

Abstract
In an effort to identify novel biallelically inactivated tumor suppressor genes (TSG) in sporadic invasive and pre-invasive non-small cell lung cancer (NSCLC) genomes, we applied a comprehensive integrated multi-‘omics approach to investigate patient matched, paired NSCLC tumor and non-malignant parenchymal tissues. By surveying lung tumor genomes for genes concomitantly inactivated within individual tumors by multiple mechanisms, and by the frequency of disruption in tumors across multiple cohorts, we have identified a putative lung cancer TSG, Eyes Absent 4 (EYA4). EYA4 is frequently and concomitantly deleted, hypermethylated and underexpressed in multiple independent lung tumor data sets, in both major NSCLC subtypes, and in the earliest stages of lung cancer. We find not only that decreased EYA4 expression is associated with poor survival in sporadic lung cancers, but EYA4 SNPs are associated with increased familial cancer risk, consistent with EYA4’s proximity to the previously reported lung cancer susceptibility locus on 6q. Functionally, we find that EYA4 displays TSG-like properties with a role in modulating apoptosis and DNA repair. Cross examination of EYA4 expression across multiple tumor types suggests a cell type-specific tumorigenic role for EYA4, consistent with a tumor suppressor function in cancers of epithelial origin. This work shows a clear role for EYA4 as a putative TSG in NSCLC.
Keywords: EYA4, two-hit, hypermethylation, tumor suppressor, TSG, non-small cell lung cancer
Introduction
Lung cancer is the leading cause of cancer mortality in the world, accounting for 1.5 million deaths each year (1). Over 80% of lung cancers are non-small cell lung cancer (NSCLC), of which adenocarcinomas (AC) and squamous cell carcinomas (SqCC) are the predominant subtypes (2). Due to late stage diagnosis and paucity of effective treatments, the five year survival for lung cancer patients is less than 15%. There remains an urgent, worldwide need for early detection markers and improved chemoprevention and therapeutic regimes for this disease.
Multiple genetic mechanisms contribute to the evolution of cancer genomes, therefore integration of data from multi-‘omics levels for an individual tumor represents a powerful approach for discovering genes selectively altered in tumors. Within individual tumor genomes, it is likely that genes selectively disrupted sustain biallelic, or ‘two-hit’, disruptions, as commonly observed with many tumor suppressor genes (TSGs) (3). Therefore we hypothesized that genes, i) sustaining frequent, high level, and two-hit gene dosage and/or DNA methylation alterations and ii) undergoing concomitant alterations at the mRNA level, would be indicative of genes selectively inactivated in lung tumors and therefore relevant to lung tumor biology.
Applying this rationale to genome-wide copy number, DNA methylation and gene expression profiles from a large panel of NSCLC tumor clinical specimens with patient-matched non-malignant parenchymal tissues, we discovered a novel putative lung cancer tumor suppressor gene Eyes Absent 4 (EYA4). EYA4, a putative oncogene in tumors of neural origin, is an atypical, dual-functioning protein phosphatase which functions in mediating DNA repair, apoptosis and innate immunity in response to DNA damage, damaged cells and viruses. Our findings suggest a dual-role for EYA4 in carcinogenesis, likely dependent on cancer cell type of origin and strongly supportive of a tumor suppressor role in lung cancer. Collectively, our findings illustrate the utility of a multi-dimensional tumor systems approach to cancer ‘omics research applied to the discovery of novel lung cancer TSGs.
Results
Few genes are inactivated by homozygous deletion in lung adenocarcinomas
We sought to investigate whether homozygous deletion (HD) is a mechanism of recurrent gene inactivation in a panel of adenocarcinoma (AC) tumors and patient matched non-malignant lung parenchyma tissues (n=77 pairs), using Affymetrix SNP 6.0 arrays. We calculate that a DNA copy number (CN) of 0.4 should represent a HD if tumor cell content is 80% in a given specimen. In our panel of 77 AC specimens, we identified only two genes, CDKN2A and CDKN2B, which were HD at CN <0.4 in more than two specimens, consistent with previous reports for these genes (4). To compensate for cytological heterogeneity within tumors, we relaxed our HD detection threshold to CN<1.0, which yielded no further HD genes (Supplementary Table S1). Therefore, we reasoned that biallelic inactivation of TSGs must occur through a combination of other mechanisms such as DNA hypermethylation and single copy loss.
EYA4 is frequently inactivated by deletion and hypermethylation in lung adenocarcinoma
To identify genes disrupted by deletion and promoter hypermethylation, we obtained genome-wide DNA methylation profiles (Illumina Infinium HumanMethylation27) for this same panel of 77 AC tumor pairs. We searched for frequent (>15%) and concurrent copy number loss and promoter hypermethylation events. We identified 114 genes that were frequently deleted and hypermethylated in the same tumor (Supplementary Table S2), which included both previously reported and novel putative lung TSGs. Integration with expression data revealed 37 genes that were significantly underexpressed, lost and hypermethylated in our cohort (indicated in Supplementary Table S2). Of these, we focused on the putative TSG EYA4, based on: the high frequency of biallelic disruption (19.5%) and significant underexpression (32.5%) in our cohort (Figure 1A–B), frequency of inactivation by multiple mechanisms in other epithelial cancers, and proximity to the lung cancer susceptibility locus at 6q23 (5–9). Overall, 67.5 % of our AC panel sustained allelic inactivation of EYA4 by either copy number loss (26%) or by promoter hypermethylation (61%) (e.g. Figure 1A). We calculated the probability of observing a 2- hit DNA level and gene expression event in a single tumor pair by multiplying the proportion of any probe we observed undergoing hypermethylation, copy number loss and underexpression alterations (10). The average proportion of each of these events occurring in our cohort of 77 AC tumor pairs, was: hypermethylation 0.0825, copy number loss 0.1614, and underexpression 0.1176. Therefore the probability of observing a 2-hit inactivating DNA level alteration and underexpression event for a single gene in a tumor sample from our cohort was 0.0016. Moreover, the probability of randomly observing the frequency for which we detect EYA4 inactivated by these mechanisms is extremely low (1.433 X 10 −22) (Supplemental Figure. S1). These findings suggest that EYA4 inactivation is strongly selected for in AC. We validated mechanistic control of EYA4 expression by DNA methylation by observing re-expression of hypermethylated EYA4 in AC cancer cells after treatment with a de-methylating agent (5-azacytidine) (Supplementary Figure. S2, Supplemental Table S3).
Figure 1. Identification of EYA4 as a frequently inactivated TSG in lung cancer.

(A) Summary of gene dosage, DNA methylation, and gene expression data for 77 tumor normal pairs. Each column represents one tumor sample, and red boxes indicate the presence of either a copy number loss, hypermethylation or underexpression alteration in a tumor relative to its matched non-malignant parenchymal profile. (B) EYA4 mRNA is significantly (p<1 x 106, paired t-test) under-expressed in AC tumors (n=83) compared to patient-matched non-malignant lung specimens (see also Figure S3). (C) EYA4 is significantly underexpressed in SqCC tumors (n=45) compared to bronchial epithelia from small airways (n=67) (p<0.0001 Wilcoxon signed rank) (D) EYA4 is significantly more methylated in promoters of SqCC tumors compared to matched non-malignant lung tissue (n=8 pairs) (p<0.02). Illumina GoldenGate probe β values were averaged for each sample and plotted as a separate dot.
EYA4 is inactivated by copy number loss and hypermethylation in both major NSCLC subtypes
We also found that EYA4 was significantly underexpressed (p < 0.0001) in a panel of 45 SqCC tumors compared to 67 histologically normal bronchial epithelia samples, and also hypermethylated (p < 0.02) in a panel of 8 SqCC tumors compared to 8 bronchial epithelia samples for which DNA methylation profiles were available (Figure 1C and D). We also applied our criteria to DNA methylation data downloaded from the recently published lung squamous Cancer Genome Atlas (TCGA) study (11). We limited our validation only to those TCGA SqCC tumors with available matched normal specimens (n = 27 tumor pairs). We found 15%–37% of these tumors had a 20% or greater methylation increase at EYA4 loci compared to their matched normal counterparts. We further examined an external set of NSCLC specimens (n=883 tumors) from 4 additional datasets and found 14% (n= 123 tumors) exhibited deletion of EYA4 (12–15). Reduced EYA4 expression was confirmed in two additional independent NSCLC cohorts for which matched normal references were available (Supplementary Figure. S3 (16, 17). Taken together, these results strongly indicate that EYA4 is disrupted in both major subtypes of NSCLC.
Mutation is not a major mechanism of EYA4 disruption
We evaluated whether DNA sequence mutations occurred in EYA4 by analyzing the Catalogue of Somatic Mutations in Cancer (COSMIC) database (http://cancer.sanger.ac.uk/cancergenome/projects/cosmic/) and also by sequencing all 20 coding exons of EYA4 in a panel of 38 AC cell lines (listed in Supplementary Table S3). COSMIC analysis revealed 20 confirmed somatic mutations (3%) in a cohort of 639 clinical NSCLC samples. In lung cancer cell lines, we identified only one likely pathogenic variant in exon 7 (EYA4:c.385G>C, p.Gly129Arg) of sample H23 that converts a Gly codon to Arg (Figure 2A). This mutation is predicted by PolyPhen2 and SIFT to be deleterious (18) based on the nature of the amino acid change and the conservation at that residue. To examine the impact of this mutation in these cells, we assessed the locus for gene dosage, loss of heterozygosity, DNA methylation, and mRNA and protein expression. These analyses revealed that EYA4 is diploid in H23, is not hypermethylated, and resides within an area of acquired uniparental disomy. This is consistent with our observation of a homozygous EYA4 mutation and moderate mRNA expression in these cells, but no detectable EYA4 protein, indicating perhaps that this mutation may impact protein stability (Figure 2B). Given the frequency of observed copy number loss (43%) or hypermethylation (40%) events affecting EYA4 relative to low frequency of EYA4 mutations (3%), we posit that sequence level mutations are not a major mechanism contributing to EYA4 disruption in lung cancer.
Figure 2. Mechanisms of two-hit EYA4 inactivation.
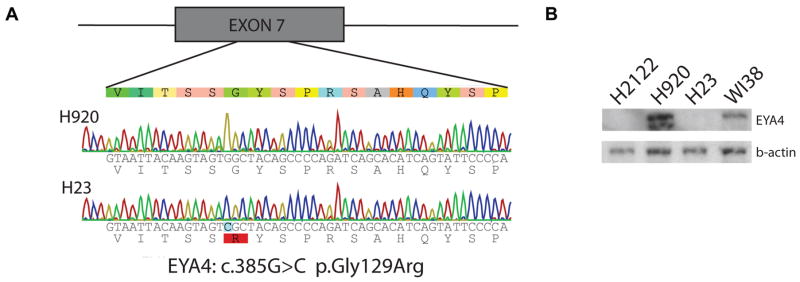
(A) Sample H23 shows a coding sequence mutation in EYA4. All exons and proximal intronic sequences were screened in 38 AC lines for mutations by Sanger sequencing. A likely pathogenic missense mutation in exon 7 was identified (EYA4:c.385G>C, p.Gly129Arg), which is shown along with wild-type H920. (B) This mutation is associated with a lack of protein production as shown by immunoblot, despite mRNA production by H23. H2122 and H920 are negative and positive controls, respectively, while WI38 is a normal fibroblast reference line. This suggests that the mutation may result in premature protein degradation.
EYA4 inactivation is an early event in lung cancer
To assess whether somatic DNA level disruptions of EYA4 occur early in tumorigenesis, we evaluated gene dosage levels in 20 carcinoma in situ (CIS) specimens. These rare samples were collected by fluorescent bronchoscopy (Figure 3A) and represent a stage of cancer development typically too early for detection using routine imaging procedures. Deletion of EYA4 was observed in 35% of these pre-invasive squamous CIS lesions, supporting loss of EYA4 as an early neoplastic event (Table 1). Transcriptome sequencing data generated in a previous study (19), revealed reduced EYA4 expression in CIS and SqCC samples compared to histologically normal bronchial epithelial cells (Figure 3B). At the level of DNA methylation, we detected EYA4 hypermethylation in 40% (n=10) of cytologically normal bronchial epithelia from patients with NSCLC, as well as in one high-risk patient with chronic-obstructive pulmonary disease (Figure 3C). Inactivation of EYA4 in the precursors of disease implicates EYA4 as an early event in lung tumorigenesis.
Figure 3. EYA4 inactivation is an early event.

(A) White light and autofluorescence bronchoscopy identifies carcinoma in situ lesions (CIS). (B) EYA4 expression was assessed by serial-analysis of gene expression in normal bronchial epithelia (n=14), CIS (n=5), and invasive SqCC (n=6) samples. EYA4 is underexpressed in the CIS group (p<0.05 compared to normal) and remains reduced in SqCC. (C) EYA4 is hypermethylated in histologically normal bronchial epithelia of NSCLC patients (n=10) compared to patients without cancer (n=5) as detected by MS-PCR, providing compelling evidence that inactivation of EYA4 is a very early event. One high-risk COPD patient is indicated (*), and positive (H1395) and negative (HCC-2935) controls are shown.
Table 1. Deletion of EYA4 locus in carcinoma in situ specimens.
DNA copy number of EYA4 was assessed by array CGH in CIS (n=20). “del” indicate clones that undergo copy number loss, “0” is neutral, and “x” are uninformative. EYA4 is frequently (35%) deleted in these very early lesions.
| Clone Name | N0654A14 | N0261J24 |
|---|---|---|
| Start (hg18 bp) | 133578312 | 133659674 |
| End (hg18 bp) | 133721268 | 133850462 |
| CIS 1 | del | del |
| CIS 2 | del | del |
| CIS 3 | del | del |
| CIS 4 | 0 | 0 |
| CIS 5 | 0 | 0 |
| CIS 6 | 0 | 0 |
| CIS 7 | 0 | x |
| CIS 8 | x | 0 |
| CIS 9 | 0 | 0 |
| CIS 10 | del | del |
| CIS 11 | 0 | 0 |
| CIS 12 | 0 | 0 |
| CIS 13 | 0 | 0 |
| CIS 14 | 0 | 0 |
| CIS 15 | del | del |
| CIS 16 | 0 | 0 |
| CIS 17 | del | del |
| CIS 18 | 0 | 0 |
| CIS 19 | del | del |
| CIS 20 | x | 0 |
EYA4 exhibits functional characteristics of a lung cancer tumor suppressor gene
Stable EYA4 knock-down (EYA4kd) of lung lymphoblastoid (HCC-1954BL) cell lines and ectopic overexpression (EYA4+) of AC cells (H2122, H2405), were established for all in vitro and in vivo assays (Supplemental Figure S4A). AC lines were chosen based on lack of detectable EYA4 protein expression (Supplemental Figure S2E), and lymphoblastoids as karyotypically normal models highly amenable to the assays performed. Notably, while more appropriate non-malignant lung models were available, including human bronchial epithelial cells (HBECs), small airway epithelial cells (SAEC), and fetal lung fibroblasts (WI-38), we found these cells unsuitable for subsequent analyses due to unselectability based on our knock-down construct (HBECs), or drastic changes to cell morphology and behavior (WI-38) upon EYA4 knock-down. EYA4 knock-down was also attempted in non-transformed SAEC lines, however loss of EYA4 in these normal epithelial cells was not tolerated and resulted in cellular senescence, as previously described (20) (data not shown).
EYA4 loss negatively impacts DNA repair and genomic instability
EYA4 is an atypical phosphatase, containing a C-terminal tyrosine (Tyr) and a recently discovered N-terminal threonine (Thr) phosphatase domain, that have distinct functions and catalytic activity conditions (21, 22). In response to double stranded breaks (DSB) EYA4 dephosphorylates the Tyr-142 residue of H2AX facilitating phosphorylation of Ser-139 of H2AX (forming γH2AX) leading to recruitment of DNA repair complex components to sites of DSB (21, 23–26). We assessed levels of γH2AX and Tyr-142 phosphorylation of H2AX in EYA4kd and control cells following DNA damage induced by irradiation. Cells lacking EYA4 accumulate markedly more and longer lasting γH2AX than controls (Figure 4A). Consistently, we observed markedly higher levels of Tyr-142 phosphorylated H2AX in EYA4kd cells in response to irradiation, compared to empty vector cells (Figure 4B–D). However, restoration of EYA4 expression in EYA4+ cancer cell models had no significant effect on γH2AX levels following irradiation (Supplementary Figure S4C–D). These results suggest that while EYA4kd in karyotypically normal cells results in increased DSB during DNA damage onslaught (as measured by γH2AX accumulation); EYA4 restoration in lung cancer cells is not sufficient to modulate γH2AX-mediated repair. This is likely due to high frequency of mutations in key DNA repair genes, such as TP53 or CDKN2A (both mutated in H2122 and H2405).
Figure 4. EYA4 promotes efficient DNA repair in karyotypically normal cells.

(A) γH2AX levels in EYA4kd cells. DNA damage response was assessed in EYA4kd cells. Normalized fluorescence intensity for γH2AX in cells at different time points following irradiation with 5 Gy show cells lacking EYA4 (square markers) accumulate markedly more γH2AX than wild type cells (round markers). Cells lacking EYA4 also demonstrate a marked reduction in the removal of γH2AX up to 6h post irradiation. (B) Western blot of the levels of Tyr-142 phosphorylated H2AX in HCC-1954BL control and EYA4kd cell lysates at 2 and 5 hours post-irradiation. Beta-actin was used as a loading control. C) Cisplatin sensitivity assays for EYA4 ectopically expressing H2405 lung adenocarcinoma cells and H2405 cells that do not express EYA4 (LacZ controls) were performed in replicates of five. Lung adenocarinoma cells (no EYA4) are more sensitive to the DNA damage inducing agent, cisplatin. p value calculated by a paired 1 tailed t-test. (D) Fraction of genome encompassed by segmental copy number alterations (a measure for genomic instability) calculated for AC tumors (n=83, BCCA) (see Methods). Genomic instability was significantly higher in tumors with low versus high EYA4 expressing tumors (p<0.05). (E) Low EYA4 expression was also significantly associated with a greater extent of genomic instability (p<0.05) in an additional, independent cohort of AC (n=193, MSKCC).
To further explore the relationship between EYA4 and DNA damage response in cancer cells, we examined the effect of a DNA damage-inducing agent, cisplatin in response to EYA4 overexpression. We observed cancer cells made to overexpress EYA4 were reproducibly more resistant to cisplatin than those without EYA4, indicating that in the absence EYA4, cancer cells appear more sensitive to DNA damage onslaught (Figure 4C). Given the observed effect of EYA4 on DNA repair and response to DNA damage, we hypothesized that tumors with reduced EYA4 expression would exhibit a greater extent of genomic instability. To test this, we compared the proportion of the genome encompassed by segmental copy number alterations in 83 lung AC tumors with high and low EYA4 gene expression (3.2 fold expression difference between groups). This comparison revealed a significant trend towards increased genomic instability in tumors with low EYA4 expression (,p=0.0413, two-tailed U-test, Figure 4D); an association further supporting a role for EYA4 in DNA damage repair. We found a similar trend in a second, independent lung AC cohort (n=193) downloaded from the Memorial Sloan Kettering Cancer Centre (14) (p=0.0027) (Figure 4E).
EYA4 loss results in decreased induction of apoptosis
Previous literature implicates EYA4 as a modulator of apoptotic response (25, 27). We tested this by fluorescence activated cell sorting (FACS) of annexin V/propidium iodide (AV/PI) stained EYA4 modulated and control cells following serum starvation. EYA4+ cancer cells displayed no differences in percentage of early or late apoptotic cells (Supplemental Figure. S4B). Since cancer cells have heavily disrupted genetic backgrounds that likely interfere with apoptotic pathways, we examined the effect of EYA4 expression on apoptosis of karyotypically normal EYA4kd lines and controls. FACS results indicated that, contrary to our cancer cell lines, EYA4kd cells displayed a marked and reproducible decrease in the numbers of early (AV+/PI-) and late (AV+/PI+) apoptotic cells compared to pLKO (26.6% ± 4.6 for control vs. 14.5% ± 1.7 for EYA4kd) (Figure 5A, 5B, 5C), indicative of a pro-apoptotic role for EYA4.
Figure 5. EYA4 promotes apoptosis and genomic stability.

(A) Apoptosis was assessed in EYA4kd cells. Annexin V binding is depicted on the X-axis and PI staining on the Y-axis. FACS analysis following serum starvation indicates abrogation of the apoptotic program in EYA4kd HCC-1954BL cells compared to control cells. (B) Empty pLKO vector control cells show more apoptotic cells (upper right quadrant) vs EYA4kd cells. (C) Triplicate FACS comparisons show significantly more apoptotic cells in the control cells (grey) than EYA4kd cells (white) (p<0.05, t-test). (D) GADD45a expression levels in serum-starved EYA4kd (white) and control cells (grey) were assessed by qRT-PCR. GADD45a expression change following 24 hr of serum starvation increases substantially in control cells, and is attenuated in EYA4kd cells (p<0.001, t-test).
Previous reports have suggested a TSG role for EYA4 may be mediated via its role as a transcriptional co-activator (5, 6, 27, 28). To identify genes correlated with EYA4 expression (and potentially activated by EYA4), profiles for the ten highest and ten lowest EYA4-expressing normal bronchial specimens were compared using the significance analysis of microarrays (SAM) algorithm (29). Twenty-eight genes potentially co-activated by EYA4 were identified (q-value percentage threshold of 5%) (Supplementary Table S4). One of the most highly correlated of these genes was GADD45a, which has a role in the apoptotic program, DNA damage, and cell cycle arrest (30). Indeed, we found that following serum starvation, GADD45a expression was attenuated in EYA4kd cells compared to control cells (Figure 5D). These results are intriguing, although not explicitly demonstrative of any mechanistic link between the two. The observed relationship between EYA4 and GADD45a expression warrants further exploration.
Restoration of EYA4 expression inhibits tumor growth in vitro and in vivo
Although restoration of EYA4 expression in two AC cell lines had little effect on DNA damage and apoptosis, we sought to determine whether EYA4 expression had any effect on anchorage-independent growth and tumor growth in vivo. Indeed, our EYA4+ AC cell lines had significantly impaired cell growth and colony formation ability as measured by soft agar colony formation (Figure 6A–B). When H2122 EYA4+ was implanted into NOD-SCID mice, EYA4 overexpression significantly impeded tumor growth (Figure 6C, Supplemental Table S5).
Figure 6. EYA4 suppresses colony growth and is widely underexpressed in epithelial malignancies.

(A) Reconstitution of EYA4 suppresses colony formation. EYA4 was overexpressed in the lung cancer cell H2122 which lacks EYA4, by stable integration of an EYA4 vector (levels indicated by corresponding immunoblot). Colony formation was significantly (p=0.00037, t-test). (B) Reconstitution of EYA4 in the lung cancer cell lines, H2405 also significantly (p=0.0153, t-test) suppressed colony formation, consistent with a TSG role for EYA4. (C) in vivo tumor growth of EYA4+ and control H2122 cells (also see Supplemental Table S5). Overexpression of EYA4 significantly (p<0.05 indicated by *) impairs tumor growth in vivo. (D) EYA4 expression in >350 cancer cells from multiple tissues (also see Supplementary Table S6). Whiskers encompass middle 90% of data points. EYA4 expression appears to be tissue-specific and higher in sarcomas and neurally-derived cancers than epithelial cancers. (E) No observed lung cancer histological subtype specific expression was observed.
EYA4 exhibits cell-type specific expression
Intriguingly, in addition to its putative TSG functions in various cancers, EYA4 has also been described as an oncogene in neural cancer (31). We evaluated cancer cell type specific expression of EYA4 in a panel of over 350 cancer cell lines encompassing a variety of cancer types (Supplementary Table S6). We detected high EYA4 expression in cancer cells derived from sarcomas, autonomic ganglia, and brain, relative to epithelial cancers such as lung, gastrointestinal, pancreatic, head and neck, and colorectal tumors (Figure 6D). These findings are consistent with a TSG role for EYA4 in lung cancer, and interestingly support a dual role (TSG or oncogenic) for EYA4 that is likely dependent on the cell type of origin. Of note, EYA4 was not differentially expressed between small cell lung cancer, which are thought to derive from pulmonary neuroendocrine cells, and NSCLC (Figure 6E).
Clinical relevance of EYA4
We performed a Mantel-Cox survival analysis in two external lung AC datasets with survival data (GSE3141) (16) and (GSE12428) (32), and found that reduced EYA4 expression was significantly and consistently associated with poor survival (Figure 7A–B), underscoring the clinical relevance of EYA4 expression to AC patient prognosis. Although EYA4 SNPs have not been previously associated with familial lung cancer risk, given the frequency of EYA4 inactivation in sporadic lung cancer, and the proximity of EYA4 to the previously refined lung cancer susceptibility locus by Bailey-Wilson and You et al. at 6q23–25, we sought to evaluate the significance of EYA4 SNPs in familial lung cancer (33, 34). A Cochrane-Armitage Trend test corrected for multiple comparisons (p<0.05), revealed a cluster of SNPs in the EYA4 gene (rs7743259, rs159420, rs35689029, rs1878551, and rs2677826) were indeed enriched in a panel of familial NSCLC cases (Figure 7C–D, Supplementary Table S7) (35).
Figure 7. EYA4 is associated with lung cancer risk and poor survival.

(A) Kaplan-Meier plot. Survival information from an external dataset (GSE3141) for the highest and lowest tertiles (of EYA4 expression) were compared using the Mantel-Cox log test. EYA4 is significantly associated with poorer prognosis (p=0.007). (B) The analysis was repeated using another external dataset (GSE12428). Low EYA4 expression is significantly associated with poor prognosis (p=0.018). (C) EYA4 SNPs are significantly associated with familial lung cancer risk. Genotype data for 6q-linked familial lung cancers (n=194) and unrelated non-cancer controls (n=217) were compared to determine whether EYA4 allelotypes associate with risk. Five SNPs depicted were significantly associated (p<0.05). Gene structure is shown in blue. (D) The allele associated with an increased risk is described as the risk allele. Demographic features, stage and histology are unavailable for the external datasets for panels A and B.
Discussion
Cancer genomes are frequently disrupted at multiple ‘omic levels - all of which may be differentially impacted by unique selective pressures occurring throughout the tumorigenic process. Within these tumor systems it is likely that genes critical to abrogating tumor development undergo biallelic (‘two-hit’) disruption, as commonly observed with many TSGs. If two-hit events occur by differing mechanisms, the frequency of alteration for that gene may be low when assessed for only one mechanism, and thus likely overlooked. However, when multiple dimensions of disruption are considered simultaneously, alteration of the two-hit gene in question may be detected at a high frequency. Therefore identification of these events in the complicated genomes of epithelial malignancies, such as lung cancer, requires the simultaneous interrogation of multiple ‘omics level datasets.
We applied such a multidimensional approach to a panel of patient-matched, paired tumor and non-malignant parenchymal samples from patients with NSCLC, and identified Eyes Absent 4 (EYA4); a gene frequently and often simultaneously disrupted by copy number loss and promoter hypermethylation in multiple clinical cohorts, representing over one thousand NSCLC tumors. Disruption of EYA4 was also detected at the level of copy number loss in SqCC precursor CIS specimens, and by promoter hypermethylation in cytologically normal small airway epithelia from patients with NSCLC. In addition to invasive lung adenocarcinoma, promoter hypermethylation of EYA4 was also recently reported in atypical adenomatous hyperplasia (AAH) and adenocarcinoma in situ (AIS) (formerly known as bronchioloalveolar carcinoma), collectively pointing to the importance of EYA4 in cancer initiation events of both major NSCLC subtypes (5, 36). Interestingly, early inactivation of EYA4 by hypermethylation has been associated with Barrett’s esophagus related tumorigenesis, sporadic and colitic neoplasia in chronic ulcerative colitis and is currently being explored as an epigenetic biomarker for colorectal and pancreatic cancer screening (6, 9, 37). Given our findings, exploration of EYA4 as a maker for early NSCLC detection is warranted.
Consistent with TSG function, reconstitution of EYA4 in NSCLC cell lines decreased soft agar colony formation and in vivo tumor growth. And while we found lower EYA4 expression associated with increased sensitivity to DNA-damaging cisplatin treatment in NSCLC cell lines, and increased proportions of genome altered in lung tumors; functional impact on both DNA repair and apoptosis was only significant when EYA4 was abrogated in karyotypically normal cell models as opposed to restored in NSCLC models. Given the multiplicity of genomic aberrations in lung cancer cell lines models, particularly affecting DNA repair and apoptotic proteins (e.g., p53, ATM, PTEN) we were not surprised that modulation of one gene failed to cause significant phenotypic changes. However, in the context of an early tumor suppressor role for EYA4, it is possible that inactivation of EYA4 could lead to increased lung cancer risk by promoting an impaired response to DNA damaging agents such as cigarette smoke.
While multiple lines of evidence in several cancers are supportive of a TSG role, EYA4 is also an over-expressed putative oncogene in tumors of neural origin (31). This discrepancy may be inherent to the unique and recently discovered dual phosphatase properties of EYA4, whereby a Tyr-phosphatase domain at the C- terminal and a Thr-phosphatase domain at the N-terminal function independently and under distinct catalytic conditions (21, 22). Mutations of the Thr-phosphatase domain, but not the Tyr-phosphatase domain, have been shown to abolish the ability of EYA4 to enhance the innate immune response to viruses and undigested dsDNA from apoptotic cells (22), and mutations to the Tyr phosphatase domain, which normally promotes DNA repair in response to DSB, are linked with neuronal developmental defects and deafness. Our findings in over 350 cancer cell lines from multiple tumor types demonstrate that EYA4 is expressed at substantially lower levels in epithelial tumors compared to sarcomas or neurally derived tumors, supporting the seemingly contradictory findings and pointing towards an either tissue-specific or dual TSG or oncogene role for EYA4 in cancer development. In the context of lung cancer, inherited or acquired EYA4 disruption could potentially result in uncontrolled cellular division, accumulation of genetic damage from genotoxic agents such as cigarette smoke, or impaired innate immunity from dsDNA released from damaged respiratory cells.
The Genetic Epidemiology of Lung Cancer Consortium (GELCC) has identified a susceptibility locus on 6q which contains the overexpressed candidate oncogene, RGS17 (33–35, 38–41). We discovered a cluster of SNPs within EYA4, immediately adjacent to this susceptibility locus, that are significantly associated with lung cancer risk in familial lung cancer cases. Patterns of EYA4 disruption in lung cancer are similar to other important TSGs involving both sporadic and familial cancers which also frequently sustain biallelic inactivation (42). For example, BRCA1/2 germline mutations and somatic promoter hypermethylation events occur in familial breast tumors, while biallelic inactivation by hypermethylation and deletion is common in sporadic breast cancer (42–44). Taken together with evidence strongly indicating selective inactivation of EYA4 in sporadic lung cancer, our findings raise the intriguing possibility of a novel familial lung cancer susceptibility gene.
In summary, the prevalence of bi-allelic inactivation of EYA4 in NSCLC, its multiple tumor suppressor functions, and association with survival in sporadic lung cancers and familial lung cancer risk suggest that EYA4 is important to NSCLC development, and may be a promising marker for early lung cancer detection. Considering its diverse biological functions and likely multi-faceted role in lung carcinogenesis, the development of appropriate in vivo models to assess the role and potential manipulation of EYA4 pathways in lung cancer are needed, albeit highly challenging.
Materials and Methods
Sample collection
Lung tumors and adjacent non-malignant tissue were obtained from freshly resected tumors and microdissected so that the tumor cell content exceeded 80%, and DNA and RNA were extracted using standard protocols. Bronchial epithelial specimens from airways ≤2 mm diameter, and biopsy of locally invasive SqCC and CIS specimens, were obtained during bronchoscopy as previously described (19). This study was approved by the Review of Ethics Board of the University of British Columbia and the British Columbia Cancer Agency.
Molecular profiling
Genomic DNA for 83 lung adenocarcinomas and matched non-malignant lung tissues from the same individuals was hybridized to Affymetrix SNP 6.0 arrays. Raw data was processed, segmented and copy number alterations called using Partek Genomic Suite Software, as previously defined (12). SNP array data for the BCCA tumors is in compliance with the MIAME guidelines and has been deposited in the Gene Expression Omnibus. DNA methylation profiling for AC tumor pairs (n=77), and cell lines (n=38) was performed using the Illumina Infinium HumanMethylation27 chip, and SqCC tumor pairs (n=8) on Illumina GoldenGate Cancer chip (Illumina, San Diego), using methods previously described, including bisulfite conversion using EZ DNA methylation-Gold kit (Zymo Research, Irvine, CA) (45). Array data were analyzed and methylation levels determined using GenomeStudio software. β = methylated signal/(methylated signal + unmethylated signal + α). β with a detection p value < 0.05 were included. Hypermethylation was defined as >20% β value difference in tumor relative to patient-matched non-malignant control. Methylation validation was performed by real time MSP as described previously using the EYA4 primers, 5′-TTGCGTAAGTGCGAGGTTGTC-3′ (forward), 5′-AACAACGACAACTTCACGTAA-3′ (reverse), and 5′-FAM TCGTTTTCGGTTTTCGCGTAA BHQ1-3′ (probe), using non-methylated MYOD1 as an internal reference standard. Standard MS-PCR was performed using primers specific to methylated and unmethylated forms of EYA4 as described previously (28). Re-expression of methylated EYA4 following de-methylation by 10 μM 5-azacytidine every two days (5 -aza, Sigma Aldrich, St. Louis) for 6 days was validated in NCI-H1395 and NCI-HCC2935 cells, otherwise cultured per ATCC directions. Gene expression profiles were generated for AC tumor-normal pairs on the Illumina WG6 microarray (Illumina, San Diego). EYA4 DNA methylation status was also validated in an external dataset downloaded from The Cancer Genome Atlas Data Portal (https://tcga-data.nci.nih.gov/tcga/) for all SqCC specimens that had methylation profiles for tumor and matched non-malignant specimens (n = 27 pairs) (46). 45 SqCC Affymetrix expression profiles (GSE3141) and 67 non-malignant bronchial epithelia samples (sigma.bccrc.ca) were retrieved from heavy current and former smokers. Publicly available Affymetrix CEL files were downloaded from NCBI Gene Expression Omnibus (GEO) (GSE3141, GSE10072, GSE12428) (16, 17, 32, 47) or from The Sanger Cell Line Project at the BROAD Institute. Where appropriate, CEL files were grouped, and RMA analysis (48) was performed using the “affy” package from Bioconductor (49). Gene expression profiles for lung AC cell lines (n=38) were obtained by Human WG-6 gene expression chip. Copy number and gene expression data (EYA4 SAGE tag: TAATTTGTGT) from CIS specimens were obtained from a previous study (GSE7898) (19). EYA4 allelotypes data of 6q-linked familial lung cancers (n=194) and unrelated non-cancer controls (n=217) was obtained from a previous study (50). qPCR validation was performed in triplicate using the primers EYA4- Hs00187965_m1, 18s - Hs99999901_s1, GADD45a - Hs00169255_m1. Protein lysates and Western blots were prepared as described (51) (EYA4, Santa Cruz sc-15106; pTyr-H2AX, Millipore 07-1590; beta-actin, Abcam ab8226). DNA sequencing was performed for EYA4 exons and proximal intronic sequences in 38 lung AC cell lines (listed in Supplemental Table S3) using previously described primers (Supplemental Table S8).
Statistical Analyses
Correlation coefficients for EYA4 DNA methylation and mRNA expression levels for 38 lung AC cell lines were obtained by Spearman tests, for each of the 8 EYA4 DNA methylation probes against the average of the 3 EYA4 expression probes (Supplemental Table S3). For genomic integrity assessment, tumors were segregated into tertiles based on EYA4 expression and the proportion of genome altered was compared in tumors with low versus high EYA4 expression using a U test. This analyses was applied to the 83 AC tumor pairs, as well as publicly available matched copy number and gene expression data from the Memorial Sloan Kettering Cancer Centre (14), for which matched copy number (Agilent 44K arrays) and gene expression data (Affymetrix U133A arrays) for 193 lung adenocarcinoma tumors were available. MSKCC copy number data was segmented using the segmentation algorithm, FACADE (52). For survival analyses, data were downloaded from NCBI GEO (47) (GSE3141, GSE12428). Highest and lowest tertiles of samples, based on EYA4 expression (probe 1561088_at), were analyzed using a Log-rank (Mantel-Cox) test. Data from Liu et al were re-analyzed to determine whether EYA4 allelotypes were associated with risk in familial lung cancers (50). The statistical significance of the association between SNP allele and disease status was assessed primarily with Cochran-Armitage trend test with 1 degree of freedom, implemented in PLINK software. Allelic odds ratios (ORs) associated with each SNP and 95% confidence intervals (CIs) were estimated.
in vitro assays
Lung AC (NCI-H2122, NCI-H2405) and lymphoblastoid (NCI-HCC1954BL) cells were obtained from ATCC (Manassas, VA). Stable knockdowns of EYA4 were performed in triplicate for all cells using lentiviral vectors (Clone Id T TRCN0000051094), and a puromycin resistance selectable marker (Open Biosystems, Huntsville, AL). Overexpression of EYA4 was performed in AC cells by transfection using Invitrogen’s Ultimate ORF with clone ID IOH57275. EYA4 was inserted by shuttle from entry vector pENTR™221 to the lentiviral destination vector pLenti6.3/V5-DEST using LR recombination reaction. Lentiviral stock were produced using Invitrogen’s ViraPower™ HiPerform™ Lentiviral expression system (Life Technologies, Carlsbad), after transfecting with lentiviral particles for 24h and Blasticidin selection for 10–14 days. Soft agar colony formation was performed as described (53). AnnexinV/Propidium iodide staining for EYAkd and controls was performed (in triplicate), following 24hr serum starvation. Cells were washed in PBS, resuspended in AnnexinV-binding buffer, propidium iodide, and FITC conjugated anti-Annexin V antibody (BD Bioscience). Apoptotic cells were counted by flow cytometry in a FACS Canto II (BD Biosciences). γH2AX kinetics in response to radiation induced DNA damage was performed as previously described(54), using 5 Gy radiation, and mouse monoclonal anti-phospho-Ser13 H2AX primary antibody (Abcam #18311, 1:4000 dilution; BD Bioscience Alexa Fluor® 647 conjugated anti-γH2AX antibody, DAPI). To account for differences in radiation-induced changes in cell cycle distribution that affect average γH2AX intensity measurements, γH2AX expression per cell was analyzed separately in G1 phase cells and results were expressed as a ratio of the signal intensity for irradiated versus unirradiated cells, and analyzed using the FACS Canto II flow cytometer. For cisplatin assays, 24 hours after seeding, cells were treated for 72h and then stained with Alamar Blue for absorbance analysis and IC50 calculation (Graph Pad Prism 6 software).
Supplementary Material
Acknowledgments
We thank Miwa Suzuki, Denise McDougal, Chad Malloff and Bradley Coe for assistance. Grant supports were from Canadian Institutes of Health Research [CIHR MOP86731, MOP77903, MOP94867], Canadian Cancer Society [CCS017076, CCS20485]; Terry Fox Foundation [20395]; NCI Early Detection Research Network [5U01 CA84971-10], Canary Foundation, NIH Genetic Epidemiology of Lung Cancer Consortium [U01CA76293]; and scholarships from CIHR (IMW, EAV, KSSE, WWL) and Vanier Canada (RC, KLT, NR).
Footnotes
Conflict of interest
The authors of this manuscript have no conflicts of interest to declare.
Supplementary Information accompanies this manuscript on the Oncogene website (http://www.nature.com/onc)
References
- 1.Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380(9859):2095–128. doi: 10.1016/S0140-6736(12)61728-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA: a cancer journal for clinicians. 2011;61(2):69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 3.Knudson AG., Jr Mutation and cancer: statistical study of retinoblastoma. Proceedings of the National Academy of Sciences of the United States of America. 1971;68(4):820–3. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nagayama K, Kohno T, Sato M, Arai Y, Minna JD, Yokota J. Homozygous deletion scanning of the lung cancer genome at a 100-kb resolution. Genes, chromosomes & cancer. 2007;46(11):1000–10. doi: 10.1002/gcc.20485. [DOI] [PubMed] [Google Scholar]
- 5.Selamat SA, Galler JS, Joshi AD, Fyfe MN, Campan M, Siegmund KD, et al. DNA methylation changes in atypical adenomatous hyperplasia, adenocarcinoma in situ, and lung adenocarcinoma. PloS one. 2011;6(6):e21443. doi: 10.1371/journal.pone.0021443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Osborn NK, Zou H, Molina JR, Lesche R, Lewin J, Lofton-Day C, et al. Aberrant methylation of the eyes absent 4 gene in ulcerative colitis-associated dysplasia. Clinical gastroenterology and hepatology : the official clinical practice journal of the American Gastroenterological Association. 2006;4(2):212–8. doi: 10.1016/j.cgh.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 7.Oster B, Thorsen K, Lamy P, Wojdacz TK, Hansen LL, Birkenkamp-Demtroder K, et al. Identification and validation of highly frequent CpG island hypermethylation in colorectal adenomas and carcinomas. International journal of cancer Journal international du cancer. 2011;129(12):2855–66. doi: 10.1002/ijc.25951. [DOI] [PubMed] [Google Scholar]
- 8.Kim YH, Lee HC, Kim SY, Yeom YI, Ryu KJ, Min BH, et al. Epigenomic analysis of aberrantly methylated genes in colorectal cancer identifies genes commonly affected by epigenetic alterations. Annals of surgical oncology. 2011;18(8):2338–47. doi: 10.1245/s10434-011-1573-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kisiel JB, Yab TC, Taylor WR, Chari ST, Petersen GM, Mahoney DW, et al. Stool DNA testing for the detection of pancreatic cancer: assessment of methylation marker candidates. Cancer. 2012;118(10):2623–31. doi: 10.1002/cncr.26558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thu KL, Radulovich N, Becker-Santos DD, Pikor LA, Pusic A, Lockwood WW, et al. SOX15 is a candidate tumor suppressor in pancreatic cancer with a potential role in Wnt/beta-catenin signaling. Oncogene. 2013 doi: 10.1038/onc.2012.595. [DOI] [PubMed] [Google Scholar]
- 11.Hammerman PS, Hayes DN, Wilkerson MD, Schultz N, Bose R, Chu A, et al. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489(7417):519–25. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thu KL, Vucic EA, Chari R, Zhang W, Lockwood WW, English JC, et al. Lung adenocarcinoma of never smokers and smokers harbor differential regions of genetic alteration and exhibit different levels of genomic instability. PloS one. 2012;7(3):e33003. doi: 10.1371/journal.pone.0033003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weir BA, Woo MS, Getz G, Perner S, Ding L, Beroukhim R, et al. Characterizing the cancer genome in lung adenocarcinoma. Nature. 2007;450(7171):893–8. doi: 10.1038/nature06358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chitale D, Gong Y, Taylor BS, Broderick S, Brennan C, Somwar R, et al. An integrated genomic analysis of lung cancer reveals loss of DUSP4 in EGFR-mutant tumors. Oncogene. 2009;28(31):2773–83. doi: 10.1038/onc.2009.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weiss J, Sos ML, Seidel D, Peifer M, Zander T, Heuckmann JM, et al. Frequent and focal FGFR1 amplification associates with therapeutically tractable FGFR1 dependency in squamous cell lung cancer. Science translational medicine. 2010;2(62):62ra93. doi: 10.1126/scitranslmed.3001451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bild AH, Yao G, Chang JT, Wang Q, Potti A, Chasse D, et al. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature. 2006;439(7074):353–7. doi: 10.1038/nature04296. [DOI] [PubMed] [Google Scholar]
- 17.Landi MT, Dracheva T, Rotunno M, Figueroa JD, Liu H, Dasgupta A, et al. Gene expression signature of cigarette smoking and its role in lung adenocarcinoma development and survival. PloS one. 2008;3(2):e1651. doi: 10.1371/journal.pone.0001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thusberg J, Olatubosun A, Vihinen M. Performance of mutation pathogenicity prediction methods on missense variants. Human mutation. 2011;32(4):358–68. doi: 10.1002/humu.21445. [DOI] [PubMed] [Google Scholar]
- 19.Lonergan KM, Chari R, Coe BP, Wilson IM, Tsao MS, Ng RT, et al. Transcriptome profiles of carcinoma-in-situ and invasive non-small cell lung cancer as revealed by SAGE. PloS one. 2010;5(2):e9162. doi: 10.1371/journal.pone.0009162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005;436(7051):725–30. doi: 10.1038/nature03918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sadatomi D, Tanimura S, Ozaki K, Takeda K. Atypical protein phosphatases: emerging players in cellular signaling. International journal of molecular sciences. 2013;14(3):4596–612. doi: 10.3390/ijms14034596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Okabe Y, Sano T, Nagata S. Regulation of the innate immune response by threonine-phosphatase of Eyes absent. Nature. 2009;460(7254):520–4. doi: 10.1038/nature08138. [DOI] [PubMed] [Google Scholar]
- 23.MacPhail SH, Banath JP, Yu TY, Chu EH, Lambur H, Olive PL. Expression of phosphorylated histone H2AX in cultured cell lines following exposure to X-rays. International journal of radiation biology. 2003;79(5):351–8. doi: 10.1080/0955300032000093128. [DOI] [PubMed] [Google Scholar]
- 24.Pignoni F, Hu B, Zavitz KH, Xiao J, Garrity PA, Zipursky SL. The eye-specification proteins So and Eya form a complex and regulate multiple steps in Drosophila eye development. Cell. 1997;91(7):881–91. doi: 10.1016/s0092-8674(00)80480-8. [DOI] [PubMed] [Google Scholar]
- 25.Clark SW, Fee BE, Cleveland JL. Misexpression of the eyes absent family triggers the apoptotic program. The Journal of biological chemistry. 2002;277(5):3560–7. doi: 10.1074/jbc.M108410200. [DOI] [PubMed] [Google Scholar]
- 26.Liu X, Sano T, Guan Y, Nagata S, Hoffmann JA, Fukuyama H. Drosophila EYA regulates the immune response against DNA through an evolutionarily conserved threonine phosphatase motif. PloS one. 2012;7(8):e42725. doi: 10.1371/journal.pone.0042725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.De Carvalho DD, Sharma S, You JS, Su SF, Taberlay PC, Kelly TK, et al. DNA methylation screening identifies driver epigenetic events of cancer cell survival. Cancer cell. 2012;21(5):655–67. doi: 10.1016/j.ccr.2012.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zou H, Osborn NK, Harrington JJ, Klatt KK, Molina JR, Burgart LJ, et al. Frequent methylation of eyes absent 4 gene in Barrett’s esophagus and esophageal adenocarcinoma. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2005;14(4):830–4. doi: 10.1158/1055-9965.EPI-04-0506. [DOI] [PubMed] [Google Scholar]
- 29.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(9):5116–21. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang X, Sun H, Danila DC, Johnson SR, Zhou Y, Swearingen B, et al. Loss of expression of GADD45 gamma, a growth inhibitory gene, in human pituitary adenomas: implications for tumorigenesis. The Journal of clinical endocrinology and metabolism. 2002;87(3):1262–7. doi: 10.1210/jcem.87.3.8315. [DOI] [PubMed] [Google Scholar]
- 31.Miller SJ, Lan ZD, Hardiman A, Wu J, Kordich JJ, Patmore DM, et al. Inhibition of Eyes Absent Homolog 4 expression induces malignant peripheral nerve sheath tumor necrosis. Oncogene. 2010;29(3):368–79. doi: 10.1038/onc.2009.360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boelens MC, van den Berg A, Fehrmann RS, Geerlings M, de Jong WK, te Meerman GJ, et al. Current smoking-specific gene expression signature in normal bronchial epithelium is enhanced in squamous cell lung cancer. The Journal of pathology. 2009;218(2):182–91. doi: 10.1002/path.2520. [DOI] [PubMed] [Google Scholar]
- 33.You M, Wang D, Liu P, Vikis H, James M, Lu Y, et al. Fine mapping of chromosome 6q23–25 region in familial lung cancer families reveals RGS17 as a likely candidate gene. Clinical cancer research : an official journal of the American Association for Cancer Research. 2009;15(8):2666–74. doi: 10.1158/1078-0432.CCR-08-2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bailey-Wilson JE, Amos CI, Pinney SM, Petersen GM, de Andrade M, Wiest JS, et al. A major lung cancer susceptibility locus maps to chromosome 6q23–25. American journal of human genetics. 2004;75(3):460–74. doi: 10.1086/423857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Amos CI, Pinney SM, Li Y, Kupert E, Lee J, de Andrade MA, et al. A susceptibility locus on chromosome 6q greatly increases lung cancer risk among light and never smokers. Cancer research. 2010;70(6):2359–67. doi: 10.1158/0008-5472.CAN-09-3096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Selamat SA, Chung BS, Girard L, Zhang W, Zhang Y, Campan M, et al. Genome-scale analysis of DNA methylation in lung adenocarcinoma and integration with mRNA expression. Genome research. 2012;22(7):1197–211. doi: 10.1101/gr.132662.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kisiel JB, Yab TC, Nazer Hussain FT, Taylor WR, Garrity-Park MM, Sandborn WJ, et al. Stool DNA testing for the detection of colorectal neoplasia in patients with inflammatory bowel disease. Alimentary pharmacology & therapeutics. 2013;37(5):546–54. doi: 10.1111/apt.12218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sellers TA, Bailey-Wilson JE, Elston RC, Wilson AF, Elston GZ, Ooi WL, et al. Evidence for mendelian inheritance in the pathogenesis of lung cancer. Journal of the National Cancer Institute. 1990;82(15):1272–9. doi: 10.1093/jnci/82.15.1272. [DOI] [PubMed] [Google Scholar]
- 39.Sellers TA, Potter JD, Bailey-Wilson JE, Rich SS, Rothschild H, Elston RC. Lung cancer detection and prevention: evidence for an interaction between smoking and genetic predisposition. Cancer research. 1992;52(9 Suppl):2694s–7s. [PubMed] [Google Scholar]
- 40.Tokuhata GK, Lilienfeld AM. Familial aggregation of lung cancer in humans. Journal of the National Cancer Institute. 1963;30:289–312. [PubMed] [Google Scholar]
- 41.Tessema M, Willink R, Do K, Yu YY, Yu W, Machida EO, et al. Promoter methylation of genes in and around the candidate lung cancer susceptibility locus 6q23–25. Cancer research. 2008;68(6):1707–14. doi: 10.1158/0008-5472.CAN-07-6325. [DOI] [PubMed] [Google Scholar]
- 42.Bell DW, Erban J, Sgroi DC, Haber DA. Selective loss of heterozygosity in multiple breast cancers from a carrier of mutations in both BRCA1 and BRCA2. Cancer research. 2002;62(10):2741–3. [PubMed] [Google Scholar]
- 43.Bijron JG, van der Groep P, van Dorst EB, Seeber LM, Sie-Go DM, Verheijen RH, et al. Promoter hypermethylation patterns in Fallopian tube epithelium of BRCA1 and BRCA2 germline mutation carriers. Endocrine-related cancer. 2011 doi: 10.1530/ERC-11-0338. [DOI] [PubMed] [Google Scholar]
- 44.Birgisdottir V, Stefansson OA, Bodvarsdottir SK, Hilmarsdottir H, Jonasson JG, Eyfjord JE. Epigenetic silencing and deletion of the BRCA1 gene in sporadic breast cancer. Breast cancer research : BCR. 2006;8(4):R38. doi: 10.1186/bcr1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bibikova M, Le J, Barnes B, Saedinia-Melnyk S, Zhou L, Shen R, et al. Genome-wide DNA methylation profiling using Infinium(R) assay. Epigenomics. 2009;1(1):177–200. doi: 10.2217/epi.09.14. [DOI] [PubMed] [Google Scholar]
- 46.Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489(7417):519–25. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barrett T, Troup DB, Wilhite SE, Ledoux P, Rudnev D, Evangelista C, et al. NCBI GEO: archive for high-throughput functional genomic data. Nucleic acids research. 2009;37:D885–90. doi: 10.1093/nar/gkn764. Database issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4(2):249–64. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 49.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome biology. 2004;5(10):R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu P, Vikis HG, Wang D, Lu Y, Wang Y, Schwartz AG, et al. Familial aggregation of common sequence variants on 15q24–25.1 in lung cancer. Journal of the National Cancer Institute. 2008;100(18):1326–30. doi: 10.1093/jnci/djn268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thu KL, Pikor LA, Chari R, Wilson IM, Macaulay CE, English JC, et al. Genetic disruption of KEAP1/CUL3 E3 ubiquitin ligase complex components is a key mechanism of NF-kappaB pathway activation in lung cancer. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer. 2011;6(9):1521–9. doi: 10.1097/JTO.0b013e3182289479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Coe BP, Chari R, MacAulay C, Lam WL. FACADE: a fast and sensitive algorithm for the segmentation and calling of high resolution array CGH data. Nucleic acids research. 2010;38(15):e157. doi: 10.1093/nar/gkq548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lockwood WW, Wilson IM, Coe BP, Chari R, Pikor LA, Thu KL, et al. Divergent genomic and epigenomic landscapes of lung cancer subtypes underscore the selection of different oncogenic pathways during tumor development. PloS one. 2012;7(5):e37775. doi: 10.1371/journal.pone.0037775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Olive PL, Banath JP. Phosphorylation of histone H2AX as a measure of radiosensitivity. International journal of radiation oncology, biology, physics. 2004;58(2):331–5. doi: 10.1016/j.ijrobp.2003.09.028. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.