

Abstract
The in vivo function status of the ubiquitin-proteasome system (UPS) in pressure overloaded hearts remains undefined. Cardiotoxicity was observed during proteasome inhibitor chemotherapy, especially in those with preexisting cardiovascular conditions; however, proteasome inhibition (PsmI) was also suggested by some experimental studies as a potential therapeutic strategy to curtail cardiac hypertrophy. Here we used genetic approaches to probe cardiac UPS performance and determine the impact of cardiomyocyte-restricted PsmI (CR-PsmI) on cardiac responses to systolic overload. Transgenic mice expressing an inverse reporter of the UPS (GFPdgn) were subject to transverse aortic constriction (TAC) to probe myocardial UPS performance during systolic overload. Mice with or without moderate CR-PsmI were subject to TAC and temporally characterized for cardiac responses to moderate and severe systolic overload. After moderate TAC (pressure gradient: ~40mmHg), cardiac UPS function was upregulated during the first two weeks but turned to functional insufficiency between 6 and 12 weeks as evidenced by the dynamic changes in GFPdgn protein levels, proteasome peptidase activities, and total ubiquitin conjugates. Severe TAC (pressure gradients >60mmHg) led to UPS functional insufficiency within a week. Moderate TAC elicited comparable hypertrophic responses between mice with and without genetic CR-PsmI but caused cardiac malfunction in CR-PsmI mice significantly earlier than those without CR-PsmI. In mice subject to severe TAC, CR-PsmI inhibited cardiac hypertrophy but led to rapidly progressed heart failure and premature death, associated with a pronounced increase in cardiomyocyte death. It is concluded that cardiac UPS function is dynamically altered, with the initial brief upregulation of proteasome function being adaptive; and CR-PsmI facilitates cardiac malfunction during systolic overload.
Keywords: ubiquitin-proteasome system, proteasome inhibition, pressure overload, cardiac hypertrophy, heart failure, transgenic mice
1. Introduction
The ubiquitin proteasome system (UPS) is responsible for the degradation of most intracellular proteins for protein quantity and quality control [1]. Proteasome-mediated degradation occurs in the interior chamber of the 20S proteasome, which is composed of an axial stack of four heptameric rings: 2 outer α rings (α1-α7) and 2 inner β rings (β1-β7). The eukaryotic proteasome possesses three peptidase activities residing in 3 distinct subunits: chymotrypsin-like (β5), trypsin-like (β2), and caspase-like (β1). Clinically used proteasome inhibitors (bortezomib and carfilzomib) target the β5 subunit, thereby inhibiting the proteasome [2].
UPS dysfunction is implicated in a variety of cardiovascular diseases [1-4], including load-dependent cardiac disorders [5-7], The pathogenic significance of impaired UPS-mediated protein degradation in cardiac hypertrophy and cardiomyopathy is underscored by the recent association of mutations in TRIM63, a ubiquitin ligase, with familial hypertrophic cardiomyopathy in humans [8]. Further characterization of the identified mutations suggests that they impair TRIM63 ubiquitin ligase activity [8]. Proteasome functional insufficiency (PFI) in cardiomyocytes has also been experimentally demonstrated to mediate the pathogenesis of cardiac proteinopathy and acute myocardial ischemia-reperfusion injury [9-11]. In pressure-overload, myocardial total ubiquitinated proteins are always increased whereas the proteasomal peptidase activities were reportedly both increased or decreased [6, 12], indicating that the extent to which the UPS is affected during the progression of pressure overload cardiac hypertrophy remains undefined. The role of the proteasome in the development and progression of pathological hypertrophy also remains elusive. Pharmacological inhibition of the proteasome was found by some to facilitate maladaptive remodeling of stressed hearts [13, 14]; however, there were also reports that systemic inhibition of the proteasome suppressed cardiac hypertrophy and attenuated maladaptive cardiac remodeling [6, 15]. These controversies prompted us to use genetic approaches in the present study.
An inverse reporter mouse model of UPS performance is used here for the first time to determine dynamic changes in myocardial UPS performance at various stages of transverse aortic constriction (TAC)-induced left ventricle (LV) systolic overload. To determine the role of proteasome dysfunction, we also utilized a cardiomyocyte-restricted proteasome inhibition (CR-PsmI) mouse model [9]. Our results show that changes in myocardial UPS function and proteasome activities are stage-dependent during systolic overload and that CR-PsmI attenuates cardiac hypertrophy only in mice subject to severe TAC but not moderate TAC and, in both cases, facilitates cardiac failure.
2. Materials and methods
2.1 Transgenic mice
GFPdgn transgenic mice, an inverse reporter of UPS performance, were developed and initially characterized as reported [16]. The creation and initial characterization of the stable transgenic mouse lines of CR-PsmI were described [9]. Briefly, this is a stable mouse line expressing a protease-disabled Myc-tagged missense mutation (T60A) of the murine β5 subunit precursor of the 20S proteasome (T60A-β5) in cardiomyocytes, under the control of an attenuated murine myh6 promoter.
2.2 Transverse aortic constriction (TAC)
TAC was performed as described [17]. The aortic arch was isolated and ligated against a 27-gauge needle for moderate TAC (mTAC, pressure gradient: ~40mmHg) or a 29-gauge needle for severe TAC (sTAC, pressure gradient: ~60mmHg). The needle was used as the constriction template and was withdrawn immediately after ligation is completed.
2.3 Left ventricular pressure-volume analysis
Left ventricular (LV) pressure-volume relationship was analyzed in mice as previously reported [9]. In brief, the mouse were anesthetized with 2% isoflorane in medical grade oxygen, intubated, and mechanically ventilated. A 1.2-F mouse pressure-volume catheter (Scisense, London, Ontario) was inserted into the LV via the right carotid artery. The animal was allowed to stabilize during steady state conditions for 10 minutes prior to data collection with a sampling rate of 1,500 Hz with Ponemah software (Data Sciences International, Valley View, OH).
2.4 Protein extraction and western blot analysis
Proteins were extracted from LV myocardium. Bicinchoninic acid (BCA) reagents (Pierce biotechnology, Rockford, IL) were used to determine protein concentrations. SDS-PAGE, immunoblotting analysis, and densitometry were performed as previously described [18]. The following primary antibodies were used: green fluorescence protein (GFP, clone B2), GAPDH (Santa Cruz Biotechnology), RPT6 (Biomol), sarcomeric α-actinin, ubiquitin (Sigma), phosphatase and tensin homolog (PTEN), Ser473-phosphorylated-Akt, total Akt, caspase 3, cleaved caspase 3 (Cell Signaling), and PSMB5 (i.e., proteasome subunit β5, customized antibody). The corresponding horseradish peroxidase-conjugated anti-mouse or anti-rabbit secondary antibodies (Santa Cruz) were used respectively.
2.5 Proteasome peptidase activity assay
Proteasome peptidase activity assays were performed as reported [19]. Snap-frozen tissues were homogenized on ice in cytosolic extraction buffer (50 mmol/L Tris-HCl pH 7.5, 250 mmol/L Sucrose, 5 mmol/L MgCl2, 0.5 mmol/L EDTA, and 1 mmol/L DTT). Samples were then centrifuged at 8,000 g for 10 minutes at 4°C. The protein concentration of the supernatant were determined by a BCA assay. Proteasome assay buffer (50 mmol/L Tris-HCl pH 7.5, 40 mmol/L KCl, 5 mmol/L MgCl2, and 1 mmol/L DTT) was added to each well of a dark 96-well plate. ATP was added to certain wells to differentiate between peptidase activities in the presence and absence of ATP: Chymotrypsin-like activity (28 μmol/L), Caspase-like (14 μmol/L), and Trypsin-like (14 μmol/L). Equal amounts of sample were loaded to each well, except for the blank wells. Proteasome inhibitors of the specific proteasome activities were applied to decipher the respective activities: Chymotrypsin-like (MG132, 20 μmol/L), Caspase-like (MG 132, 20 μmol/L), and Trypsin-like (Epoxomicin, 5 μmol/L). Specific proteasome activity fluorogenic substrates were added for chymotrypsin-like (Suc-LLVY-AMC, 18 μmol/L), caspase-like (Suc-LLE-AMC, 45 μmol/L), and trypsin-like activities (AC-RLR-AMC (Bz), 40 μmol/L). The 96-well plates were incubated in a 37°C incubator for 30, 60, 90, 120, 150, and 180 minutes. At each time point the plate was read in a plate reader (Perkin Elmer, model 2030, Waltham, MA) with an excitation wavelength of 380 nm and an emission wavelength of 460 nm. Sample readings were subtracted from the blank reading. The proteasome inhibitor suppressible portion of peptidase activity is attributed to the proteasome.
2.6 Measurement of mRNA expression of the fetal gene program
This was done using either RNA dot blot analysis (for the mTAC experiments) or Real time reverse transcription PCR (for the sTAC experiments). Total RNA was isolated from LV myocardial samples using Tri-Reagent® (Molecular Research Center, Cincinnati, OH). RNA dot blot analysis was performed as previously reported [20]. Two micrograms of total RNA were loaded to each dot of a nitrocellulose membrane using a dot blot apparatus. Transcript-specific p32-labeled radioactive oligonucleotide probes were used. The mRNA of following murine genes were assessed: atrial natriuretic peptide (NPPA), B-type natriuretic peptide (NPPB), skeletal α-actin (ACTA1), α-myosin heavy chain (MYH6), β-myosin heavy chain (MYH7), sarcoplasmic reticulum calcium ATPase 2A (ATP2A2),and phospholamban (PLN), with the housekeeping gene glyceraldehyde phosphate dehydrogenase (GAPDH) probed for loading control. The radioactive probe-bound membrane was exposed to a phosphor screen and imaged and digitized using a Typhoon 860 imager and associated software (Molecular Dynamics).
Real time reverse transcription PCR (RT-PCR) was performed as previously described [21], for quantification of myocardial mRNA levels of the fetal gene program. The High Capacity cDNA Archive Kit from Applied Biosystems (Foster City, CA) was used for cDNA synthesis from 0.1 μg of total RNA by reverse transcription. The PCR reaction used the cDNA and thermal stable AmpliTaq Gold DNA polymerase. Predesigned TaqMan primers and probes specific for NPPA, NPPB, MYH6, MYH7, and GAPDH were obtained from Applied Biosystems.
2.7 Fluorescence confocal microscopy
Ventricular myocardium from T60A-β5 tg mice was fixed with 3.8% paraformaldehyde and processed for cryosectioning, as previously described [22]. To discern cardiomyocytes, the myocardial sections were stained for desmin (Abcam, Cambridge, MA) using indirect immunofluorescence. Necrotic cells were stained with a Fluro565-conjugated anti-mouse IgG antibody (Life Technologies, Grand Island, NY). Nuclear DNA was stained with DAPI. The fluorescence staining was visualized and imaged using a multi-laser fluorescence confocal microscope (Olympus Fluoview 500). ImagePro Plus software (Media Cybernetics, Rockville, MD) was used to quantify the percentage of the amount of red fluorescence (necrosis) to green fluorescence (cardiomyocytes) [22].
2.8 Statistical analysis
All continuous variables are presented as mean±SD. Differences between two groups were evaluated for statistical significance using two-tailed Student’s t-test, unless otherwise noted. A difference among 3 or more groups, one-way ANOVA or when appropriate, 2-way ANOVA, followed by the Holm-Sidak test for pair-wise comparisons were performed. The p<0.05 is considered statistically significant.
3. Results
3.1 Dynamic changes of myocardial proteasomal activities and UPS performance during systolic overload
To determine the time course of global UPS performance changes following systolic overload, we subjected GFPdgn mice to moderate TAC (mTAC) and examined myocardial GFPdgn protein levels and proteasome peptidase activities. Myocardial GFPdgn protein levels were significantly decreased and proteasome peptidase activities were increased at 2 weeks after mTAC (Figure 1A, 1B), indicating that acute systolic overload initially upregulates myocardial UPS function. This hyper-function dwindled at 4 weeks and UPS performance returned to the sham control level at 6 weeks; proteasome hypo-function and proteasome functional insufficiency (PFI) were evidenced at 12 weeks after mTAC by significant increases in myocardial GFPdgn proteins, decreases in myocardial proteasome caspase-like and trypsin-like peptidase activities, and marked increases in the levels of total soluble and insoluble ubiquitinated proteins (Figure 1).
Figure 1. Temporal changes in myocardial UPS performance in mice subject to moderate trans-aortic constriction (mTAC).

NTG and GFPdgn TG mice were subject to mTAC at ~10 weeks of age. A, Representative images (upper panel) and pooled densitometry data of western blot analyses for LV myocardial GFPdgn at the indicated time points. GFPdgn was detected using antibodies against GFP and α-actinin (Actn) was probed for loading control. For each time point, n=4~6/group; T=TAC; S=sham. B~D, Temporal changes in myocardial 20S proteasome chymotrypsin-like (β5), trypsin-like (β2), and caspase-like (β1) peptidase activities after mTAC. AU, arbitrary unit; n=8~12/group. E and F, Western blot analyses for total ubiquitinated proteins in the 1% Triton X-100 soluble and insoluble fractions of myocardium at 12 weeks after mTAC. *p<0.05, **p<0.01 vs. Sham.
3.2 Moderate CR-PsmI does not prevent cardiac hypertrophy in response to mTAC
We created stable TG mouse lines overexpressing a peptidase-disabled mutant proteasome subunit β5 (T60A-β5) in cardiomyocytes, the first mouse model of CR-PsmI [9]. Confirming the previous report, transgenic expression of T60A-β5 drives down the expression of endogenous mature β5 (Figure 2A), thereby inhibiting myocardial proteasomal chymotrypsin-like activity by ~60% (Figure 2B). At 6 days after mTAC, T60A-β5 TG and NTG mice displayed comparable cardiac growth as revealed by gravimetric analyses (Figure 2C) and indistinguishable reactivation of the fetal genes (Figure 2D, 2E). No difference in cardiac function was detected at this early time point (data not shown). The results demonstrate that moderate CR-PsmI does not discernibly alter cardiac hypertrophic responses to mTAC.
Figure 2. CR-PsmI does not prevent cardiac hypertrophic responses to mTAC.

B> Western blot analyses for endogenous (endo) proteasome β5 subunit and Myc-tagged protease-disabled β5 (Myc-T60A-β5) in LV myocardium of T60A-β5 Tg and Ntg littermate mice. B, The impact of T60A-β5 expression on myocardial chymotrypsin-like peptidase activity. C ~ E, Effects of CR-PsmI on cardiac hypertrophic responses to mTAC. Both gravimetric analyses (C) and RNA dot blot assessment of the fetal gene program (D, E) at 6 days after mTAC show comparable hypertrophic responses between T60A-β5 Tg and Ntg mice (p>0.05). *p<0.05, **p<0.01 vs. Ntg sham.
3.3 Moderate CR-PsmI expedites cardiac malfunction in response to mTAC
By 24 weeks after mTAC, cardiac hypertrophic responses remained comparable between CR-PsmI and control mice (Figure 3A). However, the absolute values of the maximal rate of LV pressure rise (dP/dt-max) and decline (dP/dt-min) were both significantly decreased and the time constant of relaxation (Tau) was markedly increased in the T60A-β5 TG mTAC group, compared with the NTG mTAC and the sham control groups (p<0.05), indicating that chronic CR-PsmI expedites cardiac malfunction during systolic overload.
Figure 3. Effects of CR-PsmI on cardiac hypertrophy and function at 24 weeks after mTAC.
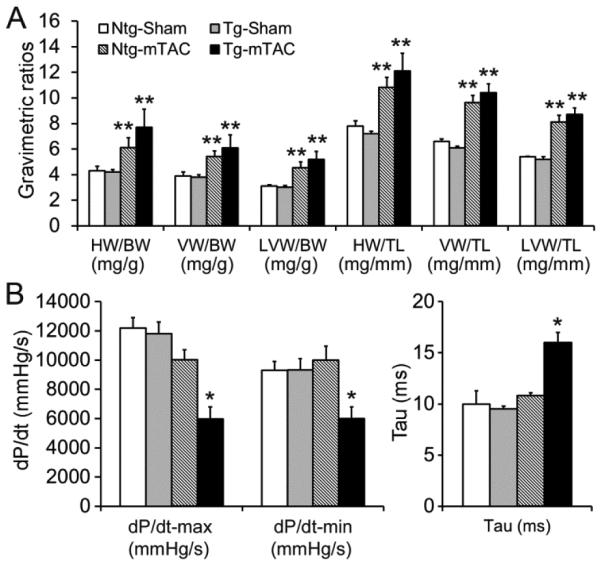
T60A-β5 Tg and Ntg littermate mice of 10-week-old were subject to mTAC or sham surgery. A, Gravimetric analyses. For each parameter shown, the difference between Ntg-mTAC and Tg-mTAC groups is not statistically significant (p>0.05). **p<0.01 vs. Ntg sham; n=8 mice/group. B, LV hemodynamics assessments. *p<0.05 vs. Ntg sham and Ntg TAC; n=5 mice/group.
3.4 Moderate CR-PsmI suppresses cardiac growth but causes cardiac failure upon sTAC
To further determine the effect of CR-PsmI on cardiac responses to a greater systolic overload, T60A-β5 TG and NTG mice were subject to sTAC or sham surgery. In NTG mice, sTAC did not significantly increase myocardial β5 and Rpt6 protein levels but induced significant increases in all three peptidase activities of the 26S proteasome at 6 days after TAC (Figure 4A~4C). These increases were apparently insufficient to meet the increased demand for protein degradation as evidenced by accumulation of UPS substrate GFPdgn at 1 and 2 weeks after sTAC (Supplemental Figure 1), indicating that cardiac PFI occurs much faster in sTAC than mTAC. sTAC did not alter T60A-β5 and endogenous β5 expression in the TG mice; consequently, the sTAC-induced increases in chymotrypsin-like activity were selectively blocked in TG mice, exacerbating sTAC-induced accumulation of ubiquitinated proteins (Figure 4D, 4E).
Figure 4. The impact of TG expression of T60A-β5 on proteasome functional changes during sTAC.

The T60A-β5 Ntg and Tg littermate mice were subject to sTAC or sham surgery. At 6 days after the surgery, the LV tissue was sampled for extraction of crude myocardial proteins. A and B, Representative images (A) and pooled densitometry data (B) of western blot analyses of the indicated proteins. C, Proteasomal peptidase activity assays. D and E, Western blot analysis for LV myocardial total ubiquitinated (Ub’n) proteins. *p<0.05, #p<0.01, ##p<0.001 vs. Ntg sham; $p<0.01; n=4 mice/group.
Gravimetric measurements at 6 days after sTAC showed that T60A-β5 TG mice displayed smaller cardiac growth than NTG mice in response to sTAC (Table 1). Notably, the sTAC-induced reactivation of various fetal genes was differentially affected by CR-PsmI. The upregulation of NPPA and NPPB was exacerbated, whereas MYH7 reactivation was attenuated, by CR-PSMI (Figure 5A). Consistent with early LV function insufficiency, lung weight to body weight or tibial length ratios were significantly higher in T60A-β5 sTAC mice than in NTG sTAC or sham control mice (Table 1).
Table 1.
Gravimetric analyses at 6 days after sTAC
| NTG Sham | T60A-β5 Sham | NTG sTAC | T60A-β5 sTAC | |
|---|---|---|---|---|
| N | 10 | 10 | 10 | 6 |
|
| ||||
| HW/BW, mg/g | 4.4 ± 0.4 | 4.6 ± 0.4 | 6.3 ± 0.6* | 5.0 ± 0.5† |
|
| ||||
| VW/BW, mg/g | 4.0 ± 0.3 | 4.1 ± 0.2 | 5.6 ± 0.4* | 4.4 ± 0.3† |
|
| ||||
| LVW/BW, mg/g | 3.1 ± 0.2 | 3.1 ± 0.2 | 4.6 ± 0.4* | 3.4 ± 0.2† |
|
| ||||
| LuW/BW, mg/g | 4.8 ± 0.3 | 5.5 ± 0.3 | 6.0 ± 0.3 | 12.8 ± 2.2*† |
|
| ||||
| LiW/BW, mg/g | 47.3 ± 10.5 | 50.7 ± 7.7 | 54.6 ± 8.3 | 53.8 ± 9.5 |
|
| ||||
| KiW/BW, mg/g | 15.0 ± 1.3 | 15.7 ± 0.7 | 16.5 ± 1.2 | 16.2 ± 3.7 |
|
| ||||
| HW/TL, mg/mm | 6.8 ± 0.3 | 6.6 ± 0.4 | 8.0 ± 0.9* | 6.6 ± 0.8† |
|
| ||||
| VW/TL, mg/mm | 6.1 ± 0.4 | 6.0 ± 0.5 | 7.0 ± 0.5* | 5.9 ± 0.7† |
|
| ||||
| LVW/TL, mg/mm | 4.7 ± 0.3 | 4.5 ± 0.4 | 5.8 ± 0.5* | 4.5 ± 0.5† |
|
| ||||
| LuW/TL, mg/mm | 7.4 ± 0.6 | 8.0 ± 0.6 | 7.5 ± 0.5 | 16.7 ± 1.5*† |
|
| ||||
| LiW/TL, mg/mm | 71.8 ± 9.7 | 73.1 ± 10.9 | 69.0 ± 7.1 | 70.6 ± 8.2 |
|
| ||||
| KiW/TL, mg/mm | 23.3 ± 4.3 | 22.7 ± 2.3 | 20.8 ± 1.3 | 21.0 ± 2.8 |
Heart Weight (HW), Ventricular Weight (VW), Left Ventricular Weight (LVW), Lung Weight (LuW), Liver Weight (LiW), Kidney Weight (KiW), Body Weight (BW), Tibial Length (TL).
p<0.05 vs. NTG Sham;
p<0.05 vs. NTG TAC.
Figure 5. Effects of CR-PsmI on cardiac responses to sTAC.

A, Changes in the mRNA expression of the indicated genes at 6 days after surgery. GAPDH was probed as a house keeping gene for RNA quantification control. *p<0.05 vs. NTG sham; $p<0.05; n=4 mice/group. B, Kaplan-Meier survival curve. n=10 mice/group; p < 0.01, log-rank test. C & D, Prevalence of cardiomyocyte necrosis. Cryosections from LV myocardium collected 6 days after sTAC were immuno-stained for mouse IgG (red) using a Fluor-568 conjugated anti-mouse IgG antibody and for desmin using rabbit primary antibodies for desmin and Fluor-488 conjugated anti-rabbit IgG secondary antibodies. Desmin- and mouse IgG positive (IgG+) cardiomyocytes are considered necrotic. Nuclear DNA was stained blue with DAPI. Representative images (C) and pooled data from 3 mice per group (D) are shown.
Analyses of LV pressure-volume relationship at 6 days after sTAC (Table 2) revealed that compared with NTG sham, LV systolic and diastolic function remained compensated in the NTG sTAC group; however, de-compensation were evidenced in the TG sTAC group. Compared with both the sham groups and the NTG sTAC group, the T60A-β5 sTAC group displayed significantly depressed ejection fraction, reduced cardiac output, and smaller maximal and minimal dP/dt (dP/dtmax, dP/dtmin), and increased Tau, indicative of significantly impairment in both systolic and diastolic function.
Table 2.
LV pressure-volume relationship at 6 days after sTAC
| NTG Sham | T60A-β5 Sham | NTG sTAC | T60A-β5 sTAC | |
|---|---|---|---|---|
| N | 6 | 6 | 6 | 6 |
|
| ||||
| HR, beats/min | 505±23.6 | 514±22.7 | 554±28.3 | 506±35.6 |
|
| ||||
| Pes, mmHg | 118.3±10.4 | 111.5±2.0 | 166.3±5.3* | 130.4±33.3 |
|
| ||||
| Ves, μl | 10.4±2.9 | 12.0±4.2 | 5.0±1.8* | 32.9±6.7*† |
|
| ||||
| Ped, mmHg | 4.1±2.0 | 3.8±2.8 | 14.8±2.1* | 10.3±1.7*† |
|
| ||||
| Ved, μl | 36.4±6.2 | 38.4±7.6 | 24.8±2.4* | 51.2±8.3*† |
|
| ||||
| SV, μl | 26.0±3.4 | 26.4±3.3 | 19.8±2.8* | 18.3±1.6* |
|
| ||||
| SW, mmHg*μl | 3072±393.0 | 2963±693.3 | 3273±396.3 | 2415±819.0† |
|
| ||||
| CO, μl/min | 13108±1346.6 | 13537±1135.4 | 10947±1541.7* | 36.00±2.7*† |
|
| ||||
| EF, % | 71.82±3.0 | 69.12±4.8 | 79.83±6.8* | 9293±1465.8*† |
|
| ||||
| dP/dtmax, mmHg/s | 11075±2020.2 | 10722±2913.3 | 12079±2029.7 | 5595±955.3*† |
|
| ||||
| dP/dtmin mmHg/s | 9911±1287.4 | 9796±1834.5 | 10468±1025.0 | 4834±1487.7*† |
|
| ||||
| Tau, ms | 8.7±1.4 | 9.2±1.3 | 10.6±1.2 | 16.9±1.8*† |
Heart Rate (HR), end-systolic pressure (Pes), end-systolic volume (Ves), end-diastolic pressure (Ped), end-diastolic volume (Ved), Stroke Volume (SV), Stroke Work (SW), Cardiac Output (CO), Ejection Fraction (EF), the maximal rate of pressure increasing (+dP/dt), the maximal rate of pressure decreasing (−dP/dt), relaxation time constant calculated by the Glantz method (Tau).
p<0.05 vs. NTG Sham;
:p0.05 vs. NTG TAC.
These data indicate that despite suppression of cardiac hypertrophy, CR-PsmI impairs cardiac functional adaptation to severe systolic overload. Consistent with the early functional deterioration, Kaplan-Meier analysis showed significantly decreased survival in the T60A-β5 sTAC group (Figure 5B, p<0.01).
3.5 Moderate CR-PsmI causes cardiomyocyte death in the heart during severe systolic overload
To explore potential factors underlying this rapid cardiac functional deterioration in the CR-PsmI sTAC mice, we examined whether CR-PsmI causes loss of cardiomyocytes via necrosis by immunofluorescence staining of myocardial sections for mouse IgG. We found that cardiomyocytes positive for mouse IgG were rare in the NTG sTAC mice but the percentage of cardiomyocytes positive for mouse IgG was significantly increased in the T60A-β5 sTAC mice (Figure 5C, 5D). IgG proteins should not be found inside cardiomyocytes but the loss of cell membrane integrity during necrosis allows IgG to enter the cell [22]. These data indicate that CR-PsmI causes cardiomyocyte necrosis during severe systolic overload.
Further examination showed that myocardial Ser-473 phosphorylated Akt (p-Akt) was significantly increased at 6 days after sTAC in Ntg mice but this increase was completely blocked by CR-PsmI. In fact, CR-PsmI even significantly increased PTEN and decreased p-Akt protein levels in the sham group. Meanwhile, myocardial PTEN protein levels were significantly higher in the T60A-β5 TG sTAC hearts than in the NTG sTAC group (Figure 6A, B). These findings indicate that CR-PsmI not only prevents compensatory Akt activation but also suppresses baseline Akt activity, an important cell survival factor. Moreover, activation of the apoptotic pathway by CR-PsmI is suggested by the finding that CR-PsmI increased the levels of the cleaved (activated) form of caspase 3 in both sham and sTAC hearts (Figure 6C, D).
Figure 6. CR-PsmI suppresses cell survival signaling and activates caspase 3.

LV crude proteins collected as described in Figure 4 were used. A and B, Representative images (A) and pooled densitometric data (B) of western blot analyses for total Akt, Ser473-phosphorylated Akt (p-Akt), and PTEN. GAPDH was probed as loading control. C and D, Representative images (C) and pooled densitometry data (D) of western blot analyses for total and cleaved (CL) caspase 3. *p<0.05, #p<0.01 vs. Ntg sham; δ, p<0.05; $, p<0.01; n=6 mice/group.
4. Discussion
Previous studies estimated changes in myocardial proteasome activities during systolic overload using in vitro peptidase activity assays but these assays are not designed to, and often cannot, assess the in vivo function status of the UPS or proteasomes [23]. Given the importance of the UPS in cellular protein quality and quantity control, a better assessment of myocardial UPS functioning under systolic overload will facilitate the research into the molecular basis of cardiac remodeling in load-dependent heart disease. Using a well-established inverse reporter mouse model of the UPS, here we unveil for the first time the highly dynamic nature of myocardial UPS performance during the progression of pressure overloaded cardiac hypertrophy. UPS performance and proteasome peptidase activities undergo a brief initial global increase (2 weeks after mTAC), which is followed by a new equilibrium (4 ~ 6 weeks) before reaching functional insufficiency (12 weeks). In a greater systolic overload setting, UPS functional insufficiency occurs much earlier (within 1 week). Moreover, we have used a genetic approach to demonstrate that moderate CR-PsmI does not produce any beneficial effects on cardiac responses to either moderate or severe systolic overload. In the mTAC setting, CR-PsmI does not display discernible effect on cardiac hypertrophic responses but rather facilitates cardiac malfunction. In the sTAC setting, CR-PsmI suppresses acutely cardiac growth but this is accompanied by increased cardiomyocyte necrosis, rapid deterioration of cardiac function, and increased mouse mortality. Our findings also support the notion that the early heightened myocardial UPS performance and proteasome activities are compensatory responses to systolic overload and that inhibition of 20S proteasomes in cardiomyocytes does not yield therapeutic benefit during systolic overload.
4.1 Dynamic changes in myocardial UPS performance during systolic overload
Previous studies on myocardial proteasome functioning during LV systolic overload were limited to the use of in vitro peptidase assays and yielded conflicting results. Depre et al. reported that myocardial proteasomal abundance and chymotrypsin-like activity in mice were remarkably increased at 5 days after TAC [6]. Their subsequent study further showed progressive increases in myocardial chymotrypsin-like activity during the first 3 weeks after TAC [15]. However, an earlier report by Tsukamoto and colleague failed to detect increases in the peptidase activities but rather showed significant decreases in all three proteasomal peptidases activities in mouse hearts at 2 and 4 weeks after TAC [12]. Here we have observed statistically significant increases in proteasomal chymotrypsin-like, trypsin-like, and caspase-like activities at 2 weeks after mTAC; the trypsin-like activity remained increased but the other two returned to levels comparable to those seen in the sham control at 4 weeks; and by 12 weeks trypsin-like and caspase-like activities were significantly decreased. Hence, it appears that myocardial proteasome peptidases are both dynamically and differentially regulated during systolic overload, resulting in a brief upregulation in the first 2 weeks, returning to normal levels between 4 and 6 weeks, and decreasing thereafter at least in caspase-like and trypsin-like activities. Interestingly, a similar pattern of differential changes in myocardial activity of the three proteasome peptidases were also observed in isoproterenol-induced cardiac hypertrophy [24]. The decrease of myocardial proteasome peptidase activities occurred before heart failure became discernible in this mTAC model, suggesting that the decreased UPS performance contributes to the development of heart failure, which is supported by some of the previous reports [12, 13].
The use of a constitutively expressed biologically inert protein reporter of the UPS (e.g., GFPdgn) allows assessment of in vivo global UPS performance. As reflected by a significant decrease in the steady state protein level of GFPdgn, myocardial UPS function shows a significant increase upon acute mTAC, which is underlined by increases in proteasome activities. This improved UPS performance is obviously a counter measure to the increased by-production of misfolded proteins during the rapid cardiac hypertrophy and is pivotal to maintaining proteostasis in the early phase of systolic overload (Figure 7). However, this increase does not last long because by 4~6 weeks after mTAC the GFPdgn protein levels return to the same level as the sham control group. Moreover, as evidenced by a significant increase in GFPdgn at 12 weeks, myocardial UPS function becomes significantly inadequate during the progression of systolic overload cardiac hypertrophy. This inadequacy is primarily caused by PFI as the total myocardial ubiquitinated proteins were significantly increased and myocardial 20S proteasomal caspase-like and trypsin-like peptidase activities were significantly decreased by this stage.
Figure 7. Central illustration.
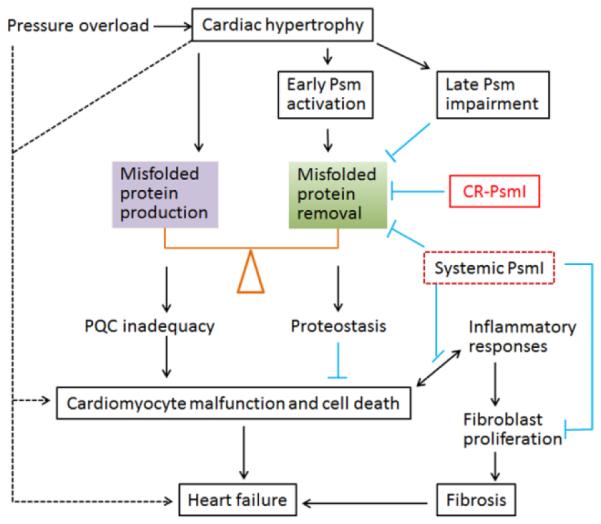
Maintaining cardiac proteostasis requires timely removal of misfolded proteins whereas increased production of misfolded proteins tilts the balance toward PQC inadequacy. In response to acute systolic overload, the heart undergoes hypertrophy, which inevitably increases protein synthesis and by-production of misfolded proteins. To counter the increased proteotoxic stress during hypertrophy, cardiomyocytes adaptively mobilizes proteasomal reserve and thereby elevates proteasome (Psm) activities and UPS performance; however, when the stress sustains, proteasome function and UPS performance are impaired by unknown factors, which diminishes the capability of cardiomyocytes to maintain proteostasis and suppresses cell survival signaling, leading to cardiomyocyte malfunction and cell death. Cardiomyocyte death, especially in the form of necrosis, triggers inflammatory responses which could further fuel cardiomyocyte death and stimulate proliferation of interstitial cells (e.g., fibroblasts), leading to fibrosis. Both loss of cardiomyocytes and fibrosis are main pathogenic factors of heart failure. CR-PsmI prevents the initial adaptive proteasomal activation and hastens UPS impairment, thereby facilitating heart failure during systolic overload. Systemic PsmI inhibits UPS-mediated proteolysis in both cardiomyocytes and non-cardiomyocyte compartments. The non-cardiac PsmI could potentially attenuate inflammatory responses and the proliferation of fibroblasts, thereby being anti-fibrotic; hence, systemic PsmI might be beneficial at the certain stage of systolic overload. The dashed lines denote other potential pathways that are directly tested by the present study.
The direct causes of increased myocardial 20S proteasome activities at the early stage and the decreased proteasome function later are not explored in the present study but post-translational modifications of the proteasome might play a role. A number of post-translational modifications can regulate proteasome activities [1, 25, 26]. Increased phosphorylation heightens proteasomal peptidase activities [24, 27, 28], whereas decreased acetylation in 20S proteasome subunits is associated with depressed proteasome proteolytic function in end-stage failing human hearts [29].
4.2 CR-PsmI facilitates cardiac failure during systolic overload
Previous studies on the impact of systemic PsmI with pharmacological inhibitors on pressure overload cardiac hypertrophy and remodeling yielded mixed results [6, 13, 15]. Most reports showed that moderate PsmI suppressed cardiac hypertrophy [30], with some even showing long term beneficial effect in pressure overload models [15]. Here we demonstrate that genetically induced moderate CR-PsmI facilitates the development of cardiac failure in both mTAC and sTAC settings although its effects on cardiac hypertrophic responses depend on the severity of systolic overload. Moderate CR-PsmI did not discernibly alter cardiac hypertrophic responses, including both cardiac growth and reactivation of fetal genes, in a commonly used mTAC model. Intriguingly, in a setting of greater systolic overload, CR-PsmI did inhibit cardiac growth but exacerbated the reactivation of NPPA and NPPB, a sign of more pronounced cardiac injury. Indeed, this cardiac growth inhibition is not beneficial because it is followed by progressive cardiac malfunction and significant increases in mortality. Consistent with impaired PQC and increased proteotoxicity, caspase activation and cardiomyocyte necrosis were significantly increased by sTAC in CR-PsmI mice, which was associated with increased PTEN and decreased Akt activation. The findings of the present study suggest that the previously reported beneficial effects of moderate systemic PsmI during pressure overload might mainly result from its effects on the non-cardiomyocyte compartment of the heart and extra-cardiac organs/systems, such as inhibition of inflammatory responses [31], suppression of fibroblast proliferation and fibrosis [32], and reduction of peripheral vascular resistance. Systemic PsmI has recently been shown to attenuate angiotensin-II induced hypertension and vascular remodeling [33]; PsmI inhibits the activation of NFκB via blocking IκB degradation [3, 31]; systemic PsmI may also inhibit fibroblast proliferation and collagen synthesis [32], thereby suppressing cardiac fibrosis. These potential benefits of systemic PsmI cannot be conferred by CR-PsmI, which might account for the differential effects on cardiac function between moderate systemic PsmI and genetic CR-PsmI (Figure 7).
Through a number of potential mechanisms, such as failure to generate critical amino acids for new polypeptide synthesis [34], hindering ribosome assembly, and modifying the activity of the elongation factor eEF1A [35], severe PsmI could inhibit protein synthesis and thereby suppress cardiac hypertrophy. However, this is at the cost of impairing degradation of misfolded proteins and increasing proteotoxic stress, leading to cardiomyocyte malfunction or cell death. This scenario is well demonstrated by both mTAC and sTAC on T60A-β5 mice. In mTAC, our genetic CR-PsmI is not severe enough to directly suppress hypertrophy but, when coupled with intrinsic PFI at the later stage, cardiac PQC is impaired, exacerbating cardiac malfunction. In sTAC, the intrinsic UPS functional inadequacy occurs much earlier, therefore the same moderate CR-PsmI can now achieve enough inhibition at an early time point to suppress cardiac protein synthesis (i.e., cardiac growth) but by the same mechanism this also quickly impairs PQC in cardiomyocytes, leading to cardiomyocyte malfunction, increased cell death and ultimately heart failure. Supporting our contention, intermittent primary PsmI caused restrictive cardiomyopathy in pigs [14] and cardiotoxicity including heart failure was frequently reported for PsmI used in clinical chemotherapy, especially in patients with preexisting cardiovascular conditions [36, 37].
4.3 Translational Implications
Patients who are implicated for proteasome inhibitor treatment should be screened for pre-existence of cardiovascular disorders, such as hypertension and aortic stenosis, and closely monitored for cardiotoxicity when receiving proteasome inhibitors as part of chemotherapy. Several reports have shown potential benefit of moderate PsmI on pressure overloaded hearts; to this end, our study suggests a further focus on searching for measures that mainly inhibit non-cardiac proteasomes.
4.4 Conclusions
Cardiac UPS function is dynamically altered during systolic overload, with the initial brief upregulation of proteasome function being adaptive; and CR-PsmI promotes cardiomyocyte death and facilitates cardiac malfunction during systolic overload.
Supplementary Material
Highlights.
Systolic overload alters dynamically myocardial proteasomal performance
Cardiac proteasome inhibition does not suppress cardiac hypertrophy responses
Genetic cardiac proteasome inhibition promotes cardiac failure in systolic overload
Cardiac proteasome inhibition promotes cardiomyocyte death during systolic overload
Acknowledgments
We thank the Imaging Core of the Division of Basic Biomedical Sciences for assistance with confocal microscopy and Ms. Andrea Jahn for maintaining mouse colonies and genotyping.
Sources of Funding:
This work was supported in part by NIH grants R01HL085629 and R01HL072166 (to X.W.) and American Heart Association grant 11PRE5730009 (to M.J.R). The Imaging Core was supported by an NIH grant 5P20RR015567.
Abbreviations
- CHF
congestive heart failure
- PsmI
proteasome inhibition
- CR-PsmI
cardiomyocyte-restricted PsmI
- LV
left ventricle
- NTG
non-transgenic
- PFI
proteasome functional insufficiency
- PQC
protein quality control
- TAC
transverse aortic constriction
- TG
transgenic
- UPS
ubiquitin-proteasome system
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure
None declared.
References
- [1].Wang X, Pattison JS, Su H. Posttranslational modification and quality control. Circ Res. 2013;112:367–81. doi: 10.1161/CIRCRESAHA.112.268706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Drews O, Taegtmeyer H. Targeting the ubiquitin-proteasome system in heart disease: the basis for new therapeutic strategies. Antioxid Redox Signal. 2014;21:2322–43. doi: 10.1089/ars.2013.5823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Willis MS, Townley-Tilson WH, Kang EY, Homeister JW, Patterson C. Sent to destroy: the ubiquitin proteasome system regulates cell signaling and protein quality control in cardiovascular development and disease. Circ Res. 2010;106:463–78. doi: 10.1161/CIRCRESAHA.109.208801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Schlossarek S, Frey N, Carrier L. Ubiquitin-proteasome system and hereditary cardiomyopathies. J Mol Cell Cardiol. 2014;71:25–31. doi: 10.1016/j.yjmcc.2013.12.016. [DOI] [PubMed] [Google Scholar]
- [5].Tsukamoto O, Minamino T, Kitakaze M. Functional alterations of cardiac proteasomes under physiological and pathological conditions. Cardiovasc Res. 2009;14:14. doi: 10.1093/cvr/cvp282. [DOI] [PubMed] [Google Scholar]
- [6].Depre C, Wang Q, Yan L, Hedhli N, Peter P, Chen L, et al. Activation of the cardiac proteasome during pressure overload promotes ventricular hypertrophy. Circulation. 2006;114:1821–8. doi: 10.1161/CIRCULATIONAHA.106.637827. [DOI] [PubMed] [Google Scholar]
- [7].Razeghi P, Baskin KK, Sharma S, Young ME, Stepkowski S, Essop MF, et al. Atrophy, hypertrophy, and hypoxemia induce transcriptional regulators of the ubiquitin proteasome system in the rat heart. Biochem Biophys Res Commun. 2006;342:361–4. doi: 10.1016/j.bbrc.2006.01.163. [DOI] [PubMed] [Google Scholar]
- [8].Chen SN, Czernuszewicz G, Tan Y, Lombardi R, Jin J, Willerson JT, et al. Human molecular genetic and functional studies identify TRIM63, encoding Muscle RING Finger Protein 1, as a novel gene for human hypertrophic cardiomyopathy. Circ Res. 2012;111:907–19. doi: 10.1161/CIRCRESAHA.112.270207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Tian Z, Zheng H, Li J, Li Y, Su H, Wang X. Genetically induced moderate inhibition of the proteasome in cardiomyocytes exacerbates myocardial ischemia-reperfusion injury in mice. Circ Res. 2012;111:532–42. doi: 10.1161/CIRCRESAHA.112.270983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Li J, Horak KM, Su H, Sanbe A, Robbins J, Wang X. Enhancement of proteasomal function protects against cardiac proteinopathy and ischemia/reperfusion injury in mice. J Clin Invest. 2011;121:3689–700. doi: 10.1172/JCI45709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Chen Q, Liu JB, Horak KM, Zheng H, Kumarapeli AR, Li J, et al. Intrasarcoplasmic amyloidosis impairs proteolytic function of proteasomes in cardiomyocytes by compromising substrate uptake. Circ Res. 2005;97:1018–26. doi: 10.1161/01.RES.0000189262.92896.0b. [DOI] [PubMed] [Google Scholar]
- [12].Tsukamoto O, Minamino T, Okada K, Shintani Y, Takashima S, Kato H, et al. Depression of proteasome activities during the progression of cardiac dysfunction in pressure-overloaded heart of mice. Biochem Biophys Res Commun. 2006;340:1125–33. doi: 10.1016/j.bbrc.2005.12.120. [DOI] [PubMed] [Google Scholar]
- [13].Tang M, Li J, Huang W, Su H, Liang Q, Tian Z, et al. Proteasome functional insufficiency activates the calcineurin-NFAT pathway in cardiomyocytes and promotes maladaptive remodelling of stressed mouse hearts. Cardiovasc Res. 2010;88:424–33. doi: 10.1093/cvr/cvq217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Herrmann J, Wohlert C, Saguner AM, Flores A, Nesbitt LL, Chade A, et al. Primary proteasome inhibition results in cardiac dysfunction. Eur J Heart Fail. 2013;15:614–23. doi: 10.1093/eurjhf/hft034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hedhli N, Lizano P, Hong C, Fritzky LF, Dhar SK, Liu H, et al. Proteasome inhibition decreases cardiac remodeling after initiation of pressure overload. Am J Physiol Heart Circ Physiol. 2008;295:H1385–93. doi: 10.1152/ajpheart.00532.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kumarapeli AR, Horak KM, Glasford JW, Li J, Chen Q, Liu J, et al. A novel transgenic mouse model reveals deregulation of the ubiquitin-proteasome system in the heart by doxorubicin. FASEB J. 2005;19:2051–3. doi: 10.1096/fj.05-3973fje. [DOI] [PubMed] [Google Scholar]
- [17].Kumarapeli AR, Su H, Huang W, Tang M, Zheng H, Horak KM, et al. Alpha B-crystallin suppresses pressure overload cardiac hypertrophy. Circ Res. 2008;103:1473–82. doi: 10.1161/CIRCRESAHA.108.180117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Su H, Li J, Menon S, Liu J, Kumarapeli AR, Wei N, et al. Perturbation of cullin deneddylation via conditional Csn8 ablation impairs the ubiquitin-proteasome system and causes cardiomyocyte necrosis and dilated cardiomyopathy in mice. Circ Res. 2011;108:40–50. doi: 10.1161/CIRCRESAHA.110.230607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Powell SR, Davies KJ, Divald A. Optimal determination of heart tissue 26S-proteasome activity requires maximal stimulating ATP concentrations. J Mol Cell Cardiol. 2007;42:265–9. doi: 10.1016/j.yjmcc.2006.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Wang X, Osinska H, Klevitsky R, Gerdes AM, Nieman M, Lorenz J, et al. Expression of R120G-alphaB-crystallin causes aberrant desmin and alphaB-crystallin aggregation and cardiomyopathy in mice. Circ Res. 2001;89:84–91. doi: 10.1161/hh1301.092688. [DOI] [PubMed] [Google Scholar]
- [21].Eyster KM, Klinkova O, Kennedy V, Hansen KA. Whole genome deoxyribonucleic acid microarray analysis of gene expression in ectopic versus eutopic endometrium. Fertil Steril. 2007;88:1505–33. doi: 10.1016/j.fertnstert.2007.01.056. [DOI] [PubMed] [Google Scholar]
- [22].Su H, Li F, Ranek MJ, Wei N, Wang X. COP9 signalosome regulates autophagosome maturation. Circulation. 2011;124:2117–28. doi: 10.1161/CIRCULATIONAHA.111.048934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Wang X, Su H, Ranek MJ. Protein quality control and degradation in cardiomyocytes. J Mol Cell Cardiol. 2008;45:11–27. doi: 10.1016/j.yjmcc.2008.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Drews O, Tsukamoto O, Liem D, Streicher J, Wang Y, Ping P. Differential regulation of proteasome function in isoproterenol-induced cardiac hypertrophy. Circ Res. 2010;107:1094–101. doi: 10.1161/CIRCRESAHA.110.222364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Cui Z, Scruggs SB, Gilda JE, Ping P, Gomes AV. Regulation of cardiac proteasomes by ubiquitination, SUMOylation, and beyond. J Mol Cell Cardiol. 2014;71:32–42. doi: 10.1016/j.yjmcc.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Gomes AV, Zong C, Edmondson RD, Li X, Stefani E, Zhang J, et al. Mapping the murine cardiac 26S proteasome complexes. Circ Res. 2006;99:362–71. doi: 10.1161/01.RES.0000237386.98506.f7. [DOI] [PubMed] [Google Scholar]
- [27].Zong C, Gomes AV, Drews O, Li X, Young GW, Berhane B, et al. Regulation of murine cardiac 20S proteasomes: role of associating partners. Circ Res. 2006;99:372–80. doi: 10.1161/01.RES.0000237389.40000.02. [DOI] [PubMed] [Google Scholar]
- [28].Ranek MJ, Terpstra EJM, Li J, Kass DA, Wang X. Protein kinase G positively regulates proteasome-mediated degradation of misfolded proteins. Circulation. 2013;128:365–76. doi: 10.1161/CIRCULATIONAHA.113.001971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wang D, Fang C, Zong NC, Liem DA, Cadeiras M, Scruggs SB, et al. Regulation of acetylation restores proteolytic function of diseased myocardium in mouse and human. Mol Cell Proteomics. 2013;12:3793–802. doi: 10.1074/mcp.M113.028332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Meiners S, Dreger H, Fechner M, Bieler S, Rother W, Gunther C, et al. Suppression of cardiomyocyte hypertrophy by inhibition of the ubiquitin-proteasome system. Hypertension. 2008;51:302–8. doi: 10.1161/HYPERTENSIONAHA.107.097816. [DOI] [PubMed] [Google Scholar]
- [31].Wang Y, Sun W, Du B, Miao X, Bai Y, Xin Y, et al. Therapeutic effect of MG-132 on diabetic cardiomyopathy is associated with its suppression of proteasomal activities: roles of Nrf2 and NF-kappaB. Am J Physiol Heart Circ Physiol. 2013;304:H567–78. doi: 10.1152/ajpheart.00650.2012. [DOI] [PubMed] [Google Scholar]
- [32].Ma Y, Chen Y, Yang Y, Chen B, Liu D, Xiong Z, et al. Proteasome inhibition attenuates heart failure during the late stages of pressure overload through alterations in collagen expression. Biochem Pharmacol. 2013;85:223–33. doi: 10.1016/j.bcp.2012.10.025. [DOI] [PubMed] [Google Scholar]
- [33].Li S, Wang X, Li Y, Kost CK, Jr., Martin DS. Bortezomib, a proteasome inhibitor, attenuates angiotensin II-induced hypertension and aortic remodeling in rats. PLoS One. 2013;8:e78564. doi: 10.1371/journal.pone.0078564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Vabulas RM, Hartl FU. Protein synthesis upon acute nutrient restriction relies on proteasome function. Science. 2005;310:1960–3. doi: 10.1126/science.1121925. [DOI] [PubMed] [Google Scholar]
- [35].Hedhli N, Depre C. Proteasome inhibitors and cardiac cell growth. Cardiovasc Res. 2010;85:321–9. doi: 10.1093/cvr/cvp226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Grandin EW, Ky B, Cornell RF, Carver J, Lenihan DJ. Patterns of cardiac toxicity associated with irreversible proteasome inhibition in the treatment of multiple myeloma. J Card Fail. 2015;21:138–44. doi: 10.1016/j.cardfail.2014.11.008. [DOI] [PubMed] [Google Scholar]
- [37].Honton B, Despas F, Dumonteil N, Rouvellat C, Roussel M, Carrie D, et al. Bortezomib and heart failure: case-report and review of the French Pharmacovigilance database. Fundam Clin Pharmacol. 2014;28:349–52. doi: 10.1111/fcp.12039. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.