

Background: GCAPs regulate photoreceptor guanylyl cyclase RetGC1 but not hormone receptor guanylyl cyclase NPRA.
Results: Mutations in RetGC1 dimerization domain disrupt GCAP1 and GCAP2 binding.
Conclusion: Met823 in dimerization domain strongly contributes to its specificity in forming GCAP binding interface.
Significance: Congenital blindness-causing mutation in the neighboring residue prohibits GCAP binding.
Keywords: calcium-binding protein, cyclic GMP (cGMP), guanylate cyclase (guanylyl cyclase), neurobiology, phototransduction, eye
Abstract
The photoreceptor-specific proteins guanylyl cyclase-activating proteins (GCAPs) bind and regulate retinal membrane guanylyl cyclase 1 (RetGC1) but not natriuretic peptide receptor A (NPRA). Study of RetGC1 regulation in vitro and its association with fluorescently tagged GCAP in transfected cells showed that R822P substitution in the cyclase dimerization domain causing congenital early onset blindness disrupted RetGC1 ability to bind GCAP but did not eliminate its affinity for another photoreceptor-specific protein, retinal degeneration 3 (RD3). Likewise, the presence of the NPRA dimerization domain in RetGC1/NPRA chimera specifically disabled binding of GCAPs but not of RD3. In subsequent mapping using hybrid dimerization domains in RetGC1/NPRA chimera, multiple RetGC1-specific residues contributed to GCAP binding by the cyclase, but the region around Met823 was the most crucial. Either positively or negatively charged residues in that position completely blocked GCAP1 and GCAP2 but not RD3 binding similarly to the disease-causing mutation in the neighboring Arg822. The specificity of GCAP binding imparted by RetGC1 dimerization domain was not directly related to promoting dimerization of the cyclase. The probability of coiled coil dimer formation computed for RetGC1/NPRA chimeras, even those incapable of binding GCAP, remained high, and functional complementation tests showed that the RetGC1 active site, which requires dimerization of the cyclase, was formed even when Met823 or Arg822 was mutated. These results directly demonstrate that the interface for GCAP binding on RetGC1 requires not only the kinase homology region but also directly involves the dimerization domain and especially its portion containing Arg822 and Met823.
Introduction
Photoreceptor retinal membrane guanylyl cyclases 1 and 2 (RetGC1 and RetGC2)2 (1–3) produce a secondary messenger of phototransduction, cGMP. RetGCs make complexes with two types of photoreceptor-specific proteins, guanylyl cyclase-activating proteins (GCAPs) (4–8) and retinal degeneration 3 protein (RD3) (9), that bind with high affinity and inhibit RetGC (10, 11). GCAPs are Mg2+/Ca2+-binding proteins (12) that control the shape of the photoresponse (13–16) through acceleration of cGMP synthesis by RetGC in the light when Ca2+ concentrations in the photoreceptor outer segment decline (17, 18). RD3 protein, which is linked to rare forms of congenital blindness (9), is required for normal accumulation of RetGC in photoreceptors and can form a very tight complex with RetGC (10), effectively blocking both RetGC catalytic activity and activation by GCAPs (11). Mutations in RetGC1 isozyme cause different forms of inherited blindness: loss-of-function mutations cause recessive Leber congenital amaurosis (LCA) (19–21), and gain-of function mutations cause dominant cone-rod degeneration 6 (CORD6) (22–25). The disease-causing mutations are found in various RetGC1 domains defined by their function and homology to other membrane guanylyl cyclases (19, 22). RetGC1 has a single transmembrane region that connects the extracellular domain (ECD) and the cytoplasmic portion that consists of the kinase homology (KHD), dimerization, and catalytic domains (2, 26). The region that includes the KHD and dimerization domain forms the binding interface for both GCAP1 and GCAP2 (27), but the precise location of the side chains that enable the docking site(s) for GCAPs on RetGC1 remains to be identified.
RetGC1 can function only as a homodimer (28, 29) in which the two catalytic subunits complement each other by supplying residues coordinating two Mg2+GTP molecules in the active site (28). The complex is also likely to contain up to two GCAP molecules, in the case of RetGC1 primarily GCAP1 and in the case of RetGC2 primarily GCAP2 (27, 30, 31). However, when compared with a hormone receptor membrane guanylyl cyclase, such as NPRA (GUCY2A) (26), the role of the dimerization domain in RetGC function appears to be rather intriguing. On the one hand, mutations affecting the coiled coil interactions of the dimerization dimer change the Ca2+ sensitivity of RetGC1 regulation by GCAP1 (23–25). On the other hand, the role of this domain as being essential for the cyclase activity (which requires the cyclase dimerization) and for binding GCAPs has been disputed (32).
Contrary to the models hypothesized elsewhere (32–34), we recently demonstrated that the docking interface(s) for both GCAP1 and GCAP2 is encoded by a region, Arg488–Arg851, containing the KHD and dimerization domain of RetGC1 (27) rather than its C terminus and transmembrane-proximal portions, making two independent docking sites for different GCAPs (33, 35). In the present study, we tested how mutations (site-directed or naturally occurring) specifically in the RetGC1 dimerization domain affected binding of GCAPs. We found that (i) RetGC1-specific residues in the dimerization domain are essential for the ability of cyclase to bind GCAP albeit not essential for the RD3 binding interface; (ii) the ability of RetGC1 to bind GCAP lacking in hormone receptor cyclase, such as NPRA, requires RetGC1-specific residues in the dimerization domain, especially Met823, the residue that does not constitute a direct contact between the RetGC1 coiled coil domains; and (iii) mutation of Met823 and LCA-linked mutation of the neighboring Arg822 destabilize the binding of GCAP1 and -2 but not the binding of RD3.
Experimental Procedures
Guanylyl Cyclase Expression and Mutagenesis
Human RetGC1 (GUCY2D) and NPRA (GUCY2A) guanylyl cyclases, either tagged with mOrange red fluorescent protein (Clontech) or untagged, were expressed in HEK293 cells as described (27). Mutagenesis was performed utilizing Thermo Scientific Phusion Flash DNA polymerase in a “splicing by overlap extension” approach (36) to produce a series of single amino acid substitutions or replacing portions of RetGC1 sequence with that of NPRA. The amplified fragments were inserted into the DraIII/ClaI sites of the modified plasmid, mOrangeRetGC1 plasmid, encoding new restriction sites in the RetGC1 without changing the amino acid sequence (27). The resultant constructs contained mOrange fluorescent tag in the extracellular segment as described in detail previously (27). Oligonucleotide primers were synthesized by Integrated DNA Technologies. Restriction endonucleases were purchased from New England Biolabs.
Immunostaining
Following 7% polyacrylamide-SDS gel electrophoresis, the samples were transferred on PVDF membrane using an Invitrogen/Life Technologies iBlot apparatus, stained with Ponceau S to mark positions of the molecular mass standards, and probed with rabbit polyclonal antibodies (37) raised against the Arg540–Asn815 human RetGC1 KHD fragment or the Met797–Ser1103 catalytic domain fragment (residues are numbered from Met1 of the leader peptide encoded by GUCY2D gene). The blots were developed using a Pierce SuperSignal Femto chemiluminescence substrate kit, and the images were taken using a Luminous FX imaging system.
GCAP and RD3 Expression
Myristoylated GCAP1 and GCAP2 were expressed from pET11d vector in BLR(DE3) Escherichia coli strain harboring N-myristoyltransferase, extracted from inclusion bodies, and purified using hydrophobic and size exclusion chromatography as described in detail previously (38–40). For the in cyto binding analysis, GCAP1 and GCAP2 were tagged at the C terminus with the SuperGlo enhanced green fluorescent protein (Clontech) and expressed from pQBIfN3 vector (Clontech) using a Promega FuGENE protocol as described previously (27, 41, 42).
Human RD3 (9) cDNA was amplified with HindIII/EcoRI restriction sites at the ends using as a template MHS1010-9206149 Thermo Scientific/Open Biosystems cDNA clone (catalog number 6140075) in high fidelity Phusion Flash polymerase chain reaction mixture and inserted into the HindIII/EcoRI sites of the pQBIfN3 expression vectors to tag RD3 with GFP at the C terminus. RD3-GFP was expressed in HEK293 cells using a FuGENE transfection reagent protocol.
Transfection for Confocal Imaging
Unless specified otherwise, HEK293 cells were transfected in a Lab-Tek 4-well cover glass chamber with 1 μg of mOrangeRetGC1 DNA/well using 3 μl/μg DNA of the FuGENE reagent as described (27) at an ∼1:100 molar ratio of GCAP-GFP- or RD3-GFP-coding plasmid versus mOrangeRetGC1-coding plasmid. Confocal images were taken after 24 h of incubation utilizing an Olympus FV1000 Spectral instrument using the respective 543- and 488-nm excitation for the red and green fluorochromes and processed using Olympus FluoView FV10-ASW software as described previously (27, 41, 42). No changes to the original images were made except for occasional minor γ correction applied to the whole image for more clear presentation in print. Quantitative analysis was performed using only original images without γ corrections. Where applicable, Pearson's correlation coefficient (PCC) for testing co-localization of RD3-GFP with mOrange-tagged RetGC1 variants was calculated using Olympus FluoView FV10-ASW software as described previously (27, 41, 42), and the statistical difference between the PCC values was tested using the analysis of variance function in Synergy KaleidaGraph 4 software applying Bonferroni post hoc processing.
RetGC1 Activity Assay
Human RetGC1 cDNA was expressed in HEK293 cells from a modified pRCCMV vector (Invitrogen) using calcium phosphate precipitation for the transfection, and the membrane fraction containing expressed RetGC1 was isolated as described in detail previously (27, 43). The activity of the cyclase was assayed using [α-32P]GTP (PerkinElmer Life Sciences) as a substrate, and the [32P]cGMP product was quantified using TLC as described previously (27, 39, 43). Briefly, the assay mixture (25 μl) incubated at 30 °C contained 30 mm MOPS-KOH (pH 7.2), 60 mm KCl, 4 mm NaCl, 1 mm DTT, 2 mm Ca2+/EGTA buffer (<10 nm [Ca2+]free), 6 mm free Mg2+ as indicated in the text, 0.3 mm ATP, 4 mm cGMP, 1 mm GTP, and 1 μCi of [α-32P]GTP. The resultant [32P]cGMP product was separated by TLC on fluorescently backed polyethyleneimine cellulose plates (Merck), developed in 0.2 m LiCl, and eluted with 2 m LiCl. For the mutants of RetGC1 converted into adenylyl cyclase by mutations in the active site (44), the assay was performed using the same protocol except that 2 mm [α-32P]ATP (PerkinElmer Life Sciences) was used as a substrate instead of GTP, and 3 mm cAMP was the internal standard to identify the product of the reaction on the TLC plate.
Computation of Coiled Coil Probability
The coiled coil structure probability for the whole intracellular segment of RetGC1 and the RetGC/NPRA chimeric protein was computed using conventional MARCOIL software, a hidden Markov model-based program (45) that predicts the existence and location of potential coiled coil domains in protein sequences based on a standard MTIDK matrix, an implementation of the method of Lupas et al. (46).
Results
LCA Mutation R822P in Dimerization Domain of RetGC1 Suppresses GCAP Binding
LCA mutation R822P (20) (residues here and further are numbered from the Met1 of the leader peptide as coded by the wild type RetGC1 cDNA; Ref. 2) disabled RetGC1 activity in vitro by blocking its binding with GCAP1 and GCAP2 (Fig. 1). GCAP complex with the cyclase is known to disintegrate upon extraction of RetGC1 from the membranes with detergents (47). Therefore, the RetGC-GCAP complex cannot be detected using conventional co-precipitation or pulldown assays. However, it can be detected using a cell-based assay in which GCAP1-GFP or GCAP2-GFP expressed in HEK293 cells displays a uniform distribution through the cytoplasm and the nucleus of the cell but becomes anchored to the membranes by the cyclase (primarily to the endoplasmic reticulum; Ref. 41) when co-expressed with either untagged or mOrange-tagged RetGC1 (27, 40–42). The tagged cyclase carrying the R822P substitution was neither able to become effectively stimulated by GCAP1 and GCAP2 (similarly to the untagged mutants; see Ref. 20) nor to alter GCAP distribution. A PCC (mean ± S.E.) value of 0.25 ± 0.04 (n = 23), which is below the co-localization criterion threshold (∼0.5; Ref. 48), effectively confirmed the lack of strong GCAP1 binding to the mutated cyclase in a sharp contrast to the normal cyclase (PCC = 0.9 ± 0.05, n = 33) (Fig. 1, B and C, and Table 1).
FIGURE 1.
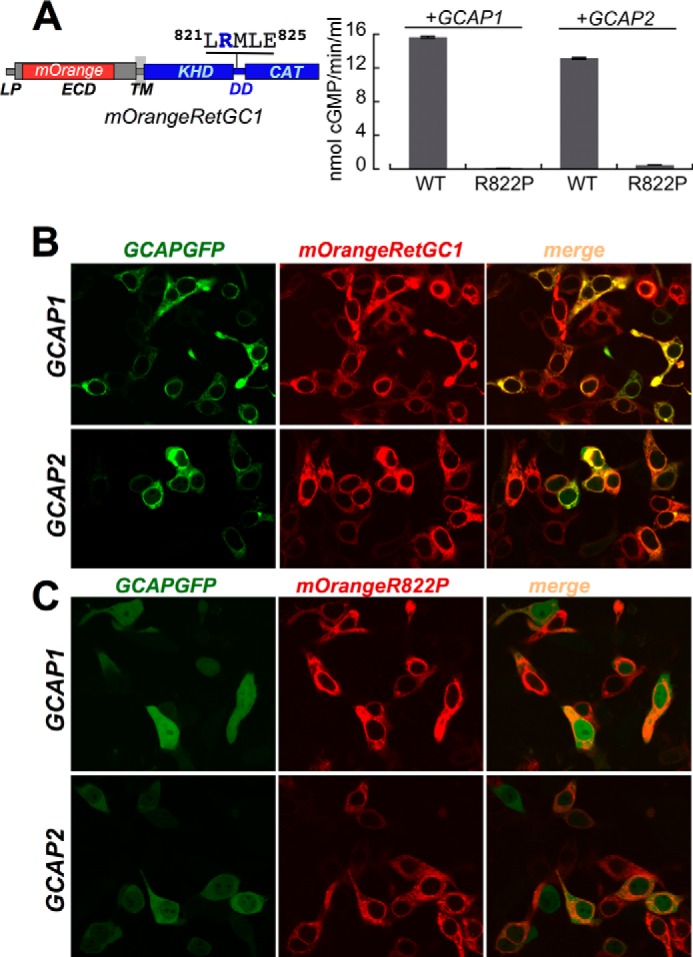
LCA mutation R822P blocks GCAP1 and GCAP2 binding to RetGC1. A, R822P RetGC1 (20) fails to become activated by GCAP1 and GCAP2. The wild type and R882P RetGC1 were labeled with the mOrange red fluorescent protein tag substituting a portion of the ECD and expressed in HEK293 cells. The membranes were reconstituted with 15 μm purified GCAP1 or GCAP2 at ≪10 nm free Ca2+ and saturating Mg2+ concentrations and analyzed as described under “Experimental Procedures”. Error bars are S.E. The schematics on the left represents the domain sequence in RetGC1 primary structure: LP, leader peptide; TM, transmembrane region; CAT, catalytic domain. All residues are numbered from Met1 of the RetGC1. B and C, in cyto GCAP binding assay (41, 42). HEK293 cells were transfected with a mixture of GCAP-GFP and either wild type (B) or R822P (C) mOrangeRetGC1-expressing plasmids at a GCAP:RetGC1 plasmid DNA ratio of ∼1:100. Transfection and confocal imaging after 24 h of incubation were performed as described under “Experimental Procedures.” PCC values from quantitative analysis have been summarized in Table 1.
TABLE 1.
Co-localization of different variants of RetGC1 with GCAP1 in HEK293 cells
The PCC values for mOrangeRetGC1 and GCAP-GFP co-expressed in HEK293 cells were determined from the confocal images of the whole cells using Olympus FluoView FV10-ASW software.
| Co-transfection | PCCa (mean ± S.E.; n cells) |
|---|---|
| GCAP1-GFP + mOrangeRetGC1 | 0.90 ± 0.05; 33 |
| GCAP1-GFP + mOrangeRetGC1 R822P | 0.25 ± 0.04; 23b |
| GCAP1-GFP + Cat1DD I | 0.30 ± 0.03; 41b |
| GCAP1-GFP + DD II | 0.39 ± 0.04; 40b |
| GCAP1-GFP + DD III | 0.91 ± 0.01; 41 |
| GCAP1-GFP + DD IV | 0.66 ± 0.03; 42b |
| GCAP1-GFP + DD V | 0.87 ± 0.01; 53 |
| GCAP1-GFP + DD VI | 0.46 ± 0.02; 27b |
| GCAP1-GFP + DD VII | 0.52 ± 0.04; 21b |
| GCAP1-GFP + DD VIII | 0.83 ± 0.01; 40 |
| GCAP1-GFP + DD IX | 0.49 ± 0.03; 21b |
| GCAP1-GFP mOrangeRetGC1 M823R | 0.55 ± 0.03; 31b |
| GCAP1-GFP mOrangeRetGC1 M823E | 0.52 ± 0.03; 32b |
| GCAP1-GFP mOrangeRetGC1 M823S | 0.89 ± 0.01; 41 |
| GCAP2-GFP mOrangeRetGC1 M823R | 0.40 ± 0.03; 28c |
| GCAP2-GFP mOrangeRetGC1 M823S | 0.87 ± 0.01; 43 |
| RD3-GFP + mOrangeRetGC1 R822P | 0.90 ± 0.01; 35 |
| RD3-GFP + DD hybrid IV | 0.88 ± 0.01; 21 |
| RD3-GFP + DD hybrid V | 0.90 ± 0.01; 23 |
| RD3-GFP + DD hybrid VI | 0.91 ± 0.01; 15 |
| RD3-GFP + mOrangeRetGC1 M823R | 0.91 ± 0.01; 44 |
a The mOrangeRetGC1 and GCAP-GFP or RD3-GFP were co-expressed in HEK293 cells, and confocal microscopy was performed as described under “Experimental Procedures.” PCC values indicating strong co-localization are highlighted in bold (note that PCC ≤ 0.5 generally means no co-localization, whereas PCC = 1.0 means co-localization of all red and green pixels in the image (48)).
b p < 0.0001 when compared with the mOrangeRetGC1; from one-way analysis of variance/Bonferroni all-pairs comparison test (α = 0.01; confidence level, 99%) processed using Synergy KaleidaGraph 4 software.
c p < 0.0001, unpaired t test.
Unlike GCAP1-GFP, RD3-GFP binding to RetGC1 in HEK293 cells remained despite the R822P substitution (Fig. 2). We found that in a fashion similar to that of GCAP1-GFP (41), RD3-GFP displayed a mostly diffuse distribution throughout the cytoplasm and the nucleus when expressed in HEK293 cells (Fig. 2A, top panel) but became anchored to the membranes when co-expressed with the cyclase and void from the nucleus (Fig. 2B). It needs to be pointed, however, that the overall pattern of expressed RD3-GFP in the conditions of our experiments was different from the punctate pattern of RD3 produced in the HEK293 nuclei (9) and/or the cytoplasm (10). Because recombinant RD3 was found to be very prone to self-aggregation (11), we tested whether the variations in the protein expression levels could explain the variability between its distribution patterns reported by different groups. We found that the dose of RD3-GFP DNA used for transfection indeed critically affected the RD3 pattern in HEK293 cells (Fig. 2A). At a low dosage of the RD3-GFP-expressing plasmid (≤0.02 μg of DNA cm−2), the distribution was mostly uniform throughout the cell (top panel) similar to that of GCAP1-GFP expressed in the absence of RetGC1 (41), whereas at the increased dosage (up to 2 μg of DNA cm−2), a distinct bright punctate staining of various sizes appeared in both the cytoplasm and the nucleus (Fig. 2A). The granules of the aggregated protein were also observed when a non-tagged RD3 was expressed and probed with anti-RD3 antibody (data not shown). Therefore, it needs to be underscored that to avoid the potential artifacts caused by RD3 self-aggregation, such as shown in the bottom three panels in Fig. 2A, we used a very low dose (≤0.02 μg of DNA cm−2) (Fig. 2A, top panel) of the RD3-GFP-expressing vector in all subsequent co-transfection experiments designed to probe for the RD3-GFP co-localization with the mOrangeRetGC1. That was the concentration at which RD3 expressed in the absence of the cyclase was distributed fairly uniformly through the cell (Fig. 2A, top panel) but acquired a sharp membrane-bound, rather than punctate, pattern through its association with the cyclase (Fig. 2, B and C) similar to that of GCAP1 (41). The R822P RetGC1 co-localized with RD3-GFP very strongly (PCC = 0.9 ± 0.01, n = 35), thus arguing that the disruption of GCAP binding by the R822P mutation was not a result of a nonspecific unfolding of the cyclase. Hence, altering RetGC1 amino acid sequence in the dimerization domain specifically disrupted the GCAP binding interface.
FIGURE 2.
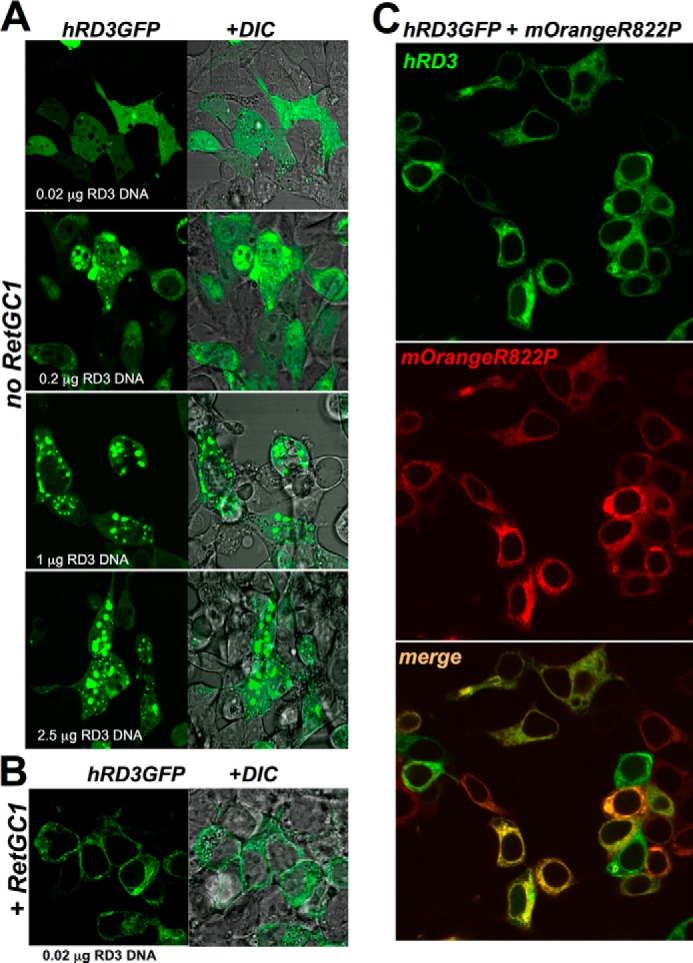
LCA mutation R822P does not block RD3 binding to RetGC1. A, uniform versus punctate pattern of human RD3-GFP localization in HEK293 cells depends on the DNA dose for transfection. RD3 was tagged at the C terminus with GFP as described under “Experimental Procedures,” and RD3-GFP-expressing plasmid was transfected in HEK293 cells using a calcium phosphate precipitation protocol and various amounts of DNA (top to bottom: 0.02, 0.2, 1, and 2.5 μg of DNA cm−2). Left, confocal image of GFP fluorescence; right, the same but superimposed over a differential interference contrast (DIC) image. Note the predominantly uniform distribution of RD3-GFP throughout the cytoplasm and the nucleus at low DNA transfection dose versus the punctate appearance of aggregates in both the cytoplasm and nucleus with the increase in DNA dosage. B, in HEK293 cells expressing low levels of RD3-GFP (0.02 μg of DNA cm−2), its pattern changes to membrane-anchored in the presence of RetGC1 (2 μg of plasmid DNA cm−2 for RetGC1). Note the difference from the pattern in the upper panel of A. C, RD3-GFP co-localizes with the R822P RetGC1. The mixture of 0.02 μg of RD3-GFP plasmid (green) and 2 μg of mOrangeRetGC1 R822P DNA (red) was transfected in HEK293 cells as described under “Experimental Procedures.” Note the difference of the pattern from that of GCAPs in Fig. 1C. The PCC values are presented in Table 1.
Replacing RetGC1 Dimerization Domain with That of NPRA Incapacitates Both GCAP1 and GCAP2 Binding
In contrast to RetGC1, the peptide hormone receptor cyclase NPRA does not bind GCAPs (27). However, a chimeric protein containing the KHD and dimerization domain from RetGC1 and catalytic domain of NPRA (construct shown in Fig. 3A and referred to hereafter as “Cat1”) binds GCAP1 and GCAP2 with high apparent affinity, even exceeding that of wild type RetGC1 (Ref. 27 and Fig. 4, A and B). To test whether or not the dimerization domain was essential for creating the GCAP binding interface on the cyclase, we used a chimera with the NPRA sequence extended to include the dimerization domain (“Cat1DD I” chimera in Fig. 3A). The Cat1DD I chimera completely failed to bind GCAP1 and GCAP2 (Fig. 3, B and C, and Table 1). However, Cat1 and Cat1DD I were both able to bind RD3 (Fig. 3D). These results indicated that the dimerization domain of NPRA cyclase did not cause unfolding of the Cat1DD I chimera but rather specifically disrupted its GCAP binding interface.
FIGURE 3.
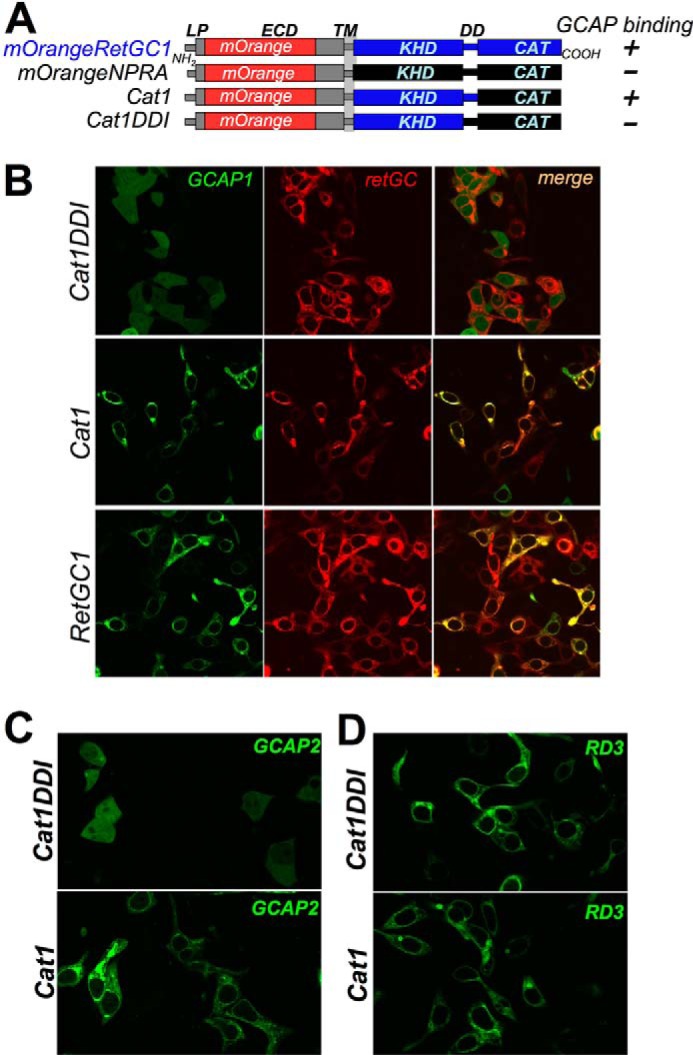
Binding of GCAPs but not RD3 by RetGC1/NPRA chimera strongly depends on RetGC1-specific amino acid sequence in dimerization domain. A, schematics representing the domain structures of RetGC1 (blue), NPRA (black), and two chimeric constructs: in one of them (27) the catalytic domain of RetGC1 (Cat1) and in the other both the catalytic domain and dimerization domain (Cat1DD I chimera) were replaced with the homologous regions from NPRA; the “+” and the “−” indicate that the construct binds or fails to bind GCAPs in the co-transfection assays, respectively. LP, leader peptide; TM, transmembrane region; CAT, catalytic domain. B and C, unlike WT or Cat1 chimera, the Cat1DD I chimera fails to anchor either GCAP1 (B) or GCAP2 (C) to the membranes in cyto. Note the diffuse pattern of GCAP-GFP spread through the cytoplasm and the nucleus. D, in contrast to GCAPs, the Cat1DD I chimera continues to anchor RD3-GFP to the membranes and prevents its diffusion to the nuclei.
FIGURE 4.
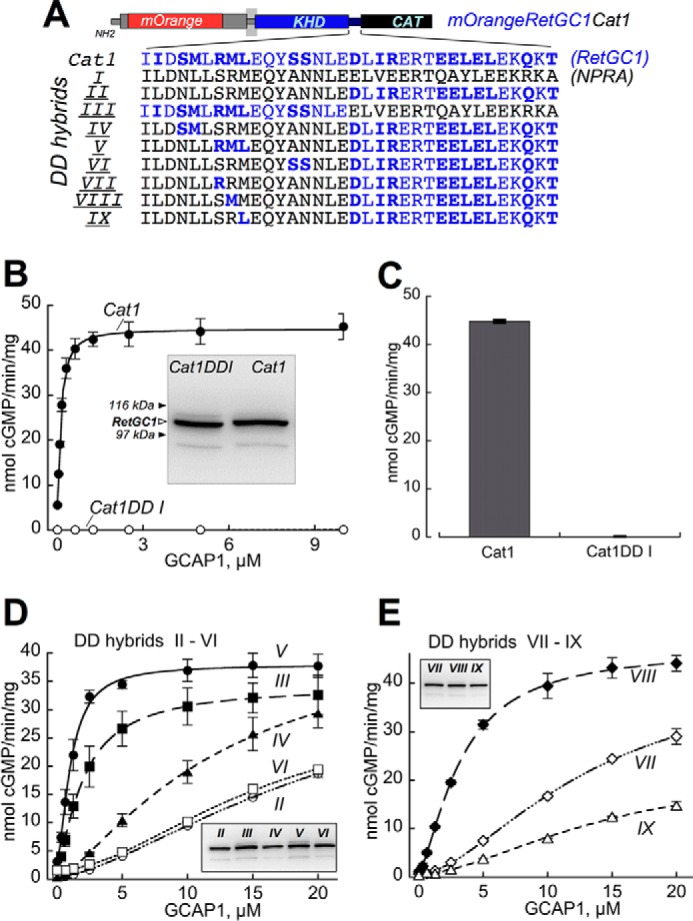
Sensitivity of RetGC1/NPRA dimerization domain hybrids to GCAP-dependent activation. A, portions of the dimerization domain coded by NPRA gene (black) in the Cat1DD I chimera were replaced by those coded by RetGC1 gene (blue). The RetGC1-specific amino acid residues are highlighted in bold (note that some residues coded by the two respective different genes are identical). CAT, catalytic domain. B and C, GCAPs activate Cat1 but fail to activate Cat1DD I. B, dose dependence of Cat1 (●) versus Cat1DD I (○) activation by GCAP1 at ≪10 nm free [Ca2+] and 6 mm free [Mg2+]. Inset, immunoblotting of the two chimeric constructs expressed in HEK293 membranes probed with anti-RetGC1 KHD antibody (37); the filled arrows indicate positions of β-galactosidase (116 kDa) and phosphorylase b (97 kDa). C, the membranes expressing Cat1 and Cat1DD I were reconstituted with 10 μm GCAP2 at ≪10 nm free [Ca2+] and 6 mm [Mg2+]. D and E, dose dependence of GCAP1 activation of the Cat1 chimeras containing NPRA/RetGC1 hybrids II–VI (D) and VII–IX (E) depicted in A. Insets, immunoblotting of 7% SDS-polyacrylamide gel loaded with 10-μl aliquots of the respective membrane fractions (molecular weight markers are not shown because of space constraints). The activities in assays shown in B–E were standardized by the protein content. The data were fitted using Synergy KaleidaGraph 4 utilizing the standard Levenberg-Marquardt algorithm of nonlinear least square routines assuming a Hill function: a = (amax − amin)/(1 + ([GCAP]/(K1/2GCAP)h) + amin where a is the activity of RetGC in the assay; amin and amax are the minimal and the maximal activities, respectively; [GCAP] is the concentration of GCAP, K1/2GCAP is the GCAP concentration required for half-maximal activation, and h is the Hill coefficient. The K1/2GCAP was low for DD V (1.1 ± 0.06 μm) and DD III (2 ± 0.03 μm) but sharply increased in DD hybrids II, IV, and VI (21.5 ± 0.70, 12.5 ± 1.2, and 15 ± 1.8, respectively). The K1/2GCAP values for DD VII, VIII, and IX hybrids in E were 15 ± 2.3, 3.2 ± 0.16, and 22 ± 12 μm, respectively (the low activity in DD hybrid IX increased the error for the K1/2GCAP extraction from the fitting). The assay conditions are described under “Experimental Procedures.” Error bars are S.E.
Consistent with the GCAP binding pattern in cyto, the hybrid cyclase containing the dimerization domain of NPRA in a striking contrast to the Cat1 chimera containing the RetGC1 dimerization domain (Fig. 4, A–C) also completely failed to undergo activation by GCAP1 and GCAP2 in vitro despite the comparable levels of expression in HEK293 membranes for both chimeras (Fig. 4B, inset). Hence, not only the LCA-related mutation but also substitution with a homologous sequence derived from a receptor membrane guanylyl cyclase dimerization domain failed to support RetGC ability to bind either GCAP.
The Part of RetGC1 Dimerization Domain Containing Met823 Has the Strongest Effect in Enabling GCAP1 and GCAP2 Binding
The prominent difference between the affinities of Cat1 and Cat1 DDI chimeric cyclases for GCAPs (Figs. 3 and 4, B and C) made it possible to probe for the regions in the dimerization domain that strongly contributed to the unique ability of RetGC1 to associate with GCAPs. The amino acid residues encoded by RetGC1 and NPRA cDNAs (Fig. 4A) were arranged in various hybrid combinations (“DD I–DD VI”) and tested for their activation by GCAP1 (Fig. 4D). Replacement of the C-proximal half of the NPRA dimerization domain with the RetGC1-specific sequence enabled activation, originally lacking in the DD I hybrid, but the apparent affinity for GCAP1 was strongly diminished (Fig. 4D, hybrid “DD II”). In contrast, preservation of the N-proximal half of the RetGC1 (hybrid “DD III”) substantially improved the apparent affinity for GCAP1. Further mapping of that region in hybrid DD III by replacing shorter RetGC1-specific clusters of amino acid residues (hybrids DD IV–VI) showed that the better dose response for the activation in the hybrid DD III compared with DD II co-segregated with a short RetGC1-specific fragment, 822RML824 (hybrid DD V).
Subsequent single amino acid substitutions restoring RetGC1-specific residues in DD II, such as S822R (hybrid “DD VII”), R822M (hybrid “DD VIII”), and M824L (hybrid “DD IX”) pointed to Met823 as the residue enhancing the affinity for GCAP1 in the N-proximal portion of the RetGC1 dimerization domain (Fig. 4, A and E). Co-localization assay (Figs. 5 and 6 and Table 1) also demonstrated that the GCAP1 binding pattern was the sharpest with hybrids DD III, V, and VIII (respective PCC values were 0.91, 0.87, and 0.83, which are well above the co-localization threshold of 0.5 (48)). Co-localization was poorly defined for the rest of the tested hybrids (respective PCC values for hybrids DD II, IV, VI, VII, and IX were 0.39, 0.66, 0.46, 0.52, and 0.49; Table 1). Importantly, even the hybrids demonstrating a weaker GCAP1 binding pattern (DD IV and VI) all bound RD3 in a manner similar to that of the GCAP1-binding hybrid DD V (Fig. 5B and Table 1) (respective PCC values for hybrids DD IV, V, and VI were 0.88, 0.90, and 0.91). Hence, the RetGC1-specific sequence in the dimerization domain was only required for maintaining the interface for GCAP but not RD3.
FIGURE 5.
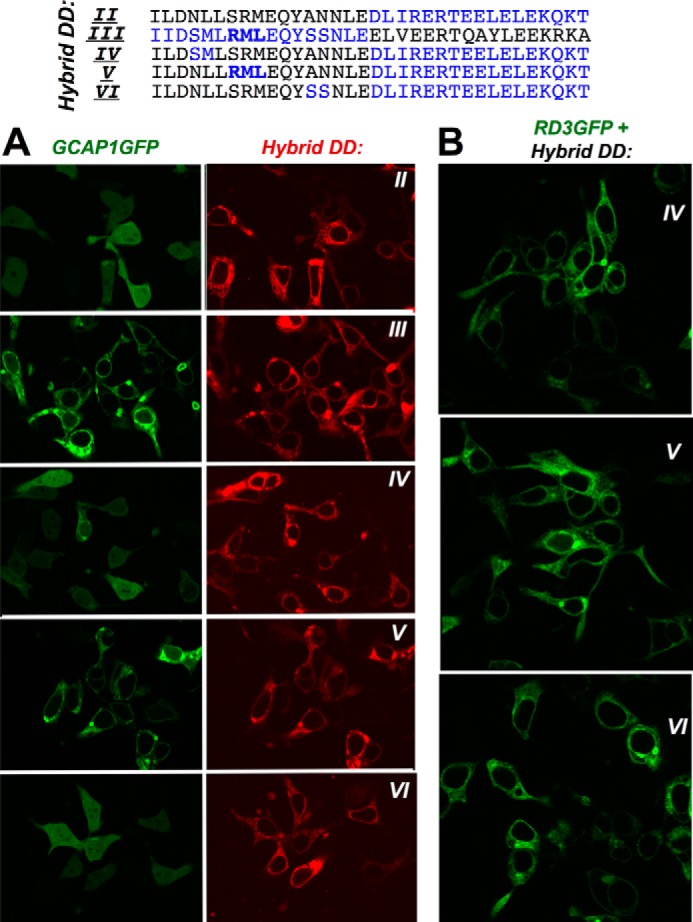
Localization of GCAP1-GFP co-expressed in HEK293 cells with RetGC1/NPRA dimerization domain hybrids II–VI. A, GCAP1-GFP was co-expressed with mOrange Cat1 chimera containing hybrids (top to bottom) DD II–VI. The DD III and DD V hybrids that harbor the RetGC1-specific 822RML824 peptide sequence (bold) retain an obvious GCAP binding pattern, whereas in the other hybrids it has been compromised. B, RD3-GFP co-expressed with (top to bottom) hybrids DD IV, V, and VI retains a normal membrane-anchored pattern regardless of the effectiveness of the hybrid in anchoring GCAP1-GFP. The PCC values for the co-localization are presented in Table 1.
FIGURE 6.
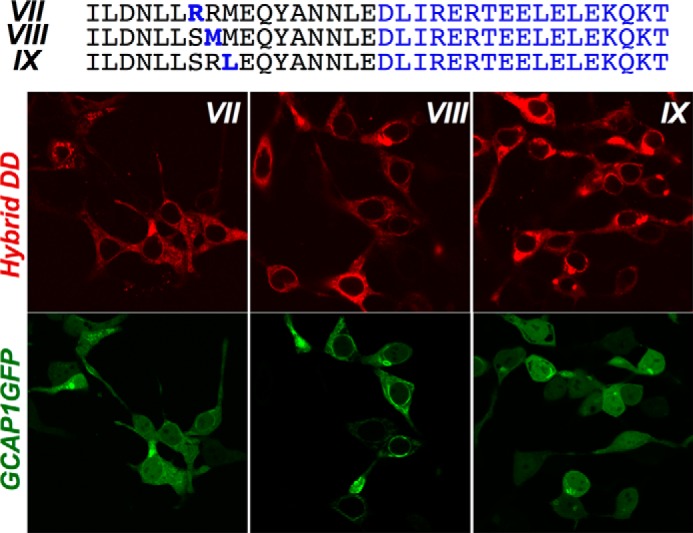
Met823 in dimerization domain enhances GCAP1-GFP co-localization with RetGC1/NPRA chimera. GCAP1-GFP was co-expressed with mOrange Cat1 chimera containing hybrids (left to right) DD VII–IX. Note that hybrid DD VIII retains a clear GCAP binding pattern, which is compromised in the hybrids lacking the Met823. The PCC values for the co-localization are presented in Table 1.
The importance of Met823 in contributing to the strength of the GCAP1 binding interface created by the RetGC1-specific residues was further tested in wild type RetGC1 by a series of single amino acid substitutions at this position. The abilities of the non-chimeric cyclase to become activated by GCAPs (Fig. 7) and to relocate GCAPs to the membranes in the co-transfection assay (Fig. 8) both tolerated substitution with a non-charged polar residue (Ser) but became compromised by the replacement of the Met823 with either negatively charged Glu or positively charged Arg (data for M823E RetGC1/GCAP2-GFP co-expression is not shown due to the space constraint in the figure). Most interestingly, the Arg residue prohibiting effective GCAP1 binding is present in the corresponding position of the dimerization domains from all peptide hormone receptor guanylyl cyclases: NPRA (GUCY2D), NPRB (GUCY2B), and NPRC (GUCY2C) (Fig. 7, top). At the same time, neither Arg823 nor Glu823 (not shown) disrupted the cyclase co-localization with RD3-GFP (Fig. 8C).
FIGURE 7.
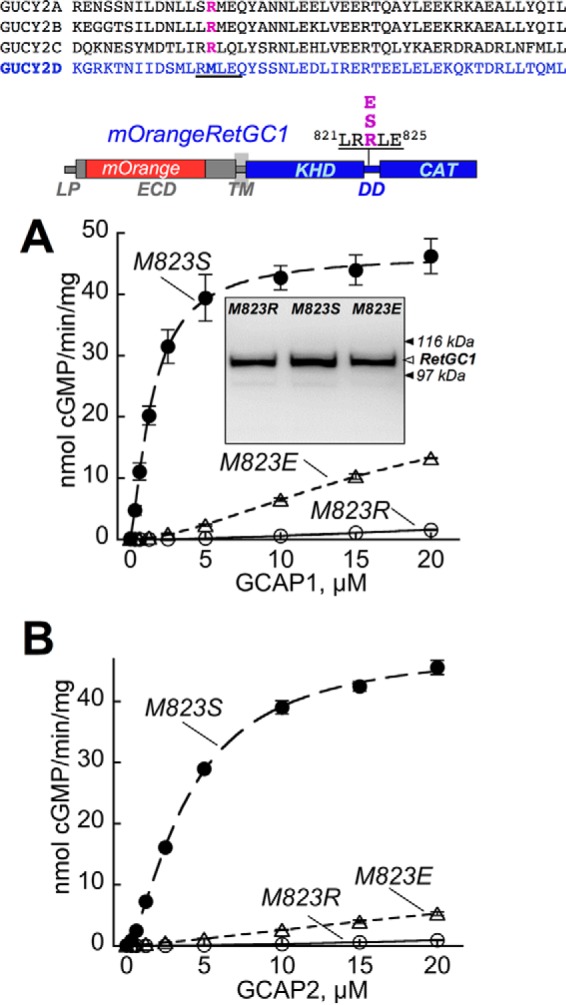
Replacement of Met823 in wild type RetGC1 with charged side chains inhibits activation by GCAP1 and GCAP2. Top, RetGC1 (GUCY2D) has Met823 (highlighted) in its dimerization domain, whereas peptide hormone receptor cyclases GUCY2A, GUCY2B, and GUCY2C all have Arg in the corresponding positions of their dimerization domains. CAT, catalytic domain. A and B, Met823 of the wild type mOrangeRetGC1 was substituted with Ser (●), Glu (▵), or Arg (○), and the mutated RetGC1 harboring the point mutation was expressed in HEK293 cells and reconstituted with either GCAP1 (A) or GCAP2 (B). The dose dependence data were fitted assuming a Hill equation. Inset in A, Western immunoblotting of the Met823 RetGC1 mutants expressed in HEK293 cells probed with anti-RetGC1 antibody raised against the catalytic domain (37); the filled arrows (▶) indicate positions of β-galactosidase (116 kDa) and phosphorylase b (97 kDa). The activities in the cyclase assay were normalized by the protein content. Error bars are S.E.
FIGURE 8.
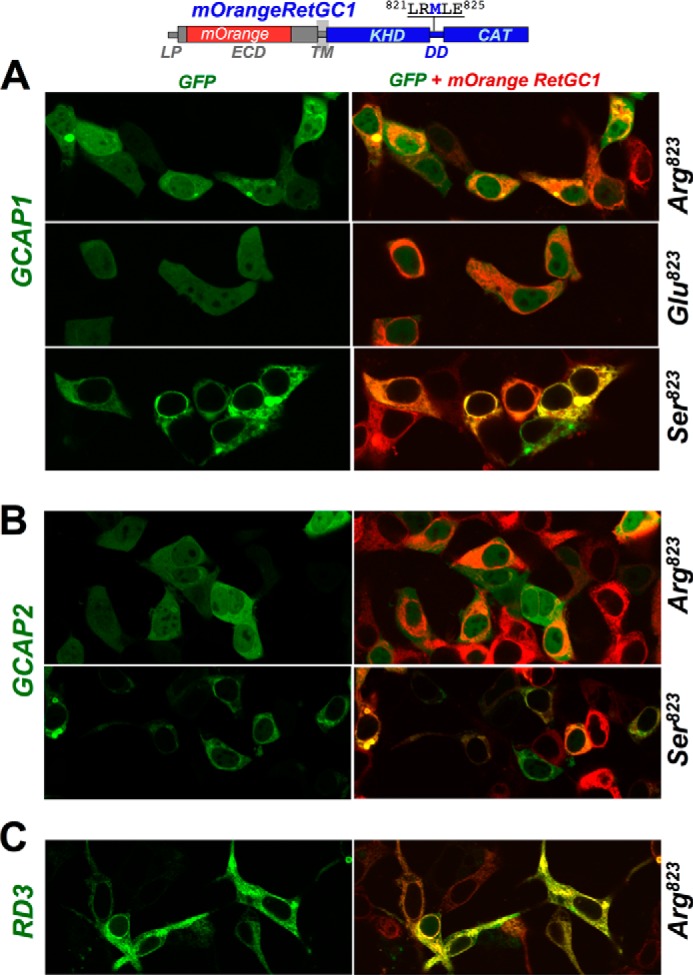
Replacement of Met823 in wild type RetGC1 dimerization domain with Arg inhibits binding of GCAP1 and GCAP2 but not of RD3. mOrangeRetGC1 (1 μg of DNA) was co-transfected in HEK293 cells with 0.01 μg of GCAP1-GFP (A), GCAP2-GFP (B), or RD3-GFP (C) DNA constructs as described under “Experimental Procedures”. The respective PCC values (mean ± S.E.) were as follows: for co-localization of GCAP1 with RetGC1 harboring M823R, M823E, and M823S substitutions, 0.55 ± 0.03, n = 31; 0.52 ± 0.03, n = 32; and 0.89 ± 0.01, n = 41; for GCAP2 co-localization with M823R and M823S RetGC1, 0.40 ± 0.03, n = 28 and 0.89 ± 0.01, n = 41; for RD3 co-localization with M823R RetGC1, 0.91 ± 0.01, n = 44 (see Table 1 for the summary of the quantitative analysis).
The Ability of the Mutated Dimerization Domains to Promote Coiled Coil Dimerization of the Cyclase
The probabilities of creating the coiled coil structure for the hybrid RetGC1/NPRA chimeras and for RetGC1 harboring point mutations were evaluated using a conventional MARCOIL computation (45, 46) in which the primary structure of the entire intracellular segment of the enzyme downstream of the transmembrane domain was tested for the likelihood of a coiled coil dimerization between two identical protein molecules (Fig. 9). All constructs tested in the present study yielded a high probability of a coiled coil dimer. The lack of the apparent correlation between the probability to create a coiled coil dimer and the extent to which they maintained GCAP binding was quite remarkable for the hybrids DD I–VI (Fig. 9A). For example, DD hybrids I, II, and V, although strikingly different in binding GCAP (see Figs. 3–5 and Table 1), all exceeded 99% probability of forming coiled coil in the dimerization domain between residues 812 and 846.
FIGURE 9.
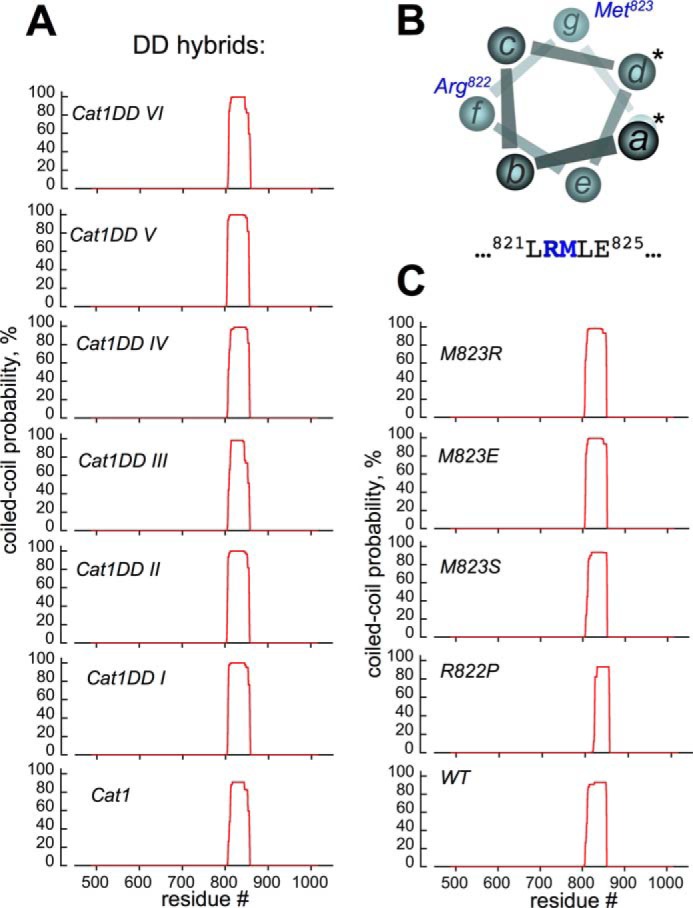
Probabilities of coiled coil dimerization remain high for RetGC1/NPRA hybrids and RetGC1 point mutants. The primary structure of the cyclase cytoplasmic domain was tested for the probability of forming a coiled coil dimer at different positions (red line) using conventional MARCOIL software utilizing the standard MTIDK matrix (45, 46); positions of the residues are numbered relative to the Met1 of the leader peptide encoded by the human GUCY2D gene. A, top to bottom, RetGC1/NPRA chimeras Cat1DD VI–I and RetGC1/NPRA chimera Cat1. B, in the first coiled coil heptad of the RetGC1, neither Arg822 nor Met823 (positions f and g, respectively) can make direct contact between two coils (positions a and d making the coiled coil contacts are marked with asterisks). The MARCOIL probability of Met823 or Arg822 to occupy the a or d position was below 0.1%. C, point mutations (top to bottom), M823R/E/S or R822P substitutions do not prevent coiled coil dimerization of the RetGC1 DD.
It needs to be noted that based on the computational analysis in the heptad I helix, which is the most N-proximal of the four heptads in the RetGC1 dimerization domain (44), neither Arg822 nor Met823 create the immediate contacts between two coils (positions “a” and “d” in the heptad shown in Fig. 9B). Instead, Met823 occupies position “g” and Arg822 occupies position “f” opposite from the points of contact between the dimerizing coils. The effect of point mutations introduced into wild type RetGC1 was tested and is shown in Fig. 9C. The point mutations failed to negate the coiled coil dimerization by the RetGC1. No evidence of the coiled coil structure being disrupted was found for the Met823 mutants, but for the R822P, the N-proximal portion of the coiled coil appeared partially destabilized: the >90% coiled coil probability region in the R822P RetGC1 spanned the residues 828–855, shorter than the 818–856 region in wild type or Met823 mutants.
Testing Dimerization of RetGC Mutants by Functional Complementation within the Cyclase Homodimer
The ability of the M823R and R822P RetGC1 to dimerize was directly verified in a biochemical assay using a method originally introduced by Ramamurthy et al. (24) by functional complementation between two RetGC molecules whose substrate binding was altered (Fig. 10). RetGC1 is a homodimer (28) whose active site is formed by two complementary subunits converting two GTP substrate molecules into two cGMP molecules (Fig. 10A). Each subunit coordinates the purine base of one GTP molecule and the ribose ring of another GTP molecule (28). Replacement of two residues, Glu925 and Cys997, coordinating the purine base (shown in Fig. 10A, yellow) with Lys and Asp, respectively, changes the substrate specificity of RetGC1 from GTP to ATP (44), thus converting RetGC1 from a guanylyl cyclase into an adenylyl cyclase (construct “GC1AC” in Fig. 10B). A different point mutation, Asp929 (shown in Fig. 10A, red) to Ala, prevents proper ribose coordination and the GTP → cGMP (or ATP → cAMP in GC1AC) catalysis and thus completely inactivates the homodimeric enzyme (24) (Fig. 10B, “GC1AC rib(−)”). Because the ECD portion of RetGC1 is not required for either catalytic activity or regulation by GCAPs (27, 37, 42), we deleted most of the ECD in both GC1AC and GC1AC rib(−) variants to distinguish them from the full-size RetGC1 by immunoblotting in the subsequent co-transfection experiments. It is worth re-emphasizing that GCAP binding to RetGC1 did not require preservation of the guanylyl cyclase active site. GCAP1-GFP became anchored to the membranes and void from the nuclei when co-expressed in with the GC1AC rib(−) cyclase mutant HEK293 cells (Fig. 10C) similarly to the wild type RetGC1 (41).
FIGURE 10.

Both M823R and R822P RetGC1 can form catalytically active dimers. A, structural model by Liu et al. (28) displays two molecules of Mg2+GTP as the substrate in the active site formed by the RetGC1 homodimer. Side chains Glu925 and Cys997 (yellow) in each subunit are required to coordinate the guanine base of one of the two GTP molecules, whereas Asp929 (red) in each subunit coordinates the ribose of another GTP molecule. B, schematics of the mutated forms of RetGC1 produced for the functional complementation analysis: GC1AC in which E925K/C997D substitutions enable binding of ATP instead of GTP in the active site and thus converting RetGC1 into an adenylyl cyclase (44) and GC1AC rib(−) in which conversion of ATP into cAMP in the active site of GC1AC is disabled by the additional D929A mutation (24) that disrupts coordination of the ribose moiety (28); a portion of the ECD is deleted between the leader peptide and the transmembrane domain in both GC1AC variants to reduce their size and make them identifiable on an immunoblot. The schematic below (modified from Ramamurthy et al. (24)) illustrates that in wild type RetGC1 the active site binds two GTP molecules (i), RetGC1/GC1AC dimer utilizes GTP and ATP as substrates in the respective portions of the active site (ii), GC1AC rib(−)/GC1AC rib(−) homodimer is inactive because either subunit fails to coordinate the ribose moiety of the substrate (iii), and a mixed RetGC1/GC1AC rib(−) dimer cannot utilize GTP because the opposite subunit fails to coordinate its ribose moiety, but it has adenylyl cyclase activity because RetGC1 subunit coordinates the ribose of ATP recognized by GC1AC rib(−) (iv). The expected properties of the active sites in various cyclase dimers are summarized in Table 2. C, E925K/C997D/D929A substitutions and partial deletion of ECD do not block anchoring of GCAP1-GFP to the membranes by GC1AC rib(−) in HEK293 cells. GCAP1-GFP was co-expressed with the wild type RetGC1 (top panel), expressed without RetGC1 (middle panel), or co-expressed with GC1AC rib(−) (bottom panel). The GCAP1-GFP fluorescence was superimposed on differential interference contrast to help identify the nucleus versus the periphery of the cells. The right portion of each panel shows a representative fluorescence profile scanned across the cells marked with a yellow asterisk in the confocal image on the left. Horizontal scale, distance in μm; vertical scale, fluorescence intensity in arbitrary units (a.u.). Note how the diffuse pattern of the GCAP1-GFP in the absence of RetGC1 changes to the membrane-associated pattern in the presence of wild type or GC1AC rib(−) cyclase; the GCAP1 accumulates in the cyclase-containing membranes on the periphery and does not diffuse to the nucleus (41). D, complementation with M823R RetGC1 enables cAMP production by GC1AC rib(−). The membrane fractions were isolated from HEK293 cells transfected with GC1AC rib(−)-, M823R RetGC1-, or wild type RetGC1-expressing vectors or from cells co-transfected with two vectors as indicated. The membranes were reconstituted with 20 μm GCAP1 in the presence of <10 nm free [Ca2+] and 6 mm [Mg2+], and [32P]cAMP synthesis (mean ± S.E., n = 3) was measured using an adenylyl cyclase assay as described under “Experimental Procedures.” Note the sharp increase in cAMP synthesis when GC1AC rib(−) was co-expressed with either wild type or M823R RetGC1. Inset, Western immunoblotting of 7% SDS-polyacrylamide gel loaded with equal aliquots of the HEK293 membrane fractions from cells transfected with the indicated expression vectors and probed with a polyclonal antibody against the catalytic domain of human RetGC1 (37). Note the different sizes of RetGC1 versus GC1AC (▷). The filled arrows (▶) indicate the positions of β-galactosidase (116 kDa), phosphorylase b (97 kDa), bovine serum albumin (66 kDa), and ovalbumin (45 kDa). E, complementation with R822P RetGC1 enables cAMP production (mean ± S.E., n = 3) by GC1AC rib(−). The conditions of the experiment were similar to that in D except that, instead of M823R, GC1AC rib(−) was co-expressed with the R822P RetGC1. Inset, immunoblotting of the membrane fractions isolated from the cells transfected with the indicated expression vectors. F, complementation with GC1AC enhances guanylyl cyclase activity of the M823R RetGC1. The membranes from the transfected cells were reconstituted with 20 μm GCAP1 in the presence of <10 nm free [Ca2+] and 6 mm [Mg2+], and [32P]cGMP synthesis (mean ± S.E., n = 3) was measured using a standard guanylyl cyclase assay protocol described under “Experimental Procedures.” Inset, immunoblotting of the membrane fractions isolated from the cells transfected with the indicated expression vectors. Error bars are S.E.
The ability of various mutants to utilize GTP versus ATP as a substrate as summarized in Table 2 predicted that GC1AC rib(−) could not display adenylyl cyclase activity unless dimerized with a subunit harboring unchanged Asp929. Indeed, when expressed alone, neither wild type nor GC1AC rib(−) RetGC1 or M823R RetGC1 was able to produce cAMP in the presence of a high concentration of GCAP1 (Fig. 10D). However, when co-expressed with the GC1AC rib(−), the M823R and the wild type both promoted well detectable adenylyl cyclase activity, which could only become possible if they dimerized with GC1AC rib(−) and complemented its ATP-binding active site with the Asp929, the essential residue (Fig. 10A) that GC1AC rib(−) was lacking. In a similar experiment, the R822P RetGC1 was also able to rescue GC1AC rib(−) adenylyl cyclase activity (Fig. 10E).
TABLE 2.
Substrate(s) in active site formed by the cyclase dimer
Conversion of the substrate highlighted in bold to the respective cyclic monophosphate was tested (Fig. 10) to probe for the presence of the corresponding functional cyclase dimer.
| RetGC1 dimer (subunit A/subunit B) | Substrate in active site (subunit A/subunit B) |
|---|---|
| mOrangeRetGC1/mOrangeRetGC1a | GTP/GTPa |
| GC1AC/GC1ACb | ATP/ATPb |
| GC1AC rib(−)/GC1AC rib(−)c | Inactive/inactivec |
| mOrangeM823R/mOrangeM823Ra | GTP/GTPa |
| mOrangeR822P/mOrangeR822Pa | GTP/GTPa |
| mOrangeM823R/GC1AC rib(−)d | Inactive/ATPd,e |
| mOrangeR822P/GC1AC rib(−)d | Inactive/ATPd,e |
| mOrangeM823R/GC1ACf | GTP/ATPf |
| mOrangeRetGC1/GC1AC rib(−)d | Inactive/ATPd,e |
a Each subunit of RetGC1 in a dimer coordinates the purine base of GTP and provides coordination of ribose and Mg2+ for the second Mg2+GTP molecule in the active site (Fig. 10A) (28).
b RetGC1 with short ECD (37) was converted to adenylyl cyclase by the E925K/C997D substitutions as in Ref. 44.
c In addition to the E925K/C997D substitutions, the GC1AC also had the D929A mutation (24), disabling coordination of ribose and eliminating catalytic activity.
d The purine base of GTP is recognized by subunit A, but the opposite subunit B fails to coordinate ribose of the GTP, whereas ATP bound through the purine base by subunit B is held in a normal fashion by the Asp929 of subunit A (28).
In a reciprocal experiment, we co-expressed GC1AC with the M823R RetGC1. Membranes isolated from the transfected HEK293 cells were then reconstituted with purified GCAP1 but this time to measure GCAP-stimulated guanylyl cyclase activity (Fig. 10F). Although GC1AC alone could not utilize GTP as a substrate (Table 2 and Ref. 44) and the M823R RetGC1 alone had very low guanylyl cyclase activity due to its poor binding of GCAP1 (see Figs. 7 and 8), the two mutants when co-expressed displayed markedly enhanced cGMP synthesis. That would only be possible if the two mutated cyclases were able to form a dimer in which a portion of the active site (from the M823R RetGC1 subunit) was able to catalyze conversion of GTP, whereas the other subunit, GC1AC, contributed its unabated GCAP1 binding to the cyclase dimer.
Discussion
RetGC remains one of the most important but least understood photoreceptor enzymes in rod and cone physiology and disease in part because the data on its three-dimensional structure and organization of the quaternary complex with GCAPs are scarce. A model of the three-dimensional structure for the RetGC1 catalytic domain based on the homology with the known structure of adenylyl cyclase (28) gives a reliable description of its active site and has been directly verified by biochemical experiments (24, 44). However, no other structural data about the rest of the molecule, including the KHD and dimerization domain, is presently available. Likewise, the structure of the Ca2+-regulated RetGC1-GCAP complex has never been established primarily because the complex could not be stabilized in the presence of a detergent (47). Evidently, regulation of RetGC1 by GCAPs is accompanied by a change in the tightness of the dimer (49), and one can reasonably suggest that changes affecting the RetGC1 homodimer could make the catalysis in the active site more or less efficient by even subtle conformational shifts imparted by the Mg2+- versus Ca2+-bound GCAPs (50, 51). However, even the exact binding site(s) for GCAPs on RetGC1 has not been unequivocally established despite several prior attempts to assign them to a particular region(s) in the RetGC1 primary structure (32, 35, 52–55). The difficulties in mapping the binding interface for GCAPs exacerbated by the lack of reliable detection of the complex in detergent solution can be effectively mitigated by applying the in cyto assay in which the formation of the complex becomes visualized by confocal microscopy using fluorescently labeled GCAPs and RetGC1 (41). This approach was effective in the recent mapping of the RetGC1 binding interface in GCAP1 (42) and in the partial mapping of the GCAP binding interface in RetGC1 (27). The latter revealed that the binding interface for both GCAPs is located in the portion of the RetGC1 molecule that includes both the KHD and the dimerization domain (27). As the first step toward more detailed mapping of the interface, we found here that the dimerization domain strongly contributes to the interface. Although some of the earlier studies pointed at the KHD as the possible part of the interface (52, 54, 55), the dimerization domain as a regulatory element in RetGC-GCAP interactions was deemed irrelevant for GCAP2 in a publication describing that GCAP2, but not GCAP1, activated recombinant RetGC1 lacking the dimerization domain (32). We find it difficult to reconcile our findings with the results described in the cited publication because in our study GCAP1 and GCAP2 regulate RetGC in a similar fashion (56), and we show here that both lost the ability to activate the cyclase when its dimerization domain was affected. Our recent study rules out a possibility that the primary binding site for GCAP1 and GCAP2 operates outside the KHD and dimerization domain of the cyclase (27). Furthermore, our experiments presented here clearly demonstrate that GCAP1 and GCAP2 both failed to activate recombinant RetGC1 not only when the entire dimerization domain was replaced by that of a peptide receptor cyclase but even when just a single amino acid residue, Arg822 or Met823, was altered. Results of RetGC1 mutagenesis combined with the GCAP-dependent activation and in cyto GCAP binding assays conducted in our study strongly argue that the dimerization domain participates in creating and/or directly regulating the GCAP1 and -2 binding interface on RetGC1. The selectivity for GCAP binding enabled by the dimerization domain of RetGC1 in comparison with that of NPRA is attributable to the RetGC1-specific amino acid residues in different portions of the domain but most strongly depends on the presence the N-proximal portion of the domain containing Met823 (Figs. 5–8). This residue is evolutionarily conserved between different species (e.g. human, mouse, and bovine) in both RetGC1 and RetGC2 (GUCY2F) in contrast to hormone receptor cyclases GUCY2A (NPRA), GUCY2B, and GUCY2C where it becomes replaced by Arg, a residue that does not support binding of GCAPs (Figs. 7 and 8). This would be consistent with the unique regulatory properties of the photoreceptor RetGC isozymes as Ca2+/GCAP-regulated enzymes in contrast to the peptide hormone receptor guanylyl cyclase (27). The hydrophilic non-charged residue in this position of the dimerization domain is required for the cyclase to enable its contact with GCAPs (Figs. 7 and 8).
The RetGC1 DD and especially Met823 are more likely to be a part of GCAP binding interface than to affect it through the dimerization of the cyclase. First, MARCOIL prediction disfavors Met823 making a direct contact between the coiled coil structures in heptad I (Fig. 9). Second, computational analysis (Fig. 9) and functional complementation assays (Fig. 10) both demonstrated that the M823R mutation, although strongly disabling GCAP binding, did not preclude RetGC1 from dimerization.
Our finding that the dimerization domain is a part of the GCAP binding interface on the cyclase agrees with earlier observations that mutations of Arg838 (23–25) or Gln847 (57) causing dominant CORD6 shift Ca2+ sensitivity of RetGC1 regulation by GCAPs. There is, however, an important difference. In contrast to Arg822 and Met823, Arg838 is one of the residues in the corresponding heptad of the RetGC1 homodimer that make immediate contact between the two coils; hence, the CORD6 mutations alter the coiled coil structure itself (24, 57). Therefore, unlike the recessive LCA mutation in Arg822 (20), which breaks the interface for GCAP and blocks its binding to the cyclase, CORD6 mutations of Arg838 only alter the relative affinities for the Mg2+-liganded versus Ca2+-liganded GCAP1 (24, 25). One can speculate that the CORD6-related change of the coiled coil structure alters the accessibility of those residues in the upstream portion of the dimerization domain that contribute to the binding interface for GCAPs and/or directly affect it. The precise mechanism of RetGC1 activation by GCAPs presently remains unclear, but considering that (i) the dimerization domain evidently contributes to the GCAP binding interface, (ii) the overall packing of the cyclase dimer is sensitive to the presence of GCAP (49), (iii) GCAPs drastically increase Vmax of the GTP conversion to cGMP in the active site (1, 56), and (iv) the active site of the cyclase requires dimerization of two catalytic domains (24, 28, 44), it is conceivable that Mg2+-liganded GCAP bound to RetGC1 via a part of the dimerization domain increases the efficiency of catalysis in the active site by altering the coiled coil structure of the dimer and hence adjusting the orientation of the two catalytic subunits relative to each other.
The R822P mutation linked to LCA (20) also implicates the RetGC1 dimerization domain as an important regulatory region. Unlike Met823, the neighboring residue Arg822 does not define the specificity of GCAP1 binding in RetGC1 versus NPRA, but its replacement with Pro evidently prevents binding (Fig. 1) and subsequent activation of the cyclase by both GCAP1 and GCAP2. A likely explanation for this is that Pro822 despite its not being at the coiled coil contact partially destabilizes the structure of the α-helix at the first heptad of the domain (Fig. 9C).
As yet another intriguing observation from our experiments, M823R RetGC1 and R822P RetGC1, which both poorly bind GCAPs, were both able to sustain the GC1AC rib(−) activation by GCAP in the functional complementation experiments (Fig. 10, D and E). At present, it remains unclear whether one or two GCAP molecules are required to fully activate the cyclase. On the one hand, at saturation by GCAP, the complex is likely to contain two GCAP molecules per two cyclase subunits (30). However, it is presently impossible to conclude whether or not the ratio could be lowered to just one GCAP per complex in the complementation assays and still remain sufficient for activation of the cyclase dimer. On the other hand, because GCAPs themselves are able to dimerize (58), it cannot be excluded that the GCAP dimer is able to regulate the cyclase homodimer even when firmly anchored by only one of the RetGC1 subunits (provided that secondary interaction required for the cyclase regulation is preserved).
The R822P is an LCA-causing mutation (20) that blocks GCAP1 binding but does not disrupt binding of RD3 (Figs. 1 and 2). This argues that (i) R822P does not cause nonspecific unfolding of the cyclase but affects specifically the GCAP binding interface on the cyclase and (ii) the interfaces for the two types of regulator proteins on RetGC1 are not identical. However, there is a strong possibility that the two interfaces can overlap in a tertiary structure of the enzyme based on the following evidence. (i) RD3 and GCAPs have opposite and mutually exclusive effects on RetGC1 activity in recombinant HEK293 cells and in native photoreceptor membranes (11), and (ii) some LCA mutations, such as R768W, evidently block binding of both GCAP1 (30) and RD3 (59) altogether. It needs to be reiterated that the R822P substitution per se does not block RD3 binding. Hence, LCA in that case is unlikely to be a result of an impaired RetGC1 maturation or trafficking to the outer segment membranes, processes in which RD3 has been implicated (10, 59). Hence, in the case of R822P, the LCA it causes is likely a result of impaired activation and/or regulation of the mutated cyclase per se.
It also needs to be emphasized that although our study demonstrates that the cyclase dimerization domain is essential for the GCAP binding, this is only a part of the binding interface formed by the dimerization domain and KHD together (27). The KHD is also an indispensable part of that interface because relative affinities of GCAPs for RetGC1 and RetGC2 depend on the KHD (52) and because deletion and point mutations in KHD can completely disable GCAP binding by RetGC1 (20, 27, 30, 54, 55) even when the dimerization domain remains unaltered. We have verified in a separate experiment that RetGC1-specific sequence of the dimerization domain alone without the KHD could not turn NPRA into a GCAP-activated enzyme (data not shown).
To conclude, our study directly demonstrates that the dimerization domain of the photoreceptor guanylyl cyclase, especially its portion surrounding Met823, is a part of the GCAP binding interface and is therefore essential for the physiological regulation of the cyclase by Ca2+. When mutated, the portion of the interface containing the cyclase dimerization domain can cause LCA by altering normal activation of the enzyme by GCAPs (20). Another portion of the GCAP binding interface that includes the KHD represents a much larger portion of the RetGC1 primary structure than the dimerization domain. Therefore, detailed functional mapping of the residues in the KHD to identify their roles in forming the binding interface for GCAPs remains a major challenge for future studies.
Author Contributions
A. M. D. conceived and coordinated the study and wrote the paper. E. V. O. and A. M. D. constructed the expression vectors for the study. I. V. P performed guanylyl cyclase assays and transfection experiments. I. V. P. and A. M. D. analyzed the data and prepared figures for publication.
This work was supported, in whole or in part, by National Institutes of Health Grant EY11522 from the NEI. This work was also supported by a Commonwealth Universal Research Enhancement formula grant from the Pennsylvania Department of Health. The authors declare that they have no conflicts of interest with the contents of this article.
- RetGC
- retinal membrane guanylyl cyclase
- CORD6
- cone-rod degeneration 6
- GCAP
- guanylyl cyclase-activating protein
- KHD
- kinase homology domain
- LCA
- Leber congenital amaurosis
- PCC
- Pearson's correlation coefficient
- RD3
- retinal degeneration 3 protein
- NPR
- natriuretic peptide receptor
- ECD
- extracellular domain
- DD
- dimerization domain.
REFERENCES
- 1. Dizhoor A. M., Lowe D. G., Olshevskaya E. V., Laura R. P., Hurley J. B. (1994) The human photoreceptor membrane guanylyl cyclase, RetGC, is present in outer segments and is regulated by calcium and a soluble activator. Neuron 12, 1345–1352 [DOI] [PubMed] [Google Scholar]
- 2. Lowe D. G., Dizhoor A. M., Liu K., Gu Q., Spencer M., Laura R., Lu L., Hurley J. B. (1995) Cloning and expression of a second photoreceptor-specific membrane retina guanylyl cyclase (RetGC), RetGC-2. Proc. Natl. Acad. Sci. U.S.A. 92, 5535–5539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yang R. B., Foster D. C., Garbers D. L., Fülle H. J. (1995) Two membrane forms of guanylyl cyclase found in the eye. Proc. Natl. Acad. Sci. U.S.A. 92, 602–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Koch K. W., Stryer L. (1988) Highly cooperative feedback control of retinal rod guanylate cyclase by calcium ions. Nature 334, 64–66 [DOI] [PubMed] [Google Scholar]
- 5. Palczewski K., Subbaraya I., Gorczyca W. A., Helekar B. S., Ruiz C. C., Ohguro H., Huang J., Zhao X., Crabb J. W., Johnson R. S., Walsh K. A., Gray-Keller M. P., Detwiller P. B., Baehr W. (1994) Molecular cloning and characterization of retinal photoreceptor guanylyl cyclase-activating protein. Neuron 13, 395–404 [DOI] [PubMed] [Google Scholar]
- 6. Dizhoor A. M., Olshevskaya E. V., Henzel W. J., Wong S. C., Stults J. T., Ankoudinova I., Hurley J. B. (1995) Cloning, sequencing, and expression of a 24-kDa Ca2+-binding protein activating photoreceptor guanylyl cyclase. J. Biol. Chem. 270, 25200–25206 [DOI] [PubMed] [Google Scholar]
- 7. Imanishi Y., Yang L., Sokal I., Filipek S., Palczewski K., Baehr W. (2004) Diversity of guanylate cyclase-activating proteins (GCAPs) in teleost fish: characterization of three novel GCAPs (GCAP4, GCAP5, GCAP7) from zebrafish (Danio rerio) and prediction of eight GCAPs (GCAP1–8) in pufferfish (Fugu rubripes). J. Mol. Evol. 59, 204–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Scholten A., Koch K.-W. (2011) Differential calcium signaling by cone specific guanylate cyclase-activating proteins from the zebrafish retina. PLoS One 6, e23117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Friedman J. S., Chang B., Kannabiran C., Chakarova C., Singh H. P., Jalali S., Hawes N. L., Branham K., Othman M., Filippova E., Thompson D. A., Webster A. R., Andréasson S., Jacobson S. G., Bhattacharya S. S., Heckenlively J. R., Swaroop A. (2006) Premature truncation of a novel protein, RD3, exhibiting subnuclear localization is associated with retinal degeneration. Am. J. Hum. Genet. 79, 1059–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Azadi S., Molday L. L., Molday R. S. (2010) RD3, the protein associated with Leber congenital amaurosis type 12, is required for guanylate cyclase trafficking in photoreceptor cells. Proc. Natl. Acad. Sci. U.S.A. 107, 21158–21163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Peshenko I. V., Olshevskaya E. V., Azadi S., Molday L. L., Molday R. S., Dizhoor A. M. (2011) Retinal degeneration 3 (RD3) protein inhibits catalytic activity of retinal membrane guanylyl cyclase (RetGC) and its stimulation by activating proteins. Biochemistry 50, 9511–9519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dizhoor A. M., Olshevskaya E. V., Peshenko I. V. (2010) Mg2+/Ca2+ cation binding cycle of guanylyl cyclase activating proteins (GCAPs): role in regulation of photoreceptor guanylyl cyclase. Mol. Cell. Biochem. 334, 117–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Burns M. E., Mendez A., Chen J., Baylor D. A. (2002) Dynamics of cyclic GMP synthesis in retinal rods. Neuron 36, 81–91 [DOI] [PubMed] [Google Scholar]
- 14. Makino C. L., Peshenko I. V., Wen X. H., Olshevskaya E. V., Barrett R., Dizhoor A. M. (2008) A role for GCAP2 in regulating the photoresponse. Guanylyl cyclase activation and rod electrophysiology in GUCA1B knock-out mice. J. Biol. Chem. 283, 29135–29143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Makino C. L., Wen X. H., Olshevskaya E. V., Peshenko I. V., Savchenko A. B., Dizhoor A. M. (2012) Enzymatic relay mechanism stimulates cyclic GMP synthesis in rod photoresponse: biochemical and physiological study in guanylyl cyclase activating protein 1 knockout mice. PLoS One 7, e47637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sakurai K., Chen J., Kefalov V. J. (2011) Role of guanylyl cyclase modulation in mouse cone phototransduction. J. Neurosci. 31, 7991–8000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gray-Keller M. P., Detwiler P. B. (1994) The calcium feedback signal in the phototransduction cascade of vertebrate rods. Neuron 13, 849–861 [DOI] [PubMed] [Google Scholar]
- 18. Woodruff M. L., Sampath A. P., Matthews H. R., Krasnoperova N. V., Lem J., Fain G. L. (2002) Measurement of cytoplasmic calcium concentration in the rods of wild-type and transducin knock-out mice. J. Physiol. 542, 843–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Stone E. M. (2007) Leber congenital amaurosis—a model for efficient genetic testing of heterogeneous disorders: LXIV Edward Jackson Memorial Lecture. Am. J. Ophthalmol. 144, 791–811 [DOI] [PubMed] [Google Scholar]
- 20. Jacobson S. G., Cideciyan A. V., Peshenko I. V., Sumaroka A., Olshevskaya E. V., Cao L., Schwartz S. B., Roman A. J., Olivares M. B., Sadigh S., Yau K. W., Heon E., Stone E. M., Dizhoor A. M. (2013) Determining consequences of retinal membrane guanylyl cyclase (RetGC1) deficiency in human Leber congenital amaurosis en route to therapy: residual cone-photoreceptor vision correlates with biochemical properties of the mutants. Hum Mol Genet. 22, 168–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Perrault I., Rozet J. M., Calvas P., Gerber S., Camuzat A., Dollfus H., Châtelin S., Souied E., Ghazi I., Leowski C., Bonnemaison M., Le Paslier D., Frézal J., Dufier J. L., Pittler S., Munnich A., Kaplan J. (1996) Retinal-specific guanylate cyclase gene mutations in Leber's congenital amaurosis. Nat. Genet. 14, 461–464 [DOI] [PubMed] [Google Scholar]
- 22. Kelsell R. E., Gregory-Evans K., Payne A. M., Perrault I., Kaplan J., Yang R. B., Garbers D. L., Bird A. C., Moore A. T., Hunt D. M. (1998) Mutations in the retinal guanylate cyclase (RETGC-1) gene in dominant cone-rod dystrophy. Hum. Mol. Genet. 7, 1179–1184 [DOI] [PubMed] [Google Scholar]
- 23. Tucker C. L., Woodcock S. C., Kelsell R. E., Ramamurthy V., Hunt D. M., Hurley J. B. (1999) Biochemical analysis of a dimerization domain mutation in RetGC-1 associated with dominant cone-rod dystrophy. Proc. Natl. Acad. Sci. U.S.A. 96, 9039–9044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ramamurthy V., Tucker C., Wilkie S. E., Daggett V., Hunt D. M., Hurley J. B. (2001) Interactions within the coiled-coil domain of RetGC-1 guanylyl cyclase are optimized for regulation rather than for high affinity. J. Biol. Chem. 276, 26218–26229 [DOI] [PubMed] [Google Scholar]
- 25. Peshenko I. V., Moiseyev G. P., Olshevskaya E. V., Dizhoor A. M. (2004) Factors that determine Ca2+ sensitivity of photoreceptor guanylyl cyclase. Kinetic analysis of the interaction between the Ca2+-bound and the Ca2+-free guanylyl cyclase activating proteins (GCAPs) and recombinant photoreceptor guanylyl cyclase 1 (RetGC-1). Biochemistry 43, 13796–13804 [DOI] [PubMed] [Google Scholar]
- 26. Garbers D. L. (1999) The guanylyl cyclase receptors. Methods 19, 477–484 [DOI] [PubMed] [Google Scholar]
- 27. Peshenko I. V., Olshevskaya E. V., Dizhoor A. M. (2015) Evaluating the role of retinal membrane guanylyl cyclase 1 (RetGC1) domains in binding guanylyl cyclase-activating proteins (GCAPs). J. Biol. Chem. 290, 6913–6924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liu Y., Ruoho A. E., Rao V. D., Hurley J. H. (1997) Catalytic mechanism of the adenylyl and guanylyl cyclases: modeling and mutational analysis. Proc. Natl. Acad. Sci. U.S.A. 94, 13414–13419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yang R. B., Garbers D. L. (1997) Two eye guanylyl cyclases are expressed in the same photoreceptor cells and form homomers in preference to heteromers. J. Biol. Chem. 272, 13738–13742 [DOI] [PubMed] [Google Scholar]
- 30. Peshenko I. V., Olshevskaya E. V., Yao S., Ezzeldin H. H., Pittler S. J., Dizhoor A. M. (2010) Activation of retinal guanylyl cyclase RetGC1 by GCAP1: stoichiometry of binding and the effect of new LCA-related mutations. Biochemistry 49, 709–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Olshevskaya E. V., Peshenko I. V., Savchenko A. B., Dizhoor A. M. (2012) Retinal guanylyl cyclase isozyme 1 is the preferential in vivo target for constitutively active GCAP1 mutants causing congenital degeneration of photoreceptors. J. Neurosci. 32, 7208–7217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Duda T., Pertzev A., Sharma R. K. (2012) Differential Ca2+ sensor guanylate cyclase activating protein modes of photoreceptor rod outer segment membrane guanylate cyclase signaling. Biochemistry 51, 4650–4657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sharma R. K., Duda T. (2014) Membrane guanylate cyclase, a multimodal transduction machine: history, present, and future directions. Front. Mol. Neurosci. 7, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Duda T., Wen X. H., Isayama T., Sharma R. K., Makino C. L. (2015) Bicarbonate modulates photoreceptor guanylate cyclase (ROS-GC) catalytic activity. J. Biol. Chem. 290, 11052–11060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Duda T., Fik-Rymarkiewicz E., Venkataraman V., Krishnan R., Koch K. W., Sharma R. K. (2005) The calcium-sensor guanylate cyclase activating protein type 2 specific site in rod outer segment membrane guanylate cyclase type 1. Biochemistry. 44, 7336–7345 [DOI] [PubMed] [Google Scholar]
- 36. Horton R. M., Pease L. R. (1991) in Directed Mutagenesis. Practical Approach. (McPherson M. J., ed) pp. 217–250, Oxford University Press, Oxford, UK [Google Scholar]
- 37. Laura R. P., Dizhoor A. M., Hurley J. B. (1996) The membrane guanylyl cyclase, retinal guanylyl cyclase-1, is activated through its intracellular domain. J. Biol. Chem. 271, 11646–11651 [DOI] [PubMed] [Google Scholar]
- 38. Olshevskaya E. V., Hughes R. E., Hurley J. B., Dizhoor A. M. (1997) Calcium binding, but not a calcium-myristoyl switch, controls the ability of guanylyl cyclase-activating protein GCAP-2 to regulate photoreceptor guanylyl cyclase. J. Biol. Chem. 272, 14327–14333 [DOI] [PubMed] [Google Scholar]
- 39. Peshenko I. V., Olshevskaya E. V., Lim S., Ames J. B., Dizhoor A. M. (2012) Calcium-myristoyl Tug is a new mechanism for intramolecular tuning of calcium sensitivity and target enzyme interaction for guanylyl cyclase-activating protein 1: dynamic connection between N-fatty acyl group and EF-hand controls calcium sensitivity. J. Biol. Chem. 287, 13972–13984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Peshenko I. V., Olshevskaya E. V., Dizhoor A. M. (2012) Interaction of GCAP1 with retinal guanylyl cyclase and calcium: sensitivity to fatty acylation. Front. Mol. Neurosci. 5, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Peshenko I. V., Olshevskaya E. V., Dizhoor A. M. (2008) Binding of guanylyl cyclase activating protein 1 (GCAP1) to retinal guanylyl cyclase (RetGC1). The role of individual EF-hands. J. Biol. Chem. 283, 21747–21757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Peshenko I. V., Olshevskaya E. V., Lim S., Ames J. B., Dizhoor A. M. (2014) Identification of target binding site in photoreceptor guanylyl cyclase-activating protein 1 (GCAP1). J. Biol. Chem. 289, 10140–10154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Peshenko I. V., Dizhoor A. M. (2007) Activation and inhibition of photoreceptor guanylyl cyclase by guanylyl cyclase activating protein 1 (GCAP-1): the functional role of Mg2+/Ca2+ exchange in EF-hand domains. J. Biol. Chem. 282, 21645–21652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tucker C. L., Hurley J. H., Miller T. R., Hurley J. B. (1998) Two amino acid substitutions convert a guanylyl cyclase, RetGC-1, into an adenylyl cyclase. Proc. Natl. Acad. Sci. U.S.A. 95, 5993–5997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Delorenzi M., Speed T. (2002) An HMM model for coiled-coil domains and a comparison with PSSM-based predictions. Bioinformatics 18, 617–625 [DOI] [PubMed] [Google Scholar]
- 46. Lupas A., Van Dyke M., Stock J. (1991) Predicting coiled coils from protein sequences. Science 252, 1162–1164 [DOI] [PubMed] [Google Scholar]
- 47. Koch K. W. (1991) Purification and identification of photoreceptor guanylate cyclase. J. Biol. Chem. 266, 8634–8637 [PubMed] [Google Scholar]
- 48. Zinchuk V, Zinchuk O. (2008) Quantitative colocalization analysis of confocal fluorescence microscopy images. Curr. Protoc. Cell Biol. Chapter 4, Unit 4.19 [DOI] [PubMed] [Google Scholar]
- 49. Yu H., Olshevskaya E., Duda T., Seno K., Hayashi F., Sharma R. K., Dizhoor A. M., Yamazaki A. (1999) Activation of retinal guanylyl cyclase-1 by Ca2+-binding proteins involves its dimerization. J. Biol. Chem. 274, 15547–15555 [DOI] [PubMed] [Google Scholar]
- 50. Peshenko I. V., Dizhoor A. M. (2004) Guanylyl cyclase-activating proteins (GCAPs) are Ca2+/Mg2+ sensors: implications for photoreceptor guanylyl cyclase (RetGC) regulation in mammalian photoreceptors. J. Biol. Chem. 279, 16903–16906 [DOI] [PubMed] [Google Scholar]
- 51. Lim S., Peshenko I., Dizhoor A., Ames J. B. (2009) Effects of Ca2+, Mg2+, and myristoylation on guanylyl cyclase activating protein 1 structure and stability. Biochemistry 48, 850–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Laura R. P., Hurley J. B. (1998) The kinase homology domain of retinal guanylyl cyclases 1 and 2 specifies the affinity and cooperativity of interaction with guanylyl cyclase activating protein-2. Biochemistry 37, 11264–11271 [DOI] [PubMed] [Google Scholar]
- 53. Sokal I, Haeseleer F., Arendt A., Adman E. T., Hargrave P. A., Palczewski K. (1999) Identification of a guanylyl cyclase-activating protein-binding site within the catalytic domain of retinal guanylyl cyclase 1. Biochemistry 38, 1387–1393 [DOI] [PubMed] [Google Scholar]
- 54. Lange C., Duda T., Beyermann M., Sharma R. K., Koch K. W. (1999) Regions in vertebrate photoreceptor guanylyl cyclase ROS-GC1 involved in Ca2+-dependent regulation by guanylyl cyclase-activating protein GCAP-1. FEBS Lett. 460, 27–31 [DOI] [PubMed] [Google Scholar]
- 55. Krylov D. M., Hurley J. B. (2001) Identification of proximate regions in a complex of retinal guanylyl cyclase 1 and guanylyl cyclase-activating protein-1 by a novel mass spectrometry-based method. J. Biol. Chem. 276, 30648–30654 [DOI] [PubMed] [Google Scholar]
- 56. Peshenko I. V., Olshevskaya E. V., Savchenko A. B., Karan S., Palczewski K., Baehr W., Dizhoor A. M. (2011) Enzymatic properties and regulation of the native isozymes of retinal membrane guanylyl cyclase (RetGC) from mouse photoreceptors. Biochemistry 50, 5590–5600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zägel P., Dell'Orco D., Koch K. W. (2013) The dimerization domain in outer segment guanylate cyclase is a Ca2+-sensitive control switch module. Biochemistry 52, 5065–5074 [DOI] [PubMed] [Google Scholar]
- 58. Olshevskaya E. V., Ermilov A. N., Dizhoor A. M. (1999) Dimerization of guanylyl cyclase-activating protein and a mechanism of photoreceptor guanylyl cyclase activation. J. Biol. Chem. 274, 25583–25587 [DOI] [PubMed] [Google Scholar]
- 59. Zulliger R., Naash M. I., Rajala R. V., Molday R. S., Azadi S. (2015) Impaired association of retinal degeneration-3 with guanylate cyclase-1 and guanylate cyclase-activating protein-1 leads to Leber congenital amaurosis-1. J. Biol. Chem. 290, 3488–3499 [DOI] [PMC free article] [PubMed] [Google Scholar]