

Abstract
Adenosine receptors (ARs) have emerged as new drug targets. The majority of data on affinity/potency and selectivity of AR ligands described in the literature has been obtained for the human species. However, preclinical studies are mostly performed in mouse or rat, and standard AR agonists and antagonists are frequently used for studies in rodents without knowing their selectivity in the investigated species. In the present study, we selected a set of frequently used standard AR ligands, 8 agonists and 16 antagonists, and investigated them in radioligand binding studies at all four AR subtypes, A1, A2A, A2B, and A3, of three species, human, rat, and mouse. Recommended, selective agonists include CCPA (for A1AR of rat and mouse), CGS-21680 (for A2A AR of rat), and Cl-IB-MECA (for A3AR of all three species). The functionally selective partial A2B agonist BAY60-6583 was found to additionally bind to A1 and A3AR and act as an antagonist at both receptor subtypes. The antagonists PSB-36 (A1), preladenant (A2A), and PSB-603 (A2B) displayed high selectivity in all three investigated species. MRS-1523 acts as a selective A3AR antagonist in human and rat, but is only moderately selective in mouse. The comprehensive data presented herein provide a solid basis for selecting suitable AR ligands for biological studies.
Electronic supplementary material
The online version of this article (doi:10.1007/s11302-015-9460-9) contains supplementary material, which is available to authorized users.
Keywords: Agonist, Antagonist, Selectivity, Species differences, Potency
Introduction
Although purinergic signaling was proposed as early as 1970, the cloning of these receptors in the early 1990s was a turning point in the acceptance of the purinergic signaling hypothesis [1]. P1 (adenosine) receptors, which are distinguished from P2 (ATP) receptors, comprise four subtypes of G protein-coupled receptors: A1, A2A, A2B, and A3 [1, 2]. All adenosine receptors (ARs) are coupled to adenylate cyclase. Whereas A1 and A3ARs inhibit adenylate cyclase, A2A and A2BARs stimulate its activity and thereby increase the intracellular concentration of cAMP [3]. The wide distribution of ARs in virtually all tissues is an indication of their significance and their promising potential as drug targets [4]. The nonselective AR antagonists, caffeine, and theophylline, which are plant-derived alkaloids, have been used as drugs since a long time for various indications, including countering of fatigue and restoration of alertness, treatment of pain (in combination with analgesics), bronchial asthma, chronic obstructive pulmonary disease (COPD), prevention of sleep apnea, and treatment of apnea in preterm infants [5]. In the past decades, many potent AR agonists and antagonists with selectivity for one of the four subtypes have been developed [5, 6]. Radioligands as well as fluorescent-labeled ligands for ARs have been prepared [7–9]. The development of new ligands targeting ARs has been facilitated by recently published crystal structures of the A2AAR bound to both, agonists and antagonists [10–13]. Several AR agonists and antagonists are currently evaluated in clinical trials or have recently been approved as diagnostic or therapeutic drugs [14, 15].
The A1AR of human, rat, and mouse consists of 326 amino acids. The percentage of amino acid sequence identity of the A1AR in the three species was determined to be as follows: human vs. rat 95 %, human vs. mouse 95 %, and rat vs. mouse 98 % (for details see supporting information). (Partial) A1AR agonists are clinically developed for cardiologic indications including arrhythmia, atrial fibrillation, and coronary and ventricular dysfunction [16, 17]. Further indications may comprise metabolic syndrome including hyperlipidemia and type II diabetes [18]. A1 antagonists have been evaluated for the treatment of congestive heart failure and renal failure [19].
The A2AAR is the largest AR subtype consisting of 412 amino acids in humans and 410 amino acids in rat and mouse. The percentage of amino acid sequence homology of A2AAR in the three species was determined to be as follows: human vs. rat 82 %, human vs. mouse 82 %, and rat vs. mouse 96 %. Adenosine (Adenoscan®) and regadenoson (Lexiscan®) are used in myocardial stress imaging acting on A2AARs of coronary blood vessels which leads to dilation and a drop in blood pressure [20, 21]. The A2AAR represents one of the most important regulators of the innate immune response. A2AARs inhibit the secretion of proinflammatory mediators by immune cells dampening inflammatory reactions [22, 23] and therefore have potential as anti-inflammatory, anti-rheumatic, and immunosuppressive drugs. Separation of their anti-inflammatory and hypotensive effects has recently been achieved by a prodrug approach [24]. A2AAR antagonists are widely investigated for their role in neurodegenerative diseases like Parkinson’s and Alzheimer’s disease [25–27]. The A2A-selective antagonist istradefylline (NOURIAST®) has recently been approved in Japan for the treatment of Parkinson’s disease, and further A2AAR antagonists are in clinical development [28, 29].
The A2BAR consists of 332 amino acids in human, rat, and mouse. The percentage of amino acid sequence homology of A2BAR in the three species was determined to be as follows: human vs. rat 86 %, human vs. mouse 87 %, and rat vs. mouse 96 %. Preclinical studies indicated a potential of A2B agonists for the treatment of atherosclerosis and hyperlipidemia [30]. A2B antagonists may be useful for the treatment of asthma, diabetes, pain, and inflammatory diseases in general, although the A2BAR has been reported to promote pro- as well as anti-inflammatory effects [6, 31–33]. Furthermore, A2B antagonists were proposed for the treatment of cancer and the prevention of metastasis [34, 35].
The A3AR consists of 318 amino acids in human, 320 amino acids in rat, and 319 amino acids in mouse. The percentage of amino acid sequence homology of A3AR in the three species was determined to be as follows: human vs. rat 73 %, human vs. mouse 73 %, and rat vs. mouse 91 %. A3AR agonists are developed for the treatment of rheumatoid arthritis, psoriasis, and other autoimmune diseases [36]. A3 antagonists may be useful for the treatment of asthma [37] or glaucoma [38].
Taking the (patho) physiological importance of ARs and their great potential as drug targets into consideration, it is obvious that the question of species differences is pivotal. Since rat and mouse are the most widely employed animals in preclinical studies, it is extremely important to know and consider potential differences in potency and selectivity of the receptor ligands that are applied as pharmacological tools, e.g., for target validation studies. Differences in affinity and potency of several AR ligands at different species have been obvious [39, 40], especially regarding the A3AR subtype. Many A3 antagonists are highly potent at the human receptor, but only weakly active or inactive at rodent A3ARs [5, 41, 42].
Since the event of receptor cloning and heterologous expression, new compounds are typically evaluated on human receptors, the ultimate target in drug development. However, the first in vivo studies are performed in rodents. Very often, it appears difficult to choose the most suitable subtype-selective AR agonist or antagonist for a specific preclinical in vivo study or to judge the results of such investigations, due to the lack of knowledge about potential species differences regarding the various ligands. This knowledge is essential for tool compounds that are commonly utilized in experimental pharmacology of ARs in order to avoid jumping to false conclusions about their effects and the AR subtypes involved.
Surprisingly, we discovered that for many AR agonists and antagonists, that are commonly applied in in vitro and in vivo studies, comprehensive data at all subtypes of human, rat, and mouse ARs are lacking. Especially data for mouse receptors are sparse despite the fact that mice represent the most abundant animal models in drug research. In fact, for many compounds described in the literature, only data at human ARs have been published. Nevertheless, such ligands are frequently used in mouse or rat models without knowing but just assuming that their profiles were identical across species. In the present study, we therefore investigated the most frequently applied standard AR agonists and antagonists in radioligand binding assays at human, mouse, and rat A1, A2A, A2B, and A3AR. Our goal was to determine the compounds’ affinities and to calculate their selectivity within each species. Based on the results, suitable ligands for each of the investigated species can then be recommended.
Experimental section
Chemicals and compounds
The AR ligands, caffeine, theophylline, NECA, R-PIA, and MRS-1523 were commercially available from Sigma Aldrich. CADO was commercially available from TCI. CCPA, CGS-21680, Cl-IB-MECA, IB-MECA, DPCPX, ZM-241835, and MRS-1745 were commercially available from TOCRIS. BAY60-6583 was kindly provided by Dr. Thomas Krahn (Bayer Healthcare). The following compounds were synthesized in our laboratory according to published procedures: PSB-36 [43], PSB-63 [43], PSB-601 [44], PSB-1115 [45], PSB-0788 [46], PSB-603 [46], istradefylline (KW-6002) [47], and preladenant (SCH-240814) [48].
Molecular biology
The cDNAs of rat A1 (rA1), rA2A, and mouse A3 (mA3) were purchased from OriGeneInc. (Rockville, USA) and subcloned into the vector pQCXIN. The cDNAs of the mA1, mA2A, and mA2B receptor were obtained from ImaGenesInc. (Rockville, USA) and subcloned into the vector pLXSN. All cDNAs were amplified by PCR (initial denaturation for 4 min at 94 °C; 25 cycles of 1 min 94 °C, 1 min 66 °C, and 2 min 72 °C; final elongation for 10 min at 72 °C) using the primers listed in supplemental Table 1 adding the desired restriction sites necessary for the insertion into the vectors.
Table 1.
K D values of the employed radioligands at the four adenosine receptor subtypes
| Radioligand | K D value ± SEM (nM)a | ||
|---|---|---|---|
| Human A1AR | Rat A1AR | Mouse A1AR | |
| [3H]CCPA | 0.60 [49] | 0.21 [50] | 0.610 ± 0.060 |
| [3H]DPCPX | 3.86 [49] | 0.28 [51] | 0.504 ± 0.054 |
| Human A2AAR | Rat A2AAR | Mouse A2AAR | |
| [3H]CGS-21680 | 32 [49] | 15.5 [41] | 17.0 ± 1.7 |
| [3H]MSX-2 | 7.29 [52] | 8.0 [5] | 12.1 ± 1.3 |
| Human A2BAR | Rat A2BAR | Mouse A2BAR | |
| [3H]PSB-603 | 0.403 [53] | 0.457 ± 0.051 | 4.09 ± 0.29 |
| Human A3AR | Rat A3AR | Mouse A3AR | |
| [3H]NECA | 6.18 [49] | 48.6 ± 2.5 | 15.1 ± 2.1 |
aData are means ± SEM of three independent assays performed in duplicates; values without SEM are taken from the literature
The cDNAs of rA2B and rA3 were amplified from genomic DNA of PC12 cells. Genomic DNA was isolated using the PurelinkTM genomic DNA mini kit (Invitrogen, Darmstadt, Germany), and both exons of the rA2B and rA3 genes were amplified separately using the primers listed in supplemental Table 2 with the following PCR protocol: initial denaturation for 4 min at 94 °C; 40 cycles of 1 min 94 °C, 1 min gradient of 49–62 °C, and 1 min 72 °C; final elongation for 10 min at 72 °C.
Table 2.
K i values of standard AR ligands at A1 adenosine receptors
| A1 Adenosine receptor affinity | |||
|---|---|---|---|
| Human | Rat | Mouse | |
| Agonists | K i ± SEM (nM) vs. [3H]CCPAa | ||
| NECA | 14 [49] | 5.1 [54] | 2.49 ± 0.15 |
| CADO | 1.39 [3] | 6.7 [54] | 2.79 ± 0.04 |
| CCPA | 0.83 [49] | 7.43 [55] | 0.269 ± 0.064 |
| R-PIA | 2.04 [49] | 1.2 [56] | 0.587 ± 0.070 |
| CGS-21680 | 289 [49] | 1800 [56] | 961 ± 108 |
| BAY60-6583 | 387 ± 70 | 514 ± 57 | 351 ± 19 |
| IB-MECA | 51 [57] | 54 [58] | 16.4 ± 1.7 |
| CI-IB-MECA | 220 [57] 115 [3] |
820 [59] | 46.2 ± 3.6 |
| Antagonists | K i ± SEM (nM) vs. [3H]DPCPXa | ||
| Caffeine | 44,900 [60] 10,700 [61] |
18,800 [46] 41,000 [62] |
50,700 ± 5800 |
| Theophylline | 6200 [46] | 8500 [46] | 14,200 ± 6000 |
| CGS-15943 | 3.5 [56] | 6.4 [56] | 1.15 ± 0.16 |
| DPCPX | 3.0 [63] | 0.50 [64] | 0.413 ± 0.064 |
| PSB-36 | 0.7 [5] | 0.124 [60] | 1.58 ± 0.18 |
| PSB-63 | 90.6 [5] | 16.9 [5] | 2370 ± 475 |
| CPT | 23.7 ± 2.4 | 24 [5] 6.3 [65] |
7.41 ± 0.41 |
| Istradefylline | 841 [5] | 230 [46] 150 [66] |
438 ± 93 |
| ZM-241385 | 774 [56] | 684 ± 63 | 236 ± 36 |
| Preladenant | >1000 [56] | >1000 | 462 ± 118 |
| PSB-601 | 2070 [44] | 260 [44] | 123 ± 25 |
| PSB-603 | >10,000 [46] | >10,000 [46] | 42.4 ± 4.2 |
| PSB-1115 | >10,000 [46] | 2200 [45] | 591 ± 63 |
| PSB-0788 | 2240 [46] | 386 [46] | 118 ± 21 |
| MRS-1754 | 403 [67] | 16.8 [67] | 1.45 ± 0.42 |
| MRS-1523 | >10,000 [56] | 15,600 [68] | >10,000 |
aData are means ± SEM of three independent assays performed in duplicates. The values without SEM are taken from the literature as indicated
The two exons of rA2B were inserted into the vector pLXSN by sequential ligation. In brief, in a second PCR reaction, the desired restriction sites were added into the 5′ end of exon 1 and the 3′ end of exon 2 using the primer pairs f-rA2B-EcoRI/r-rA2B-Exon1 and f-rA2B-Exon2/r-rA2B-BamHI applying the protocol for cloning described above. The digested vector and both exons were then ligated using T4 ligase for 1 h at room temperature followed by heat inactivation of the ligase for 10 min at 65 °C. The remaining blunt ends of the two exons were phosphorylated using the T4 protein kinase N for 45 min at 37 °C followed by heat inactivation of the enzyme for 15 min at 55 °C. The second ligation was carried out at 16 °C overnight.
The two exons of rA3 were combined by overlap extension PCR, and the final product was cloned into pQCXIN as described above. In brief, the exons were amplified in two separate PCR reactions with the primer pairs f-rA3/r-rA3-Overlap-Ex1 and f-rA3-Overlap-Ex2/r-rA3, respectively, (see supporting information Table 3) using the following protocol: initial denaturation for 4 min at 94 °C; 35 cycles of 1 min 94 °C, 1 min gradient of 49–57 °C, and 1 min 72 °C; final elongation for 10 min at 72 °C. Both products were mixed and, without adding additional primers, the following PCR protocol was run: initial denaturation for 4 min at 94 °C; 10 cycles of 1 min 94 °C, 1 min 49 °C, and 1 min 72 °C; final elongation for 5 min at 72 °C. The primers f-rA3-EcoRI and r-rA3-XhoI (see supporting information Table 2) were then added, and the final product, which was used for cloning, was obtained by 35 cycles of the same PCR protocol.
Table 3.
K i values of standard AR ligands at A2A adenosine receptors
| A2A Adenosine receptor affinity | |||
|---|---|---|---|
| Human | Rat | Mouse | |
| Agonists | K i ± SEM (nM) vs. [3H]CGS-21680a | ||
| NECA | 20 [56] | 9.7 [54] | 43.4 ± 13.2 |
| CADO | 180 [3] | 76 [54] | 59.7 ± 2.2 |
| CCPA | 2270 [49] | 640 [55] | 988 ± 189 |
| R-PIA | 860 [49] | 220 [56] | 683 ± 58 |
| CGS-21680 | 27 [49] | 19 [56] | 13.7 ± 4.5 |
| BAY60-6583 | >10,000 | >10,000 | >10,000 |
| IB-MECA | 2900 [57] | 948 ± 395 | 326 ± 121 |
| CI-IB-MECA | 5360 [69] 2100 [58] |
470 [59] | 290 ± 57 |
| Antagonists | K i ± SEM (nM) vs. [3H]MSX-2a | ||
| Caffeine | 23,400 [60] 9560 [5] |
43,000 [62] 32,500 [46] |
11,100 ± 2000 |
| Theophylline | 4200 [46] 1710 [5] |
25,000 [46] | 5770 ± 340 |
| CGS-15943 | 1.2 [56] | 3.3 [70] | 0.177 ± 0.059 |
| DPCPX | 129 [5] | 157 [46] | 263 ± 71 |
| PSB-36 | 980 [5] | 552 [60] | 697 ± 208 |
| PSB-63 | 34,500 [5] | 22,000 [5] | >10,000 |
| CPT | 147 ± 23 | 488 ± 132 | 744 ± 65 |
| Istradefylline | 26.4 ± 5.9 | 2.2 [66] 5.15 [46] |
6.83 ± 0.62 |
| ZM-241385 | 1.6 [56] | 1.25 ± 0.44 | 0.554 ± 0.072 |
| Preladenant | 0.9 [56] | 0.986 ± 0.014 | 0.241 ± 0.025 |
| PSB-601 | 484 [44] | 93.7 [44] | 598 ± 39 |
| PSB-603 | >10,000 [46] | >10,000 [46] | >10,000 |
| PSB-1115 | 3790 ± 520 | 24,000 [45] | >10,000 |
| PSB-0788 | 333 [46] | 1730 [46] | 235 ± 14 |
| MRS-1754 | 503 [67] | 612 [67] | >10,000 |
| MRS-1523 | 3660 [56] | 2050 [68] | >10,000 |
aData are means ± SEM of three independent assays performed in duplicates. The values without SEM values are taken from the literature
The GP+envAM12 cell line, derived from mouse fibroblasts, was used as a packaging cell line. Then the cDNAs were introduced into Chinese hamster ovary (CHO) cells via retroviral transfection. A cotransfection with a vector encoding the glycoprotein G of the vesicular stomatitis virus (VSV-G) was conducted in order to expand the host range of the retrovirus. Thus, stably transfected cell lines with the desired receptors were established.
Membrane preparation
Membranes of CHO cells expressing the A1, A2A, A2B, or A3 receptor were prepared by scratching the cells off the previously frozen cell culture dishes in ice-cold hypotonic buffer (5 mM TRIS, 2 mM EDTA, pH 7.4). The cell suspension was homogenized on ice for 20 s with an Ultra-Turrax and centrifuged for 10 min (4 °C) at 1000g in order to get rid of the cell organelles. The supernatant was subsequently centrifuged for 60 min at 48,000g in order to collect the protein pellets. The supernatant was then discarded, and the obtained membrane pellets were resuspended in 0.1 mL per dish of 50 mM TRIS buffer, pH 7.4. Aliquots of the membrane preparation (1 mL each) were stored at −80 °C until used.
Saturation binding assays
Saturation assays were carried out to determine the KD values of the radiolabeled ligands. Three independent experiments, each in duplicates, were performed for the determination of the KD values. [3H]CCPA and [3H]DPCPX (specific activity, 58 and 93 Ci/mmol, respectively; GE Healthcare, Buckinghamshire, UK) were employed at the mA1AR as an agonist and antagonist radioligand, respectively. [3H]CGS-21680 (specific activity, 41 Ci/mmol; PerkinElmer, Massachusetts, USA) and [3H]MSX-2 (specific activity, 84 Ci/mmol; GE Healthcare, Buckinghamshire, UK) were used at the mA2AAR as an agonist and antagonist radioligand, respectively. [3H]PSB-603 (specific activity, 73 Ci/mmol; GE Healthcare, Buckinghamshire, UK) was employed at the mA2B and rA2BAR as an antagonist RL. [3H]NECA (specific activity, 15.9 Ci/mmol; PerkinElmer, Massachusetts, USA) was used at the mA3 and rA3AR as an agonist RL. All saturation assays were carried out in a total volume of 400 μl, with the exception of A2B assays, which were performed in a total volume of 1000 μl. The radioligand stock solutions were prepared using 50 mM TRIS buffer, pH 7.4 with the exception of the [3H]NECA stock solutions, which were prepared using 50 mM TRIS, 1 mM EDTA, 10 mM MgCl2 buffer of pH 8.25. For A1, A2A, and A3AR, the saturation assays were performed by adding 190 μl of the buffer, 10 μl DMSO (total binding), or 10 μl of a specific ligand in DMSO for the non-specific binding (CADO or DPCPX at a final concentration of 10 μM for agonists, or antagonists at A1AR, respectively; NECA or CGS-15943 at a final concentration of 50 μM or 10 μM for agonists or antagonists at A2AAR, respectively; R-PIA at a final concentration of 100 μM for A3AR). The corresponding membrane preparation (100 μl) and 100 μl of the suitable radioligand solution were added. Both saturation and competition assays were performed in the presence of 2 U/ml of adenosine deaminase (ADA).
The saturation assays for the A2BAR were performed by adding 775 μl of the buffer, 25 μl of DMSO (total binding), or 25 μl of DPCPX dissolved in DMSO at a final concentration of 10 μM for non-specific binding, 100 μl of the corresponding membrane preparation, and 100 μl of the radioligand solution.
The incubation was done at room temperature, and the time was 90 min (for agonists) or 60 min (for antagonists) at A1ARs, 30 min (for agonists) and 60 min (for antagonists) at A2AARs, 75 min at A2BARs for both agonists and antagonists, and 180 min at A3ARs for both agonists and antagonists.
Competition binding assays
Competition binding assays were performed using the radioligands mentioned above previously characterized in saturation binding assays. The final concentrations were as follows: [3H]CCPA, 1 nM; [3H]DPCPX, 0.4 nM; [3H]CGS-21680, 5 nM; [3H]MSX-2, 1 nM; [3H]PSB-603, 0.3 nM; and [3H]NECA, 10 nM. All assays except for the A2B assays were performed in a final volume of 400 μl containing 190 μl of 50 mM TRIS buffer, pH 7.4, 10 μl of the test compound in DMSO, 100 μl of membrane preparation, and 100 μl of the radioligand. The A2B assays were performed in a final volume of 1000 μl containing 775 μl of the 50 mM TRIS buffer, pH 7.4, 25 μl of the test compound in DMSO, 100 μl of membrane preparation, and 100 μl of the radioligand. Non-specific binding was determined using the compounds CADO or DPCPX, NECA or CGS15943, DPCPX, and R-PIA, at final concentration of 10, 50 or 10, 10, and 100 μM for A1, A2A, A2B, and A3ARs, respectively.
The incubation was performed at room temperature for 90 min (agonists) and 60 min (antagonists) at A1ARs, 30 min (agonists) and 60 min (antagonists) at A2AARs, 75 min at A2BAR for both agonists and antagonists, and 180 min at A3AR for both agonists and antagonists. After the incubation, the assay mixture was filtered through GF/B glass fiber filters using a Brandel harvester (Brandel, Gaithersburg, MD, USA). Filters were washed four times (3–4 ml each) with ice-cold 50 mM Tris–HCl buffer, pH 7.4 (for A1, A2A, and A3 assays). Filters in the A2B assays were washed four times (3–4 mL each) with ice-cold 50 mM Tris–HCl buffer, pH 7.4 containing 0.1 % bovine serum albumin (BSA) in order to reduce non-specific binding. Then filters were transferred to scintillation vials, incubated for 9 h with 2.5 mL of scintillation cocktail (Beckmann Coulter), and counted in a liquid scintillation counter (Tricarb 2700TR) with a counting efficiency of ~53 %. Three separate experiments in duplicates were performed for each assay.
ß-arrestin recruitment assay
The β-arrestin recruitment assay is based on the detection of the interaction of a GPCR with β-arrestin by β-galactosidase fragment complementation (β-arrestin Path Hunter assay, DiscoverX, Fremont, CA, USA). The GPCR of interest is fused to the Prolink tag, the N-terminal part of β-galactosidase, and β-arrestin is fused to the enzyme acceptor, which is β-galactosidase lacking the first 41 amino acids. Upon receptor activation, β-arrestin is recruited to the receptor which leads to the complementation of both β-galactosidase fragments [71]. The activity of the functional β-galactosidase is measured by chemiluminescence using commercial Path Hunter detection reagents.
Recruitment of β-arrestin molecules to the respective receptor was detected by using β-galactosidase fragment complementation technology (β-arrestin Path Hunter assay, DiscoverX, Fremont, CA, USA). CHO cells stably expressing the respective receptor were seeded in a volume of 90 μL into a 96-well plate and incubated at a density of 20,000 cells/well in the provided plating reagent for 24 h at 37 °C. After the incubation, test compounds were diluted in PBS buffer containing 10 % DMSO and 0.1 % BSA and added to the cells in a volume of 10 μL, followed by incubation for 90 min at 37 °C. For the determination of baseline luminescence, PBS buffer (containing 10 % DMSO, 0.1 % BSA) in the absence of test compound was used. During the incubation period, the detection reagent was prepared. For the determination of β-arrestin recruitment, the provided detection reagent was used according to the supplier’s protocol. After the addition of 50 μL/well detection reagent to the cells, the plate was incubated for an additional 60 min at room temperature. Finally, luminescence was determined in a luminometer (TopCount NXT, Packard/Perkin-Elmer).
For the determination of antagonistic properties of tested compounds, the assay was performed as described for agonists except that the test compounds were added to the cells in a volume of 5 μL/well 60 min prior to addition of the agonist (100 nM CCPA final concentration, corresponding to EC80, for A1AR; 30 nM Cl-IB-MECA final concentration, corresponding to EC80, for A3AR). Data were obtained from three independent experiments performed in duplicate. Data were analyzed using Graph Pad Prism, version 4.02 (San Diego, CA, USA).
cAMP accumulation in CHO cells
Cells were removed from a confluent 175-cm2 flask, transferred into a 50-ml Falcon tube, and centrifuged at 200g, 4 °C for 5 min. After removal of the supernatant, the cell pellet was resuspended in DMEM-F12 medium. The cell suspension (500 μl, ∼200,000 cells per well) was transferred to 24-well plates and incubated for 24 h at 37 °C. After removal of the culture medium, cells were washed HBSS buffer and then incubated with 230 μl HBSS buffer for 2 h at 37 °C. The phosphodiesterase inhibitor Ro20-1724 (final concentration 40 μM) dissolved in 100 % HBSS buffer was added to each well, and the cells were incubated for 10 min. For assaying antagonists, 12.5 μl of antagonist was added to the cells which were subsequently incubated for 10 min, and thereafter 12.5 μl of agonist was added and the cells were incubated again for 10 min. For assaying agonists, 25 μl of the agonist was added and the cells were incubated for 20 min. After incubation with the agonist, cAMP production was stimulated by the addition of forskolin (final concentration 10 μM) for 10 min; the final DMSO concentration did not exceed 1.4 %. The reaction was stopped by the removal of the reaction buffer followed by the addition of a hot lysis buffer (250 μl; 90 °C; 4 mM EDTA, 0.01 % Triton X-100; Sigma, Munich, Germany). The 24-well plates were kept at room temperature for 5 min and then kept at −20 °C. For competition binding experiments, 50 μl of the cell lysates were transferred into 2.5-ml tubes. [3H]cAMP (30 μl) (3 nM final concentration) in lysis buffer and 40 μl of cAMP-binding protein in the same buffer (75 μg protein per vial) were added. Total binding was determined with 50 μl of lysis buffer, 30 μl of [3H]cAMP solution, and 40 μl of cAMP-binding protein containing solution. Nonspecific filter binding was determined with 90 μl of lysis buffer and 30 μl of [3H]cAMP solution. For a cAMP standard curve, 50 μl of known cAMP concentrations were used instead of 50 μl of cell lysate. After an incubation time of 1 h on ice, the assay mixture was filtered through GF/B glass fiber filters using a Brandel harvester. Filters were washed three times with ice-cold 50 mM Tris–HCl buffer, pH 7.4. Then filters were transferred into mini vials, incubated for 9 h with 2.5 ml of scintillation cocktail (LumaSafe Plus; PerkinElmer Life and Analytical Sciences), and counted in a liquid scintillation counter (Tricarb 2700TR; PerkinElmer Life and Analytical Sciences) with a counting efficiency of ∼53 %.
Data analysis and statistics
The specific binding, which is described as the radioligand binding to a ligand-specific binding site of the respective receptor, was calculated as the difference between total and nonspecific binding. The specific binding was used to determine the KD and Ki values. The KD values were determined by the one-site binding function. The Ki values were calculated from the IC50 values, the concentration of the radioligand [L] and the equilibrium dissociation constant KD of the radioligand used (Ki = IC50/(1 + [L]/KD)) based on the equation of Cheng and Prusoff (1973). The data for the functional assays were analyzed using sigmoidal dose–response function with variable slope. The results are given as mean ± SEM. For all functional assays, sigmoidal curves were obtained. Data from the binding and ß-arrestin assays were analyzed using GraphPad Prism 6.01 (San Diego, CA, USA).
Results
For the evaluation of a selection of the most frequently used standard AR agonists and antagonists at human (h), rat (r), and mouse (m) A1, A2A, A2B, and A3 ARs recombinant CHO cell lines were employed. Cells expressing the human AR subtypes were previously prepared [46, 49], and data for standard AR ligands at the human receptors had already been reported in the literature. Cell lines expressing the rat and the mouse AR subtypes were established in the present study.
Cloning and expression of adenosine receptors
The cloning of the following receptor genes was conducted: rA1, rA2A, rA2B, rA3, mA1, mA2A, mA2B, and mA3. Sequence homology of the cloned receptors with the published receptor sequences was controlled, and no mutation was found (results not shown). Stable CHO cell lines were generated by retroviral transfection.
Saturation binding assays
In order to allow the calculation of Ki values from competition binding assays, KD values of the employed radioligands have to be known. Therefore, we determined those KD values that had not been previously described. Saturation binding assays at the mA1AR were carried out with the agonist radioligand [3H]CCPA and the antagonist radioligand [3H]DPCPX (see Figs. 1a, b). The determined KD values were 0.610 nM for [3H]CCPA and 0.504 nM for [3H]DPCPX. Similar Bmax values of 1150 and 1280 fmol/mg were determined with [3H]CCPA and [3H]DPCPX, respectively, indicating a high expression level in the mA1 CHO cell line.
Fig. 1.
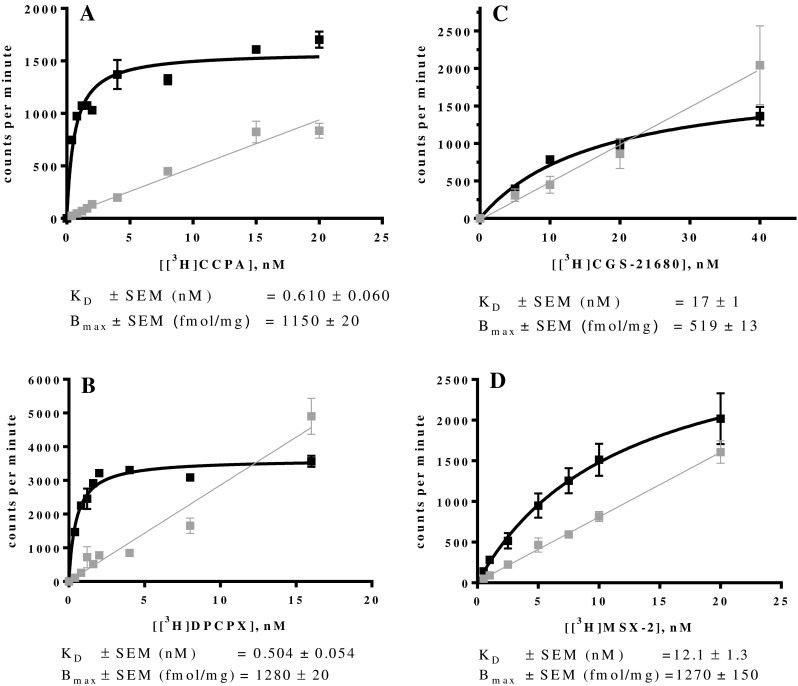
Saturation binding assays at ARs stably expressed in recombinant CHO cells: a mA1AR using [3H]CCPA; b mA1AR using [3H]DPCPX; c mA2AAR using [3H]CGS-21680; d mA2AAR using [3H]MSX-2. Data are means of three independent saturation assays each performed in duplicates. Curves represent specific binding and gray lines represent non-specific binding. Curves and lines were obtained by plotting the counts per minute against increasing concentrations of the radioligand
Saturation binding assays at mA2AARs were performed using the agonist radioligand [3H]CGS-21680 and the antagonist radioligand [3H]MSX-2 (see Figs. 1c, d). The KD value for [3H]MSX-2 determined at the mA2A was 12.1 nM, which is in accordance with results from an autoradiography study using mouse striatum [53]. The Bmax value of 211 fmol/mg was not high, but similar to that determined by Klotz et al. for the human A2AAR expressed in CHO cells [49]. To address this problem of only moderate expression of the A2AAR, the retroviral transfection was repeated and considerably higher specific binding was observed for the new batch indicating a higher expression level. Saturation assays using [3H]CGS-21680 at mA2AARs provided a KD value of 17.0 nM. The determined Bmax value of 519 fmol/mg was high enough to efficiently conduct competition radioligand binding assays. It is noteworthy to mention that the expression level of the receptor in the pooled cell line deteriorated quickly after some cell passages. This indicates the need to pick single clones in order to obtain a monoclonal cell line, which can exhibit a high expression level over a long period of time.
Due to the lack of a highly potent agonist A2BAR radioligand, for A2BARs, only the antagonist radioligand [3H]PSB-603 was employed, and its KD value was determined at mA2B and rA2BARs (Figs. 2a, b). The determined KD values were 4.09 nM for the mA2BAR and 0.457 nM for the rat A2BAR. The KD value for the rA2BAR is similar to the previously determined KD value of 0.403 nM for this radioligand for the human A2BAR [53]. Interestingly, the KD value for the mA2BAR was ca. 10-fold higher than that determined for the human and rat orthologues. Borrmann et al. [46] had reported a KD value of 0.35 nM for PSB-603 at the mA2BAR determined by homologous competition, which is typically less accurate than saturation binding. The Bmax value for the mA2B cell line was determined to be 1480 fmol/mg indicating a high expression level. For the rA2BAR cell line, a Bmax value of 408 fmol/mg protein was found, which was lower that for the mA2BAR, but still adequate to perform competition radioligand binding assays.
Fig. 2.
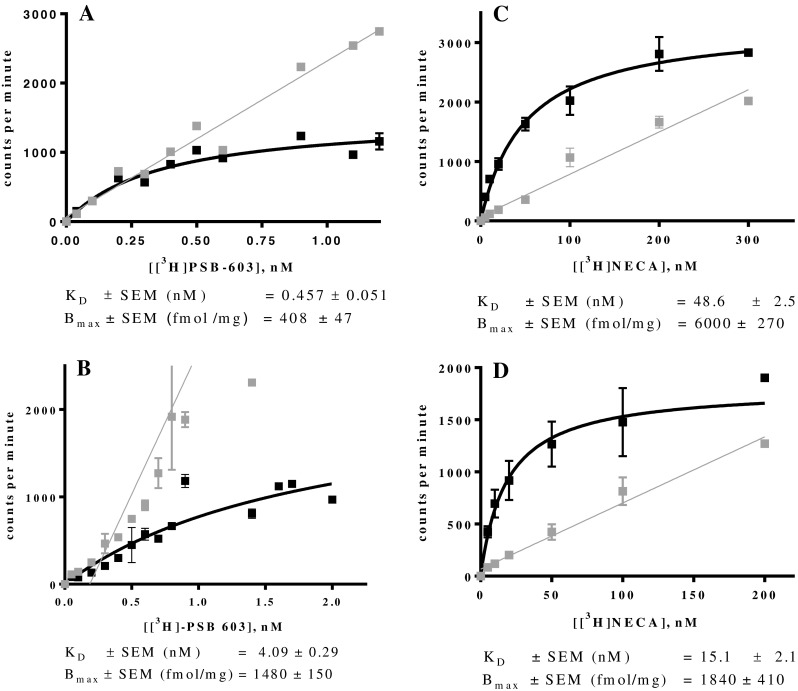
Saturation binding assays at ARs stably expressed in recombinant CHO cells: a rA2BAR using [3H]PSB-603; b mA2BAR using [3H]PSB-603; c rA3AR using [3H]NECA; d mA3AR using [3H]NECA. Data are means of three independent saturation assays each performed in duplicates. Curves represent the specific binding and were obtained by plotting the counts per minute against increasing concentrations of the radioligand
The non-specific agonist radioligand [3H]NECA was used for labeling A3ARs. The determined KD values were 48.6 nM for rat and 15.1 nM for mouse A3ARs (Figs. 2c, d). The determined Bmax values of 6000 fmol/mg protein for the rat and 1840 fmol/mg for the mouse A3AR indicated high expression levels of both receptor subtypes. Scatchard transformations of all conducted saturation experiments were calculated (see Supporting Information, Fig. S1 and S2).
The results of the saturation binding assays are summarized in Table 1.
Competition binding assays
A set of eight agonists and 16 antagonists was selected and investigated at the four AR subtypes of the three species, human, rat, and mouse. We picked the most frequently used standard ligands. Further criteria included general availability and subtype-selectivity, although we additionally investigated some non-selective standard ligands, such as NECA, CADO, and caffeine. Figure 3 shows the chemical structures of the selected agonists as well as their AR-subtype selectivity. Structures of the investigated antagonists along with their selectivity are depicted in Fig. 4.
Fig. 3.
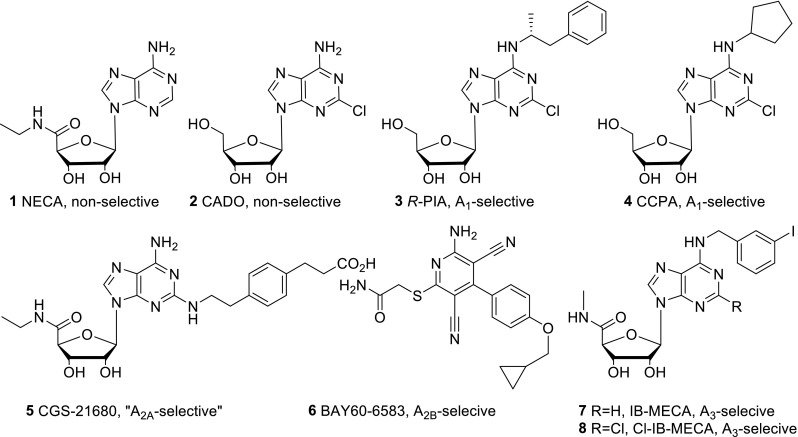
Structures and selectivity of the investigated adenosine receptor agonists
Fig. 4.
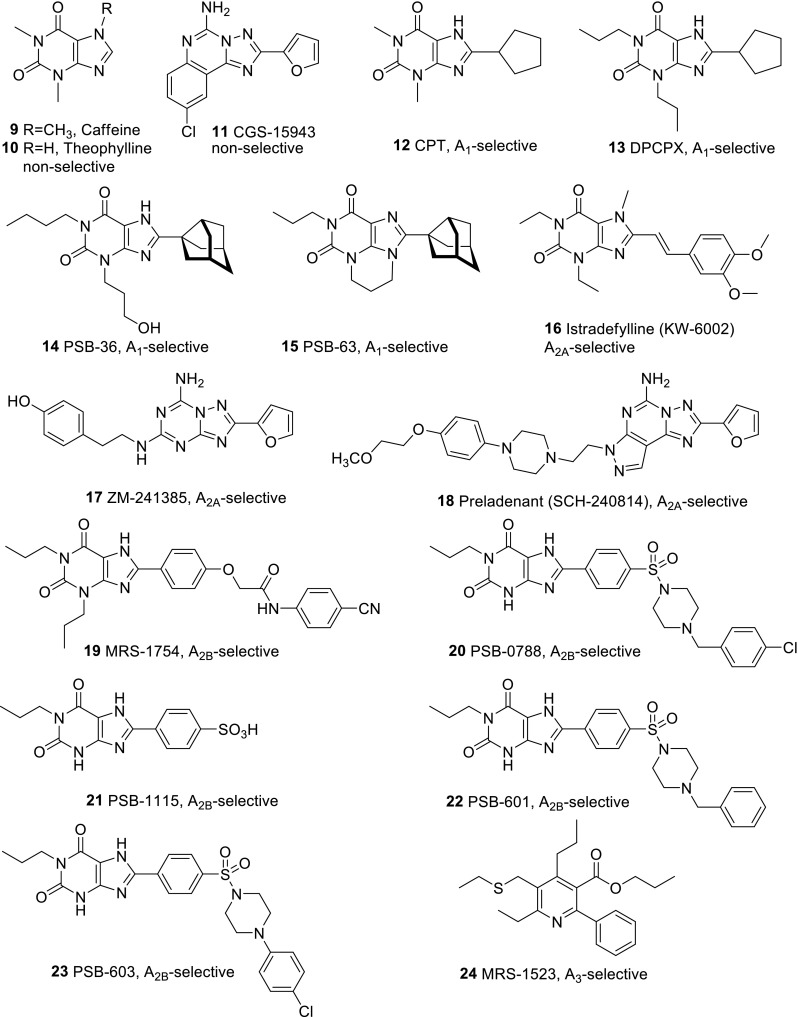
Structures and selectivity of the investigated adenosine receptor antagonists
Before conducting the assays, a systematic search was carried out to find literature Ki values for these agonists and antagonists. Data at human ARs for almost all of the selected compounds had previously been published. Our aim was to determine those Ki values that had not been previously reported; therefore, we focused our studies on the rodent receptors. The compounds were initially tested at a high concentration of 10 or 1 μM, depending on solubility, and if the Ki value was expected to be below 10 μM, full concentration-inhibition curves were recorded. In all cases, three independent experiments were performed, each in duplicates.
A1 adenosine receptor affinities
Affinities at human and rat A1ARs were available for most of the investigated standard ligands; however, affinities for the mouse A1AR had to be determined. In Table 2, literature data and the additional newly determined values are collected.
CCPA was the most potent agonist at mouse and human A1ARs displaying subnanomolar affinities. Only at the rat A1AR, R-PIA was somewhat more potent than CCPA.
The most potent A1 agonist at human and rat A1ARs was the xanthine derivative PSB-36 displaying subnanomolar affinities. However, at the mA1AR, DPCPX was fourfold more potent than PSB-36. Interestingly, some of the most potent compounds at the mA1AR were the non-selective antagonist CGS-15943 and the A2B-selective antagonist MRS-1754 with Ki values around 1 nM. MRS-1754 was weaker at the rat and much weaker at the human A1AR.
It is also noteworthy that the A1-selective antagonists PSB-36 and PSB-63 (see Fig. 4) displayed significantly lower affinity at the mouse as compared to the rat A1AR. Their affinity for the human A1AR, especially that of PSB-63, was also lower at the mouse A1AR in comparison with the human A1AR. These antagonists were clearly most potent at the rat A1AR. The potent and selective A2BAR antagonist PSB-603 was relatively potent at the mA1AR.
A2A adenosine receptor affinities
After determination of the KD values of [3H]CGS-21680 and [3H]MSX-2 at the mouse A2AAR, it was possible to conduct competition binding assays and thereafter to compare the results obtained in the different species (see Table 3).
NECA and CGS-21680 were the most potent A2A agonists in all three species. Both nucleosides showed similar A2A affinity. Only at the mA2A receptor NECA was somewhat less potent. The A2B partial agonist BAY60-6583 was not active at A2AARs of the three species.
The selective A2A antagonists preladenant, ZM-241385 and istradefylline, and the non-selective CGS-15943 were the most potent A2A antagonists at all three species. Preladenant and CGS-15943 were several-fold more potent at the mouse as compared to the rat and human A2AAR.
A2B adenosine receptor affinities
A suitable agonist radioligand for A2BARs is currently not available. Therefore, agonists as well as antagonists were investigated using the antagonist radioligand [3H]PSB-603. It should be kept in mind that the affinity of the agonists may be underestimated when using an antagonist radioligand [72]. Results of competition binding assays at A2BARs are presented in Table 4.
Table 4.
K i values of standard AR ligands at A2B adenosine receptors
| A2B Adenosine receptor affinity | |||
|---|---|---|---|
| Human | Rat | Mouse | |
| Agonists | K i ± SEM (nM) vs. [3H]PSB-603a | ||
| NECA | 1890 [46] | 1110 ± 240 | 656 ± 79 |
| CADO | 21,400 [56] 33,900 [73] |
4080 ± 1150 | 6450 ± 860 |
| CCPA | 18,800 [56] | 6160 ± 170 | 25,300 ± 10,200 |
| R-PIA | 150,000 [56] | 19,000 [56] | 28,300 [40] 19,000 [56] |
| CGS-21680 | >10,000 [49] | >10,000 [56] | >10,000 |
| BAY60-6583 | 114 [74] | 100 ± 5 | 136 ± 19 |
| IB-MECA | 11,000 [49] | 3340 ± 220 | 12,400 [40] |
| CI-IB-MECA | >10,000 [56] >100,000 [5] |
1210 ± 40 | 44,300 ± 8600 |
| Antagonists | K i ± SEM (nM) vs. [3H]PSB-603a | ||
| Caffeine | 33,800 [46] | 30,000 [56] | 23,000 ± 3800 |
| Theophylline | 7850 [46] 74,000 [56] |
15,100 [46] | 24,300 ± 7300 |
| CGS-15943 | 32.4 [40] | 1200 [70] | 15.0 ± 4.3 |
| DPCPX | 51 [5] | 186 [75] | 86.2 [5] |
| PSB-36 | 187 [5] | 350 ± 97 | 704 ± 91 |
| PSB-63 | 3190 [5] | >10,000 | >10,000 |
| CPT | 710 [76] 902 [5] |
1520 ± 170 | 2800 ± 406 |
| Istradefylline | >10,000 [56] | 5940 ± 750 | 3590 ± 270 |
| ZM-241385 | 75 [56] | 373 ± 40 | 31.3 [46] |
| Preladenant | >1000 [56] | >1000 | >1000 |
| PSB-601 | 3.6 [44] | 3.25 ± 0.23 | 2.48 ± 0.43 |
| PSB-603 | 0.553 [46] | 0.355 ± 0.034 | 0.265 ± 0.091 |
| PSB-1115 | 53.4 [77] | 3140 ± 290 | 1940 ± 250 |
| PSB-0788 | 0.393 [46] | 2.12 ± 0.29 | 1.90 ± 0.36 |
| MRS-1754 | 1.97 [67] | 12.8 [56] | 3.12 ± 0.60 |
| MRS-1523 | >10,000 [56] | >10,000 | >10,000 |
aData are means ± SEM of three independent assays performed in duplicates. The values without SEM values are taken from the literature
BAY60-6583 showed the highest affinity of all investigated agonists at A2BARs of all three species. NECA was several-fold weaker but still one of the most potent A2B agonists. The higher affinity of BAY60-6583 as compared to NECA may be due to the fact that it is a partial agonist and therefore may show higher affinity than a full agonist when tested versus an antagonist radioligand. Both compounds did not display species differences.
The most potent A2B antagonists were PSB-603 with subnanomolar affinity at all three species, and its analog PSB-0788, which was similarly potent at the human, but somewhat less potent at rat and mouse A2B receptors. The A1 and A2A antagonists DPCPX, PSB-36, and ZM-241385 also showed relatively high affinity for A2BARs.
A3 adenosine receptor affinities
The agonist radioligand [3H]NECA was used in competition binding assays for the determination of agonist as well as antagonist affinities since an antagonist radioligand for rodent A3ARs is currently not available. The frequently applied A3-selective antagonist radioligand [3H]PSB-11 [78] only binds to human A3ARs with high affinity but not to rat or mouse A3ARs. Binding data at human, rat, and mouse A3ARs are collected in Table 5.
Table 5.
K i values of standard AR ligands at A3 adenosine receptors
| A3 Adenosine receptor affinity | |||
|---|---|---|---|
| Human | Rat | Mouse | |
| Agonists | K i ± SEM (nM) vs. [3H]NECA | ||
| NECA | 25 [79] | 113 [80] | 13.2 ± 1.9 |
| CADO | 523 [56] | 1890 [80] | 130 ± 9 |
| CCPA | 43 [49] | 237 [80] | 15.6 ± 1.6 |
| R-PIA | 33 [79] | 158 [80] | 9.98 ± 0.89 |
| CGS-21680 | 67 [49] | 584 [80] | 93.0 ± 9.1 |
| BAY60-6583 | 223 ± 93 | 2750 ± 330 | 3920 ± 890 |
| IB-MECA | 1.8 [57] 0.19 [5] |
1.1 [59] | 0.219 ± 0.017 |
| CI-IB-MECA | 1.4 [69] | 0.33 [59] | 0.18 [6] |
| Antagonists | Ki ± SEM (nM) vs. [3H]NECA | ||
| Caffeine | 13,300 [5] | >100,000 [56] | >100,000 |
| Theophylline | 52,300 [46] 22,300 [5] |
100,000 [46] | >100,000 |
| CGS-15943 | 35 [56] | 1270 ± 170 | 2970 ± 200 |
| DPCPX | 243 [64] | >10,000 [43] 43,000 [5] |
>10,000 |
| PSB-36 | 2300 [56] | 6500 [56] | >10,000 |
| PSB-63 | >10,000 [5] | >10,000 | >10,000 |
| CPT | 100,000 [5] | >10,000 | >10,000 |
| Istradefylline | 4470 [64] | >10,000 | >10,000 |
| ZM-241385 | 743 [56] | >10,000 | >10,000 |
| Preladenant | >1000 [56] | >1000 | >1000 |
| PSB-601 | >1000 [44] | >10,000 | >10,000 |
| PSB-603 | >10,000 [46] | >10,000 | >10,000 |
| PSB-1115 | >10,000 [46] | >10,000 | >10,000 |
| PSB-0788 | >1000 [46] | >10,000 | >10,000 |
| MRS-1754 | 570 [46] | >10,000 | >10,000 |
| MRS-1523 | 18.9 [70] | 113 [70] 519 [70] |
1980 ± 150 |
aData are means ± SEM of three independent assays performed in duplicates. The values without SEM values are taken from the literature
Cl-IB-MECA and IB-MECA were the most potent A3AR agonists displaying affinity in the nanomolar to sub-nanomolar range. However, many of the non-A3-selective agonists also showed relatively high A3-affinity. Interestingly, the partial A2B agonist BAY60-6583 displayed relatively high affinity for A3ARs (e.g., Ki hA3 223 nM), similar to its significant affinity determined for A1ARs (e.g., Ki mA1AR 355 nM). Rosentreter et al. had demonstrated functional selectivity of BAY60-6583 for the human A2BAR against other human AR subtypes overexpressed in CHO cells [81]. Van der Hoeven et al. confirmed the functional selectivity of BAY60-6583 at mA2BAR using cAMP assays, showing an EC50 value of 2.83 nM at mA2BAR and no activation of the other mAR subtypes at concentrations of up to 1 μM [82]. This, together with our data, suggests that BAY60-6583 has a high selectivity only in functional assays but not in binding studies. BAY60-6583 has recently been shown to act as a partial agonist at A2BARs [83]. It may be an antagonist at other AR subtypes, which could be an explanation for the fact that BAY60-6583 was found to lack activation of human A1, A2A, and A3 AR subtypes, but showed affinity in binding studies. In order to investigate this possibility, β-arrestin assays at hA1 and hA3ARs were conducted. Our results showed no agonism of BAY60-6583 in ß-arrestin translocation assays using β-galactosidase complementation technology (data not shown). However, in the same assays, BAY60-6583 inhibited hA1 receptor activation by CCPA (100 nM, corresponding to its EC80) with an IC50 value of 7400 ± 2190 nM; it also blocked hA3AR activation by Cl-IB-MECA (30 nM, corresponding to its EC80) with an IC50 value of 6700 ± 830 nM (see Fig. 5). The corresponding Ki values for BAY60-6583 were 3190 nM (hA1) and 5630 nM (hA3). Since both, hA1 and hA3, are Gi-coupled receptors [56], we additionally performed cAMP accumulation assays after stimulating the cells with forskolin. The effects of 10 μM BAY60-6583 on the concentration-response curves of the agonist NECA were determined at both receptor subtypes. As shown in Fig. 6, BAY60-6583 induced a rightward shift of the NECA curves at both receptors with Kb values of 347 nM (hA1AR) and 652 nM (hA3AR). The Kb values from cAMP studies were consistent with the data obtained in radioligand binding studies (see Tables 2 and 5). Ki values determined in β-arrestin arrestin assays were about 10-fold higher. These findings proved that BAY60-6583 can act as a moderately potent antagonist at hA1 and hA3ARs inhibiting Gi- as well as β-arrestin-mediated signaling.
Fig. 5.
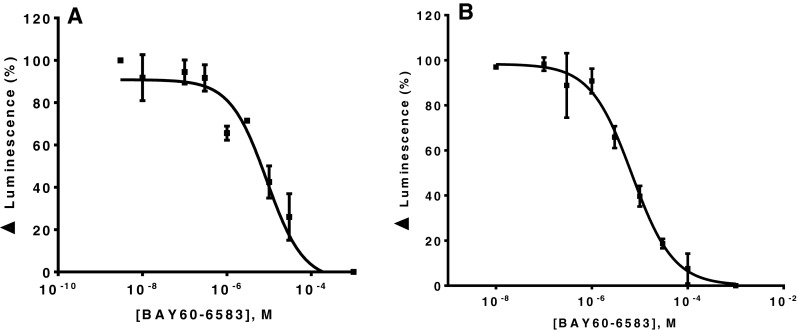
Concentration-dependent antagonistic effect of BAY60-6583 at a hA1AR (IC50 = 7.40 ± 2.19 μM) and b hA3AR (IC50 = 6.70 ± 0.83 μM). The hA1AR was activated by CCPA (100 nM), the hA3AR was activated by Cl-IB-MECA (30 nM). The employed agonist concentrations corresponded to their EC80 values. Receptor-induced ß-arrestin translocation was measured by using a ß-galactosidase complementation assay. Values represent means of three independent experiments. K i value BAY60-6583 at hA1 is 3190 ± 780 nM, K i value BAY60-6583 at hA3 is 5630 ± 700 nM
Fig. 6.
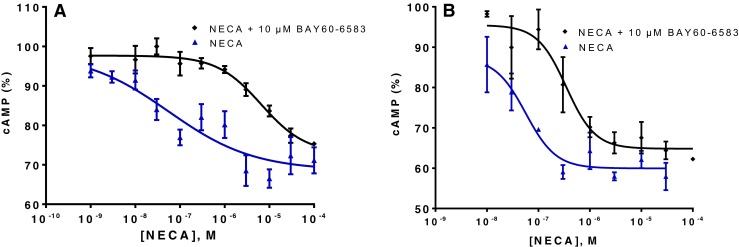
Antagonistic effects of BAY60-6583 at the Gi-coupled hA1 and hA3 ARs determined in cAMP accumulation assays; a at the hA1AR a rightward shift of the curve for the agonist NECA was observed; the EC50 was shifted from 56.5 ± 6.7 nM in the absence of BAY60-6583 to 6060 ± 780 nM in the presence of 10 μM BAY60-6583; b at the hA3AR, a rightward shift of the curve for NECA was also observed from an EC50 of 56.4 ± 7.3 nM in the absence of BAY60-6583 to an EC50 of 348 ± 25 nM in the presence of 10 μM BAY 60–6583. Cells were stimulated with forskolin at a final concentration of 10 μM. Calculated K b values were 0.347 ± 0.032 μM (hA1) and 0.652 ± 0.058 μM (hA3)
The development of potent antagonists that are similarly potent at human as at rodent A3ARs seems to be a formidable challenge. The only antagonists that showed potency at A3ARs of all three species were MRS-1523 followed by the non-selective CGS-15934. This could be, at least partially, attributed to the aforementioned significant sequence differences observed for this AR subtype between human and rodent receptors.
The high affinity of the agonist NECA and the lacking affinity, for example, of the antagonist caffeine indicate the importance of the ribose moiety for the binding of ligands to the A3AR. A medicinal chemical approach to develop A3AR antagonists that are active across species has recently been successfully pursued by Jacobson and coworkers who modified the ribose moiety so that the affinity stayed unaltered but the efficacy disappeared [84].
Correlations between species
Ki values determined for the same compounds in different species were compared, and correlation coefficients were calculated (see Supporting Information for more details). Results are shown in Table 6 and Figs. S7 and S8 (Supporting Information). It was found that A2AARs and A2BARs showed the best correlation among the three species, whereas, as expected, A3ARs exhibited the least correlation due to larger differences in receptor sequence.
Table 6.
Correlation coefficients of pK i values of the investigated compounds at human, rat, and mouse adenosine receptors
| Correlation coefficient | ||||
|---|---|---|---|---|
| A1AR | A2AAR | A2BAR | A3AR | |
| Human vs. mouse | 0.74 | 0.88 | 0.90 | 0.52 |
| Human vs. rat | 0.80 | 0.83 | 0.82 | 0.83 |
| Rat vs. mouse | 0.76 | 0.84 | 0.83 | 0.73 |
Discussion
The aim of this study was to determine the affinities of standard agonists and antagonists in the field of AR research at all four receptor AR subtypes of the three species that are of major interest in drug research: human, rat, and mouse. Those data should then lead to recommendations as to which agonist and which antagonist to choose as the most appropriate one for in vitro and in vivo studies in a certain species. A compound was considered selective for one AR subtype versus another if it showed an at least 100-fold preference for one receptor over the others. Based on the collected data, we selected the preferred agonists and antagonists for each AR subtype as discussed below.
The A1 agonist CCPA
The potency of CCPA is in the nanomolar range at the A1AR subtype in all three species, making this compound one of the most potent agonists at this AR subtype. The selectivity versus A2A and A2BARs was high (ca. 10,000-fold). The compound was also selective versus A3ARs in rat and mouse, but not highly selective in humans (40-fold) as illustrated in Fig. 6. Regarding both, potency and selectivity, CCPA seems to be a better choice than R-PIA for use as an A1AR agonist in preclinical studies.
The A1 antagonist PSB-36
PSB-36 is a very potent A1AR antagonist with Ki values in the nanomolar range in all three species. The selectivity for the A1AR was also very high versus the other AR subtypes (at least 400-fold) as shown in Fig. 7. This makes PSB-36 a powerful, recommended tool compound for blocking the A1AR subtype in experimental pharmacological approaches in rat, mouse, and human. It is by far superior to the frequently used A1 antagonist DPCPX, which shows some affinity for A2A and A2BARs in the three species as well as measurable affinity for the hA3AR.
Fig. 7.

pKi values of the A1 agonist CCPA and the A1 antagonist PSB-36 at human, rat, and mouse AR subtypes; number sign indicates the highest tested concentration was 10 μM and K i value is >10 μM
The A2A agonist CGS-21680
CGS-21680 was the only selective A2A agonist, which was included in the present study. It showed good affinity for the A2AAR subtype of the three investigated species. A2A-selectivity was high versus the A2BAR, but lower versus the other receptor subtypes. In humans, the A2A selectivity vs. A3 was very low (2.5-fold) and moderate vs. A1 (10-fold). The highest selectivity was observed in rat (>30-fold). CGS-21680 is one of the best available A2A agonists, but it is only moderately selective. The compound displayed selectivity versus the A1AR subtype only in rat. It was not selective versus A3ARs in the three species as shown in Fig. 8. Thus, CGS-21680 is not a very selective A2A agonist and should therefore be used with care. A study by Linden et al., however, indicated that binding data obtained in recombinant cells might underestimate the potency of A2AAR agonists due to poor coupling of the receptor to Gs protein [85]. Therefore, CGS21680 and other A2A agonists may display higher selectivity in vivo. However, based on results from binding studies, it is concluded that A2A agonists with high selectivity across species are currently not available.
Fig. 8.

pK i values of the A2A agonist CGS-21680 and the A2A antagonist preladenant; (number sign) highest tested concentration was 10 μM and K i value is >10 μM, (section sign) highest tested concentration was 1 μM and K i value is >1 μM
The A2A antagonist preladenant
Preladenant showed the highest potency and selectivity among the investigated A2A antagonists, it was superior to istradefylline and ZM-241385. This non-xanthine compound, which was evaluated in a phase III clinical trial as an anti-Parkinsonian drug, but was recently discontinued, may be employed as a very effective antagonist for investigating the A2AAR subtype in experimental pharmacology. Its affinity is in the (sub) nanomolar range and its selectivity was at least ca. 1000-fold versus all other AR subtypes in the three investigated species (see Fig. 8). The A2AAR antagonist MSX-2 was not included in this study. However, it is a potent, selective, and widely employed antagonist (Ki values at A2AAR are 5.38 and 8.04 nM in human and rat, respectively). In spite of that, its selectivity versus the rA1AR (112-fold) is lower than that of preladenant [6].
The A2B agonist BAY60-6583
BAY60-6585, which was recently characterized as a partial agonist at A2BARs [83], shows moderate affinity for the A2BAR. It showed the best A2B-selectivity profile among the investigated compounds. However, in radioligand binding studies, BAY60-6583 only displayed selectivity versus the A2AAR subtype. It was not selective versus A1 and A3ARs in the three species as shown in Fig. 8. However, BAY60-6583 was shown to be functionally selective in cAMP assays, where it only activated A2BAR, but not the other subtypes. We have now shown that BAY60-6583 is a moderately potent antagonist at both hA1 and hA3AR. Hence, the use of BAY60-6583 as a “selective A2B agonist” is questionable, and the results obtained may be confounded on the one hand by partial agonism at A2BARs resulting in A2B receptor blockade in the presence of high adenosine concentrations and A2B receptor stimulation in the absence of adenosine, and on the other hand by a blockade of the other AR subtypes. A potent and selective full A2BAR agonist is still urgently needed.
The A2B antagonist PSB-603
The high selectivity and affinity of PSB-603 for human and rat A2BARs was clearly confirmed. Interestingly, PSB-603 showed an affinity in the micromolar range for the mA1AR. Despite that moderate affinity for the mA1AR, PSB-603 can still be considered as one of the most suitable antagonists for studying A2BARs due to its high affinity and selectivity in all three species. Surprisingly, both BAY60-6583 and PSB-603 showed relatively high affinity for the mouse A1AR (Fig. 9). PSB-603 was found to be superior to the related A2B antagonist PSB-0788 because the latter exhibited higher affinity for A2AARs than PSB-603 and was therefore less selective.
Fig. 9.

pK ivalues of the A2B partial agonist BAY60-6583 and the A2B antagonist PSB-603 at the four adenosine receptor subtypes; (number sign) highest tested concentration was 10 μM and K i value is >10 μM
The A3 agonist Cl-IB-MECA
Cl-IB-MECA was superior to IB-MECA concerning both affinity and selectivity. Its affinity for A3ARs was in the nanomolar range, and its selectivity versus the A2A and A2BAR subtypes was at least 1000-fold. The affinity for the A1AR subtype was found to be moderate. Thus, Cl-IB-MECA can be considered as a highly potent and selective A3AR agonist in humans, rat, and mouse (see Fig. 10).
Fig. 10.

pK i values of the A3 agonist Cl-IB-MECA and the A3 antagonist MRS-1523 at the four adenosine receptor subtypes; (number sign) highest tested concentration was 10 μM and K i value is >10 μM
The A3 antagonist MRS-1523
Most of the potent A3 antagonists that have been described show a large preference for the human A3 receptor and only low or lacking affinity for the rodent A3ARs. MRS-1523 is one of the few compounds that had been described to block not only human but also rodent A3ARs. Our data show, however, that the compounds’ affinity for rat and especially for mouse A3ARs is significantly lower than that for the human A3AR. Affinity for A1 and A2BARs was found to be low. However, somewhat higher affinities were observed for A2AARs in human and rat. This leads to the conclusion that MRS-1523 is not a suitable A3 antagonist for studies in mice. Its selectivity in rat is also moderate (e.g., fourfold against A2AARs). It can only be unreservedly recommended as an A3 antagonist in humans. But for the human species, much better, more potent, and selective antagonists, e.g., PSB-10, PSB-11, MRE3005-F20, and MRE3008-F20, are available [6]
Conclusions
In this study, we aimed at investigating the potency and selectivity of selected AR ligands at the four receptor subtypes in human, rat, and mouse using radioligand binding studies. The investigated ligands included 8 agonists and 16 antagonists, which have been widely used in pharmacological and preclinical studies. To address this aim, all rat and mouse AR subtypes were cloned and expressed in CHO cells. The KD values of [3H]CCPA and [3H]DPCPX at mA1, [3H]CGS-21680 and [3H]MSX-2 at mA2A, [3H]PSB-603 at mA2B and rA2B, and [3H]NECA at mA3 and rA3 were determined. Ki values of the ligands were measured, and a comparison of their affinities and selectivities was conducted to determine the best compound at each receptor subtype. CCPA is the best A1 agonist of the investigated selection of compounds although its selectivity versus the A3 subtype is not sufficient. PSB-36 represents the most suitable A1 antagonist. CGS-21680 is the best A2A agonist of the tested compounds but its potency and especially its selectivity are moderate. The A2A antagonist preladenant shows high affinity and an impressive selectivity. Results obtained for the moderately potent A2B partial agonist BAY60-6583 demonstrated the urgent need to develop better A2BAR agonists. However, the A2B antagonist PSB-603 proved to be a very powerful pharmacological tool for studying A2B receptors concerning both affinity and selectivity. Cl-IB-MECA appears to be a potent and selective agonist at the A3 subtype. MRS-1523 was found to be the best A3 antagonist among the investigated compounds for use in rodents, although it is far from being ideal for studies in rats and especially in mice. In contrast, for investigations at human A3ARs, much more potent and selective antagonists are available, including PSB-10 and PSB-11, but those compounds are inactive at rodent A3ARs. The present study clearly demonstrates that tool compounds have to be comprehensively characterized in the appropriate species before they are used as pharmacological tools in in vitro and in vivo studies. Moreover, concentrations used in in vitro and in vivo studies have to be carefully chosen based on the compounds’ affinity and selectivity.
Electronic supplementary material
Below is the link to the electronic supplementary material.
(PDF 180 kb)
Contributor Information
Mohamad Wessam Alnouri, Email: wessam.alnouri@uni-bonn.de.
Stephan Jepards, Email: stephan.jepards@uni-bonn.de.
Alessandro Casari, Email: alessandro.casari@studenti.unipd.it.
Anke C. Schiedel, Email: schiedel@uni-bonn.de
Sonja Hinz, Email: shinz@uni-bonn.de.
Christa E. Müller, Phone: +49 228 732301, Email: christa.mueller@uni-bonn.de
References
- 1.Burnstock G. Purinergic signalling: Its unpopular beginning, its acceptance and its exciting future. Bioessays. 2012;34:218–225. doi: 10.1002/bies.201100130. [DOI] [PubMed] [Google Scholar]
- 2.Burnstock G, Verkhratsky A. Evolutionary origins of the purinergic signaling system. Acta Physiol (Oxf) 2009;195:415–447. doi: 10.1111/j.1748-1716.2009.01957.x. [DOI] [PubMed] [Google Scholar]
- 3.Fredholm BB, IJzerman AP, Jacobson KA, Klotz KN, Linden J. International union of pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev. 2001;53:527–552. [PMC free article] [PubMed] [Google Scholar]
- 4.Jacobson KA, Balasubramanian R, Deflorian F, Gao ZG. G protein-coupled adenosine (P1) and P2Y receptors: ligand design and receptor interactions. Purinergic Signal. 2012;8:419–436. doi: 10.1007/s11302-012-9294-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Müller CE, Jacobson KJ. Xanthines as adenosine receptor antagonists. Handb Exp Pharmacol. 2011;200:151–199. doi: 10.1007/978-3-642-13443-2_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Müller CE, Jacobson KA. Recent developments in adenosine receptor ligands and their potential as novel drugs. Biochim Biophys Acta. 2011;1808:1290–1308. doi: 10.1016/j.bbamem.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heisig F, Gollos S, Freudenthal SJ, El-Tayeb A, Iqbal J, Müller CE. Synthesis of BODIPY derivatives substituted with various bioconjugatable linker groups: a construction kit for fluorescent labeling of receptor ligands. J Fluoresc. 2014;24:213–230. doi: 10.1007/s10895-013-1289-4. [DOI] [PubMed] [Google Scholar]
- 8.Stoddart LA, Vernall AJ, Denman JL, Briddon SJ, Kellam B, Hill SJ. Fragment screening at adenosine-A(3) receptors in living cells using a fluorescence-based binding assay. Chem Biol. 2012;19:1105–1115. doi: 10.1016/j.chembiol.2012.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kozma E, Jayasekara PS, Squarcialupi L, Paoletta S, Moro S, Federico S, Spalluto G, Jacobson KA. Fluorescent ligands for adenosine receptors. Bioorg Med Chem Lett. 2013;23:26–36. doi: 10.1016/j.bmcl.2012.10.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xu F, Wu H, Katritch V, Han GW, Jacobson KA, Gao ZG, Cherezov V, Stevens RC. Structure of an agonist-bound human A2A adenosine receptor. Science. 2011;332:322–327. doi: 10.1126/science.1202793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EY, Lane JR, Ijzerman AP, Stevens RC. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science. 2008;322:1211–1217. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu W, Chun E, Thompson AA, Chubukov P, Xu F, Katritch V, Han GW, Roth CB, Heitman LH, IJzerman AP, Cherezov V, Stevens RC. Structural basis for allosteric regulation of GPCRs by sodium ions. Science. 2012;337:232–236. doi: 10.1126/science.1219218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doré AS, Robertson N, Errey JC, Ng I, Hollenstein K, Tehan B, Hurrell E, Bennett K, Congreve M, Magnani F, Tate CG, Weir M, Marshall FH. Structure of the adenosine A(2A) receptor in complex with ZM241385 and the xanthines XAC and caffeine. Structure. 2011;19:1283–1293. doi: 10.1016/j.str.2011.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Lera RM, Lim YH, Zheng J. Adenosine A2A receptor as a drug discovery target. J Med Chem. 2014;57:3623–3650. doi: 10.1021/jm4011669. [DOI] [PubMed] [Google Scholar]
- 15.Chen JF, Eltzschig HK, Fredholm BB. Adenosine receptors as drug targets-what are the challenges? Nat Rev Drug Discov. 2013;12:265–286. doi: 10.1038/nrd3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Albrecht-Küpper BE, Leineweber K, Nell PG. Partial adenosine A1 receptor agonists for cardiovascular therapies. Purinergic Signal. 2012;8:91–99. doi: 10.1007/s11302-011-9274-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liao Y, Lin L, Lu D, Fu Y, Bin J, Xu D, Kitakaze M. Activation of adenosine A1 receptor attenuates tumor necrosis factor-α induced hypertrophy of cardiomyocytes. Biomed Pharmacother. 2011;65:491–495. doi: 10.1016/j.biopha.2011.06.008. [DOI] [PubMed] [Google Scholar]
- 18.Faulhaber-Walter R, Jou W, Mizel D, Li L, Zhang J, Kim SM, Huang Y, Chen M, Briggs JP, Gavrilova O, Schnermann JB. Impaired glucose tolerance in the absence of adenosine A1 receptor signaling. Diabetes. 2011;60:2578–2587. doi: 10.2337/db11-0058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Teerlink JR, Iragui VJ, Mohr JP, Carson PE, Hauptman PJ, Lovett DH, Miller AB, Piña IL, Thomson S, Varosy PD, Zile MR, Cleland JG, Givertz MM, Metra M, Ponikowski P, Voors AA, Davison BA, Cotter G, Wolko D, Delucca P, Salerno CM, Mansoor GA, Dittrich H, O’Connor CM, Massie BM. The safety of an adenosine A(1)-receptor antagonist, rolofylline, in patients with acute heart failure and renal impairment: findings from PROTECT. Drug Saf. 2012;35:233–244. doi: 10.2165/11594680-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 20.Nair PK, Marroquin OC, Mulukutla SR, Khandhar S, Gulati V, Schindler JT, Lee JS. Clinical utility of regadenoson for assessing fractional flow reserve. JACC Cardiovasc Interv. 2011;4:1085–1092. doi: 10.1016/j.jcin.2011.07.011. [DOI] [PubMed] [Google Scholar]
- 21.Cheng AS, Pegg TJ, Karamitsos TD, Searle N, Jerosch-Herold M, Choudhury RP, Banning AP, Neubauer S, Robson MD, Selvanayagam JB. Cardiovascular magnetic resonance perfusion imaging at 3-tesla for the detection of coronary artery disease: a comparison with 1.5-tesla. J Am Coll Cardiol. 2007;49:2440–2449. doi: 10.1016/j.jacc.2007.03.028. [DOI] [PubMed] [Google Scholar]
- 22.Lappas CM, Rieger JM, Linden J. A2A adenosine receptor induction inhibits IFN-gamma production in murine CD4+ T cells. J Immunol. 2005;174:1073–1080. doi: 10.4049/jimmunol.174.2.1073. [DOI] [PubMed] [Google Scholar]
- 23.Haskó G, Kuhel DG, Chen JF, Schwarzschild MA, Deitch EA, Mabley JG, Marton A, Szabó C. Adenosine inhibits IL-12 and TNF-[alpha] production via adenosine A2A receptor-dependent and independent mechanisms. FASEB J. 2000;14:2065–2074. doi: 10.1096/fj.99-0508com. [DOI] [PubMed] [Google Scholar]
- 24.Flögel U, Burghoff S, van Lent PL, Temme S, Galbarz L, Ding Z, El-Tayeb A, Huels S, Bönner F, Borg N, Jacoby C, Müller CE, van den Berg WB, Schrader J. Selective activation of adenosine A2A receptors on immune cells by a CD73-dependent prodrug suppresses joint inflammation in experimental rheumatoid arthritis. Sci Transl Med. 2012;4:146ra108. doi: 10.1126/scitranslmed.3003717. [DOI] [PubMed] [Google Scholar]
- 25.Armentero MT, Pinna A, Ferré S, Lanciego JL, Müller CE, Franco R. Past, present and future of A(2A) adenosine receptor antagonists in the therapy of Parkinson’s disease. Pharmacol Ther. 2011;132:280–299. doi: 10.1016/j.pharmthera.2011.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Salamone JD, Collins-Praino LE, Pardo M, Podurgiel SJ, Baqi Y, Müller CE, Schwarzschild MA, Correa M. Conditional neural knockout of the adenosine A(2A) receptor and pharmacological A(2A) antagonism reduce pilocarpine-induced tremulous jaw movements: studies with a mouse model of parkinsonian tremor. Eur Neuropsychopharmacol. 2013;23:972–977. doi: 10.1016/j.euroneuro.2012.08.004. [DOI] [PubMed] [Google Scholar]
- 27.Jerónimo-Santos A, Batalha VL, Müller CE, Baqi Y, Sebastião AM, Lopes LV, Diógenes MJ. Impact of in vivo chronic blockade of adenosine A2A receptors on the BDNF-mediated facilitation of LTP. Neuropharmacology. 2014;83:99–106. doi: 10.1016/j.neuropharm.2014.04.006. [DOI] [PubMed] [Google Scholar]
- 28.Jenner P. Istradefylline, a novel adenosine A2A receptor antagonist, for the treatment of Parkinson’s disease. Expert Opin Investig Drugs. 2005;14:729–738. doi: 10.1517/13543784.14.6.729. [DOI] [PubMed] [Google Scholar]
- 29.Dungo R, Deeks ED. Istradefylline: first global approval. Drugs. 2013;73:875–882. doi: 10.1007/s40265-013-0066-7. [DOI] [PubMed] [Google Scholar]
- 30.Koupenova M, Johnston-Cox H, Vezeridis A, Gavras H, Yang D, Zannis V, Ravid K. A2B adenosine receptor regulates hyperlipidemia and atherosclerosis. Circulation. 2012;125:354–363. doi: 10.1161/CIRCULATIONAHA.111.057596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Michael S, Warstat C, Michel F, Yan L, Müller CE, Nieber K. Adenosine A(2A) agonist and A(2B) antagonist mediate an inhibition of inflammation-induced contractile disturbance of a rat gastrointestinal preparation. Purinergic Signal. 2010;6:117–124. doi: 10.1007/s11302-009-9174-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Figler RA, et al. Links between insulin resistance, adenosine receptors and inflammatory markers in mice and humans. Diabetes. 2011;60:669–679. doi: 10.2337/db10-1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Haskó G, Csóka B, Németh ZH, Vizi ES, Pacher P. A2B adenosine receptors in immunity and inflammation. Trends Immunol. 2009;30:263–270. doi: 10.1016/j.it.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xiang HJ, Chai FL, Wang DS, Dou KF. Downregulation of the adenosine a2b receptor by RNA interference inhibits hepatocellular carcinoma cell growth. ISNR Oncol. 2011;2011:875684–875687. doi: 10.5402/2011/875684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cekic C, Sag D, Li Y, Theodorescu D, Strieter RM, Linden J. Adenosine A2B receptor blockade slows growth of baldder and breast tumors. J Immunol. 2012;188:198–205. doi: 10.4049/jimmunol.1101845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Silverman MH, Strand V, Markovits D, Nahir M, Reitblat T, Molad Y, RosnerI RM, Mader R, Adawi M, Caspi D, Tishler M, Langevitz P, Rubinow A, Friedman J, Green L, Tanay A, Ochaion A, Cohen S, Kerns WD, Cohn I, Fishman-Furman S, Farbstein M, Yehuda SB, Fishman P. Clinical evidence for the utilization of A3 adenosine receptor as a target to treat rheumatoid arthritis: data from phase II clinical trial. J Rheumatol. 2008;35:41–48. [PubMed] [Google Scholar]
- 37.Koscsó B, Csóka B, Pacher P, Haskó G. Investigational A3 adenosine receptor targeting agents. Expert Opin Investig Drugs. 2011;20:757–768. doi: 10.1517/13543784.2011.573785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang Z, Do CW, Avila MY, Peterson-Yantorno K, Stone RA, Gao ZG, Joshi B, Besada P, Jeong LS, Jacobson KA, Civan MM. Nucleosine-derived antagonists to A3 adenosine receptors lower mouse intraocular pressure and act across species. Exp Eye Res. 2010;90:146–154. doi: 10.1016/j.exer.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Müller CE, Scior T. Adenosine receptors and their modulators. Pharm Acta Helv. 1993;68:77–111. doi: 10.1016/0031-6865(93)90012-U. [DOI] [PubMed] [Google Scholar]
- 40.Auchampach JA, Kreckler LM, Wan TC, Maas JE, van der Hoeven D, Gizewski E, Narayanan J, Maas GE. Characterization of the A2Badenosinereceptor from mouse, rabbit, and dog. J Pharmacol Exp Ther. 2009;329:2–13. doi: 10.1124/jpet.108.148270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Müller CE, Stein B. Adenosine receptor antagonists: Structure and potential therapeutic applications. Curr Pharmaceut Design. 1996;2:501–530. [Google Scholar]
- 42.X-d J, von Lubitz D, Olah ME, Stiles GL, Jacobson KA. Species differences in ligands affinity at central A3 adenosine receptors. Drug Develop Res. 1994;33:51–59. doi: 10.1002/ddr.430330109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Weyler S, Fülle F, Diekmann M, Schumacher B, Hinz S, Klotz KN, Müller CE. Improving potency, selectivity, and water solubility of adenosine A1 receptor antagonists: xanthines modified at position 3 and related pyrimido[1,2,3-cd] purinediones. Chem Med Chem. 2006;1:891–902. doi: 10.1002/cmdc.200600066. [DOI] [PubMed] [Google Scholar]
- 44.Yan L, Bertarelli DC, Hayallah AM, Meyer H, Klotz KN, Müller CE. A new synthesis of sulfonamides by aminolysis of p-nitrophenylsulfonates yielding potent and selective adenosine A2B receptor antagonists. J Med Chem. 2006;49:4384–4391. doi: 10.1021/jm060277v. [DOI] [PubMed] [Google Scholar]
- 45.Müller CE, Shi D, Manning M, Jr, Daly JW. Synthesis of paraxanthine analogs (1,7-disubstituted xanthines) and other xanthinesunsubstituted at the 3-position: structure-activity relationships at adenosine receptors. J Med Chem. 1993;36:3341–3349. doi: 10.1021/jm00074a015. [DOI] [PubMed] [Google Scholar]
- 46.Borrmann T, Hinz S, Bertarelli DC, Li W, Florin NC, Scheiff AB, Müller CE. 1-alkyl-8-(piperazine-1-sulfonyl) phenylxanthines: development and characterization of adenosineA2B receptor antagonists and a new radioligand with subnanomolar affinity and subtype specificity. J Med Chem. 2009;52:3994–400. doi: 10.1021/jm900413e. [DOI] [PubMed] [Google Scholar]
- 47.Hockemeyer J, Burbiel JC, Müller CE. Multigram-scale syntheses, stability, and photoreactions of A2A adenosine receptor antagonists with 8-styrylxanthine structure: Potential drugs for Parkinson’s disease. J Org Chem. 2004;69:3308–3318. doi: 10.1021/jo0358574. [DOI] [PubMed] [Google Scholar]
- 48.Neustadt BR, Hao J, Lindo N, Greenlee WJ, Stamford AW, Tulshian D, Ongini E, Hunter J, Monopoli A, Bertorelli R, Foster C, Arik L, Lachowicz J, Ng K, Feng KI. Potent, selective, and orally active adenosine A2A receptor antagonists: Arylpiperazine derivatives of pyrazolo[4,3-e]-1,2,4-triazolo[1,5-c] pyrimidines. Bioorg Med Chem Lett. 2007;5:1376–1380. doi: 10.1016/j.bmcl.2006.11.083. [DOI] [PubMed] [Google Scholar]
- 49.Klotz KN, Hessling J, Hegler J, Owman C, Kull B, Fredholm BB, Lohse MJ. Comparative pharmacology of human adenosine receptor subtypes—Characterization of stably transfected receptors in CHO cells. Naunyn Schmiedebergs Arch Pharmacol. 1998;357:1–9. doi: 10.1007/PL00005131. [DOI] [PubMed] [Google Scholar]
- 50.Klotz KN, Lohse MJ, Schwabe U, Cristalli G, Vittori S, Grifantini M. 2-Chloro-N6-[3H] cyclopentyladenosine ([3H]CCPA)- a high affinity agonist radioligand for A1 adenosine receptors. Naunyn Schmiedebergs Arch Pharmacol. 1989;340:679–683. doi: 10.1007/BF00717744. [DOI] [PubMed] [Google Scholar]
- 51.Schumacher B (2002) Identifizierung, Struktur-Wirkungsbeziehungen und Aktivitätsprofile neuer Adenosinrezeptor-Liganden, University of Bonn
- 52.Reith U (2001) Native und rekombinante humane Adenosinrezeptoren: Charakterizierung, Interaktion mit pflanzlichen Inhaltsstoffen und Suche nach neuen Leitstrukturen, University of Bonn
- 53.Sihver W, Schulze A, Wutz W, Stüsgen S, Olsson RA, Bier D, Holschbach MH. Autoradiographiccomparison of in vitro binding characteristics of various tritiated adenosine A2A receptor ligands in rat, mouse and pig brain and first ex vivo results. Eur J Pharmacol. 2009;616:107–114. doi: 10.1016/j.ejphar.2009.06.025. [DOI] [PubMed] [Google Scholar]
- 54.Daly JW, Padgett WL, Secunda SI, Thompson RD, Olsson RA. Structure-activity relationships for 2-substituted adenosines at A1 and A2 adenosine receptors. Pharmacology. 1993;46:91–100. doi: 10.1159/000139033. [DOI] [PubMed] [Google Scholar]
- 55.Vittori S, Lorenzen A, Stannek C, Costanzi S, Volpini R, IJzerman AP, Kunzel JK, Cristalli G. N-cycloalkyl derivatives of adenosine and 1-deazaadenosine as agonists and partial agonists of the A(1) adenosine receptor. J Med Chem. 2000;43:250–260. doi: 10.1021/jm9911231. [DOI] [PubMed] [Google Scholar]
- 56.Fredholm BB, IJzerman AP, Jacobson KA, Linden J, Müller CE. International union of basic and clinical pharmacology. LXXXI. Nomenclature and classification of adenosine receptors-an update. Pharmacol Rev. 2011;63:1–34. doi: 10.1124/pr.110.003285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tchilibon S, Joshi BV, Kim SK, Duong HT, Gao ZG, Jacobson KA. (N)-methanocarba 2,N6-disubstituted adenine nucleosides as highly potent and selective A3 adenosine receptor agonists. J Med Chem. 2005;6:1745–1758. doi: 10.1021/jm049580r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Müller CE. adenosine receptor ligands-recent developments part I. Agonists. Curr Med Chem. 2001;7:1269–1288. doi: 10.2174/0929867003374101. [DOI] [PubMed] [Google Scholar]
- 59.Jacobson KA. Adenosine A3 receptors: Novel ligands and paradoxical effects. Trends Pharmacol Sci. 1998;5:184–191. doi: 10.1016/S0165-6147(98)01203-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Abo-Salem OM, Hayallah AM, Bilkei-Gorzo A, Filipek B, Zimmer A, Müller CE. Antinociceptive effects of novel A2B adenosine receptor antagonists. J Pharmacol Exp Ther. 2004;1:358–366. doi: 10.1124/jpet.103.056036. [DOI] [PubMed] [Google Scholar]
- 61.Kenneth AK, Ijzerman AP, Linden J. 1,3-dialkylxanthine derivatives having high potency as antagonists at human A2B adenosine receptors. Drug Devel Res. 1999;47:45–53. doi: 10.1002/(SICI)1098-2299(199905)47:1<45::AID-DDR6>3.0.CO;2-U. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Grahner B, Winiwarter S, Lanzner W, Müller CE. Synthesis and structure-activity relationships of deazaxanthines: Analogs of potent A1- and A2-adenosine receptor antagonists. J Med Chem. 1994;37:1526–1534. doi: 10.1021/jm00036a019. [DOI] [PubMed] [Google Scholar]
- 63.Bulicz J, Bertarelli DC, Baumert D, Fülle F, Müller CE, Heber D. Synthesis and pharmacology of pyrido [2,3-d] pyrimidinediones bearing polar substituents as adenosine receptor antagonists. Bioorg Med Chem. 2006;14:2837–2849. doi: 10.1016/j.bmc.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 64.Massip S, Guillon J, Bertarelli D, Bosc JJ, Léger JM, Lacher S, Bontemps C, Dupont T, Müller CE, Jarry C. Synthesis and preliminary evaluation of new 1- and 3-[1-(2-hydroxy-3-phenoxypropyl)] xanthines from 2-amino-2-oxazolines as potential A1 and A2A adenosine receptor antagonists. Bioorg Med Chem. 2006;14:2697–2719. doi: 10.1016/j.bmc.2005.11.050. [DOI] [PubMed] [Google Scholar]
- 65.Klotz KN, Vogt H, Tawfik-Schlieper H. Comparison of A1 adenosine receptors in brain from different species by radioligand binding and photoaffinity labelling. Naunyn Schmiedebergs Arch Pharmacol. 1991;343:196–201. doi: 10.1007/BF00168610. [DOI] [PubMed] [Google Scholar]
- 66.Kieć-Kononowicz K, Drabczyńska A, Pękala E, Michalak B, Müller CE, Schumacher B, Karolak-Wojciechowska J, Duddeck H, Rockitt S, Wartchow R. New developments in A1 and A2A adenosine receptor antagonists. Pure Appl Chem. 2001;73:1411–1420. [Google Scholar]
- 67.Kim YC, Ji X, Melman N, Linden J, Jacobson KA. Anilide derivatives of an 8-phenylxanthine carboxylic congener are highly potent and selective antagonists at human A(2B) adenosine receptors. J Med Chem. 2000;43:1165–1172. doi: 10.1021/jm990421v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Akkari R, Burbiel JC, Hockemeyer J, Müller CE. Recent progress in the development of adenosine receptor ligands as antiinflammartory drugs. Curr Top Med Chem. 2006;6:1375–1379. doi: 10.2174/15680266106061375. [DOI] [PubMed] [Google Scholar]
- 69.Gao ZG, Blaustein JB, Gross AS, Melman N, Jacobson KA. N6-Substituted adenosine derivatives: selectivity, efficacy, and species differences at A3 adenosine receptors. Biochem Pharmacol. 2003;65:1675–1684. doi: 10.1016/S0006-2952(03)00153-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Müller CE. A3 adenosine receptor antagonsits. Mini Rev Med Chem. 2001;1:417–427. doi: 10.2174/1389557510101040417. [DOI] [PubMed] [Google Scholar]
- 71.Olson KR, Eglen RM. Beta-galactosidase complementation: a cell-based luminescent assay platform for drug discovery. Assay Drug Dev Technol. 2007;5:137–144. doi: 10.1089/adt.2006.052. [DOI] [PubMed] [Google Scholar]
- 72.Müller CE, Maurinsh J, Sauer R. Binding of [3H]MSX-2 (3-(3-hydroxypropyl)-7-methyl-8-(m-methoxystyryl)-1-propargylxanthine) to rat striatal membranes—a new, selective antagonist radioligand for A(2A) adenosine receptors. Eur J Pharm Sci. 2000;10:259–265. doi: 10.1016/S0928-0987(00)00064-6. [DOI] [PubMed] [Google Scholar]
- 73.Bertarelli DC, Diekmann M, Hayallah AM, Rüsing D, Iqbal J, Preiss B, Verspohl EJ, Müller CE. Characterization of human and rodent native and recombinant adenosine A(2B) receptors by radioligand binding studies. Purinergic Signal. 2006;2:559–571. doi: 10.1007/s11302-006-9012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Seibt BF, Schiedel AC, Thimm D, Hinz S, Sherbiny FF, Müller CE. The second extracellular loop of GPCRs determines subtype-selectivity and controls efficacy as evidenced by loop exchange study at A2 adenosine receptors. Biochem Pharmacol. 2013;85:1317–1329. doi: 10.1016/j.bcp.2013.03.005. [DOI] [PubMed] [Google Scholar]
- 75.Kim SA, Marshall MA, Melman N, Kim HS, Müller CE, Linden J, Jacobson KA. Structure-activity relationships at human and rat A2B adenosine receptors of xanthine derivatives substituted at the 1-, 3-, 7-, and 8-positions. J Med Chem. 2002;45:2131–2138. doi: 10.1021/jm0104318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bruns RF, Lu GH, Pugsley TA. Characterization of the A2 adenosine receptor labeled by [3H]NECA in rat striatal membranes. Mol Pharmacol. 1986;29:331–346. [PubMed] [Google Scholar]
- 77.Hayallah AM, Sandoval-Ramírez J, Reith U, Schobert U, Preiss B, Schumacher B, Daly JW, Müller CE. 1,8-disubstituted xanthine derivatives: Synthesis of potent A2B-selective adenosine receptor antagonists. J Med Chem. 2002;45:1500–1510. doi: 10.1021/jm011049y. [DOI] [PubMed] [Google Scholar]
- 78.Müller CE, Diekmann M, Thorand M, Ozola V. [(3)H]8-Ethyl-4-methyl-2-phenyl-(8R)-4,5,7,8-tetrahydro-1H-imidazo [2,1-i]-purin-5-one ([(3)H]PSB-11), a novel high-affinity antagonist radioligand for human A(3) adenosine receptors. Bioorg Med Chem Lett. 2002;12:501–503. doi: 10.1016/S0960-894X(01)00785-5. [DOI] [PubMed] [Google Scholar]
- 79.Salvatore CA, Jacobson MA, Taylor HE, Linden J, Johnson RG. Molecular cloning and characterization of the human A3 adenosine receptor. Proc Natl Acad Sci U S A. 1993;90:10365–10369. doi: 10.1073/pnas.90.21.10365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.van Galen PJ, van Bergen AH, Gallo-Rodriguez C, Melman N, Olah ME, IJzerman AP, Stiles GL, Jacobson KA. A binding site model and structure-activity relationships for the rat A3 adenosine receptor. Mol Pharmacol. 1994;45:1101–1111. [PMC free article] [PubMed] [Google Scholar]
- 81.Rosentreter U, Henning R, Bauser M, Krämer T, Vaupel A, Hübsch W, Dembowsky K, Salcher-Schraufstätter O, Stasch J P, Krahn T, Perzborn E (2006) Substituted 2-thio-3,5-dicyano-4-aryl-6-aminopyridines and the use thereof. 1–273
- 82.van der Hoeven D, Wan TC, Gizewski ET, Kreckler LM, Maas JE, Van Orman J, Ravid K, Auchampach JA. A role for the low-affinity A2B adenosine receptor in regulating superoxide generation by murine neutrophils. J Pharmacol Exp Ther. 2011;338:1004–1012. doi: 10.1124/jpet.111.181792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hinz S, Lacher SK, Seibt BF, Müller CE. BAY60-6583 acts as a partial agonist at adenosine A2B receptors. J Pharmacol Exp Ther. 2014;349:427–436. doi: 10.1124/jpet.113.210849. [DOI] [PubMed] [Google Scholar]
- 84.Jacobson KA. Structure-based approaches to ligands for G-protein-coupled adenosine and P2Y receptors, from small molecules to nanoconjugates. J Med Chem. 2013;56:3749–3767. doi: 10.1021/jm400422s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Murphree LJ, Marshall MA, Rieger JM, MacDonald TL, Linden J. Human A(2A) adenosine receptors: High-affinity agonist binding to receptor-G protein complexes containing Gbeta(4) Mol Pharmacol. 2002;61:455–462. doi: 10.1124/mol.61.2.455. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 180 kb)