

Abstract
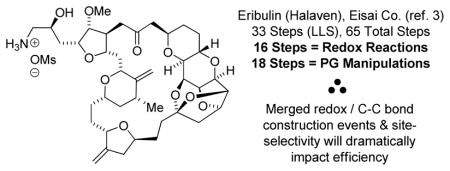
(+)-Zincophorin methyl ester is prepared in 13 steps (longest linear sequence). A bidirectional redox-triggered double anti-crotylation of 2-methyl-1,3-propane diol directly assembles the triketide stereopolyad spanning C4-C12, significantly enhancing step-economy and enabling construction of (+)-zincophorin methyl ester in nearly half the steps previously required.
Polyketides derived from soil bacteria are estimated to account for roughly 20% of the top-selling small molecule drugs,1 yet less than 5% of soil bacteria are amenable to culture.2 As methods for bacterial culture improve, the use of polyketides in human medicine will surely increase, as will the need for concise manufacturing routes to these stereochemically complex structures and their functional analogues. Presently, all commercial polyketides, with the single exception of Eribulin,3 are prepared by fermentation or semi-synthesis. Although de novo chemical synthesis can deliver otherwise inaccessible structural variants, routes that are reliant upon current technologies for acyclic stereocontrol via stepwise bond construction are especially lengthy, diminishing prospects for commercial application.
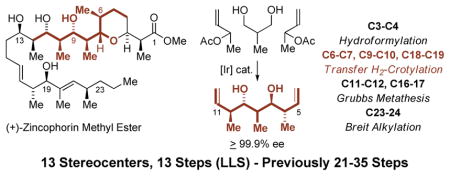
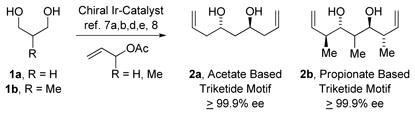
We have developed a suite of catalytic methods for polyketide construction wherein lower alcohols are directly transformed to higher alcohols in a stereo- and site-selective fashion.4 In such processes, hydrogen transfer from alcohols to π-unsaturated reactants triggers pairwise generation of carbonyl-organometal species en route to products of addition. These merged redox-construction events5 bypass discrete alcohol-to-aldehyde redox reactions and, because they may be deployed in a site-selective manner,6 streamline or eliminate the use of protecting groups. Most importantly, such redox-triggered carbonyl additions enable transformations and strategies beyond those accessible via conventional carbanion chemistry. Indeed, as borne out in total syntheses of roxaticin,7a bryostatin 7,7b trienomycins A and F,7c cyanolide A,7d and 6-deoxyerythronolide B,7e application of these methods have availed a “step-function increase” in efficiency – in each case, the synthetic route was significantly more concise than in any prior approach.4b These studies brought to light an especially powerful protocol for the direct assembly of acetate- or propionate-based triketide stereopolyads 2a or 2b involving the bidirectional enantioselective double allylation8a or anti-crotylation8b of 1,3-diols 1a or 1b, respectively (eq. 1).8
 |
(eq. 1) |
The iconic polyketide ionophore antibiotic (+)-zincophorin (Figure 1),9 which possesses potent (≤ 1 ppm) in vivo activity against gram positive bacteria,9c,10 including Clostridium coelchii, presented an opportunity to further assess the impact of direct triketide stereopolyad generation across diverse polyketide families. (+)-Zincophorin and its methyl ester have been the subject of five total syntheses.11,12,13 The shortest route previously reported is 21 steps (LLS).11g Here, we report a “more ideal” total synthesis of (+)-zincophorin methyl ester in 13 steps (LLS) based on direct triketide stereopolyad generation via two-directional double anti-crotylation of 2-methyl-1,3-propane diol 1b.
Figure 1.

(+)-Zincophorin and (+)-Zincophorin Methyl Ester and Summary of Prior Total Syntheses.a
aFor graphical summaries of prior total syntheses, see Supporting Information. Longest Linear Sequence (LLS); Total Steps (TS).
Retrosynthetically, (+)-zincophorin methyl ester was envisioned to arise via convergent assembly of Fragments A and B via stereoselective carbonyl addition in accordance with the merged Felkin-Anh and Evans models,14 followed by oxocarbenium ion addition to install the terminal monoketide Scheme 1. Retrosynthesis of (+)-Zincophorin Methyl Ester. moiety using a chiral propionate enolate.11g,16 Fragment A is prepared in 8 steps from (+)-tert-butyl D-lactate 3. Key C-C bond formations include Breit’s method for the stereospecific substitution of α-hydroxy ester triflates with Grignard reagents to create the C22 stereocenter,15 stereoselective Wittig olefination,17 which defines the geometry of the trisubstituted olefin, and direct redox-triggered anti-crotylation of allylic alcohol 5 to form the C18-C19 stereodiad.18 The synthesis of Fragment B takes advantage of the two-directional double anti-crotylation of 2-methyl-1,3-propane diol 1b to form adduct 2b,7e,8b which directly establishes the triketide stereopolyad spanning C6-C10. Cross-metathesis is used to introduce the C12-C13 carbon atoms of allyl alcohol 8, and hydroformylation is used to forge the C3-C4 bond of Fragment B.
Scheme 1.

Retrosynthesis of (+)-Zincophorin Methyl Ester.
The synthesis of Fragment A (Scheme 2) begins with the conversion of (+)-tert-butyl D-lactate 3 to the corresponding triflate, which upon exposure to n-propylmagnesium chloride in the presence of substoichiometric quantities of zinc chloride delivers the product of substitution with inversion of stereochemistry.15 Reduction of the ester mediated by lithium aluminum hydride delivers alcohol 4.19 Swern oxidation of 4 followed by Wittig olefination of the chiral α-stereogenic aldehyde provides an α,β-unsaturated ester, which is subjected to DIBAL reduction to form the previously reported allylic alcohol 5.17 Direct redox-triggered anti-crotylation18 of allylic alcohol 5 forms the C18-C19 bond, delivering the homoallylic alcohol 6 with good levels of catalyst-directed diastereoselectivity (5.5:1 dr). Cross-metathesis of compound 6 with 4-iodo-1-butene occurred uneventfully.20 Minor diastereomers generated in the formation of compound 6 are easily separated at this stage. Conversion of the C19 hydroxyl to the TES-ether completes the synthesis of Fragment A in 8 steps from (+)-tert-butyl D-lactate.
Scheme 2.

Synthesis of Fragment A via Direct anti-Crotylation of Allylic Alcohol 5.a
aYields are of material isolated by silica gel chromatography. Enantiomeric excess was determined by chiral GLC. See Supporting Information for further experimental details.
To construct Fragment B (Scheme 3), diol 1b is subjected to two-directional double anti-crotylation followed by iodoetherification to deliver 7.7e,8b Iodoetherification defines the chirotopic nonstereogenic center of diol 2b at C8, and serves to differentiate the terminal olefin moieties. The pseudo-C -symmetric diol 2b is produced as a single enantiomer due to Horeau’s principle,21 that is, the minor enantiomer of the intervening mono-adduct is converted to the pseudo-meso-diastereomer.22 In the conversion of diol 1b to adduct 2b, it was found that use of α-methyl allyl acetate prepared via acetylation of the corresponding alcohol using triethylamine rather than pyridine as base gave the best results. Whereas attempted cross-metatheses of 7 with allyl acetate or cis-butene diol di-acetate suffered from competing olefin isomerization, the Stewart-Grubbs catalyst enabled conversion of 7 to the allylic acetate in 81% yield.23 Bernet-Vasella cleavage24 of the iodoether in methanol solvent occurs with concomitant loss of the acetate. Subsequent formation of the acetonide delivers the allylic alcohol 8, which is converted to a single diastereomeric epoxide using the Sharpless protocol.25 Reaction of the epoxide with Gilman’s reagent delivers compound 9,26 which incorporates the stereoheptad spanning C6-C12. Hydroformylation of 9 using a XantPhos modified rhodium catalyst27 provides the linear aldehyde, which upon exposure to methanol in the presence of substoichiometric p-toluenesulfonic acid delivers the pyran 10 as a mixture of diastereomers at the anomeric position. The major diastereomeric pyran 10 could be separated by flash chromatography and was converted to the tris(triethylsilyl) ether. Exposure to Swern oxidation conditions results in cleavage of the primary TES-ether and formation of the aldehyde Fragment B.28
Scheme 3.

Synthesis of Fragment B via Two-Directional Double anti-Crotylation of 2-Methyl-1,3-Propane Diol 1b.a
aYields are of material isolated by silica gel chromatography. See Supporting Information for further experimental details.
The union of Fragments A and B is achieved through lithiation of the primary alkyl iodide Fragment A and subsequent addition to the aldehyde Fragment B.29 Synergistic 1,2- and 1,3-stereoinduction effects14 were anticipated to enforce highly diastereoselective addition. However, under standard reaction conditions using HMPA as additive, the adduct 11 formed as a 1:1 ratio of C13 diastereomers. HMPA was necessary to facilitate Li-halogen exchange of Fragment A, as its omission resulted in tert-butylation of Fragment B. This was not the case using 4-iodo-1-butene, which underwent addition to Fragment B in ether in 70% yield to furnish 2:1 ratio of C13 diastereomers. The diastereomeric ratio did not change upon introduction of HMPA, suggesting chelation control was not operative, however, an improved 99% yield was observed. The Cram-Reetz and Evans polar models14 assume the C11-OSiEt3 moiety should predominantly populate a conformation wherein the C11-OSiEt3 bond dipole cancels the C13 formyl bond dipole. It appears the negative inductive effect of the highly oxygenated C10-C3 moiety erodes this conformational bias, leading to diminished diastereoselectivities (Figure 2). Hence, modification of the organolithium reagent by chiral 1,2-diamines was investigated as a means of amplifying stereoselectivity.30 Using tetramethylcyclohexane diamine, a 3:1 molar ratio of diastereomers was obtained, which could be separated by flash chromatography. With compound 11 in hand, installation of the terminal C3 monoketide moiety was achieved using a chiral propionate enolate,11g,16 providing the trans-pyran as a single diastereomer. Subsequent methanolysis of the thiazole thione from the crude reaction mixture delivered (+)-zincophorin methyl ester, which was identical in all respects to literature. In this way, (+)-zincophorin methyl ester, which incorporates 13 stereocenters, was prepared 13 steps (LLS) with 4 C – C bonds formed using hydrogenative coupling protocols.
Figure 2.
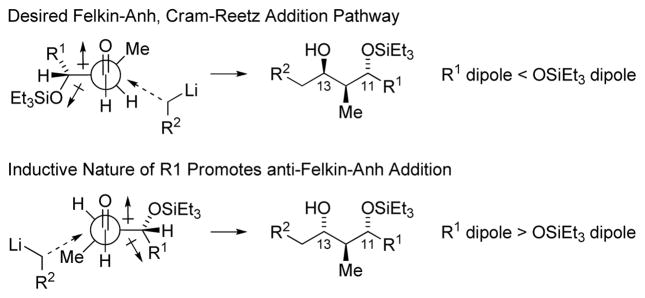
Merged 1,2- and 1,3-Stereoinduction Model.a
aR1 = C10-C3 of Fragment B, R2 = C15-C25 of Fragment A.
Despite enormous progress in synthetic methods development, the vast majority of de novo chemical syntheses remain distant from the Hendricksonian ideal.31 This is principally due to (a) the separation of redox and skeletal construction events, and (b) the persistent requirement of protecting groups. Both deficiencies may be addressed through the design of catalytic methods that merge redox and C-C bond formation events,5 especially transformations that may be deployed in a site-selective manner, and the new strategies that such methods evoke. In the present study, hydrogenative couplings that exploit alcohol-to-carbonyl oxidation as a driver for carbanion generation,4 are used to directly generate triketide stereopoly-ads that would otherwise require lengthy multi-step syntheses. As demonstrated here and in prior work,7 these methods have availed a “step-function” change in efficiency across diverse contexts, bringing us one step closer to the Hendricksonian ideal.31 More immediately, the concise nature of the present route to (+)-zincophorin methyl ester will enable access to material that will allow for a more complete investigation into its biological properties; studies which are currently underway.
Supplementary Material
Scheme 4.
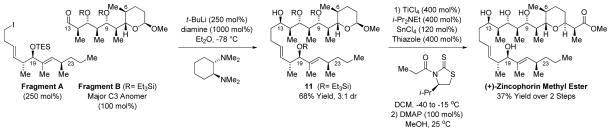
Union of Fragment A and Fragment B and Total Synthesis of (+)-Zincophorin Methyl Ester.a
aYields are of material isolated by silica gel chromatography. See Supporting Information for further experimental details.
Acknowledgments
The Robert A. Welch Foundation (F-0038), the NIH-NIGMS (RO1-GM093905) and the University of Texas Center for Green Chemistry and Catalysis are acknowledged for partial support of this research.
Footnotes
Supporting Information Available: Experimental procedures and spectral data. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Reviews: O’Hagan D. The Polyketide Metabolites. Ellis Horwood; Chichester: 1991. Rohr J. Angew Chem Int Ed. 2000;39:2847. doi: 10.1002/1521-3773(20000818)39:16<2847::aid-anie2847>3.0.co;2-0.Rimando AM, Baerson SR. ACS Symp Ser. 2007;955:282.Newman DJ, Cragg GM. J Nat Prod. 2007;70:461. doi: 10.1021/np068054v.Cragg GM, Grothaus PG, Newman DJ. Chem Rev. 2009;109:3012. doi: 10.1021/cr900019j.
- 2.Sait M, Hugenholtz P, Janssen PH. Environ Microbiol. 2002;4:654. doi: 10.1046/j.1462-2920.2002.00352.x. and references cited therein. [DOI] [PubMed] [Google Scholar]
- 3.Eribulin (halaven), a truncated analogue of the marine polyketide halichondrin B and FDA approved treatment for metastatic breast cancer, requires 65 steps for its preparation. Over half these steps are protecting group (PG) manipulations and redox reactions. Eribulin is 200 times more potent than taxol, which compensates for its relatively high manufacturing costs Yu MJ, Zheng W, Seletsky BM. Nat Prod Rep. 2013;30:1158. doi: 10.1039/c3np70051h.
- 4.For selected reviews on enantioselective redox-triggered carbonyl addition from the alcohol oxidation level, see: Ketcham JM, Shin I, Montgomery TP, Krische MJ. Angew Chem Int Ed. 2014;53:9142. doi: 10.1002/anie.201403873.Dechert-Schmitt AMR, Schmitt DC, Gao X, Itoh T, Krische MJ. Nat Prod Rep. 2014;31:504. doi: 10.1039/c3np70076c.
- 5.For a review on redox-economy, see: Burns NZ, Baran PS, Hoffmann RW. Angew Chem Int Ed. 2009;48:2854. doi: 10.1002/anie.200806086.
- 6.For site-selective C-C coupling of unprotected diols and higher polyols, see: Dechert-Schmitt AMR, Schmitt DC, Krische MJ. Angew Chem Int Ed. 2013;52:3195. doi: 10.1002/anie.201209863.Shin I, Wang G, Krische MJ. Chem Eur J. 2014;20:13382. doi: 10.1002/chem.201404065.Wang G, Franke J, Ngo CQ, Krische MJ. J Am Chem Soc. 2015;137:7915. doi: 10.1021/jacs.5b04404.
- 7.(a) Han SB, Hassan A, Kim IS, Krische MJ. J Am Chem Soc. 2010;132:15559. doi: 10.1021/ja1082798. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lu Y, Woo SK, Krische MJ. J Am Chem Soc. 2011;133:13876. doi: 10.1021/ja205673e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Del Valle DJ, Krische MJ. J Am Chem Soc. 2013;135:10986. doi: 10.1021/ja4061273. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Waldeck AR, Krische MJ. Angew Chem Int Ed. 2013;52:4470. doi: 10.1002/anie.201300843. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Gao X, Woo SK, Krische MJ. J Am Chem Soc. 2013;135:4223. doi: 10.1021/ja4008722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Lu Y, Kim IS, Hassan A, Del Valle DJ, Krische MJ. Angew Chem Int Ed. 2009;48:5018. doi: 10.1002/anie.200901648. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gao X, Han H, Krische MJ. J Am Chem Soc. 2011;133:12795. doi: 10.1021/ja204570w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.For the isolation and structure determination of (+)-zincophorin, also known as M144255 or griseocholin, see: Gräfe U, Schade W, Roth M, Radics L, Incze M, Ujszászy K. J Antibiot. 1984;37:836. doi: 10.7164/antibiotics.37.836.Radics L. J Chem Soc, Chem Commun. 1984:599.Brooks HA, Gardner D, Poyser JP, King TJ. J Antibiot. 1984;37:1501. doi: 10.7164/antibiotics.37.1501.Radics L, Kajtár-Peredy M. J Chem Soc, Perkin Trans 2. 1986:1471.Tonew E, Tonew M, Graefe U, Zopel P. Pharmazie. 1988;43:717.Scharfenberg-Pfeiffer D, Czugler M. Pharmazie. 1991;46:781.
- 10.For a review of the antibacterial properties of polyether ionophores, see: Kevin DA, II, Meujo DAF, Hamann MT. Expert Opin Drug Discov. 2009;4:109. doi: 10.1517/17460440802661443.
- 11.For total syntheses of (+)-zincophorin and its methyl ester, see: Danishefsky SJ, Selnick HG, DeNinno MP, Zelle RE. J Am Chem Soc. 1987;109:1572.Danishefsky SJ, Selnick HG, Zelle RE, DeNinno MP. J Am Chem Soc. 1988;110:4368.Defosseux M, Blanchard N, Meyer C, Cossy J. Org Lett. 2003;5:4037. doi: 10.1021/ol035177n.Defosseux M, Blanchard N, Meyer C, Cossy J. J Org Chem. 2004;69:4626. doi: 10.1021/jo0496042.Cossy J, Meyer C, Defosseux M, Blanchard N. Pure Appl Chem. 2005;77:1131.Komatsu K, Tanino K, Miyashita M. Angew Chem Int Ed. 2004;43:4341. doi: 10.1002/anie.200460434.Harrison TJ, Ho S, Leighton JL. J Am Chem Soc. 2011;133:7308. doi: 10.1021/ja201467z.Godin F, Mochirian P, St-Pierre G, Guindon Y. Tetrahedron. 2015;71:709.
- 12.For a review highlighting synthetic approaches to (+)-zincophorin and its methyl ester, see: Song Z, Lohse AG, Hsung RP. Nat Prod Rep. 2009;26:560. doi: 10.1039/b821450f.
- 13.For selected formal syntheses and fragment syntheses, see: Balestra M, Wittman MD, Kallmerten J. Tetrahedron Lett. 1988;29:6905.Cywin CL, Kallmerten J. Tetrahedron Lett. 1993;34:1103.Marshall JA, Palovich MR. J Org Chem. 1998;63:3701.Chemler SR, Roush WR. J Org Chem. 1998;63:3800. doi: 10.1021/jo0267908.Song Z, Hsung RP. Org Lett. 2007;9:2199. doi: 10.1021/ol070791a.Sabitha G, Srinivas R, Yadav JS. Synthesis. 2011:1484.Cooksey JP. Org Biomol Chem. 2013;11:5117. doi: 10.1039/c3ob40894a.Yadav JS, Gyanchander E, Das S. Tetrahedron Lett. 2014;55:3996.
- 14.(a) Leitereg TJ, Cram DJ. J Am Chem Soc. 1968;90:4011. [Google Scholar]; (b) Anh NT, Eisenstein O, Lefour JM, Dau ME. J Am Chem Soc. 1973;95:6146. [Google Scholar]; (c) Anh NT, Eisenstein O. Nouv J Chim. 1977;1:61. [Google Scholar]; (d) Reetz MT, Kesseler K, Jung A. Tetrahedron Lett. 1984;25:729. [Google Scholar]; (e) Evans DA, Duffy JL, Dart MJ. Tetrahedron Lett. 1994;35:8537. [Google Scholar]; (f) Evans DA, Dart MJ, Duffy JL, Yang MG, Livingston AB. J Am Chem Soc. 1995;117:6619. [Google Scholar]; (g) Evans DA, Dart MJ, Duffy JL, Yang MG. J Am Chem Soc. 1996;118:4322. [Google Scholar]
- 15.Studte C, Breit B. Angew Chem Int Ed. 2008;47:5451. doi: 10.1002/anie.200800733. [DOI] [PubMed] [Google Scholar]
- 16.(a) Cosp A, Romea P, Talavera P, Urpí F, Vilarrasa J, Font-Bardia M, Solans X. Org Lett. 2001;3:615. doi: 10.1021/ol0070177. [DOI] [PubMed] [Google Scholar]; (b) Larrosa I, Romea P, Urpí F, Balsells D, Vilarrasa J, Font-Bardia M, Solans X. Org Lett. 2002;4:4651. doi: 10.1021/ol0270226. [DOI] [PubMed] [Google Scholar]; (c) Larrosa I, Romea P, Urpí F. Org Lett. 2006;8:527. doi: 10.1021/ol052900w. [DOI] [PubMed] [Google Scholar]
- 17.The conversion of alcohol 4 to allylic alcohol 5 via Swern oxidation followed by Wittig olefination has been described: Lister T, Perkins MV. Aust J Chem. 2004;57:787.
- 18.For iridium catalyzed carbonyl crotylation from the alcohol oxidation level employing α-methyl allyl acetate as the crotyl donor, see: Kim IS, Han SB, Krische MJ. J Am Chem Soc. 2009;131:2514. doi: 10.1021/ja808857w.Gao X, Townsend IA, Krische MJ. J Org Chem. 2011;76:2350. doi: 10.1021/jo200068q.
- 19.Alcohol 4 is a known compound: Chen H-Y, McDonald FE. J Am Chem Soc. 2006;128:4568. doi: 10.1021/ja061082f. and reference 17.
- 20.(a) Nicolaou KC, Bulger PG, Sarlah D. Angew Chem Int Ed. 2005;44:4490. doi: 10.1002/anie.200500369. [DOI] [PubMed] [Google Scholar]; (b) Fürstner A. Chem Commun. 2011;47:6505. doi: 10.1039/c1cc10464k. [DOI] [PubMed] [Google Scholar]
- 21.Vigneron JP, Dhaenens M, Horeau A. Tetrahedron. 1973;29:1055.For a historical review, see: Heller D, Drexler H-J, Fischer C, Buschmann H, Baumann W, Heller B. Angew Chem Int Ed. 2000;39:495. doi: 10.1002/(sici)1521-3773(20000204)39:3<495::aid-anie495>3.0.co;2-g.
- 22.(a) Kogure T, Eliel EL. J Org Chem. 1984;49:576. [Google Scholar]; (b) Midland MM, Gabriel J. J Org Chem. 1985;50:1143. [Google Scholar]; (c) Poss CS, Schreiber SL. Acc Chem Res. 1994;27:9. [Google Scholar]
- 23.(a) Stewart IC, Ung T, Pletnev AA, Berlin JM, Grubbs RH, Schrodi Y. Org Lett. 2007;9:1589. doi: 10.1021/ol0705144. [DOI] [PubMed] [Google Scholar]; (b) Stewart IC, Douglas CJ, Grubbs RH. Org Lett. 2008;10:441. doi: 10.1021/ol702624n. [DOI] [PubMed] [Google Scholar]
- 24.(a) Bernet B, Vasella A. Helv Chim Acta. 1979;62:1990. [Google Scholar]; (b) Bernet B, Vasella A. Helv Chim Acta. 1979;62:2400. [Google Scholar]; (c) Bernet B, Vasella A. Helv Chim Acta. 1984;67:1328. [Google Scholar]
- 25.Katsuki T, Sharpless KB. J Am Chem Soc. 1980;102:5974. [Google Scholar]
- 26.(a) Gilman H, Jones RG, Woods LA. J Org Chem. 1952;17:1630. [Google Scholar]; (b) Nagaoka H, Kishi Y. Tetrahedron. 1981;37:3873. [Google Scholar]
- 27.For impact of large bite-angle ligands on regioselectivity in hydroformylation, see reference (a). For seminal use of XantPhos in linear regioselective hydroformylation, see reference (b): Casey CP, Whiteker GT, Melville MG, Petrovich LM, Gavney JA, Jr, Powel DR. J Am Chem Soc. 1992;114:5535.Kranenburg M, van der Burgt YEM, Kamer PCJ, van Leeuwen PWNM, Goubitz KG, Fraanje J. Organometallics. 1995;14:3081.
- 28.Rodríguez A, Nomen M, Spur BW, Godfroid JJ. Tetrahedron Lett. 1999;40:5161. [Google Scholar]
- 29.(a) Wittig G, Pockels U, Dröge H. Chem Ber. 1938;71:1903. [Google Scholar]; (b) Gilman H, Langham W, Jacoby AL. J Am Chem Soc. 1939;61:106. [Google Scholar]; (c) Bailey WF, Punzalan ER. J Org Chem. 1990;55:5404. [Google Scholar]
- 30.Goldfuss B. Top Organomet Chem. 2003;5:21. [Google Scholar]
- 31.“The ideal synthesis creates a complex skeleton… in a sequence only of successive construction reactions involving no intermediary refunctionalizations, and leading directly to the structure of the target, not only its skeleton but also its correctly placed functionality.” Hendrickson JB. J Am Chem Soc. 1975;97:5784.Shin I, Montgomery TP, Krische MJ. Aldrichim Acta. 2015;47:15.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.