

The presence and breadth of two histone modifications associated with active genes correlate to changes in gene expression during leaf aging, supporting senescence-related chromatin structural changes.
Abstract
The genome-wide abundance of two histone modifications, H3K4me3 and H3K9ac (both associated with actively expressed genes), was monitored in Arabidopsis (Arabidopsis thaliana) leaves at different time points during developmental senescence along with expression in the form of RNA sequencing data. H3K9ac and H3K4me3 marks were highly convergent at all stages of leaf aging, but H3K4me3 marks covered nearly 2 times the gene area as H3K9ac marks. Genes with the greatest fold change in expression displayed the largest positively correlated percentage change in coverage for both marks. Most senescence up-regulated genes were premarked by H3K4me3 and H3K9ac but at levels below the whole-genome average, and for these genes, gene expression increased without a significant increase in either histone mark. However, for a subset of genes showing increased or decreased expression, the respective gain or loss of H3K4me3 marks was found to closely match the temporal changes in mRNA abundance; 22% of genes that increased expression during senescence showed accompanying changes in H3K4me3 modification, and they include numerous regulatory genes, which may act as primary response genes.
Leaf senescence is the final nutritive stage of leaf development, in which the organ that provided sugars to the growing plant undergoes a controlled degradation process that recycles many of the nutrients located in the protein-rich photosynthetic apparatus. Leaf senescence is regulated by developmental age and can be accelerated by adverse environmental conditions, such as drought, nutrient deprivation, and heat stress. Ethylene, jasmonic acid, and salicylic acid promote, whereas cytokinin prevents leaf senescence (Lim et al., 2007a). Numerous microarray studies have cataloged major changes in gene expression during leaf senescence, and a large overlap with defense responses has been noted (Guo et al., 2004; van der Graaff et al., 2006; Breeze et al., 2011; Guo and Gan, 2012).
Toward understanding the mechanisms that regulate gene expression during senescence, numerous studies have shown that transcription factors, such as WRKY53 (Hinderhofer and Zentgraf, 2001; Miao et al., 2004; Miao and Zentgraf, 2010), ORESARA1/NAC2 (Oh et al., 1997; Matallana-Ramirez et al., 2013), and ETHYLENE-INSENSITIVE3 (Li et al., 2013b), promote leaf senescence as well as many others listed in the Leaf Senescence Database (Liu et al., 2011). In addition, mutants that affect chromatin structure have been shown to affect senescence; however, phenotypes are often pleiotropic (Lim et al., 2007b; Wu et al., 2008; Ay et al., 2009; Humbeck, 2013).
In a previous study, changes in two euchromatin histone modifications, trimethylation of Lys-4 in Histone H3 (H3K4me3; Zhang et al., 2009) and trimethylation of Lys-27 in Histone H3 (H3K27me3; Zhang et al., 2007), were measured on a genome-wide scale by chromatin immunoprecipitation Illumina sequencing (ChIP-seq) in young and old leaves (Brusslan et al., 2012). This study did not have accompanying gene expression data, and published microarray data (van der Graaff et al., 2006) were used to compare gene expression and changes in histone methylation status. Even with these limitations, a correlation between the H3K4me3 mark and gene expression was apparent. Genes that displayed a gain in the H3K4me3 mark were most highly represented by genes expressed to a greater extent in older leaves, and the opposite distribution was observed for genes that lost the H3K4me3 mark. Genes that lost the H3K27me3 mark were expressed in older leaf tissue, but overall gene numbers were low, and a gain of the H3K27me3 mark for down-regulated genes was rarely observed.
H3K4me3 marks are localized to actively transcribed genes, but they become associated with chromatin after RNA polymerase II binding (Kim et al., 2008; Malapeira et al., 2012) and thus, are considered to be marks for transcribed genes. However, interaction between the H3K4me3 mark and TAF3, a TFIID subunit of the preinitiation complex, has been observed in human cells (Lauberth et al., 2013), suggesting that the H3K4me3 mark plays a role during the initiation of transcription. In addition, the Arabidopsis (Arabidopsis thaliana) trithorax1 (ATX1) methyltransferase was found to be important for recruiting TATA binding protein and RNA polymerase II to promoters. Interestingly, this activity was independent of the ATX1 methyltransferase activity, which was necessary for elongation of transcription (Ding et al., 2012). The extent of H3K4me3 modifications was recently shown to be greatest for cell identity genes, and in young leaves, genes with the greatest breadth of H3K4me3 coverage were enriched for photosynthesis (Benayoun et al., 2014).
A second well-studied histone mark is acetylation of Lys-9 of Histone H3 (H3K9ac; Zhou et al., 2010). Changes in H3K9 acetylation have been shown to correlate with changes in gene expression during deetiolation and after UV-B treatment (Charron et al., 2009; Schenke et al., 2014). The timing of H3K9ac gain or loss closely paralleled gene expression changes for the circadian clock components and response to drought stress (Kim et al., 2012; Malapeira et al., 2012), or it occurred after genes had been activated in the case of flowering (Adrian et al., 2010). Thus, the exact role of H3K9ac in gene activation is not yet defined, and it likely plays numerous roles.
In this study, both of these well-established histone marks (H3K4me3 and H3K9ac), which positively correlate to gene expression, have been measured on a genome-wide scale using ChIP-seq along with accompanying RNA sequencing (RNA-seq) data at different stages of leaf aging. Most (78%) senescence up-regulated genes (SURGs) were premarked with H3K4me3 and H3K9ac, and most (85%) of the down-regulated genes retained both marks, even when gene expression had dropped to low levels. However, the breadth of the H3K4me3 and H3K9ac marks was positively related to gene expression. For a subset of genes (22% of up-regulated genes and 15% of down-regulated genes), a strong relationship between temporal changes in gene expression and gain/loss of the H3K4me3 mark was observed during leaf aging. By identifying genes that show combined changes in gene expression and histone marks, we have produced a list of genes that includes many known to play important roles in senescence as well as potentially unique players in this important biological process.
RESULTS
RNA-Seq Gene Expression Analysis
RNA-seq was performed on mature, fully expanded rosette leaf tissue at four time points: 29, 35, 42, and 57 d (Supplemental Fig. S1). These time points were chosen to encompass the steps of leaf senescence, which commences soon after the development of the inflorescence (Breeze et al., 2011). Three independently sampled replicates were subject to RNA-seq using multiplex single-end Illumina sequencing (Supplemental Table S1). Replicates sampled on each day show a high degree of similarity, with the 29-d samples being the most distinct (Fig. 1A), allowing the use of replicates to assess variation for confident differential expression analysis as well as subsequent pooling of the replicate read count for the calculation of reads per kilobase of transcript for million mapped reads (RPKM) values. The initial data analysis identified genes that showed significant changes in expression between adjacent time points (≥2-fold change in expression, P value ≤ 0.05): 29 to 35, 35 to 42, and 42 to 57 d. Confirming the distinct separation of the 29-d replicates and those of the other time points seen in the dendrogram of the clustering results (Fig. 1A), the largest number of genes was up- (Fig. 1B) or down-regulated (Fig. 1C) during the first time interval, indicating that major changes in gene expression occur before the visual manifestations of leaf aging: the loss of chlorophyll. The greatest overlap between time intervals was between 35 and 42 d and between 42 and 57 d for up-regulated genes and between 29 and 35 d and between 35 and 42 d for down-regulated genes (Fig. 1, B and C). A hypergeometric test (Johnson et al., 1992) revealed a significant overlap between all intervals for up-regulated genes (P value ≤ 5E-04), indicating that, although most genes are fully up-regulated by 35 d, a significant proportion continues to be more gradually up-regulated across time points (Fig. 1B). The down-regulated genes only showed a significant overlap across the first two intervals (P value < 1.1E-81), indicating that a largely distinct set of genes decreases from 42 to 57 d (Fig. 1C).
Figure 1.

Gene expression differences during leaf senescence. A, Pearson correlation matrix of gene expression data [log2(read counts + 1)] from all RNA-seq libraries. The darker red boxes indicate a higher correlation. Dendrograms were generated by hierarchically clustering samples based on correlation values transformed into distance values (1 − r). B, Genes with significant (≥2-fold, P ≤ 0.05) increases in expression between adjacent ages are shown in the Venn diagram. C, Genes with significant (≥2-fold, P ≤ 0.05) decreases in expression between adjacent ages are shown in the Venn diagram. Box plots for SURGs (D), SDRGs (E), and non-SURG or -SDRG (F) gene expression. RNA-seq RPKM data (log2 scale) for genes are shown. Boxes represent first to third quartiles or interquartile range (IQR). Whiskers extend to the most extreme data points but no more than ±1.5 times the IQR from the box, beyond which outliers are plotted individually. Notches extend to ±1.58 IQR/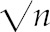 (Chambers et al., 1983). G, The abundance of three gene families for different thresholds used to classify SURGs and SDRGs is shown. bHLH, Basic helix-loop-helix.
(Chambers et al., 1983). G, The abundance of three gene families for different thresholds used to classify SURGs and SDRGs is shown. bHLH, Basic helix-loop-helix.
We sought to generate a high-confidence set of SURGs and senescence down-regulated genes (SDRGs) by requiring that they showed significant changes in expression (≥2-fold, P ≤ 0.05) in two of six pairwise comparisons (29–35, 29–42, 29–57, 35–42, 35–57, and 42–57 d). This analysis permits the inclusion of genes with significant changes in expression in just one interval; for example, the distinct group of genes that is down-regulated between 42 and 57 d (Fig. 1C) will be listed in the 29 to 57, 35 to 57, and 42 to 57 d pairwise comparisons. To remove genes with low expression, we also required that they have RPKM values (after merging replicate data sets) above the median RPKM value (0.764 for 29 d, 0.911 for 35 d, 0.790 for 42 d, and 0.752 for 57 d) at the time of higher expression. Figure 1, D and E shows the robust up- and down-regulation of expression in the SURGs and SDRGs, respectively. As was generally the case, the biggest changes in expression occurred between 29 and 35 d, but the respective upward and downward trends persisted for the duration of the time course. In contrast to the SURGs and SDRGs, the expression distributions of all other genes show no trend, indicating that the classification was reasonable (Fig. 1F). Figure 1G shows that setting the threshold at two of six pairwise comparisons resulted in a fair estimate of gene expression changes that represented a good compromise between overly stringent and more lenient criteria. WRKY transcription factor genes, usually associated with senescence and thus, representing a likely false positive, were observed in the down-regulated category when only one of six pairwise comparisons was the threshold; conversely, small up-regulated auxin (SAUR) genes, down-regulated in senescence and representing a likely false positive, were seen at increasing numbers in the up-regulated category when only one of six pairwise comparisons was the threshold. Genes encoding basic helix-loop-helix transcription factors showed no preference for up- or down-regulation during leaf aging, and the numbers of these genes became more plentiful as the threshold decreased.
The selection procedure described above resulted in 1,432 SURGs (plus 11 pseudo-genes and 6 transposable element genes) and 964 SDRGs (plus 4 pseudo-genes and 5 transposable element genes; Supplemental Data Sets S1 and S2). Small numbers of transposable element genes, mostly retroposons, and pseudo-genes were both up- and down-regulated during leaf senescence. This contrasts to animal cells, where a general increase in expression and mobility of transposable element genes has been observed in older somatic cells (De Cecco et al., 2013; Li et al., 2013a). A gene ontology (GO) analysis was performed on this list of SURGs and SDRGs, and enriched biological processes with false discovery rates below 1% are shown in Table I. Genes related to defense, jasmonic acid, and transport were enriched in SURGs as expected (Guo et al., 2004; van der Graaff et al., 2006; Breeze et al., 2011). In addition, enrichment for indole glucosinolate synthesis genes suggested a role for these secondary metabolites during senescence (Wang et al., 2013). SDRGs were enriched for photosynthesis and growth-related processes, such as response to auxin stimulus, response to light stimulus, response to gibberellin, lipid biosynthesis, and cell wall organization.
Table I. GO enrichment for SURGs and SDRGs.
Enriched GO biological processes and false discovery rates (FDR in percent) are reported.
| Biological Process | Gene Count | FDR |
|---|---|---|
| UP_GO biological process: two of six pairwise comparisons | ||
| Defense response | 146 | 1.5E-20 |
| Protein amino acid phosphorylation | 147 | 1.5E-19 |
| Response to organic substance | 128 | 2.3E-08 |
| Glucosinolate metabolic process | 19 | 2.8E-08 |
| Jasmonic acid metabolic process | 12 | 1.7E-04 |
| Indole-derivative metabolic process | 14 | 8.8E-04 |
| Multidrug transport | 17 | 0.010 |
| Aromatic compound biosynthetic process | 28 | 0.307 |
| Response to oxidative stress | 33 | 0.741 |
| Chitin catabolic process | 8 | 0.767 |
| DOWN_GO biological process | ||
| Response to auxin stimulus | 49 | 3.8E-12 |
| Cell wall organization | 34 | 1.1E-05 |
| Response to light stimulus | 43 | 3.5E-04 |
| Regulation of cellular component size | 24 | 0.068 |
| Lipid biosynthetic process | 34 | 0.093 |
| Photosynthesis | 19 | 0.207 |
| Microtubule-based movement | 12 | 0.217 |
| Response to gibberellin | 15 | 0.280 |
ChIP-Seq Analysis for H3K4me3 and H3K9ac
Nuclei were prepared from the same tissue used in RNA-seq, and ChIP-seq was performed using an antibody that recognized H3K4me3. A second set of tissue was grown and harvested at 30, 34, and 42 d for H3K9ac chromatin immunoprecipitation (ChIP) libraries. Although the days were not identical, the developmental stages were similar to the first three time points of the first experiment. ChIP and input sequencing reads are summarized in Supplemental Table S2. Figure 2A shows two typical gene-rich regions on chromosomes 1 and 5 with H3K4me3 and H3K9ac read counts. The peaks of read counts are most commonly located in the 500-bp region downstream of the transcription start site (TSS; Fig. 2B). Regions of significant H3K4me3 and H3K9ac ChIP enrichment were determined (“Materials and Methods”). Approximately 1.82 Mb of the 115-Mb genome were found to be significantly enriched for one or the other of two histone marks relative to input, with the marks coinciding across 1.42 Mb (78%; Fig. 2C). A cooperative interaction between H3K4me3 and H3K9ac modifications is suggested by the high degree of colocalization, which has been noted previously (Kim et al., 2008, 2012; Anzola et al., 2010; Jaskiewicz et al., 2011; Roudier et al., 2011; Malapeira et al., 2012).
Figure 2.

Coincidence of H3K4me3 and H3K9ac marks. A, ChIP-seq reads from two gene-rich regions of the Arabidopsis genome (upper, Chromosome 1 and lower, chromosome 5) are displayed using Integrated Genome Viewer. Data from 29 (H3K4me3) and 30 d (H3K9ac) are displayed in pairs of ChIP and input reads. The scale for H3K4me3 ChIP and input is 0 to 120, whereas the scale for H3K9ac ChIP and input is 0 to 60. Gene tracks with exons and introns are shown in blue below the read tracks. B, At5g45340 (the boxed gene in A) is shown in a zoomed-in view. The peaks for both histone modifications are colocalized to the 5′ ends of the gene. At5g45340 is transcribed from right to left and encodes an abscisic acid hydroxylase. C, ChIP reads were compared with input, and peaks were called as described in “Materials and Methods” for each histone modification. The Venn diagram shows the overlap between peaks comprising at least two sequential 100-bp bins exhibiting significant ChIP counts for each histone modification across all time points.
Changes in Histone Marks during Leaf Senescence
To determine if the levels of the two histone marks changed significantly during leaf senescence, six pairwise comparisons were made between ChIP read counts at four different time points for the H3K4me3 mark, and three pairwise comparisons were made at three different time points for H3K9ac read counts. Based on these pairwise comparisons, a region was defined as displaying a consistent and significant gain or loss of histone marks during the course of the experiment if at least three of six pairwise comparisons for H3K4me3 showed significant [−log(P value) ≥ 6] changes or at least two of three comparisons for H3K9ac showed significant [−log(P value) ≥ 6] changes, thereby ruling out spurious changes. We used these more stringent thresholds for the histone marks relative to gene expression [two of six pairwise comparisons and −log(P value) ≥ 1.3], because the lower requirement for leaf material allowed us to produce replicate RNA-seq libraries (n = 3; Fig. 1A), which in turn, allowed for greater confidence in the differential expression results. Representative regions displaying a gain in H3K4me3 or H3K9ac are shown in Figure 3. Figure 3A shows At5g13080 that encodes WRKY75, a transcription factor known to be important in regulating leaf senescence (Li et al., 2012). ChIP and input read counts in progressively older leaves are displayed at different time points, and the increase in H3K4me3 is clearly seen. The region that shows significant gain is indicated in the K4_GAIN 3 of 6 track, and three of six comparisons (29–35, 29–42, and 29–57 d) show −log(P values) ≥ 6. H3K9ac is enriched at the WRKY75 locus but does not show a change in abundance. In Figure 3B, the gain in H3K9ac marks can be seen for At1g66760, which encodes a member of the multidrug and toxic compound extrusion (MATE) transporter family. The K9_GAIN 2 of 3 track displays regions with −log(P values) ≥ 6 in two of three comparisons (30–34 and 30–42 d). This gene has consistent levels of the H3K4me3 mark.
Figure 3.

Histone modifications in leaves from differently aged plants. A, Integrated Genome Viewer (IGV) tracks for ChIP and input reads for the WRKY75 gene (At5g13080) that show a significant increase in H3K4me3 marks (K4_GAIN 3 of 6 track). The read count tracks show ChIP and input reads for the numbers of days of plant growth for each histone modification as indicated. Data from younger leaves are darker green, whereas data from older leaves are more yellow. The read range is 0 to 120 for H3K4me3 reads and 0 to 60 for H3K9ac reads. The criteria for significance are −log(P ) ≥ 6 in three of six pairwise comparisons, and P values for six pairwise comparisons are shown below (K4_GAIN 29–35 to K4_GAIN 42–57; P scale is 0–10). B, IGV tracks are the same as in A but for one member of the MATE Efflux transporter family (At1g66760), which shows a significant increase in H3K9ac marks. Significant changes in acetylation were identified as those that showed −log(P ) ≥ 6 in two of three comparisons (K9_GAIN 2 of 3 track). K9_GAIN 30 to 34 to K9_GAIN 34 to 42 tracks show −log(P ) for three pairwise comparisons on a 0 to 10 scale.
Overall, 564 genes showed a significant gain of H3K4me3 within the region from the TSS to 500 bp downstream during leaf aging; 222 genes were associated with a significant loss of H3K4me3 over the same period. Of these genes, 56% (315 of 564) that gained the H3K4me3 mark were SURGs, and 63% (139 of 222) that lost the H3K4me3 mark were SDRGs. This shows a high positive correlation between significant changes in these histone modifications and gene expression. For H3K9ac marks, the numbers of genes associated with a consistent significant gain (128) and loss (150) were lower than for H3K4me3 marks, in part because of one less time point and reduced read counts compared with H3K4me3. Furthermore, the correlations with gene expression were not as high: 20% (26 of 128) for gain of the acetylation mark and up-regulation and 17% (26 of 150) for loss of the acetylation mark and down-regulation. In fact, of 128 genes that significantly gained the H3K9ac mark, 51 (40%) showed no upward or downward trend in expression during the course of leaf senescence, and 59 of 150 genes (39%) that significantly lost H3K9ac had unchanged gene expression.
Considering SURGs as a whole, 22% (315 of 1,432) gained H3K4me3, whereas only 2% (26 of 1,432) gained the H3K9ac mark. Similarly, 14% (139 of 964) of SDRGs displayed a loss of the H3K4me3 mark, with 3% (26 of 964) showing H3K9ac loss. Intrigued by these relatively low proportions, we first studied the 1,117 SURGs (1,432 − 315 genes) and 825 SDRGs (964 − 139 genes) that were not associated with H3K4me3 acquisition or loss. Average H3K4me3 and H3K9ac read counts ±2,500 bp from the TSS for these genes are displayed in Figure 4. These SURGs were marked with H3K4me3 at the earliest time point, and a slight increase in marks was apparent; however, counts remained below the whole-genome time point average (Fig. 4A). Interestingly, H3K9ac counts increased with gene expression and were above the whole-genome time point average at 42 d (Fig. 4B). Increasing gene expression RPKM values are shown in Figure 4C. These two histone modifications were present before the first harvest and before the significant increase in expression captured by the RNA-seq time course experiments, but for these genes, only the H3K9ac mark more closely reflected gene expression. For this subset of SDRGs, there is a decrease in H3K4me3 modification levels that takes place in parallel with decreased mRNA abundance; however, there was only a moderate enrichment for this mark (approximately the same as on average), and the losses were not significant by our criteria, with a peak still evident at 57 d, when RPKM levels were low (Fig. 4, D and F). For H3K9ac, the modification levels hover below the whole-genome time point average and do not correspond to changes in gene expression (Fig. 4E). The decreasing gene expression profiles for the SDRGs not associated with a significant decrease in histone mark levels are shown in Figure 4F. Thus, for SDRGs, a decrease in H3K4me3, albeit not a significant one, still accompanies reduced gene expression, but both marks are retained, even in older tissue.
Figure 4.

H3K4me3 and H3K9ac histone marks in SURGs and SDRGs that were not identified as K4-SURGs or K4-SDRGs. Histone modification profiles for 1,117 SURGs are shown as lines for H3K4me3 (A) and H3K9ac (B), excluding 315 K4-SURGs that had significant and consistent changes in H3K4me3 marks. The average counts per 100-bp bins for all genes at each time point are shown by dashed lines. C, The gene expression values are shown for this group of 1,117 genes below the histone modification profiles. Histone profiles for 825 SDRGs are shown as lines for H3K4me3 (D) and H3K9ac (E), excluding 139 K4-SDRGs. (F) The gene expression values are shown for this group of 825 genes below the histone modification profiles. The P values for pairwise comparisons of gene expression in C are all significant: 1.33E-62, 29 to 35 d; 2.54E-106, 29 to 42 d; 2.53E-134, 29 to 57 d; 2.92E-04, 35 to 42 d; 2.64E-09, 35 to 57 d; and 0.009, 42 to 57 d. The P values for pairwise comparisons of gene expression in F are:5.49E-57, 29 to 35 d; 3.47E-79, 29 to 42 d; 3.99E-89, 29 to 57 d; 3.81E-07, 35 to 42 d; 2.46E-10, 35 to 57 d; and 0.34, 42 to 57 d. Box plot representations are described in Figure 1E.
Next, we considered those genes for which significant and consistently changing histone modifications and gene expression were identified (Supplemental Data Set S3). The 315 genes with increases in both H3K4me3 and mRNA levels (K4-SURGs) include NAC- and WRKY-domain transcription factors, numerous classes of receptor-like protein kinases, late embryogenesis-abundant dehydration factors, MATE efflux transporters, and RING/U-box proteins. The number of genes with increased H3K9ac and mRNA levels (K9-SURGs) is much smaller (26), and these include the WRKY41 transcription factor known to play a role in defense. Abundant gene classes within the 139 K4-SDRGs (genes with a decrease in both H3K4me3 and mRNA) include tubulin, gibberellin response, and cell wall extension; 3 YUCCA (auxin biosynthesis) and 13 SAUR-like (auxin response) genes show decreased expression accompanied by a loss in H3K4me3 marks. The 26 K9-SDRGs include the Photosystem I subunit K gene encoding a PSI subunit as well as 1 SAUR-like gene. The intersection of K4- and K9-SURGs was only 9 genes, whereas that for K4- and K9-SDRGs was 11 genes (Supplemental Data Set S3).
Temporal Patterns in Histone Mark Acquisition and Loss
Our time course allowed a comparative analysis of temporal changes in gene expression and histone modifications. Regions that showed significant gain or loss for each histone modification were subject to k-means clustering to produce three groups with different temporal patterns. Figure 5 shows the clustering for H3K4me3 acquisition (Fig. 5A) and how gene expression for the genes in each cluster coincides with the trend seen in the average histone mark profiles (Fig. 5B). The average H3K4me3 profiles for each cluster of genes at each time point are shown for the region −2,500 to +2,500 bp in relation to the TSS, and the mean levels for all genes at each time point are denoted with dashed lines in Figure 5B. In all cases, H3K4me3 peaks are centered approximately 400 bp downstream from the TSS, as seen before. A rapid increase in H3K4 trimethylation is observed between 29 and 35 d for cluster 1, and this is reflected in the accompanying gene expression box plot (Fig. 5B, cluster 1). The more gradual increase in H3K4me3 marks for cluster 2 is also mirrored in a more gradual increase in gene expression for genes in cluster 2, and the more subtle increases in H3K4me3 marks are accompanied by a smaller fold increase in gene expression for cluster 3 (Fig. 5B, clusters 2 and 3). The P values for all pairwise comparisons (after correction for multiple testing) can be viewed in Table II, and in all but one case, differences in gene expression are significant. The one case in which gene expression did not differ significantly (42–57 d for cluster 1) showed a concomitant negligible gain in H3K4 trimethylation. K-means clustering was also used to partition regions exhibiting a loss of H3K4me3 into three groups reflecting different temporal patterns of diminution (Fig. 6A). As with the gain of H3K4me3 marks, the magnitude and timing of loss in histone marks and decreases in gene expression mirror one another for all three clusters (Fig. 6B). For all clusters, the decrease in gene expression was not significant between 42 and 57 d (Table II), and H3K4me3 marks are at nearly identical levels. These correlations support an active and finely tuned role for the H3K4me3 mark in regulating gene expression during leaf aging for a subset of genes.
Figure 5.

k-means clustering for H3K4me3 gain peaks and correlation to gene expression. A, Peaks with a significant gain in H3K4me3 marks were subjected to k-means clustering to generate three cluster groups with different temporal trends of acquisition. B, The mean counts per 100-bp bin were calculated for genes associated with peaks in each cluster (lines) as well as all genes at each time point (dashed lines) for clusters 1 to 3. Data are shown for the region comprising −2,500 to +2,500 bp in relation to the TSS. RNA-seq data for genes that coincided with each peak are shown in the box plots for each cluster. Box plot representations are described in Figure 1E.
Table II. P values associated with pairwise comparisons of gene expression.
Accompanying H3K4me3 gain cluster data are shown in Figure 5, and loss cluster data are shown in Figure 6. Accompanying H3K9ac gain cluster data are shown in Supplemental Figure S2, and loss cluster data are shown in Supplemental Figure S4. Pairwise comparisons are listed at the top. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001.
| Histone Modification | 29 to 35 | 29 to 42 | 29 to 57 | 35 to 42 | 35 to 57 | 42 to 57 |
|---|---|---|---|---|---|---|
| H3K4me3 | d | |||||
| GAIN_cluster 1 | 4.68E-23*** | 3.1E-31*** | 4.84E-36*** | 0.008** | 2.14E-5*** | 0.062 |
| GAIN_cluster 2 | 1.47E-17*** | 1.55E-25*** | 6.79E-30*** | 0.003** | 1.41E-6*** | 0.036* |
| GAIN_cluster 3 | 1.23E-15*** | 3.76E-26*** | 6.2E-32*** | 7.37E-4*** | 1.71E-8*** | 0.008** |
| LOSS_cluster 1 | 4.57E-6*** | 2.46E-9*** | 9.6E-11*** | 0.201 | 0.017* | 0.204 |
| LOSS_cluster 2 | 4.22E-14*** | 2.07E-19*** | 5.47E-22*** | 0.006** | 2.43E-4*** | 0.312 |
| LOSS_cluster 3 | 3.59E-9*** | 7.14E-14*** | 1.66E-17*** | 0.015* | 1.4E-4*** | 0.232 |
| H3K9ac | ||||||
| GAIN_cluster 1 | 0.139 | 0.11 | 1 | |||
| GAIN_cluster 2 | 1 | 1 | 1 | |||
| GAIN_cluster 3 | 0.024* | 0.01* | 1 | |||
| LOSS_cluster 1 | 0.742 | 0.124 | 0.789 | |||
| LOSS_cluster 2 | 0.371 | 0.039* | 0.371 | |||
| LOSS_cluster 3 | 1 | 1 | 1 | |||
Figure 6.

k-means clustering for H3K4me3 loss peaks and correlation to gene expression. A, The same as in Figure 5A but for regions exhibiting a significant loss of H3K4me3. B, H3K4me3 marks and gene expression data are displayed as described in Figure 5B.
A corresponding analysis was performed for peaks that gained the H3K9ac histone mark, which revealed less distinct temporal trends. Of 33 genes comprising the H3K9ac gain cluster 3, a significant increase in gene expression was observed during leaf aging between 29 and 35 d and between 29 and 42 d (Table II; Supplemental Fig. S2B, cluster 3). The remaining two k-means clusters showed an upward trend when gene expression was compared with histone modifications; however, changes in gene expression were not significant (Table II; Supplemental Fig. S2B, clusters 1 and 2). Gene clusters 1 and 2 showed increased acetylation approximately 1.5 kb upstream of the TSS, which was most apparent at 34 and 42 d, and colocalized with the H3K4me3 mark (Supplemental Fig. S3).
Peaks that lost the H3K9ac mark were also subject to k-means clustering (Supplemental Fig. S4A), and cluster-associated gene expression showed a similar downward trend for all three clusters, but again, changes in gene expression were significant for only one pairwise comparison (29–42 d, cluster 2; Table II; Supplemental Fig. S4B). In the case of this one significant interval, the mean H3K9ac counts at 30 and 42 d were almost identical (Supplemental Fig. S4B, cluster 2). These data suggest that H3K9ac marks do not dynamically correspond to gene expression during leaf senescence as do the H3K4me3 marks, most strikingly for down-regulated genes. This is well illustrated by the H3K9ac profiles for 314 K4-SURGs (Fig. 7). K4-SURGs significantly increased expression during leaf senescence; the H3K4me3 profiles start well below the time point average and consistently increase during leaf senescence, whereas the H3K9ac profiles are on a par with the time point average at 30 and 34 d and only show an increase at 42 d.
Figure 7.

Average H3K4me3 and H3K9ac read count profiles for 387 K4-SURGs. Histone modification profiles as described in Figure 5B for 315 K4-SURGs. Both histone marks are shown for K4-SURGs. H3K4me3 marks increase consistently throughout leaf aging starting at a low level, whereas H3K9ac marks start at the whole-genome time point average and only increase at 42 d.
GO enrichment analysis was performed for the k-means gene clusters. H3K4me3_Gain cluster 2 showed a dramatic increase in gene expression and enrichment for multidrug transport, and indole glucosinolate biosynthesis was observed, whereas the genes in H3K4me3 gain cluster 3 with a smaller increase in expression were enriched for immune response and secondary metabolic process. For H3K4me3 loss cluster 2, which showed a dramatic decrease in expression, response to hormone stimulus and plant-type cell wall modification were enriched, whereas the less steep decline in expression observed in H3K4me3 loss cluster 3 was also enriched for response to hormone stimulus as well as response to light stimulus and developmental growth involved in morphogenesis. The remaining clusters showed no significant enriched GO biological process clusters. GO enrichment is summarized in Supplemental Data Set S4.
The Breadth of Histone Marks Correlates with Gene Expression during Leaf Senescence
A recent study by Benayoun et al. (2014) showed that the extent of H3K4me3 marks was greatest for genes that impart cell identity. In leaves, for instance, a high proportion of genes with the broadest H3K4me3 coverage was assigned the photosynthesis GO term. With this in mind, the extent to which each gene was covered by the two respective histone modifications was determined. Using 100-bp bins that exhibited a significant enrichment in ChIP-seq read counts relative to input [−log(P value ≥ 4)], we calculated coverage in terms of both absolute base pairs and the proportion of the gene to normalize for length (TSS to transcription termination site ±500 bp to allow for potential spreading of the marks beyond the confines of the gene body). Considering the absolute extent of significant H3K4me3 enrichment at 42 d, the top 5% of genes had at least 1,500 bp of coverage by the mark (Fig. 8A). As a proportion of the gene length, the top 5% of genes with the highest coverage had at least 50% of their lengths significantly enriched for H3K4me3 (Fig. 8B). In contrast, genes in the 95th percentile of H3K9ac coverage at 42 d showed significant ChIP-to-input enrichment over at least 900 bp (Fig. 8C). Normalizing for gene length, the top 5% of genes with the broadest H3K9ac enrichment had at minimum 32% of their gene bodies covered (Fig. 8D). Similar overall coverage distributions were observed for the respective marks at all leaf ages (data not shown), with H3K9ac observed to cover only 60% to 65% of the region covered by H3K4me3.
Figure 8.

Breadth of histone modifications and gene expression during leaf senescence. The breadth of H3K4me3 modifications for 42-d samples are shown according to base pairs covered (A) or percentage of gene covered (B). The breadth of H3K9ac modifications for 42-d samples are shown according to base pair covered (C) or percentage of gene covered (D). Red dashed vertical lines delineate the top 95th percentile. E, The percentage change in H3K4me3 gene coverage from 29 to 57 d is plotted for gene expression bins. Genes are placed in bins according to the fold change in gene expression over the same time interval. Gene counts per bin are given in parentheses. For example, the bin labeled 0.5 includes 2,712 genes that display a log2 increase in gene expression greater than 0.5 to less than or equal to 1.0. The box plot representations are as described in Figure 1E. The overall Pearson correlation between percentage change in gene coverage and fold change in gene expression was 0.47 (upper left corner), with a 95% confidence interval of 0.463 to 0.481 (P < 2.2E-16). F, The percentage change in H3K9ac gene coverage from 30 to 42 d is plotted similarly to A, except that the expression time interval is from 29 to 42 d to better correspond to the H3K9ac time interval. The overall Pearson correlation in this case was 0.20, with a 95% confidence interval of 0.185 to 0.207 (P < 2.2E-16).
Enrichment for biological process GO terms was performed for genes with the top 5% broadest coverage for both modifications. Photosynthesis was enriched for H3K4me3 breadth for all time points but was not enriched at any time for genes with the broadest H3K9ac coverage. The 57-d H3K4me3 time point showed enrichment for response to bacterium (defense genes expressed during senescence). Regulation of transcription was enriched in 30- and 34-d H3K9ac samples but not enriched in any of the H3K4me3 samples, suggesting that H3K9ac coverage breadth may control regulatory loci. A summary of the enrichment analysis is found in Supplemental Data Set S4.
To better illustrate the association between the breadth of the histone marks and the expression levels of genes involved in leaf senescence, genes were placed into different bins based on their log2 fold change in expression across four time points used for the H3K4me3 analysis and three time points used for the H3K9ac analysis. The change in the proportion of histone mark coverage was determined across the corresponding time points for each bin, and their distributions are plotted in Figure 8, E and F. A positive correlation between fold change in gene expression and breadth of histone marks was apparent, with the correlation being higher for H3K4me3 marks (r = 0.47) than for H3K9ac marks (r = 0.20). The strength of the relationship between gene expression and H3K4me3 gene coverage was further illustrated by the dendrograms for correlation matrices made for the RNA-seq data and the H3K4me3 coverage data (Supplemental Fig. S5).
DISCUSSION
The abundance of H3K4me3 and H3K9ac histone modifications changes at specific loci during numerous developmental transitions in plants (Kim et al., 2008, 2009a, 2012; Charron et al., 2009; Adrian et al., 2010; Anzola et al., 2010; van Dijk et al., 2010; Brusslan et al., 2012; Malapeira et al., 2012; López-González et al., 2014; Molitor et al., 2014; Schenke et al., 2014), and thus, it is likely that the final stage of leaf development, leaf senescence, would be accompanied by changes in these two well-characterized histone modifications that positively correlate with gene expression. Leaves were harvested from progressively more senescent plants and subject to RNA-seq and ChIP-seq, so that a direct correlation between gene expression and abundance and breadth of histone modifications could be performed. Our data showed a strong correlation between the breadth of both histone marks and gene expression. Differing amounts of H3K4me3 marks correlated well to gene expression; however, a weaker trend was observed between gene expression and the abundance of H3K9ac marks.
Our harvest protocol maximized material to allow for both nuclei and RNA extraction and used multiple as well as larger leaves (approximately leaves 12–15) than leaf 7, which was used in an extensive transcriptome analysis of developmental senescence (Breeze et al., 2011). RPKM values for 18 senescence marker genes and the corresponding cluster number previously assigned are shown in Supplemental Table S3. Genes from cluster U1 were expressed early in both experiments, and genes from clusters U2 and U3 mostly showed the greatest fold induction in the first time interval (29–35 d), whereas genes from the U5 to U7 clusters showed highest expression at 57 d, the final time point for the RNA-seq data. Although this study did not have the temporal refinement of the extensive transcriptome work, the intervals are generally equivalent, and trends should translate among studies.
The signal strength for the H3K4me3 peaks was higher than that of the H3K9ac peaks. A recent analysis of multiple chromatin marks shows weaker peaks for numerous acetylation marks compared with H3K4me3 marks, suggesting that acetylation marks are less pronounced than H3K4me3 modifications in Arabidopsis (Sequeira-Mendes et al., 2014). The high coincidence of H3K4me3 and H3K9ac marks (Fig. 2) has been noted previously (Kim et al., 2008, 2010, 2012; Anzola et al., 2010; Roudier et al., 2011; Malapeira et al., 2012) and supports the validity of the H3K9ac data sets. Although the coincidence of these two modifications has been noted before, our observation that the extent of H3K9ac coverage per gene was about 60% of the H3K4me3 coverage level is unique. For most genes, modifications centered +400 bp from the TSS, but for genes identified in the H3K9ac_GAIN_clusters 1 and 2, modifications that peaked at approximately −1,200 bp from the TSS were also observed for both marks (Supplemental Fig. S3). The combination of the two marks in the promoter region might contribute to an upward trend in gene expression; however, neither of these H3K9ac_GAIN clusters exhibited significantly increased gene expression (Table II). Promoter methylation for H3K4me3_GAIN genes did increase with plant age, but no clear peak was identified, and the magnitude of the change was much less compared with the increase that centered at +400 bp from the TSS. Thus, for these genes, the role of promoter histone modifications is unclear.
The breadth of histone mark coverage was found to correlate well with gene expression, especially for H3K4me3 (Fig. 8; Supplemental Fig. S5). Genes with the greatest fold change in expression had the largest corresponding changes in gene coverage by the mark. These data support the fact that the extent of gene coverage, as well as the peak intensity, of these histone marks is important for gene regulation. Because this study is based on an ensemble of cells, the increase in the total histone mark signal at active genes could result from an increasing number of cells up-regulating the genes involved in the senescence process over time.
A similar experiment coupling expression to the H3K4me3 mark was performed on rice (Oryza sativa) subject to drought stress (Zong et al., 2013). Results of this study corresponded to our work, noting that genes with a significant change in the H3K4me3 mark and gene expression (609 rice genes) were directionally correlated, such that gain of the H3K4me3 mark and up-regulation occurred in 89% of cases and loss of the H3K4me3 mark and down-regulation occurred in 90% of cases. Identification of significantly changed H3K4me3 marks was less precise, because only two conditions, control and drought, were studied, and thus, the significant change in three of six (or two of three) pairwise comparisons that were done for this work was not possible. The observation that, of 4,387 rice genes with differential H3K4me3 modification levels, many were not expressed (45%) or had no change in expression during drought (40%) might be caused by an overestimation of genes with differential H3K4me3 marks.
The relatively few genes that showed significant changes in H3K4me3 (786 genes) and H3K9ac (278 genes) marks are surprising. Our work showed that only a small proportion of SURGs (22%) and SDRGs (15%) had changes in the H3K4me3 mark, and an even smaller percentage (2% for SURGs and 3% for SDRGs) had changes in the H3K9ac mark. This can be partially explained by the stringent criteria used to call regions of significant change, but raising the P value resulted in the inclusion of genes that did not show convincing gains/losses when raw data were viewed. Additionally, many SURGs already had both histone marks before the first leaf harvest; however, levels were below the whole-genome average for H3K4me3 and did not change significantly (Fig. 4). For these genes, H3K9ac levels showed an increasing trend that peaked above whole-genome time point averages, indicating that small changes in this mark do accompany changes in gene activation. SDRGs that did not show a significant loss in H3K4 trimethylation over the time points studied did display a decreasing trend in H3K4me3 marks; however, they remained above the genome-wide average, and H3K9ac marks for these genes were low during the entire time course. Thus, for the bulk of up-regulated genes, low levels of H3K4me3 marks were present before the first time point, and H3K9ac showed an increasing trend. For the bulk of down-regulated genes, the opposite was observed: low levels of H3K9ac were present, whereas H3K4me3 levels showed a decreasing trend. This reciprocal relationship between the two marks has not been noted previously, and it is worth additional study.
Pathogen infiltration of Arabidopsis leaves was found to result in increased expression of WRKY70, Pathogenesis-related1 (PR1) and Thionin12.1 (TH12.1). WRKY70 was found to have differential H3K4me3 and be a direct target of the ATX1 histone methyltransferase, whereas PR1 and TH12.1 had constant levels of H3K4me3 marks and were targets of WRKY70 (Alvarez-Venegas et al., 2007). The pathogen infiltration study suggested that changing H3K4me3 may occur for primary response regulatory genes but not for secondary response genes. In agreement with this observation, our list of SURGs that displayed a gain of H3K4me3 (K4-SURGs) includes four WRKY and six NAC transcription factors. Of these WRKY and NAC genes, mutant analysis has shown that WRKY75 (Li et al., 2012), NAC016 (Kim et al., 2013), and NAC019 (Guan et al., 2014) are positive regulators of senescence, whereas WRKY54 (Besseau et al., 2012) is a negative regulator of senescence. Thus, numerous important primary regulators of senescence do display a concomitant change in H3K4me3 marks and gene expression. A small number of regulatory genes displayed changes in H3K9ac as leaf senescence progressed. K9-SURGs include WRKY41, which plays a protective role during defense (Higashi et al., 2008), and nonrace-specific disease resistance1/hairpin induced1, a mitogen-activated protein kinase gene that is salicylic acid responsive during senescence (Zheng et al., 2004).
Of 175 K4-SDRGs that displayed a loss of H3K4me3 marks and decreased gene expression, 13 encoded YUCCA- and SAUR-like genes involved in auxin biosynthesis (Mashiguchi et al., 2011; Stepanova et al., 2011; Won et al., 2011) and response, respectively (Spartz et al., 2012, 2014). Auxin synthesis sets in motion major changes in gene expression and physiology (Guilfoyle and Hagen, 2007), and the down-regulation of auxin synthesis and signaling can be considered a primary event in the progression of senescence. Although reports of the role of auxin in leaf senescence conflict (Quirino et al., 1999; Okushima et al., 2005; Hou et al., 2013; Jiang et al., 2014), our molecular data suggest that auxin action is down-regulated during the progression of leaf senescence.
A well-characterized senescence regulator, WRKY53, showed a constant high level of H3K4me3 and H3K9ac marks in our study (Supplemental Fig. S6A), thus indicating that not all important regulators were identified as K4-SURGs. RPKM levels for WRKY53 increased from 42.6 to 548.1 between 29 and 35 d, indicating that a large change in expression (13-fold) did occur during the time points included in this study. WRKY53 is one example of the many genes that are marked before significant up-regulation of mRNA levels. WRKY53 expression is down-regulated by Whirly1 (Miao et al., 2013), which may explain the coincidence of low transcript levels and high levels of H3K4me3 marks. Examples of posttranscriptional regulation mediated by small RNAs have been identified during leaf senescence (Kim et al., 2009a, 2009b; Li et al., 2013b; Thatcher et al., 2015) and may also explain some of the inconsistencies between H3K4me3 marks and gene expression. Senescence associated gene12 (SAG12), a molecular marker for senescence, was up-regulated 3,300-fold between 29 and 57 d (0.022–72.5 RPKM) and showed increased levels of H3K4me3 marks 250 to 650 bp upstream of the TSS but no clear H3K9ac marks (Supplemental Fig. S6B), suggesting that H3K4me3 modifications in the promoter can be associated with changes in gene expression. This differed from our previous study, in which SAG12 was not marked by H3K4me3 (Brusslan et al., 2012). The other gene that was reported to be strongly up-regulated that did not have H3K4me3 marks, At1g73220, was devoid of both marks in this study (Supplemental Fig. S6C); however, expression levels were low for this gene and only reached median RPKM levels at 57 d. Because four time points were included in this more comprehensive work, these data have more certainty than our previous data.
During leaf senescence, changes in H3K4me3 marks reflected changes in gene expression to a much greater extent than changes in H3K9ac marks. This is, in part, because of the slightly different ages of the H3K9ac ChIP-seq data set compared with the RNA-seq data set. However, the criteria for a significant change required changes that covered two of three intervals for H3K9ac; thus, the differences in leaf harvest dates (29 versus 30 d and 35 versus 34 d) for the two marks were minimized by the nature of the analysis. However, a small number of genes that was specifically up- or down-marked between 29 and 35 d but not between the shorter interval of 30 to 34 d would be missed. In drought stress, H3K9ac and H3K4me3 marks increased in parallel for three drought-responsive genes (Kim et al., 2008). During rehydration, expression of the drought-responsive genes was down-regulated, and the H3K9ac marks were rapidly lost, whereas the H3K4me3 marks were retained and decreased at a rate similar to mRNA levels (Kim et al., 2012). The drought study was done on a timescale of hours, whereas this developmental senescence study was performed on a timescale of days; however, in both, H3K4me3 marks paralleled gene expression. It should be noted, however, that H3K4me3 marks can change rapidly, which has been observed for clock genes, in which both H3K9ac and H3K4me3 marks cycle for 24 h each (Malapeira et al., 2012). Sampling was always performed at the same time each harvest day, and therefore, diurnal cycles would not interfere with our study.
In this study, 387 K4-SURGs were noted to have average H3K9ac levels at the earliest time points that increased by 42 d, unlike the H3K4me3 marks, which at first, were substantially below average and greatly increased by the end of the time course (Fig. 7). These data suggest that H3K9ac modifications are present before H3K4 trimethylation for the K4-SURGs. In animal systems, evidence supporting K4 trimethylation of acetylated H3 has been published (Bradbury et al., 2005; Nightingale et al., 2007; Taverna et al., 2007), and it is possible that the H3K9 acetylation serves as a template for the gain of H3K4me3 marks during leaf senescence.
Interestingly, for K4-SURGs that display the largest gain in H3K4me3 marks between the first two time points (29–35 d), additional H3K9ac acquisition, above average levels, only occurs at a subsequent stage between 34 and 42 d (Fig. 7). Furthermore, those genes that gain H3K9ac (K9-SURGs) already show elevated levels of H3K4me3 and show no further increase in trimethylation (Supplemental Fig. S3), indicating that H3K4me3 acquisition occurred sometime before that of H3K9ac for these genes. Non-K4-SURGs/SDRGs showed a reciprocal relationship, with one mark remaining at a basal low level, whereas the other showed an increasing/decreasing trend (Fig. 4).
CONCLUSION
Gene expression and H3K4me3 and H3K9ac marks were monitored at a genome-wide level at four stages of leaf senescence. These two histone marks have been found to positively correlate to gene expression in many organisms. Most genes were premarked with the two modifications at levels below the whole-genome average before up-regulation. Most down-regulated genes remained marked, even when expression levels substantially dropped. A subset of genes (22% of up-regulated genes and 14% of down-regulated genes), including known regulatory loci, showed a strong correlation between gene expression and H3K4me3 marks. H3K9ac coverage per gene was approximately 60% of that for H3K4me3, and the percentage of coverage for both histone marks positively correlated to the fold change in aging-related gene expression. These studies bolster support for a role of chromatin modification in the process of leaf aging.
MATERIALS AND METHODS
Plant Growth, Nuclei Isolation, ChIP, ChIP Library Preparation, RNA Isolation, and RNA-Seq Library preparation
Plants were grown as described (Brusslan et al., 2012), with the exceptions that the light intensity was 30 µmol photons m−2 s−1 at 22°C and the diurnal cycle was 20 h of light and 4 h of dark. The low light intensity was chosen, because the leaves of older plants grown at standard light intensities (120–150 μmol photons m−2 s−1 for 16 h is recommended by the Arabidopsis Biological Resource Center) became purple and stressed; to compensate for the low light intensity, a longer light period of 20 h was used. Mature, fully expanded rosette leaves were harvested as shown by the pink arrows in Supplemental Figure S1. Newly emerging leaves at the center of the rosette were not harvested, and petioles were trimmed, such that harvested tissue contained leaf blades only. No inflorescence tissue was harvested. The first harvest was performed after the vegetative-to-reproductive transition, when incipient bolts (0.5–3 cm) had developed (29-d-old plants; 29 d). Mature rosette leaf tissue was then harvested at 35 d, when bolts had lengthened and secondary bolts were forming, 42 d, when siliques were present on primary and secondary bolts, and 57 d, when about 50% of siliques were brown and dry and chlorophyll loss was evident throughout the rosette leaves. Isolation of nuclei and ChIP were performed as previously described, with the exception that the final pellet was resuspended in 1 mL of cold nuclei lysis buffer and 2 mL of cold ChIP dilution buffer. The H3K9ac antibody (07-352) and H3K4me3 antibody (17-678) were purchased from Millipore. The H3K4me3 ChIP library was prepared for sequencing as described previously (Brusslan et al., 2012), and the H3K9ac library was prepared using the Illumina TruSeq ChIP Sample Preparation Kit-Set A. RNA was isolated using Trizol Reagent (Life Technologies), and RNA was prepared for sequencing using the Illumina TruSeq RNA Sample Prep Kit v2-SetA. Sequencing was performed at the University of California, Los Angeles using an Illumina HiSeq2000 to produce 50-bp single reads from both the ChIP-seq and RNA-seq libraries.
RNA-Seq Data Processing
RNA-seq reads were mapped to The Arabidopsis Information Resource 10 (TAIR10) reference genome and gene annotation using tophat-2.0.4 (Trapnell et al., 2009) calling bowtie-0.12.8 (Langmead et al., 2009) using the following parameters:
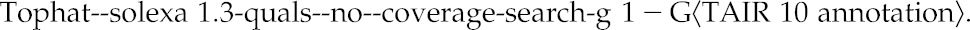 |
HTSeq (Anders et al., 2014) was used to perform gene counts, and DESeq2 (Love et al., 2014) was used for differential analysis. Intersection and union analyses of gene lists were performed using the Virtual Plant Web site (Katari et al., 2010).
ChIP-Seq Data Processing
ChIP-seq reads were mapped to the TAIR10 reference genome using bowtie-0.12.8 (Langmead et al., 2009) using the following parameters:
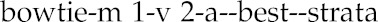 |
Regions of significant H3K4me3 and H3K9ac ChIP enrichment were determined by comparing the numbers of ChIP with input reads within 100-bp bins across the genome. Bins showing −log(P value) ≥ 6 by the Poisson test were deemed to be significantly enriched. To ensure an even higher level of confidence in these peaks, we required that at least two sequential 100-bp bins show significant ChIP enrichment when comparing the locations of the two marks (Fig. 2). Pearson correlations and significance values were calculated using R’s cor.test function (R Core Team).
ChIP-Seq Differential Analysis
Pairwise comparisons were made between ChIP signals at each time point for each histone mark using a Poisson test to determine regions of significant difference [−log(P value) ≥ 6]. Based on these pairwise comparisons, a region was defined as displaying a consistent gain or loss of histone marks during the course of the experiment if changes were observed in at least three of six pairwise comparisons for H3K4me3 and two of three pairwise comparisons for H3K9ac.
k-Means Clustering
For each mark, only regions displaying a consistent significant gain or loss of the mark during the course of the experiment were considered (“Materials and Methods, ChIP-seq Differential Analysis”). In each of four cases (significant K4me3 gain, K4me loss, K9ac gain, and K9ac loss), −log(P value) results from the ChIP versus input Poisson test (see above) were grouped by k-means clustering (k = 3) using the kmeans function in the R package (R Core Team). Genes were assigned to these peaks if the peaks overlapped the TSS to TSS + 500-bp region of the genes. In some cases, multiple regions from a single gene were assigned to different clusters.
Gene Coverage
For each mark, the coverage of each gene was determined as the proportion of 100-bp bins from TSS − 500 bp to transcription termination site + 500 bp that exhibited a significant enrichment in ChIP-seq read counts relative to input [−log(P value) ≥ 4]. A lower threshold was chosen in this case, because we were comparing immunoprecipitate (IP) with input as opposed to IP versus IP, which has the potential for containing more false positives; also, this helped to better capture the continuity in the breadth of coverage of significant histone mark deposition along the gene body. The top 5% of genes with the highest proportion of coverage at each time point were analyzed for absolute length of coverage and percentage of gene coverage.
Sequence data from this article can be found in the GenBank/EMBL data libraries under accession numbers GSE67776, GSE67777, and GSE67778.
Supplemental Data
The following supplemental materials are available.
Supplemental Figure S1. Arabidopsis rosettes at harvest dates.
Supplemental Figure S2. k-means clustering for H3K9ac gain peaks and correlation to gene expression.
Supplemental Figure S3. Average H3K4me3 and H3K9ac read count profiles for genes associated with H3K9ac_GAIN clusters 1 and 2.
Supplemental Figure S4. k-means clustering for H3K9ac loss peaks and correlation to gene expression.
Supplemental Figure S5. Correlation matrices and dendrograms for RNA-seq RPKMs and H3K4me3 gene coverage.
Supplemental Figure S6. H3K4me3 and H3K9ac marks for three genes.
Supplemental Table S1. Read and alignment summary for RNA-seq libraries.
Supplemental Table S2. Read and alignment summary for ChIP-seq libraries.
Supplemental Table S3. Gene expression comparison between this study and the previously reported leaf 7 time course.
Supplemental Data Set 1. List of senescence up- and down-regulated genes.
Supplemental Data Set 2. RPKM values for SURGs, SDRGs, and all other genes in which one time point was above the median RPKM value.
Supplemental Data Set 3. List of K4-SURGs, K9-SURGs, K4- and K9-SURGs, K4-SDRGs, K9-SDRGs, and K4- and K9-SDRGs.
Supplemental Data Set 4. GO biological process enrichment.
Supplementary Material
Glossary
- ChIP
chromatin immunoprecipitation
- ChIP-seq
chromatin immunoprecipitation sequencing
- GO
gene ontology
- RNA-seq
RNA sequencing
- SDRG
senescence down-regulated gene
- SURG
senescence up-regulated gene
- TAIR10
The Arabidopsis Information Resource 10
- TSS
transcription start site
Footnotes
This work was supported by the National Institutes of Health (grant no. 1SC2GM092281) and the University of California, Los Angeles (Whitcome Predoctoral Training Program to G.B. and Dissertation Year Fellowship to G.B.).
Articles can be viewed without a subscription.
References
- Adrian J, Farrona S, Reimer JJ, Albani MC, Coupland G, Turck F (2010) cis-Regulatory elements and chromatin state coordinately control temporal and spatial expression of FLOWERING LOCUS T in Arabidopsis. Plant Cell 22: 1425–1440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Venegas R, Abdallat AA, Guo M, Alfano JR, Avramova Z (2007) Epigenetic control of a transcription factor at the cross section of two antagonistic pathways. Epigenetics 2: 106–113 [DOI] [PubMed] [Google Scholar]
- Anders S, Pyl PT, Huber W (2015) HTSeq: a Python framework to work with high-throughput sequencing data. Bioinformatics 31: 166–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anzola JM, Sieberer T, Ortbauer M, Butt H, Korbei B, Weinhofer I, Müllner AE, Luschnig C (2010) Putative Arabidopsis transcriptional adaptor protein (PROPORZ1) is required to modulate histone acetylation in response to auxin. Proc Natl Acad Sci USA 107: 10308–10313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ay N, Irmler K, Fischer A, Uhlemann R, Reuter G, Humbeck K (2009) Epigenetic programming via histone methylation at WRKY53 controls leaf senescence in Arabidopsis thaliana. Plant J 58: 333–346 [DOI] [PubMed] [Google Scholar]
- Benayoun BA, Pollina EA, Ucar D, Mahmoudi S, Karra K, Wong ED, Devarajan K, Daugherty AC, Kundaje AB, Mancini E, et al. (2014) H3K4me3 breadth is linked to cell identity and transcriptional consistency. Cell 158: 673–688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besseau S, Li J, Palva ET (2012) WRKY54 and WRKY70 co-operate as negative regulators of leaf senescence in Arabidopsis thaliana. J Exp Bot 63: 2667–2679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradbury CA, Khanim FL, Hayden R, Bunce CM, White DA, Drayson MT, Craddock C, Turner BM (2005) Histone deacetylases in acute myeloid leukaemia show a distinctive pattern of expression that changes selectively in response to deacetylase inhibitors. Leukemia 19: 1751–1759 [DOI] [PubMed] [Google Scholar]
- Breeze E, Harrison E, McHattie S, Hughes L, Hickman R, Hill C, Kiddle S, Kim YS, Penfold CA, Jenkins D, et al. (2011) High-resolution temporal profiling of transcripts during Arabidopsis leaf senescence reveals a distinct chronology of processes and regulation. Plant Cell 23: 873–894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brusslan JA, Rus Alvarez-Canterbury AM, Nair NU, Rice JC, Hitchler MJ, Pellegrini M (2012) Genome-wide evaluation of histone methylation changes associated with leaf senescence in Arabidopsis. PLoS ONE 7: e33151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers J, Cleveland W, Kleiner B, Tukey P (1983) Graphical Methods for Data Analysis. Wadsworth International Group, Belmont, CA [Google Scholar]
- Charron JBF, He H, Elling AA, Deng XW (2009) Dynamic landscapes of four histone modifications during deetiolation in Arabidopsis. Plant Cell 21: 3732–3748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Cecco M, Criscione SW, Peterson AL, Naeretti N, Sedivy JM, Kreiling JA (2013) Transposable elements become active and mobile in the genomes of aging mammalian somatic tissues. Aging 5: 867–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y, Ndamukong I, Xu Z, Lapko H, Fromm M, Avramova Z (2012) ATX1-generated H3K4me3 is required for efficient elongation of transcription, not initiation, at ATX1-regulated genes. PLoS Genet 8: e1003111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Q, Yue X, Zeng H, Zhu J (2014) The protein phosphatase RCF2 and its interacting partner NAC019 are critical for heat stress-responsive gene regulation and thermotolerance in Arabidopsis. Plant Cell 26: 438–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilfoyle TJ, Hagen G (2007) Auxin response factors. Curr Opin Plant Biol 10: 453–460 [DOI] [PubMed] [Google Scholar]
- Guo Y, Cai Z, Gan S (2004) Transcriptome of Arabidopsis leaf senescence. Plant Cell Environ 27: 521–549 [Google Scholar]
- Guo Y, Gan SS (2012) Convergence and divergence in gene expression profiles induced by leaf senescence and 27 senescence-promoting hormonal, pathological and environmental stress treatments. Plant Cell Environ 35: 644–655 [DOI] [PubMed] [Google Scholar]
- Higashi K, Ishiga Y, Inagaki Y, Toyoda K, Shiraishi T, Ichinose Y (2008) Modulation of defense signal transduction by flagellin-induced WRKY41 transcription factor in Arabidopsis thaliana. Mol Genet Genomics 279: 303–312 [DOI] [PubMed] [Google Scholar]
- Hinderhofer K, Zentgraf U (2001) Identification of a transcription factor specifically expressed at the onset of leaf senescence. Planta 213: 469–473 [DOI] [PubMed] [Google Scholar]
- Hou K, Wu W, Gan SS (2013) SAUR36, a SMALL AUXIN UP RNA gene, is involved in the promotion of leaf senescence in Arabidopsis. Plant Physiol 161: 1002–1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humbeck K. (2013) Epigenetic and small RNA regulation of senescence. Plant Mol Biol 82: 529–537 [DOI] [PubMed] [Google Scholar]
- Jaskiewicz M, Conrath U, Peterhänsel C (2011) Chromatin modification acts as a memory for systemic acquired resistance in the plant stress response. EMBO Rep 12: 50–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Liang G, Yang S, Yu D (2014) Arabidopsis WRKY57 functions as a node of convergence for jasmonic acid- and auxin-mediated signaling in jasmonic acid-induced leaf senescence. Plant Cell 26: 230–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson NL, Kotz S, Kemp AW (1992) Univariate Discrete Distributions. Wiley, New York [Google Scholar]
- Katari MS, Nowicki SD, Aceituno FF, Nero D, Kelfer J, Thompson LP, Cabello JM, Davidson RS, Goldberg AP, Shasha DE, et al. (2010) VirtualPlant: a software platform to support systems biology research. Plant Physiol 152: 500–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DH, Doyle MR, Sung S, Amasino RM (2009a) Vernalization: winter and the timing of flowering in plants. Annu Rev Cell Dev Biol 25: 277–299 [DOI] [PubMed] [Google Scholar]
- Kim JH, Woo HR, Kim J, Lim PO, Lee IC, Choi SH, Hwang D, Nam HG (2009b) Trifurcate feed-forward regulation of age-dependent cell death involving miR164 in Arabidopsis. Science 323: 1053–1057 [DOI] [PubMed] [Google Scholar]
- Kim JM, To TK, Ishida J, Matsui A, Kimura H, Seki M (2012) Transition of chromatin status during the process of recovery from drought stress in Arabidopsis thaliana. Plant Cell Physiol 53: 847–856 [DOI] [PubMed] [Google Scholar]
- Kim JM, To TK, Ishida J, Morosawa T, Kawashima M, Matsui A, Toyoda T, Kimura H, Shinozaki K, Seki M (2008) Alterations of lysine modifications on the histone H3 N-tail under drought stress conditions in Arabidopsis thaliana. Plant Cell Physiol 49: 1580–1588 [DOI] [PubMed] [Google Scholar]
- Kim JM, To TK, Nishioka T, Seki M (2010) Chromatin regulation functions in plant abiotic stress responses. Plant Cell Environ 33: 604–611 [DOI] [PubMed] [Google Scholar]
- Kim YS, Sakuraba Y, Han SH, Yoo SC, Paek NC (2013) Mutation of the Arabidopsis NAC016 transcription factor delays leaf senescence. Plant Cell Physiol 54: 1660–1672 [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauberth SM, Nakayama T, Wu X, Ferris AL, Tang Z, Hughes SH, Roeder RG (2013) H3K4me3 interactions with TAF3 regulate preinitiation complex assembly and selective gene activation. Cell 152: 1021–1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Prazak L, Chatterjee N, Gruninger S, Krug L, Theodorou D, Dubnau J (2013a) Activation of transposable elements during aging and neuronal decline in Drosophila. Nat Neurosci 16: 529–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Peng J, Wen X, Guo H (2012) Gene network analysis and functional studies of senescence-associated genes reveal novel regulators of Arabidopsis leaf senescence. J Integr Plant Biol 54: 526–539 [DOI] [PubMed] [Google Scholar]
- Li Z, Peng J, Wen X, Guo H (2013b) Ethylene-insensitive3 is a senescence-associated gene that accelerates age-dependent leaf senescence by directly repressing miR164 transcription in Arabidopsis. Plant Cell 25: 3311–3328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim PO, Kim HJ, Nam HG (2007a) Leaf senescence. Annu Rev Plant Biol 58: 115–136 [DOI] [PubMed] [Google Scholar]
- Lim PO, Kim Y, Breeze E, Koo JC, Woo HR, Ryu JS, Park DH, Beynon J, Tabrett A, Buchanan-Wollaston V, et al. (2007b) Overexpression of a chromatin architecture-controlling AT-hook protein extends leaf longevity and increases the post-harvest storage life of plants. Plant J 52: 1140–1153 [DOI] [PubMed] [Google Scholar]
- Liu X, Li Z, Jiang Z, Zhao Y, Peng J, Jin J, Guo H, Luo J (2011) LSD: a leaf senescence database. Nucleic Acids Res 39: D1103–D1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-González L, Mouriz A, Narro-Diego L, Bustos R, Martínez-Zapater M, Jarillo JA, Piñeiro M (2014) Chromatin-Dependent Repression of the Arabidopsis Floral Integrator Genes Involves Plant Specific PHD-Containing Proteins. Plant Cell 26: 3922–3938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-Seq data with DESeq2. Genome Biol 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malapeira J, Khaitova LC, Mas P (2012) Ordered changes in histone modifications at the core of the Arabidopsis circadian clock. Proc Natl Acad Sci USA 109: 21540–21545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashiguchi K, Tanaka K, Sakai T, Sugawara S, Kawaide H, Natsume M, Hanada A, Yaeno T, Shirasu K, Yao H, et al. (2011) The main auxin biosynthesis pathway in Arabidopsis. Proc Natl Acad Sci USA 108: 18512–18517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matallana-Ramirez LP, Rauf M, Farage-Barhom S, Dortay H, Xue GP, Dröge-Laser W, Lers A, Balazadeh S, Mueller-Roeber B (2013) NAC transcription factor ORE1 and senescence-induced BIFUNCTIONAL NUCLEASE1 (BFN1) constitute a regulatory cascade in Arabidopsis. Mol Plant 6: 1438–1452 [DOI] [PubMed] [Google Scholar]
- Miao Y, Jiang J, Ren Y, Zhao Z (2013) The single-stranded DNA-binding protein WHIRLY1 represses WRKY53 expression and delays leaf senescence in a developmental stage-dependent manner in Arabidopsis. Plant Physiol 163: 746–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao Y, Laun T, Zimmermann P, Zentgraf U (2004) Targets of the WRKY53 transcription factor and its role during leaf senescence in Arabidopsis. Plant Mol Biol 55: 853–867 [DOI] [PubMed] [Google Scholar]
- Miao Y, Zentgraf U (2010) A HECT E3 ubiquitin ligase negatively regulates Arabidopsis leaf senescence through degradation of the transcription factor WRKY53. Plant J 63: 179–188 [DOI] [PubMed] [Google Scholar]
- Molitor AM, Bu Z, Yu Y, Shen WH (2014) Arabidopsis AL PHD-PRC1 complexes promote seed germination through H3K4me3-to-H3K27me3 chromatin state switch in repression of seed developmental genes. PLoS Genet 10: e1004091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nightingale KP, Gendreizig S, White DA, Bradbury C, Hollfelder F, Turner BM (2007) Cross-talk between histone modifications in response to histone deacetylase inhibitors: MLL4 links histone H3 acetylation and histone H3K4 methylation. J Biol Chem 282: 4408–4416 [DOI] [PubMed] [Google Scholar]
- Oh SA, Park JH, Lee GI, Paek KH, Park SK, Nam HG (1997) Identification of three genetic loci controlling leaf senescence in Arabidopsis thaliana. Plant J 12: 527–535 [DOI] [PubMed] [Google Scholar]
- Okushima Y, Mitina I, Quach HL, Theologis A (2005) AUXIN RESPONSE FACTOR 2 (ARF2): a pleiotropic developmental regulator. Plant J 43: 29–46 [DOI] [PubMed] [Google Scholar]
- Quirino BF, Normanly J, Amasino RM (1999) Diverse range of gene activity during Arabidopsis thaliana leaf senescence includes pathogen-independent induction of defense-related genes. Plant Mol Biol 40: 267–278 [DOI] [PubMed] [Google Scholar]
- Roudier F, Ahmed I, Bérard C, Sarazin A, Mary-Huard T, Cortijo S, Bouyer D, Caillieux E, Duvernois-Berthet E, Al-Shikhley L, et al. (2011) Integrative epigenomic mapping defines four main chromatin states in Arabidopsis. EMBO J 30: 1928–1938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenke D, Cai D, Scheel D (2014) Suppression of UV-B stress responses by flg22 is regulated at the chromatin level via histone modification. Plant Cell Environ 37: 1716–1721 [DOI] [PubMed] [Google Scholar]
- Sequeira-Mendes J, Aragüez I, Peiró R, Mendez-Giraldez R, Zhang X, Jacobsen SE, Bastolla U, Gutierrez C (2014) The functional topography of the Arabidopsis genome is organized in a reduced number of linear motifs of chromatin states. Plant Cell 26: 2351–2366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spartz AK, Lee SH, Wenger JP, Gonzalez N, Itoh H, Inzé D, Peer WA, Murphy AS, Overvoorde PJ, Gray WM (2012) The SAUR19 subfamily of SMALL AUXIN UP RNA genes promote cell expansion. Plant J 70: 978–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spartz AK, Ren H, Park MY, Grandt KN, Lee SH, Murphy AS, Sussman MR, Overvoorde PJ, Gray WM (2014) SAUR inhibition of PP2C-D phosphatases activates plasma membrane H+-ATPases to promote cell explansion in Arabidopsis. Plant Cell 26: 2129–2142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepanova AN, Yun J, Robles LM, Novak O, He W, Guo H, Ljung K, Alonso JM (2011) The Arabidopsis YUCCA1 flavin monooxygenase functions in the indole-3-pyruvic acid branch of auxin biosynthesis. Plant Cell 23: 3961–3973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taverna SD, Ueberheide BM, Liu Y, Tackett AJ, Diaz RL, Shabanowitz J, Chait BT, Hunt DF, Allis CD (2007) Long-distance combinatorial linkage between methylation and acetylation on histone H3 N termini. Proc Natl Acad Sci USA 104: 2086–2091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thatcher SR, Burd S, Wright C, Lers A, Green PJ (2015) Differential expression of miRNAs and their target genes in senescing leaves and siliques: insights from deep sequencing of small RNAs and cleaved target RNAs. Plant Cell Environ 33: 188–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL (2009) TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25: 1105–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Graaff E, Schwacke R, Schneider A, Desimone M, Flügge UI, Kunze R (2006) Transcription analysis of Arabidopsis membrane transporters and hormone pathways during developmental and induced leaf senescence. Plant Physiol 141: 776–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dijk K, Ding Y, Malkaram S, Riethoven JJ, Liu R, Yang J, Laczko P, Chen H, Xia Y, Ladunga I, et al. (2010) Dynamic changes in genome-wide histone H3 lysine 4 methylation patterns in response to dehydration stress in Arabidopsis thaliana. BMC Plant Biol 10: 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Bouwmeester K, van de Mortel JE, Shan W, Govers F (2013) A novel Arabidopsis-oomycete pathosystem: differential interactions with Phytophthora capsici reveal a role for camalexin, indole glucosinolates and salicylic acid in defence. Plant Cell Environ 36: 1192–1203 [DOI] [PubMed] [Google Scholar]
- Won C, Shen X, Mashiguchi K, Zheng Z, Dai X, Cheng Y, Kasahara H, Kamiya Y, Chory J, Zhao Y (2011) Conversion of tryptophan to indole-3-acetic acid by TRYPTOPHAN AMINOTRANSFERASES OF ARABIDOPSIS and YUCCAs in Arabidopsis. Proc Natl Acad Sci USA 108: 18518–18523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu K, Zhang L, Zhou C, Yu CW, Chaikam V (2008) HDA6 is required for jasmonate response, senescence and flowering in Arabidopsis. J Exp Bot 59: 225–234 [DOI] [PubMed] [Google Scholar]
- Zhang X, Bernatavichute YV, Cokus S, Pellegrini M, Jacobsen SE (2009) Genome-wide analysis of mono-, di- and trimethylation of histone H3 lysine 4 in Arabidopsis thaliana. Genome Biol 10: R62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Clarenz O, Cokus S, Bernatavichute YV, Pellegrini M, Goodrich J, Jacobsen SE (2007) Whole-genome analysis of histone H3 lysine 27 trimethylation in Arabidopsis. PLoS Biol 5: e129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng MS, Takahashi H, Miyazaki A, Hamamoto H, Shah J, Yamaguchi I, Kusano T (2004) Up-regulation of Arabidopsis thaliana NHL10 in the hypersensitive response to Cucumber mosaic virus infection and in senescing leaves is controlled by signalling pathways that differ in salicylate involvement. Planta 218: 740–750 [DOI] [PubMed] [Google Scholar]
- Zhou J, Wang X, He K, Charron JBF, Elling AA, Deng XW (2010) Genome-wide profiling of histone H3 lysine 9 acetylation and dimethylation in Arabidopsis reveals correlation between multiple histone marks and gene expression. Plant Mol Biol 72: 585–595 [DOI] [PubMed] [Google Scholar]
- Zong W, Zhong X, You J, Xiong L (2013) Genome-wide profiling of histone H3K4-tri-methylation and gene expression in rice under drought stress. Plant Mol Biol 81: 175–188 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.