

A crystallographic cocktail screening campaign against 680 fragments revealed a binding hotspot in the structure of T. cruzi histidyl-tRNA synthetase.
Keywords: fragment-based crystallographic cocktail screening, structure-guided drug discovery, inhibitor of histidyl-tRNA synthetase, trypanosomiasis, reactive fragment
Abstract
American trypanosomiasis, commonly known as Chagas disease, is a neglected tropical disease caused by the protozoan parasite Trypanosoma cruzi. The chronic form of the infection often causes debilitating morbidity and mortality. However, the current treatment for the disease is typically inadequate owing to drug toxicity and poor efficacy, necessitating a continual effort to discover and develop new antiparasitic therapeutic agents. The structure of T. cruzi histidyl-tRNA synthetase (HisRS), a validated drug target, has previously been reported. Based on this structure and those of human cytosolic HisRS, opportunities for the development of specific inhibitors were identified. Here, efforts are reported to identify small molecules that bind to T. cruzi HisRS through fragment-based crystallographic screening in order to arrive at chemical starting points for the development of specific inhibitors. T. cruzi HisRS was soaked into 68 different cocktails from the Medical Structural Genomics of Pathogenic Protozoa (MSGPP) fragment library and diffraction data were collected to identify bound fragments after soaking. A total of 15 fragments were identified, all bound to the same site on the protein, revealing a fragment-binding hotspot adjacent to the ATP-binding pocket. On the basis of the initial hits, the design of reactive fragments targeting the hotspot which would be simultaneously covalently linked to a cysteine residue present only in trypanosomatid HisRS was initiated. Inhibition of T. cruzi HisRS was observed with the resultant reactive fragments and the anticipated binding mode was confirmed crystallographically. These results form a platform for the development of future generations of selective inhibitors for trypanosomatid HisRS.
1. Introduction
American trypanosomiasis, also known as Chagas disease, is a neglected tropical disease caused by the protozoan parasite Trypanosoma cruzi. The disease is endemic in Latin America (Coura & Viñas, 2010 ▸). Based on data from the Global Burden of Disease Study 2010, 7.5 million cases of the disease were estimated for the year 2010, resulting in 0.55 million lost disability-adjusted life years (Hotez et al., 2014 ▸). Chagas disease is most commonly transmitted through the blood-feeding vector triatomines (kissing bugs), although nonvector transmissions are also prevalent (Andrade et al., 2014 ▸). The only two drugs that are currently used for treatment, benznidazole and nifurtimox, have well documented side effects. The discovery and development of new treatments for the disease has been challenging owing to a number of issues (Chatelain, 2015 ▸). For example, despite promising in vitro and in vivo pre-clinical data, two recent drug candidates, posaconazole and E1224, a prodrug of ravuconazole, have failed in their respective clinical trials, appearing to be unable to maintain sustained efficacy after the end of treatment (Chatelain, 2015 ▸). Therefore, in the search for a safe and efficacious treatment of Chagas disease, a pipeline of new compounds against different and novel targets must be pursued.
A group of well validated antiparasitic drug targets consists of the aminoacyl-tRNA synthetases (aaRSs; Pham et al., 2014 ▸; Kalidas et al., 2014 ▸; Shibata et al., 2011 ▸, 2012 ▸; Merritt et al., 2010 ▸). AaRSs are a group of essential enzymes involved in protein synthesis. Typically, a specific aaRS recognizes and charges a specific amino acid to its cognate tRNA. The amino acid charged onto its tRNA will subsequently be incorporated into the growing polypeptide chain through recognition by the tRNA of the anticodon on the mRNA at the ribosome (Ibba & Söll, 2000 ▸). In general, aminoacylation of tRNA occurs in two steps: (i) the formation of an active aminoacyl-adenylate intermediate after recognition of the amino acid and ATP and (ii) the transfer of the aminoacyl group to the terminal ribose 2′-OH or 3′-OH of the acceptor stem of the cognate tRNA. Perturbation of the aminoacylation reaction in any of these steps results in the disruption of protein translation, thereby killing the pathogens (Pham et al., 2014 ▸; Kalidas et al., 2014 ▸; Shibata et al., 2011 ▸, 2012 ▸; Merritt et al., 2010 ▸).
Similar to other well validated drug targets, a structure-based approach to the drug-discovery process can be very useful. Therefore, we and others have determined the crystal structures of aaRSs of mostly pathogenic organisms in complex with inhibitors to guide the drug-design and drug-discovery process. These complexes include those of methionyl-tRNA synthetase (MetRS; Koh et al., 2012 ▸; Koh, Kim et al., 2014 ▸), TyrRS (Larson et al., 2011 ▸; Qiu et al., 2001 ▸), IleRS (Silvian et al., 1999 ▸; Nakama et al., 2001 ▸), PheRS (Evdokimov et al., 2008 ▸; Abibi et al., 2014 ▸), LeuRS (Rock et al., 2007 ▸; Seiradake et al., 2009 ▸; Chopra et al., 2013 ▸; Hernandez et al., 2013 ▸; Palencia et al., 2012 ▸; Hu et al., 2013 ▸), ThrRS (Teng et al., 2013 ▸), LysRS (Khan et al., 2014 ▸) and ProRS (Son et al., 2013 ▸; Zhou et al., 2013 ▸). Many other inhibitors of aaRSs have been reported, although structures in complex with their targets are not available (for a recent review, see Gadakh & Van Aerschot, 2012 ▸). Starting points for these inhibitors varied from natural substrates to natural products and synthetic compounds identified by high-throughput screens. In many cases, determination of protein•ligand complex structures elucidates the binding mode of ligands and this information can guide subsequent optimization of inhibitors (see, for example, Phillips & Rathod, 2010 ▸).
We have previously determined structures of T. cruzi HisRS (TcHisRS) in complex with histidine (His) and histidyl-adenosine monophosphate (HAMP) (i.e. TcHisRS•His and TcHisRS•HAMP, where the symbol ‘•’ represents ‘forming a noncovalent complex with’), ligand-free T. brucei HisRS (TbHisRS; Merritt et al., 2010 ▸) and human cytosolic HisRS in complex with His (Hs-cHisRS•His; Koh, Wetzel et al., 2014 ▸). The resolution limits of the TcHisRS•His, TcHisRS•HAMP, TbHisRS and Hs-cHisRS•His structures are 1.80, 3.05, 2.85 and 2.84 Å, respectively. We observed and reported substantial differences between the structures of trypanosomatid HisRS and their human ortholog in the active site. Two putative selectivity pockets were identified based on comparisons of these HisRS structures in complex with various natural substrates (Koh, Wetzel et al., 2014 ▸). The pockets, named ‘pocket F’ (observed in ligand-free TbHisRS) and ‘pocket H’ (observed in TcHisRS•His), are adjacent and are connected to the histidine-binding and ATP-binding pockets of the respective HisRS. Therefore, we hypothesize that targeting these pockets, which are observed only in trypanosomatid HisRS and not in Hs-cHisRS, should impart selectivity to inhibitors that would extend into the substrate pockets and compete with substrate binding. Considering the feasibility of obtaining high-resolution structures of TcHisRS•His, we report here a crystallographic fragment screening approach as a focused search for chemical starting points to target pocket H or other possible selectivity pockets in TcHisRS.
Over the last two decades, fragment-based drug discovery (FBDD) has become a major approach in drug-discovery projects (Zartler, 2014 ▸; Davis & Erlanson, 2013 ▸; Baker, 2013 ▸). A primary screen in FBDD can be performed using a large variety of biophysical methods, although surface plasmon resonance (SPR), nuclear magnetic resonance (NMR) and differential scanning fluorimetry (thermal shift) are among the most popular (Davis & Erlanson, 2013 ▸). Crystallography, in contrast, is mostly used as a secondary approach in FBDD after a primary screening method has filtered out a large number of fragments in the library. The application of crystallography to FBDD by soaking crystals in a mixture of fragments as cocktails was initially explored in the early 1990s (Verlinde et al., 1997 ▸, 2009 ▸). Subsequently, better designs of cocktail libraries and improvements in crystallographic software and hardware such as synchrotron radiation consolidated the technique as an important tool in FBDD (Nienaber et al., 2000 ▸; Patel et al., 2014 ▸). The use of crystallography as a primary fragment screen has several advantages, mostly because it enables direct observation of the binding modes of fragment hits. As a result, the rate of false-positive hits is low. It allows rapid structure-based design of subsequent generations of compounds. It also provides immediate feedback on the possibilities of growing, linking and/or merging fragments, and in addition helps with efficient and accurate clustering, prioritization and development of hits. Here, we report the results of using TcHisRS•His crystals in a crystallographic fragment screen and the initial modifications of bound fragments that have excellent potential for imparting selectivity in future generations of inhibitors.
2. Materials and methods
2.1. Fragment cocktails
The details of the construction of the 68 cocktails have been described previously (Verlinde et al., 2009 ▸). Briefly, under the Medical Structural Genomics of Pathogenic Protozoa (MSGPP) project, 680 compounds were selected based on a number of criteria (number of halogens < 2, molecular weight < 200, clogP < 3, number of rotatable bonds < 3, number of rings ≥ 1, without reactive groups). These compounds were split into 68 cocktails of ten compounds each with maximum shape diversity. Each cocktail contains 10 mM of each individual fragment in 100% DMSO.
2.2. Cloning, expression and purification of HisRS
The cloning, expression and purification of TcHisRS (Merritt et al., 2010 ▸) and Hs-cHisRS (Koh, Wetzel et al., 2014 ▸) have been described previously. Briefly, the nucleotide sequence corresponding to amino-acid residues 45–478 of TcHisRS was cloned into the AVA0421 vector by ligation-independent cloning (LIC). AVA0421 is a LIC-ready, pET-14b-modified expression vector with an N-terminal hexahistidine tag and a modified human rhinovirus 3C protease recognition site (Mehlin et al., 2006 ▸; Choi et al., 2011 ▸). The protein was expressed in Escherichia coli BL21 (DE3) host cells in autoinduction medium (Studier, 2005 ▸) and was purified using an Ni–NTA affinity column. For TcHisRS, the eluted protein, without cleavage of the expression tag, was further purified on a Mono S cation-exchange column (GE Healthcare Life Sciences, USA) at pH 6.8 before a final size-exclusion step using a Superdex 200 column (GE Healthcare Life Sciences, USA). For Hs-cHisRS, the expression tag of the eluted protein was cleaved using 3C protease before purification by size exclusion using a Superdex 200 column (GE Healthcare Life Sciences, USA). The eluted proteins were concentrated to 12 mg ml−1 and flash-frozen for storage. The final protein buffer consisted of 25 mM HEPES, 500 mM NaCl, 2 mM DTT, 5% glycerol, 0.025% NaN3 at pH 7.0.
2.3. Crystallization and soaks
2.3.1. Crystallization
TcHisRS•His tends to crystallize in multiple crystal forms, often in the same drop, under the previously reported crystallization condition (0.2 M lithium sulfate or ammonium sulfate, 26% PEG 3350, 0.1 M bis-tris pH 5.5, 1 mM TCEP; Merritt et al., 2010 ▸). We found that only two of those crystal forms, forms I and II, diffracted to sufficiently high resolutions (better than 2.5 Å) for crystallographic fragment screening. Initial efforts towards crystallization were hindered by the presence of other crystal forms. The inconsistency was attributed to the weak buffering capacity of bis-tris at pH 5.5. Therefore, 0.1 M sodium citrate buffer at pH 4.8–5.3 was used to replace the bis-tris, resulting in the elimination of other weakly diffracting crystal forms.
2.3.2. Cocktail soaks
Simultaneous cryoprotection and fragment soaks were carried out in a single soaking solution containing the crystallization reservoir supplemented with 10% ethylene glycol and 15% cocktail solution, giving final concentrations of 1.5 mM of each fragment in 15% DMSO. Crystals were soaked in the solution for 0.5–2 min before flash-cooling in liquid nitrogen for data collection.
2.3.3. Single soaks
When a new density feature was determined as a result of a particular cocktail soak, the putative hit was selected from the list of fragments present in the cocktail. A new stock solution for the putative hit was prepared at 1 M and this single fragment was soaked into crystals to confirm the identity of a hit. Subsequent structure refinements were performed using data collected from single soaks.
2.4. Data collection, structure determination and refinement
All data, except for those from crystals soaked with Chem 1698, were collected in-house using a MicroMax-007 HF rotating-anode generator (Rigaku) equipped with VariMax HF (Osmic) optics and a Saturn 994 (Rigaku) CCD detector at a wavelength of 1.54 Å. Data from crystals soaked with Chem 1698 were collected on Stanford Synchrotron Radiation Lightsource (SSRL) beamline 12-2 at a wavelength of 1 Å and were integrated with either XDS (Kabsch, 2010 ▸) or HKL-2000 (Otwinowski & Minor, 1997 ▸) and scaled with AIMLESS (Winn et al., 2011 ▸).
Data collected from two different forms of crystals, forms I and II (see §2.3.1), were used to determine structures of TcHisRS•His. The form I crystal is the previously reported form of TcHisRS•His crystals (PDB entry 3lc0, space group C2, unit-cell parameters a = 90, b = 119, c = 66 Å, β = 133°; Merritt et al., 2010 ▸), but is here reported in space group I2 (unit-cell parameters a = 65, b = 119, c = 66 Å, β = 93°) as we use the crystal setting that results in the smaller β angle, as recommended by the IUCr convention. It has one copy of the protein in the asymmetric unit. Form II with space group C2 (unit-cell parameters a = 90, b = 119, c = 94 Å, β = 91°) is related to form I but with a strong non-origin peak in the native Patterson, indicating translational noncrystallographic symmetry, the result of two similarly oriented copies of the protein in one asymmetric unit. Structures were solved by molecular replacement using Phaser (McCoy et al., 2007 ▸). TcHisRS forms dimers in solution, which correspond to dimers with large interfaces generated through crystallographic twofold symmetry in the crystals. Structures of both form I and II crystals soaked in 15% DMSO, representing ‘reference’ structures without fragments, were also determined.
New density features in fragment-soaked crystals that are not owing to changes in protein conformation and solvent ligands such as DMSO were identified after an initial round of limited refinement. Data collected from crystals soaked with a single fragment were used for refinement and deposited in the Protein Data Bank. Model building/rebuilding was performed in Coot (Emsley & Cowtan, 2004 ▸). Refinement was carried out with REFMAC5 (Murshudov et al., 2011 ▸), with translational/libration/screw (TLS) groups identified by the TLS Motion Determination server (Painter & Merritt, 2006 ▸). In form II crystals, global noncrystallographic symmetry restraints were applied during refinement. Throughout the model-building process, the MolProbity structure-validation server (Chen et al., 2010 ▸) and the wwPDB validation server (http://wwpdb.org/ValidationPDFNotes.html) were used to guide model building. The real-space R-factor (RSR) values and local ligand-density fit (LLDF) scores generated by the wwPDB validation server were used to judge the quality of density fit of fragments. The final crystallographic data-collection and refinement statistics are given in Table 1 ▸. Figures were created and rendered with PyMOL (DeLano, 2002 ▸). Sequence alignment was performed using ClustalW (Larkin et al., 2007 ▸) and rendered with ESPript (Robert & Gouet, 2014 ▸).
Table 1. Crystallographic data-collection and refinement statistics.
Values in parentheses are for the highest resolution shell.
| Fragment | Chem 79 | Chem 84 | Chem 89 | Chem 744 | Chem 148 | Chem 145 | Chem 149 | Chem 166 | Chem 256 |
|---|---|---|---|---|---|---|---|---|---|
| PDB code | 4yp0 | 4ypf | 4yrc | 4yrq | 4yrf | 4yre | 4yrg | 4yri | 4yrj |
| Data collection | |||||||||
| Space group | I2 | I2 | I2 | I2 | I2 | C2 | I2 | I2 | I2 |
| Unit-cell parameters | |||||||||
| a () | 64.8 | 64.7 | 65.2 | 65.1 | 64.4 | 89.9 | 64.6 | 64.4 | 64.9 |
| b () | 118.9 | 119.1 | 119.4 | 119.3 | 118.8 | 118.6 | 119.4 | 118.7 | 119.3 |
| c () | 65.9 | 66.1 | 65.7 | 66.1 | 66.0 | 93.5 | 66.1 | 66.1 | 66.2 |
| () | 92.9 | 92.6 | 93.6 | 92.9 | 92.4 | 91.3 | 92.6 | 92.7 | 92.6 |
| Resolution () | 34.02.10 (2.162.10) | 36.02.20 (2.272.20) | 37.32.10 (2.162.10) | 27.82.05 (2.112.05) | 29.02.20 (2.272.20) | 33.22.25 (2.322.25) | 37.12.15 (2.222.15) | 36.92.00 (2.052.00) | 29.82.30 (2.382.30) |
| R merge | 0.053 (0.502) | 0.061 (0.581) | 0.062 (0.544) | 0.055 (0.336) | 0.044 (0.374) | 0.113 (0.563) | 0.077 (0.693) | 0.047 (0.325) | 0.084 (0.658) |
| R p.i.m. | 0.035 (0.402) | 0.041 (0.432) | 0.048 (0.430) | 0.037 (0.270) | 0.031 (0.282) | 0.097 (0.504) | 0.047 (0.473) | 0.032 (0.259) | 0.051 (0.410) |
| Observed reflections | 83516 (3729) | 72379 (4875) | 68265 (3335) | 87563 (4470) | 66337 (4517) | 116068 (10338) | 97263 (5496) | 87865 (2993) | 80135 (6859) |
| Unique reflections | 27636 (1725) | 24924 (1966) | 27892 (1663) | 31011 (2186) | 24799 (2066) | 45938 (4261) | 26213 (1800) | 31161 (1444) | 21706 (1972) |
| Mean I/(I) | 15.7 (1.8) | 15.1 (1.8) | 9.8 (1.7) | 10.6 (2.4) | 18.0 (2.2) | 8.6 (1.9) | 14.0 (1.8) | 18.5 (2.6) | 8.7 (1.7) |
| Multiplicity | 3.0 (2.2) | 2.9 (2.5) | 2.4 (2.0) | 2.8 (2.0) | 2.7 (2.2) | 2.5 (2.4) | 3.7 (3.1) | 2.8 (2.1) | 3.7 (3.5) |
| Completeness (%) | 95.2 (72.4) | 98.4 (90.2) | 95.5 (69.5) | 98.2 (89.6) | 98.6 (96.3) | 99.0 (99.5) | 96.4 (76.5) | 93.2 (58.5) | 97.2 (89.2) |
| CC1/2 | 0.999 (0.690) | 0.999 (0.740) | 0.997 (0.722) | 0.998 (0.799) | 0.999 (0.864) | 0.993 (0.669) | 0.998 (0.643) | 0.999 (0.859) | 0.997 (0.681) |
| Refinement | |||||||||
| Resolution () | 34.02.10 | 36.02.20 | 37.32.10 | 27.82.05 | 29.02.20 | 32.82.25 | 30.02.15 | 30.02.00 | 29.82.30 |
| Reflections used | 26205 | 23616 | 26457 | 29417 | 23499 | 43599 | 24843 | 29566 | 20575 |
| R work/R free | 0.188/0.226 | 0.201/0.243 | 0.184/0.219 | 0.184/0.218 | 0.212/0.261 | 0.200/0.226 | 0.199/0.237 | 0.190/0.228 | 0.205/0.245 |
| No. of atoms | |||||||||
| Protein | 3208 | 3122 | 3291 | 3178 | 3130 | 6418 | 3151 | 3154 | 3197 |
| His | 11 | 11 | 11 | 11 | 11 | 22 | 11 | 11 | 11 |
| Other solvent ligands | 22 | 27 | 26 | 17 | 27 | 61 | 27 | 34 | 27 |
| Water | 154 | 95 | 146 | 203 | 152 | 336 | 138 | 199 | 101 |
| Average B factors (2) | |||||||||
| Protein | 46.5 | 55.6 | 53.8 | 41.2 | 57.7 | 32.4 | 59.2 | 36.6 | 60.9 |
| His | 24.8 | 33.0 | 32.7 | 23.2 | 32.8 | 17.0 | 34.6 | 20.3 | 40.1 |
| Other solvent ligands | 57.2 | 58.2 | 54.7 | 47.3 | 68.0 | 41.0 | 57.0 | 38.4 | 73.4 |
| Water | 40.7 | 41.0 | 43.9 | 39.9 | 48.1 | 28.5 | 41.3 | 36.1 | 46.1 |
| R.m.s. deviations | |||||||||
| Bond lengths () | 0.006 | 0.007 | 0.007 | 0.007 | 0.006 | 0.07 | 0.007 | 0.007 | 0.006 |
| Bond angles () | 1.10 | 1.13 | 1.15 | 1.09 | 1.07 | 1.14 | 1.10 | 1.16 | 1.04 |
| Ramachandran plot† | |||||||||
| Favored (%) | 98 | 98 | 98 | 98 | 96 | 99 | 98 | 99 | 98 |
| Outliers (%) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Fragments‡ | |||||||||
| No. of atoms | 11/11 | 11 | 12/12 | 12 | 8 | 9/9/9/9 | 9/9 | 9/9 | 9 |
| Average B factors (2) | 43.0/42.9 | 51.9 | 56.4/41.3 | 48.8 | 90.0 | 33.0/26.3/35.5/27.8 | 43.8/44.7 | 27.7/32.3 | 46.7 |
| LLDF§ | 1.75/0.97 | 1.85 | 0.15/1.67 | 2.77 | 1.29 | 1.29/1.29/1.48/1.48 | 0.70/0.70 | 0.41/0.41 | 0.38 |
| RSR¶ | 0.19/0.19 | 0.10 | 0.13/0.23 | 0.22 | 0.21 | 0.09/0.09/0.10/0.10 | 0.11/0.11 | 0.12/0.12 | 0.12 |
| Fragment | Chem 262 | Chem 260 | Chem 443 | Chem 475 | Chem 707 | Chem 491 | Chem 1691 | Chem 1698 | Chem 1781 |
|---|---|---|---|---|---|---|---|---|---|
| PDB code | 4yrl | 4yrk | 4yrm | 4yrn | 4yrp | 4yro | 4yrr | 4yrs | 4yrt |
| Data collection | |||||||||
| Space group | I2 | C2 | C2 | C2 | C2 | I2 | C2 | I2 | I2 |
| Unit-cell parameters | |||||||||
| a () | 64.8 | 89.9 | 90.1 | 90.3 | 90.2 | 64.6 | 90.0 | 66.5 | 65.4 |
| b () | 118.7 | 118.7 | 119.5 | 119.2 | 119.2 | 118.3 | 118.4 | 119.7 | 119.3 |
| c () | 66.1 | 93.7 | 94.6 | 94.2 | 94.3 | 66.1 | 93.8 | 66.8 | 66.3 |
| () | 92.5 | 91.5 | 91.0 | 91.3 | 91.4 | 92.5 | 91.2 | 94.2 | 93.4 |
| Resolution () | 29.02.30 (2.382.30) | 35.82.20 (2.272.20) | 36.02.30 (2.382.30) | 36.42.25 (2.322.25) | 33.02.20 (2.272.20) | 33.92.50 (2.602.50) | 36.82.30 (2.382.30) | 37.82.75 (2.902.75) | 37.42.05 (2.112.05) |
| R merge | 0.048 (0.536) | 0.100 (0.552) | 0.067 (0.471) | 0.100 (0.536) | 0.078 (0.500) | 0.063 (0.435) | 0.101 (0.822) | 0.054 (0.729) | 0.054 (0.461) |
| R p.i.m. | 0.043 (0.483) | 0.070 (0.427) | 0.045 (0.326) | 0.069 (0.375) | 0.068 (0.440) | 0.050 (0.345) | 0.061 (0.533) | 0.030 (0.402) | 0.041 (0.368) |
| Observed reflections | 45045 (3867) | 136941 (6971) | 137111 (11967) | 136922 (12182) | 101869 (6979) | 39544 (4430) | 161495 (14058) | 53548 (8173) | 79897 (4533) |
| Unique reflections | 21307 (1910) | 48253 (3335) | 44048 (4279) | 46858 (4286) | 49246 (3909) | 16909 (1931) | 43573 (4228) | 13023 (1938) | 31152 (2171) |
| Mean I/(I) | 15.1 (1.9) | 9.7 (1.6) | 12.9 (2.3) | 9.8 (2.1) | 9.1 (1.7) | 14.4 (2.2) | 12.6 (1.9) | 17.0 (2.0) | 15.6 (1.9) |
| Multiplicity | 2.1 (2.0) | 2.8 (2.1) | 3.1 (2.8) | 2.9 (2.8) | 2.1 (1.8) | 2.3 (2.3) | 3.7 (3.3) | 4.1 (4.2) | 2.6 (2.1) |
| Completeness (%) | 96.5 (89.2) | 96.9 (77.7) | 99.2 (98.3) | 99.2 (99.3) | 97.6 (89.8) | 98.4 (99.2) | 99.8 (98.6) | 96.2 (97.6) | 98.0 (87.7) |
| CC1/2 | 0.999 (0.719) | 0.996 (0.671) | 0.999 (0.857) | 0.996 (0.713) | 9.1 (1.7) | 0.997 (0.804) | 0.998 (0.685) | 0.999 (0.732) | 0.998 (0.74) |
| Refinement | |||||||||
| Resolution () | 28.42.30 | 30.02.20 | 29.92.30 | 30.02.20 | 29.12.20 | 30.02.50 | 36.152.30 | 37.82.75 | 29.12.05 |
| Reflections used | 20198 | 45798 | 41781 | 47387 | 46700 | 16036 | 41316 | 12361 | 29548 |
| R work/R free | 0.199/0.240 | 0.199/0.227 | 0.214/0.242 | 0.205/0.240 | 0.221/0.252 | 0.186/0.231 | 0.217/0.245 | 0.181/0.237 | 0.185/0.226 |
| No. of atoms | |||||||||
| Protein | 3161 | 6133 | 6401 | 6405 | 6291 | 3254 | 6269 | 3237 | 3217 |
| His | 11 | 22 | 22 | 22 | 22 | 11 | 22 | 11 | 11 |
| Other solvent ligands | 27 | 49 | 35 | 65 | 57 | 31 | 49 | 0 | 9 |
| Water | 97 | 291 | 276 | 452 | 246 | 99 | 225 | 30 | 217 |
| Average B factors (2) | |||||||||
| Protein | 61.8 | 36.0 | 46.2 | 38.6 | 39.0 | 62.2 | 45.9 | 44.6 | 39.0 |
| His | 36.2 | 19.6 | 23.5 | 18.8 | 21.9 | 35.2 | 24.4 | 57.1 | 21.5 |
| Other solvent ligands | 69.5 | 49.8 | 60.6 | 46.4 | 49.4 | 65.3 | 58.0 | 47.7 | |
| Water | 44.4 | 31.5 | 35.3 | 32.9 | 30.3 | 36.0 | 35.8 | 58.9 | 36.6 |
| R.m.s. deviations | |||||||||
| Bond lengths () | 0.007 | 0.007 | 0.007 | 0.007 | 0.007 | 0.007 | 0.007 | 0.007 | 0.007 |
| Bond angles () | 1.11 | 1.14 | 1.124 | 1.12 | 1.08 | 1.18 | 1.08 | 1.11 | 1.15 |
| Ramachandran plot† | |||||||||
| Favored (%) | 97 | 98 | 98 | 99 | 99 | 97 | 98 | 97 | 98 |
| Outliers (%) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Fragments‡ | |||||||||
| No. of atoms | 9 | 9/9 | 8/8/8/8 | 8/8 | 9/9/9/9 | 9 | 14/14/14/14 | 15 | 16 |
| Average B factors (2) | 58.7 | 32.9/36.0 | 42.5/46.0/39.9/43.0 | 27.2/25.5 | 43.6/56.2/48.9/55.8 | 51.3 | 47.2/57.4/50.6/55.0 | 76.6 | 32.3 |
| LLDF§ | 1.30 | 2.43/0.18 | 1.21/1.21/0.90/0.90 | 2.15/2.32 | 2.05/2.05/0.73/0.73 | 1.78 | 0.54/0.54/1.65/1.74 | 0.12 | 0.51 |
| RSR¶ | 0.22 | 0.11/0.13 | 0.20/0.20/0.19/0.19 | 0.07/0.08 | 0.19/0.19/0.17/0.17 | 0.10 | 0.18/0.18/0.24/0.24 | 0.18 | 0.15 |
Ramachandran plot statistics as reported by MolProbity (Chen et al., 2010 ▸).
When more than one fragment is present owing to alternative conformations, extra copies in the asymmetric unit and/or a second binding site, the values for each are separated by a forward slash.
Local ligand density fit as reported by the wwPDB validation report.
Real-space R value as reported by the wwPDB validation report.
2.5. Differential thermal fluorimetry
The thermal shift assay was performed as described previously (Shibata et al., 2011 ▸, 2012 ▸) using 0.5 mg ml−1 TcHisRS, 1 mM fragments and 10% DMSO with or without 1 mM histidine. The assays were repeated three times independently.
2.6. Compound synthesis
Unless otherwise stated, all chemicals were purchased from commercial suppliers and were used without further purification. The final purity of all compounds was determined by analytical LCMS with a Phenomenex Onyx Monolithic C18 column (4.6 × 100 mm). The products were detected by UV at 220 nm. All compounds were determined to be >95% pure by this method. The mass spectra were recorded with an Agilent Ion Trap Mass Spectrometer. NMR spectra were recorded on either a Bruker 500 MHz spectrometer or a Bruker 300 MHz spectrometer at ambient temperature. Chem 1691 (Batt et al., 2003 ▸), Chem 1697 (Ratnakar et al., 2012 ▸) and Chem 1698 (Guerry et al., 2003 ▸) are known compounds. Details of the synthesis and characterization data of Chem 1781 can be found in the Supporting Information.
2.7. Aminoacylation assays
Fragments were tested in the TcHisRS aminoacylation assay using the methodology described previously (Pedro-Rosa et al., 2015 ▸; Shibata et al., 2011 ▸) with assay conditions optimized for TcHisRS. Fragments (three serial dilutions starting at 2 mM assayed in triplicate) were pre-incubated for 30 min at room temperature with 13 mM MgCl2, 15 mM KCl, 50 mM HEPES–KOH pH 7.5, 0.1 mM ATP, 0.1 U ml−1 pyrophosphatase (Sigma–Aldrich), 0.2 mM spermine, 0.1 mg ml−1 bovine serum albumin, 2.5 mM dithiothreitol, 2% DMSO, 418 nM [3H]-l-histidine (47.9 Ci mmol−1) and 3 nM T. cruzi HisRS in 75 µl total reaction volume per well assayed in plates with Durapore membranes (Millipore). The reaction was started by adding 800 µg ml−1 bulk brewer’s yeast tRNA (Roche) and was incubated at room temperature for 120 min. Reactions were quenched by adding 100 µl ice-cold 10% trichloroacetic acid per well, precipitated for approximately 10 min at −20°C and then washed three times with 300 µl ice-cold 10% trichloroacetic acid per well using a 96-well plate vacuum manifold. The plates were dried, 25 µl scintillation fluid was added per well and the plates were read on a scintillation plate reader. Percentage inhibition was calculated using a high control (wells without inhibitors) and a low control (wells without bulk brewer’s yeast tRNA). IC50 values were calculated from sigmoidal dose–response curves in GraphPad Prism 3.0.
Fragments that had >50% inhibition at a concentration of 2 mM against TcHisRS were then tested against the human mitochondrial HisRS and the human cytosolic HisRS in three serial dilutions starting at 2 mM assayed in triplicate. Either human mitochondrial HisRS (10 nM) or human cytosolic HisRS (2 nM) was pre-incubated for 30 min at room temperature with 6 mM MgCl2, 25 mM KCl, 50 mM HEPES–KOH pH 7.6, 0.2 mM ATP, 0.1 U ml−1 pyrophosphatase (Sigma–Aldrich), 0.2 mM spermine, 0.1 mg ml−1 bovine serum albumin, 2.5 mM dithiothreitol, 2% DMSO and 418 nM [3H]-l-histidine (47.9 Ci mmol−1) in 75 µl total reaction volume per well assayed in plates with Durapore membranes (Millipore). The reaction was started by adding 400 µg ml−1 bulk brewer’s yeast tRNA (Roche) and was incubated at room temperature for 120 min. Reactions were quenched by adding 100 µl ice-cold 10% trichloroacetic acid per well and were processed as described above.
3. Results
3.1. Cocktail soaks and identification of hits
Of the total of 68 cocktails used to soak TcHisRS•His crystals, 15 showed the presence of possible hits as judged by the appearance of new density in the difference map after a round of limited refinement without placement of any fragment in density. Therefore, 22% of the cocktails contained putative hits. Single soaks of all of the candidate fragments identified as possible hits were subsequently performed to confirm the identity of the bound fragments. As a result, 15 fragments, one from each of the cocktails mentioned above, were confirmed as hits. Data sets from these 15 single soaks were used to refine the structure (Table 1 ▸). This produced a final confirmed hit rate of 2.2% based on a total of 680 fragments present in the library. All of the hits contained a single six-membered or double aromatic ring system. Many of the fragments were substituted with halogen atoms. All 15 hits are listed in Table 2 ▸ with their chemical structures. Two additional fragments also produced weak densities in the single soaks. These two fragments were not from unique cocktails but from cocktails that each also had a primary candidate hit identified. This implies that at least in some cocktails more than a single fragment can occupy the same binding site. No follow-up was performed for these two weakly binding fragments.
Table 2. Chemical structures and activities of fragment hits.
| T m (C) | |||||
|---|---|---|---|---|---|
| Fragment | Chemical structure | Without His | With His | IC50 (mM) | Inhibition at 2mM (%) |
| Chem 79 |
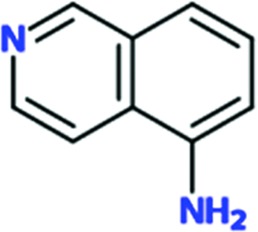
|
0.3 | 0.1 | ND | ND |
| Chem 84 |
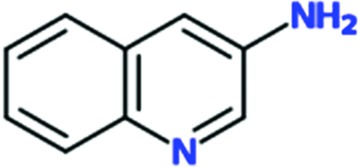
|
0.8 | 1.6 | >2 | 20 |
| Chem 89 |
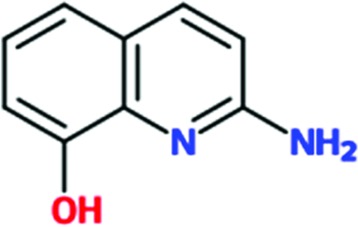
|
0.4 | 0.5 | >2 | 39 |
| Chem 744 |
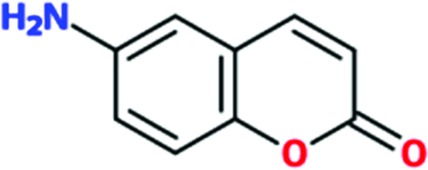
|
0.6 | 0.1 | ND | ND |
| Chem 148 |
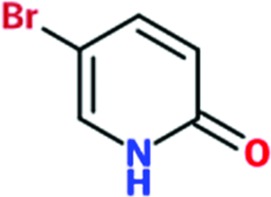
|
0.4 | 0.1 | ND | ND |
| Chem 145 |
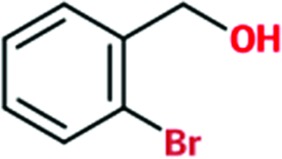
|
0.3 | 0.1 | ND | ND |
| Chem 149 |
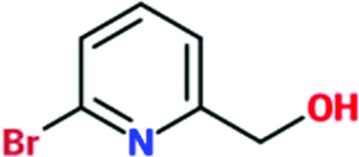
|
0.2 | 0.2 | >2 | 21 |
| Chem 166 |
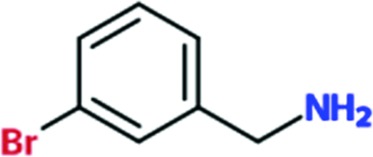
|
0.3 | 0.1 | ND | ND |
| Chem 256 |
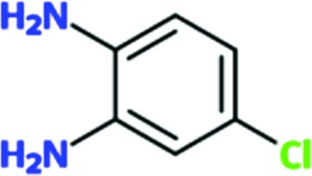
|
0.1 | 0.1 | ND | ND |
| Chem 262 |
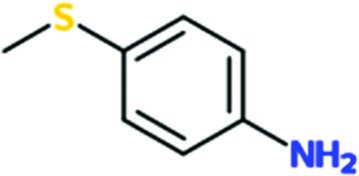
|
0.3 | 0.1 | ND | ND |
| Chem 260 |
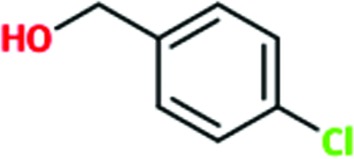
|
0.4 | 0.5 | ND | ND |
| Chem 443 |
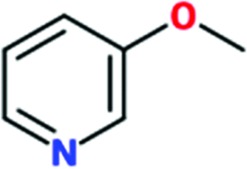
|
0.3 | 0.1 | ND | ND |
| Chem 475 |
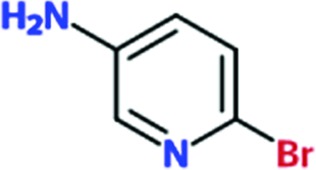
|
0.4 | 0 | ND | ND |
| Chem 707 |
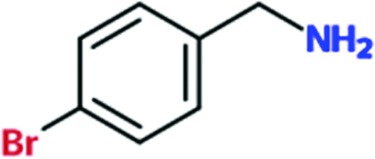
|
0.2 | 0.4 | ND | ND |
| Chem 491 |
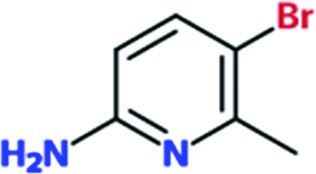
|
0.4 | 0.4 | ND | ND |
For all of the structures solved, either from cocktail soaks or single soaks, the maps clearly show the presence of histidine (Fig. 1 ▸). The fragment hits can be confidently identified since (i) all cocktail hits can be deconvoluted using single soaks and (ii) there are concomitant changes in the conformation of the side chains of two neighboring protein residues, Arg156 and His168. The changes in these side chains and their interactions with all fragments will be discussed in detail in §3.2. However, interpretation of the difference map to fit the fragments is sometimes challenging, presumably owing to complications such as weak affinity, low fragment solubility and the possibility of multiple binding modes (Fig. 1 ▸ and Supplementary Fig. S1). As a result, some of the fragments are fitted with alternative binding modes to fully account for the observed density (Figs. 1 ▸ c and 1 ▸ d). The quality of the fit to densities varies among fragments. The average B factors of fragments are typically higher than that of histidine, indicating weaker binding than the amino acid (Table 1 ▸). Nonetheless, the fit to density is good for most fragments, as indicated by the maps (Fig. 1 ▸ and Supplementary Fig. S1), real-space R factor (RSR) values and local ligand-density fit (LLDF) scores generated by the wwPDB validation server (Read et al., 2011 ▸; Table 1 ▸). The presence of electron-rich atoms such as sulfur, chlorine and bromine in the fragments sometimes helped in guiding the fit.
Figure 1.

Examples of electron densities of fragments and histidine bound to TcHisRS. σA-weighted F obs − F calc electron-density maps calculated by omitting histidine and fragments are shown in green (positive) and red (negative) contours at the 3σ level, and σA-weighted 2F obs − F calc electron densities are shown in light blue at the 1.0σ level. Histidine and fragments are depicted as ball-and-stick models, while TcHisRS is shown in ribbon representation. The bound fragments shown are from structures determined from individual soaks with (a) Chem 262, (b) Chem 84, (c) Chem 166 and (d) Chem 145. For chemical formulae, see Table 2 ▸. For some fragments, such as those shown in (c) and (d), the densities can only be fully accounted for by modeling alternative binding modes of the fragment.
3.2. Binding site and interactions
All 15 fragments bind to the same site in the TcHisRS•His structure, with Fig. 2 ▸(a) schematically depicting the major features of the binding site. The fragment-binding site is a narrow groove formed between the side chains of Arg156 (top) and His168 (bottom), close to the bound histidine. The shortest distance between two atoms from any of the fragments and histidine ranges from 5.2 to 6.4 Å. The aromatic cores of the fragments are typically sandwiched in a groove between the two side chains through stacking interactions, akin to slotting a piece of paper into a document sleeve. The narrow sleeve restricts the space available for binding of fragments in two of their three directions. As a result there is little variation in the orientations of the core of the fragments when viewed laterally (i.e. along an axis parallel to the stacking planes; Fig. 2 ▸ b). The only exception is Chem 707. Part of this fragment extends away at an angle from the center of the binding groove. The density around the extended part of fragment Chem 707 is quite poor, possibly reflecting flexibility outside the binding groove (Supplementary Figs. S1s and S1t).
Figure 2.

Characteristics of the fragment-binding site. (a) Schematic representation of the major features and orientation of the fragment-binding site. (b, c) Stereo pairs of the fragment-binding site showing all 15 fragment hits (ball-and-stick models) identified by the screens in two different views. All structures are superimposed using their protein structures (gray ribbon). The bound histidine and the side chains of Arg156 and His168 from the TcHisRS•His•Chem 84 complex are depicted as a representative of the positions of these residues in the entire set of structures. (d) Stereo pair showing the histidine and all side chains within 4.5 Å radius of the bound fragment in the TcHisRS•His•Chem 84 complex. The side chains at both sides of the ring of the fragment, an essential feature of the fragment-binding site, are highlighted in yellow.
If the fragments are viewed down the axis perpendicular to the stacking planes, vastly diverse orientations of the fragments can be observed (Fig. 2 ▸ c). In addition, multiple binding modes of a single fragment with different orientations but keeping its ring system in the canonical stacking plane can be identified in six of the hits. This variation is most likely owing to a lack of distinctly shaped lateral boundaries at the edges of the binding sleeve that would have selected for particular features of a fragment or a group of fragments. Instead, the most common lateral interaction, occurring in nine fragments, is a hydrogen bond between the carbonyl O atom of Pro125 and an amine group of the fragment on the left side. As a result, variation in relative orientations among these nine fragments is mainly owing to different positions of the amine group in the fragments. In the remaining six of the hits, which are not hydrogen-bonded to Pro125, five are bound with alternative binding modes. Only one hit, Chem 79, forms a hydrogen bond to Pro125 yet displays alternative binding modes. Other hydrogen bonds between protein and hits are also observed, but none appear to have such a dominant effect on ligand orientation as this amine group.
Since all fragments are bound to the same binding site, one of them, Chem 84 (3-aminoquinoline), will be used as the prototypical fragment to further describe the binding site (Fig. 2 ▸ d). Chem 84 binds in proximity to the histidine in the active site, facing the carboxylate group of the bound substrate. The shortest distance between the histidine and Chem 84 is about 6.2 Å. As mentioned above, the fragment-binding pocket can be described as a ‘document sleeve’. The side chains of Arg156 and His168 stack with the aromatic core of the fragment as the top and bottom ‘leaves’ of the slot occupied by Chem 84. The backbone atoms of residues 154CWR156 and 166REH168 form the ‘spine’ of the document sleeve. The sleeve is closed at the other two ends by residues Ile106 and Pro125 on the left side and Arg165 on the right side. In the case of Chem 84, the 3-amino group makes a hydrogen bond to the carbonyl O atom of Pro125 at 2.5 Å on the left side (Fig. 2 ▸ d). As described above, this is the hydrogen bond that is observed in nine of the fragment hits. The right side of the sleeve is formed mainly by Arg165 and leads to the M2 loop (159AITRG163), which is disordered in all of the current structures (Fig. 2 ▸ a). In fact, the M2 loop is typically disordered in histidine-bound structures of HisRS and is only ordered when ATP or HAMP is bound (Åberg et al., 1997 ▸; Arnez et al., 1995 ▸, 1997 ▸; Merritt et al., 2010 ▸). As a result of the flexibility in the M2 loop, there appears to be more room on the right side than the left side.
Remarkably, the binding site for fragments is not present in the TcHisRS•His structures before the fragments are bound (Figs. 3 ▸ a and 3 ▸ b). As stated in §3.1, the appearance of new density features after cocktail and single soaks are always accompanied by changes in two protein side chains. The guanidinium group of Arg156 swings ‘upwards’, away from His168, by an average of 5.4 Å owing to a rotation mainly about the Cβ—Cγ bond into a planar structure. Concomitantly, the imidazole ring of His168 rotates about its Cβ—Cγ bond by around 150° (Fig. 3 ▸ c). Binding of Chem 84 in the fragment-binding pocket is stabilized by stacking interactions at an optimum distance of around 3.5–3.8 Å between the fragment and the planar group on both sides.
Figure 3.

Motions of TcHisRS upon binding fragments. Superpositions of the (a) TcHisRS•His (dark gray) and (b) TcHisRS•His•Chem 84 (light gray) structures in surface representation showing the absence of the binding pocket before the fragment is bound. The histidine and side chains of Arg156 and His168 are shown as ball-and-stick models. (c) The binding pocket is formed owing to movements of Arg156 and His168 as indicated by the arrows as shown by the superimposed structures of TcHisRS•His (dark gray C atoms) and TcHisRS•His•Chem 84 (light gray C atoms). (d) Superimposed structures of TcHisRS•His•Chem 84 and TcHisRS•HAMP show that HAMP binding is incompatible with fragment binding owing to steric clashes, as exemplified by the close proximity of Chem 84 (cyan C atoms) and HAMP (dark green C atoms).
Superposition of the TcHisRS•His•Chem 84 structure with the TcHisRS•HAMP structure shows that the fragment-binding site is in close proximity to the HAMP-binding site (Fig. 3 ▸ d). Specifically, multiple atoms of Chem 84 are within 2.2 Å of the amine group of the adenine ring. In addition, the binding of HAMP requires the His168 imidazole to remain in its original orientation for stacking with the adenine ring, while Arg156 moves by ∼6 Å towards the histidine and the α-phosphate of HAMP to form hydrogen bonds. Both movements would not be compatible with the binding modes of the fragment hits, in which Arg156 and His168 are locked into another position by stacking interactions with fragments. Therefore, the fragments would most likely compete with the binding of either ATP or the product HAMP, or both, resulting in inhibition.
3.3. Secondary binding site
One fragment, Chem 89, appears to also occupy a second, likely lower affinity, site (Supplementary Fig. S1d). Density for Chem 89 at this secondary site only appears in the single soak experiment where the concentration of the fragment is 10× higher than during the cocktail soak. The binding site is in close proximity to the peptide linker between the catalytic domain and the anticodon-binding domain of the TcHisRS•His structure. This secondary binding site is at least 30 Å away from the active site and is also not at the HisRS dimer interface, and hence is unlikely to be relevant for inhibitor-design purposes (Supplementary Fig. S2).
3.4. Activity assays
We performed thermal shift assays using all 15 hit compounds. No significant thermal stabilization of TcHisRS can be observed at a concentration of 1 mM. Since the crystallographic screens were performed with TcHisRS•His, the thermal shift assay was repeated in the presence of 1 mM histidine. Chem 84 is the only hit which reliably increases the T m of the TcHisRS•His complex, by about 1.6°C (Table 2 ▸). We also tested three hits for inhibition in the aminoacylation assay. None, including Chem 84, showed sufficient inhibition to allow the estimation of an IC50 value. However, at the highest concentration of fragments tested (2 mM), Chem 84, Chem 89 and Chem 149 showed inhibitions of 20, 39 and 21%, respectively (Table 2 ▸). The lack of thermal shift and limited inhibitory effects probably reflect low binding affinities of the fragment hits, which is not uncommon in the initial stages of FBDD.
3.5. Fragment growing and tethering
Chem 84 was chosen for follow-up to explore the possibility of growing the fragment to improve its interactions with TcHisRS. Three analogs of Chem 84 were synthesized, extending the molecule from its 3-amino group towards histidine. The intention was to try to extend Chem 84 into the immediate proximity of the histidine, thereby arriving at a compound with the opportunity, in a next step, of linking Chem 84 to a histidine-like moiety occupying both the fragment-binding and histidine-binding pockets. One of the analogs, Chem 1691, could be successfully soaked into TcHisRS•His crystals. Surprisingly, the binding mode of Chem 1691 is very different from that of Chem 84 (Fig. 4 ▸ a). In the case of Chem 1691 an acetyl group was attached to the 3-amino group of Chem 84, changing this group into an acetamide (Table 3 ▸). Based on the binding mode of Chem 84, the 3-amino group was expected to retain its ability as a hydrogen-bond donor to the carbonyl O atom of Pro125 while extending the acetyl group towards the histidine, which is about 6.3 Å away. Yet, in the binding mode observed for Chem 1691 the fragment flips around such that the 3-acetamide group points away from the histidine into the solvent. In addition, alternative binding modes can be built into the density, again indicating a lack of specific interactions with TcHisRS. No shift in the T m of TcHisRS was observed when adding Chem 1691, and the level of inhibition observed in the aminoacylation assay was similar to that of the parent fragment Chem 84 (Tables 2 ▸ and 3 ▸).
Figure 4.

The development of reactive fragments targeting Cys365 of TcHisRS. (a) The initial fragment hit Chem 84 (cyan C atoms) was developed into Chem 1691 (purple; two alternative binding modes superimposed). The 3-acetamide group of Chem 1691 is close to Cys365 (yellow surface). (b) Based on Chem 1691, the reactive fragments Chem 1697, Chem 1698 and Chem 1781 (pink) were modified to target Cys365 (yellow), thereby tethering fragments to TcHisRS. The structure of TcHisRS–Chem 1781•His shown indicates that the acrylamide group of Chem 1781 forms a covalent bond to the thiol group of Cys365 as designed. (c) and (d) are two different views of σA-weighted F obs − F calc electron-density maps omitting the coordinates of histidine, Chem 1781 and the side chain of Cys365. Contours shown are green (positive) and red (negative) at the 3σ level in the difference map and light blue at the 1.0σ level in the σA-weighted 2F obs − F calc electron-density map.
Table 3. Chemical structures and activities of fragments developed based on Chem 84.
| TcHisRS | Hs-cHisRS | Hs-mHisRS | |||||
|---|---|---|---|---|---|---|---|
| Fragment | Chemical structure | IC50 (mM) | Inhibition at 2mM (%) | IC50 (mM) | Inhibition at 2mM (%) | IC50 (mM) | Inhibition at 2mM (%) |
| Chem 1691 |
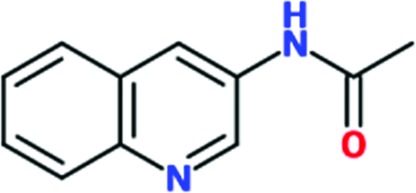
|
>2 | 20 | ND | ND | ND | ND |
| Chem 1697 |
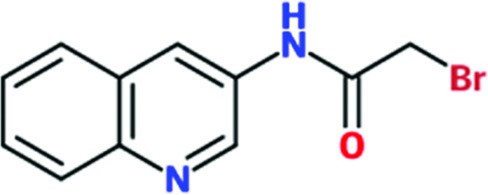
|
0.80 | 88 | 0.90 | 91 | 1.0 | 88 |
| Chem 1698 |
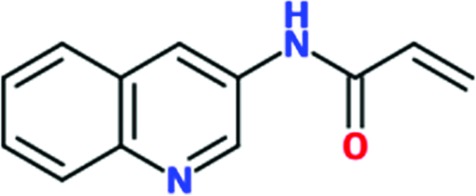
|
0.85 | 69 | >2 | 29 | >2 | 25 |
| Chem 1781 |
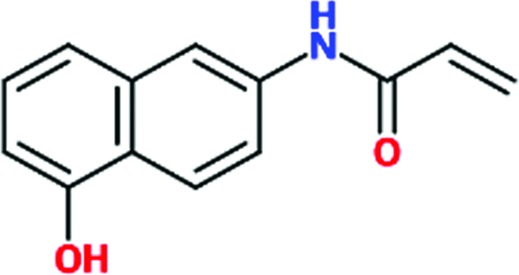
|
1.65 | 69 | >2 | 31 | >2 | 37 |
Interestingly, the new binding mode observed for Chem 1691 revealed close proximity of the 3-acetamide group to Cys365 (Fig. 4 ▸ a). The distances between the last non-H atoms of Chem 1691 and Cys365 are between 3.7 and 4.3 Å. A comparison of all TcHisRS•His•fragment structures showed two possible Cys365 side-chain rotamers, sometimes both in the same structure. Considering the low affinity of fragments and the inconsistency in binding modes, Cys365 was targeted for the design of reactive fragments to help to tether fragments to TcHisRS. The slight flexibility of the Cys365 side chain possibly allows a small adjustment of angles for reactions between the thiol group and the fragment. Tethering fragments to TcHisRS will reduce uncertainty in fragment binding modes, improve affinity and provide opportunities to explore new interactions by growing the fragment.
Three reactive fragments carrying an electrophile on the acetamide group of Chem 1691 were synthesized (Table 3 ▸). Unfortunately, attempts to co-crystallize TcHisRS–Chem 1697•His were unsuccessful. However, structures of the TcHisRS–Chem 1781•His (Fig. 4 ▸ b) and TcHisRS–Chem 1698•His (Supplementary Fig. S1x) complexes were determined. Both Chem 1698 and Chem 1781 have an acrylamide moiety replacing the acetamide group in Chem 1691. The aromatic core in Chem 1691 and Chem 1698 was changed from a quinoline to a 5-hydroxynaphthalene in Chem 1781 (Table 3 ▸). The difference electron density of both Chem 1781 and Chem 1698 showed the reactive fragments bound in the fragment-binding site with continuous density towards the thiol group of Cys365 (Figs. 4 ▸ c and 4 ▸ d and Supplementary Fig. S1x). A TcHisRS–Chem 1781•His crystal produced a data set with higher resolution (2.05 Å) than that of TcHisRS–Chem 1698•His (2.75 Å), and hence the former will be the focus of the discussion from here onwards. Density around the acrylamide group of Chem 1781 and the thiol group of Cys365 can only be sufficiently accounted for by the formation of a covalent bond through Michael addition, consistent with our design. Only a single binding mode was observed for Chem 1781 tethered to Cys365. The 5-hydroxy substitution on the naphthalene group of Chem 1781 points to the open space in the binding site. Despite the formation of a covalent bond between the reactive fragments and TcHisRS, no significant changes in T m were observed. However, the inhibitory activities of the reactive fragments are improved over that of the parent fragment. Chem 1697, Chem 1698 and Chem 1781 showed IC50 values of 0.8, 0.85 and 1.65 mM, respectively, while the parent compounds Chem 84 and the first-generation analog Chem 1691 showed only approximately 20% inhibition at the highest concentration tested (2 mM). Chem 1697 showed IC50 values of 0.9 and 1.0 mM against human cytosolic HisRS (Hs-cHisRS) and mitochondrial HisRS (Hs-mHisRS), while Chem 1698 and Chem 1781 do not inhibit the human HisRS enzymes sufficiently to be able to calculate IC50 values (i.e. they are greater than 2 mM).
4. Discussion
By comparing the structures of TcHisRS•His and Hs-cHisRS•His, we have recently identified a ‘pocket H’ in the TcHisRS•His structure that could potentially impart selectivity to inhibitors of the parasite enzyme (Fig. 5 ▸ a; Koh, Wetzel et al., 2014 ▸). A crystallographic fragment screen was carried out to search for small molecules that target this pocket. However, despite identifying at least 15 fragment hits that bind to TcHisRS•His, none of them binds to pocket H. Instead, all of the fragments are bound to a new pocket that was not previously present in the TcHisRS•His structure and is located right next to, and partially overlaps with, pocket H. Interestingly, pocket H remains visible in the fragment-bound structures. The fragment-binding pocket extends into pocket H (Fig. 5 ▸ b). Unfortunately, the two pockets appear to be positioned perpendicular to each other along their widest planes, posing synthetic challenges to extend fragments into molecules that fully occupy both the fragment-binding pocket and pocket H.
Figure 5.

Two pockets near the histidine substrate-binding site. (a) ‘Pocket H’ (the yellow dashed line indicates its boundaries) was identified previously (Koh, Wetzel et al., 2014 ▸) as a pocket that exists only in TcHisRS•His when compared with Hs-cHisRS•His. (b) The fragment hits identified in this study are bound to a new fragment-binding pocket (green dashed outline) that was not present in the TcHisRS•His structure but is located right next to, and partially overlaps with, pocket H (yellow dashed line). (c) Neither pocket H nor the fragment-binding pocket is present in the Hs-cHisRS•His structure shown, revealing opportunities for the development of inhibitors selective for TcHisRS targeting these two pockets.
The new fragment-binding pocket has the potential to be a selectivity pocket. A comparison shows that the fragment-binding pocket is not present in the homologous human structure, Hs-cHisRS•His (Fig. 5 ▸ c). We postulate that it will be more difficult for the fragment hits to bind to Hs-cHisRS•His compared with TcHisRS•His. Arg157 and Phe171 of Hs-cHisRS are analogous to Arg156 and His168 in TcHisRS (Figs. 6 ▸ a and 6 ▸ b). Although in principle Arg157 and Phe171 in the human enzyme are capable of forming stacking interactions with fragment hits as observed in TcHisRS, a major conformational difference between Arg157 in Hs-cHisRS and Arg156 in TcHisRS may reduce the likelihood of this occurring in Hs-cHisRS. As noted earlier, Arg157 in Hs-cHisRS•His adopts a drastically different conformation compared with Arg156 of TcHisRS•His (Koh, Wetzel et al., 2014 ▸). The guanidinium group of Arg157 of Hs-cHisRS•His forms a hydrogen bond to histidine, while Arg156 of TcHisRS•His is around 6.5 Å away from the position of Arg157, placing Arg156 ∼8.5 Å away from the closest carboxylate atom in the bound histidine (Fig. 6 ▸ a). It is this difference that is mainly responsible for the presence of pocket H only in the TcHisRS•His structure. We hypothesize that, having engaged in a hydrogen bond to histidine, it will be energetically less favorable for Arg157 of Hs-cHisRS•His, compared with Arg156 of TcHisRS•His, to realise the conformational changes that would be required for stacking with the current fragment hits (Fig. 6 ▸ a). As a result, the fragment-binding pocket is less likely to be present or formed in Hs-cHisRS•His.
Figure 6.

Comparison of binding pockets near the bound histidine substrate in trypanosomatid and mammalian HisRS. (a) The movement of Arg156 and His168 from their respective positions in TcHisRS•His (dark gray) into their new positions in TcHisRS–Chem 1781•His (light gray) is required for the binding of Chem 1781 (pink), as indicated by the black arrows. The amino acids Arg157 and Phe171 of Hs-cHisRS (cyan) are analogous to Arg156 and His168 in TcHisRS. The hypothetical corresponding movements in the human enzyme, especially of Arg157 (gray dashed arrow labeled with a question mark), to accommodate the binding of a fragment is less likely, since Arg157 is engaged in a hydrogen bond to the substrate histidine (3.0 Å; dashed line). Superposition of the structures also shows that Glu387 in Hs-cHisRS corresponds to Cys365 in TcHisRS. (b) Sequence alignment of trypanosomatid HisRSs and mammalian HisRSs shows conservation of Arg156 in all of the trypanosomatid species aligned (pink star). In contrast, His168 in the trypanosomatids is a Phe in mammals (marked by a second pink star). (c) The same alignment indicates that Cys365 (marked by a pink triangle and with a yellow background in the trypanosomatids) is conserved among trypanosomatid HisRSs but is a Glu in mammalian HisRSs. UniProt accession numbers for the sequences are listed. Tcru, T. cruzi; Tbru, T. brucei; Tcon, T. congolense; Tviv, T. vivax; Lmex, Leishmania mexicana; Ldon, L. donovani; Lbra, L. braziliensis; Lmaj, L. major; Linf, L. infantum; Hsap-cyt, Homo sapiens, cytosolic; Hsap-mit, H. sapiens, mitochondrial; Btau, Bos taurus; Mmus, Mus musculus; Cfer, Camelus ferus; Oari, Ovis aries; Fcat, Felis catus; Ecab, Equus caballus; Cfam, Canis familiaris.
A compelling case for selectivity can be made for the reactive fragments. Structural and sequence alignments revealed that Cys365, the target residue for the reactive fragments, is analogous to Glu387 in Hs-cHisRS (Fig. 6 ▸ c). Without a nucleophile at the equivalent position, reactive fragments such as Chem 1698 should not be able to form a covalent bond to Hs-cHisRS, unlike in TcHisRS. In fact, when comparing a broad family of HisRS sequences, Cys365 appears to only occur in trypanosomal HisRS, including T. cruzi, T. brucei and Leishmania spp. Humans and other animals that are commonly infected by these parasites all have a conserved Glu387 instead of Cys365 (Fig. 6 ▸ c). Therefore, future generations of fragments and inhibitors may utilize the reactive fragments as the starting point. When tested, acrylamide-based reactive fragments appear to indeed be more selective for inhibition of TcHisRS than human enzymes (Table 3 ▸). However, results for enzyme inhibition in this case have to be considered with caution since the low affinities of the reactive fragments necessitate the use of extremely high concentrations of fragments, which may result in nonspecific modification of the HisRS enzymes during the assays. For TcHisRS, the specific targeting of Cys365 was validated by the crystal structures.
The idea of tethering fragment hits, at least in the initial stages of fragment screening and optimization, has previously been explored with considerable success (Erlanson et al., 2000 ▸; Miller et al., 2013 ▸; Nonoo et al., 2012 ▸; Kathman et al., 2014 ▸). Eventually, the reactive group targeting Cys365 may or may not be removed after good future reactive leads can be found. Retaining the Cys365 targeting group to arrive at an irreversible inhibitor will be an attractive option for selectivity reasons. However, it could also be argued that the resulting inhibitor may be more susceptible to resistance arising from a single-point mutation of Cys365 in the target enzyme. In any case, the difference between Cys365 and Glu387 is large enough that more chemical options can be explored to impart selectivity even without covalent interactions. Therefore, the current fragment-binding pocket remains an interesting selectivity pocket for future inhibitors of trypanosomatid HisRS.
Compared with our previous efforts in fragment cocktail-based crystallographic screens in the MSGPP consortium (Verlinde et al., 2009 ▸; Bosch et al., 2006 ▸), the current screening campaign covers the largest library size (680 fragments) and produced the largest number of hits (15 hits). Overall, the hit rate is 2.2%, which is comparable with the reported rate of other crystallographic screens (Hartshorn et al., 2005 ▸). For example, two very successful screens reported recently against HIV reverse transcriptase (Bauman, Patel, Dharia et al., 2013 ▸) and influenza virus endonuclease (Bauman, Patel, Baker et al., 2013 ▸) had hit rates of 4.4 and 1.0%, respectively. These hit rates are also within the usual range of rates reported for other fragment-screening methods (Larsson et al., 2011 ▸).
The example that we are reporting here illustrates the advantages of a crystallographic cocktail screen. Here, the fragment screen allowed the discovery of a previously unobserved binding hotspot, which is nearly impossible to identify through virtual screening since a previously unobserved conformational change occurred. The general lack of positive signals in thermal shifts and functional assays also suggest that many, if not all, of the current hits would be missed by these types of screening methods. In addition, the ability to directly observe the binding modes of hits and their environment allows the efficient and accurate use of information to improve the fragment hits. In this case, observing the binding mode of Chem 1691 near Cys365 immediately set our path towards demonstration of the inhibitory effect and the selectivity potential of reactive fragments. This provides a foundation towards the development of future generations of molecules that have the potential to selectively inhibit HisRS in trypanosomes. As pointed out in a recent review, the use of X-ray crystallography as a primary fragment screen is underappreciated (Patel et al., 2014 ▸). The results reported here support the idea that crystallographic fragment screening has specific advantages over other screening methods. With recent advances in crystallization, automated data collection, processing and density identification, one might argue that primary screens using pre-formed crystals should be more regularly performed in FBDD projects.
Supplementary Material
PDB reference: Trypanosoma cruzi HisRS, complex with Chem 79, 4yp0
PDB reference: complex with Chem 84, 4ypf
PDB reference: complex with Chem 89, 4yrc
PDB reference: complex with Chem 145, 4yre
PDB reference: complex with Chem 148, 4yrf
PDB reference: complex with Chem 149, 4yrg
PDB reference: complex with Chem 166, 4yri
PDB reference: complex with Chem 256, 4yrj
PDB reference: complex with Chem 262, 4yrl
PDB reference: complex with Chem 260, 4yrk
PDB reference: complex with Chem 443, 4yrm
PDB reference: complex with Chem 475, 4yrn
PDB reference: complex with Chem 491, 4yro
PDB reference: complex with Chem 707, 4yrp
PDB reference: complex with Chem 744, 4yrq
PDB reference: complex with Chem 1691, 4yrr
PDB reference: complex with Chem 1698, 4yrs
PDB reference: complex with Chem 1781, 4yrt
Supporting Information.. DOI: 10.1107/S1399004715007683/dz5371sup1.pdf
Acknowledgments
We thank Stewart Turley, Frank Zucker and Jonathan Kay for providing support for the X-ray data-collection, database management and computing environment at the Biomolecular Structure Center of the University of Washington. We also thank the staff of Stanford Synchrotron Radiation Lightsource for support during data collection. The research reported in this publication was supported by National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award Nos. R56AI084004, RO1AI084004 and R01AI097177. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- Åberg, A., Yaremchuk, A., Tukalo, M., Rasmussen, B. & Cusack, S. (1997). Biochemistry, 36, 3084–3094. [DOI] [PubMed]
- Abibi, A., Ferguson, A. D., Fleming, P. R., Gao, N., Hajec, L. I., Hu, J., Laganas, V. A., McKinney, D. C., McLeod, S. M., Prince, D. B., Shapiro, A. B. & Buurman, E. T. (2014). J. Biol. Chem. 289, 21651–21662. [DOI] [PMC free article] [PubMed]
- Andrade, D. V., Gollob, K. J. & Dutra, W. O. (2014). PLoS Negl. Trop. Dis. 8, e3010. [DOI] [PMC free article] [PubMed]
- Arnez, J. G., Augustine, J. G., Moras, D. & Francklyn, C. S. (1997). Proc. Natl Acad. Sci. USA, 94, 7144–7149. [DOI] [PMC free article] [PubMed]
- Arnez, J. G., Harris, D. C., Mitschler, A., Rees, B., Francklyn, C. S. & Moras, D. (1995). EMBO J. 14, 4143–4155. [DOI] [PMC free article] [PubMed]
- Baker, M. (2013). Nature Rev. Drug Discov. 12, 5–7. [DOI] [PubMed]
- Batt, A. R., Evans, D. M., Pitt, G. R. W., Roe, M. B. & Rooker, D. P. (2003). Patent WO/2003/078398.
- Bauman, J. D., Patel, D., Baker, S. F., Vijayan, R. S., Xiang, A., Parhi, A. K., Martinez-Sobrido, L., LaVoie, E. J., Das, K. & Arnold, E. (2013). ACS Chem. Biol. 8, 2501–2508. [DOI] [PMC free article] [PubMed]
- Bauman, J. D., Patel, D., Dharia, C., Fromer, M. W., Ahmed, S., Frenkel, Y., Vijayan, R. S., Eck, J. T., Ho, W. C., Das, K., Shatkin, A. J. & Arnold, E. (2013). J. Med. Chem. 56, 2738–2746. [DOI] [PMC free article] [PubMed]
- Bosch, J. et al. (2006). J. Med. Chem. 49, 5939–5946. [DOI] [PubMed]
- Chatelain, E. (2015). J. Biomol. Screen. 20, 22–35. [DOI] [PubMed]
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Choi, R., Kelley, A., Leibly, D., Nakazawa Hewitt, S., Napuli, A. & Van Voorhis, W. (2011). Acta Cryst. F67, 998–1005. [DOI] [PMC free article] [PubMed]
- Chopra, S., Palencia, A., Virus, C., Tripathy, A., Temple, B. R., Velazquez-Campoy, A., Cusack, S. & Reader, J. S. (2013). Nature Commun. 4, 1417. [DOI] [PubMed]
- Coura, J. R. & Viñas, P. A. (2010). Nature (London), 465, S6–S7. [DOI] [PubMed]
- Davis, B. J. & Erlanson, D. A. (2013). Bioorg. Med. Chem. Lett. 23, 2844–2852. [DOI] [PubMed]
- DeLano, W. (2002). http://www.pymol.org.
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Erlanson, D. A., Braisted, A. C., Raphael, D. R., Randal, M., Stroud, R. M., Gordon, E. M. & Wells, J. A. (2000). Proc. Natl Acad. Sci. USA, 97, 9367–9372. [DOI] [PMC free article] [PubMed]
- Evdokimov, A. G. et al. (2008). J. Struct. Biol. 162, 152–169. [DOI] [PubMed]
- Gadakh, B. & Van Aerschot, A. (2012). Exp. Opin. Ther. Patents, 22, 1453–1465. [DOI] [PubMed]
- Guerry, P., Kellenberger, J. L. & Blanchard, S. (2003). Patent WO/2003/072588.
- Hartshorn, M. J., Murray, C. W., Cleasby, A., Frederickson, M., Tickle, I. J. & Jhoti, H. (2005). J. Med. Chem. 48, 403–413. [DOI] [PubMed]
- Hernandez, V. et al. (2013). Antimicrob. Agents Chemother. 57, 1394–1403. [DOI] [PMC free article] [PubMed]
- Hotez, P. J. et al. (2014). PLoS Negl. Trop. Dis. 8, e2865.
- Hu, Q.-H., Liu, R.-J., Fang, Z.-P., Zhang, J., Ding, Y.-Y., Tan, M., Wang, M., Pan, W., Zhou, H.-C. & Wang, E.-D. (2013). Sci. Rep. 3, 2475. [DOI] [PMC free article] [PubMed]
- Ibba, M. & Söll, D. (2000). Annu. Rev. Biochem. 69, 617–650. [DOI] [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Kalidas, S., Cestari, I., Monnerat, S., Li, Q., Regmi, S., Hasle, N., Labaied, M., Parsons, M., Stuart, K. & Phillips, M. A. (2014). Eukaryot. Cell, 13, 504–516. [DOI] [PMC free article] [PubMed]
- Kathman, S. G., Xu, Z. & Statsyuk, A. V. (2014). J. Med. Chem. 57, 4969–4974. [DOI] [PMC free article] [PubMed]
- Khan, S., Sharma, A., Belrhali, H., Yogavel, M. & Sharma, A. (2014). J. Struct. Funct. Genomics, 15, 63–71. [DOI] [PubMed]
- Koh, C. Y., Kim, J. E., Shibata, S., Ranade, R. M., Yu, M., Liu, J., Gillespie, J. R., Buckner, F. S., Verlinde, C. L. M. J., Fan, E. & Hol, W. G. J. (2012). Structure, 20, 1681–1691. [DOI] [PMC free article] [PubMed]
- Koh, C. Y., Kim, J. E., Wetzel, A. B., de van der Schueren, W. J., Shibata, S., Ranade, R. M., Liu, J., Zhang, Z., Gillespie, J. R., Buckner, F. S., Verlinde, C. L. M. J., Fan, E. & Hol, W. G. J. (2014). PLoS Negl. Trop. Dis. 8, e2775. [DOI] [PMC free article] [PubMed]
- Koh, C. Y., Wetzel, A. B., de van der Schueren, W. J. & Hol, W. G. J. (2014). Biochimie, 106, 111–120. [DOI] [PMC free article] [PubMed]
- Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., McGettigan, P. A., McWilliam, H., Valentin, F., Wallace, I. M., Wilm, A., Lopez, R., Thompson, J. D., Gibson, T. J. & Higgins, D. G. (2007). Bioinformatics, 23, 2947–2948. [DOI] [PubMed]
- Larson, E. T., Kim, J. E., Castaneda, L. J., Napuli, A. J., Zhang, Z., Fan, E., Zucker, F. H., Verlinde, C. L. M. J., Buckner, F. S., Van Voorhis, W. C., Hol, W. G. J. & Merritt, E. A. (2011). J. Mol. Biol. 409, 159–176. [DOI] [PMC free article] [PubMed]
- Larsson, A., Jansson, A., Åberg, A. & Nordlund, P. (2011). Curr. Opin. Chem. Biol. 15, 482–488. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Mehlin, C. et al. (2006). Mol. Biochem. Parasitol. 148, 144–160. [DOI] [PubMed]
- Merritt, E. A., Arakaki, T. L., Gillespie, J. R., Larson, E. T., Kelley, A., Mueller, N., Napuli, A. J., Kim, J., Zhang, L., Verlinde, C. L. M. J., Fan, E., Zucker, F., Buckner, F. S., Van Voorhis, W. C. & Hol, W. G. J. (2010). J. Mol. Biol. 397, 481–494. [DOI] [PMC free article] [PubMed]
- Miller, R. M., Paavilainen, V. O., Krishnan, S., Serafimova, I. M. & Taunton, J. (2013). J. Am. Chem. Soc. 135, 5298–5301. [DOI] [PMC free article] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Nakama, T., Nureki, O. & Yokoyama, S. (2001). J. Biol. Chem. 276, 47387–47393. [DOI] [PubMed]
- Nienaber, V. L., Richardson, P. L., Klighofer, V., Bouska, J. J., Giranda, V. L. & Greer, J. (2000). Nature Biotechnol. 18, 1105–1108. [DOI] [PubMed]
- Nonoo, R. H., Armstrong, A. & Mann, D. J. (2012). ChemMedChem, 7, 2082–2086. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Painter, J. & Merritt, E. A. (2006). J. Appl. Cryst. 39, 109–111.
- Palencia, A., Crépin, T., Vu, M. T., Lincecum, T. L. Jr, Martinis, S. A. & Cusack, S. (2012). Nature Struct. Mol. Biol. 19, 677–684. [DOI] [PMC free article] [PubMed]
- Patel, D., Bauman, J. D. & Arnold, E. (2014). Prog. Biophys. Mol. Biol. 116, 92–100. [DOI] [PMC free article] [PubMed]
- Pedro-Rosa, L. et al. (2015). J. Biomol. Screen. 20, 122–130. [DOI] [PMC free article] [PubMed]
- Pham, J. S., Dawson, K. L., Jackson, K. E., Lim, E. E., Pasaje, C. F., Turner, K. E. & Ralph, S. A. (2014). Int. J. Parasitol. Drugs Drug Resist. 4, 1–13. [DOI] [PMC free article] [PubMed]
- Phillips, M. A. & Rathod, P. K. (2010). Infect. Disord. Drug Targets, 10, 226–239. [DOI] [PMC free article] [PubMed]
- Qiu, X. et al. (2001). Protein Sci. 10, 2008–2016. [DOI] [PMC free article] [PubMed]
- Ratnakar, S. J., Viswanathan, S., Kovacs, Z., Jindal, A. K., Green, K. N. & Sherry, A. D. (2012). J. Am. Chem. Soc. 134, 5798–5800. [DOI] [PMC free article] [PubMed]
- Read, R. J. et al. (2011). Structure, 19, 1395–1412. [DOI] [PMC free article] [PubMed]
- Robert, X. & Gouet, P. (2014). Nucleic Acids Res. 42, W320–W324. [DOI] [PMC free article] [PubMed]
- Rock, F. L. et al. (2007). Science, 316, 1759–1761. [DOI] [PubMed]
- Seiradake, E., Mao, W., Hernandez, V., Baker, S. J., Plattner, J. J., Alley, M. R. & Cusack, S. (2009). J. Mol. Biol. 390, 196–207. [DOI] [PubMed]
- Shibata, S., Gillespie, J. R., Kelley, A. M., Napuli, A. J., Zhang, Z., Kovzun, K. V., Pefley, R. M., Lam, J., Zucker, F. H., Van Voorhis, W. C., Merritt, E. A., Hol, W. G. J., Verlinde, C. L. M. J., Fan, E. & Buckner, F. S. (2011). Antimicrob. Agents Chemother. 55, 1982–1989. [DOI] [PMC free article] [PubMed]
- Shibata, S., Gillespie, J. R., Ranade, R. M., Koh, C. Y., Kim, J. E., Laydbak, J. U., Zucker, F. H., Hol, W. G. J., Verlinde, C. L. M. J., Buckner, F. S. & Fan, E. (2012). J. Med. Chem. 55, 6342–6351. [DOI] [PMC free article] [PubMed]
- Silvian, L. F., Wang, J. & Steitz, T. A. (1999). Science, 285, 1074–1077. [PubMed]
- Son, J., Lee, E. H., Park, M., Kim, J. H., Kim, J., Kim, S., Jeon, Y. H. & Hwang, K. Y. (2013). Acta Cryst. D69, 2136–2145. [DOI] [PubMed]
- Studier, F. W. (2005). Protein Expr. Purif. 41, 207–234. [DOI] [PubMed]
- Teng, M., Hilgers, M. T., Cunningham, M. L., Borchardt, A., Locke, J. B., Abraham, S., Haley, G., Kwan, B. P., Hall, C., Hough, G. W., Shaw, K. J. & Finn, J. (2013). J. Med. Chem. 56, 1748–1760. [DOI] [PubMed]
- Verlinde, C. L. M. J. et al. (2009). Curr. Top. Med. Chem. 9, 1678–1687. [DOI] [PMC free article] [PubMed]
- Verlinde, C. L. M. J., Kim, H., Bernstein, B. E., Mande, S. C. & Hol, W. G. J. (1997). Structure-Based Drug Design, edited by P. Veerapandian, pp. 365–394. New York: Marcel Dekker.
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Zartler, E. R. (2014). ACS Med. Chem. Lett. 5, 952–953. [DOI] [PMC free article] [PubMed]
- Zhou, H., Sun, L., Yang, X.-L. & Schimmel, P. (2013). Nature (London), 494, 121–124. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: Trypanosoma cruzi HisRS, complex with Chem 79, 4yp0
PDB reference: complex with Chem 84, 4ypf
PDB reference: complex with Chem 89, 4yrc
PDB reference: complex with Chem 145, 4yre
PDB reference: complex with Chem 148, 4yrf
PDB reference: complex with Chem 149, 4yrg
PDB reference: complex with Chem 166, 4yri
PDB reference: complex with Chem 256, 4yrj
PDB reference: complex with Chem 262, 4yrl
PDB reference: complex with Chem 260, 4yrk
PDB reference: complex with Chem 443, 4yrm
PDB reference: complex with Chem 475, 4yrn
PDB reference: complex with Chem 491, 4yro
PDB reference: complex with Chem 707, 4yrp
PDB reference: complex with Chem 744, 4yrq
PDB reference: complex with Chem 1691, 4yrr
PDB reference: complex with Chem 1698, 4yrs
PDB reference: complex with Chem 1781, 4yrt
Supporting Information.. DOI: 10.1107/S1399004715007683/dz5371sup1.pdf