

Abstract
The existence of genetic variation for resistance in host populations is assumed to be essential to the spread of an emerging virus. Models predict that the rate of spread slows down with the increasing frequency and higher diversity of resistance alleles in the host population. We have been using the experimental pathosystem Arabidopsis thaliana—tobacco etch potyvirus (TEV) to explore the interplay between genetic variation in host's susceptibility and virus diversity. We have recently shown that TEV populations evolving in A. thaliana ecotypes that differ in susceptibility to infection gained within-host fitness, virulence and infectivity in a manner compatible with a gene-for-gene model of host–parasite interactions: hard-to-infect ecotypes were infected by generalist viruses, whereas easy-to-infect ecotypes were infected by every virus. We characterized the genomes of the evolved viruses and found cases of host-driven convergent mutations. To gain further insights in the mechanistic basis of this gene-for-gene model, we have generated all viral mutations individually as well as in specific combinations and tested their within-host fitness effects across ecotypes. Most of these mutations were deleterious or neutral in their local ecotype and only a very reduced number had a host-specific beneficial effect. We conclude that most of the mutations fixed during the evolution experiment were so by drift or by selective sweeps along with the selected driver mutation. In addition, we evaluated the ruggedness of the underlying adaptive fitness landscape and found that mutational effects were mostly multiplicative, with few cases of significant epistasis.
Keywords: adaptive mutations, epistasis, experimental evolution, host-range expansion, virus evolution, virus–plant interaction
1. Introduction
Characterizing the molecular basis of adaptation has been a central topic in modern evolutionary biology [1]. Compared with more complex organisms, this goal has proved to be relatively easy to achieve in RNA viruses owing to their small genomic sizes, which facilitates their full sequencing, the simplicity of the genotype-to-phenotype map, that allows performing functional analyses for many mutations, and the availability of reverse-genetic techniques to assess the effect of mutations individually or in given combinations. A common observation of such type of studies has been the identification of cases of convergent molecular evolution in independently evolved lineages [2–7]. This phenomenon is not only restricted to laboratory evolution experiments. It is also a relatively widespread observation among natural viral populations facing similar challenges, e.g. parallel changes are frequent in Human immunodeficiency virus type 1 clones isolated from patients treated with certain antiviral drugs, often following a common order of appearance [8–12]. Subsequent substitutions may confer increasing levels of drug resistance or, alternatively, may compensate for deleterious pleiotropic effects of earlier mutations [13–15]. Likewise, molecular convergences have been observed in the VPg cistron of Potato virus Y populations that acquired the ability to break the resistance conferred by the pvr2 resistance gene of pepper [16–18]. This gene encodes for the eIF(iso)4G, whose correct interaction with VPg is essential for completing the virus replication cycle [16–18]. Convergent evolution at the molecular level is not controversial as long as it can be reconciled with the neutralist or the selectionist theories. The neutral theory suggests that convergences are simply accidents, whereas within the framework of selectionism, there are two qualifications for convergences. The first explanation considers convergences as being adaptive and the result of organisms facing the same or similar environments with a few alternative pathways of adaptation, as expected for highly compacted genomes. Second, given large enough population size and mutation rate (i.e. several beneficial mutations coexisting at any given time point) to make clonal interference an important evolutionary factor, the same mutations are expected to be fixed and in a more or less predictable order [19]. Another consequence of these pervasive genomic convergences is that although RNA viruses often respond quickly to strong selective pressures, their long-term evolutionary plasticity might be less impressive.
In recent years, we have been interested in exploring the molecular basis of virus adaptation to hosts that differ in their susceptibility to infection. Host populations with low genetic diversity in resistance-related loci show higher infection prevalence than populations with great diversity [20]. Since viruses evolve much faster than their multicellular hosts, the chances of generating escape mutants while replicating in a permissive host genotype are high, hence challenging the viability of host populations formed by individuals resistant only to the wild-type virus [21]. As a matter of fact, this process leads to local adaptation of parasites, where they have higher fitness in their current local host but lower in any foreign alternative host [22,23]. Local adaptation to a particular host genotype reduces the odds of successful transmission to a different one, thus minimizing the chances of epidemic spread. Despite this effect, too much diversity in susceptibility will allow infection by a wider range of parasites [24]. In consequence, the long-term outcome of the interplay between host and virus populations depends on the degree of genetic diversity of both contenders. The interaction between host genotypes and parasite genotypes has been modelled in the context of two different approaches. On the one hand, there is the gene-for-gene (GFG) model, in which a parasite genotype can infect all host genotypes and a universally susceptible host genotype exists [25]. Resistance occurs when a host ‘resistance’ gene is matched by at least one parasite ‘avirulence’ gene. Polymorphism in infectivity and resistance can be maintained only if virulence pays a cost. Infection matrices are, hence, nested. On the other hand, the matching-allele (MA) model is based on self- versus non-self-recognition systems in invertebrates. Infection is only possible if the parasite possess all alleles that match those of the host [26]. In this case, polymorphisms in infectivity and resistance are maintained by negative frequency-dependent selection and infection matrices are modular.
To tackle the effect of host genetic diversity for susceptibility to infection on the genetic composition and evolutionary dynamics of viral populations, we have chosen a model pathosystem formed by Tobacco etch virus (TEV; genus Potyvirus, family Potyviridae) and the experimental host Arabidopsis thaliana L. [7,27–30]. A. thaliana ecotypes vary in susceptibility to TEV [31]: some allow long-distance movement from inoculated to non-inoculated leaves while other support replication in inoculated leaves but do not allow for systemic movement. Susceptibility depends on the Restricted TEV Movement (RTM) multigenic system composed of the RTM1, RTM2 and RTM3 loci [31–37]. The presence of dominant alleles at all three loci is necessary for resistance; homozygous recessive mutations at any of the three loci result in systemic infection [34,35]. Agudelo-Romero et al. [27] performed an evolution experiment in which TEV was adapted to the susceptible ecotype Ler-0 by serial passages. The ancestral TEV systemically infected Ler-0 plants, although the infection was asymptomatic. After 17 passages, the resulting strain, TEV-At17, fixed six point mutations, improved its accumulation ca 44-fold and induced severe symptoms. Comparative transcriptomics showed further differences between evolved and ancestral viruses: TEV-At17 downregulated developmental and metabolic processes, innate immunity and responses to abiotic stresses and to infection. Lalić et al. [29] showed that TEV-At17 systemically infected ecotypes that were resistant to the ancestral TEV. Furthermore, infectivity, viral load and severity of symptoms varied among ecotypes. Hillung et al. [30] compared the effect of TEV-At17b (a sequence variant of TEV-At17) infection on the transcriptome of the five ecotypes listed in table 1, finding differences in the way they perceived and responded to infection. The ecotypes could be classified into two groups according to their response to virus infection. In the first group, Ei-2 and Wt-1 upregulated genes involved in abiotic stresses and in the construction of new tissues: these ecotypes developed strong symptoms and, additionally, Wt-1 accumulated high viral loads. The second group (Ler-0, St-0 and Di-2) has mainly upregulated defence genes. Within this group, only Ler-0 developed severe symptoms as well as a very high viral load. St-0 and Di-2 developed mild symptoms, though Di-2 also accumulated high TEV-At17b loads. Finally, to evaluate the extent in which further evolution into a given plant ecotype (local host) conditioned the fitness of TEV-At17 derivatives into alternative ecotypes (foreign hosts), we performed the evolution experiments described by Hillung et al. [7] on the ecotypes listed in table 1. After 15 serial passages, we found that evolved viruses improved their within-host fitness, and showed higher infectivity and stronger virulence in their local hosts. We found that some ecotypes (e.g. St-0, Wt-1) were more permissive to infection than others (e.g. Ei-2, Di-2), and that viruses evolved in permissive hosts were more specialist than viruses evolved in more stringent hosts that became generalist, as predicted by a GFG model of infection.
Table 1.
List of A. thaliana ecotypes used in this study and the corresponding resistance phenotype according to the allelic combination at each RTM loci relative to the ancestral TEV.
| ecotype | origin | genotype | phenotype |
|---|---|---|---|
| Di-2 | France | RTM1/RTM1 RTM2/RTM2 RTM3/RTM3 | resistant |
| Wt-1 | Germany | RTM1/RTM1 RTM2/RTM2 RTM3/RTM3 | resistant |
| Ei-2 | Germany | rtm1/rtm1 RTM2/RTM2 RTM3/RTM3 | sensitive |
| Ler-0 | Germany | rtm1/rtm1 RTM2/RTM2 RTM3/RTM3 | sensitive |
| St-0 | Sweden | RTM1/RTM1 RTM2/RTM2 rtm3/rtm3 | sensitive |
The genome of evolved lineages was sequenced and we found that all lineages except Ler-0/2 contained at least one mutation, 43 being synonymous and 36 non-synonymous. Eight mutations appeared in multiple independent lineages (table 2). Three of these non-unique mutations were exclusive to Ei-2 lineages (C2116U, G3639A and G6420A). Mutation C795U was shared by St-0 lineages. Mutation G1272U was shared by Di-2 and St-0 lineages. Mutation A9240G was common to lineages Ler-0/1, St-0/1 and Wt-1/2. Very interestingly, these six convergent mutations were synonymous. Cases of convergent non-synonymous mutations were also observed: mutation C2912A was shared by lineages Di-2/1 and St-0/1, mutation C8636U was shared by all Di-2, all Ler-0 evolved lineages and by Wt-1/3, and lineages Ei-2/2 and Ei-2/3 each had a nucleotide substitution affecting the same codon at the CP cistron but resulting in different amino acid replacements. Convergent mutations are usually taken as an evidence of positive selection. Convergence at non-synonymous sites is usually explained as a consequence of identical selective pressures and the existence of limited accessible adaptive pathways. However, explaining convergences at synonymous sites is more problematic. Convergent synonymous mutations are a very common observation in evolving populations of RNA viruses [2,3,5,6,12] and studies analysing the mutational landscapes of RNA viruses have shown significant fitness effects associated to silent mutations [38–41]. Altogether, these observations support the notion that, at least for RNA genomes, equating synonymous substitution with neutral substitution is not always a valid premise. In our previous study, we provided some evidence supporting that selection for mRNA to protein translational efficiency might, in part, explain convergent synonymous mutations [7].
Table 2.
List of mutant TEV genotypes created for this study. All mutations were introduced in the TEV-At17b background [7].
| mutation | affected cistron | lineages in which mutations were observed |
|---|---|---|
| G1272U (synonymous) | HC-Pro | Di-2/1, Di-2/3, St-0/2, Wt-1/1 |
| C8636U (S2831L) | CP | Di-2/1, Di-2/2, Di-2/3, Ler-0/1, Ler-0/3, Wt-1/3 |
| A9240G (synonymous) | CP | Ler-0/1, St-0/2, Wt-1/2 |
| G1272U/C8636U | HC-Pro/CP | Di-2/1, Di-2/2, Di-2/3 |
| G1272U/A9240G | HC-Pro/CP | St-0/2 |
| C8636U/A9240G | CP/CP | Ler-0/1 |
| C2116U (synonymous) | HC-Pro | — |
| G3639A (synonymous) | CI | — |
| G6420A (synonymous) | NIaPro | — |
| C2116U/G3639A | HC-Pro/CI | — |
| C2116U/G6420A | HC-Pro/NIaPro | — |
| G3639A/G6420A | CI/NIaPro | — |
| C2116U/G3639A/G6420A | HC-Pro/CI/NIaPro | Ei-2/1, Ei-2/2, Ei-2/3 |
| C795U (synonymous) | P1 | St-0/1, St-0/3 |
In this article, we focus our attention on the analysis of the adaptive value of convergent mutations observed during the TEV-At17b adaptation to ecotypes of A. thaliana that differ in their susceptibility to infection. More specifically, we want to evaluate the fitness effects in local and foreign hosts of convergent mutations and of their combinations. Are convergent mutations only beneficial in the local host wherein they arose or do they confer a host-independent benefit? Do mutations appear together in the same genotype because they contribute epistatically to the fitness benefit? Likewise, are mutations that do not appear together involved in reciprocal sign epistasis? To tackle these questions, we selected a set of six convergent mutations found by Hillung et al. [7] and previously classified as adaptive mutations (table 2). Five of these mutations were synonymous substitutions while only one was non-synonymous. These mutations were chosen on the basis of their coexistence in certain genotypes and apparent incompatibility with others. In total, we have created 14 genotypes, seven containing single mutations, six containing pairwise combinations of these mutations and one triple mutant. Some of these combinations were observed in the evolved lineages (G1272U/C8636U, G1271U/A9240G, C8638U/A9240G and C2116U/G3639A/G6420A; table 2), while others were not so, though they may have a transient existence in the evolving viral populations prior to the time at which they were sampled (C2116U/G3639A, C2116U/G6420A and G3639A/G6420A; table 2). In particular, we created these non-observed combinations to explore the local topography of the adaptive fitness landscape and its dependence on the host ecotype (i.e. environment). For each genotype, we have evaluated within-host fitness in the local host as well as in the four alternative foreign hosts.
2. Material and methods
(a). Plant ecotypes
The five A. thaliana ecotypes listed in table 1 were chosen for this study. According to their genetic makeup at the RTM loci, Ei-2, Ler-0 and St-0 are susceptible to infection with the ancestral tobacco-isolated TEV, whereas Di-2 and Wt-1 are not. All were susceptible to the Arabidopsis-adapted TEV-At17b isolate [29,30]. Plants were maintained in a BSL-2 greenhouse at 16 L : 8 D and 24°C : 20°C day : night.
(b). Site-directed mutagenesis and production of infectious RNAs
The mutant genotypes listed in table 2 were generated by site-directed mutagenesis of a plasmid containing a cDNA of the TEV-At17b isolate, except synonymous substitution C6666U [7,30]. Mutagenesis was done using the PfuTurbo® DNA polymerase (Stratagene), as described in [42], and following the manufacturer's instructions using the pairs of mutagenic primers listed in electronic supplementary material, table S1. The uniqueness of each mutation was confirmed by sequencing an 800 bp fragment encompassing the mutated nucleotide. Sequencing was done by GenoScreen (Lille, France) using BIGDYE v. 3.1 in a 96-capillary ABI3730XL sequencing system (Applied Biosystems) and the most appropriated primers described in [27].
Infectious 5′-capped RNA of each genotype was obtained by in vitro transcription after BglII linearization of the corresponding plasmid as described in [42] and using the SP6 mMESSAGE mMACHINE kit (Ambion Inc.) following the manufacturer's instructions. All RNAs were inoculated into Nicotiana benthamiana Domin plants to produce infectious viral particles. Infected N. benthamiana leafs were collected, homogenized and their viral load was quantified as described in §2c.
(c). Virus genomic RNA purification and quantification
Symptomatic whole plants were collected, ground into fine powder using a mortar and a pestle in liquid N2 and stored at −80°C.
RNA extraction from 100 mg tissue per plant was performed using InviTrap® Spin Plant RNA Mini Kit (Invitek) following the manufacturer's instructions. The concentration of total plant RNA extracts was adjusted to 50 ng µl−1 for each sample and the quantification of viral load was done by absolute real-time RT-qPCR using an external standard [43] as described elsewhere [29]. Amplifications were done using an ABI StepOnePlus™ real-time PCR System (Applied Biosystems) as follows: 5 min at 42°C, 10 s at 95°C following 40 cycles of 5 s at 95°C and 34 s at 60°C. Quantifications were performed in triplicate for each sample. Quantification results were examined using StepOne software v. 2.2.2 (Applied Biosystems).
(d). Evaluation of within-host fitness and epistasis
Previously quantified sap from infected N. benthamiana was diluted to the same viral concentration in all samples with buffer containing 50 mM sodium borate (pH 8.0) and 5 mM EDTA and was used for inoculation of A. thaliana plants. Two leaves from 21 day old A. thaliana plants were rub-inoculated with 10 µl of infectious sap and 10% carborundum (100 mg ml−1). The inoculation of ecotypes was done in three temporal blocks, maintaining constant conditions of plant growth and inoculum. Between 1 and 10 (median 5) infected plants from each ecotype–mutant combination were collected 21 days post-inoculation (dpi) and processed as described in §2c. Total RNA was extracted and quantified by RT-qPCR as described in §2c.
A Malthusian growth rate per day was computed as m = 1/t log(Qt/Q0), where Qt is the number of TEV genomes/100 ng of total RNA quantified at t dpi by RT-qPCR. Relative within-host fitness was calculated as Wx,h = exp(mx,h − mTEV−At17b,h), where mx,h and mTEV−At17b,h are the Malthusian growth rates of the viral isolate x and of the TEV-At17b isolate (estimated for the corresponding experimental block), respectively, evaluated in the same host h. Within-host fitness was evaluated for each mutant on the five different A. thaliana ecotypes.
Epistasis among a pair of mutations x and y was evaluated as ɛxy,h = W00,hWxy,h − Wx0,hW0y,h, where W00,h, Wxy,h, Wx0,h, and W0y,h are, respectively, the fitness of the ancestral TEV-At17b, of the double mutant, and of each single mutant [44], all evaluated in host ecotype h. In those cases for which epistasis turned out to be significant, we further proceeded to determine its type: magnitude, sign or reciprocal sign. To do so, we have employed the inequality conditions derived by Poelwijk et al. [45].
All statistical analyses described in §3 have been done using IBM SPSS v. 21.
3. Results and discussion
(a). Exploration of within-host fitness data
Table 3 shows the within-host fitness values estimated for each genotype on each alternative host ecotype. For nine mutant genotypes, significant differences in within-host fitness have been observed among host ecotypes (Kruskal–Wallis test, p ≤ 0.046). However, by applying Fisher's combined probability test we can conclude that, overall, differences among host ecotypes exist for the entire dataset (χ2 = 107.167, 28 d.f., p < 0.001). It is interesting, however, to explore in more detail the five genotypes that performed equally well across all host ecotypes. Mutation G1272U was originally observed in lineages evolved in Di-2, St-0 and Wt-1; thus it likely had a host-independent fitness effect, though in all cases the effect was small and not significant compared to the performance of the TEV-At17b ancestral genotype on each host (Mann–Whitney test, p ≥ 0.05 in all cases). This is consistent with our previous observation that St-0 and Wt-1 were the most permissive hosts that selected for more specialist viruses [7]. Double-mutant G1272U/A9240G, detected in lineage St-0/2, had a significant deleterious effect in all ecotypes except in the foreign host Wt-1, thus suggesting that it swept to high frequency in lineage St-0/2 either because it was linked to other beneficial mutations or, alternatively, because it was fixed throughout a strong bottleneck event. The other three genotypes with homogeneous effects across hosts were C2116U, C2116U/G6420A and G3639A/G6420A. None of these genotypes was observed in the evolving lineages but were created ex profeso to analyse the epistasis among these three mutations in the triple mutant genotype C2116U/G3639A/G6420A observed in all Ei-2 lineages.
Table 3.
Relative within-host fitness of each viral genotype measured on every A. thaliana ecotype. Values are reported as average ±1 s.e.m. (number of replicates). Asterisks indicate cases significantly different from TEV-At17b (Mann–Whitney test; FDR correction for multiple tests). Shadowed cells indicate the local host in which the mutation was detected. Last column shows the p-value of Kruskal–Wallis tests for differences in relative within-host fitness among ecotypes for each viral genotype.
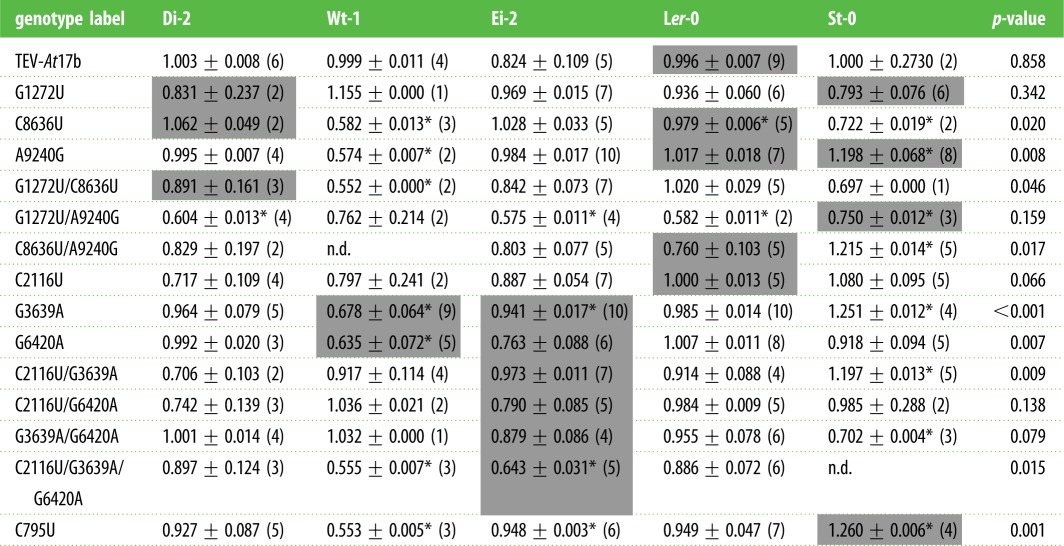 |
Three mutations provide host-specific within-host fitness advantages. Mutation C8636U, observed in four host ecotypes (table 2), only provided a significant beneficial effect in the non-permissive ecotype Ler-0. Mutation A9240G, identified in the most permissive host ecotypes (table 2), provided a significant beneficial effect only in St-0 and was significantly deleterious in Wt-1 (table 4). Mutation C795U, observed in two of the St-0 lineages (table 2), conferred a significant beneficial effect in St-0 and was significantly deleterious in Ei-2 and Wt-1 (table 3).
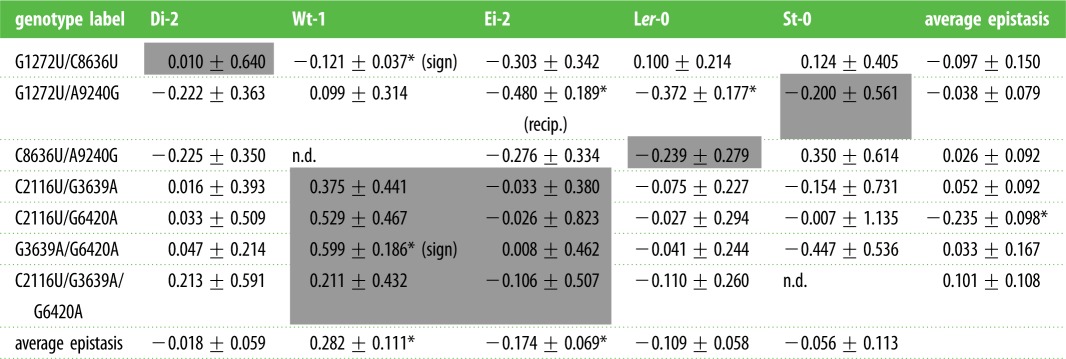
Table 4.
Epistasis coefficients (±1 s.d.) evaluated for all double and triple mutants shown in figure 2. Significant cases are indicated with asterisks (z-test; FDR correction for multiple tests). Shadowed cells indicate the local host in which the mutation was detected. Sign epistasis refers to cases in which the sign of the fitness effect depends on the genetic background. Reciprocal (recip.) sign epistasis means that the sign of the fitness effect of a mutation is conditional upon the state of another locus and vice versa. Last row shows the significance test for the average epistasis (±1 s.e.m.) among genotypes and the last column shows the significance test for average epistasis among hosts (t-test).
 |
(b). Within-host fitness effects are greater in local than in foreign hosts
The above discussion (§3a) shows a complex interaction between the within-host fitness of each TEV genotype and the ecotype in which it is evaluated. If TEV genotypes were as well adapted to their local hosts as to the average foreign host, we would expect a positive correlation between these two measures of within-host fitness. Furthermore, in the hypothetical case of a perfect match in fitness among local and foreign hosts, the slope of the linear regression of fitness in the average foreign host on the local host(s) would be equal to unity. Deviations from the unity slope can be taken as indicative of fitness effects depending on the ecotype wherein they have been evaluated. A flatter-than-unity slope would indicate that the stronger the effects on the local host, the disproportionally weaker they may be in the foreign ones, thus suggesting that mutations contribute to specialism rather than to generalism. By contrast, a steeper-than-unity slope would indicate that the stronger the effect in the local host, the disproportionally stronger in the foreign ones they may be. To test this hypothesis, we first computed the median within-host fitness across all foreign hosts. In those cases where more than one local host existed, we also computed the median fitness. Figure 1 shows the relationship between within-host fitness in local and foreign hosts. The solid line represents the above null hypothesis of identical fitness effects across hosts. The dashed line corresponds to the actual best linear regression model. The slope of this regression equation is 0.177 ± 0.199 (±1 s.e.m.), a value that is significantly smaller than the null expectation of slope unity (t = −4.116, 13 d.f., p = 0.001).
Figure 1.
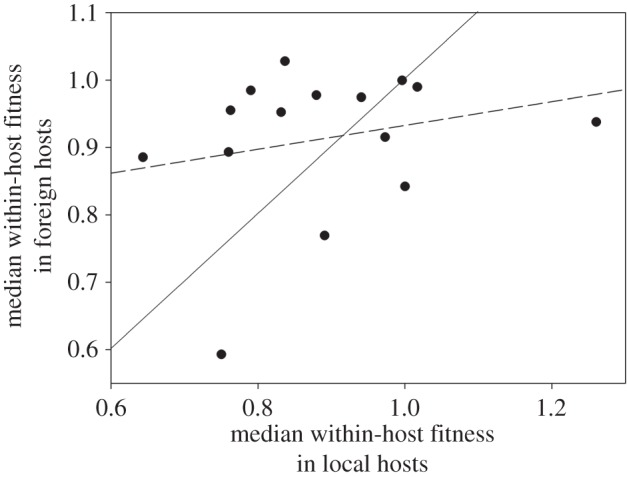
Relationship between within-host fitness in the local host and the median within-host fitness across all foreign hosts. The solid line represents the null hypothesis of equal effects in all hosts. The dashed line represents the linear model that best fits the data.
Figure 1 also shows that genotypes can be classified into two categories with respect to the null expectation (solid line). For the eight genotypes above the line, fitness effects on the average foreign host are greater than expected given their effect on the local host ecotype. For the six genotypes below the line, fitness effects on the average foreign host are smaller than expected relative to their value on the local host ecotype. After thoughtful consideration of the properties of the genotypes at each side of the diagonal, we discovered that those below the diagonal were mostly obtained in the most permissive host ecotypes (as defined in [7]: Ler-0, St-0 and Wt-1; table 2: six out of eight instances), whereas the reverse is true for those genotypes above the diagonal (Di-2 and Ei-2; table 2: four out of 16 cases). Indeed, a Fisher's exact test shows that this difference is significant (p = 0.032). Therefore, we provide further support to the conclusion of Hillung et al. [7] that more permissive hosts have selected for viral genotypes that are highly specialized for infecting their local host yet poorly capable of infecting more restrictive foreign hosts. By contrast, restrictive local hosts have selected for viral genotypes that are generalists successfully infecting every type of foreign host ecotype. This nested pattern of interactions is predicted by a GFG model of virus–plant interactions [25,46]. Nested infection networks are expected if a hierarchy of resistance among hosts and infection ability among viruses exists [46]. A GFG mechanism implies that mutations increasing fitness in the new local host exist that do not pay a fitness cost in Ler-0; thus the set of hosts that an isolate can infect are subsets of each other.
Very few experiments have tested whether the effects of adaptive mutations remain beneficial across a set of different environments or are environment-specific. In a pioneering study, Ostrowski et al. [47] found that mutations fixed in Escherichia coli lineages adapted to glucose as the only carbon source were also beneficial in five other carbon sources, concluding that positive pleiotropism was a norm for the bacterium and that local adaptation would not limit future evolution. In sharp contrast, evolution experiments with RNA viruses have shown that mutations expanding host range generally come with a cost in the original host [6,7,23,48–51], concluding that negative pleiotropism shall be the norm for these microparasites. The results reported here expand our previous observations [7,23,51] and give further support to this conclusion.
(c). Patterns of epistasis and the ruggedness of adaptive landscapes
Owing to their compact genomes and overlapping reading frames, epistasis is pervasive in RNA viruses (reviewed in [52]). The direction, magnitude and prevalence of epistasis is central to understanding many properties of virus genetic systems, such as reassortment of segments and recombination [53], complementation during co-infection [54], ploidy [55], phenotypic plasticity [56], robustness [57], the ruggedness of adaptive landscapes [45], or attempting to mechanistically explain dynamic processes such as the accumulation of mutations in finite populations upon transmission bottlenecks [58], and ultimately the origin of new viral species by an interruption of genetic exchange among populations [59].
In our previous work [7], we made two observations that pointed towards a possible role of epistasis and its dependence on the host ecotype. First, we found that mutations G1272U, C8636U and A9240G were common to lineages evolved in all ecotypes except Ei-2 (table 2), but their pairwise combinations were always exclusive to a given host ecotype (table 2): G1272U/C8636 in all Di-2-evolved lineages, G1272U/A9240G in lineage St-0/2 and C8636U/A9240G in lineage Ler-0/1. The second interesting observation involved mutations C2116U, G3639A and G6420A, their three pairwise combinations and the triple mutant. While the triple mutant was observed in all Ei-2-evolved lineages, the intermediate genotypes were observed neither in Ei-2 nor in other ecotypes. Henceforth, the observed sets of co-occurring mutations suggest that epistatic interactions played a prominent role in the genetic architecture of host adaptation. To assess this hypothesis, and more importantly the dependence of epistasis on the host ecotype, we constructed all the mutants shown in the genotypic network shown in figure 2. Table 4 shows all the epistasis values estimated. Looking at individual genotypes (rows in table 4), we found great variation in the magnitude and sign of epistasis: not a single combination of mutations had positive or negative epistasis across all host ecotypes. On average, only C2116U/G6420A had a significant negative epistasis across host ecotypes. Moving to plant ecotypes (columns in table 4), the situation is similar: in all ecotypes we found cases of positive and negative epistasis in TEV. In Ei-2 and Ler-0, all but one case were of negative epistasis; by contrast, in Wt-1 positive epistasis was the most common. Finally, looking at individual combinations of mutations, only four cases have rendered significant epistasis. The combination of mutations G1272U/C8636U had a significantly negative epistasis coefficient in the foreign host Wt-1. The combination of mutations G1272U/A9240G was involved in significant negative epistatic interactions in the foreign hosts Ei-2 and Ler-0. Finally, the combination of mutations C3639A/G6420A was involved in significant positive epistasis in the foreign host Wt-1. In all other cases, we failed to detect a significant deviation from the null hypothesis of multiplicative fitness effects.
Figure 2.
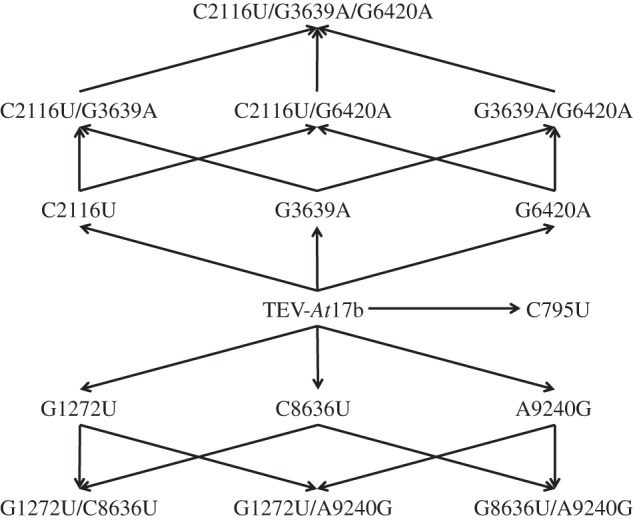
Genotypic network connecting the different mutants used in this study and for which fitness and epistasis have been evaluated across five host ecotypes. Arrows connect genotypes that are one mutational step away.
The significant negative interactions between genotypes G1272U/C8636U and C3639A/G6420A were of the sign epistasis type in Wt-1, thus suggesting the existence of local perturbations in the vicinity of these mutations in an otherwise smooth fitness landscape, although these perturbations are not strong enough as to create a deep valley. It is interesting that two out of two significant cases of sign epistasis reported are for the Wt-1 ecotype, suggesting that this host may impose some constraints that generate a certain degree of ruggedness in the adaptive landscape. The shape of the interaction between mutations G1272U/A9240G depends on the host ecotype where it is evaluated. It is of the reciprocal sign epistasis type in Ei-2, thus depicting a maximally rugged local surface around these mutations, with the existence of two local peaks; this may explain why this double mutant was never observed in Ei-2-evolved lineages. By contrast, this pair of mutations shows magnitude epistasis when evaluated in Ler-0, indicating a downward change in the local curvature of the fitness surface in the vicinity of these mutations but without creating local peaks.
All together, these analyses suggest that the topography of the fitness landscapes determining the evolution of TEV-At17b is mostly smooth, although some ridges and valleys arise in Wt-1 and Ei-2, respectively. This dependence of virus landscape topology on the environmental conditions has been previously reported for TEV [60] and ssDNA bacteriophage ID11 [61]. Therefore, rather than thinking in a fix fitness surface, we should better consider a highly dynamic and flexible adaptive surface: the seascape [62].
(d). Some concluding remarks
This theme issue seeks to provide fresh insights into the within-host dynamics of parasites and illustrate how ecology and evolution may contribute to better understanding infection. From an evolutionary perspective, within-host RNA virus population dynamics result from the complex interplay of multiple factors: mutation and recombination rates that generate new strains, competition or complementation between these strains, their interplay with the host's defences, etc. Differences in fitness among strains rely on the core of these complex interactions and on the outcome of the infection. The motivation for this study was to evaluate the effect that putative beneficial mutations may have on the within-host fitness of evolving TEV-At17b populations; especially the effect of synonymous mutations or combinations of them. To our surprise, we found that most of these mutations were deleterious or neutral in their local hosts and only a very low number had a host-specific beneficial effect. Therefore, we conclude that most of the mutations fixed during the evolution experiment were so by drift or by selective sweeps along with the selected driver beneficial mutation.
To evaluate the combined effect of several mutations, we constructed two empirical fitness landscapes. We found that the sign and magnitude of epistasis was variable among mutations and among hosts, in agreement with previous observations made with TEV [60,63]. However, an important difference between these previous studies and the results reported here must be brought forward: in previous studies with random mutations, average epistasis was positive and pervasively of reciprocal sign. In this study, mutational effects were mostly multiplicative, with very few instances of significant epistasis. Indeed, the two significant cases of sign epistasis (G1272U/A9240G measured in Ler-0 and G1272U/C8636U evaluated in Wt-1) involved a beneficial and a deleterious mutation and rendered a deleterious genotype. In these two cases, epistasis excluded the spontaneous evolution of these genotypes within the Ler-0 and Wt-1 ecotypes, since after first fixation of the beneficial mutation, the second deleterious mutation will necessarily generate low fitness individuals that will be outcompeted during within-host competition.
Following the definitions given in Remold [64], mutations G1272U and C2116U have no pleiotropic effect, as they produced no differences in within-host fitness across ecotypes. All other five mutations had a sign pleiotropic effect, i.e. the sign of a mutation's effect on within-host fitness depends on the actual ecotype. Likewise, most of the multiple mutants analysed classify under the non-epistatic sign pleiotropy category, which always results in an obligatory cost of generalism [64]. Double-mutant C2116U/G6420A belongs to the non-epistatic no pleiotropy category, which always results in no-cost generalism [64]. Finally, mutations G1272U/A9240G and G1272U/C8636U are of the sign epistatic sign pleiotropy case. Under this scenario, populations may achieve either specialism or no-cost generalism, depending on the ecotype in which they evolved [64]. A scenario in which, depending on the ecotype wherein viral populations evolve, selection may favour genetic combination producing no-cost generalists, costly generalists or even specialists, explains the evolution of a GFG model of virus–plant interaction.
In conclusion, this study illustrates the necessity of performing reverse-genetic and functional analyses of mutations observed in evolving viral populations before concluding they are effectively beneficial and responsible for driving the observed phenotypic changes.
Supplementary Material
Acknowledgements
We thank Francisca de la Iglesia and Paula Agudo for excellent technical assistance, and our laboratory mates for productive discussions and useful suggestions.
Data accessibility
All data necessary to reproduce the analyses reported in this article have been presented in tables 3 and 4.
Authors' contributions
J.H. performed all the experiments described in this study. J.M.C. participated in experimental design and supervised part of the experimental work. S.F.E. conceived the study, supervised the experimental work, analysed the data and wrote the manuscript. All authors read and approved the final version of the article.
Competing interests
We have no competing interests.
Funding
This work was supported by grant BFU2012-30805 from Spain Ministry of Economy and Competitiveness (MINECO), grant PROMETEOII/2014/021 from Generalitat Valenciana, the European Commission 7th Framework Programme (FP7-ICT-2013.9.6 FET Proactive: Evolving Living Technologies) EvoEvo project and the Santa Fe Institute to S.F.E. J.M.C. was supported by a JAE-doc postdoctoral contract from CSIC. J.H. was recipient of a predoctoral contract from MINECO.
References
- 1.Lewontin R. 2000. Population genetics: problems, foundations, and historical perspectives. In Evolutionary genetics: from molecules to morphology (eds Krimbas CB, Singh RS.), pp. 5–23. Cambridge, UK: Cambridge University Press. [Google Scholar]
- 2.Bull JJ, Badgett MR, Wichman HA, Huelsenbeck JP, Hillis DM, Gulati A, Ho C, Molineux IJ. 1997. Exceptional convergent evolution in a virus. Genetics 147, 1497–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wichman HA, Badgett MR, Scott LA, Boulianne CM, Bull JJ. 1999. Different trajectories of parallel evolution during viral adaptation. Science 285, 422–424. ( 10.1126/science.285.5426.422) [DOI] [PubMed] [Google Scholar]
- 4.Fares MA, Moya A, Escarmís C, Baranowski E, Domingo E, Barrio E. 2001. Evidence for positive selection in the capsid protein-coding region of the foot-and-mouth disease virus (FMDV) subjected to experimental passage regimens. Mol. Biol. Evol. 18, 10–21. ( 10.1093/oxfordjournals.molbev.a003715) [DOI] [PubMed] [Google Scholar]
- 5.Cuevas JM, Elena SF, Moya A. 2002. Molecular basis of adaptive convergence in experimental populations of RNA viruses. Genetics 162, 533–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Remold SK, Rambaut A, Turner PE. 2008. Evolutionary genomics of host adaptation in vesicular stomatitis virus. Mol. Biol. Evol. 25, 1138–1147. ( 10.1093/molbev/msn059) [DOI] [PubMed] [Google Scholar]
- 7.Hillung J, Cuevas JM, Valverde S, Elena SF. 2014. Experimental evolution of an emerging plant virus in host genotypes that differ in their susceptibility to infection. Evolution 9, 2467–2480. ( 10.1111/evo.12458) [DOI] [PubMed] [Google Scholar]
- 8.Larder BA, Coates KE, Kemp SD. 1991. Zidovudine-resistant human immunodeficiency virus selected by passage in cell culture. J. Virol. 65, 5232–5236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boucher CAB, O'Sullivan E, Mulder JW, Ramautarsing C, Kellam P, Darby G, Lange JMA, Goudsmit J, Larder BA. 1992. Ordered appearance of zidovudine resistance mutations during treatment of 18 human immunodeficiency virus-positive subjects. J. Infect. Dis. 165, 105–110. ( 10.1093/infdis/165.1.105) [DOI] [PubMed] [Google Scholar]
- 10.Kellam P, Boucher CAB, Tijnagel JMGH, Larder BA. 1994. Zidovudine treatment results in the selection of human immunodeficiency virus type 1 variants whose genotypes confer increasing levels of drug resistance. J. Gen. Virol. 75, 341–351. ( 10.1099/0022-1317-75-2-341) [DOI] [PubMed] [Google Scholar]
- 11.Condra JH, et al. 1996. Genetic correlates of in vivo viral resistance to indinavir, a human immunodeficiency virus type 1 protease inhibitor. J. Virol. 70, 8270–8276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martínez-Picado J, et al. 2000. Antiretroviral resistance during successful therapy of HIV type 1 infection. Proc. Natl Acad. Sci. USA 97, 10 948–10 953. ( 10.1073/pnas.97.20.10948) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Molla A, et al. 1996. Ordered accumulation of mutations in HIV protease confers resistance to ritonavir. Nat. Med. 2, 760–766. ( 10.1038/nm0796-760) [DOI] [PubMed] [Google Scholar]
- 14.Martínez-Picado J, Savara AV, Sutton L, D'Aquila RT. 1999. Replicative fitness of protease inhibitor-resistant mutants of Human immunodeficiency virus type 1. J. Virol. 73, 3744–3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nijhuis M, Schuurman R, de Jong D, Rickson EJ, Gustchina E, Albert J, Schipper P, Gulnik S, Boucher CA. 1999. Increased fitness of drug resistant HIV-1 protease as a result of acquisition of compensatory mutations during suboptimal therapy. AIDS 13, 2349–2359. ( 10.1097/00002030-199912030-00006) [DOI] [PubMed] [Google Scholar]
- 16.Ayme V, Petit-Pierre J, Souche S, Palloix A, Moury B. 2007. Molecular dissection of the potato virus Y VPg virulence factor reveals complex adaptations to the pvr2 resistance allelic series in pepper. J. Gen. Virol. 88, 1594–1601. ( 10.1099/vir.0.82702-0) [DOI] [PubMed] [Google Scholar]
- 17.Charron C, Nicolaï M, Gallois JL, Robaglia C, Moury B, Palloix A, Caranta C. 2008. Natural variation and functional analyses provide evidence for coevolution between plant eIF4E and potyviral VPg. Plant J. 54, 56–68. ( 10.1111/j.1365-313X.2008.03407.x) [DOI] [PubMed] [Google Scholar]
- 18.Moury B, Charron C, Janzac B, Simon V, Gallois JL, Palloix A, Caranta C. 2014. Evolution of plant eukaryotic initiation factor 4E (eIF4E) and Potyvirus genome-linked viral protein (VPg): a game of mirrors impacting resistance spectrum and durability. Infect. Genet. Evol. 27, 472–480. ( 10.1016/j.meegid.2013.11.024) [DOI] [PubMed] [Google Scholar]
- 19.Gerrish PJ, Lenski R. 1998. The fate of competing beneficial mutations in an asexual population. Genetica 102, 127–144. ( 10.1023/A:1017067816551) [DOI] [PubMed] [Google Scholar]
- 20.Altermatt F, Ebert D. 2008. Genetic diversity of Daphnia magna populations enhances resistance to parasites. Ecol. Lett. 11, 918–928. ( 10.1111/j.1461-0248.2008.01203.x) [DOI] [PubMed] [Google Scholar]
- 21.Martínez F, Lafforgue G, Morelli MJ, González-Candelas F, Chua NH, Daròs JA, Elena SF. 2012. Ultradeep sequencing analysis of population dynamics of virus escape mutants in RNAi-mediated resistant plants. Mol. Biol. Evol. 29, 3297–3307. ( 10.1093/molbev/mss135) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kaltz O, Shykoff JA. 1998. Local adaptation in host-parasite systems. Heredity 81, 361–370. ( 10.1046/j.1365-2540.1998.00435.x) [DOI] [Google Scholar]
- 23.Bedhomme S, Lafforgue G, Elena SF. 2012. Multihost experimental evolution of a plant RNA virus reveals local adaptation and host-specific mutations. Mol. Biol. Evol. 29, 1481–1492. ( 10.1093/molbev/msr314) [DOI] [PubMed] [Google Scholar]
- 24.Van Baalen M, Beekman M. 2006. The costs and benefits of genetic heterogeneity in resistance against parasites in social insects. Am. Nat. 167, 568–577. ( 10.1086/501169) [DOI] [PubMed] [Google Scholar]
- 25.Flor HH. 1956. The complementary genetic systems in flax and flax rush. Adv. Genet. 8, 29–54. ( 10.1016/S0065-2660(08)60498-8) [DOI] [Google Scholar]
- 26.Frank SA. 1993. Specificity versus detectable polymorphism in host–parasite genetics. Proc. R. Soc. Lond. B 254, 191–197. ( 10.1098/rspb.1993.0145) [DOI] [PubMed] [Google Scholar]
- 27.Agudelo-Romero P, Carbonell P, Pérez-Amador MA, Elena SF. 2008. Virus adaptation by manipulation of host's gene expression. PLoS ONE 3, e2397 ( 10.1371/journal.pone.0002397) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Agudelo-Romero P, Carbonell P, de la Iglesia F, Carrera J, Rodrigo G, Jaramillo A, Pérez-Amador MA, Elena SF. 2008. Changes in gene expression profile of Arabidopsis thaliana after infection with tobacco etch virus. Virol. J. 5, 92 ( 10.1186/1743-422X-5-92) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lalić J, Agudelo-Romero P, Carrasco P, Elena SF. 2010. Adaptation of tobacco etch potyvirus to a susceptible ecotype of Arabidopsis thaliana capacitates it for systemic infection of resistant ecotypes. Phil. Trans. R. Soc. B 65, 1997–2008. ( 10.1098/rstb.2010.0044) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hillung J, Cuevas JM, Elena SF. 2012. Transcript profiling of different Arabidopsis thaliana ecotypes in response to tobacco etch potyvirus infection. Front. Microbiol. 3, 229 ( 10.3389/fmicb.2012.00229) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mahajan SK, Chisholm ST, Whitham SA, Carrington JC. 1998. Identification and characterization of a locus (RTM1) that restricts long-distance movement of tobacco etch virus in Arabidopsis thaliana. Plant J. 14, 177–186. ( 10.1046/j.1365-313X.1998.00105.x) [DOI] [PubMed] [Google Scholar]
- 32.Whitham SA, Yamamoto ML, Carrington JC. 1999. Selectable viruses and altered susceptibility mutants in Arabidopsis thaliana. Proc. Natl Acad. Sci. USA 96, 772–777. ( 10.1073/pnas.96.2.772) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Whitham SA, Anderberg RJ, Chisholm ST, Carrington JC. 2000. Arabidopsis RTM2 gene is necessary for specific restriction of tobacco etch virus and encodes an unusual small heat shock-like protein. Plant Cell 12, 569–582. ( 10.1105/tpc.12.4.569) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chisholm ST, Mahajan SK, Whitham SA, Yamamoto ML, Carrington JC. 2000. Cloning of the Arabidopsis RTM1 gene, which controls restriction of long-distance movement of tobacco etch virus. Proc. Natl Acad. Sci. USA 97, 489–494. ( 10.1073/pnas.97.1.489) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chisholm ST, Parra MA, Anderberg RJ, Carrington JC. 2001. Arabidopsis RTM1 and RTM2 genes function in phloem to restrict long-distance movement of tobacco etch virus. Plant Physiol. 127, 1667–1675. ( 10.1104/pp.010479) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cosson P, et al. 2010. RTM3, which controls long-distance movement of potyviruses, is a member of a new plant gene family encoding a MEPRIN and TRAF homology domain-containing protein. Plant Physiol. 154, 222–232. ( 10.1104/pp.110.155754) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cosson P, Sofer L, Schurdi-Levraud V, Revers F. 2010. A member of a new plant gene family encoding a MEPRIN and TRAF homology (MATH) domain-containing protein is involved in restriction of long distance movement of plant viruses. Plant Signal. Behav. 5, 1321–1323. ( 10.4161/psb.5.10.13244) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sanjuán R, Moya A, Elena SF. 2004. The distribution of fitness effects caused by single-nucleotide substitutions in an RNA virus. Proc. Natl Acad. Sci. USA 101, 8396–8401. ( 10.1073/pnas.0400146101) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Carrasco P, de la Iglesia F, Elena SF. 2007. Distribution of fitness and virulence effects caused by single-nucleotide substitutions in tobacco etch virus. J. Virol. 81, 12 979–12 984. ( 10.1128/JVI.00524-07) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cuevas JM, Domingo-Calap P, Sanjuán R. 2012. The fitness effects of synonymous mutations in DNA and RNA viruses. Mol. Biol. Evol. 29, 17–20. ( 10.1093/molbev/msr179) [DOI] [PubMed] [Google Scholar]
- 41.Acevedo A, Brodsky L, Andino R. 2013. Mutational and fitness landscapes of an RNA virus revealed through population sequencing. Nature 505, 686–690. ( 10.1038/nature12861) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carrasco P, Daròs JA, Agudelo-Romero P, Elena SF. 2007. A real-time RT-PCR assay for quantifying the fitness of tobacco etch virus in competition experiments. J. Virol. Meth. 139, 181–188. ( 10.1016/j.jviromet.2006.09.020) [DOI] [PubMed] [Google Scholar]
- 43.Pfaffl MV. 2001. Quantification strategies in real-time PCR. In A-Z of quantitative PCR (ed. Bustin SA.), pp. 87–112. La Jolla, CA: International University Line. [Google Scholar]
- 44.Kouyos RD, Silander OK, Bonhoeffer S. 2007. Epistasis between deleterious mutations and the evolution of recombination. Trends Ecol. Evol. 6, 308–315. ( 10.1016/j.tree.2007.02.014) [DOI] [PubMed] [Google Scholar]
- 45.Poelwijk FJ, Tanase-Nicolas S, Kiviet DJ, Tans SJ. 2011. Reciprocal sign epistasis is a necessary condition for multi-peaked fitness landscapes. J. Theor. Biol. 272, 141–144. ( 10.1016/j.jtbi.2010.12.015) [DOI] [PubMed] [Google Scholar]
- 46.Flores CO, Meyer JR, Valverde S, Farr L, Weitz JS. 2011. Statistical structure of host-phage interactions. Proc. Natl Acad. Sci. USA 108, e288–e297. ( 10.1073/pnas.1101595108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ostrowski EA, Rozen DE, Lenski RE. 2005. Pleiotropic effects of beneficial mutations in Escherichia coli. Evolution 59, 2343–2352. ( 10.1111/j.0014-3820.2005.tb00944.x) [DOI] [PubMed] [Google Scholar]
- 48.Turner PE, Elena SF. 2000. Cost of host radiation in an RNA virus. Genetics 156, 1465–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Duffy S, Turner PE, Burch CL. 2006. Pleiotropic cost of niche expansion in the RNA bacteriophage φ6. Genetics 172, 751–757. ( 10.1534/genetics.105.051136) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ferris MT, Joyce P, Burch CL. 2007. High frequency of mutations that expand the host range of an RNA virus. Genetics 176, 1013–1022. ( 10.1534/genetics.106.064634) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Agudelo-Romero P, de la Iglesia F, Elena SF. 2008. The pleiotropic cost of host-specialization in tobacco etch potyvirus. Infect. Genet. Evol. 8, 806–814. ( 10.1016/j.meegid.2008.07.010) [DOI] [PubMed] [Google Scholar]
- 52.Elena SF, Solé RV, Sardanyés J. 2010. Simple genomes, complex interactions: epistasis in RNA viruses. Chaos 20, 026106 ( 10.1063/1.3449300) [DOI] [PubMed] [Google Scholar]
- 53.De Visser JAGM, Elena SF. 2007. The evolution of sex: empirical insights into the roles of epistasis and drift. Nat. Rev. Genet. 8, 139–149. ( 10.1038/nrg1985) [DOI] [PubMed] [Google Scholar]
- 54.Bagheri HC, Wagner GP. 2004. Evolution of dominance in metabolic pathways. Genetics 168, 1716–1735. ( 10.1534/genetics.104.028696) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kondrashov AS, Crow JF. 1991. Haploidy or diploidy: which is better. Nature 351, 314–315. ( 10.1038/351314a0) [DOI] [PubMed] [Google Scholar]
- 56.Lalić J, Cuevas JM, Elena SF. 2011. Effect of host species on the distribution of mutational fitness effects for an RNA virus. PLoS Genet. 7, e1002378 ( 10.1371/journal.pgen.1002378) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Elena SF, Carrasco P, Daròs JA, Sanjuán R. 2006. Mechanisms of genetic robustness in RNA viruses. EMBO Rep. 7, 168–173. ( 10.1038/sj.embor.7400636) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kondrashov AS. 1994. Muller's ratchet under epistatic selection. Genetics 136, 1469–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Coyne JA. 1992. Genetics and speciation. Nature 355, 511–515. ( 10.1038/355511a0) [DOI] [PubMed] [Google Scholar]
- 60.Lalić J, Elena SF. 2012. Epistasis between mutations is host-dependent for an RNA virus. Biol. Lett. 9, 20120396 ( 10.1098/rsbl.2012.0396) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Caudle SB, Miller CR, Rokyta DR. 2013. Enviroment determines epistatic patterns for an ssDNA virus. Genetics 196, 267–279. ( 10.1534/genetics.113.158154) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mustonen V, Lässing M. 2009. From fitness landscapes to seascapes: non-equilibrium dynamics of selection and adaptation. Trends Genet. 25, 111–119. ( 10.1016/j.tig.2009.01.002) [DOI] [PubMed] [Google Scholar]
- 63.Lalić J, Elena SF. 2012. Magnitude and sign epistasis among deleterious mutations in a positive-sense plant RNA virus. Heredity 109, 71–77. ( 10.1038/hdy.2012.15) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Remold SK. 2012. Understanding specialism when the jack of all trades can be the master of all. Proc. R. Soc. B 279, 4861–4869. ( 10.1098/rspb.2012.1990) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data necessary to reproduce the analyses reported in this article have been presented in tables 3 and 4.