

Abstract
Topobiology posits that morphogenesis is driven by differential adhesive interactions among heterogeneous cell populations. This paradigm has been revised to include force-dependent molecular switches, cell and tissue tension, and reciprocal interactions with the microenvironment. It is now appreciated that tissue development is executed through conserved decision-making modules that operate on multiple length scales from the molecular and subcellular level through to the cell and tissue level and that these regulatory mechanisms specify cell and tissue fate by modifying the context of cellular signaling and gene expression. Here, we discuss the origin of these decision-making modules and illustrate how emergent properties of adhesion-directed multicellular structures sculpt the tissue, promote its functionality, and maintain its homeostasis through spatial segregation and organization of anchored proteins and secreted factors and through emergent properties of tissues, including tension fields and energy optimization.
Morphogenesis is the process whereby a complex living system is created from individual components that are systemically developed to yield a functionally stable unit with a defined form and function. As proposed by Edelman and colleagues (1), topobiology is the process that sculpts and maintains differentiated tissues and is acquired by the energetically favored segregation of cells through heterologous cellular interactions. That “tissue affinity” is the primary morphogenetic driver was first demonstrated by Townes and Holtfreter, who showed that disaggregated amphibian cells self-organize into tissue structures with distinct cell fates (2). This concept was confirmed by the identification of cell adhesion molecules, which facilitate the assembly of multiprotein “signaling modules” that mediate, integrate, and stabilize multicellular interactions (3). Phenotypic cues mediated through gradients of secreted “soluble” factors such as fibroblast growth factor, transforming growth factor–β (TGFβ), and Wnt also control tissue patterning by activating genetic programs such as HOX gene clusters, thereby inducing and maintaining cellular identity and directing higher-order tissue architecture (4). Tensile forces also govern the self-organization of heterologous cellular interactions during embryogenesis and modulate tissue movements in development by altering the activity of critical transcriptional regulators such as twist, implicating physical cues as key morphometric integrators (5, 6). Indeed, composition and topology of the extracellular matrix (ECM) stroma, which is secreted and modified by cells as they develop, changes throughout morphogenesis and directly regulates cell and tissue fate by inducing signaling within cells through specific matrix adhesion receptors to modify cytoskeletal organization and cell shape (7). Soluble factors such as hepatocyte growth factor and TGFβ also modulate cell fate either by directly destabilizing multicellular tissue organization through Rho guanosine triphosphatase (GTPase)–dependent actomyosin contractility (8) or by changing ECM composition and posttranslational processing through altered transcription to stiffen the matrix (9). Thus, while morphogenesis might depend upon cell adhesion, it is orchestrated by a highly coordinated series of events that are initiated by soluble factors that activate cellular signaling at the adhesions and that are integrated by mechanical cues operating at the molecular, cellular, and tissue level. Here, we discuss how topo-biological cues are arranged from the molecular to the organism level based on the repetitive use of basic conserved “decision-making modules” (Fig. 1 and Table 1). We describe how these decision-making modules not only orchestrate rapid and highly adaptive changes in non-structured masses of cells as they mature into highly defined tissues and organs but also are dynamic—displaying exquisite sensitivity to mechanical cues and undergoing reciprocal state transitions that permit the fine tuning of the organism. Finally, we speculate how emergent properties of organized multicellular tissues dictate specialized functions and modulate the functional integrity of cell and tissue fate so that altered expression, organization, or structure of any of these decision-making modules will alter cell and tissue architecture and perturb homeostasis and ultimately lead to disease.
Fig. 1.
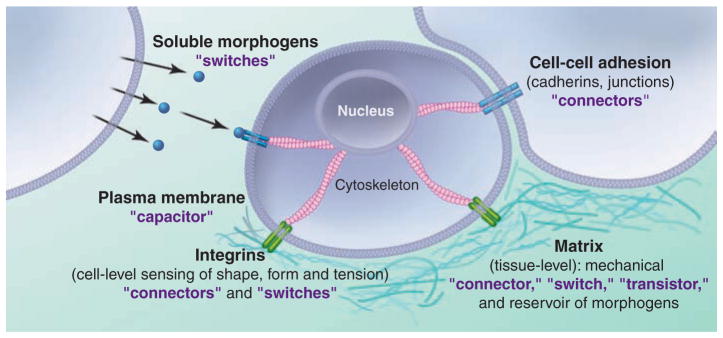
Basic biological modules operate in tissues at multiple length scales. Variations and repetitions of the critical biological modules through many length scales and systems allow the formation and maintenance of increasingly complex multicellular structures with highly evolved functions. Different elements can “connect” one cell to its neighbor by homophilic receptors such as cadherins. Other connectors, such as integrins, mechanically link cells to the extracellular matrix, a three-dimensional (3D) scaffold to which different cell types can adhere. This mechanical connection allows the contraction or cell shape change of one cell to be transmitted by matrix fibrils through the cytoskeleton to a cohort of cells embedded in the same matrix, amplifying small perturbations to cause the matrix to act as a “transistor.” Upon matrix binding, conformational changes within integrin adhesions recruit adapter proteins, which modify the cytoskeleton and act as individual switches to control adhesion, migration, and the like. Cytokine stimulation can also act as a switch, turning on and off to fine-tune cellular behavior. The plasma membrane with its intracellular recycling and storage compartment consists of a reservoir of receptors, the dynamic reshuffling of which controls the degree of signaling by acting as a capacitor. Complex interactions and repetitions of these modules through various length scales is the critical mechanism controlling morphogenesis and form.
Table 1.
Other examples of basic biological modules that could regulate form and function.
| Module | Type | Major concept | References |
|---|---|---|---|
| Switch | Soluble morphogen | Spatial modulation of growth factor receptors in oogenesis | (66, 67) |
| Morphogen regulation of PCP | (68, 69) | ||
| Focal adhesion | Force-dependent signaling of adhesion complexes | (70, 71) | |
| Connector | Cell-cell binding | Adherens junctions and β-catenin in nonmetazoans | (72) |
| Cell sorting and segregation scales with cadherin levels | (73) | ||
| Peripheral myosin regulates cell intercalation | (74) | ||
| Focal adhesion | Molecular clutch hypothesis for focal adhesions | (75, 76) | |
| Capacitor | Plasma membrane | Lipid raft-induced membrane curvature | (77) |
| Transistor | ECM | Storage of growth factors to guide tissue development and direct cancer progression | (78, 79) |
Phenotypic Complexity Through Modular Sensors, Transistors, and Amplifiers
The first niche requirement for a multicellular organism is the development of cell-cell adhesions, which act as “connector” modules that define which cells will adhere to each other as they segregate. These modules are also the nucleation point for signaling molecules and cytoskeletal elements that regulate cell and tissue shape and function, giving them “switchlike” properties. A myriad of cell-cell adhesion molecules have evolved and have gained increasing complexity to facilitate cell-cell adhesion based on conserved components consisting of cytoplasmic, transmembrane, and 3 to 5 repeated extracellular domains, such as in neural cell adhesion molecules, that homotypically bind to each other. Evolution of these repeated domains has precisely set cell-cell spacing and has also regulated the amount of force that the bond can resist (10). Nevertheless, as exemplified by the ability of classic cadherins to link the actin cytoskeleton and adjacent cells, the major function of these modules is to mediate the efficient segregation of heterogeneous cell populations into distinct entities (11). This task is achieved by constant actomyosin-mediated pushing and pulling and the initiation of signaling that optimizes connections among neighboring cells and leads to phenomena such as cell compaction, as occurs at the blastula stage during embryogenesis. Thus, cell compaction is determined by the strength and number of connectors expressed on the cell surface and is likely dictated by the tension induced at the cellular and tissue levels. For example, cortical tension enhances the strength of cell adhesion in zebrafish such that the distinct germ layers display differing adhesion strengths (6). Assuming equal module density, the number of engaged connectors and the overall energy dynamics of the system will enable cells to determine whether they are sitting within or at the periphery of a given cell mass and will dictate the ultimate stability, size, and shape of the multicellular structure. Cellular rearrangements and coordinated tissue migration are also guided by the extracellular milieu of the tissue. Thus, the assembled ECM at the exterior of the blastula provides a qualitatively different anchorage site for the actin cytoskeleton that permits the differentiation of cell-cell from cell-ECM interactions (12). Integrins, which comprise the best characterized class of cell-ECM adhesion molecules, are heterodimeric transmembrane proteins that upon activation bind to specific ECM sites. After their binding, integrins recruit a host of structural and signaling modules, such as talin and Rho GTPases, respectively, to the plasma membrane, thereby responding to matrix tension and reciprocally exerting contractility at the cell periphery (13, 14). Similar conserved modules occur in cell-cell adhesions, where structural and signaling modules act to hold cells together and communicate with the transcriptional apparatus in the nucleus to which the cellular cytoskeleton is tethered. Although they contain similar modules, integrin-matrix adhesions segregate from cadherins to define multicellular properties such as cell and tissue polarity. Thus, mice lacking β1 integrin fail to deposit ECM (e.g., laminin) at the blastula surface, leading to developmental arrest after implantation (15) that can be rescued by coating the blastula with purified laminin (16). In this manner, coordinated and dynamic interactions between cell-cell and cell-ECM are thought to direct multicellular tissue development. Nevertheless, how these events are executed and integrated at the tissue level is poorly understood and remains an area of intense investigation.
Phenotype Is Dominant over Genotype: Clues from the Evolution of Cell-Cell Interactions
Reciprocal and dynamic cell-cell and cell-ECM adhesion communication is essential for multicellular tissue morphogenesis and homeostasis. Consistently, mechanisms intersecting at different length scales have evolved to facilitate this dialogue. These mechanisms act locally at adhesions through competitive associations between conserved signaling complexes and function globally to efficiently transmit information from the cellular to the tissue level by directed cytoskeletal remodeling and cellular and tissue tension. For instance, blastula assembly is followed by blastocoel cavity formation and the assembly of a fibrillar fibronectin matrix, both of which are regulated by the integrin-linked kinase (ILK)/pinch/parvin complex (17). Consistently, blastocoel formation fails in ILK-null embryos (18), and inhibition of fibronectin-integrin interactions inhibits the epithelial-mesenchymal transition that is critical for gastrulation (19). Although the processes occur at dramatically different length scales, both require specific gene expression (e.g., Rho GTPase) and activation to drive tension-dependent processes—for example, cell-cell adhesion maturation and focal adhesion assembly—and act by initiating actin remodeling (3).
Cell-cell and cell-ECM modules share many conserved features; however, they have also evolved fundamental differences that optimize environmental responses and permit fine-tuning of the multicellular organism throughout its life span. Whereas adherens junctions are tightly regulated by receptor number and density to maximize structural variability, integrins evolved to transduce environmental cues, thereby maximizing survival advantage and adaptability for the organism (Fig. 1). Indeed, homotypic adhesion systems appeared in primitive organisms such as the Dictyostelium fruiting body to maintain its integrity through aggregation. Dictyostelium use at least two independent homotypic adhesion systems that are related to metazoan adhesion molecules (20): the Cadherin super family member DdCAD-1 and the immunoglobulin-like domain protein gp80. These connector modules have weak interactions that allow dynamic rearrangement to cluster, sort, stream migrate, and maintain the rigidity of the organism. A fundamental feature of these early adhesion molecules is their enrichment at fillipodial extensions, which, together with actin, form transient spot adhesions required for their initial clustering. These molecules are then rapidly replaced with adhesion plaque proteins such as the glycosylphosphatidylinositol-anchored protein gp80, which establishes more stable cell contacts that act as storage modules through associations with lipid-rich membrane micro domains. Although similar principles operate in higher organisms to facilitate multicellular integrity, the nature of the adhesions has become more complex so that spot-like junctions have been extended into beltlike structures such as those found in adherens and in occluding, tight, and septate junctions in higher organisms (21). A common framework for these diverse junctional complexes is that they all are organized into large complexes made up of highly clustered modules that, through adaptor proteins, i.e., “switch” modules, initiate signaling cascades, and act as connector modules to strengthen the cytoskeletal link in response to increasing tension. Whether adhesion or signaling function came first for this class of cell adhesion complexes is still a matter of contention, but it is now clear that similar downstream signaling components are used by both cadherins and junctional complexes. Yet, while the modular nature of these switches is undisputed, the molecular and physical factors that regulate their function remain unknown.
Phenotype Is Dominant over Genotype: Clues from the Evolution of Cell-Matrix Interactions
Modern heterodimeric integrins developed to link the onset of multicellular structures with the appearance of a stable form in metazoans such as Dictyostelium discoideum, where they developed as specialized sensory modules to regulate adhesion, survival, and phagocytosis (22, 23). These primitive integrin-like proteins called “sib receptors” contain several conserved motifs identical to β integrins, in addition to having tandem NPXY repeat motifs in the cytoplasmic tail (22), suggesting that sib is a cation-dependent, low-affinity receptor for exposed acidic residues in extracellular proteins. Sib also appears to act as a mechanical connector to the actin cytoskeleton through the recruitment of FERM domain–containing proteins such as talin (22). In addition to a mechanical role, sib’s recruitment into the phagocytic cup, as with integrin clustering in the membrane for metazoans, likely serves as an important signaling transistor for prey recognition and feeding stimulation (13). Thus, integrins evolved to permit organisms to respond rapidly to biochemical and physical cues from their microenvironment with specialized features that include adhesion to substrate, active mobility, and detection and capture of prey by phagocytosis (Fig. 2) (24–39).
Fig. 2.

Functional evolution of adhesion-dependent form and function, from bacteria to vertebrates. Although the mechanisms for replication are directly linked to the multiplication and management of the genetic information, the capacity to form complex multilayer organisms is likely based on the evolutionary advantage to adhere to new environments and survive in potentially hostile environments. Although bacteria and fungi use rather simple strategies to create multicellular structures, the evolution of “hunters,” such as amoeba, introduced new dynamic and controllable cell-cell and cell-substrate adhesion systems, such as integrins, allowing the capture of prey and formation of complex multicellular structures. In parallel, the evolution from polysaccharide- or cellulose-based to protein-based extracellular scaffold increased the versatility of cell-to-substrate adhesion systems. Interestingly, the emergence of integrin α/β-heterodimers correlates with the appearance of metazoans (dashed line), indicating that the intracellular perception of the extracellular scaffold is critical to the stable generation of form and function. Details can be found in (24–39).
A second distinguishing feature of integrin-matrix adhesion modules is their ability to function as molecular switches through adaptor molecules that are activated to initiate a cascade of downstream events that amplify the original signal. When compared with adenosine triphosphate–driven signaling enzymes, such as kinases that are typical of this type of switch module, the function of adapter proteins in cell adhesions may, at least at first glance, seem neither similar nor switchlike. However, many adapter proteins bind to both a cytoskeletal protein and an integrin-based adhesion protein to form a complex, where stability of the adapter protein is greatly increased by the initial binding reaction and subsequent change in conformation, for example, vinculin (40). Structural changes, such as those brought on by forces imposed on the adapter protein (41), could liberate additional protein-protein interaction sites in a cooperative manner, and the immediate influx of new binding opportunities for signaling molecules could switch on previously dormant cell behaviors and alter properties like cell shape. Another efficient way to create a switch module for adhesion is to limit protein-protein interactions by immobilizing individual binding partners to a surface. Adsorption-limited diffusion of the binding partners restricts the conformational changes that would inactivate the connector module in examples that range from the rapid rise of phosphatidylinositol biphosphate in the plasma membrane that stabilizes the integrin/talin complex (42) to the regulation of cell motility by ECM sheets like basement membrane (43). For protein interactions, receptor-like protein tyrosine phosphatase–α binding to αv integrin acts as a switch to form the nucleation site for a focal adhesion. One characteristic aspect of this switch is its response to the external application of force, resulting in new protein-protein binding sites, such as with the Src family kinase Fyn (44). In fact, integrin receptors themselves react in response to force by increasing their binding affinities to ligands through conformational changes (45), resulting in the formation of a catch bond that holds under force and gets released in the absence of force (46). An analogy to an old fashioned “finger trap” is perhaps insightful: force-dependent integrin extension acts to increase affinity, much like the pulling force by fingers stuck in the trap further tightens the trap on the fingers. Given their shape and functional differences with lower organisms as well as cell-cell proteins, this is perhaps suggestive of the evolutionary force behind integrin-driven tissue morphogenesis, which relies heavily on its binding partner, the ECM, to aid in the drive to undergo morphogenesis.
These integrin features enable the organism to discriminate noise from critical external cues as well as ensure a quick response to these stimuli, both necessary elements required for multicellular organisms to maintain their survival advantage in a rapidly changing environment. Accordingly, cells use both cadherins and integrins to assemble into multicellular tissues, to distinguish and rapidly respond to external cues, and to amplify the signal to launch an appropriate and coordinated response. Tissues achieve this task by a series of evolutionarily conserved modules that are initiated through the adhesion sensor, transduced through a series of molecular switches, and propagated through amplifiers (21, 47, 48) (Fig. 3).
Fig. 3.
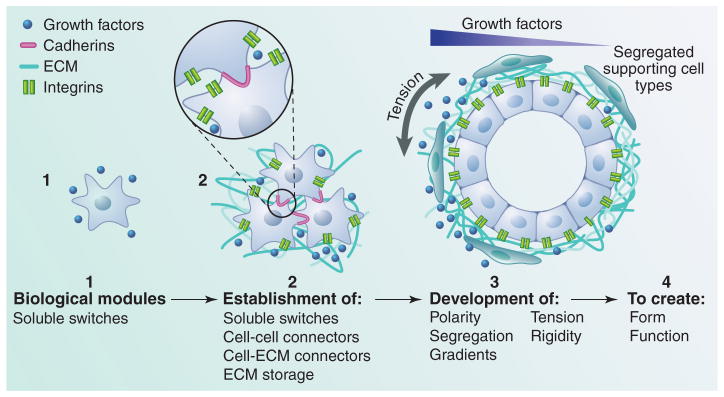
Tensional homeostasis and emergent properties of multicellular systems. At a single-cell level, filopodial projections probe the cellular environment while cells secrete and ingest growth factors that act as switches to turn on behaviors. With the onset of multicellular aggregates, adhesive “connectors,” for example, cadherins, form and link the lamellopodia to a lattice of individual actin filaments within the cell, permitting adhesion to and migration on surfaces or dense fibrillar network. Whereas these structures can contribute to cell sheets such as an epithelial cell layer adhered to a basement membrane, the development of segregated adhesion structures establishes cell polarity, morphogen gradients signal to cells to regulate cell coordinates within the body plan, and actomyosin-based contractions allow cells to integrate within a 3D environment in a particular structure. Within this framework, the continuous contraction against a compliant ECM maintains tensional homeostasis to create form and function through the incorporation of all of these conserved modules.
Signaling in Context: Emergent Properties of Complex Systems
Multicellular organisms require stable adhesion between neighboring cells and coordination of cell behaviors through cell-cell signaling to develop shape and compartmentalize function into tissues. The first coordinated event to occur in metazoa is gastrulation, which imparts a body pattern. Given the level of reorganization required to establish the resulting form (49), it is evolutionarily advantageous to ensure tighter regulation and spatial arrangement of proliferative ectodermal cells covering the embryo versus motile, involuting cells during gastrulation. The impetus to rearrange and expand the multiple layers of tissue, however, may be due to the microtubule organizing center having competing roles in motility and cell division (50). As a result, metazoans employ additional cell-cell adhesion-based mechanisms to control the identity and spatial distribution of differentiated cells, including cell polarity, tension, and morphogen gradients, rather than relying on proliferation alone. Cell polarity refers to the asymmetric distribution of cell constituents and organelles, and, if coordinated through cell-cell connectors, cell orientation can effect tissue-wide polarity, known as planar cell polarity (PCP), using a highly conserved set of polarity protein complexes (51). This behavior is likely to have arisen from unicellular organisms that would distribute unequally damaged cell components to bypass senescence (52). The advent of stable connector modules such as adherens or tight junctions further contributed to the development of apical-basal membrane segregation, permitting the establishment of cell sheets as well as outside and inside separation of an organism. Orientated cell division and polarized cell shape changes, such as those seen in the convergence and extension phase of gastrulation, can also be used to rotate the body axis out of the plane of a tissue, to contribute to the differential spatial orientation of cells, and to establish anisotropic mechanical properties (49). The latter of these characteristics can establish differences in ECM properties by secretion or cross-linking, setting up spatially controlled matrix topography and elasticity, both of which are known regulators of differentiation (53, 54). Interplay among polarity, ECM, and adhesions has also been shown in mammary acini, where increased matrix elasticity altered tensional homeostasis, perturbed tissue polarity, and promoted a malignant phenotype (55). Polarity, however, should be thought of not just in terms of how it modulates matrix and restricts secretion but also how it changes the context of signaling; for example, loss of Scribble in mammary epithelia can block morphogenesis and induce dysplasia by disrupting cell polarity and inhibiting apoptosis (56). Cells organized into polarized tissue structures respond very differently to external signaling cues than do cell sheets or isolated cells (57). In fact, by forcing a polarized tissue structure on both normal and teratocarcinoma cells, the signaling milieu that the cells inhabit can give rise to animals that retain tumor cells but exhibit no detectable tumor phenotype (58). These observations argue that additional emergent properties arise in polarized tissue structures that regulate cell and tissue behavior.
In addition to polarity, positional information within the organism and tissue also need to be programmed after initial cell segregation. Body plan axis and length, for example, are regulated by morphogen gradients, where local signal concentrations define the coordinates for each cell, based on source distance (59). Progenitor cell phenotype can be regulated by these gradients, where cells from one location transplanted to another will express the phenotype of the new niche (4). In fact, nodal gradients in the developing embryo even modulate the development of tension in the blastula (6). Mechanoregulation of homeostasis by morphogen gradients likely continues through gastrulation, the formation of organs, and internal assembly, because evidence shows that they can direct cells to stop proliferating to maintain size (60), as well as cease migration (11) once cells are appropriately segregated.
Morphogen gradients are not always present in nonstereotyped organs such as the heart or mammary acini, where homeostasis is maintained by a balance between matrix compliance and cell tension in a manner that may parallel proposed intracellular tension balances, such as with the concept of tensegrity (61). This argues for a set of newly emerging properties that can shape tissue and organ level form and function. Recent evidence implicates tension as such a regulator, not only to shape cell form as previously shown but also to control tissue formation (62). As mammary acini secrete milk proteins, this generates outward pressure on the cells, tensing their adhesive modules and forcing them into a spherical structure, which maximizes their surface area to volume ratio so that they hold as much fluid as possible while at the same time minimizing the energetics of the system to promote stability. When tension is misregulated at this length scale, as with constitutively active Rho, it can shift the acinar force balance to compromise morphogenesis and integrity and induce a cancer-like phenotype. As a disease parallel to this, breast cancer is characterized by increased matrix stiffness and cell contractility, altered rheology, and changes in cell shape and tissue architecture (55). In addition to these force-induced changes in cell and tissue behavior, matrix stiffness and elevated cell tension stimulate excessive fibronectin production that compromises tissue integrity and perturbs tissue polarity (63), illustrating how cell and matrix tension operate at multiple length scales to influence malignancy. Conversely, normal acinar morphology can be a powerful tumor suppressor, preventing expression of the malignant phenotype even in cells with a multitude of genomic alterations, including amplifications in key oncogenes (64). Heart looping also appears to be force sensitive, such that a threefold gradient in matrix deposition corresponds to a similar gradient in stiffness for the inner versus outer curvature of the heart tube. As hemodynamic forces differentially press against the softer basal wall, it induces cell shape changes that create the looped form of the embryonic heart (65). Altered differences in matrix gradients could easily upset how cells at the outer curve extend by changing the cell tension that cells along the curve can generate (7). The underpinnings of tension-driven regulation may rest in clarifying why changing membrane tension or curvature induced by matrix or shape perturbations can exert such a profound effect on the lineage specification of stem cells (14, 54). Although tissue development and homeostasis clearly require reciprocal cross-talk between the cell and its extracellular matrix mediated through dynamic adhesion interactions (64), scaling up these cell-ECM changes to tissue level behaviors, much like has been done with morphogen gradients and polarity, will greatly advance our understanding of these modules and their economies of scale.
Unresolved Issues?
Although emergent properties of multicellular tissues and signaling modules clearly regulate processes such as gastrulation and acini formation, it is not clear how these morphogenetic events shape an organ, tissue, or cell at large distances where diffusion of morphogens may be limited, direct cell-cell contacts are out of range, and the matrix is discontinuous. Do processes that sculpt multicellular tissues and maintain homeostasis at short length scales and acute time frames operate similarly in the organism at larger length scales in complex tissues or organ systems with multiple cell types and variable chronologies? For some of the open developmental questions posed above, modular analogies (Fig. 1) perhaps improve our ability to describe the toolbox of tissue homeostasis and clarify the circuitry rules required to use them, i.e., combining connectors in series. However, much of our circuitry diagram is missing, and key integrators that operate at multilength scales and that retain the molecular memory necessary for the long-term viability and adaptability demanded of complex organisms have yet to be identified. The solution to understanding how this enormous task is achieved likely rests on discovering new modules and alternate signaling paradigms. Nevertheless, it is clear that without including a comprehensive description of all the environmental players and clarifying their interrelations—for example, matrix, cadherins, and integrins—an explanation of the origin of form and function will remain incomplete.
Acknowledgments
We apologize to the many authors whose work is not cited due to space limitations. This work was supported by grants NIH R01-CA078731, Department of Defense Era of Hope Breast Cancer Research Grant W81XWH-05-1-330, Department of Energy A107165, and California Institute for Regenerative Medicine RS1-00449 to V.M.W.; American Heart Association 0865150F to A.J.E.; the Australian NHMRC, CCV, Prostate Cancer Foundation of Australia; the AICR UK and NHMRC Career Development Award to P.O.H.; and the Swiss National Science Foundation and Nevus Outreach to B.W.-H.
References and Notes
- 1.Edelman GM. Topobiology: An Introduction to Molecular Embryology. Basic Books; New York: 1988. [DOI] [PubMed] [Google Scholar]
- 2.Townes P, Holtfreter J. J Exp Zool. 1955;12853 [Google Scholar]
- 3.Brouzes E, Farge E. Curr Opin Genet Dev. 2004;14:367. doi: 10.1016/j.gde.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 4.Mariani FV, Martin GR. Nature. 2003;423:319. doi: 10.1038/nature01655. [DOI] [PubMed] [Google Scholar]
- 5.Desprat N, Supatto W, Pouille PA, Beaurepaire E, Farge E. Dev Cell. 2008;15:470. doi: 10.1016/j.devcel.2008.07.009. [DOI] [PubMed] [Google Scholar]
- 6.Krieg M, et al. Nat Cell Biol. 2008;10:429. doi: 10.1038/ncb1705. [DOI] [PubMed] [Google Scholar]
- 7.Nelson CM, et al. Proc Natl Acad Sci USA. 2005;102:11594. doi: 10.1073/pnas.0502575102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Rooij J, Kerstens A, Danuser G, Schwartz MA, Waterman-Storer CM. J Cell Biol. 2005;171:153. doi: 10.1083/jcb.200506152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heinemeier KM, et al. J Physiol. 2007;582:1303. doi: 10.1113/jphysiol.2007.127639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnson CP, Fujimoto I, Perrin-Tricaud C, Rutishauser U, Leckband D. Proc Natl Acad Sci USA. 2004;101:6963. doi: 10.1073/pnas.0307567100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gumbiner BM. Cell. 1996;84:345. doi: 10.1016/s0092-8674(00)81279-9. [DOI] [PubMed] [Google Scholar]
- 12.Rozario T, Dzamba B, Weber GF, Davidson LA, Desimone DW. Dev Biol. 2008;327:386. doi: 10.1016/j.ydbio.2008.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miyamoto S, Akiyama SK, Yamada KM. Science. 1995;267:883. doi: 10.1126/science.7846531. [DOI] [PubMed] [Google Scholar]
- 14.McBeath R, Pirone DM, Nelson CM, Bhadriraju K, Chen CS. Dev Cell. 2004;6:483. doi: 10.1016/s1534-5807(04)00075-9. [DOI] [PubMed] [Google Scholar]
- 15.Stephens LE, et al. Genes Dev. 1995;9:1883. doi: 10.1101/gad.9.15.1883. [DOI] [PubMed] [Google Scholar]
- 16.Li S, et al. J Cell Biol. 2002;157:1279. doi: 10.1083/jcb.200203073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li S, et al. J Cell Sci. 2005;118:2913. doi: 10.1242/jcs.02422. [DOI] [PubMed] [Google Scholar]
- 18.Sakai T, et al. Genes Dev. 2003;17:926. doi: 10.1101/gad.255603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Darribere T, Schwarzbauer JE. Mech Dev. 2000;92:239. doi: 10.1016/s0925-4773(00)00245-8. [DOI] [PubMed] [Google Scholar]
- 20.Siu CH, Harris TJ, Wang J, Wong E. Semin Cell Dev Biol. 2004;15:633. doi: 10.1016/j.semcdb.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 21.Magie CR, Martindale MQ. Biol Bull. 2008;214:218. doi: 10.2307/25470665. [DOI] [PubMed] [Google Scholar]
- 22.Cornillon S, et al. EMBO Rep. 2006;7:617. doi: 10.1038/sj.embor.7400701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hughes AL. J Mol Evol. 2001;52:63. doi: 10.1007/s002390010134. [DOI] [PubMed] [Google Scholar]
- 24.Ryder C, Byrd M, Wozniak DJ. Curr Opin Microbiol. 2007;10:644. doi: 10.1016/j.mib.2007.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li F, Palecek SP. Microbiology. 2008;154:1193. doi: 10.1099/mic.0.2007/013789-0. [DOI] [PubMed] [Google Scholar]
- 26.Halme A, Bumgarner S, Styles C, Fink GR. Cell. 2004;116:405. doi: 10.1016/s0092-8674(04)00118-7. [DOI] [PubMed] [Google Scholar]
- 27.Eichinger L, et al. Nature. 2005;435:43. doi: 10.1038/nature03481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cornillon S, Froquet R, Cosson P. Eukaryot Cell. 2008;7:1600. doi: 10.1128/EC.00155-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bukahrova T, et al. J Cell Sci. 2005;118:4295. doi: 10.1242/jcs.02557. [DOI] [PubMed] [Google Scholar]
- 30.King N, et al. Nature. 2008;451:783. doi: 10.1038/nature06617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Exposito JY, et al. J Biol Chem. 2008;283:28226. doi: 10.1074/jbc.M804573200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pancer Z, Kruse M, Muller I, Muller WE. Mol Biol Evol. 1997;14:391. doi: 10.1093/oxfordjournals.molbev.a025775. [DOI] [PubMed] [Google Scholar]
- 33.Brower DL, Brower SM, Hayward DC, Ball EE. Proc Natl Acad Sci USA. 1997;94:9182. doi: 10.1073/pnas.94.17.9182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Srivastava M, et al. Nature. 2008;454:955. doi: 10.1038/nature07191. [DOI] [PubMed] [Google Scholar]
- 35.Heino J, Huhtala M, Kapyla J, Johnson MS. Int J Biochem Cell Biol. 2009;41:341. doi: 10.1016/j.biocel.2008.08.021. [DOI] [PubMed] [Google Scholar]
- 36.Knack BA, et al. BMC Evol Biol. 2008;8:136. doi: 10.1186/1471-2148-8-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brown NH. Matrix Biol. 2000;19:191. doi: 10.1016/s0945-053x(00)00064-0. [DOI] [PubMed] [Google Scholar]
- 38.Whittaker CA, et al. Dev Biol. 2006;300:252. doi: 10.1016/j.ydbio.2006.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tucker RP, Chiquet-Ehrismann R. Int J Biochem Cell Biol. 2009;41:424. doi: 10.1016/j.biocel.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 40.Chen H, Cohen DM, Choudhury DM, Kioka N, Craig SW. J Cell Biol. 2005;169:459. doi: 10.1083/jcb.200410100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Johnson CP, Tang HY, Carag C, Speicher DW, Discher DE. Science. 2007;317:663. doi: 10.1126/science.1139857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cluzel C, et al. J Cell Biol. 2005;171:383. doi: 10.1083/jcb.200503017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Paulsson M. Crit Rev Biochem Mol Biol. 1992;27:93. doi: 10.3109/10409239209082560. [DOI] [PubMed] [Google Scholar]
- 44.von Wichert G, et al. J Cell Biol. 2003;161:143. doi: 10.1083/jcb.200211061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhu J, et al. Mol Cell. 2008;32:849. doi: 10.1016/j.molcel.2008.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Friedland JC, Lee MH, Boettiger D. Science. 2009;323:642. doi: 10.1126/science.1168441. [DOI] [PubMed] [Google Scholar]
- 47.Bowers-Morrow VM, Ali SO, Williams KL. Biol Rev Camb Philos Soc. 2004;79:611. doi: 10.1017/s1464793103006389. [DOI] [PubMed] [Google Scholar]
- 48.King N, Hittinger CT, Carroll SB. Science. 2003;301:361. doi: 10.1126/science.1083853. [DOI] [PubMed] [Google Scholar]
- 49.Keller R, Davidson LA, Shook DR. Differentiation. 2003;71:171. doi: 10.1046/j.1432-0436.2003.710301.x. [DOI] [PubMed] [Google Scholar]
- 50.King N. Dev Cell. 2004;7:313. doi: 10.1016/j.devcel.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 51.Lawrence PA, Struhl G, Casal J. Nat Rev Genet. 2007;8:555. doi: 10.1038/nrg2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Macara IG, Mili S. Cell. 2008;135:801. doi: 10.1016/j.cell.2008.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dalby MJ, et al. Nat Mater. 2007;6:997. doi: 10.1038/nmat2013. [DOI] [PubMed] [Google Scholar]
- 54.Engler AJ, Sen S, Sweeney HL, Discher DE. Cell. 2006;126:677. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 55.Paszek MJ, et al. Cancer Cell. 2005;8:241. doi: 10.1016/j.ccr.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 56.Zhan L, et al. Cell. 2008;135:865. doi: 10.1016/j.cell.2008.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Weaver VM, et al. Cancer Cell. 2002;2:205. doi: 10.1016/s1535-6108(02)00125-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mintz B, Illmensee K. Proc Natl Acad Sci USA. 1975;72:3585. doi: 10.1073/pnas.72.9.3585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jaeger J, et al. Nature. 2004;430:368. doi: 10.1038/nature02678. [DOI] [PubMed] [Google Scholar]
- 60.Rogulja D, Rauskolb C, Irvine KD. Dev Cell. 2008;15:309. doi: 10.1016/j.devcel.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ingber DE. Sci Am. 1998;278:48. doi: 10.1038/scientificamerican0198-48. [DOI] [PubMed] [Google Scholar]
- 62.Nelson CM, Bissell MJ. Annu Rev Cell Dev Biol. 2006;22:287. doi: 10.1146/annurev.cellbio.22.010305.104315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Williams CM, Engler AJ, Slone RD, Galante LL, Schwarzbauer JE. Cancer Res. 2008;68:3185. doi: 10.1158/0008-5472.CAN-07-2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Weaver VM, et al. J Cell Biol. 1997;137:231. doi: 10.1083/jcb.137.1.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zamir EA, Srinivasan V, Perucchio R, Taber LA. Ann Biomed Eng. 2003;31:1327. doi: 10.1114/1.1623487. [DOI] [PubMed] [Google Scholar]
- 66.Shvartsman SY, Muratov CB, Lauffenburger DA. Development. 2002;129:2577. doi: 10.1242/dev.129.11.2577. [DOI] [PubMed] [Google Scholar]
- 67.Strutt H, Strutt D. Curr Biol. 2003;13:1451. doi: 10.1016/s0960-9822(03)00545-1. [DOI] [PubMed] [Google Scholar]
- 68.Amonlirdviman K, et al. Science. 2005;307:423. doi: 10.1126/science.1105471. [DOI] [PubMed] [Google Scholar]
- 69.Weber U, Pataki C, Mihaly J, Mlodzik M. Dev Biol. 2008;316:110. doi: 10.1016/j.ydbio.2008.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.del Rio A, et al. Science. 2009;323:638. doi: 10.1126/science.1162912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Geiger B, Spatz JP, Bershadsky AD. Nat Rev Mol Cell Biol. 2009;10:21. doi: 10.1038/nrm2593. [DOI] [PubMed] [Google Scholar]
- 72.Grimson MJ, et al. Nature. 2000;408:727. doi: 10.1038/35047099. [DOI] [PubMed] [Google Scholar]
- 73.Foty RA, Steinberg MS. Dev Biol. 2005;278:255. doi: 10.1016/j.ydbio.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 74.Bertet C, Sulak L, Lecuit T. Nature. 2004;429:667. doi: 10.1038/nature02590. [DOI] [PubMed] [Google Scholar]
- 75.Chan CE, Odde DJ. Science. 2008;322:1687. doi: 10.1126/science.1163595. [DOI] [PubMed] [Google Scholar]
- 76.Gardel ML, et al. J Cell Biol. 2008;183:999. doi: 10.1083/jcb.200810060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Baumgart T, Hess ST, Webb WW. Nature. 2003;425:821. doi: 10.1038/nature02013. [DOI] [PubMed] [Google Scholar]
- 78.Small AR, et al. J Theor Biol. 2008;252:593. doi: 10.1016/j.jtbi.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 79.Wang X, Harris RE, Bayston LJ, Ashe HL. Nature. 2008;455:72. doi: 10.1038/nature07214. [DOI] [PubMed] [Google Scholar]