild-type (Figure 3A). Western blot analysis indicated that levels of the Rad50-46, -47, and -48 were unaltered. These results were confirmed by reciprocal co-immunoprecipitations (Figure S3) and by yeast 2-hybrid analysis assessing the Rad50-Mre11 interaction (data not shown). Hence, both hook dimerization and the Mre11-Rad50 interaction are destabilized in rad50hook mutant cells.
Figure 3. Mre11 complex integrity and DSB repair functions in rad50hook mutant cells.
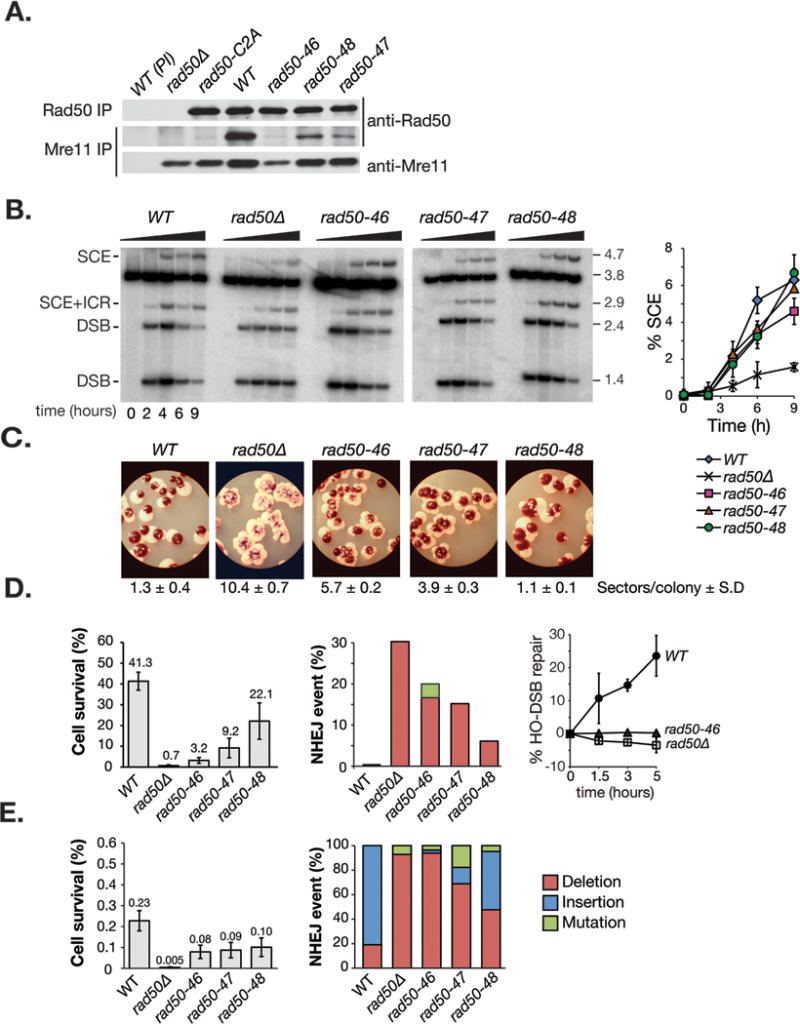
A. Mre11 complex integrity in wild-type and rad50hook mutants assessed by co-immunoprecipitation and western blot with Rad50 or Mre11 antisera. Pre-immune antibodies (PI) were included as negative controls. B. Physical assessment of HO-induced sister chromatid recombination (SCR) as previously described (Hohl et al., 2011). Representative southern blots are shown. Right panel: Quantification of the 4.7 kb band specific for SCR relative to the total DNA. Error bars denote standard deviation from three independent experiments. C. Spontaneous ade2 heteroallelic mitotic recombination in ade2-n/ade2-l-Scel heterozygot diploids. White sectors (ADE2) within >200 red colonies (ade2) were scored. The average number of white sectors per colony for each genotype is given. Standard deviations correspond to 5-7 diploids analyzed. D. Cell survival (left panel) and NHEJ repair events (middle panel) after acute (2 hours) HO DSB induction at the MAT locus. Error prone NHEJ events detected in rad50 mutants were categorized as insertion, deletion or point mutation and are given in Figure S4A. Right panel: NHEJ repair kinetics for wild-type, rad50Δ and rad50-46. Cells were cultivated for 1.5, 3 or 5 hours in glucose medium following HO induction (2 hours) and repair was monitored by quantitative PCR with primers flanking the HO site. Error bars denote standard deviation from at least three independent experiments. E. Cell survival upon chronic HO induction. Repair junctions were analyzed and sequences are given in Figure S4B. Error bars denote standard deviation from at least three experiments.
DSB repair in hook mutants
The changes in Mre11 complex stability notwithstanding, the rad50hook mutants did not exhibit pronounced MMS sensitivity (Figure 1C), suggesting that rad50-46, -47, and -48 mutants were proficient in sister chromatid based homologous recombination (SCR). We used a plasmid-based SCR assay described previously to address that interpretation (Cortes-Ledesma and Aguilera, 2006; González-Barrera et al., 2003; Hohl et al., 2011). Briefly, a HO-DSB is created during S-phase and the disappearance of the 2.4 kb HO-DSB restriction fragment and appearance of a 4.7 kb SCR repair product can be monitored by Southern blotting with a LEU2 specific probe (Figure 3B). In a time course over 9 hours, SCR products accumulated in all rad50hook mutants to similar levels (rad50-46, 4.6 ± 0.7%; rad50-47, 5.9 ± 0.1%; rad50-48, 6.7 ± 1.0%), as in wild-type (6.3 ± 0.6%), albeit with slightly delayed kinetics, whereas rad50Δ defective in SCR showed only residual repair products (1.6 ± 0.02%; Figure 3B, quantification on right). Hence, as suggested by the MMS resistance, rad50-46, -47, and -48 mutants are largely proficient in SCR.
The reduced SCR kinetics observed might account for its partial damage sensitivity at higher MMS doses (Figure 1C and Figure S1), and for the fact that rad50hook mutations were associated with a mild hyper recombination phenotype. Spontaneous heteroallelic recombination between the ade2 heteroalleles, ade2-n and ade2-ISceI, (Huang and Symington, 1994), evident as white ADE2 sectors within an otherwise red ade2 colony, was only mildly increased in rad50hook mutants. rad50Δ showed an average of >10 sectors per colony and sectoring decreased from rad50-46 (5.7 ± 0.2), to rad50-47 (3.9 ± 0.5) to wild-type levels (1.3 ± 0.4) in rad50-48 (1.1 ± 0.1) (Figure 3C).
We showed previously that truncation of the coiled coil domain exerted a similarly mild effect on SCR. In that setting, we observed a strong defect in NHEJ, leading us to propose a model wherein structural alterations within the coiled coils would lead to alterations in the disposition of globular domain and thereby affect resident functions such as NHEJ and Tel1 activation (Hohl et al., 2011).
Therefore, we examined the repair of a single HO DSB in a strain lacking a homologous template (Moore and Haber, 1996). Survival after short (2 hours) HO-induction led to 41.3% survival in wild-type cells, compared to 0.7% in rad50Δ. The rad50hook mutants exhibited NHEJ defects, with of 3.2%, 9.2%, and 22.1% viability of rad50-46, -47, and -48 respectively (Figure 3D), compared to 41% in wild type cells. In all rad50hook mutants, the frequency of imprecise rejoining was increased (Figure 3D, middle and Figure S4A). Quantitative PCR to measure rejoining of the HO site was consistent with cell survival outcomes; negligible rejoining was evident at 5 hours post HO-induction in rad50-46 and rad50Δ cells (Figure 3D, right panel).
Similar defects were evident upon chronic HO-induction. Although cell survival was close to wild types, DNA sequence analysis of NHEJ junctions revealed that the rad50-46, -47, and -48 repair products predominantly exhibited the rad50Δ deletion phenotype (Moore and Haber, 1996), characterized by small insertion (mostly +CA) events (Figure 3E, right panel; NHEJ junctions are listed in Figure S4B).
These data show that NHEJ phenotypes of the rad50hook mutants are essentially identical to those imparted by truncation of the coiled coil domain (Hohl et al., 2011), and support the view that hook mutations impair the alignment of DNA ends within the globular domain during DSB repair (Williams et al., 2008), resulting in a increased fractions of imprecise repair events at the concomitant expense of reducing precise NHEJ events.
Hook mutants are defective in checkpoint functions
The Mre11 complex is a DSB sensor that activates the ATM/Tel1 pathway, which in budding yeast governs DNA damage responses (DDR) and telomere maintenance (Stracker and Petrini, 2011; Tsukamoto et al., 2001; Usui et al., 2001). As a result, Mre11 complex deficiency is associated with short telomeres (Ritchie and Petes, 2000). Telomere length was determined by Southern blotting of freshly dissected (rad50hook/RAD50) spores propagated for 10, 30 and 50 generations (Figure 4A). rad50-46 telomere length was indistinguishable from rad50Δ, but marginally longer in rad50-47 and rad50-48. These data suggested an underlying defect in Tel1 activation.
Figure 4. rad50hook mutants are defective in telomere maintenance and Tel1 checkpoint signaling.
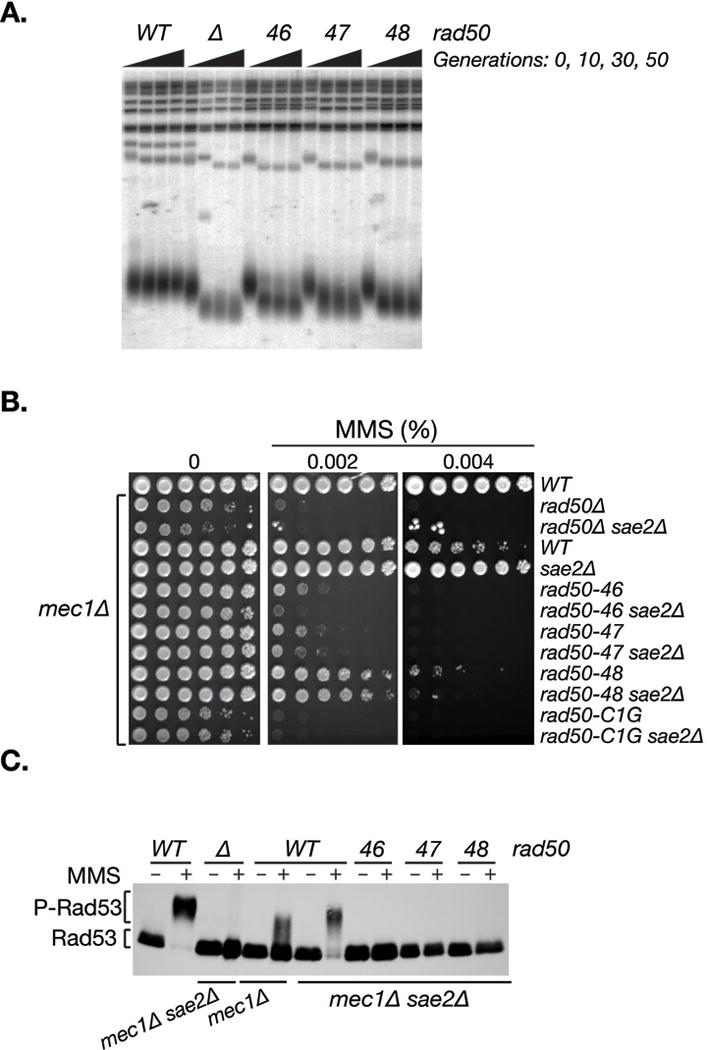
A. Telomere lengths of wild-type and rad50hook mutants after 10, 30 and 50 generations of growth at 30°C. Heterozygote diploids (RAD50/ rad50hook) were included as 0 generations of growth. B. Cell survival of rad50hook mutants in a Mec1-deficient background. All strains below the wild-type strain (top row) were mec1Δ sml1Δ. C. Tel1-dependent Rad53 phosphorylation in Mec1-deficient cells upon MMS treatment (+) assessed by western blot. The migration levels of the non-phosphorylated (Rad53) and phosphorylated form (P-Rad53) are indicated. Rad50 genotypes are given above the blot and the Mec1 and Sae2 status (either wild-type or deleted) below.
We have previously shown that Mec1-deficiency is partially suppressed in sae2Δ cells in a Tel1 and Mre11 complex dependent manner—the TM pathway, culminating in hyper-phosphorylation of the Rad53 effector kinase (Usui et al., 2001). The TM pathway thus offers a sensitive means to query the ability of Mre11 complex variants to effect Tel1 activation. In Rad50 proficient cells, in which the TM pathway is operative (Usui et al., 2001), sae2Δ suppressed mec1Δ MMS sensitivity (Figure 4B) and restored Rad53 phosphorylation (Figure 4C) to essentially wild-type levels. This suppression was absent in rad50hook mutants; the MMS sensitivity of rad50-46 mec1Δ and rad50-47 mec1Δ was indistinguishable from rad50Δ mec1Δ and rad50-C1G mec1Δ, whereas rad50-48 mec1Δ was slightly less sensitive but unaltered by sae2Δ. Rad53 activation, as inferred from phosphorylation, was absent in all three mutants (Figure 4C). These data demonstrate that the rad50hook alleles separate Mre11 complex role in DSB repair from its role in Tel1 activation and provide further evidence of the interdependence of the hook and globular domains of the Mre11 complex.
rad50hook mutations and DSB end processing
Meiosis is a specialized program, in which a DSB created by the topoisomerase II like enzyme, Spo11, induces recombination between homologous chromosomes to establish a physical linkage essential for proper chromosome segregation (Borde and de Massy, 2013). The Mre11 complex is required for Spo11-dependent DSB formation, for removal of Spo11 from the DSB end, and for timely repair of meiotic DSBs (Cherry et al., 2007; Keeney, 2008). Sporulation efficiency and viability were severely impaired in rad50-46 (1.3%, 4.3%) and rad50-47 (5.7%, 50.6%), whereas rad50-48 (63%, 88%) was closer to wild-type (81%, 96.8%) in both respects (Figure 5A).
Figure 5. Meiotic DSBs processing is impaired in rad50-46 and rad50-47 but not rad50-48.
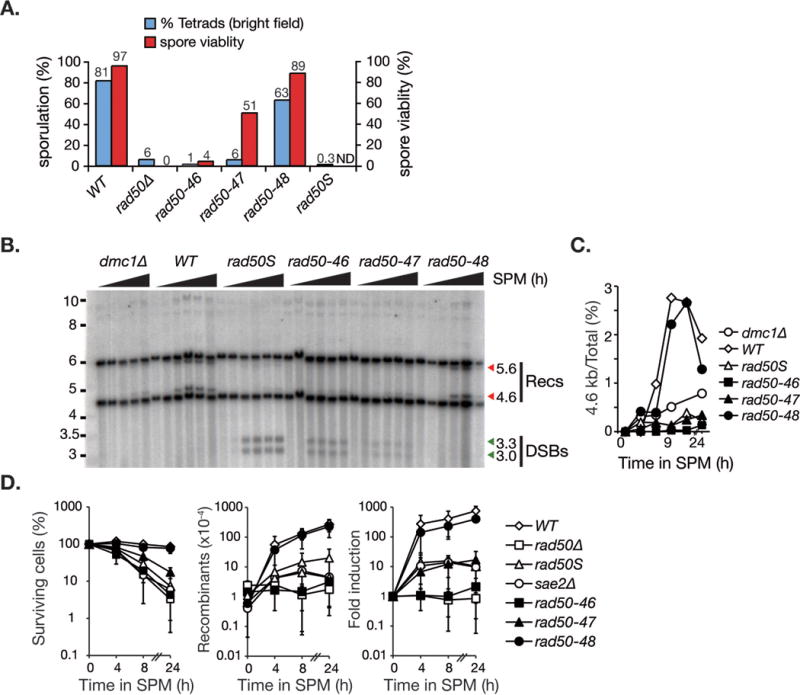
A. Sporulation efficiency (in blue) and spore viability (in red) of the indicated genotypes were assessed in SK1 homozygot diploid cells after 48 hours sporulation. The sporulation efficiency was calculated as the percentage of asci among total number of cells (% tetrads) and graphically illustrated (% sporulation, y-axis on the left). Spore viability was determined by tetrad dissection of at least 30 tetrads (% spore viability, y-axis on the right). Spore viability for rad50S/S was not determined (ND). B. Meiotic DSB formation and repair by meiotic recombination at the HIS4-LEU2 hotspot by southern blot. The migration level of the crossover recombinant fragments (Recs, recombinants; 5.6 kb and 4.6 kb, red arrows) above and below the parental band (Hunter and Kleckner, 2001; Schwacha and Kleckner, 1997) and the unprocessed 3.3 kb and 3.0 kb meiotic double strand break fragments (DSB, green arrows) are denoted. Cells were cultivated in sporulation media (SPM) for 0, 3, 6, 9 and 24 (dmc1Δ) or for 0, 3, 6, 9, 12 and 24 hours (WT, rad50S, rad50-46, rad50-47 and rad50-48). C. Relative signal intensities of the 4.6 kb recombinant fragment versus total counts per lane is shown on the right.
The sporulation defects in rad50-46 and -47 were associated with a failure to process meiotic DSBs (Figure S5), phenocopying rad50S, mre11-3 and sae2Δ which are deficient in removal of Spo11 covalently attached to the DSB end (Alani et al., 1990; Cao et al., 1990; Hunter and Kleckner, 2001; Keeney and Kleckner, 1995; Prinz et al., 1997). As expected, recombination at the HIS4LEU2 hotspot assessed by Southern blotting (Hunter and Kleckner, 2001; Schwacha and Kleckner, 1997) was essentially undetectable in rad50-46 and rad50-47 over a 24 hour time course, phenocopying dmc1Δ (the meiotic rad51 ortholog) and rad50S (Figure 5C, quantification on right), both deficient in the initiation of meiotic recombination. Genetic assessment of recombination frequencies was also consistent with the interpretation that rad50-46 and rad50-47 are largely deficient in initiation of meiotic recombination (Figure S5B).
The removal of Spo11 from DSB ends is partially dependent on the Mre11 nuclease (Moreau et al., 1999), and the data above raised the possibility that this function was impaired in rad50hook mutants. As Mre11 nuclease function also promotes the resolution of topoisomerase I adducts in vegetatively growing cells (Foster et al., 2011; Hartsuiker et al., 2009; Mimitou and Symington, 2010; Morales et al., 2008), we measured CPT sensitivity in rad50-46, -47, and -48 cells to assess the effect of rad50hook mutations on Mre11 nuclease function. In contrast to MMS and HU, rad50-46 was acutely sensitive to CPT, exhibiting at least 100 fold greater sensitivity to 5 μM CTP than either sae2Δ or mre11-3 (Figure 1C and Figure S1).
Consistent with the interpretation that the Rad50 hook domain influences Mre11 nuclease function, DNA end resection of an HO-induced DSB at the MAT locus assessed by alkaline electrophoresis and southern blot analysis in G2-arrested cells (Mantiero et al., 2007) was impaired in rad50-46 cells (Figure 6A), however not to the extent as observed in rad50Δ cells (Ivanov et al., 1994). The DSB end resection delays of rad50-47 and rad50-48 were slightly milder than the one of rad50-46 (data not shown).
Figure 6. The Mre11 nuclease function is critical for rad50-46 DSB end resection and damage survival.
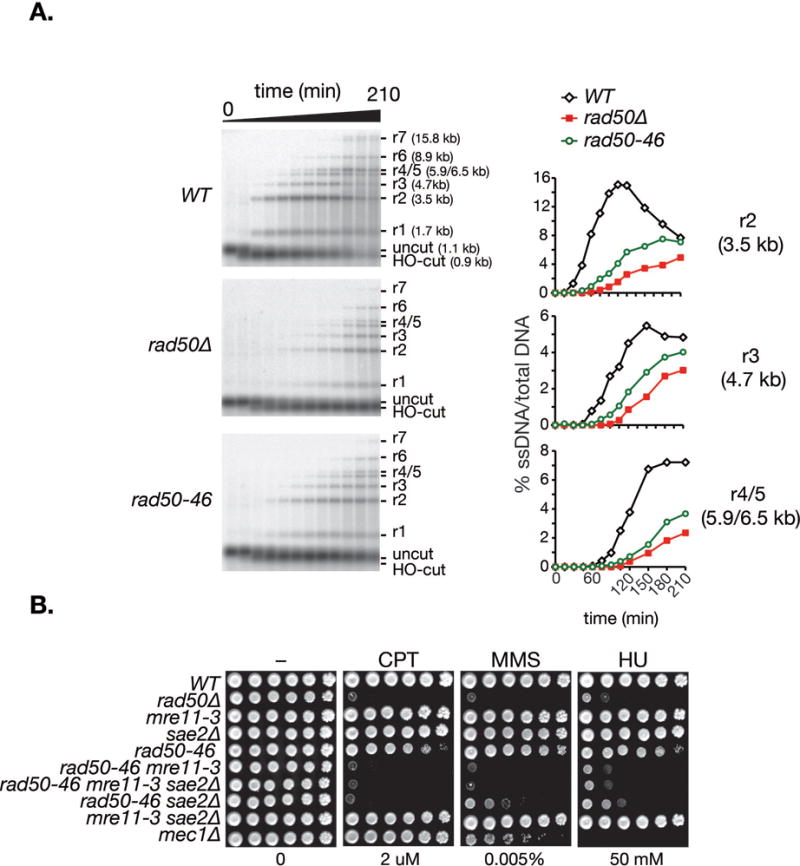
A. HO-DSB end resection in G2 arrested cells at 30°C in a time course over 210 minutes in wild-type, rad50-46 and rad50Δ cells. 5′-to-3′ end resection products of an HO-DSB are detectable with an ssRNA probe after alkaline gel electrophoresis of SspI-digested DNA. The migration levels of HO-uncut (1.1 kb), HO-cut (0.9 kb), and resection fragments r1 (1.7 kb), r2 (3.5 kb), r3 (4.7 kb), r4 (5.9 kb), r5 (6.5 kb), r6 (8.9 kb) and r7 (15.8 kb) from the HO-site (HO) are indicated. Right panel: Quantification of the r2, r3 and r4+r5 fragments of the southern blots shown. Similar results were obtained in three experiments. Note that the blots and quantifications for wild-type and rad50Δ were previously published elsewhere (Hohl et al., 2011). B. Damage survival of rad50-46 is dependent on Mre11 nuclease function and Sae2.
The extreme sensitivity of rad50-46 to CPT and the pronounced effect on DSB resection suggested that the effect of that mutation extended beyond Mre11 nuclease function. This interpretation was confirmed in rad50-46 sae2Δ and rad50-46 mre11-3 double mutants. sae2Δ, and to a greater extent mre11-3 double mutants, essentially phenocopied rad50Δ in the response to CPT, MMS or HU (Figure 6B). Consistently, loss of Sae2 also profoundly sensitized rad50-47 and rad50-48 cells to MMS and CPT (data not shown). Collectively, these data indicate that the hook domain influences Mre11 complex nuclease function and suggest that in rad50-46 (and other rad50hook mutants), it may pose an impediment to the Exo1 and Dna2, the resection machinery functioning downstream of Mre11 (Mimitou and Symington, 2009).
Intragenic rad50-46 suppressors within hook, coiled coil and globular domain
The rad50hook mutants described herein impair NHEJ, Tel1 activation, and DSB end processing, all of which appear to be mediated by the globular domain of the Mre11 complex. Mechanistic insight regarding this issue came from the identification of intragenic suppressors of the rad50-46 phenotype.
Four spontaneous chromosomal suppressors arose in the course of strain propagation. DNA sequencing of the rad50-46 ORF in the apparent revertants revealed that three of the four were intragenic; two contained a N873I mutation within the coiled coil domain, and one changed the Y688E codon to Y688K, converting rad50-46 to a mutant alike rad50-48 (S685R Y688R). The ability of N873I to suppress rad50-46 supported the idea that alterations in hook structure are transduced via the coiled coil domains, and manifest in the globular domain. Additional suppressors were obtained via random mutagenesis of the rad50-46 ORF from amino acid G78 to S1075 on a centromeric plasmid transformed into rad50Δ cells (see Figure 7A). Plasmids were recovered from MMS resistant colonies, and re-confirmed in a second rad50Δ transformation (Figure 7B and Figure S6). Eighteen additional suppressors were obtained, ten of which reverted one or both of the rad50-46 mutations. Of the eight remaining, five were within the hook domain (I680V/T, K700Q/R, L703F), one within the antiparallel coiled coil domain apposing N873I (N607Y), and two within the globular domain (P168S, S193F) (Figure 7A and Figure S6).
Figure 7. Isolation and phenotypic analysis of rad50-46 intragenic suppressors.
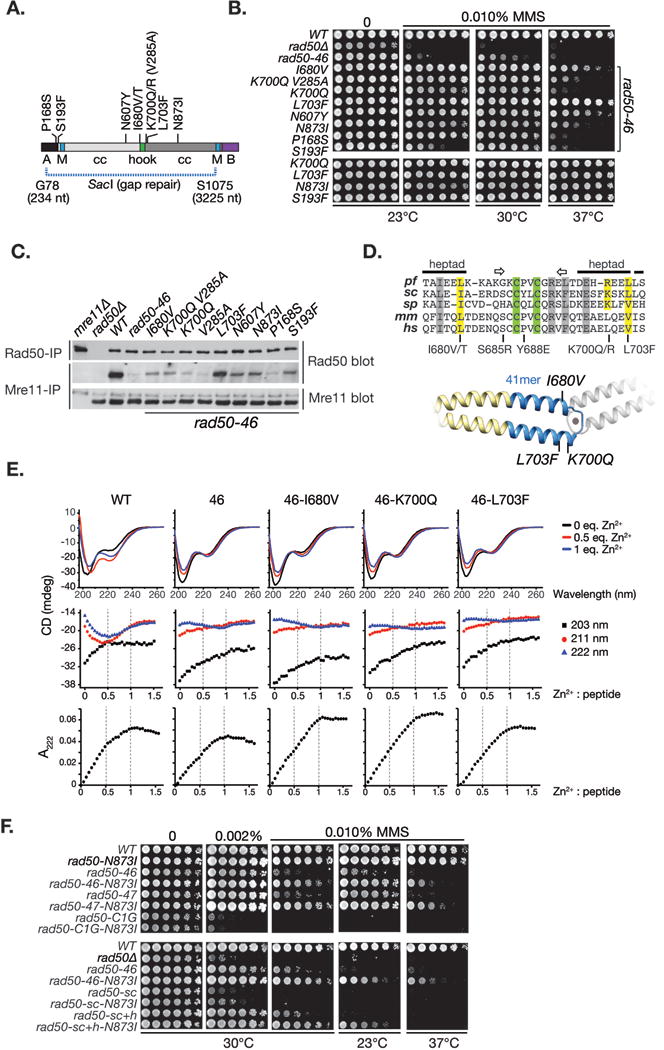
A. Schematic illustration of the genetic screen to isolate intragenic suppressors of rad50-46 MMS sensitivity at 23°C. SacI digested plasmid Ycp50-RAD50 (from nucleotide 234-3225, corresponding to amino acid G78 – S1075) was gap repaired in rad50Δ cells with a rad50-46 PCR mutagenized repair template. Cells containing the gap repaired plasmid were plated on selective media containing 0.1% MMS and grown at 23°C. Plasmid were recovered from survivor colonies, re-transformed in rad50Δ cells, and suppression was confirmed on MMS plates. Sequenced rad50-46 suppressors are given. B. rad50-46 suppressors were re-assessed by spot test on plates with 0.1% MMS, either incubated at 23°C, 30°C or 37°C. Some of the rad50-46 suppressors were also assessed in context of the wild-type hook domain (bottom 4 strains). C. Mre11 complex integrity of rad50-46 suppressor mutants was assessed by co-immunoprecipitation and western blot with Rad50 or Mre11 antisera as indicated. D. Top: Sequence alignment of the central hook domain flanked by one coiled coil heptad repeat. The conserved zinc-cysteines are highlighted in green, rad50-46 suppressor mutation in yellow and partially conserved residues in grey. Arrows denote a structurally conserved inverted beta-sheet. pf, P. furiosus; sc, S. cerevisiae; sp, S. pombe; mm, M. musculus; hs, H. sapiens. Bottom: I-tasser structure prediction of the 41 amino acid (highlighted in blue) hook dimer peptide. The approximate position of the I680V, K700Q and L703F hook suppressor mutations are denoted. E. Circular dichroism spectra and UV spectroscopy of wild-type and rad50-46 hook peptides without and with hook suppressors. F. MMS spot test to assess suppression by N873I of other hook and coiled coil mutants. Plates were incubated either at 23°C, 30°C or 37°C.
We examined the effect of suppressor mutations on the interaction between Mre11 and Rad50-46 (Figure 7C). The L703F allele restored the interaction, consistent with its pronounced effect on MMS resistance (Figure 7B). Mre11 interaction was also partially improved in I680V, K700Q V285A, N607Y, N873I and S193F, but suppression of MMS survival was not strictly correlated with restoration of Mre11 interaction, as exemplified by K700Q and P168S. That was also true for the partial suppression of the rad50-46 telomere and meiotic defects (Figure S7).
We considered two mechanistic possibilities for the observed suppression, both of which are based on the premise that changes in secondary structure were the underlying basis of rad50-46, -47, and -48 phenotypes. In one scenario, the suppressor mutations would alter the coiled coil secondary structure and thereby normalize the Rad50hook Zn2+ coordination and dimerization behavior. In the alternative scenario, manifestation of secondary structure changes distal to the hook domain mutations would be normalized in the suppressors without normalizing Zn2+ coordination behavior of the mutant.
To address these hypotheses, peptides (41-mer) spanning the hook domain and the proximal coiled coil region were synthesized. These peptides were large enough to accommodate the rad50-46 alterations in combination with I680V, K700Q or L703F suppressor mutations (Figure 7D). Changes in secondary structure induced by Zn2+ binding were assessed by circular dichroism (CD). CD titrations of the wild-type peptide exhibited characteristic inflections in ellipticity at 203, 211, and 222 nm, indicating preferential formation of the 2:1 peptide : Zn2+ complex. The CD spectra for the wild-type recorded at 2:1 and 1:1 peptide : Zn2+ ratios indicate substantial conformational changes upon Zn2+ binding (Figure 7E). Conformational changes upon Zn2+ binding of the Rad50-46 hook peptides were much less pronounced. No significant changes in the shape of CD and UV (LMCT) titration curves were observed in absence or presence of the suppressor mutations (Figure 7E) and the CD spectra at 2:1 and 1:1 peptide:Zn2+ ratios were only slightly altered by the suppressor mutations (Figure 7E, top and middle). Also, zinc binding was largely unaltered in absence or presence of suppressor mutations (Figure 7E, bottom). Consistently, in yeast 2-hybrid analysis assessing hook homodimerization, the I680V, K700Q or L703F suppressors failed to significantly alleviate reporter activation of rad50-46 hook peptides (Figure S2D).
These data suggest that suppression of rad50-46 was more likely the effect of secondary structural changes in the coiled coils distal to the hook domain than an effect on Zn2+ binding characteristics of the rad50-46 hook. This interpretation is also consistent with the observation that four of the eight suppressors fell within the coiled coil and globular domains.
If rad50-46 suppression reflected compensation for structural perturbations distal to the hook domain, we reasoned that mutations, which suppress rad50-46, would modify other mutations that impart similar alterations. Thus the ability of the N837I rad50-46 suppressor to suppress rad50-47 was assessed. The same mutation was also introduced in the rad50-C1G cysteine mutant and rad50sc and rad50sc+h, in which the coiled coil domain is truncated without or with the hook domain present respectively (Hohl et al., 2011). The N873I mutation suppressed rad50-47 and rad50sc+h (Figure 7F), indicating that suppression is not specific to rad50-46. In contrast, N873I failed to suppress rad50-C1G and rad50sc, indicating that suppression requires a functional hook domain.
In summary, the behavior of rad50hook mutants suggests that the geometry of the hook and coiled coil domain strongly influences the stability and the structural disposition of Mre11 complex globular domain. These outcomes would account for the myriad defects in functions specified by domains within the globular domain.
DISCUSSION
The Rad50 hook domain is an invariant feature of the Mre11 complex, present in all Rad50 orthologs identified. Deletion of the Rad50 hook domain phenocopies complete deficiency in any member of the complex (Hopfner et al., 2002; Stracker and Petrini, 2011). This observation does not address the possibility that only a subset of Mre11 complex functions are directly influenced by the hook domain, and others indirectly. It does appear that hook-mediated dimerization is critical, as replacement with an artificial dimerization cassette is sufficient to restore most Mre11 complex function (Lee et al., 2013; Wiltzius et al., 2005). We reasoned that hypomorphic mutations affecting the Rad50 hook domain that leave it otherwise intact would provide a means to reveal which functions are acutely dependent on the hook domain. In this study, we examined three rad50hook alleles (rad50-46, -47, and -48) affecting residues flanking the invariant cysteines of the S. cerevisiae Rad50 hook domain.
Biochemical, biophysical and genetic analyses indicate that hook-mediated dimerization is not abolished, but was compromised by these mutations. Consistent with the view that dimerization occurred in rad50hook mutants, double strand break repair by SCR was largely intact. This was illustrated by the response of the mutants to clastogens, wherein rad50-46 was most sensitive, but markedly more resistant than the rad50Δ or rad50-C1G mutants (Figure 1C). Plasmid based physical analyses of SCR over a 9 hour time course also supported the interpretation that SCR was relatively normal in the rad50hook mutants, although occurring with reduced kinetics in rad50-46 and rad50-47 (Figure 3B).
We have proposed previously that SCR requires bridging of sister chromatids via Rad50 hook domain dimerization (Hohl et al., 2011; Hopfner et al., 2002; Wiltzius et al., 2005). That model must be reconsidered in light of the data presented here, as it is contradicted by the fact that impaired hook-dependent dimerization in rad50hook mutants had only a modest impact on SCR. We consider two possibilities to account for this apparent discrepancy. Atomic force and electron microscopy show that dimerization via the hook domain can occur between Rad50 molecules within a dimeric assembly (i.e., intra-complex), or between Rad50 molecules in separate dimeric assemblies (i.e., inter-complex) (Figure 1B) (Lee et al., 2013; Moreno-Herrero et al., 2005; Williams and Tainer, 2005). Bridging of sister chromatids would presumably require inter-complex dimerization. Hence, our data may suggest that the rad50hook alleles impair intra-complex dimerization but have a relatively small impact on inter-complex interactions (Figure 1B). A second possibility is based on the observation that the Mre11 complex is abundantly and avidly associated with chromatin during S phase (Mirzoeva and Petrini, 2003). In this scenario, the increased valency of hook domain interfaces may compensate for the fact that the stability of each individual interface is weakened. Further compensation in this regard may come from cohesin, which is recruited to stalled replication forks and sites of DNA damage (Tittel-Elmer et al., 2012; Unal et al., 2004). It may be that inter-complex interactions, though labile in the mutants, are sufficiently long-lived to foster cohesin recruitment, compensating for reduction in hook-mediated bridging. In this view, the importance of SCR bridging via intercomplex interactions would be minimal.
Apart from SCR, the rad50hook mutations affected a broad range of Mre11 complex functions. The non-SCR functions affected are primarily attributed to protein domains lying within the globular domain. Based on the fact that the hook domain is so far distal to the globular domain, we propose that these outcomes reflect the potential of the hook domain to exert long range influence on Mre11 complex secondary structure, including the stability and disposition of the globular domain. It is important to consider, unlike the hook point mutations described here, that complete loss of the hook domain, either by proteolytic cleavage or by genetic deletion does not disrupt Mre11-Rad50 interaction (Hohl et al., 2011; Wiltzius et al., 2005). Hence, it is the structure of the hook, rather than its presence or absence that leads to transduction of structural information between the hook and globular domains via the coiled coils.
This potential was first suggested by the observation that mutation of the invariant cysteines disrupted the interaction between Mre11 and Rad50 (Hopfner et al., 2002), and further indicated by the impact of the rad50hook alleles on the Rad50-Mre11 interaction shown here (Figure 3A). Such an influence was also suggested by the observation that SCR was largely proficient in the rad50sc+h mutant, which harbors a truncation of the coiled coil domain abutting the hook domain, also conferred defects in NHEJ, telomere maintenance, and meiotic DSB formation (Hohl et al., 2011). The fact that rad50hook and the rad50sc+h mutations are suppressed by mutations within the coiled coil and globular domains also strongly supports the idea that changes in secondary structure underlie rad50-46 (as well as rad50-47 and rad50-48) phenotypes, and that the suppressors compensated in some way for those aberrant secondary structures. Collectively, the data suggest that rad50hook alleles are not simply hypomorphic. Rather, they provide separations of function that reveal the long range influences of the hook domain.
Tel1 activation defects are common to all rad50hook (Figure 4) and rad50coils mutations (Hohl et al., 2011). Recent biochemical analyses of the human Mre11 complex provide a potential mechanistic basis for this observation (Lee et al., 2013). The data suggest that the primary functional significance of intra-complex hook-mediated dimerization, which is compromised in the rad50hook alleles, is to promote assembly of Rad50 and Mre11 dimers within the globular domain, facilitating ATP binding and hydrolysis critical for ATM (and likely Tel1) activation (Lee et al., 2013).
In this regard, rad50-48 was unique. This mutant was essentially wild-type in all respects but defective in Tel1 activation. Nevertheless, it was indistinguishable from rad50-46 and -47 mutant peptides with respect to impaired dimerization inferred from LMCT (Figure 2B) and yeast 2-hybrid analysis (Figure S2C). Hence, in this instance, impaired dimerization was circumscribed, and linked only to defective Tel1 activation, consistent with the view that intra-complex dimerization is required for this Mre11 complex function (Lee et al., 2013). The broader phenotypic effects of rad50-46 and -47 likely reflect that those mutations impart more extensive secondary structural alterations. Supporting this view, the severity of rad50hook phenotypes is correlated with the extent to which interaction between Rad50 and Mre11 is compromised; most in rad50-46, least in rad50-48 (Figure 3A).
The effect of the hook domain on Tel1 activation is conserved. We established rad50-46 and rad50-47 mouse mutants, and found that ATM activation was abolished in Rad5046/Δ cells (Roset et al., 2014). Notably, neither homozygote was viable at either the cellular or organismal level, but both alleles exerted a pronounced dominant negative effect. Rad5046/+ mice were prone to liver cancer, and exhibited myriad abnormalities, whereas Rad5047/+ mice were inviable, indicating that the Rad5047 allele had a dominant lethal effect. These data indicate that the influence of the hook domain interface is conserved and can be exerted across the interface to alter the function of wild-type interacting partners (Roset et al., 2014).
Collectively these data demonstrate that the dimerization state of the Rad50 hook domain exerts diverse influences on Mre11 complex function. The influence appears to be manifest, at least in part as structural modulation of the globular domain. On this basis, we propose that the transitions between the open and closed forms of the Mre11 complex described for the globular domain (Lim et al., 2011; Williams et al., 2011; Wyman et al., 2011) are interdependent with transitions in the mode of hook-mediated dimerization. Together, these large scale structural transitions of the complex likely hold the key(s) to the regulation of its diverse functions in the DDR.
EXPERIMENTAL PROCEDURES
Yeast strains and manipulations
Yeast strains used in this study were in the W303 (RAD5), DBY745 or SK1 background and are listed in Table S1. Details of yeast manipulations, plasmid and yeast strain constructions are specified in the Supplemental Experimental Procedures or are available upon request. P values were calculated using the two tailed Wilcoxon rank sum test.
Fluorescence anisotropy, UV titration and CD spectroscopy
Biophysical experiments were performed as previously described (Kochanczyk et al., 2013) and further information are provided in the Supplemental Experimental Procedures.
Other Yeast methods
DNA damage survival assays, yeast extract preparations, co-immunoprecipitation (CoIP), sister chromatid recombination, heteroallelic recombination, nonhomologous end-joining, telomere length and HO-DSB end resection were assessed essentially as previously described (Hohl et al., 2011) with minor modifications specified in the Supplemental Experimental Procedures section.
Supplementary Material
Mre11 nuclease, NHEJ, and Tel1/ATM activation depend on the Rad50 hook domain.
Rad50 hook domain mutation effects separation of checkpoint and repair functions.
Mutations in the coiled coil domain suppress defects in the Rad50 hook domain.
The Rad50 hook domain is partially dispensable for sister chromatid recombination.
Hohl and colleagues show that the Rad50 hook domain, a Zn2+ dependent homodimerization interface, influences DNA repair and checkpoint functions of the Mre11 complex. They provide evidence that these influences reflect long range conformational changes in distal regions of the complex controlled by the Rad50 hook domain.
Acknowledgments
This article is dedicated to the memory of Marlis Hohl.
We are grateful to Jim Haber (Brandeis University), Lorraine Symington (Columbia University), Scott Keeney (Memorial Sloan Kettering Cancer Center) for yeast strains, reagents and technical support. We thank current and former members of the Petrini laboratory, Tom Kelly and Neil Hunter for critical reading of the manuscript and insightful comments. This work was supported by GM56888 (J.H.J.P.), PBZH33-112756 and PA0033-117484 from the Swiss National Science Foundation and the Eugen and Elisabeth Schellenberg Foundation (M.H.), BFU2010-16372 and Consolider CSD2007-0015 grants from the Spanish Ministry of Economy and Competitiveness (A.A.), 2012/07/E/NZ1/01894 from National Science Centre and FG1/2010, F1/2010/P/2013 from the Foundation for Polish Science (A.K.) and DI2011 031341 from Polish Ministry of Science and Higher Education (T.K.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
MH, TK and CT performed experiments and analyzed the data. JHJP designed research in consultation with AA and AK. JHJP and MH wrote the manuscript.
References
- Al-Ahmadie H, Iyer G, Hohl M, Asthana S, Inagaki A, Schultz N, Hanrahan AJ, Scott SN, Brannon AR, McDermott GC, et al. Synthetic lethality in ATM-deficient RAD50-mutant tumors underlie outlier response to cancer therapy. Cancer discovery. 2014 doi: 10.1158/2159-8290.CD-14-0380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alani E, Padmore R, Kleckner N. Analysis of wild-type and rad50 mutants of yeast suggests an intimate relationship between meiotic chromosome synapsis and recombination. Cell. 1990;61:419–436. doi: 10.1016/0092-8674(90)90524-i. [DOI] [PubMed] [Google Scholar]
- Borde V, de Massy B. Programmed induction of DNA double strand breaks during meiosis: setting up communication between DNA and the chromosome structure. Current opinion in genetics & development. 2013;23:147–155. doi: 10.1016/j.gde.2012.12.002. [DOI] [PubMed] [Google Scholar]
- Bressan DA, Baxter BK, Petrini JH. The Mre11-Rad50-Xrs2 protein complex facilitates homologous recombination-based double-strand break repair in Saccharomyces cerevisiae. Molecular and cellular biology. 1999;19:7681–7687. doi: 10.1128/mcb.19.11.7681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L, Alani E, Kleckner N. A pathway for generation and processing of double-strand breaks during meiotic recombination in S. cerevisiae. Cell. 1990;61:1089–1101. doi: 10.1016/0092-8674(90)90072-m. issn: 0092-8674. [DOI] [PubMed] [Google Scholar]
- Cherry SM, Adelman CA, Theunissen JW, Hassold TJ, Hunt PA, Petrini JH. The Mre11 complex influences DNA repair, synapsis, and crossing over in murine meiosis. Curr Biol. 2007;17:373–378. doi: 10.1016/j.cub.2006.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes-Ledesma F, Aguilera A. Double-strand breaks arising by replication through a nick are repaired by cohesin-dependent sister-chromatid exchange. EMBO reports. 2006;7:919–926. doi: 10.1038/sj.embor.7400774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jager M, Trujillo KM, Sung P, Hopfner KP, Carney JP, Tainer JA, Connelly JC, Leach DR, Kanaar R, Wyman C. Differential arrangements of conserved building blocks among homologs of the Rad50/Mre11 DNA repair protein complex. Journal of molecular biology. 2004;339:937–949. doi: 10.1016/j.jmb.2004.04.014. [DOI] [PubMed] [Google Scholar]
- Deshpande RA, Williams GJ, Limbo O, Williams RS, Kuhnlein J, Lee JH, Classen S, Guenther G, Russell P, Tainer JA, et al. ATP-driven Rad50 conformations regulate DNA tethering, end resection, and ATM checkpoint signaling. The EMBO journal. 2014;33:482–500. doi: 10.1002/embj.201386100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duursma AM, Driscoll R, Elias JE, Cimprich KA. A role for the MRN complex in ATR activation via TOPBP1 recruitment. Molecular cell. 2013;50:116–122. doi: 10.1016/j.molcel.2013.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster SS, Balestrini A, Petrini JH. Functional interplay of the Mre11 nuclease and Ku in the response to replication-associated DNA damage. Molecular and cellular biology. 2011;31:4379–4389. doi: 10.1128/MCB.05854-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Barrera S, Cortés-Ledesma F, Wellinger RE, Aguilera A. Equal Sister Chromatid Exchange Is a Major Mechanism of Double-Strand Break Repair in Yeast. Molecular cell. 2003;11:1661–1671. doi: 10.1016/s1097-2765(03)00183-7. [DOI] [PubMed] [Google Scholar]
- Hamman BD, Oleinikov AV, Jokhadze GG, Traut RR, Jameson DM. Dimer/monomer equilibrium and domain separations of Escherichia coli ribosomal protein L7/L12. Biochemistry. 1996;35:16680–16686. doi: 10.1021/bi9624189. [DOI] [PubMed] [Google Scholar]
- Hartsuiker E, Neale MJ, Carr AM. Distinct requirements for the Rad32(Mre11) nuclease and Ctp1(CtIP) in the removal of covalently bound topoisomerase I and II from DNA. Molecular cell. 2009;33:117–123. doi: 10.1016/j.molcel.2008.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohl M, Kwon Y, Galvan SM, Xue X, Tous C, Aguilera A, Sung P, Petrini JH. The Rad50 coiled-coil domain is indispensable for Mre11 complex functions. Nature structural & molecular biology. 2011;18:1124–1131. doi: 10.1038/nsmb.2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopfner KP. ATP puts the brake on DNA double-strand break repair: A new study shows that ATP switches the Mre11-Rad50-Nbs1 repair factor between signaling and processing of DNA ends. BioEssays : news and reviews in molecular, cellular and developmental biology. 2014;36:1170–1178. doi: 10.1002/bies.201400102. [DOI] [PubMed] [Google Scholar]
- Hopfner KP, Craig L, Moncalian G, Zinkel RA, Usui T, Owen BA, Karcher A, Henderson B, Bodmer JL, McMurray CT, et al. The Rad50 zinc-hook is a structure joining Mre11 complexes in DNA recombination and repair. Nature. 2002;418:562–566. doi: 10.1038/nature00922. [DOI] [PubMed] [Google Scholar]
- Huang KN, Symington LS. Mutation of the gene encoding protein kinase C 1 stimulates mitotic recombination in Saccharomyces cerevisiae. Molecular and cellular biology. 1994;14:6039–6045. doi: 10.1128/mcb.14.9.6039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter N, Kleckner N. The single-end invasion: an asymmetric intermediate at the double-strand break to double-holliday junction transition of meiotic recombination. Cell. 2001;106:59–70. doi: 10.1016/s0092-8674(01)00430-5. [DOI] [PubMed] [Google Scholar]
- Ivanov EL, Sugawara N, White CI, Fabre F, Haber JE. Mutations in XRS2 and RAD50 delay but do not prevent mating-type switching in Saccharomyces cerevisiae. Mol Cell Biol. 1994;14:3414–3425. doi: 10.1128/mcb.14.5.3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeney S. Spo11 and the Formation of DNA Double-Strand Breaks in Meiosis. Genome dynamics and stability. 2008;2:81–123. doi: 10.1007/7050_2007_026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeney S, Kleckner N. Covalent protein-DNA complexes at the 5′ strand termini of meiosis-specific double-strand breaks in yeast. Proc Natl Acad Sci USA. 1995;92:11274–11278. doi: 10.1073/pnas.92.24.11274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochanczyk T, Jakimowicz P, Krezel A. Femtomolar Zn(II) affinity of minimal zinc hook peptides–a promising small tag for protein engineering. Chemical communications. 2013;49:1312–1314. doi: 10.1039/c2cc38174e. [DOI] [PubMed] [Google Scholar]
- Lammens K, Bemeleit DJ, Mockel C, Clausing E, Schele A, Hartung S, Schiller CB, Lucas M, Angermuller C, Soding J, et al. The Mre11:Rad50 structure shows an ATP-dependent molecular clamp in DNA double-strand break repair. Cell. 2011;145:54–66. doi: 10.1016/j.cell.2011.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Mand MR, Deshpande RA, Kinoshita E, Yang SH, Wyman C, Paull TT. Ataxia telangiectasia-mutated (ATM) kinase activity is regulated by ATP-driven conformational changes in the Mre11/Rad50/Nbs1 (MRN) complex. The Journal of biological chemistry. 2013;288:12840–12851. doi: 10.1074/jbc.M113.460378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim HS, Kim JS, Park YB, Gwon GH, Cho Y. Crystal structure of the Mre11-Rad50-ATPgammaS complex: understanding the interplay between Mre11 and Rad50. Genes & development. 2011;25:1091–1104. doi: 10.1101/gad.2037811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantiero D, Clerici M, Lucchini G, Longhese MP. Dual role for Saccharomyces cerevisiae Tel1 in the checkpoint response to double-strand breaks. EMBO reports. 2007;8:380–387. doi: 10.1038/sj.embor.7400911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimitou EP, Symington LS. DNA end resection: many nucleases make light work. DNA repair. 2009;8:983–995. doi: 10.1016/j.dnarep.2009.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimitou EP, Symington LS. Ku prevents Exo1 and Sgs1-dependent resection of DNA ends in the absence of a functional MRX complex or Sae2. The EMBO journal. 2010;29:3358–3369. doi: 10.1038/emboj.2010.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzoeva OK, Petrini JH. DNA replication-dependent nuclear dynamics of the Mre11 complex. Mol Cancer Res. 2003;1:207–218. [PubMed] [Google Scholar]
- Moore JK, Haber JE. Cell cycle and genetic requirements of two pathways of nonhomologous end-joining repair of double-strand breaks in Saccharomyces cerevisiae. Mol Cell Biol. 1996;16:2164–2173. doi: 10.1128/mcb.16.5.2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales M, Liu Y, Laiakis EC, Morgan WF, Nimer SD, Petrini JH. DNA damage signaling in hematopoietic cells: a role for Mre11 complex repair of topoisomerase lesions. Cancer Res. 2008;68:2186–2193. doi: 10.1158/0008-5472.CAN-07-2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau S, Ferguson JR, Symington LS. The nuclease activity of Mre11 is required for meiosis but not for mating type switching, end joining, or telomere maintenance. Molecular and cellular biology. 1999;19:556–566. doi: 10.1128/mcb.19.1.556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Herrero F, de Jager M, Dekker NH, Kanaar R, Wyman C, Dekker C. Mesoscale conformational changes in the DNA-repair complex Rad50/Mre11/Nbs1 upon binding DNA. Nature. 2005;437:440–443. doi: 10.1038/nature03927. [DOI] [PubMed] [Google Scholar]
- Prinz S, Amon A, Klein F. Isolation of COM1, a new gene required to complete meiotic double-strand break-induced recombination in Saccharomyces cerevisiae. Genetics. 1997;146:781–795. doi: 10.1093/genetics/146.3.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie KB, Petes TD. The Mre11p/Rad50p/Xrs2p complex and the Tel1p function in a single pathway for telomere maintenance in yeast. Genetics. 2000;155:475–479. doi: 10.1093/genetics/155.1.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojowska A, Lammens K, Seifert FU, Direnberger C, Feldmann H, Hopfner KP. Structure of the Rad50 DNA double-strand break repair protein in complex with DNA. The EMBO journal. 2014 doi: 10.15252/embj.201488889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roset R, Inagaki A, Hohl M, Brenet F, Lafrance-Vanasse J, Lange J, Scandura JM, Tainer JA, Keeney S, Petrini JH. The Rad50 hook domain regulates DNA damage signaling and tumorigenesis. Genes & development. 2014;28:451–462. doi: 10.1101/gad.236745.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwacha A, Kleckner N. Interhomolog bias during meiotic recombination: meiotic functions promote a highly differentiated interhomolog-only pathway. Cell. 1997;90:1123–1135. doi: 10.1016/s0092-8674(00)80378-5. [DOI] [PubMed] [Google Scholar]
- Shiotani B, Nguyen HD, Hakansson P, Marechal A, Tse A, Tahara H, Zou L. Two distinct modes of ATR activation orchestrated by Rad17 and Nbs1. Cell reports. 2013;3:1651–1662. doi: 10.1016/j.celrep.2013.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiff T, Reis C, Alderton GK, Woodbine L, O’Driscoll M, Jeggo PA. Nbs1 is required for ATR-dependent phosphorylation events. The EMBO journal. 2004 doi: 10.1038/sj.emboj.7600504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stracker TH, Petrini JH. The MRE11 complex: starting from the ends. Nat Rev Mol Cell Biol. 2011;12:90–103. doi: 10.1038/nrm3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stracker TH, Roig I, Knobel PA, Marjanovic M. The ATM signaling network in development and disease. Frontiers in genetics. 2013;4:37. doi: 10.3389/fgene.2013.00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tittel-Elmer M, Lengronne A, Davidson MB, Bacal J, Francois P, Hohl M, Petrini JH, Pasero P, Cobb JA. Cohesin association to replication sites depends on rad50 and promotes fork restart. Molecular cell. 2012;48:98–108. doi: 10.1016/j.molcel.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukamoto Y, Taggart AK, Zakian VA. The role of the Mre11-Rad50-Xrs2 complex in telomerase-mediated lengthening of Saccharomyces cerevisiae telomeres. Curr Biol. 2001;11:1328–1335. doi: 10.1016/s0960-9822(01)00372-4. [DOI] [PubMed] [Google Scholar]
- Unal E, Arbel-Eden A, Sattler U, Shroff R, Lichten M, Haber JE, Koshland D. DNA damage response pathway uses histone modification to assemble a double-strand break-specific cohesin domain. Molecular cell. 2004;16:991–1002. doi: 10.1016/j.molcel.2004.11.027. [DOI] [PubMed] [Google Scholar]
- Usui T, Ogawa H, Petrini JH. A DNA damage response pathway controlled by Tel1 and the Mre11 complex. Molecular cell. 2001;7:1255–1266. doi: 10.1016/s1097-2765(01)00270-2. [DOI] [PubMed] [Google Scholar]
- van der Linden E, Sanchez H, Kinoshita E, Kanaar R, Wyman C. RAD50 and NBS1 form a stable complex functional in DNA binding and tethering. Nucleic Acids Res. 2009;37:1580–1588. doi: 10.1093/nar/gkn1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams GJ, Williams RS, Williams JS, Moncalian G, Arvai AS, Limbo O, Guenther G, SilDas S, Hammel M, Russell P, et al. ABC ATPase signature helices in Rad50 link nucleotide state to Mre11 interface for DNA repair. Nature structural & molecular biology. 2011;18:423–431. doi: 10.1038/nsmb.2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RS, Moncalian G, Williams JS, Yamada Y, Limbo O, Shin DS, Groocock LM, Cahill D, Hitomi C, Guenther G, et al. Mre11 dimers coordinate DNA end bridging and nuclease processing in double-strand-break repair. Cell. 2008;135:97–109. doi: 10.1016/j.cell.2008.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RS, Tainer JA. A nanomachine for making ends meet: MRN is a flexing scaffold for the repair of DNA double-strand breaks. Molecular cell. 2005;19:724–726. doi: 10.1016/j.molcel.2005.07.006. [DOI] [PubMed] [Google Scholar]
- Wiltzius JJ, Hohl M, Fleming JC, Petrini JH. The Rad50 hook domain is a critical determinant of Mre11 complex functions. Nature structural & molecular biology. 2005;12:403–407. doi: 10.1038/nsmb928. [DOI] [PubMed] [Google Scholar]
- Wyman C, Lebbink J, Kanaar R. Mre11-Rad50 complex crystals suggest molecular calisthenics. DNA repair. 2011;10:1066–1070. doi: 10.1016/j.dnarep.2011.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.