

Summary
MicroRNA (miRNA)-dependent regulation of gene expression confers robustness to cellular phenotypes and controls responses to extracellular stimuli. Although a single miRNA can regulate expression of hundreds of target genes, it is unclear whether any of its distinct biological functions can be due to the regulation of a single target. To explore in vivo the function of a single miRNA-mRNA interaction, we mutated the 3′ UTR of a major miR-155 target SOCS1 to specifically disrupt its regulation by miR-155. We found that under physiologic conditions and during autoimmune inflammation or viral infection some immunological functions of miR-155 were fully or largely attributable to the regulation of SOCS1, whereas others could be accounted only partially or not at all by this interaction. Our data suggest that the role of a single miRNA-mRNA interaction is cell type- and biological context-dependent.
Introduction
The microRNA (miRNA) mediated posttranscriptional regulation of gene expression features prominently during differentiation of cells of the immune system and their responses to stimulation as revealed by miRNA gene targeting in mice (Baltimore et al., 2008). The ability of a given miRNA to bind and control its targets is determined by perfect complementarity of the “seed” region at positions 2–7 in the 5′ end of the miRNA to the 3′UTR of the target mRNA followed by Argonaute (Ago) protein-containing RNA-induced silencing complex (RISC) mediated target inhibition (Bartel, 2004). This attribute enabled computational prediction of thousands of miRNA targets based on changes in transcript and protein levels induced upon genetic perturbation of miRNAs and their confirmation using in vitro reporter assays (Bartel, 2009). A single miRNA binds and inhibits expression of hundreds of targets overwhelmingly on a small scale of two-fold or less. The characteristically small range variation of multiple targets imparted by a given miRNA and their frequent enrichment in the same or related molecular pathways strongly suggest that regulation of a single target is unlikely to account for a particular biological manifestation of the individual miRNA activity with exception of targets with a highly pronounced gene dose effect (Xiao et al., 2007). However, the vast majority of functional studies of miRNAs in mice ascribed their specific biological effects to changes in expression of a single target. The “gold standard” argument in these studies has been reversal of a phenotype when a miRNA deficiency is combined with a target deficiency or knockdown, and when target overexpression leads to a roughly similar phenotype. However, constitutive down-regulation or absence of the target and its overexpression can exert multiple effects beyond those resulting from dynamic miRNA-mediated regulation of the target transcript in a physiological context. Thus, given the complexity of miRNA-mediated regulation of gene expression, it has been difficult to explore the biological significance of a single miRNA-mRNA interaction in vivo.
We sought to address this question by mutating the binding site for a given miRNA in the 3′ UTR of its target and studying cells of the immune system with their aforementioned highly dynamic features. We reasoned that a desired experimental model to address this question should satisfy the following requirements for a chosen miRNA-target mRNA pair: 1) both should be expressed in multiple immune cell types and have pronounced effects on their function supported by genetic evidence; 2) expression of both should be modulated in the course of the immune response; 3) the mRNA 3′UTR should have a single miRNA “seed” target sequence whose mutation annuls regulation. miR-155, whose expression is induced upon activation of multiple types of immune cells, with diverse functions under physiologic conditions as well as in the course of the immune response, is an ideal candidate to serve this purpose (Vigorito et al., 2013). Consistent with its pleiotropic effects, miR-155 was shown to recruit Ago complexes to ~200 seeds or ~1.5% of all miRNA bound sites in the transcriptome of activated T cells (Loeb et al., 2012). Previous analysis of mutations of the miR-155 target seed sequence in Aicda (AID) and Spi1 (PU.1) demonstrated that a single target can account for a specific miR-155 function in B cells (Dorsett et al., 2008; Lu et al., 2014; Teng et al., 2008). Here, we chose to explore a role for miR-155 dependent regulation of suppressor of cytokine signaling 1 (SOCS1), as it is expressed in multiple immune cell types in an inducible manner and serves as a pivotal regulator of many cytokine signaling pathways (Ilangumaran et al., 2004; Yoshimura et al., 2007). Several studies including our own implicated miR155-regulation of SOCS1 in multiple complex phenotypes controlled by miR-155. The miR-155-dependent repression of SOCS1 appeared essential for competitive fitness of Foxp3+ regulatory T (Treg) cells, for Th17 cells generation and dendritic cells (DCs) function during experimental autoimmune encephalomyelitis (EAE) induction, and for CD8+ and NK cell responses during viral infection (Dudda et al., 2013; Lu et al., 2009; Murugaiyan et al., 2011; O’Connell et al., 2010; Zawislak et al., 2013). The latter findings were contradicted by a recent study that was unable to identify a role for miR-155-dependent repression of SOCS1 in CD8+ T cell responses to viral infection (Gracias et al.). These results illustrate the aforementioned difficulties in mechanistic understanding of miRNA biological function.
To investigate the biological significance of a single miRNA-mRNA interaction in vivo, we generated mice with mutations specifically disrupting the interaction between miR-155 and the SOCS1 gene and selectively abolishing its regulation by miR-155. Using these mice, we found that some biological phenotypes associated with the function of miR-155 can indeed be attributed to the regulation of SOCS1, whereas this same interaction can be dispensable in different cellular and biological contexts. Our data suggest that the relevance of a single miRNA-mRNA interaction is context- and cell-type dependent and highlight the complexity of miRNA regulatory networks.
Results
Genetic disruption of the miR-155-mediated repression of SOCS1 in mice
Our previous studies suggested that miR-155-mediated repression of SOCS1 confers Treg cell competitive fitness (Lu et al., 2009). This conclusion was supported by the observation of a comparable reduction in Treg cell numbers upon deletion of miR-155 or overexpression of SOCS1 (Lu et al., 2009). The transgenic overexpression of SOCS1 in T cells precludes, however, stringent mechanistic analysis of the contribution of miR-155-mediated SOCS1 regulation to controlling Treg cell homeostasis and other phenotypes associated with miR-155 deficiency (Androulidaki et al., 2009; Lu et al., 2009; O’Connell et al.). This approach also does not rule out the possibility that a broader spectrum of miR-155 targets besides SOCS1 could be involved in the observed effects. To directly test the impact of SOCS1 regulation by miR-155, and to explore to what extent the regulation of a single target alone can account for miRNA function, we generated mice harboring mutations in the putative miR-155 target site in the 3′ UTR of the SOCS1 gene (SOCS1KI) (Fig. S1). SOCS1 protein amounts were increased in T cells isolated from SOCS1KI mice when compared to their wild-type (WT) littermate controls to a degree similar to that observed in miR-155-deficient T cells (Fig. 1A, B). This result suggested that the mutations introduced in the miR-155 target site in the 3′ UTR of SOCS1 resulted in the ablation of miR-155-dependent SOCS1 repression in SOCS1KI mice. Moreover, retroviral overexpression of miR-155 was able to reduce the elevated SOCS1 protein levels in miR-155-deficient, but not in SOCS1KI T cells (Fig. 1C). In contrast, overexpression of miR-150, used as a control in this experiment, was able to similarly reduce amounts of its target Myb in both miR-155-deficient and SOCS1KI T cells (Xiao et al., 2007). Finally, the de-repression of SOCS1 resulting from introduced 3′ UTR mutations was miR-155 specific as overexpression of miR-19, another miRNA capable of -targeting SOCS1 (Pichiorri et al., 2008; Simpson et al., 2014), in SOCS1KI, miR-155-deficient or -sufficient T cells resulted in comparable reductions in the SOCS1 protein expression (Fig. S2). As a consequence, in Treg cells isolated from SOCS1KI mice we were able to observe reduced activation of Stat5, whose phosphorylation is negatively regulated by SOCS1, consistent with our previous finding of increased amounts of SOCS1 and attenuated IL-2 signaling in miR-155-deficient Treg cells (Lu et al., 2009) (Fig. 1D). Together, these results demonstrate that miR-155-dependent regulation of SOCS1 is abolished in SOCS1KI mice.
Figure 1. De-repression of SOCS1 in mice harboring mutations in the miR-155 target site in the 3′ UTR of the Socs1 gene.
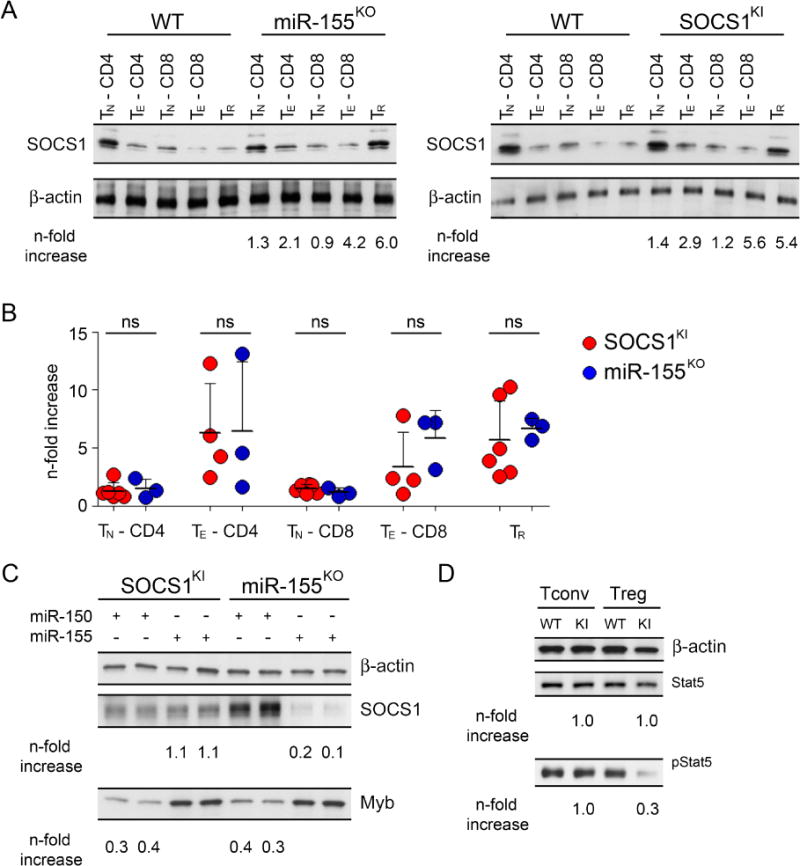
(A, B) Western blot analysis of SOCS1 amounts in different immune cell subsets isolated from miR-155KO and SOCS1KI mice. (n = 3–5) (C) Retroviral miR-155 or control miR-150 vectors equipped with a GFP reporter were expressed in T cells from miR-155KO and SOCS1KI mice. GFP+ cells were sorted 4 days after retroviral transduction and the amounts of SOCS1 and Myb proteins were assessed. (D) Immunoblot analysis of total and phospho-Stat5 (pStat5) in SOCS1KI Treg and Tconv cells. Densitometric expression values of SOCS1, Myb, total Stat5 or pStat5 normalized based on β-actin expression values and fold changes are shown below the corresponding lanes. The data are representative of two independent experiments (n = 2–4). See also Figure S1–S2.
miR-155-mediated SOCS1 regulation confers Treg cell competitive fitness
In contrast to reduced Treg cell numbers in miR-155 deficient mice, the Treg cell population was not diminished in size in SOCS1KI mice in comparison to WT littermates, despite increased SOCS1 protein amounts and reduced Stat5 activation in SOCS1KI Treg cells (Fig. 2). Therefore, de-repression of additional miR-155 targets in Treg cells or other cell types might directly or indirectly account for the aforementioned Treg cell deficiency in miR-155KO mice. Accordingly, the diminished Treg cell population was not fully restored upon SOCS1 ablation in Treg cells in miR-155KO mice (Fig. S3), although the Treg cell-restricted SOCS1 deficiency did increase Treg cell numbers in miR-155-sufficient mice in agreement with our previous report (Lu et al., 2009),
Figure 2. SOCS1KI mice did not exhibit reduced Treg cell numbers.
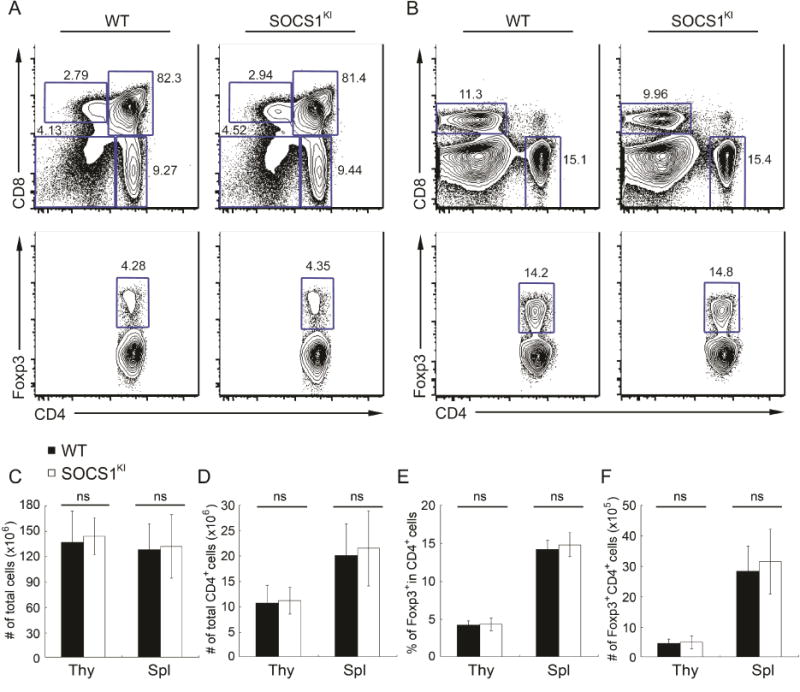
FACS analysis of (A) thymus and (B) spleen in 6~8 week old SOCS1KI mice and wild-type littermates. Percentages of different thymocyte and splenocyte subsets are shown. (C–F) Cellularity of the thymus and spleen and the proportion and absolute numbers of thymis and splenic Foxp3+CD4+ T cells in SOCS1KI and WT mice are shown. The data are shown as mean ± SD and are representative of four independent experiments (n = 4–8). See also Figure S3.
In order to examine the Treg cell autonomous role of miR-155-mediated SOCS1 repression in controlling Treg cell numbers, we transferred WT bone marrow (BM) precursor cells from WT mice mixed with BM cells from SOCS1KI or miR-155KO mice at a 1:1 ratio into irradiated T cell-deficient recipients as described previously (Lu et al., 2009). Four months after BM transfer, we observed a comparable 30%–50% reduction in the frequencies of SOCS1KI and miR-155KO Treg cells within the thymic and peripheral CD4+ T cell subsets as compared to the corresponding WT cell subsets in the control mixed chimeras (Fig. 3A, B). Thus, these experiments suggested a non-redundant cell-intrinsic role for miR-155-mediated SOCS1 repression in conferring competitive fitness to Treg cells (Lu et al., 2009). Moreover, we also detected substantial decreases in the splenic SOCS1KI CD4+ and CD8+ T cell subsets similar to those in miR-155KO compartments in comparison to their wild-type counterparts (Fig. 3C). These results provided an experimental proof for the previously suggested role for miR-155-dependent SOCS1-repression in the regulation of peripheral effector T (Teff) cell numbers in competitive settings (Lu et al., 2009).
Figure 3. miR-155-mediated SOCS1 regulation is critical to maintain normal Treg and Tconv cell numbers in a competitive setting.
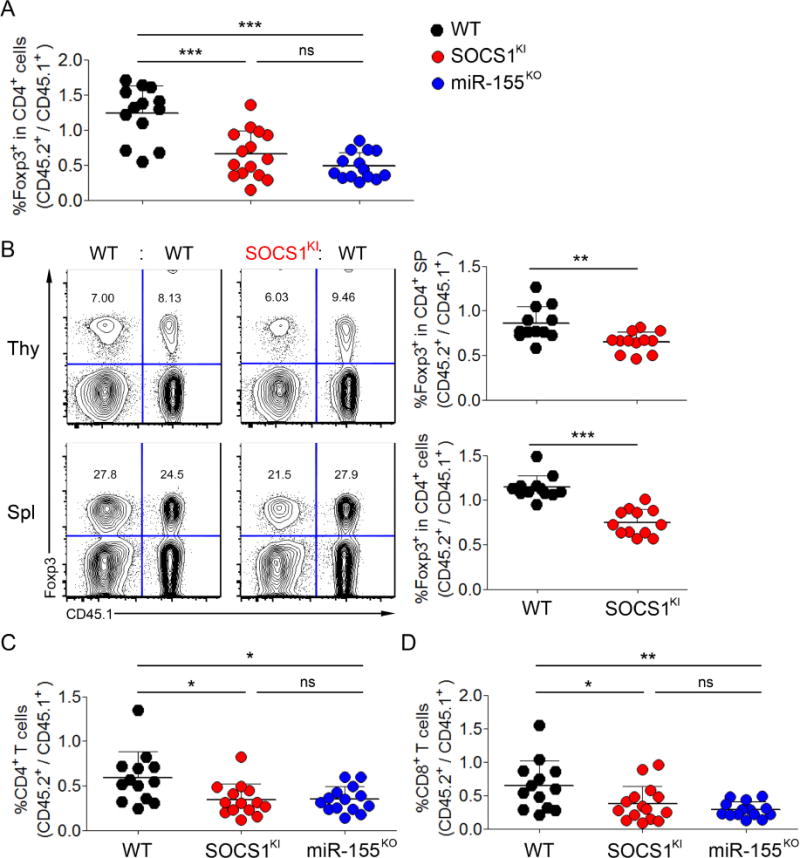
(A)Ratios of frequencies of CD45.1−Foxp3+ and CD45.1+Foxp3+ Treg cells within each donor-derived CD4+ T cell population from peripheral blood lymphocytes 100 days after BM transfer. (B) Treg cell frequencies within each donor-derived T cell population from both the thymus and spleen 120 days after BM transfer. (C) Ratios of frequencies of thymic and splenic CD45.1-Foxp3+ and CD45.1+Foxp3+ Treg cells within each donor-derived CD4+ T cell population 120 days after BM transfer. Ratios of frequencies of CD45.1−Foxp3− and CD45.1+Foxp3−(D) CD4+ and (E) CD8+ Tconv cells within each donor-derived compartment from peripheral blood lymphocytes 100 days after BM transfer. Each symbol represents an individual mouse, and the data are shown as mean ± SD.
Intermediate EAE severity in SOCS1KI in comparison to WT and miR-155KO mice
Previously, two studies have demonstrated a role for miR-155 in driving inflammatory responses during EAE (Murugaiyan et al.; O’Connell et al.). Impaired Th1 and Th17 responses as well as defective pro-inflammatory cytokine production by DCs were suggested to result in the reduced EAE observed in miR-155-deficient mice. While miR-155 could promote EAE through modulation of multiple targets and pathways, SOCS1 repression by miR-155 was proposed in both studies as a potential molecular mechanism behind the aforementioned phenomena due to a well established role of SOCS1 in regulating autoimmune responses (Alexander et al., 1999; Evel-Kabler et al., 2006; Hanada et al., 2003; Kinjyo et al., 2002; Marine et al., 1999). However, direct genetic evidence supporting the role of miR-155-mediated SOCS1 repression in EAE was lacking. To directly address this issue, we first induced EAE in SOCS1KI mice to investigate how loss of miR-155-mediated SOCS1 repression could impact autoimmune disease progression. As shown in Fig. 4A, mice with miR-155 deficiency exhibited an attenuated disease phenotype upon EAE induction, in agreement with previous studies (Murugaiyan et al.; O’Connell et al.). On the other hand, we observed an intermediate disease phenotype in SOCS1KI mice as compared to the severe or attenuated disease observed in WT littermates or miR-155KO mice, respectively (Fig. 4A). A markedly reduced disease severity could already be detected in SOCS1KI mice on day 17 of EAE induction compared to WT controls where the differences between SOCS1KI and miR-155KO mice were insignificant (Fig. 4A). Consistent with this finding, frequencies of IFNγ+ and IL-17+ CD4+ Teff cells in the brain of SOCS1KI mice were lower than those observed in WT control mice, but higher than in miR-155KO mice (Fig. 4B). The intermediate EAE disease phenotype as well as IFNγ and IL-17 responses in SOCS1KI mice harboring miR-155 resistant SOCS1 alleles were in line with the notion that a complex phenotype conferred by a given miRNA can depend on multiple targets of this miRNA.
Figure 4. Intermediate EAE disease phenotype in SOCS1KI mice compared to WT and miR-155KO mice.
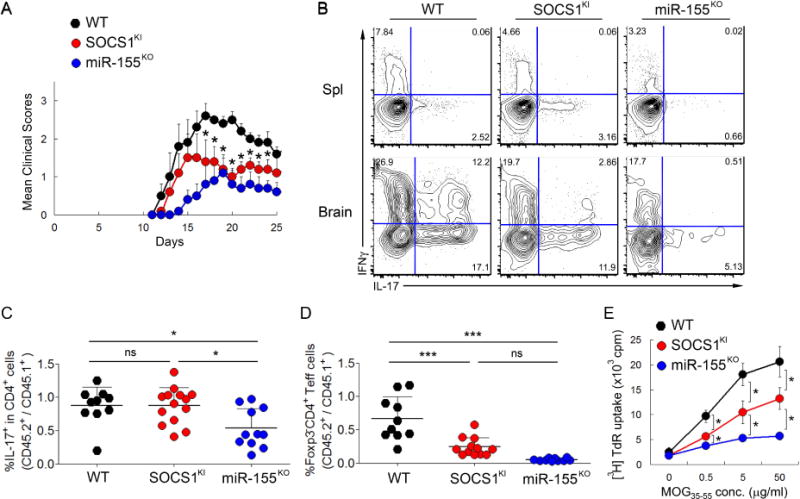
(A)EAE was induced in SOCS1KI, miR-155KO and WT control mice. Their disease severity was scored regularly based upon clinical symptoms. The data are shown as mean clinical scores ± SD and are pooled from 2 independent experiments (n = 10). (B) Frequency of IL-17+ and IFNγ+ T cells in spleen and brain from WT, SOCS1KI and miR-155KO mice induced with EAE. (C) Ratios of frequencies of CD45.1−IL-17+ and CD45.1+IL-17+ cells within each donor-derived CD4+ T cell population in the brain. (D) Ratios of frequencies of CD45.1−Foxp3−CD4+ and CD45.1+Foxp3−CD4+ Teff cells within each donor-derived compartment in the brain. Each symbol represents an individual mouse, and the data are shown as mean ± SD. (E) In vitro proliferative responses of WT, SOCS1KI and miR-155KO splenic CD4+ T cells after restimulation with indicated concentrations of MOG35–55 peptide. T cells were isolated on day 25 after EAE induction and cultured for 72 hours. Their proliferation was measured by 3[H] thymidine incorporation. The data are shown as mean cpm ± SD and are representative of 4 independent experiments (n = 8–12). See also Figure S4–S5.
Since both SOCS1 and miR-155 regulate immune responses in both T cells and non-T cell populations, it was possible that miR-155-mediated SOCS1 repression in T cells as well as other immune cell types could impact the outcome of autoimmune and inflammatory diseases (Murugaiyan et al.; O’Connell et al.). Indeed, it was suggested that miR-155 could promote the generation of inflammatory T cells and associated autoimmunity through modulating the production of pro-inflammatory cytokines by DCs via targeting SOCS1 (Murugaiyan et al.; O’Connell et al.). However, unlike DCs isolated from miR-155KO mice, SOCS1KI and WT DCs produced similar amounts of pro-inflammatory cytokines upon LPS stimulation (Fig. S4 and data not shown). To examine the T cell intrinsic role of miR-155-mediated SOCS1 repression in EAE, we induced EAE in mixed BM chimeras as previously described (Murugaiyan et al.; O’Connell et al.). Despite a similar EAE severity among all three groups of mixed BM chimeras (BM transplant composition: CD45.1+ WT + WT; CD45.1+ WT + SOCS1KI; CD45.1+ WT + miR-155KO), the frequency of IL-17-producing CD4+ Teff cells within miR155-deficient CD45.2+CD4+ Teff cells in the brain was severely reduced as compared to CD45.1+ miR-155-sufficient counterparts in agreement with previous studies (Fig. 4C). In contrast, we did not detect any difference in the frequency of IL-17-producing CD4+ Teff cells within the SOCS1KI and WT Teff cell subset in the brain. These results indicate that miR-155-mediated SOCS1 repression is not required for the differentiation of Th17 cells (Fig. 4C). Consistent with this finding, naive CD4+ T cells isolated from SOCS1KI and control mice exhibited a comparable capacity to differentiate into Th17 cells in vitro, whereas Th17 differentiation of miR-155-deficient T cells was impaired, as previously reported (Murugaiyan et al.; O’Connell et al.), (Fig. S5). Thus, it is likely that the diminished competitive fitness of Teff cells in the absence of miR-155-mediated regulation of SOCS1 contributes to the attenuated EAE progression and diminished Th17 cell numbers in the brains of SOCS1KI mice (Fig. 3C and 4A). Indeed, further analysis showed a marked reduction of both SOCS1KI and miR155KO CD4+ Teff cells as compared to the corresponding WT control populations in the mixed BM chimeras (Fig. 4D). Thus, the absolute numbers, but not the frequency of encephalitogenic SOCS1KI CD4+ effector cells were reduced. In agreement with this finding, MOG35–55 peptide-specific proliferative in vitro response of splenic CD4+ T cells isolated from SOCS1KI mice subjected to EAE induction was intermediate in magnitude in comparison to those of WT and miR-155-deficient T cells (Fig. 4E). These results suggested that the ameliorated autoimmune disease in miR-155-deficient mice could be in part attributed to impaired Teff cell homeostasis resulting from the lack of regulation of SOCS1 expression. However, the latter did not noticeably contribute to the defective Th17 cell differentiation and the altered cytokine production by DC associated with miR-155 deficiency.
SOCS1 regulation by miR-155 is dispensable for T cell responses to LCMV infection
Recent studies have demonstrated a severe defect in antiviral responses of miR-155-deficient T cells. To test whether miR-155-dependent modulation of SOCS1 played an important role in antiviral T cell responses, three groups of mixed BM chimeras described above were infected with LCMV Armstrong. On day 7 of infection we analyzed LCMV-specific T cell responses and compared the proportion of responding cells derived from either CD45.2+ WT, miR-155KO, or SOCS1KI BM to the corresponding CD45.1+ marked WT control cells in each individual mouse. We observed similar proliferative activity of SOCS1KI or WT CD4+Foxp3−Teff cells (~30% dividing cells based on expression of Ki-67), while the proliferative potential of miR-155-deficient Teff cells was severely reduced (Fig. 5A). Consistent with a SOCS1 regulation-independent role of miR-155 in antiviral CD4+ Teff responses, SOCS1KI and WT T cells showed a comparable frequency of activated CD44+CD62L− (Fig. 5B) and IFNγ-producing LCMV- GP61-specific CD4+ T cells (Fig. 5C). In contrast, the numbers of activated and LCMV-GP61-specific miR-155KO CD4+ T cells were greatly reduced as compared to the corresponding WT controls (Fig. 5A–C). SOCS1KI and WT control Treg cells expanded to a comparable degree whereas only very few Treg cells were detectable among miR-155-deficient CD4+ T cells (data not shown). CD8+ T cells specific for LCMV-GP33 or LCMV–NP396 peptides were identified by MHC class I tetramer staining and peptide-specific IFNγ production. Similar to antiviral CD4+ T cells, the responses of SOCS1KI and WT control CD8+ T cells were comparable, whereas miR-155KO CD8+ T cells exhibited a severe defect in response to LCMV (Fig. 5D and E). The observation that the induction of miR-155 in CD8+ T cells depends on TCR signal strength raised the possibility that the requirement of miR-155-dependent repression of SOCS1 may depend on TCR affinity, and that low-, but not high-affinity SOCS1KI CD8+ T cells may be able to respond to viral infection (Dudda et al., 2013). To exclude this possibility, we generated WT, SOCS1KI and miR-155KO P14 TCR transgenic (tg) mice. Naive CD8+ T cells expressing LCMV-GP33-specific TCR were isolated from either miR-155KO or SOCS1KI CD45.2+ P14 TCR tg mice and co-transferred with allelically marked CD45.1+CD45.2+ WT P14 TCR T cells into CD45.1+ hosts that were subsequently infected with LCMV Armstrong. Both SOCS1KI and WT control P14 T cells showed robust expansion by d7 of infection, whereas miR-155KO cells were strongly outnumbered by their WT control population (Fig. 5F). Thus, the response of both CD4+ and CD8+ T cells to acute viral infection was independent of miR-155-dependent regulation of SOCS1.
Figure 5. SOCS1 regulation by miR-155 is dispensable for acute antiviral T cell responses.
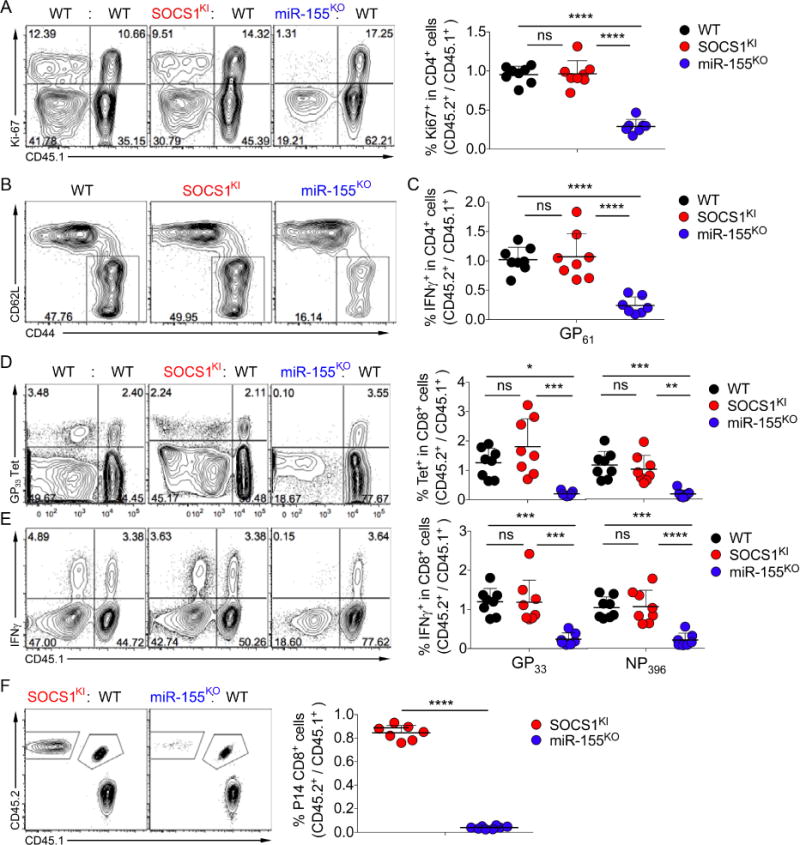
Mixed BM chimeras were infected with LCMV Armstrong and splenic T cells were analyzed by flow cytometry on d7 of infection. (A) Expression of Ki67 in Foxp3− CD4+ Teff cells. Depicted are representative FACS plots and ratios of the proportions of Ki67+ cells among CD45.2+ CD4+ WT, SOCS1KI or miR155KO cells versus CD45.1+ CD4+ wt controls. (B) Percentage of activated CD44+ CD62L− Foxp3− CD4+ Teff cells. (C) Ratios of IFNγ producing CD4+ Teff cells upon stimulation with LCMV-GP61 peptide. (D, E) Percentage and ratios of LCMV-GP33 and –NP396 specific tetramer binding (D) and IFNγ production of splenic CD8+ T cells (E). (F) CD45.2+ miR-155KO or SOCS1KI and CD45.1+CD45.2+ naïve CD8+ P14 TCR-tg T cells (104 each) were co-transferred into CD45.1+ WT hosts prior to infection with LCMV Armstrong. Splenic T cells were analyzed on d8 pi. The data are shown as mean ± SD and are pooled from 2 independent experiments (n = 7).
Cell type-dependent role of miR-155-dependent regulation of SOCS1 during MCMV infection
Besides CD8+ and CD4+ T cells, NK cells play a major role in antiviral immunity. To explore a role for SOCS1 regulation by miR-155 in NK vs. CD8+ T cell responses to the same viral pathogen we used mouse cytomegalovirus (MCMV) infection in SOCS1KI, miR-155KO and WT mice. In addition, these experiments allowed us to assess CD8+ T cell responses in a model of acute viral infection different from LCMV. MCMV encodes the m157 glycoprotein that serves as a cognate ligand for activating NK cell receptor Ly49H (Arase et al., 2002), and numerous CD8 T cell epitopes with distinct response kinetics (Snyder et al., 2008). First, we assessed antiviral responses of SOCS1KI CD8+ T cells by infecting mixed bone-marrow chimeras with MCMV and analyzing antigen-specific responses using MCMV-M45 tetramers. Similar to acute LCMV infection, we found that antigen-specific WT and SOCS1KI CD8+ T cells were generated with a comparable efficiency, while the numbers of antigen-specific miR-155KO CD8+ T cells were strongly reduced (Fig. 6A). Next, we analyzed the virus-specific expansion of Ly49H+ NK cells, which we have previously found to be strongly miR-155-dependent (Zawislak et al., 2013). miR-155KO NK cells have increased SOCS1 mRNA during MCMV infection. However, our previous study implied that additional miR-155-targets besides SOCS1 could account for the defect of antiviral NK cells. In addition, miR-155 has been proposed to regulate NK cell functions by modulating the expression of multiple signaling molecules including SHIP1 (Sullivan et al., 2013; Trotta et al., 2013). To test whether the miR-155-mediated regulation of SOCS1 alone impacts the expansion of antiviral NK cells, splenic NK cells from WT, SOCS-1KI or miR155KO were co-transferred with congenically-distinct marked WT control NK cells into mice deficient for Ly49H. Following MCMV infection, expansion of adoptively transferred Ly49H+ NK cells was analyzed on d7 post infection. SOCS1KI and miR-155KO NK cells were outnumbered by ~5–10-fold by WT control cells present within the same mouse (Fig. 6B). Thus, the inability of miR-155 to regulate SOCS1 in the SOCS1KI NK cells closely resembled the defect of complete absence of miR-155 in NK cells during MCMV infection. This surprising result revealed that the SOCS1 regulation by miR-155 is crucial for the expansion of virus-specific NK cells, but not CD8+ T cells during MCMV infection. On the other hand, we failed to observe significant effect of not only deficiency in SOCS1 regulation by miR-155, but also miR-155 itself on the numbers and responses to Nippostrongylus brasiliensis infection of type 2 innate lymphoid cells expressing increased amounts of miR155 in comparison to other ILC subsets (data not shown and http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE37448).
Figure 6. Cell type-specific role of miR-155-mediated regulation of SOCS1 during acute viral infection.
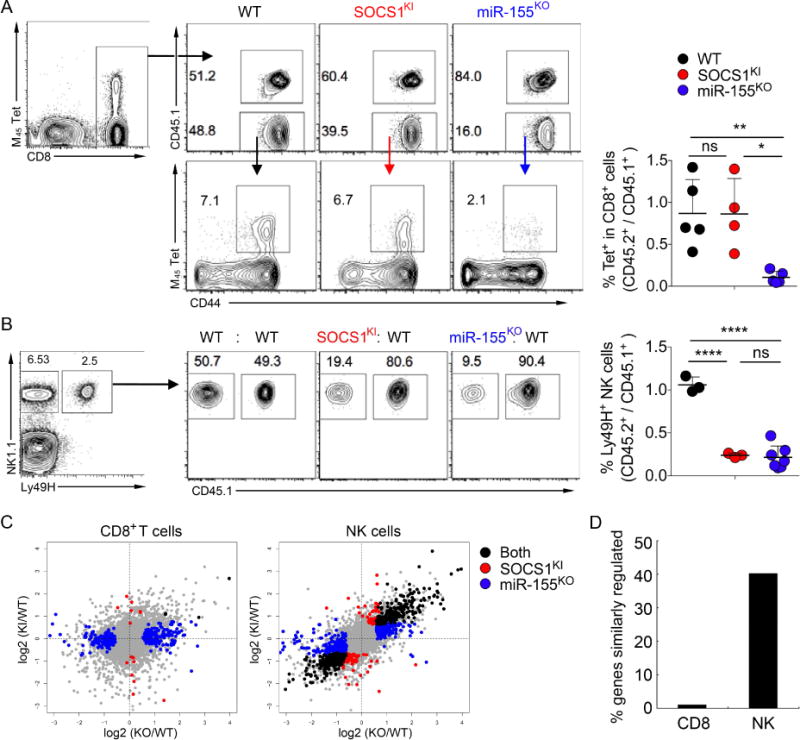
Mixed BM chimeras were infected with MCMV and splenic T cells were analyzed on d7 pi. (A) Percentage and ratios of MCMV-M45 specific tetramer binding of splenic CD8+ T cells. (B) CD45.2+ miR-155KO or SOCS1KI and CD45.1+ wt NK cells (2×105 each) were co-transferred into Ly49H-deficient hosts prior to infection with MCMV. Ratios of transferred Ly49H+ NK cells were analyzed in the spleen on d7 pi. The data are shown as mean ± SD and are representative of 3 independent experiments (n = 3–5). (C) Scatterplots depict log fold changes of gene expression in SOCS1KI and miR-155KO versus WT CD8+ T cells and Ly49H+ NK cells. Highlighted dots represent genes with significant log fold changes in SOCS1KI (red) or miR-155KO (blue) or both genotypes (black). (D) Bar graphs depict the percentage of genes similarly regulated in both SOCS1KI and miR-155KO from all the genes that were significantly changed in miR-155KO in indicated cell types. Sequencing data represent analysis of 3 biological replicates.
In order to quantify the number of genes whose expression was directly or indirectly regulated through the binding of miR-155 to the SOCS1 3′UTR in these two different cell types we performed RNA-seq analysis of gene expression. To compare virus-specific WT, miR-155KO and SOCS1KI CD8 and NK cells in mice with an intact WT myeloid and non-myeloid compartment, we performed adoptive transfers of WT, SOCS1KI and miR-155KO OVA-specific OT-1 TCR transgenic CD8+ T cells similar to those used for NK cells (Fig. 6B). Naïve OT-1 CD8+ T cells were adoptively transferred into CD45.1+ wt mice and then FACS-sorted for mRNA preparation on d4 post infection with MCMV-OVA. Adoptively transferred SOCS1KI and wt OT-1 T cells showed comparable expansion on d7 of infection whereas the expansion of miR-155KO OT-1 was strongly reduced (data not shown) in full agreement with our analysis of polyclonal antiviral CD8+ T cell responses (Fig 6A). For comparison of virus specific NK and CD8+ T cells, we adoptively transferred WT, SOCS1KI and miR-155KO NK cells as described above and FACS-purified Ly49H+ cells for mRNA preparation on d4 of infection. We found that only a small fraction (<2%) of the genes differentially regulated in miR-155KO versus wt CD8+ T cells were also dysregulated in SOCS1KI CD8+ T cells while expression of the majority of genes was not significantly altered (Fig 6C, D). These results suggest that the miR-155-SOCS1 axis has rather minor effects for the global gene expression in antiviral CD8+ T cells. In contrast, a large fraction (>40%) of the genes were similarly regulated in both miR-155KO and SOCS1KI NK cells (Fig. 6C, D), suggesting that miR-155-mediated regulation of SOCS1 plays a major role in miR-155-dependent regulated gene expression in antiviral responses of NK cells. This cell-type specific difference in the dependence of global gene expression on the miR-155-SOCS1 axis translates into a strict requirement of SOCS1 regulation by miR-155 for the expansion of virus-specific NK cells, but not CD8+ T cells during MCMV infection.
Context-dependent role of miR-155-SOCS1 interaction for antiviral CD8+ T cells
Our experiments showed a distinct requirement for a single miRNA-target interaction for responses of adaptive and innate cytotoxic lymphocytes to acute viral infection. Next, we wanted to test a requirement for the regulation of SOCS1 by miR-155, which was dispensable for CD8 T cell responses to acute LCMV infection, in the context of chronic infection. To address this question, we transferred SOCS1KI and congenically marked WT control P14 TCR tg CD8+ T cells into recipient mice that were subsequently infected with LCMV Armstrong or clone 13 to induce an acute and resolving or a chronically persisting viral infection, respectively. In agreement with the experiments described above, both SOCS1KI and WT T cells showed similar robust expansion on d8 of LCMV Armstrong infection (Fig. 5F and 7A). On d8 of LCMV clone 13 infection, however, SOCS1KI T cells were underrepresented when compared to WT control cells and were gradually lost over the course of persistent infection (Fig. 7A and B). miR-155KO P14 T cells exhibited a severe expansion defect during acute LCMV infection and were almost undetectable on d8 of chronic LCMV infection (Fig. S6). Thus, the miR-155-mediated regulation of SOCS1 expression was superfluous for the acute expansion of CD8+ T cells, but was indispensable for the maintenance of the antiviral T cell response during chronic infection.
Figure 7. Context-dependent role of miR-155-SOCS1 interaction for antiviral CD8+ T cells.
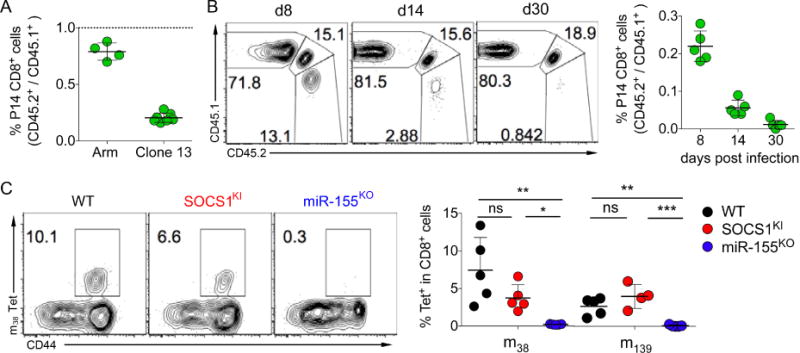
(A, B) CD45.2+ miR-155KO or SOCS1KI and CD45.1+ P14 TCR-tg T cells (104 each) were co-transferred into CD45.1+CD45.2+ WT hosts prior to infection with LCMV Armstrong (Arm) or clone 13. (A) Ratios of P14 T cells in the spleen on d8 pi. (B) Kinetic analysis of P14 T cells in peripheral blood of LCMV clone 13 infected mice. (C) Mixed BM chimeras were infected with MCMV and the percentage and ratios of MCMV-m38 and –m139 specific tetramer binding of splenic CD8+ T cells was analyzed on d40 pi. The data are shown as mean ± SD and are representative of 2–3 independent experiments (n = 4–7). See also Figure S6–S7.
Persistent LCMV clone 13 infection is characterized by continuous viral replication and chronic T cell stimulation. In contrast, MCMV persistence is achieved through latency without continuous viremia. During latency, some viral genes are intermittently transcribed and T cells specific for the respective gene products are intermittently stimulated and undergo progressive expansion and terminal differentiation, a process termed “memory inflation” (Snyder et al., 2008). SOCS1KI T cells were able to respond to latent viral infection and underwent normal memory inflation. We detected comparable frequencies of SOCS1KI and WT control T cells specific for “inflationary” MCMV antigens m38 and m139 (Fig. 7C). Both SOCS1KI and WT inflated T cells underwent terminal effector differentiation characterized by a KLRG-1hi CD127lo phenotype (Fig. S7). In contrast, m38 and m139 specific miR-155KO T cells were barely detectable (Fig. 7C). Therefore, the T cell response to acute and latently persisting viral infection was independent of miR-155-dependent regulation of SOCS1, but was indispensable for the antiviral T cell response during chronic persistent infection.
Together, our analysis of regulation of SOCS1 by miR-155 revealed its distinct impact on numbers and function of T cell subsets and NK cells in physiologic settings and in settings of autoimmunity and viral infection. These studies provide the first genetic evidence that a single miRNA-mRNA interaction can account for complex biological phenotypes in a cell-type and biological context dependent manner.
Discussion
miR-155 is one of the most prominent miRNA operating in the immune system. Many miR-155 targets were identified and their regulation by miR-155 has been proposed to affect a wide range of immunological processes (Dorsett et al., 2008; Dudda et al., 2013; O’Connell et al., 2009; Rodriguez et al., 2007; Teng et al., 2008; Vigorito et al., 2007; Zawislak et al., 2013). Conventional genetic approaches such as the germ-line or cell-type restricted ablation of individual miRNAs have been helpful in identifying major phenotypes associated with miRNA function, but are unable to reveal the role of individual miRNA-mRNA interactions. While prior work relying on the overexpression, knockdown or complete ablation of specific miRNA targets have provided correlative evidence for the biological role of their regulation, conflicting data emerged from these studies. Direct testing of the biological significance of individual miRNA-mRNA interactions requires the genetic disruption of miRNA binding to a specific target. Such approaches have previously been used to explore the regulation of AID and PU.1 by miR-155 in the B cell lineage (Dorsett et al., 2008; Lu et al., 2014; Teng et al., 2008). However, these studies did not address a role of a given miRNA dependent regulation in different biological and cellular settings. Through side-by-side comparison of mice harboring the SOCS1 allele uncontrollable by miR-155 and mice lacking miR-155, we isolated the effect of a miRNA on one of its targets and demonstrated the biological significance of a single target repression in miRNA-mediated regulation of the immune response during autoimmunity and infection.
Previous studies showed that the expression of miR-155 in Treg cells is required to maintain normal Treg cell numbers both in the thymus and periphery (Kohlhaas et al., 2009; Lu et al., 2009). The Treg cell phenotype observed in miR-155KO mice was at least in part attributed to attenuated IL-2 signaling in the absence of miR-155-mediated regulation of SOCS1 (Lu et al., 2009). Indeed, our experiments unequivocally demonstrated the Treg cell-intrinsic role of miR-155-mediated SOCS1 repression in conferring Treg cell competitive fitness. However, in contrast to the reduced Treg cell subset observed in miR-155KO mice, Treg cell numbers were unaffected in SOCS1KI mice. These results indicate that, in contrast to the non-redundant role of miR155-mediated regulation of SOCS1 in competitive settings, additional targets contribute to the miR-155 dependent Treg cell maintenance in non-competitive settings. This notion is also supported by the observation that even a complete ablation of the SOCS1 gene in Treg cells does not rescue the diminished Treg cell population size in miR-155-deficient mice. Moreover, the altered function of miR-155-deficient non-Treg cell populations could also contribute to the impaired Treg cell development and maintenance in miR-155-deficient mice. Finally, our observation of normal Treg cell numbers in SOCS1KI mice despite reduced pStat5 activation suggests that a somewhat relaxed demand for IL-2 signaling enabled normal Treg cell numbers in a non-competitive setting. It has been recently reported that a population of CD44hiCD62LloCCR7lo Treg cells relies on ICOS signaling rather than IL-2 for their maintenance in vivo (Smigiel et al., 2014). Thus, it is also possible that the latter Treg cell subset is maintained at the expense of Treg cells that are more sensitive to diminished IL-2 signaling in SOCS1KI mice.
Several studies have demonstrated a pivotal role of miR-155 in EAE through promoting pathogenic Th17 responses (Murugaiyan et al., 2011; O’Connell et al., 2010; Zhang et al., 2014). Reduced numbers of effector Th17 cells in miR-155-deficient mice were shown to be largely due to the T cell-intrinsic lack of miR-155-mediated gene regulation. Additionally, impaired production of pro-inflammatory cytokines by miR-155-deficient DCs could also contribute to the attenuated EAE progression in miR-155-deficient mice (Murugaiyan et al., 2011; O’Connell et al., 2010). Although the exact molecular mechanisms accounting for these phenotypes are currently unknown, the loss of miR-155-mediated SOCS1 repression has been proposed to be responsible for defective DC functions (Murugaiyan et al., 2011; O’Connell et al., 2010). Moreover, miR-155-mediated SOCS1 repression has also been shown to promote Th17 differentiation through modulation of JAK-STAT pathway in a T cell-autonomous manner (Yao et al., 2012).
However, we did not observe any alteration in cytokine production by DCs nor did we detect impaired Th17 responses of T cells upon the selective abrogation of SOCS1 regulation by miR-155. These results clearly suggested that SOCS1 repression by miR-155 is dispensable for the aforementioned biological responses. Instead, we found miR-155-mediated SOCS1 regulation contributed to EAE pathogenesis likely through maintaining the size of effector T cell populations particularly at the site of autoimmune inflammation. Collectively, our results indicate that de-repression of SOCS1 together with other miR-155 targets is required to render miR-155KO mice largely resistant to EAE.
Recent studies have demonstrated a severe defect of miR-155-deficient T cells in response to viral infection. The vigorous up-regulation of miR-155 observed upon T cell activation enables the expansion and accumulation of virus-specific Teff cells in the acute phase of infection. It was suggested that miR-155 moderately affected transcripts of a number of target genes associated with type I interferon signaling (Gracias et al., 2013). The cumulative effect of the miR-155-mediated down-regulation of these targets was proposed to counteract the anti-proliferative effect of type I interferons during infection. In contrast, another study attributed the defect of antiviral responses of miR-155-deficient T cells to increased SOCS1 mRNA levels and showed that the overexpression of SOCS1 results in a phenocopy of miR-155-deficiency, i.e. inhibition of the expansion of LCMV-specific T cells (Dudda et al., 2013). Our analysis of SOCS1KI mice indisputably showed that the responses of both CD8+ and CD4+ T cells to acute viral infection were independent of miR155-mediated regulation of SOCS1. During chronic persistent viral infection, however, the maintenance of virus-specific CD8+ T cells required the modulation of SOCS1-levels by miR-155. SOCS1KI T cells were progressively lost during the chronic phase of infection. These results suggest that increased SOCS1 levels are detrimental during continuous T cell stimulation by viral antigen or the altered inflammatory environment generated upon persisting viral infection., SOCS1KI T cells could respond to both acute and latent viral infection and undergo memory inflation during latent MCMV infection. While we failed to detect a role for the miR-155-mediated regulation of SOCS1 expression in CD8+ T cell responses during acute viral infection, this mechanism proved to be critical for the expansion of virus-specific NK cells in the context of the same infection. Moreover, the fact that in infected miR-155KO or SOCS1KI mice a large proportion of genes was similarly regulated in NK cells, but not CD8+ T cells supported our in vivo observations. In addition, these findings raised an interesting question as to whether distinct cytokine signaling strength is required for activating the downstream transcriptional programs in CD8+ T cells and NK cells. Alternatively, it is also possible that the miR-155-mediated SOCS1 regulation is redundant in CD8+ T cells, but not in NK cells due to the presence of other CD8+ T cell-specific miR-155 targets that could play a compensatory role in regulating cytokine-driven responses. While the mechanisms determining cell-type-specific regulation of miRNA/target interactions remain to be further investigated at a biochemical level, our side-by-side comparison of the impact of miR-155 deficiency and of the inability of miR-155 to regulate SOCS1 on the response of the two major cytotoxic lymphocyte subsets revealed a context-dependent and cell-type specific role of a single miRNA-target RNA interaction during acute and latent versus persistent chronic viral infection and highlight the complex and dynamic character of these interactions.
The SOCS1KI mouse model enabled us to directly test the role of an individual miRNA/target interaction in different biological and cellular contexts. Nevertheless, it was possible that the mutation of the SOCS1 3′UTR may also affect potential binding of miRNAs other than miR-155, or the recruitment of RNA binding proteins that are required for SOCS1 regulation by other miRNAs. While we found that in both SOCS1KI and miR-155KO CD4+ T cells the mutated 3′UTR can still be regulated by another SOCS1-targeting miRNA, miR-19, we cannot completely rule out this seemingly unlikely scenario. Another possibility is that the modulation of one target site may potentially also affect the abundance of a specific miRNA and thereby influence the regulation of other competing miRNA targets indirectly. A recent study demonstrated that modulation of miRNA target abundance is unlikely to cause significant effects on gene expression as target repression can only be released after many competing target sites (≥1.5 × 105 target sites per cell) were introduced (Denzler et al., 2014). Our previous unbiased analysis of miR-155 targets using HITS-CLIP showed that any single of the most abundant miR-155 targets is bound by less than 5–10% of total miR155 pool (Loeb et al., 2012). Thus, the extent of regulation of other miR-155 targets cannot be perturbed in a meaningful way by excluding regulation of any single miR-155 target (data not shown). Moreover, considering the relatively low expression level of SOCS1 mRNA with only one miR-155 binding site compared to the level of miR-155, one of the highly expressed miRNAs in activated lymphocytes (Bosson et al., 2014), we do not anticipate that such effects significantly contributed to the phenotypes observed in our study. Finally, as complex miRNA-dependent cellular phenotypes may require the coordinate repression of multiple target genes by a given miRNA, our finding that miR-155-mediated SOCS1 regulation plays an indispensable role in specific cellular and biological contexts does not exclude the possible functional involvement of other miR-155 targets.
In summary, our findings highlight that for related biological functions a given miRNA-target mRNA interaction can be critical as de-repression of an individual target can abolish miRNA-dependent phenotypes. In other cases, such an interaction can only have a partial role or might even be fully dispensable depending on the cellular and physiological context.
Experimental Procedures
Detailed experimental procedures available as online supplemental information
Animals
The targeting construct harboring mutations in the miR-155 target site in the 3′ UTR of the SOCS1 gene was generated using recombineering (http://redrecombineering.ncifcrf.gov/) on a B6 genetic background. miR-155KO and C57BL/6J mice were purchased from the Jackson Laboratories. SOCS1fl and Klra8KO mice were kindly provided by A. Yoshimura and S. Vidal, respectively.
Retroviral Transduction and Western Blot Analyses
To overexpress miR-155 and control miR-150 in T cells, CD4+CD25−CD62Lhi naive T cells isolated from indicated mice were first activated by plate-bound CD3 and CD28 antibodies (2 μg/ml each) and IL-2 (50U) for one day and retrovirally transduced with miR-155-expressing or control miR-150-expressing pMDH-PGK-EGFP plasmids (Lu et al., 2009). Four days after initial retroviral transduction, cells were harvested and sorted for GFP expression. FACS-sorted GFP+ miR-155 (or miR-150, or miR-19a/b) overexpressing cells as well as different T cell subsets from indicated mice were lysed and subjected to western blot analysis as described previously (Lu et al., 2009). SOCS1, Myb, Stat5, phospho-Stat5 and β-actin (loading control) were visualized with monoclonal antibodies ab62584 (Abcam), #1–1 (Upstate), #9363, #9359 (both from Cell Signaling Technology) and AC-74 (Sigma), correspondingly. Protein quantitation was performed with NIH Image J software (http://rsb.info.nih.gov/ij/).
Generation of Mixed Bone-Marrow Chimeras
TCRβδKO were irradiated (950cGy) and rested over night. The next day bone marrow cells were isolated from femurs and tibias of CD45.2+ WT, miR-155KO or SOCS1KI mice and CD45.1+ WT mice and depleted of CD90+ and NK1.1+ cells (Dynabeads Flow Comp). A 1:1 mixture of mutant or CD45.2+ WT control and CD45.1+ WT cell populations (2.5×106 each) were i.v. injected into recipients. Mice were kept on neomycin water for 3 weeks.
EAE
Mice were immunized s.c. with 100μg MOG35–55 emulsified with complete Freund’s adjuvant plus 200ng pertussis toxin in 500ul PBS. Animals were scored 3 times a week for disease symptoms as described previously (O’Connell et al., 2010).
Virus Infections
Mice were infected with cell culture-grown LCMV Armstrong (2×105 pfu i.p.) or LCMV clone 13 (2×106 pfu i.v.). Salivary gland preparations of MCMV (1×104 PFU) were injected i.p. as described previously (Zawislak et al., 2013).
Adoptive Cell Transfer
NK cells were negatively enriched using NK cell isolation Kit (Miltenyi) (Sun et al., 2009). TCR-tg T cells were stained with surface antibodies and then CD8+ CD44- naïve cells were sorted on a FACS AriaII cell sorter (BD Biosciences). Cells were washed and 104 TCR-tg T cells or 2×105 NK cells were transferred by i.v. injection 4–12h prior to infection.
Quantification of Antigen-Specific T cell responses
Splenocytes were stained with multimers for LCMV-GP33, LCMV-NP396, MCMV-M45, -m38 and -m139 or OVA257 produced from biotinylated monomers provided by the NIH Tetramer facility. For cytokine production, splenocytes were briefly incubated with indicated peptides and Brefeldin A (Sigma-Aldrich) and then stained for intracellular cytokine production. For live/dead-discrimination propidium iodide or LIVE/DEAD fixable yellow dye (Molecular Probes) was used. Cells were analyzed on a LSRII cytometer (BD Biosciences) and analyzed with FlowJo software (Treestar).
Gene Expression Profiling Analysis
For gene expression profiling analysis, CD45.2+ miR-155KO or SOCS1KI or WT OT-1 TCR-tg CD8+ T cells or NK cells were adoptively transferred into CD45.1+ WT or Ly49H-deficient hosts, respectively, prior to infection with MCMV-OVA or MCMV. OT-1 CD8+ T cells or Ly49H+ NK cells were FACS-sorted on d4 post infection and poly-A RNA-sequencing was performed using 3 biological replicates for each cell type analyzed using an Illumina HiSeq2500 platform. RNA-sequencing data are available from NCBI under accession number GSE68511.
Statistical Analysis
All statistical analyses were performed using GraphPad Prism5 software. Results are expressed as means +/− standard deviation. Differences between individual groups were analyzed for statistical significance using the one-way ANOVA *=p<0.05, **=p<0.01, ***=p<0.001, ns= not significant.
Supplementary Material
Acknowledgments
This work was supported by NIH grants AI089935, AI103646, AI108651 (LFL), AI085034, AI100874 (JCS) and AI034206 (AYR). GG is supported by an Irvington Fellowship of the Cancer Research Institute and the DFG Emmy Noether programme. JCS is a Searle Scholar. AYR is an investigator with the Howard Hughes Medical Institute. We thank the technical services provided by the Transgenic Mouse Model Core Facility of the National Core Facility Program for Biotechnology, National Science Council and the Gene Knockout Mouse Core Laboratory of National Taiwan University Center of Genomic Medicine. We would also like to thank Alice Kamphorst and Rafi Ahmed for providing P14 T cells, and Andrea Ventura for providing miR-19a/b retroviral plasmids. The authors declare no competing financial interests.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
LFL, GG, AYR conceived and designed the experiments. LFL, GG, AC, JPH, PDB, LLL, CLZ, SC performed the experiments. LFL, GG, JPH, YL analyzed the data. LFL, ISY, JCS, CSL, SWL established and contributed reagents/materials/analysis tools. LFL, GG, AYR wrote the paper.
References
- Alexander WS, Starr R, Fenner JE, Scott CL, Handman E, Sprigg NS, Corbin JE, Cornish AL, Darwiche R, Owczarek CM, et al. SOCS1 is a critical inhibitor of interferon gamma signaling and prevents the potentially fatal neonatal actions of this cytokine. Cell. 1999;98:597–608. doi: 10.1016/s0092-8674(00)80047-1. [DOI] [PubMed] [Google Scholar]
- Androulidaki A, Iliopoulos D, Arranz A, Doxaki C, Schworer S, Zacharioudaki V, Margioris AN, Tsichlis PN, Tsatsanis C. The kinase Akt1 controls macrophage response to lipopolysaccharide by regulating microRNAs. Immunity. 2009;31:220–231. doi: 10.1016/j.immuni.2009.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arase H, Mocarski ES, Campbell AE, Hill AB, Lanier LL. Direct recognition of cytomegalovirus by activating and inhibitory NK cell receptors. Science. 2002;296:1323–1326. doi: 10.1126/science.1070884. [DOI] [PubMed] [Google Scholar]
- Baltimore D, Boldin MP, O’Connell RM, Rao DS, Taganov KD. MicroRNAs: new regulators of immune cell development and function. Nat Immunol. 2008;9:839–845. doi: 10.1038/ni.f.209. [DOI] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosson AD, Zamudio JR, Sharp PA. Endogenous miRNA and target concentrations determine susceptibility to potential ceRNA competition. Molecular cell. 2014;56:347–359. doi: 10.1016/j.molcel.2014.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denzler R, Agarwal V, Stefano J, Bartel DP, Stoffel M. Assessing the ceRNA hypothesis with quantitative measurements of miRNA and target abundance. Molecular cell. 2014;54:766–776. doi: 10.1016/j.molcel.2014.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorsett Y, McBride KM, Jankovic M, Gazumyan A, Thai TH, Robbiani DF, Di Virgilio M, San-Martin BR, Heidkamp G, Schwickert TA, et al. MicroRNA-155 suppresses activation-induced cytidine deaminase-mediated Myc-Igh translocation. Immunity. 2008;28:630–638. doi: 10.1016/j.immuni.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudda JC, Salaun B, Ji Y, Palmer DC, Monnot GC, Merck E, Boudousquie C, Utzschneider DT, Escobar TM, Perret R, et al. MicroRNA-155 is required for effector CD8+ T cell responses to virus infection and cancer. Immunity. 2013;38:742–753. doi: 10.1016/j.immuni.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evel-Kabler K, Song XT, Aldrich M, Huang XF, Chen SY. SOCS1 restricts dendritic cells’ ability to break self tolerance and induce antitumor immunity by regulating IL-12 production and signaling. J Clin Invest. 2006;116:90–100. doi: 10.1172/JCI26169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gracias DT, Stelekati E, Hope JL, Boesteanu AC, Doering TA, Norton J, Mueller YM, Fraietta JA, Wherry EJ, Turner M, Katsikis PD. The microRNA miR-155 controls CD8(+) T cell responses by regulating interferon signaling. Nature immunology. 2013;14:593–602. doi: 10.1038/ni.2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanada T, Yoshida H, Kato S, Tanaka K, Masutani K, Tsukada J, Nomura Y, Mimata H, Kubo M, Yoshimura A. Suppressor of cytokine signaling-1 is essential for suppressing dendritic cell activation and systemic autoimmunity. Immunity. 2003;19:437–450. doi: 10.1016/s1074-7613(03)00240-1. [DOI] [PubMed] [Google Scholar]
- Ilangumaran S, Ramanathan S, Rottapel R. Regulation of the immune system by SOCS family adaptor proteins. Semin Immunol. 2004;16:351–365. doi: 10.1016/j.smim.2004.08.015. [DOI] [PubMed] [Google Scholar]
- Kinjyo I, Hanada T, Inagaki-Ohara K, Mori H, Aki D, Ohishi M, Yoshida H, Kubo M, Yoshimura A. SOCS1/JAB is a negative regulator of LPS-induced macrophage activation. Immunity. 2002;17:583–591. doi: 10.1016/s1074-7613(02)00446-6. [DOI] [PubMed] [Google Scholar]
- Kohlhaas S, Garden OA, Scudamore C, Turner M, Okkenhaug K, Vigorito E. Cutting edge: the Foxp3 target miR-155 contributes to the development of regulatory T cells. J Immunol. 2009;182:2578–2582. doi: 10.4049/jimmunol.0803162. [DOI] [PubMed] [Google Scholar]
- Loeb GB, Khan AA, Canner D, Hiatt JB, Shendure J, Darnell RB, Leslie CS, Rudensky AY. Transcriptome-wide miR-155 binding map reveals widespread noncanonical microRNA targeting. Molecular cell. 2012;48:760–770. doi: 10.1016/j.molcel.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu D, Nakagawa R, Lazzaro S, Staudacher P, Abreu-Goodger C, Henley T, Boiani S, Leyland R, Galloway A, Andrews S, et al. The miR-155-PU.1 axis acts on Pax5 to enable efficient terminal B cell differentiation. The Journal of experimental medicine. 2014;211:2183–2198. doi: 10.1084/jem.20140338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu LF, Thai TH, Calado DP, Chaudhry A, Kubo M, Tanaka K, Loeb GB, Lee H, Yoshimura A, Rajewsky K, Rudensky AY. Foxp3-dependent microRNA155 confers competitive fitness to regulatory T cells by targeting SOCS1 protein. Immunity. 2009;30:80–91. doi: 10.1016/j.immuni.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marine JC, Topham DJ, McKay C, Wang D, Parganas E, Stravopodis D, Yoshimura A, Ihle JN. SOCS1 deficiency causes a lymphocyte-dependent perinatal lethality. Cell. 1999;98:609–616. doi: 10.1016/s0092-8674(00)80048-3. [DOI] [PubMed] [Google Scholar]
- Murugaiyan G, Beynon V, Mittal A, Joller N, Weiner HL. Silencing microRNA-155 ameliorates experimental autoimmune encephalomyelitis. Journal of immunology. 2011;187:2213–2221. doi: 10.4049/jimmunol.1003952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell RM, Chaudhuri AA, Rao DS, Baltimore D. Inositol phosphatase SHIP1 is a primary target of miR-155. Proc Natl Acad Sci U S A. 2009;106:7113–7118. doi: 10.1073/pnas.0902636106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell RM, Kahn D, Gibson WS, Round JL, Scholz RL, Chaudhuri AA, Kahn ME, Rao DS, Baltimore D. MicroRNA-155 promotes autoimmune inflammation by enhancing inflammatory T cell development. Immunity. 2010;33:607–619. doi: 10.1016/j.immuni.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichiorri F, Suh SS, Ladetto M, Kuehl M, Palumbo T, Drandi D, Taccioli C, Zanesi N, Alder H, Hagan JP, et al. MicroRNAs regulate critical genes associated with multiple myeloma pathogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:12885–12890. doi: 10.1073/pnas.0806202105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez A, Vigorito E, Clare S, Warren MV, Couttet P, Soond DR, van Dongen S, Grocock RJ, Das PP, Miska EA, et al. Requirement of bic/microRNA-155 for normal immune function. Science. 2007;316:608–611. doi: 10.1126/science.1139253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson LJ, Patel S, Bhakta NR, Choy DF, Brightbill HD, Ren X, Wang Y, Pua HH, Baumjohann D, Montoya MM, et al. A microRNA upregulated in asthma airway T cells promotes TH2 cytokine production. Nature immunology. 2014;15:1162–1170. doi: 10.1038/ni.3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smigiel KS, Richards E, Srivastava S, Thomas KR, Dudda JC, Klonowski KD, Campbell DJ. CCR7 provides localized access to IL-2 and defines homeostatically distinct regulatory T cell subsets. The Journal of experimental medicine. 2014;211:121–136. doi: 10.1084/jem.20131142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder CM, Cho KS, Bonnett EL, van Dommelen S, Shellam GR, Hill AB. Memory inflation during chronic viral infection is maintained by continuous production of short-lived, functional T cells. Immunity. 2008;29:650–659. doi: 10.1016/j.immuni.2008.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan RP, Fogel LA, Leong JW, Schneider SE, Wong R, Romee R, Thai TH, Sexl V, Matkovich SJ, Dorn GW, 2nd, et al. MicroRNA-155 tunes both the threshold and extent of NK cell activation via targeting of multiple signaling pathways. Journal of immunology. 2013;191:5904–5913. doi: 10.4049/jimmunol.1301950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun JC, Beilke JN, Lanier LL. Adaptive immune features of natural killer cells. Nature. 2009;457:557–561. doi: 10.1038/nature07665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng G, Hakimpour P, Landgraf P, Rice A, Tuschl T, Casellas R, Papavasiliou FN. MicroRNA-155 is a negative regulator of activation-induced cytidine deaminase. Immunity. 2008;28:621–629. doi: 10.1016/j.immuni.2008.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trotta R, Chen L, Costinean S, Josyula S, Mundy-Bosse BL, Ciarlariello D, Mao C, Briercheck EL, McConnell KK, Mishra A, et al. Overexpression of miR-155 causes expansion, arrest in terminal differentiation and functional activation of mouse natural killer cells. Blood. 2013;121:3126–3134. doi: 10.1182/blood-2012-12-467597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigorito E, Kohlhaas S, Lu D, Leyland R. miR-155: an ancient regulator of the immune system. Immunological reviews. 2013;253:146–157. doi: 10.1111/imr.12057. [DOI] [PubMed] [Google Scholar]
- Vigorito E, Perks KL, Abreu-Goodger C, Bunting S, Xiang Z, Kohlhaas S, Das PP, Miska EA, Rodriguez A, Bradley A, et al. microRNA-155 regulates the generation of immunoglobulin class-switched plasma cells. Immunity. 2007;27:847–859. doi: 10.1016/j.immuni.2007.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao C, Calado DP, Galler G, Thai TH, Patterson HC, Wang J, Rajewsky N, Bender TP, Rajewsky K. MiR-150 controls B cell differentiation by targeting the transcription factor c-Myb. Cell. 2007;131:146–159. doi: 10.1016/j.cell.2007.07.021. [DOI] [PubMed] [Google Scholar]
- Yao R, Ma YL, Liang W, Li HH, Ma ZJ, Yu X, Liao YH. MicroRNA-155 modulates Treg and Th17 cells differentiation and Th17 cell function by targeting SOCS1. PloS one. 2012;7:e46082. doi: 10.1371/journal.pone.0046082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura A, Naka T, Kubo M. SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol. 2007;7:454–465. doi: 10.1038/nri2093. [DOI] [PubMed] [Google Scholar]
- Zawislak CL, Beaulieu AM, Loeb GB, Karo J, Canner D, Bezman NA, Lanier LL, Rudensky AY, Sun JC. Stage-specific regulation of natural killer cell homeostasis and response against viral infection by microRNA-155. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:6967–6972. doi: 10.1073/pnas.1304410110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Cheng Y, Cui W, Li M, Li B, Guo L. MicroRNA-155 modulates Th1 and Th17 cell differentiation and is associated with multiple sclerosis and experimental autoimmune encephalomyelitis. Journal of neuroimmunology. 2014;266:56–63. doi: 10.1016/j.jneuroim.2013.09.019. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.