

Abstract
Cleft palate is a common birth defect in humans. Therefore, understanding the molecular genetics of palate development is important from both scientific and medical perspectives. Lhx6 and Lhx8 encode LIM homeodomain transcription factors, and inactivation of both genes in mice resulted in profound craniofacial defects including cleft secondary palate. The initial outgrowth of the palate was severely impaired in the mutant embryos, due to decreased cell proliferation. Through genome-wide transcriptional profiling, we discovered that p57Kip2 (Cdkn1c), encoding a cell cycle inhibitor, was up-regulated in the prospective palate of Lhx6−/−;Lhx8−/− mutants. p57Kip2 has been linked to Beckwith–Wiedemann syndrome and IMAGe syndrome in humans, which are developmental disorders with increased incidents of palate defects among the patients. To determine the molecular mechanism underlying the regulation of p57Kip2 by the Lhx genes, we combined chromatin immunoprecipitation, in silico search for transcription factor-binding motifs, and in vitro reporter assays with putative cis-regulatory elements. The results of these experiments indicated that LHX6 and LHX8 regulated p57Kip2 via both direct and indirect mechanisms, with the latter mediated by Forkhead box (FOX) family transcription factors. Together, our findings uncovered a novel connection between the initiation of palate development and a cell cycle inhibitor via LHX. We propose a model in which Lhx6 and Lhx8 negatively regulate p57Kip2 expression in the prospective palate area to allow adequate levels of cell proliferation and thereby promote normal palate development. This is the first report elucidating a molecular genetic pathway downstream of Lhx in palate development.
Introduction
Craniofacial abnormalities are a major class of birth defects in humans. For example, cleft palate affects 1 in ∼1000 births, and it can lead to serious physical (eating difficulty, ear infection) and socio-psychological (speech, self-esteem) problems (1–5). The current treatment for cleft palate often requires repeat surgeries and lengthy therapies by a team of multidisciplinary professionals (6–8), imposing significant burden to the patients, families and the society. To devise novel and improved strategies against the cleft palate defect, it is essential to gain detailed knowledge on the molecular mechanisms underlying normal and abnormal palate development (palatogenesis).
Mice provide an excellent model system for studying palatogenesis because the process is highly conserved between mice and humans (9–11). In both organisms, the orofacial region develops from the embryonic facial primordia known as the frontonasal prominence and the first pharyngeal arch, which are bulges of largely neural crest-derived mesenchyme covered with epithelium (12,13). The first pharyngeal arch gives rise to the jaw, and it is further divided into the maxillary arch (prospective upper jaw) and the mandibular arch (prospective lower jaw). The primary palate forms from the fusion of the frontonasal prominence and maxillary arches at the rostral end of the face, around embryonic day (E) 10.5 in mice (mouse gestation is 19 days). The secondary palate develops more caudally, from the medial side of the maxillary arches. It first appears as a bilateral outgrowth of the palatal anlagen, called palatal shelves, on either side of the tongue at ∼E11.5. Subsequently, the palatal shelves elongate vertically, elevate themselves into a horizontal position above the tongue (∼E14), grow toward each other and fuse at the midline at ∼E16.5 to complete the palatogenesis (9–11). This process is under tight genetic regulation, and perturbation in any of the steps can lead to cleft palate.
Studies of mouse mutants have identified a large number of genes regulating various stages of palatogenesis (10,11). These include components of major signaling pathways in development, such as FGF, BMP, SHH, TGFβ, WNT and Notch pathways, and transcription factor genes such as Dlx1/2, Msx1, Msx2, Osr2, Shox2, Pax9, Mn1, Tbx22 and Irf6. Many of these genes have also been linked to syndromic and/or non-syndromic cleft palate in humans (14–17). However, the molecular genetics of palatogenesis are incompletely understood. In particular, little is known about the factors that regulate the initiation of palatogenesis following the establishment of the facial primordia (10,11).
LIM domain homeodomain (LIM-HD) proteins are a family of transcription factors characterized by two cysteine-rich LIM domains for protein–protein interactions and a homeodomain for binding DNA (18). Mammals have 12 LIM-HD proteins (ISL1, ISL2, LHX1-LHX6, LHX8, LHX9, LMX1A and LMX1B), which can be classified into 6 paralogous pairs based on sequence homology (18). Data mainly from the studies on LHX3 have indicated that LHX proteins function in multimeric complexes (18,19). The LIM domains of an LHX protein bind to the LIM-interaction domain of LIM domain binding protein 1 (LDB1), a ubiquitously expressed co-factor. Because LDB1 also has a self-dimerization domain, a tetramer of two LDB1 and two LHX proteins forms. If more than one types of LHX proteins are expressed in a cell, the LHX–LDB1 complexes can contain different combinations of LHX proteins, which may result in transcriptional complexes with different DNA target specificity.
While many Lhx genes are involved in tissue patterning and cell differentiation during embryonic development, a pair of paralogues, Lhx6 and Lhx8, plays the most prominent role in the developing face (20–22). The two genes are expressed in the oral mesenchyme of the first pharyngeal arch from ∼E9.5, and subsequently in the developing palate and teeth (20,21). Inactivating Lhx6 in mice (Lhx6−/−) caused reduction of the palatal processes of maxilla bone without overt cleft (23,24). Lhx8−/− mutants had cleft secondary palate with 60% penetrance, and this phenotype was attributed to the deficiency in the horizontal growth of the palatal shelves after elevation (21). Importantly, simultaneous inactivation of Lhx6 and Lhx8 (Lhx6−/−;Lhx8−/−) led to fully penetrant cleft secondary palate (22), indicating that the two genes play crucial yet overlapping roles in palatogenesis. However, no molecular and cellular details of this phenotype have been reported to date. In humans, LHX8 has been linked to an increased risk of cleft palate and a familial case of cleft lip (23,25,26).
Here, we describe a detailed pathology of the cleft palate defect of Lhx6−/−;Lhx8−/− mutants. We discovered that the ablation of Lhx6 and Lhx8 severely impaired the outgrowth of the palatal shelves from the onset of the palatogenesis, and this was attributed to decreased cell proliferation in the prospective palate area of the mutant maxillary arches. Furthermore, we characterized the molecular mechanism underlying the function of LHX6 and LHX8 in promoting palatal growth, which is to repress a cell cycle inhibitor gene p57Kip2 in the maxillary arches.
Results
Lhx6−/−;Lhx8−/− mutants exhibit deficient outgrowth of the palatal shelves from the onset of palatogenesis
While a previous study reported that mice missing three or four copies of Lhx6 and Lhx8 (Lhx6−/−;Lhx8+/−, Lhx6+/−;Lhx8−/−, Lhx6−/−;Lhx8−/−) had fully penetrant cleft secondary palate (22), details of this phenotype were not described. Therefore, we examined the morphology of the palate in Lhx6 and Lhx8 compound mutants throughout palatogenesis (E11.5–E16.5) to determine which step was regulated by the Lhx genes.
In Lhx6−/−;Lhx8−/− mutants, the palatal defect was apparent from the initial stage of the palatogenesis (E11.5) as severe hypoplasia of the palatal shelves at the posterior region of the maxillary arches (Fig. 1E, H and I). The mutant palatal shelves remained small at subsequent stages and failed to elevate above the tongue (Fig. 1N, Q, V and Y). Eventually, they were retracted to the lateral wall of the oral cavity (Fig. 1Z and c). The palatal defect of Lhx6−/−;Lhx8−/− mutants was less pronounced at an anterior level than at a posterior level (Fig. 1A, D, J, M, R and U). However, the cleft extended along the entire length of the secondary palate in the mutants (Fig. 1U, Y and C) (22).
Figure 1.

Morphological analysis of the palate defect caused by inactivation of Lhx6 and Lhx8. Coronal sections of the head of mouse embryos were stained by cresyl violet to visualize tissue morphology. Only the right half of the face is shown in A–H and J–Q. The dotted lines in E and H indicate the boundary of the palatal shelf used for the area measurements shown in I. The error bars in I are standard deviations of the data from three embryos per genotype. **P < 0.01 from Student's t-test. Bidirectional arrows in N–P indicate the vertical length and width of each palatal shelf. PS, palatal shelf; To, tongue. Scale bar = 0.25 mm.
The phenotypes of Lhx6−/−;Lhx8+/− and Lhx6+/−;Lhx8−/− mutants provided additional insights into the function of Lhx6 and Lhx8 in palate development. In these mutants, the size of the palatal shelves at E11.5 and E13.5 appeared to be in between those of wild types and Lhx6−/−;Lhx8−/− mutants (Fig. 1E–H, N–Q), suggesting that the dosage of the LHX proteins is important for palatal growth. The next step, elevation of the palatal shelves, was impaired in both Lhx6−/−;Lhx8+/− and Lhx6+/−;Lhx8−/− embryos (Fig. 1S, T, W, X and b). Although the mutant palatal shelves were eventually positioned above the tongue by E16.5 (Lhx6−/−; Lhx8+/−) or E18.5 (Lhx6+/−;Lhx8−/−) (Fig. 1A; data not shown), they remained widely separated at birth (data not shown).
Ablation of Lhx6 and Lhx8 leads to decreased cell proliferation in the maxillary arch
To determine the mechanism underlying the regulation of palatal outgrowth by Lhx6 and Lhx8, we performed a series of cellular and molecular analyses on Lhx6−/−;Lhx8−/− mutants. Because the hypoplasia of the mutant palatal shelves was evident from E11.5, we focused the investigation on E10.5 maxillary arches to uncover causative changes.
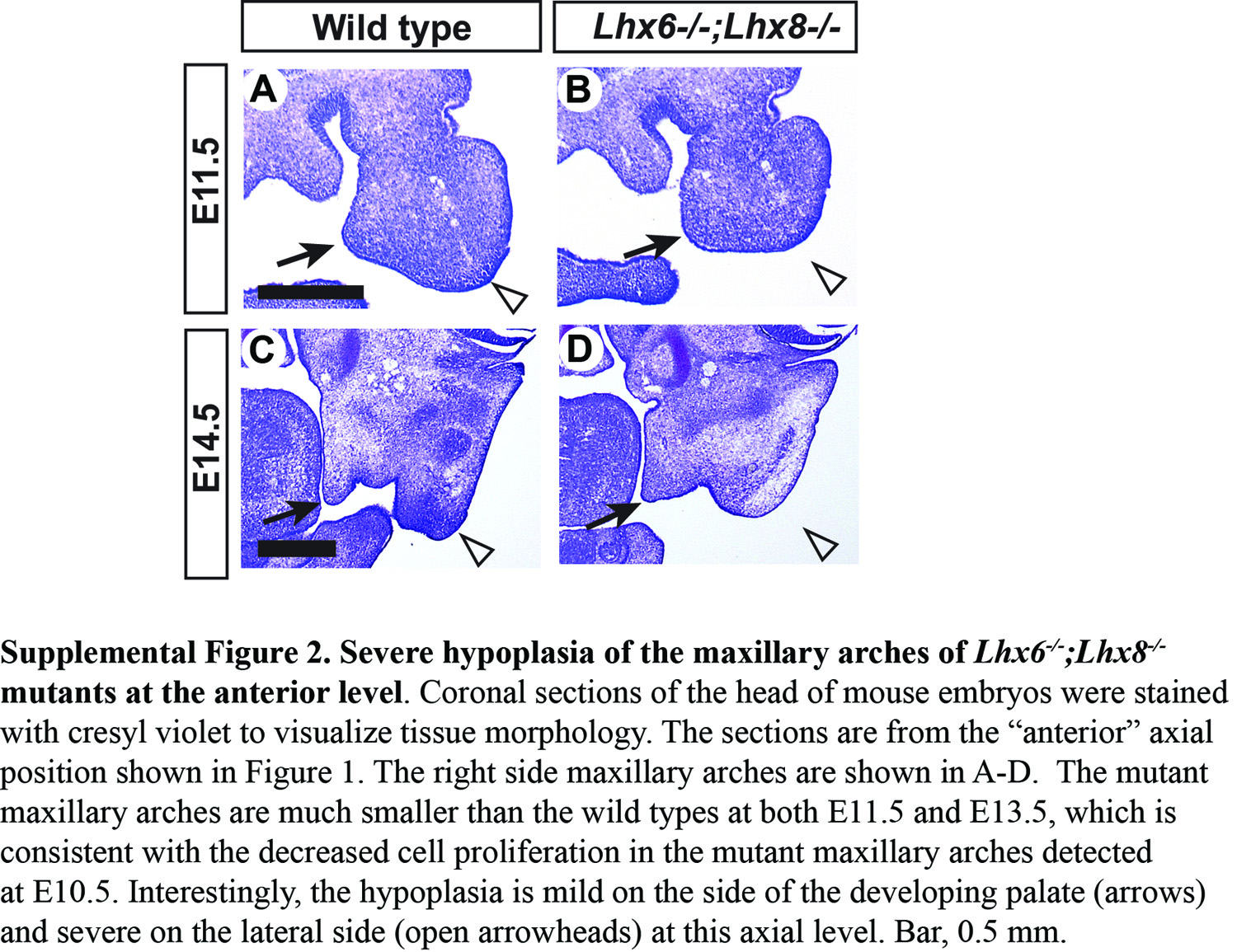
We examined apoptosis and cell proliferation in the normal expression domain of Lhx6 and Lhx8 within the maxillary arch (demarcated in Fig. 2A and B), using cleaved caspase-3 and phospho-histone H3 as respective markers. The apoptosis appeared comparable between Lhx6−/−;Lhx8−/− mutant and wild-type maxillary arches, with very few apoptotic cells detected in both groups (Supplementary Material, Fig. S1). On the other hand, cell proliferation was severely reduced in the Lhx-mutant maxillary arches, and this was consistent along the entire antero-posterior axis (Fig. 2A–C). While we noted earlier that the growth deficiency in the Lhx6−/−;Lhx8−/−-mutant palatal shelves was relatively mild at the anterior level (Fig. 1), the maxillary arch as a whole was severely hypoplastic at this position (Supplementary Material, Fig. S2). Therefore, our cell proliferation results were consistent with the morphological phenotype. Combining the above data, we concluded that the palatal growth defect of Lhx6−/−;Lhx8−/− mutants was attributable to the decrease in cell proliferation in the maxillary arches.
Figure 2.
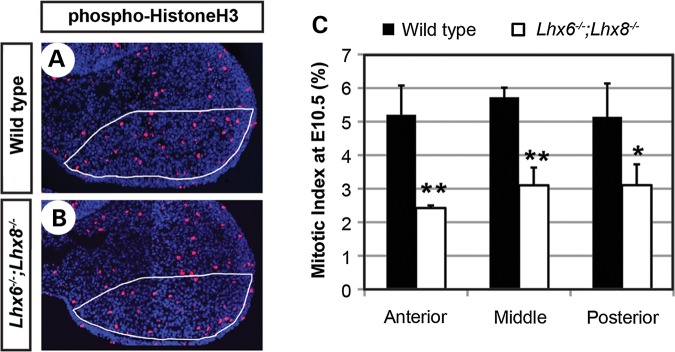
Reduced cell proliferation in the maxillary arches of Lhx6−/−;Lhx8−/− mutants. (A and B) Coronal sections of the head of E10.5 embryos stained for nuclei (blue) and phospho-Histone H3 (red). Only the right maxillary arch is shown. The white lines demarcate an approximate area of Lhx6 and Lhx8 expression during normal development, from which the mitotic indices in C were measured. (C) The comparison of mitotic indices between wild-type and Lhx6−/−;Lhx8−/− mutant embryos at three positions along the antero-posterior axis of the maxillary arch. The error bars are standard deviations of the data from three embryos per genotype. **P < 0.01, *P < 0.05 from Student's t-test.
Genome-wide transcriptional profiling identifies the genes that are differentially expressed between Lhx6−/−;Lhx8−/− mutant and wild-type maxillary arches
Because Lhx6 and Lhx8 encode transcription factors, the regulation of cell proliferation by these genes can only be indirect, through changes in the expression of other genes. Therefore, we performed genome-wide transcriptional profiling to identify the genes whose expression was altered in Lhx6−/−;Lhx8−/− mutant maxillary arches. We used laser-capture microdissection to precisely excise the Lhx6 and Lhx8 expression domain from E10.5 maxillary arches (Fig. 3A–C). RNA samples from three Lhx6−/−;Lhx8−/− mutant and three littermate wild-type embryos of matching stage and sex were amplified and hybridized onto Affymetrix Mouse Genome 430 2.0 arrays (Fig. 3D). From this experiment, we obtained a list of 212 genes that were up-regulated (93 genes) or down-regulated (119 genes) more than 1.5-fold in Lhx6−/−;Lhx8−/− mutants compared with wild types (= Lhx-regulated genes; Supplementary Material, Table S1). A gene ontology analysis using the Database for Annotation, Visualization and Integrated Discovery (DAVID) (27,28) revealed that two categories of molecular function were most noticeably over-represented in the Lhx-regulated genes, i.e. transcriptional regulation (red in Fig. 3E) and extracellular matrix binding (blue in Fig. 3E).
Figure 3.

Genome-wide transcriptional profiling of Lhx6−/−;Lhx8−/− mutant and wild-type maxillary arches. (A–C) Coronal sections of the head of E10.5 wild-type embryos showing the right maxillary arch. (A and B) The sections were processed by RNA in situ hybridization. (C) The section was subject to laser-capture microdissection as demarcated by the red line to harvest the tissue for transcriptional profiling. (D) Summary of the transcriptional profiling experiment. (E) Result of the gene ontology (GO) analysis of Lhx-regulated genes using DAVID (27,28).
A cell cycle inhibitor p57Kip2 is overexpressed in the maxillary arches of Lhx6−/−;Lhx8−/− mutants
The parsimonious explanation for the decreased cell proliferation in Lhx6−/−;Lhx8−/− mutants would be that the LHX proteins directly activate or repress the expression of the gene(s) encoding cell cycle regulators. To investigate this hypothesis, we searched the list of the Lhx-regulated genes for any of the 126 genes compiled under ‘Cell Cycle’ in Kyoto Encyclopedia of Genes and Genomes Pathway Database (29). This search identified two genes, Gadd45g (Growth arrest and DNA-damage-inducible 45 gamma) and p57Kip2 (=Cdkn1c, Cyclin-dependent kinase inhibitor 1C; Mouse Genome Informatics) (Supplementary Material, Fig. S3). GADD45G is one of the three GADD45 family proteins; they inhibit cyclin/cyclin-dependent kinase (CDK) complex at the G2-M checkpoint of the cell cycle, in addition to playing various roles in apoptosis and DNA repair (30–32). p57Kip2 was originally identified as an inhibitor of the cyclin/CDK complex at G1–S transition and has been studied extensively in relation to cancer (33–36). Furthermore, mutations of p57Kip2 have been linked to syndromes of developmental defects in humans (see Discussion for details).
Our microarray data showed up-regulation of Gadd45g and p57Kip2 in Lhx6−/−;Lhx8−/− mutant maxillary arches compared with wild types, 3.3-fold and 1.7-fold, respectively. The up-regulations were confirmed by section RNA in situ hybridization (Fig. 4A–D) and reverse transcription followed by quantitative real-time PCR (qPCR) (data not shown). Interestingly, the overexpression of p57Kip2 was most pronounced in the oral-medial domain of the maxillary arch (Fig. 4A and B, boxed areas), whereas the overexpression of Gadd45g was in the oral-lateral domain (Fig. 4C and D, arrows). Because the palatal shelves develop from the oral-medial side of the maxillary arches, we reasoned that the up-regulation of p57Kip2 would be more relevant to the palatal growth defect of Lhx6−/−;Lhx8−/− mutants than the up-regulation of Gadd45g. Therefore, we investigated the Lhx − p57Kip2 genetic pathway in detail in the context of palate development.
Figure 4.

Repression of p57Kip2 by Lhx6 and Lhx8 in the palate area of the maxillary arches. (A–D, G–I) Coronal sections of the head of E10.5 (A–D) or E11.5 (G–I) embryos processed by RNA in situ hybridization. The right half of the face is shown. The boxes in A and B highlight the strong up-regulation of p57Kip2 in the prospective palate area. The arrows in C and D point to the up-regulation of Gadd45g in the oral-lateral domain of Lhx6−/−;Lhx8−/− mutant maxillary arches. (E, F and J) Coronal sections of the head of E10.5 (E and F) or E11.5 (J) embryos stained for nuclei (blue) and p57Kip2 protein (red). E and F are equivalent to the boxed areas in A and B. DM, dental mesenchyme.
Immunofluorescence for p57Kip2 protein at E10.5 confirmed the result of in situ hybridization (Fig. 4E and F); p57Kip2 was clearly detected in the prospective palate area of Lhx6−/−;Lhx8−/− mutants, whereas it was minimally present in the same area of the wild-type embryos. Also, we found that p57Kip2 was only moderately up-regulated in Lhx6−/− and Lhx8−/− single mutants, indicating that the two Lhx genes co-regulate p57Kip2 (Supplementary Material, Fig. S4). At E11.5, the wild-type expression of p57Kip2 had intensified in some parts of the first pharyngeal arch (Fig. 4I and J). However, it was largely excluded from the areas with strong Lhx6 and Lhx8 expression, namely, the palatal shelves and the dental mesenchyme of the molars (Fig. 4G–J). These striking complementary patterns of expression between p57Kip2 and the Lhx genes further suggested that the repression of p57Kip2 by LHX6 and LHX8 in the oral mesenchyme likely has functional significance for normal development of the face.
LHX binds to a genomic region at the 5′ end of p57Kip2 and acts as a transcriptional repressor
Next, we wanted to determine whether p57Kip2 is directly regulated by LHX6 and LHX8, through the binding of LHX proteins to a cis-regulatory element(s) of p57Kip2. To obtain comprehensive data on the binding of LHX to the genome during craniofacial development, we performed chromatin immunoprecipitation (ChIP) followed by high-throughput sequencing (seq) from E11.5 maxillary arches using anti-LHX6 antibody (Fig. 5A). This experiment identified 6560 genomic regions showing enrichment of LHX6 binding (=LHX6 peaks) (see Supplementary Material, Table S2 for the genomic coordinates of the LHX6 peaks).
Figure 5.

Identification of an LHX target cis-regulatory element in p57Kip2 locus through LHX6 ChIP-seq. (A) Summary of the ChIP-seq experiment. The dotted line demarcates the maxillary arch. (B) Distribution of the LHX6 peaks relative to the nearest TSS was analyzed using GREAT (37). (C) Comparison of the percentage of the genes associated with LHX6 peaks among the whole-genome gene set, which comprises 20 221 well-annotated genes selected by GREAT, and among the 212 Lhx-regulated genes from our transcriptional profiling. (D and E) Results of the gene ontology analysis of the LHX6 peaks by GREAT. Top ten most enriched terms in the categories of ‘Expression’ and ‘Phenotype’ are listed. The entries that are relevant to craniofacial development are in red. (F) Comparison of the number of LHX6 peaks within 10 kb of the TSS of Lhx-regulated genes and random genes, shown as the average number per gene. The error bar is a 95% confidence interval. *Z score >1.98, or P < 0.05. (G) LHX6 ChIP-seq result at the p57Kip2 locus is visualized by IGB (38). The y-axis corresponds to the fragment density derived from the Illumina sequencing result (see Materials and Methods for details). The orange bar indicates the LHX6 peak determined by MACS peak-finding algorithm (39). The red box (sil) corresponds to p57Kip2_sil used in H–K. Shown at the bottom are the Genomic Evolutionary Rate Profiling (GERP) scores of the region taken from UCSC Genome Browser (40,41). (H and I) ChIP-qPCR from E11.5 maxillary arch cells for p57Kip2_sil and an NC region (NC) located 10 kb away. The error bars are standard deviations from triplicates, and P-values were calculated between the fold enrichments of NC and p57Kip2_sil. *P < 0.05, **P < 0.01. (J and K) Luciferase reporter assay in primary culture of maxillary arch cells from E11.5 embryos. The relative luciferase activity from the minimal promoter (minP) was not significantly different (n.s., P > 0.05) whether co-transfected with an LHX expression vector or empty pCIG as a control. In contrast, the relative luciferase activity from p57Kip2_sil reporter was reduced upon co-transfection of LHX6-pCIG (J) or LHX8-pCIG (K) compared with empty pCIG. The error bars are standard deviations from triplicates. *P < 0.05.
The LHX6 peaks were highly concentrated near transcription start sites (TSSs), with 43% of them within 5 kb from a TSS (Fig. 5B). We identified genes that were ‘associated’ with LHX6 peaks using the criteria that the gene's TSS is the nearest to the peak and is <1000 kb away. We found that 4377 genes out of 20 221 genes (=22%) in the whole-genome gene set were associated with at least 1 LHX6 peak, whereas 94 out of 212 Lhx-regulated genes (=44%) were associated with LHX6 peak(s) (Fig. 5C). A gene ontology analysis showed that the LHX6 peaks were significantly associated with the genes expressed in the face and regulating craniofacial development (Fig. 5D and E). We also compared the number of LHX6 peaks within 10 kb of the TSS, and the average number was significantly higher for Lhx-regulated genes than for random genes (Fig. 5F). So far, we have performed ChIP-qPCR for eight of the LHX6 peaks (three described below, and data not shown), and for seven of them, the result from the ChIP-seq was confirmed.
One of the LHX6 peaks was associated with p57Kip2. This peak (p57Kip2_sil) encompassed the promoter, the first exon, the first intron and a part of the second exon (Fig. 5G). p57Kip2_sil, and in fact the entire p57Kip2 locus, was moderately conserved across different species, with Genomic Evolutionary Rate (GERP) score (40,41) of <2.3 (the score of >2 is considered evolutionarily constrained, with the maximum possible score of 4.14) (Fig. 5G). While we did not find the consensus LHX-binding motif (TAATTA) (42) in p57Kip2_sil, it had two generic homeodomain-binding motifs, TAAT (43). ChIP-qPCR confirmed that p57Kip2_sil was enriched in LHX6 ChIP DNA over IgG mock ChIP DNA (Fig. 5H). A negative control (NC) region 10 kb away from p57Kip2_sil also showed some enrichment in LHX6 ChIP, possibly reflecting low-affinity nonspecific background binding of anti-LHX6 antibody. However, the fold enrichment of anti-LHX6/IgG was significantly higher for p57Kip2_sil than the NC region (Fig. 5H), indicating specific interaction between LHX6 and p57Kip2_sil. We also confirmed the binding of LHX8 to p57Kip2_sil (Fig. 5I). Because ChIP-grade anti-LHX8 antibody was not available, we expressed triple FLAG-tagged LHX8 in primary culture of dissociated maxillary arch cells (PMACs) by transient transfection and used anti-FLAG antibody for ChIP. We previously verified that this primary culture system was a reliable surrogate for the in vivo maxillary arches for investigating transcription regulation (44). FLAG::LHX8 was expressed from pCIG (45), which is a β-actin promoter-driven expression vector with an internal ribosome entry site followed by the coding sequence of green fluorescence protein (GFP). From this ChIP experiment, the fold enrichment of the NC region (anti-FLAG/IgG) was consistently <1, which is likely because the nonspecific background bindings of the normal IgG were eliminated in the affinity-purified FLAG antibody. Importantly, the fold enrichment of p57Kip2_sil (anti-FLAG/IgG) was significantly higher than that of the NC region (Fig. 5I).
To determine whether the binding of LHX to p57Kip2_sil has a functional consequence, we cloned p57Kip2_sil into a plasmid containing a minimal promoter followed by the luciferase coding sequence for in vitro reporter assays. PMACs from E11.5 wild-type embryos were co-transfected with the luciferase reporter plasmid and pCIG containing the coding sequence for the LHX proteins (Lhx6-pCIG, Lhx8-pCIG). pCIG without an insert in the first cistron (empty pCIG) was used as a control. We found that the overexpression of LHX6 or LHX8 significantly repressed the reporter expression from p57Kip2_sil, whereas it had no effect on the expression from the minimal promoter (Fig. 5J and K). This result suggested that LHX6 and LHX8 directly regulated p57Kip2 via p57Kip2_sil, which acts as an LHX-responsive transcriptional silencer in the maxillary arch cells.
Overexpression of p57Kip2 in primary maxillary arch cells inhibits cell proliferation
Although the function of p57Kip2 as a cell cycle inhibitor has been well established in the context of cancer, it had not been directly examined during craniofacial development. Therefore, we tested whether p57Kip2 can affect cell cycle in the developing face using PMACs. The cells were taken from E10.5 wild-type embryos and plated in chamber slides (Fig. 6A). Each chamber was transfected with pCIG, or pCIG containing p57Kip2-coding sequence (p57Kip2-pCIG), and the proliferating cells were labeled with 5-bromo-2-deoxyuridine (BrdU) overnight (Fig. 6A). Immunofluorescence against p57Kip2 confirmed its overexpression in p57Kip2-pCIG-transfected cells (data not shown). Importantly, when compared with the cells transfected with empty pCIG (GFP+ cells in Fig. 6B–D), p57Kip2-pCIG-transfected cells (GFP+ cells in Fig. 6E–G) showed significantly reduced percentage of BrdU labeling (Fig. 6H, ‘Transfected cells’). This result demonstrated that the overexpression of p57Kip2 inhibited the proliferation of maxillary arch cells. There was no difference in BrdU incorporation rates between the untransfected cells in the empty pCIG and p57Kip2-pCIG chambers (GFP− cells in Fig. 6B–G), confirming that the effect of p57Kip2 was cell-autonomous (Fig. 6H, ‘Untransfected’).
Figure 6.

Inhibition of cell proliferation upon overexpression of p57Kip2 in PMACs. (A) A schema of the experiment. The maxillary arches were dissected from E10.5 embryos following the dotted line (left picture), and the dissociated cells were plated in chamber slides (right picture). (B–G) Representative images of dual immunofluorescence of the cells transfected with empty pCIG (B–D) or p57Kip2-pCIG (E–G), showing GFP channel only (B and E), BrdU channel only (C and F) or overlay of the two along with DAPI channel for nuclei (D and G). Note that some of the GFP+ cells in B are also labeled with BrdU as shown in C and D (white arrows), indicating their proliferation, but none of the GFP+ cells in E are co-labeled with BrdU in E and F. (H) Quantitative comparison of the percentage of proliferating cells between the chambers transfected with empty pCIG and those transfected with p57Kip2-pCIG. The error bars are standard deviations from triplicates. ***P < 0.001.
Multiple FOX transcription factor genes are up-regulated in Lhx6−/−;Lhx8−/− maxillary arches
From the list of Lhx-regulated genes, it was highly noticeable that five members of Forkhead box (FOX) transcription factor family genes, Foxc1, Foxd1, Foxd2, Foxp1 and Foxp2, were up-regulated in Lhx6−/−;Lhx8−/− mutants (Fig. 7A). FOX transcription factors have a wide range of functions during development (46). Importantly, Foxp1 and Foxp2 have been shown to regulate p57Kip2 in the context of hair follicle, lung and heart (47–49). We performed literature search for over 100 of the Lhx-regulated genes in PubMed (http://www.ncbi.nlm.nih.gov/pubmed), and the Fox genes were the only ones for which we found evidence to be regulators of p57Kip2. Therefore, we considered a possibility that LHX6 and LHX8 might regulate p57Kip2 indirectly via FOX transcription factors in addition to directly by binding to p57Kip2_sil.
Figure 7.

Up-regulation of multiple Fox genes in Lhx6−/−;Lhx8−/− maxillary arches. (A) Summary of the changes in the expression of Fox genes discovered from transcriptional profiling of wild-type and Lhx6−/−;Lhx8−/− mutant maxillary arches (see Fig. 4). (B–M) Coronal sections of the head of E10.5 wild-type and Lhx6−/−;Lhx8−/− mutant embryos processed by RNA in situ hybridization. Only the right maxillary arch is shown in each panel. The dotted lines indicate the border between the maxillary arch and cephalic paraxial mesoderm. The sections in L and M were hybridized with the probes against all five Fox genes in A mixed at an equal concentration. Arrows, prospective palate area.
RNA in situ hybridization confirmed the up-regulation of the five Fox genes in Lhx6−/−;Lhx8−/− mutant maxillary arches, with each Fox gene showing a unique pattern of overexpression (Fig. 7B–K). We then pooled the hybridization probes to compare the combined expression of the five Fox genes between the wild-type and Lhx6−/−;Lhx8−/− embryos. We found that the strongest up-regulation of the Fox genes in the Lhx6−/−;Lhx8−/− mutant maxillary arches was in the oral-medial domain (Fig. 7L and M, arrows), which is the prospective palate area, resembling the up-regulation pattern of p57Kip2 (Fig. 4A and B). On the other hand, the expression of the Fox genes in Lhx6−/− or Lhx8−/− single mutants showed minimal change compared with wild types, indicative of functional redundancy between LHX6 and LHX8 in repressing the Fox genes (Supplementary Material, Fig. S4). Taken together, these observations supported the idea that the up-regulation of the Fox genes in Lhx6−/−;Lhx8−/− maxillary arches might have contributed to the strong increase in p57Kip2 expression.
LHX proteins bind to a genomic region within Foxp1 and act as a transcriptional repressor
To test whether a genetic pathway existed connecting Lhx, Fox and p57Kip2, we investigated whether the Fox genes were direct targets of LHX6 and LHX8. Our ChIP-seq showed 1–5 LHX6 peaks associated with each of the five Fox genes from Figure 7 (data not shown). We focused on Foxp1 because there was more evidence for the regulation of p57Kip2 by Foxp1 than by any other Fox genes (47–49). According to NCBI Reference Sequence Database, mouse Foxp1 has three isoforms using different promoters, with the longest isoform extending over 500 kb of the genome. Among the five LHX6 peaks associated with Foxp1, one peak was located upstream of the shortest isoform and an intron of the other isoforms (Foxp1_sil, Fig. 8A). Other LHX6 peaks were outside of all the Foxp1 isoforms; the closest one was located ∼4 kb upstream of the longest isoform (4 U in Fig. 8A) whereas the rest were > 50 kb away from either end of Foxp1 (data not shown). We performed ChIP-qPCR validation for LHX6 binding to Foxp1_sil and 4 U, the two peaks closest to the TSS's of Foxp1. This confirmed that Foxp1_sil, but not 4 U, was significantly enriched in LHX6 ChIP DNA (Fig. 8B, data not shown). Foxp1_sil was bound by LHX8 as well according to ChIP-qPCR assay (Fig. 8C).
Figure 8.

Identification of an LHX target cis-regulatory element in Foxp1 locus. (A) LHX6 ChIP-seq result at the Foxp1 locus visualized by IGB. The dotted box (sil) corresponds to Foxp1_sil used in B–F. The GERP scores from UCSC Genome Browser are shown at the bottom. (B and C) ChIP-qPCR from E11.5 maxillary arch cells for Foxp1_sil and an NC region (NC) located 23 kb away. The error bars are standard deviations from triplicates. P-values were calculated between the fold enrichments of NC and Foxp1_sil. *P < 0.05, **P < 0.01. (D) Multiz Alignments of the Foxp1_sil region from UCSC Genome Browser (50). (E and F) Luciferase reporter assay in PMACs from E11.5 embryos. The relative luciferase activity from Foxp1_sil reporter was significantly reduced when co-transfected with an LHX expression vector in comparison with when co-transfected with empty pCIG. The error bars are standard deviations from triplicates. *P < 0.05, **P < 0.01.
The sequence of Foxp1_sil showed strong evolutionary conservation (maximum GERP score of the region is 3.63) (Fig. 8A and D), a feature often found in cis-regulatory elements (51). Foxp1_sil contained no consensus LHX-binding motif TAATTA, but it had six TAAT homeodomain-binding motifs. To determine the functional significance of LHX binding to Foxp1_sil, we cloned it into a luciferase reporter plasmid and performed a co-transfection experiment in PMACs, as described earlier for p57Kip2_sil (Fig. 5). Overexpression of LHX6 or LHX8 significantly repressed reporter expression from Foxp1_sil (Fig. 8E and F), which indicated that the negative regulation of Foxp1 by LHX6 and LHX8 in the maxillary arches could be direct.
FOX transcription factors induce the expression of p57Kip2 in maxillary arch cells
Next, we tested the regulatory relationship between the FOX proteins and p57Kip2 in the maxillary arch cells. We transfected PMACs from E10.5 wild-type embryos with FOXP1 expression vector (Foxp1-pCIG) or empty pCIG and detected p57Kip2 by immunofluorescence (Fig. 9A–F). Over 90% of the Foxp1-pCIG-transfected cells (GFP+ cells in Fig. 9D–F) expressed p57Kip2, whereas <10% of the empty pCIG-transfected cells (GFP+ cells in Fig. 9A–C) did (Fig. 9G, ‘Transfected Cells’). This striking difference indicated that FOXP1 efficiently induced p57Kip2 expression in the maxillary arch cells. The untransfected cells from empty pCIG and Foxp1-pCIG chambers (GFP− cells in Fig. 9A–F) showed similar percentages of p57Kip2 labeling (Fig. 9G, ‘Untransfected cells’), consistent with the cell-autonomous action of FOXP1. In the same assay, FOXD1 was also able to activate p57Kip2 in the maxillary arch cells although it was not as potent as FOXP1 (Fig. 9H).
Figure 9.
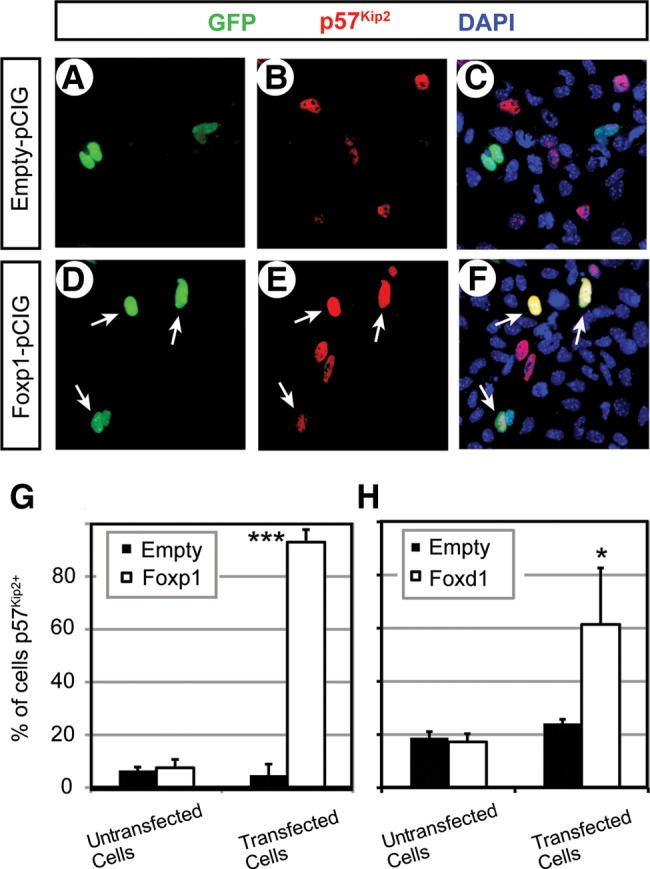
Induction of p57Kip2 expression by FOX proteins in the PMACs. (A–F) Representative images of dual immunofluorescence of the cells transfected with empty pCIG (A–C) or Foxp1-pCIG (D–F), showing GFP channel only (A and C), p57Kip2 channel only (B and E) or overlay of the two along with DAPI channel for nuclei (C and F). Note that none of the GFP+ cells in A expressed p57Kip2, but three out of the four GFP+ cells in D expressed p57Kip2 (arrows in D–F). (G and H) Quantitative comparison of the percentage of p57Kip2+ cells between the chambers transfected with empty pCIG and those transfected with Foxp1-pCIG (G) or Foxd1-pCIG (H). The error bars are standard deviations from triplicates. ***P < 0.001, *P < 0.05.
Taken together, our data point to the FOX proteins as intermediaries between LHX and p57Kip2, which activate p57Kip2 expression in the maxillary arches if their own expression is not repressed by LHX6 and LHX8.
FOXP1 activates transcription from the genomic regions in and around p57Kip2
To elucidate the mechanism underlying the regulation of p57Kip2 by FOX proteins, we examined the sequence near p57Kip2 for potential binding sites for FOX. Using rVISTA2.0 (52), we searched 10 kb upstream to 10 kb downstream of p57Kip2 for FOX-binding motifs (RYMAAYA) (46) that were conserved between mice and humans. We found four FOX motifs, two of which were in an exon (not shown), one in the second intron and one located 2 kb upstream of p57Kip2 TSS (Fig. 10A and B). Notably, the FOX motif in the intron 2 was also conserved in lower vertebrates such as platypus and lizard (Fig. 10B).
Figure 10.

Identification of FOX target cis-regulatory elements in p57Kip2 locus. (A) UCSC Genome Browser view of p57Kip2 locus in the mouse genome. enh1 and enh2 are candidate enhancers of p57Kip2 used in C. (B) Sequence alignments showing the conservation of the FOX-binding motifs in the species indicated on the left, generated by Clustal Omega (53). (C) Luciferase reporter assay in PMACs from E11.5 embryos. The relative luciferase activity from the minimal promoter was not significantly different (P > 0.05) when co-transfected with Foxp1–pCIG or empty pCIG as a control. In contrast, the relative luciferase activities from p57Kip2_enh1 and p57Kip2_enh2 reporters were increased upon co-transfection of FOXP1 expression vector. The error bars are standard deviations from triplicates. **P < 0.01, ***P < 0.001. (D) ChIP-qPCR from E11.5 maxillary arch cells for p57Kip2_enh1, p57Kip2_enh2 and an NC region (NC) located 19 kb upstream of p57Kip2_enh2. The error bars are standard deviations from triplicates. The P-values were calculated for the differences in fold enrichment between each enhancer and NC. *P < 0.05. (E) A model for the mechanism underlying the regulation of the initial outgrowth of the palatal shelves by Lhx6 and Lhx8.
To determine the potential role of the two non-coding regions that contain the FOX motifs in mediating the activation of p57Kip2 by FOX, we cloned a 300-bp fragment of the intron 2 (p57Kip2_enh1, Fig. 10A) and a 500-bp fragment of the p57Kip2 upstream region (p57Kip2_enh2, Fig. 10A) into a luciferase reporter plasmid. With these reporter constructs, we performed a co-transfection experiment in E11.5 PMACs as in Figures 5 and 8. The overexpression of FOXP1 enhanced the reporter expression from p57Kip2_enh1 and p57Kip2_enh2, but not from the minimal promoter (Fig. 10C). We also investigated physical binding of FOXP1 to p57Kip2_enh1 and p57Kip2_enh2. Because the maxillary arches of wild-type embryos have very limited endogenous expression of Foxp1 (Fig. 7J), we transfected PMACs with Foxp1-pCIG and used these cells for ChIP-qPCR. The fold enrichment (FOXP1 ChIP/IgG ChIP) for p57Kip2_enh1 and p57Kip2_enh2 was significantly higher than that of an NC region located ∼20 kb away (Fig. 10D). These results indicated that FOX proteins likely activate p57Kip2 directly in the maxillary arches.
Discussion
In the current study, we report that the function of Lhx6 and Lhx8 is essential for the initial outgrowth of the palatal shelves from the maxillary arches. Based on the results presented here, we propose a model (Fig. 10E) that LHX6 and LHX8 repress the expression of a cell cycle inhibitor gene p57Kip2 in the prospective palate area of the maxillary arches, thereby allowing an adequate level of cell proliferation necessary for the outgrowth of the palatal shelves. The regulation of p57Kip2 by LHX uses both direct and indirect mechanisms, with the latter mediated by FOX transcription factors.
This work is novel for multiple reasons: (i) it is the first to identify direct targets of LHX6 and LHX8 in a comprehensive manner through the combination of genome-wide transcriptional profiling and ChIP-seq in any tissue. (ii) It is the first to identify direct targets of any transcription factor in a comprehensive manner through the combination of genome-wide transcriptional profiling and ChIP-seq in the context of craniofacial development. (iii) It is the first to demonstrate the regulatory relationship between Lhx and p57Kip2, and between Lhx and Fox genes. This information can provide insights into the gene regulatory network in other organs where the above genes are expressed. (iv) Most importantly, it is the first to elucidate a molecular genetic pathway downstream of Lhx in palate development.
Normal development of the palate requires multiple steps of tissue growth and morphogenesis over several days in mice and several weeks in humans, and perturbation in any of these steps can lead to the failure in palatogenesis manifested as clefting. Mouse and human genetics studies have identified a large number of genes that are important for palatogenesis. Furthermore, from the detailed analyses of the mutant mouse models of cleft palate, the cellular and molecular mechanisms underlying the process are beginning to be elucidated (10,11). However, most of the mouse cleft palate models exhibited defects after the vertical elongation of the palatal shelves (E13.5 and later), and thus the information has remained sparse on the regulation of the early steps (10,11). Therefore, Lhx6−/−;Lhx8−/− mutants made a valuable model providing insights into the genetic regulation at the initiation of palatogenesis.
According to the notion that the LHX proteins act as two LHX-two LDB1 tetramers, the oral mesenchyme cells can have three types of complexes, i.e. those containing two of LHX6, two of LHX8, or one each of LHX6 and LHX8. However, for the location (developing palate) and the stage (initiation of palatogenesis, E10.5–E11.5) examined in the current study, we believe that all of the LHX6/8–LDB1 complexes play essentially identical functions, based on the following lines of evidence. First, LHX6 and LHX8 have very similar amino acid sequences, with their homeodomains and LIM domains showing 95 and 74% identity, respectively (20). Furthermore, a systematic analysis of protein–DNA binding using microarrays found that the top affinity nucleotide motifs for the homeodomains of LHX6 and LHX8 were the same (42). Second, the early stages of palate development (E11–E13) were severely disrupted in Lhx6−/−;Lhx8−/− mutants, but unaffected in Lhx6−/− and Lhx8−/− mutants, indicating functional redundancy between the two Lhx genes. Third, we examined the expression of LHX target genes p57Kip2 and Foxp1 in Lhx6−/− and Lhx8−/− single mutants as well as in Lhx6−/−;Lhx8−/− mutants and found that the two genes were co-regulated by Lhx6 and Lhx8. In addition, we demonstrated that both LHX6 and LHX8 proteins physically bound to the LHX-regulated silencers near p57Kip2 and Foxp1 and that both LHX proteins were able to repress gene expression through these silencers. Therefore, LHX6 and LHX8 appear to regulate the same targets to ensure the initial outgrowth of the palate.
p57Kip2 belongs to a Cip/Kip family of cyclin/CDK inhibitors, which includes p21Cip1 (=Cdkn1a) and p27Kip1 (=Cdkn1b) (34–36). Among the three members, p57Kip2 is unique in that it shows tissue-specific expression patterns in embryos and adults, whereas the others are ubiquitously expressed (33,36,54). Although initially identified as a cell cycle inhibitor, p57Kip2 can also regulate other cellular processes such as apoptosis, migration and differentiation depending on the context (55–57). Furthermore, gene knockout experiments in mice showed that the function of p57Kip2 was important for normal development of multiple organ systems (55,58). The various defects resulting from the deletion of p57Kip2 could be classified into three types of abnormalities at the cellular level: (i) increase in cell proliferation leading to hyperplasia (adrenal gland) (58), (ii) impaired cell differentiation, either due to the direct regulation of the differentiation program by p57Kip2 or as an indirect consequence of cells continuing proliferation at the expense of differentiation (endochondral skeleton, neurons of the brain and skeletal muscle) (55,58–60) and (iii) increase in apoptosis triggered by the inappropriate entry into the cell cycle by the mutant cells (lens, palate) (55,58). In fact, ∼50% of the mice lacking p57Kip2 developed cleft palate, which was explained by the increased apoptosis in the mutant palatal shelves late in palatogenesis (E14.5), i.e. during their horizontal growth after the elevation (55). On the other hand, Lhx6−/−;Lhx8−/− mutants present a case where overexpression of p57Kip2 just before the onset of palatogenesis is linked to cleft palate, through the inhibition of cell proliferation and the impaired outgrowth of the palatal shelves. As with many other examples of developmental regulators, it appears that too much or too little p57Kip2 can both be deleterious, requiring tight control of its expression for normal development.
In line with the last point, in humans, both loss-of-function and gain-of-function mutations of p57Kip2 are linked to congenital disorders, Beckwith–Wiedemann syndrome and IMAGe syndrome (intrauterine growth restriction, metaphyseal dysplasia, adrenal hypoplasia congenita and genital anomalies), respectively (61–65). Beckwith–Wiedemann syndrome is characterized by prenatal or postnatal overgrowth and predisposition to certain cancers (61–63), whereas IMAGe syndrome is an undergrowth disorder (65–67). Interestingly, cleft palate was found in some patients for both syndromes (61–63,66,67), although the molecular and cellular etiology is likely distinct.
While our findings indicate that the repression of p57Kip2 is an important function of Lhx6 and Lhx8 in promoting normal palate development, it is unlikely to be the only role of the Lhx genes in this context. Given the large number of the Lhx-regulated genes identified from our transcriptional profiling, the phenotype of Lhx6−/−;Lhx8−/− mutants probably result from the combined effects of the altered expression of multiple genes. In addition, although the severe early defect in Lhx6−/−;Lhx8−/− mutants masks any requirement of the Lhx genes at later steps of palate development, they may have additional roles at these steps because the Lhx genes continue to be expressed in the palate at least until birth (20,21).
As mentioned earlier, evidence for the regulation of p57Kip2 by Foxp genes has previously been reported in other organ systems. For example, inactivation of Foxp1 in the embryonic heart led to down-regulation of p57Kip2 (48). Foxp2−/−;Foxp1+/− mutants suffered impaired lung development with reduced cell proliferation, and p57Kip2 was found to be up-regulated in the mutant lung (47). In the hair follicle, ablation of Foxp1 caused precocious proliferation of the stem cells, which was attributed to the severe down-regulation of p57Kip2 (49). In the current study, the overexpression of FOXP1 induced the expression of p57Kip2 in the maxillary arch cells. Together, these results point to the existence of a Foxp–p57Kip2 regulatory module that is recurrent in organogenesis, although the direction of the regulation (activation or repression of p57Kip2) is determined by the context. Normally, this module remains dormant in the prospective/nascent palate as there is little expression of Foxp1 and Foxp2 here. We speculate that in Lhx6−/−;Lhx8−/− mutants, the up-regulation of the Fox genes in the maxillary arches triggers the Foxp–p57Kip2 module, which contributes to the palate defect of the Lhx mutants. The three previous studies that connected Foxp and p57Kip2 did not provide any evidence that the regulation might be direct. We identified potential FOX-binding cis-regulatory elements in p57Kip2 locus in the genome and found that they responded to FOXP1 by activating reporter expression in the maxillary arch cells. Therefore, it is possible that FOXP1 and FOXP2 directly regulate p57Kip2 via these regions in other organs as well.
Materials and Methods
Animals
All the experiments involving animals were performed with the approval from New York University Institutional Animal Care and Use Committee. Lhx6 and Lhx8 mutant mouse lines have been described previously (21,68). Lhx6−/−, Lhx8−/− and Lhx6−/−;Lhx8−/− mutant embryos were obtained from crosses between double heterozygote (Lhx6+/−;Lhx8+/−) males and females. The embryos were genotyped by PCR using DNA from the tail. Lhx6+/−;Lhx8+/+ and Lhx6+/+;Lhx8+/− animals were indistinguishable from wild types (Lhx6+/+;Lhx8+/+). Therefore, the littermates with any of these three genotypes were used as controls and referred to as ‘wild type’, to distinguish them from the controls for experimental conditions. The embryos were stage-matched by counting the number of tail somites.
Cresyl violet staining of the head sections and morphometric analysis of the palatal shelves
Frozen sections were prepared and stained with cresyl violet as described (24) to visualize tissue morphology. The area of the palatal shelf was measured from the photographs of cresyl violet-stained sections using the ImageJ program. Two sections of the palatal shelves, from the antero-posterior level just posterior to the molars and cutting through the optic nerves, were measured from each embryo, and measurements from three mutants and three wild types were used for statistical analysis (Student's t-test).
RNA in situ hybridization and immunofluorescence on tissue sections
Section RNA in situ hybridization was performed as described (24) using digoxigenin-labeled probes. The templates for some probes were obtained as a plasmid from other researchers (Lhx6, Lhx8 and Foxc1) (20,69) or purchased from a company (Gadd45g, OriGene, Inc.). The templates for the rest were PCR amplified from a cDNA clone (Foxp1, Foxp2 and p57Kip2) or tail genomic DNA of wild-type CD-1 mice (Foxd1 and Foxd2) introducing a T3 RNA polymerase site in the reverse primer (see Supplementary Material, Table S3 for the sequences of all the primers used in this study). The cDNA clones were obtained from another laboratory (Foxp1 and Foxp2) (70) or purchased from a company (p57Kip2, OriGene, Inc.).
Immunofluorescence on frozen sections was performed as described (24) using the following antibodies: rabbit anti-phospho-histone H3 antibody (Millipore), rabbit anti-cleaved caspase 3 antibody (Cell Signaling Technology) and rabbit anti-p57Kip2 antibody (Santa Cruz Biotech). DAPI was used to stain all the nuclei.
Analysis of cell proliferation and apoptosis in the maxillary arches
Mitotic and apoptotic cells were detected by staining the sections for phospho-histone H3 and cleaved caspase 3, respectively, as previously described (24). Mitotic indices were calculated as described (24) from the Lhx6 and Lhx8 expression domain in the maxillary arches demarcated in Figure 2. Three embryos per genotype, six sections of the maxillary arches per embryo, covering the anterior, middle and posterior positions, were analyzed. Student's t-test was used to determine whether the difference between the genotypes was statistically significant.
Laser-capture microdissection, transcriptional profiling with microarrays and gene ontology analysis of the result from transcriptional profiling
The head of E10.5 embryos was collected and embedded in Optimal Cutting Temperature resin (Tissue-Tek) by flash freezing on dry ice. The frozen sections were collected on polyethylene naphthalate membrane slides (Leica). Leica LMD6000 Laser Micro-Dissection System was used to cut out the normal expression domain of Lhx6 and Lhx8 in the maxillary arch mesenchyme, as shown in Figure 3. The tissue was collected from the entire antero-posterior extent of the maxillary arches. Total RNA was extracted using RNeasy Micro Kit (Qiagen). Subsequent steps of transcriptional profiling were performed by the New York University Genome Technology Center, beginning with the amplification of RNA by Ovation Nano Amplification system (NuGen). RNA samples from three wild-type and three Lhx6−/−;Lhx8−/− mutant embryos, all somite count- and sex-matched (females), were analyzed with Affymetrix GeneChip Mouse Genome 430 2.0 arrays. The raw data from the arrays will be available from Gene Expression Omnibus (GEO) database as ‘.CEL’ files.
A list of Lhx-regulated genes were generated from the microarray result based on the following criteria: fold change in the average expression between wild types and Lhx6−/−;Lhx8−/− mutants is >1.5, the difference is statistically significant (P < 0.05) and the average intensity of the probe signal is >100 for wild-type and/or mutant samples. The resulting list of 212 genes was used for a gene ontology analysis with DAVID (27,28).
LHX6 ChIP-Seq
The maxillary arches were dissected from 47 of E11.5 CD-1 wild-type embryos in phosphate-buffered saline and snap-frozen on dry ice. The tissue was then sent to Active Motif, Inc. (Carlsbad, CA) for FactorPath™ service, which included chromatin preparation, ChIP-seq for LHX6, and bioinformatics analysis of the sequencing result to identify LHX6-enriched genomic regions (peaks). Twenty micrograms of the maxillary arch chromatin was used for ChIP with a rabbit polyclonal anti-LHX6 antibody (71). Libraries for Illumina sequencing (Hi-Seq) were prepared from ChIP DNA and input chromatin as described (72), and the sequence reads from each sample were aligned to the mouse genome (NCBI Build 37, mm 9) using BWA algorithm (73). The aligned sequence tags were extended in silico at the 3′-ends into 150-bp fragments, and the histograms of the fragment density along the genome were stored in BAR (Binary Analysis Results) files and visualized using Integrated Genome Browser (IGB) (38). LHX6 peaks were identified using MACS peak-finding algorithm (39) with the following parameters: band width = 150, model fold = 8,24, P-value cutoff = 1E-7. The coordinates of the all the peaks are listed in Supplementary Material, Table S2. The raw data will be deposited in the GEO database as BAR files.
Statistical analyses of LHX6 ChIP-seq peaks
Genomic Regions Enrichment of Annotations Tool (GREAT) v. 2.0.2 (37) was used to determine the distribution of the LHX6 peaks relative to the nearest TSS, to identify genes that are associated with the LHX6 peaks (association rule = single nearest gene, maximum extension 1 Mb) and to analyze the gene ontology terms for the genes associated with the peaks. The comparison between the number of LHX6 peaks around the Lhx-regulated genes and around 10 sets of the same number of randomly selected genes was performed as described (44). The values from the 10 sets of random genes were averaged, and the 95% confidence interval was calculated using the formula in Microsoft Excel. Z score was calculated by Z = (x − μ)/σ, in which x is the value from the Lhx-regulated gene set, μ is the average value from the control gene sets and σ is the standard deviation from the control gene sets.
Culture and transfection of primary maxillary arch cells
PMACs were prepared and transfected as described (44).
Constructs used for transfection
The full-length open-reading frames (ORFs) of the genes were obtained from other researchers (human LHX8) (74), mouse Lhx6 (75), mouse Foxp1 (70) or purchased from a company (mouse p57Kip2 and mouse Foxd1, OriGene, Inc.). The ORFs were cloned into pCIG for protein expression (45). For FLAG::LHX8-pCIG, the coding sequence of LHX8 was first cloned into p3xFLAG-CMV-10 expression vector (3xFLAG on the N terminal side; Sigma) (74) and then moved to pCIG with the tag.
For luciferase reporter assays, we used pGL4.23 vector (Promega), which contains the coding sequence of firefly luciferase downstream of a minimal promotor, after eliminating a fortuitous consensus FOX-binding motif near the promoter by PCR-based mutagenesis (TGTTGGT→TCCTGGT). The putative cis-regulatory elements were cloned from the tail genomic DNA of CD-1 wild-type mice by PCR, except for p57Kip2_sil. p57Kip2_sil could not be amplified by PCR presumably because of its high GC content, and thus it was synthesized in vitro (Integrated DNA Technologies, Inc.). The genomic coordinates of all the cis-regulatory elements used in this work are listed in Supplementary Material, Table S3. To control for the variability in transfection efficiency, a plasmid constitutively expressing Renilla luciferase (pGL4.73, Promega) was included in all the transfections for a luciferase assay.
All the inserts were verified by sequencing after cloning.
ChIP-qPCR
For LHX6 ChIP-qPCR, the maxillary arches were dissected from 35 to 40 CD-1 wild-type embryos at E11.5. The chromatin was cross-linked with formaldehyde and sonicated with Bioruptor (Diagenode) into 0.5- to 1-kb fragments. The rest of the chromatin preparation and immunoprecipitation steps were performed with ChIP-IT High Sensitivity kit (Active Motif). Four micrograms of anti-LHX6 antibody was used for immunoprecipitation, and the same amount of rabbit IgG was used for mock ChIP. DNA was purified from the immunoprecipitated chromatin using the ChIP-IT High Sensitivity kit and subject to qPCR. ChIP-qPCR results were calculated as fold enrichment of a sequence in LHX6 ChIP DNA compared with in IgG mock ChIP DNA, and the fold enrichment was further compared between the LHX6 target sequence and a negative control sequence in the vicinity. The negative control sequences were selected from a genomic region devoid of LHX6 ChIP-seq peaks and LHX consensus binding motifs (see Supplementary Material, Table S3 for the genomic coordinates).
For FLAG::LHX8 ChIP-qPCR, PMACs were prepared from 14 to 20 of E11.5 CD-1 wild-type embryos and transiently transfected with FLAG::LHX8-pCIG plasmid. The cells were harvested 2 days after the transfection, and the chromatin was prepared as described earlier. Mouse monoclonal anti-FLAG M2 antibody (Sigma) was used for immunoprecipitation, and normal mouse IgG was used for mock ChIP. Subsequent steps were performed as described for LHX6 ChIP-qPCR.
FOXP1 ChIP-qPCR was performed in the same way as FLAG::LHX8 ChIP-qPCR, by transfecting the maxillary arch cells from E11.5 CD-1 wild-type embryos with Foxp1-pCIG plasmid. Mouse anti-FOXP1 antibody (Active Motif) was used for immunoprecipitation, and normal mouse IgG was used for mock ChIP. qPCR was performed for FOX target regions and an NC region in the vicinity that was devoid of FOX-binding motifs (see Supplementary Material, Table S3 for the coordinates).
All the ChIP-qPCR experiments were repeated at least three times, and the results were combined for a statistical analysis.
Analysis of cell proliferation in PMACs
The cells from E10.5 maxillary arches were plated in chamber slides (Lab-Tek) and transfected with p57Kip2-pCIG or empty pCIG. BrdU (Life Technologies) was added to the culture medium the following day. After overnight (16 h) of labeling, the cells were fixed with 4% paraformaldehyde for 1.5 h, treated with 2N HCl for 20 min at 37°C and processed for immunofluorescence of BrdU following the same protocol as tissue sections (rat anti-BrdU antibody, Abcam). DAPI was used to stain all the nuclei. From each chamber, 160–430 cells were examined, and transfected cells (GFP+) and untransfected cells were counted separately to calculate the percentages of BrdU-labeled cells. p57Kip2-pCIG and empty pCIG were each transfected into four chambers, and the results were combined for a statistical analysis.
Analysis of p57Kip2 induction in PMACs
The cells from E10.5 maxillary arches were plated in chamber slides and transfected with Foxp1-pCIG or empty pCIG. Forty-eight hours after the transfection, the cells were fixed with 4% paraformaldehyde and processed for immunofluorescence for p57Kip2. The percentages of p57Kip2-expressing cells were calculated as described above for BrdU-labeled cells. 400–1100 cells were examined from each chamber, and the results from 4 chambers transfected with Foxp1-pCIG (or empty pCIG) were combined for a statistical analysis. The same procedures were performed using Foxd1-pCIG in place of Foxp1-pCIG.
Luciferase reporter assay in PMACs
The cells from E11.5 maxillary arches were transfected with pGL4.23 (with or without a putative cis-regulatory element), pGL4.73 and a transcription factor-pCIG or empty pCIG as a control. After 48 h, the cells were lysed and analyzed by the Dual Luciferase Reporter Assay System (Promega). The ratio between the firefly luciferase and Renilla luciferase was calculated (=relative Luc activity) and presented as a fold change in the relative Luc activity caused by the transcription factor-pCIG compared with the empty pCIG control. The experiments were performed in triplicates, and the statistical significance was determined by Student's t-test. For all the reporter assays presented here, we obtained consistent results from at least three separate rounds of experiments.
Supplementary Material
Funding
This work was supported by National Institute of Dental and Craniofacial Research, National Institutes of Health (R00 DE019486 to J.J.).
Supplementary Material
Acknowledgements
We thank Dr Jean-Pierre Saint-Jeannet and his laboratory members for helpful discussions and sharing equipment.
Conflict of Interest statement. None declared.
References
- 1.Chetpakdeechit W., Hallberg U., Hagberg C., Mohlin B. (2009) Social life aspects of young adults with cleft lip and palate: grounded theory approach. Acta Odontol. Scand., 67, 122–128. [DOI] [PubMed] [Google Scholar]
- 2.Severens J.L., Prahl C., Kuijpers-Jagtman A.M., Prahl-Andersen B. (1998) Short-term cost-effectiveness analysis of presurgical orthopedic treatment in children with complete unilateral cleft lip and palate. Cleft Palate Craniofac J., 35, 222–226. [DOI] [PubMed] [Google Scholar]
- 3.Mildinhall S. (2012) Speech and language in the patient with cleft palate. Front Oral Biol., 16, 137–146. [DOI] [PubMed] [Google Scholar]
- 4.Miller C.K. (2011) Feeding issues and interventions in infants and children with clefts and craniofacial syndromes. Semin Speech Lang., 32, 115–126. [DOI] [PubMed] [Google Scholar]
- 5.Genisca A.E., Frias J.L., Broussard C.S., Honein M.A., Lammer E.J., Moore C.A., Shaw G.M., Murray J.C., Yang W., Rasmussen S.A. et al. (2009) Orofacial clefts in the National Birth Defects Prevention Study, 1997–2004. Am. J. Med. Genet. A, 149A, 1149–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Magee W.P. Jr, Vander Burg R., Hatcher K.W. (2010) Cleft lip and palate as a cost-effective health care treatment in the developing world. World J. Surg., 34, 420–427. [DOI] [PubMed] [Google Scholar]
- 7.Oberg K.C., Krisch W.M., Hardesty R.A. (1993) Prospectives in cleft lip and palate repair. Clin. Plast. Surg., 20, 815–821. [PubMed] [Google Scholar]
- 8.Wellens W., Vander Poorten V. (2006) Keys to a successful cleft lip and palate team. B-ENT, 2 Suppl. 4, 3–10. [PubMed] [Google Scholar]
- 9.Ferguson M.W. (1988) Palate development. Development, 103 suppl., 41–60. [DOI] [PubMed] [Google Scholar]
- 10.Bush J.O., Jiang R. (2012) Palatogenesis: morphogenetic and molecular mechanisms of secondary palate development. Development, 139, 231–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gritli-Linde A. (2007) Molecular control of secondary palate development. Dev. Biol., 301, 309–326. [DOI] [PubMed] [Google Scholar]
- 12.Minoux M., Rijli F.M. (2010) Molecular mechanisms of cranial neural crest cell migration and patterning in craniofacial development. Development, 137, 2605–2621. [DOI] [PubMed] [Google Scholar]
- 13.Helms J.A., Schneider R.A. (2003) Cranial skeletal biology. Nature, 423, 326–331. [DOI] [PubMed] [Google Scholar]
- 14.Dixon M.J., Marazita M.L., Beaty T.H., Murray J.C. (2011) Cleft lip and palate: understanding genetic and environmental influences. Nat. Rev. Genet., 12, 167–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vieira A.R. (2008) Unraveling human cleft lip and palate research. J. Dent. Res., 87, 119–125. [DOI] [PubMed] [Google Scholar]
- 16.Marazita M.L. (2012) The evolution of human genetic studies of cleft lip and cleft palate. Annu. Rev. Genomics Hum. Genet., 13, 263–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rahimov F., Jugessur A., Murray J.C. (2012) Genetics of nonsyndromic orofacial clefts. Cleft Palate Craniofac. J., 49, 73–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hobert O., Westphal H. (2000) Functions of LIM-homeobox genes. Trends Genet, 16, 75–83. [DOI] [PubMed] [Google Scholar]
- 19.Kadrmas J.L., Beckerle M.C. (2004) The LIM domain: from the cytoskeleton to the nucleus. Nat. Rev. Mol. Cell Biol., 5, 920–931. [DOI] [PubMed] [Google Scholar]
- 20.Grigoriou M., Tucker A.S., Sharpe P.T., Pachnis V. (1998) Expression and regulation of Lhx6 and Lhx7, a novel subfamily of LIM homeodomain encoding genes, suggests a role in mammalian head development. Development, 125, 2063–2074. [DOI] [PubMed] [Google Scholar]
- 21.Zhao Y., Guo Y.J., Tomac A.C., Taylor N.R., Grinberg A., Lee E.J., Huang S., Westphal H. (1999) Isolated cleft palate in mice with a targeted mutation of the LIM homeobox gene lhx8. Proc. Natl Acad. Sci. USA, 96, 15002–15006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Denaxa M., Sharpe P.T., Pachnis V. (2009) The LIM homeodomain transcription factors Lhx6 and Lhx7 are key regulators of mammalian dentition. Dev. Biol., 333, 324–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nikopensius T., Kempa I., Ambrozaityte L., Jagomagi T., Saag M., Matuleviciene A., Utkus A., Krjutskov K., Tammekivi V., Piekuse L. et al. (2011) Variation in FGF1, FOXE1, and TIMP2 genes is associated with nonsyndromic cleft lip with or without cleft palate. Birth Defects Res. A Clin. Mol. Teratol., 91, 218–225. [DOI] [PubMed] [Google Scholar]
- 24.Jeong J., Cesario J., Zhao Y., Burns L., Westphal H., Rubenstein J.L. (2012) Cleft palate defect of Dlx1/2-/- mutant mice is caused by lack of vertical outgrowth in the posterior palate. Dev. Dyn., 241, 1757–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vieira A.R., Avila J.R., Daack-Hirsch S., Dragan E., Felix T.M., Rahimov F., Harrington J., Schultz R.R., Watanabe Y., Johnson M. et al. (2005) Medical sequencing of candidate genes for nonsyndromic cleft lip and palate. PLoS Genet., 1, e64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yildirim Y., Kerem M., Koroglu C., Tolun A. (2014) A homozygous 237-kb deletion at 1p31 identified as the locus for midline cleft of the upper and lower lip in a consanguineous family. Eur. J. Hum. Genet., 22, 333–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang da W., Sherman B.T., Lempicki R.A. (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc., 4, 44–57. [DOI] [PubMed] [Google Scholar]
- 28.Huang da W., Sherman B.T., Lempicki R.A. (2009) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucl. Acids Res., 37, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ogata H., Goto S., Sato K., Fujibuchi W., Bono H., Kanehisa M. (1999) KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucl. Acids Res., 27, 29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takekawa M., Saito H. (1998) A family of stress-inducible GADD45-like proteins mediate activation of the stress-responsive MTK1/MEKK4 MAPKKK. Cell, 95, 521–530. [DOI] [PubMed] [Google Scholar]
- 31.Vairapandi M., Balliet A.G., Hoffman B., Liebermann D.A. (2002) GADD45b and GADD45 g are cdc2/cyclinB1 kinase inhibitors with a role in S and G2/M cell cycle checkpoints induced by genotoxic stress. J. Cell Physiol., 192, 327–338. [DOI] [PubMed] [Google Scholar]
- 32.Tamura R.E., de Vasconcellos J.F., Sarkar D., Libermann T.A., Fisher P.B., Zerbini L.F. (2012) GADD45 proteins: central players in tumorigenesis. Curr. Mol. Med., 12, 634–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matsuoka S., Edwards M.C., Bai C., Parker S., Zhang P., Baldini A., Harper J.W., Elledge S.J. (1995) p57KIP2, a structurally distinct member of the p21CIP1 Cdk inhibitor family, is a candidate tumor suppressor gene. Genes Dev., 9, 650–662. [DOI] [PubMed] [Google Scholar]
- 34.Borriello A., Caldarelli I., Bencivenga D., Criscuolo M., Cucciolla V., Tramontano A., Oliva A., Perrotta S., Della Ragione F. (2011) p57(Kip2) and cancer: time for a critical appraisal. Mol. Cancer Res., 9, 1269–1284. [DOI] [PubMed] [Google Scholar]
- 35.Pateras I.S., Apostolopoulou K., Niforou K., Kotsinas A., Gorgoulis V.G. (2009) p57KIP2: “Kip”ing the cell under control. Mol. Cancer Res., 7, 1902–1919. [DOI] [PubMed] [Google Scholar]
- 36.Besson A., Dowdy S.F., Roberts J.M. (2008) CDK inhibitors: cell cycle regulators and beyond. Dev. Cell, 14, 159–169. [DOI] [PubMed] [Google Scholar]
- 37.McLean C.Y., Bristor D., Hiller M., Clarke S.L., Schaar B.T., Lowe C.B., Wenger A.M., Bejerano G. (2010) GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol., 28, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nicol J.W., Helt G.A., Blanchard S.G. Jr, Raja A., Loraine A.E. (2009) The Integrated Genome Browser: free software for distribution and exploration of genome-scale datasets. Bioinformatics, 25, 2730–2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W. et al. (2008) Model-based analysis of ChIP-Seq (MACS). Genome Biol., 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Davydov E.V., Goode D.L., Sirota M., Cooper G.M., Sidow A., Batzoglou S. (2010) Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput. Biol., 6, e1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cooper G.M., Stone E.A., Asimenos G., Program N.C.S., Green E.D., Batzoglou S., Sidow A. (2005) Distribution and intensity of constraint in mammalian genomic sequence. Genome Res., 15, 901–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Berger M.F., Badis G., Gehrke A.R., Talukder S., Philippakis A.A., Pena-Castillo L., Alleyne T.M., Mnaimneh S., Botvinnik O.B., Chan E.T. et al. (2008) Variation in homeodomain DNA binding revealed by high-resolution analysis of sequence preferences. Cell, 133, 1266–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Laughon A. (1991) DNA binding specificity of homeodomains. Biochemistry, 30, 11357–11367. [DOI] [PubMed] [Google Scholar]
- 44.Landin Malt A., Cesario J.M., Tang Z., Brown S., Jeong J. (2014) Identification of a face enhancer reveals direct regulation of LIM homeobox 8 (Lhx8) by Wingless-Int (WNT)/beta-catenin signaling. J. Biol. Chem., 289, 30289–30301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Megason S.G., McMahon A.P. (2002) A mitogen gradient of dorsal midline Wnts organizes growth in the CNS. Development, 129, 2087–2098. [DOI] [PubMed] [Google Scholar]
- 46.Carlsson P., Mahlapuu M. (2002) Forkhead transcription factors: key players in development and metabolism. Dev. Biol., 250, 1–23. [DOI] [PubMed] [Google Scholar]
- 47.Shu W., Lu M.M., Zhang Y., Tucker P.W., Zhou D., Morrisey E.E. (2007) Foxp2 and Foxp1 cooperatively regulate lung and esophagus development. Development, 134, 1991–2000. [DOI] [PubMed] [Google Scholar]
- 48.Zhang Y., Li S., Yuan L., Tian Y., Weidenfeld J., Yang J., Liu F., Chokas A.L., Morrisey E.E. (2010) Foxp1 coordinates cardiomyocyte proliferation through both cell-autonomous and nonautonomous mechanisms. Genes Dev., 24, 1746–1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Leishman E., Howard J.M., Garcia G.E., Miao Q., Ku A.T., Dekker J.D., Tucker H., Nguyen H. (2013) Foxp1 maintains hair follicle stem cell quiescence through regulation of Fgf18. Development, 140, 3809–3818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Blanchette M., Kent W.J., Riemer C., Elnitski L., Smit A.F., Roskin K.M., Baertsch R., Rosenbloom K., Clawson H., Green E.D. et al. (2004) Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res., 14, 708–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Visel A., Rubin E.M., Pennacchio L.A. (2009) Genomic views of distant-acting enhancers. Nature, 461, 199–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Loots G.G., Ovcharenko I. (2004) rVISTA 2.0: evolutionary analysis of transcription factor binding sites. Nucl. Acids Res., 32, W217–W221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sievers F., Wilm A., Dineen D., Gibson T.J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Soding J. et al. (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol., 7, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee M.H., Reynisdottir I., Massague J. (1995) Cloning of p57KIP2, a cyclin-dependent kinase inhibitor with unique domain structure and tissue distribution. Genes Dev., 9, 639–649. [DOI] [PubMed] [Google Scholar]
- 55.Yan Y., Frisen J., Lee M.H., Massague J., Barbacid M. (1997) Ablation of the CDK inhibitor p57Kip2 results in increased apoptosis and delayed differentiation during mouse development. Genes Dev., 11, 973–983. [DOI] [PubMed] [Google Scholar]
- 56.Tury A., Mairet-Coello G., DiCicco-Bloom E. (2011) The cyclin-dependent kinase inhibitor p57Kip2 regulates cell cycle exit, differentiation, and migration of embryonic cerebral cortical precursors. Cereb. Cortex, 21, 1840–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hirata M., Kugimiya F., Fukai A., Ohba S., Kawamura N., Ogasawara T., Kawasaki Y., Saito T., Yano F., Ikeda T. et al. (2009) C/EBPbeta Promotes transition from proliferation to hypertrophic differentiation of chondrocytes through transactivation of p57. PLoS One, 4, e4543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang P., Liegeois N.J., Wong C., Finegold M., Hou H., Thompson J.C., Silverman A., Harper J.W., DePinho R.A., Elledge S.J. (1997) Altered cell differentiation and proliferation in mice lacking p57KIP2 indicates a role in Beckwith-Wiedemann syndrome. Nature, 387, 151–158. [DOI] [PubMed] [Google Scholar]
- 59.Joseph B., Wallen-Mackenzie A., Benoit G., Murata T., Joodmardi E., Okret S., Perlmann T. (2003) p57(Kip2) cooperates with Nurr1 in developing dopamine cells. Proc. Natl Acad. Sci. USA, 100, 15619–15624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang P., Wong C., Liu D., Finegold M., Harper J.W., Elledge S.J. (1999) p21(CIP1) and p57(KIP2) control muscle differentiation at the myogenin step. Genes Dev., 13, 213–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cohen M.M., Jr (2005) Beckwith-Wiedemann syndrome: historical, clinicopathological, and etiopathogenetic perspectives. Pediatr. Dev. Pathol., 8, 287–304. [DOI] [PubMed] [Google Scholar]
- 62.Romanelli V., Belinchon A., Benito-Sanz S., Martinez-Glez V., Gracia-Bouthelier R., Heath K.E., Campos-Barros A., Garcia-Minaur S., Fernandez L., Meneses H. et al. (2010) CDKN1C (p57(Kip2)) analysis in Beckwith-Wiedemann syndrome (BWS) patients: genotype-phenotype correlations, novel mutations, and polymorphisms. Am. J. Med. Genet. A, 152A, 1390–1397. [DOI] [PubMed] [Google Scholar]
- 63.Choufani S., Shuman C., Weksberg R. (2010) Beckwith-Wiedemann syndrome. Am. J. Med. Genet. C Semin. Med. Genet., 154C, 343–354. [DOI] [PubMed] [Google Scholar]
- 64.Arboleda V.A., Lee H., Parnaik R., Fleming A., Banerjee A., Ferraz-de-Souza B., Delot E.C., Rodriguez-Fernandez I.A., Braslavsky D., Bergada I. et al. (2012) Mutations in the PCNA-binding domain of CDKN1C cause IMAGe syndrome. Nat. Genet., 44, 788–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Balasubramanian M., Sprigg A., Johnson D.S. (2010) IMAGe syndrome: Case report with a previously unreported feature and review of published literature. Am. J. Med. Genet. A, 152A, 3138–3142. [DOI] [PubMed] [Google Scholar]
- 66.Lienhardt A., Mas J.C., Kalifa G., Chaussain J.L., Tauber M. (2002) IMAGe association: additional clinical features and evidence for recessive autosomal inheritance. Horm. Res., 57 Suppl. 2, 71–78. [DOI] [PubMed] [Google Scholar]
- 67.Vilain E., Le Merrer M., Lecointre C., Desangles F., Kay M.A., Maroteaux P., McCabe E.R. (1999) IMAGe, a new clinical association of intrauterine growth retardation, metaphyseal dysplasia, adrenal hypoplasia congenita, and genital anomalies. J. Clin. Endocrinol. Metab., 84, 4335–4340. [DOI] [PubMed] [Google Scholar]
- 68.Choi G.B., Dong H.W., Murphy A.J., Valenzuela D.M., Yancopoulos G.D., Swanson L.W., Anderson D.J. (2005) Lhx6 delineates a pathway mediating innate reproductive behaviors from the amygdala to the hypothalamus. Neuron, 46, 647–660. [DOI] [PubMed] [Google Scholar]
- 69.Kume T., Deng K.Y., Winfrey V., Gould D.B., Walter M.A., Hogan B.L. (1998) The forkhead/winged helix gene Mf1 is disrupted in the pleiotropic mouse mutation congenital hydrocephalus. Cell, 93, 985–996. [DOI] [PubMed] [Google Scholar]
- 70.Shu W., Yang H., Zhang L., Lu M.M., Morrisey E.E. (2001) Characterization of a new subfamily of winged-helix/forkhead (Fox) genes that are expressed in the lung and act as transcriptional repressors. J. Biol. Chem., 276, 27488–27497. [DOI] [PubMed] [Google Scholar]
- 71.Vogt D., Hunt R.F., Mandal S., Sandberg M., Silberberg S.N., Nagasawa T., Yang Z., Baraban S.C., Rubenstein J.L. (2014) Lhx6 directly regulates Arx and CXCR7 to determine cortical interneuron fate and laminar position. Neuron, 82, 350–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Labhart P., Karmakar S., Salicru E.M., Egan B.S., Alexiadis V., O'Malley B.W., Smith C.L. (2005) Identification of target genes in breast cancer cells directly regulated by the SRC-3/AIB1 coactivator. Proc. Natl Acad. Sci. USA, 102, 1339–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li H., Durbin R. (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics, 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Flandin P., Zhao Y., Vogt D., Jeong J., Long J., Potter G., Westphal H., Rubenstein J.L. (2011) Lhx6 and Lhx8 coordinately induce neuronal expression of Shh that controls the generation of interneuron progenitors. Neuron, 70, 939–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kimura N., Ueno M., Nakashima K., Taga T. (1999) A brain region-specific gene product Lhx6.1 interacts with Ldb1 through tandem LIM-domains. J. Biochem., 126, 180–187. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}