

Abstract
A gain-of-function mutation (T635A) in the transient receptor potential (TRP) channel TRPC3 results in abnormal channel gating and causes cerebellar ataxia in the dominant Moonwalker (Mwk) mouse mutant. However, the underlying molecular and structural mechanisms are unclear. Here, we used a combined approach of computational modeling and functional characterization of proposed TRPC3 mutants. Our findings support a mechanism by which the hydrogen bonding capability of threonine 635 plays a significant role in maintaining a stable, closed state channel. This capability is lost in the Mwk mutant, suggesting a structural basis for the disease-causing phenotype in the Mwk mouse.
The transient receptor potential (TRP) family of ion channels is a large family of cation channels of diverse functionality. Members of the TRP family have a wide range of sensitivities, including temperature, membrane stretch, and internal Ca2+ concentration, and are involved in numerous physiological functions.1,2 With a wide variety of functions, the N- and C-termini of TRP channels also exhibit a wide variety of structural domains. However, the structural core of the TRP channels consisting of a homotetramer of six transmembrane α-helices, topologically similar to the voltage-gated K+ channels, remains constant throughout all subtypes of the channel family.
Of the seven subfamilies of the TRP channel family, the “canonical” TRPC subfamily is found in the largest variety of species, including the first species in which TRP channels were found, Drosophila melanogaster. The TRPC subfamily has six members, which can be divided into two groups by sequence identity: TRPC1, -4, and -5 and TRPC2, -3, and -6 with 41 and 69% sequence identity, respectively. The physiological and pathological roles of the different TRPC channels are beginning to emerge. Genetic mouse models have revealed a role for TRPC3 in the cerebellum.3−5 Moreover, TRPC3 is part of a key signaling pathway that is affected in cerebellar ataxia in mice and humans,6 and recently, the first functionally pathogenic variant in the human TRPC3 gene was identified in a patient with adult-onset cerebellar ataxia7 (OMIM entry 616410). Interestingly, not only the loss but also a dominant gain-of-function mutation in TRPC3 causes cerebellar ataxia. The ataxic Moonwalker (Mwk) mouse harbors a single nonsynonymous point mutation in exon 7 of the Trpc3 gene, resulting in a threonine-to-alanine amino acid change (T635A according to UniprotKB entry B1ATV3) in the cytoplasmic S4–S5 linker region of the TRPC3 channel protein (Figure 1A) and in altered channel gating.5 The molecular and structural mechanisms by which the Mwk mutation leads to abnormal TRPC3 channel gating remain to be characterized.
Figure 1.

Structural overview and sequence alignments of TRPC3 and the S4–S5 linker region. (A) Schematic of the six-transmembrane TRPC3 channel protein, in which the six transmembrane helices, S1–S6 (numbered cylinders), are preceded by a coiled coil domain (gray box) and an ankyrin repeat domain (black diamonds) and followed by the TRP domain (checkered box), another predicted coiled coil domain (gray box), and a predicted CIRB domain (light gray box). The Mwk mutation (T635A) resides within the S4–S5 linker region and is indicated by a yellow circle. The sequence alignment of the wild-type and mutant mouse TRPC3 S4–S5 linker region is shown below. Figure adapted from ref (6). (B) Sequence alignment of the S4–S5 linker regions of several TRP channels and Kv1.2. The position of the Mwk mutation is highlighted in yellow. Other residues found to have significant effects on channel function when mutated are colored red (see the text for details). (C) Structure of apo TRPV1 (PDB entry 3J5P) with mutants in the S4–S5 linker region found to have significant effects on TRP channel function highlighted with red dots. (D) Sequence conservation diagram of the S4 transmembrane helix and the S4–S5 linker region of Kv and TRP channels showing that the threonine mutated in the Mwk mouse is surprisingly more conserved in Kv channels than in TRP channels. The analysis includes Kv’s 1.x–10.x for a total of 40 sequences analyzed for Kv channels, and all representatives of the TRPC, TRPM, TRPV, TRPA, TRPP, and TRPML subfamilies for a total of 28 sequences analyzed for TRP channels. Figures made using WebLogo and MAFFT sequence alignments of all Kv and TRP channels identified in ref (38).
Interestingly, gain-of-function mutations in the S4–S5 linker region have also been described in other TRP channels, highlighting the importance of this region for TRP channel function. Recently, the mutation of a glycine to a serine nearer to S4 in this region of TRPC4 and TRPC5 was shown to result in constitutively active channels.8 Similarly, a gain-of-function mutant of the yeast TRP channel (YVC1) and diseases linked to mutants of TRPV3 and TRPV4 and TRPML3 have been seen in the same region9−12 (Figure 1B). With the recently published high-resolution structure of TRPV1,13,14 the first of its kind of any TRP channel, it is clearer than ever that this disease-implicated region is in fact the structurally and functionally relevant S4–S5 linker domain that links the pore domain to the other transmembrane helices of the channel (Figure 1C).
In this study, we used a combined approach of computational modeling and functional characterization of proposed mutants to investigate the mechanistic basis of the Mwk mutation in the S4–S5 linker of TRPC3. Previously, a phosphorylation-related mechanism was proposed.5 However, our findings described here favor a mechanism in which the hydrogen bonding capability of residue 635 plays a significant role in maintaining a stable, closed state channel. This capability is lost in the Mwk T635A mutant, thereby providing a structural basis for the gain-of-function disease phenotype in the Mwk mouse.
Experimental Procedures
Plasmids
The FLAG-tagged TRPC3 plasmid has been described previously.5 Point mutations were introduced by site-directed mutagenesis (Agilent Technologies). All constructs were verified by Sanger sequencing. The HA-NFAT1(1–460)-GFP expression construct was obtained from Addgene.
Cell Culture, Transfections, and Cellular Assays
Neuro-2a cells (American Type Culture Collection) were grown under standard conditions. Transfections were performed using Fugene 6 Transfection Reagent (Promega) according to the manufacturer’s instructions. For cell death assays, cells transfected with FLAG-TRPC3 constructs were fixed after 24 h with 4% paraformaldehyde and subjected to indirect immunofluorescence using an antibody to FLAG (1:500, Sigma). Cell survival and death were assessed on the basis of cell integrity and the morphology of the nucleus as determined using the DNA dye DAPI (Vector Laboratories). Cell counts were conducted in a blinded manner, and 50–100 cells were counted per condition. Cells were acquired from three independent experiments. For NFAT translocation assays, cells transfected with FLAG-TRPC3 constructs alongside GFP-tagged NFAT were fixed after 18 h with 4% paraformaldehyde and subjected to indirect immunofluorescence using antibodies to FLAG (1:500, Sigma), GFP (1:500, Clontech), and the DNA dye DAPI (Vector Laboratories). Cytoplasmic and nuclear localization of GFP-NFAT were scored in 50–100 FLAG-positive cells. Cells were acquired from three independent experiments. Data were analyzed using GraphPad Prism.
Biotinylation of Cerebellar Slice Cultures
Cerebellar organotypic slice cultures were prepared as described previously,5 and biotinylation experiments were conducted as described previously15 using Sulfo-NHS-SS-Biotin (Thermo Scientific) and StrepTactin sepharose (GE Healthcare). Protein lysates prepared with RIPA buffer (Thermo Scientific) were subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) followed by immunoblotting with anti-TRPC3 (1:200, Alomone) and anti-Actin (1:1000, Abcam) antibodies. Antibody binding was detected by enhanced chemoluminescence (ECL; GE Healthcare).
Homology Modeling
The S1–S6 regions of mouse TRPC3 and rat TRPV1 have a sequence identity of only ∼19%, despite both being in the TRP channel family. Additional methods were therefore employed to ensure an optimal sequence alignment, such as transmembrane helix prediction, disorder prediction, and glycosylation site data. Transmembrane helix prediction was performed with TMHMM,16 GlobPlot,17 PSIPRED,18 and Jpred19 to aid confirmation of helix placements in alignments with the TRPV1 sequence. In initial alignments, the S1 and S4–S6 alignments were consistent and robust to changes in alignment methods and to slight variations in sequence. In contrast, the S2–S4 alignment was less robust. Thus, even in the final alignment, the S2 helix included some gaps. However, this is unlikely to have a major influence on our results, as our primary focus is on the S4–S5 linker and S6 helix regions of the protein. To inform our alignment, previously published data on the placement of extracellular and intracellular loops from glycosylation site data were used.20 Additionally, both the Regional Order Neural Network21 (RONN, http://www.strubi.ox.ac.uk/RONN) and disEMBL22 (http://dis.embl.de) servers predicted a sequence of Q568–P578 between the S3 and S4 helices of mouse TRPC3 to be disordered, informing the removal of this region (continuing to P593) from the final MAFFT23 alignment used to build the homology model on the rat TRPV1 structure.
In total, five templates were used in homology modeling: the Kv 1.2/2.1 chimera structure (PDB entry 2R9R), the TRPV1 apo structure (PDB entry 3J5P), the TRPV1 apo structure (PDB entry 3J5P) with the Kv 1.2/2.1 channel chimera structure for the S4–S5 linker, the TRPV1 DkTx- and RTX-bound structure (PDB entry 3J5Q) with the Kv 1.2/2.1 channel chimera structure for the S4–S5 linker, and the TRPV1 capsaicin-bound structure (PDB entry 3J5R) with the Kv 1.2/2.1 channel chimera structure for the S4–S5 linker. One hundred models of mTRPC3 were made with MODELLER version 9.1024 on each of these templates (generating a total of 500 models). All of these models were made using the same sequence alignment, and none of them included the Q568–P593 region. Modeling required the use of both the multichain and the multitemplate protocols in MODELLER, allowing all four chains of the homotetrameric mTRPC3 to have similar conformations through symmetry restraints. The top models according to the MODELLER scoring function were chosen to examine a possible mechanistic basis for the Mwk gain-of-function phenotype. Potential hydrogen bonding partners were determined after adding hydrogens and according to the Baker–Hubbard definition25 as implemented in MDTraj.26 Final model quality was assessed with PROCHECK.27 Additional assessment of the localization and orientation of the S4–S5 in the bilayer was conducted via insertion of the top atomistic models into a coarse-grained template using CG2AT.28 The template was created via a 100-ns coarse-grained self-assembly simulation, in which a POPC bilayer was allowed to form around a coarse-grained model of the mTRPC3 using the MARTINI force field29 within Gromacs version 4.5.5.30
Results
Similar TRPC3 Localization and Calcium Influx in Wild-Type and Mwk Neuronal Cells
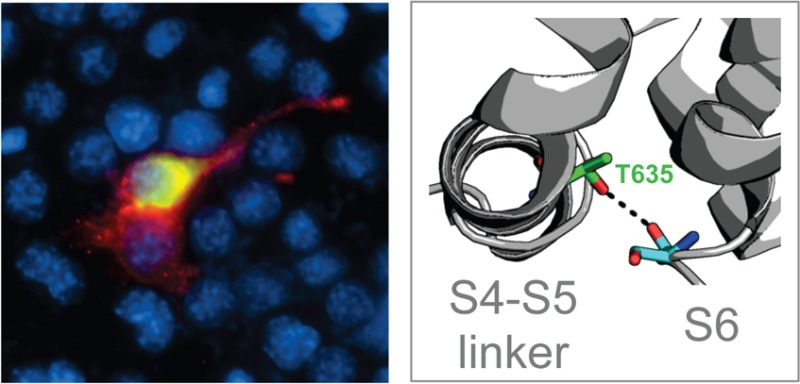
The mutated Mwk TRPC3 channel is expressed at levels similar to that of wild-type TRPC3 in the mouse cerebellum.5 Furthermore, we found in biotinylation experiments that equal amounts of TRPC3 were localized to the cell surface in cerebellar slice cultures from both wild-type and Mwk mice (Figure 2A), suggesting that the Mwk mutation does not alter the normal expression pattern of TRPC3 at the plasma membrane in Purkinje cells of the cerebellum. Electrophysiological recordings have shown that Mwk Purkinje cells are profoundly impaired in their intrinsic and evoked electrophysiological properties consistent with a gain-of-function mutation in TRPC3.3,5 Moreover, Mwk (T635A) but not wild-type TRPC3 robustly induced cell death upon overexpression in mouse neuronal cell lines, including NSC-345 and Neuro-2a cells (Figure 2B). This is consistent with the Mwk mutation being a gain-of-function mutation, which renders the channel in a more open conformation, thereby allowing nonrestricted influx of calcium into the transfected cells causing cell death. To demonstrate that the overexpression of mutant TRPC3 increases calcium influx, we co-expressed a GFP-tagged version of the calcium-sensitive transcription factor NFAT alongside TRPC3. Under normal conditions, inactive NFAT is phosphorylated and resides in the cytoplasm; however, upon calcium influx, NFAT is dephosphorylated by calcineurin and translocates to the nucleus where it activates gene expression.31 Indeed, we found that overexpression of mutant but not wild-type TRPC3 significantly increased the nuclear localization of GFP-tagged NFAT, indicative of increased calcium signaling downstream of mutant TRPC3 (Figure 2C,D). Consistent with these in vitro findings, increased calcium signaling is also evident in vivo in the Mwk cerebellum.32
Figure 2.

The gain-of-function Mwk mutation in TRPC3 causes increased cell death and calcium signaling in neuronal cells. (A) Equal amounts of TRPC3 are expressed at the cell surface in the mouse cerebellum. Biotinylated cerebellar slice cultures from wild-type (WT) and Mwk mice were lysed and subjected to pull-down experiments using streptavidin beads, followed by immunoblotting for TRPC3 and actin. Abbreviations: I, input; S, supernatant; B, pellet (biotinylated fraction). (B) Overexpression of Mwk (T635A) but not wild-type (WT) TRPC3 significantly induced cell death in mouse Neuro-2a cells (mean ± SEM; n = 3; ANOVA followed by Bonferroni’s post hoc test; p < 0.005). V denotes vector control. (C and D) Overexpression of Mwk (T635A) but not wild-type (WT) TRPC3 causes significant nuclear localization of co-expressed GFP-tagged NFAT (mean ± SEM; n = 3; ANOVA followed by Bonferroni’s post hoc test; p < 0.001). Cells were fixed 24 h after transfection and subjected to indirect immunofluorescence using antibodies against FLAG, GFP, and the DNA dye DAPI. Cells transfected with mutant TRPC3 have a more rounded appearance because of their impending cell death. (E) The in vitro gain-of-function phenotype of Mwk TRPC3 is not likely to be mediated by phosphorylation. Overexpression of the phospho-mimic T635D mutation but also of the control T635N mutation did not induce cell death in mouse Neuro-2a cells (mean ± SEM; n = 3; ANOVA followed by Bonferroni’s post hoc test). (F) Overexpression of the phospho-mimic T635D mutation but also of the control T635N mutation did not cause significant nuclear translocation of GFP-NFAT (mean ± SEM; n = 3; ANOVA followed by Bonferroni’s post hoc test).
Functional Characterization of TRPC3 Phosphometic Residues
One proposed explanation for the abnormal TRPC3 channel gating observed in the Mwk mouse was the loss of an inhibitory phosphorylation by protein kinase C γ (PKCγ).5 PKCγ has been shown to inhibit TRPC3 channel activity in overexpression experiments in heterologous cell lines.33−36 Moreover, the mutated threonine 635 in Mwk TRPC3 was shown to be phosphorylated by PKCγ in an in vitro kinase assay.5 To assess the possible effect of phosphorylation at threonine 635 on cell survival and calcium signaling, we expressed a TRPC3 mutant in which this threonine was replaced with aspartate (T635D) to mimic phosphorylation at this site. We found that overexpression of the TRPC3 T635D mutant did not cause cell death or an increased level of calcium signaling, consistent with the hypothesis that phosphorylation at this site is inhibitory (Figure 2E,F). However, to our surprise, we also did not detect any effect on cell death or calcium signaling when overexpressing a TRPC3 mutant, in which threonine 635 was replaced with asparagine (T635N), which is neither a phospho-mimic nor itself phosphorylated (Figure 2E,F). We therefore sought an alternative explanation for the observed functional consequences of the TRPC3 Mwk mutation.
TRPV1 and Kv Channel Structures Provide Initial Clues
The striking sequence identity between the S4–S5 linker region of mouse TRPC3 and that of Kv channels encouraged us to examine a wide variety of structural and functional data available for the transmembrane region of Kv channels. Indeed, the corresponding threonine of the Kv1.2/2.1 chimera structure (PDB entry 2R9R, a proposed open state structure) points inward toward the S6 helix, suggestive of a key role in determining channel open state probability. However, in Kv channels, it is thought that the threonine corresponding to the mTRPC3 T635 (T320 according to UniprotKB entry P63142) fits inside a groove in the S6 helix along with a conserved isoleucine.37 The same threonine to alanine mutation that induces the Mwk phenotype in TRPC3 produces faster desensitizing Kv channels. This suggests that the alanine mutation stabilizes the closed state in Kv channels, instead of destabilizing it as in mouse TRPC3, hinting that the mechanism may not be conserved.
Previously, a number of structures of voltage-gated potassium (Kv) channels were used as templates for modeling studies aimed at understanding TRPC channel structure–function relationships. While the overall sequence identity of Kv and TRP channels is poor,38 the S4–S5 linker region is surprisingly similar between TRPCs and Kv channels (Figure 1B). Even more striking is the fact that the threonine mutated in the Mwk mouse is more highly conserved in Kv channels than in TRP channels (Figure 1D). However, while Kv channel structures provide a good template for the S4–S5 linker, the pore-lining S6 helices of Kv and TRP channels differ substantially. For example, the Kv channel has the PVP motif, which causes a functionally significant kink in the helix, but this is not present in TRP channels. The recently determined high-resolution structures of TRP channels make the role of the S4–S5 linker in TRP channels clearer.13,14,39 In addition, the TRPV1 structure provided the first confirmation that the proposed homology to the Kv channel structure was in fact correct: despite a low sequence identity, the structural similarity between the TRPV1 structure and previous structures of voltage-gated channels is significant (∼1 Å root-mean-square deviation for the tetrameric transmembrane domain). Notably, the intracellular portion of the channel, especially the region of S6 near the S4–S5 linker, is crucial to gating. This can be seen explicitly in the conformational difference between the ligand-bound (open) and apo (closed) states of the recently determined structures of TRPV1.14
Modeling the Mwk TRPC3 Channel
With this evidence in hand, the Mwk mutation in the S4–S5 linker could disturb intramolecular interactions within TRPC3 that are important for the gating mechanism of the channel. To test this hypothesis, we built homology models of the mouse TRPC3 transmembrane region based on available high-resolution structures of homotetrameric 6TM ion channels (see Experimental Procedures). First, the high-resolution apo structure of TRPV1 was used on its own, and then the Kv 1.2/2.1 chimera structure was also included. A multitemplate combination consisting mostly of the apo TRPV1 structure, but including the linker region of the Kv channel chimera structure, was then devised. The full tetrameric transmembrane domain, including the TRP box motif, was modeled. The final sequence identity was 17% to the Kv 1.2/2.1 chimera sequence and 21% to the TRPV1 sequence (Figure 3A,B).
Figure 3.

Homology modeling reveals potential for hydrogen bonding. (A) Sequence alignment of mouse TRPC3 to rat TRPV1 and to the S4–S5 linker of the Kv 1.2/2.1 chimera used to create the homology models. A cut of 21 amino acids (indicated by double lines) was made at the end of S3 to facilitate modeling. Transmembrane helices are indicated by black boxes with the corresponding label for each S1–S6 transmembrane helix above. Coloring is according to the Clustal coloring scheme. The TRPC3 Mwk mutation in the S4–S5 linker region is indicated by a double-lined box. Arrows indicate residues highlighted in other panels of the figure. Image made with JalView. (B) Top-ranked (according to MODELLER score) mouse TRPC3 homology model on the TRPV1 apo structure (PDB entry 3J5P) with the Kv 1.2/2.1 channel chimera structure for the S4–S5 linker shown from a top view and a side view. Coloring indicates separate chains of the tetrameric channel. The S1–S4 helices of the purple chain have been removed for the sake of clarity in the side view figure. (C) Homology models of TRPC3 indicate that the threonine 635 (in green) mutated in the Mwk mouse has the potential for hydrogen bonding with the end of helix S6. The first quadrant shows an overlap of four models and the resulting T635 orientation. The remaining quadrants show three possible hydrogen bonding partners: S735 (cyan), Y736 (magenta), and the adjacent R634 (yellow). (D) Overexpression of the TRPC3 mutants Mwk (T635A) and T635V significantly induced cell death in mouse Neuro-2a cells (mean ± SEM; n = 3; ANOVA followed by the Newman–Keuls post hoc test; p < 0.001 and p < 0.05). (E) Overexpression of the TRPC3 mutants Mwk (T635A) and T635V caused significant nuclear translocation of GFP-NFAT (mean ± SEM; n = 3; ANOVA followed by the Newman–Keuls post hoc test; p < 0.0001). (F) Overexpression of the TRPC3 mutants S735A and Y736F did not induce cell death upon overexpression in Neuro-2a cells (mean ± SEM; n = 3; ANOVA followed by the Newman–Keuls post hoc test). See also Figure S2 of the Supporting Information. (G) Other possible hydrogen bonding partners on the S6 helix (indicated as wheat mesh) were not seen as being viable for experimental validation as they pointed away from the site in question. Building homology models using the capsaicin-bound open state TRPV1 structure, however, indicated a further possible hydrogen bonding interaction with N726 (red) on helix S6. This interaction is with an adjacent subunit, and not the same subunit as with residues predicted with models based on the apo TRPV1. N726 is 10 residues toward the center of the membrane along S6 compared to other residues suspected to interact with T635.
The resulting models were broadly reasonable (Figure S1 of the Supporting Information) and, upon their insertion into a POPC bilayer of the S4–S5 helix, proved to be amphipathic and parallel to the bilayer plane, while residue T635 was not in a position likely to interact with lipids. In all of the resulting models of mTRPC3, the T635 residue in the S4–S5 linker pointed inward, toward the pore domain, and in some models within the ensemble, hydrogen bonds to residues at the end of helix S6 were seen (Figure 3C). These observations were consistent with the experimental results that showed that replacing threonine 635 with the polar amino acid aspartate or asparagine produced the wild-type phenotype (Figure 2), suggesting that the wild-type phenotype was propagated by the hydrogen bonding capabilities of all three of these amino acids.
Evidence of Hydrogen Bonding and Search for a Partner
To further test whether a substitution that would abrogate the hydrogen bonding would behave like the Mwk mutation, we replaced the threonine residue at position 635 with valine (T635V), a nonpolar amino acid that is isosteric with threonine. Upon overexpression in neuronal cells, the TRPC3 T635V mutant caused increased calcium influx and cell death, similar to the Mwk TRPC3 mutant T635A (Figure 3D,E). Although overall the cellular phenotype was not as strong as for the Mwk mutation, this result supports the suggestion that the loss of a critical hydrogen bond contributes to the Mwk gain-of-function phenotype.
Once this phenotype was established, we undertook further investigation of the specific hydrogen bonds seen in the models. Indeed, serine 735 and tyrosine 736 at the end of helix S6 emerged as possible hydrogen bonding partners (Figure 3C, full description available in Tables S1–S3 of the Supporting Information). However, some indication of possible hydrogen bonds with backbone carbonyls at this end of S6 was also seen, but only in initial models based on the Kv 1.2/2.1 chimera structure (Table S3 of the Supporting Information). Moreover, as this type of interaction is less tractable to address via mutagenesis experiments, this initial finding was not pursued experimentally.
To experimentally address the viability of the proposed hydrogen bonding partners, tyrosine 736 was mutated to phenylalanine (Y736F) and subjected to cell death assays in neuronal cells. However, overexpression of the TRPC3 Y736F mutant did not result in a Mwk-like phenotype in these assays (Figure 3F). Similarly, serine 735 was replaced with nonpolar residues (alanine, valine, or leucine) and overexpressed in neuronal cells (Figure 3F and Figure S2 of the Supporting Information). These nonpolar mutants behaved like wild-type TRPC3 and did not mimic the gain-of-function Mwk phenotype in vitro. These results indicate that neither of the proposed residues’ side chains interacts as a hydrogen bonding partner with T635. As an alternative explanation, the channel might be able to compensate for the loss of hydrogen bonding.
While S735 and Y736 were immediate candidates, the other possible hydrogen bonding side chains along S6 were excluded because of their location on the wrong side of the helix to facilitate interactions (Figure 3G). This encourages a view that, as seen in some models, the threonine could be hydrogen bonding to a backbone carbonyl to stabilize the closed state channel in this region of S6. However, upon construction of models of mouse TRPC3 based on the two ligand-bound structures of TRPV1, an unexpected tentative interaction with N726 on the S6 helix, which is 10 residues closer to the center of the membrane, was seen with an adjacent subunit, rather than of the same subunit as had been seen for the previous potential partners discussed. However, this was seen only upon visual inspection and not supported by more rigorous Baker–Hubbard determination of hydrogen bonding (see the Supporting Information). Alternatively, it is possible that the T635 hydrogen bond partner sought is to other residues on the S4–S5 linker, which was seen in models but not investigated experimentally because they were not expected to result in the large effect seen in the Mwk mouse (Figure 3C).
Discussion
In this study, through a combination of modeling and functional experiments, we have investigated a possible mechanistic basis for the Mwk mouse disease phenotype, which is caused by the mutation of TRPC3’s threonine 635 to an alanine (T635A). We have shown that the Mwk gain-of-function phenotype most likely arises because of the loss of the hydrogen bonding capabilities of threonine 635. Together, our findings suggest that the ataxic phenotype in the Mwk mouse is the result of the loss of this hydrogen bond, thereby destabilizing the closed state TRPC3 and resulting in a constitutively active channel.
While it is not clear currently which residue forms a hydrogen bonding interaction with threonine 635, our studies have narrowed the possibilities for these interactions. The possibilities of S735 and Y736 presented by homology modeling were investigated experimentally with negative results. This leaves four possibilities remaining: (1) One of these is a hydrogen bonding partner, but the channel has some way of compensating for its loss. (2) The T635 side chain hydrogen bonds to a backbone carbonyl not tractable to investigate via simple mutagenesis experiments. (3) The hydrogen bonding partner is adjacent on the S4–S5 linker and was not investigated experimentally. (4) Hydrogen bonding is in fact with an adjacent subunit with a residue such as N726. Alternatively, it is conceivable if somewhat unlikely that the mTRPC3 channel adopts a conformation in this region very different from that seen in the TRPV1 and Kv structures that were used as templates.
Because the threonine to valine mutant (T635V) recapitulated the Mwk phenotype to a lesser extent than the Mwk threonine to alanine mutant (T635A) upon overexpression in neuronal cells, it is possible that there are other steric interactions at play that help to stabilize the closed state channel further. This supports the proposal that removing the hydrogen bonding partner is not entirely sufficient to reproduce the Mwk phenotype, as such a mutation could leave these steric interactions intact and other compensatory mechanisms might then result in no significant change in phenotype.
In looking at models of mTRPC3, we were most interested in hydrogen bonding that might stabilize the closed state structures through hydrogen bonding to T635; therefore, the apo TRPV1 structure was the focus as a model template. However, models based on the ligand-bound TRPV1 structures resulted in a new suggestion (N726) as a possible interaction partner for T635. Because the sequences and functional properties of TRPV1 and TRPC3 are quite different, it is possible that the open state structures of TRPV1 and TRPC3 do not correspond. Additionally, the interaction with N726 is seen for the TRPC3 model on the TRPV1 capsaicin-bound structure, which has an outer pore more closed than that of the RTX/DkTx-bound structure. Thus, this model could indeed correspond to a closed state TRPC3, indicating a real possibility that N726 is a viable bonding partner.
Also of note is the peculiar sequence identity between the S4–S5 linker of TRPC3 and Kv channels. This similarity adds another interesting feature to the question of Kv–TRP structure–function similarities, as it is still unclear if the similar topology of voltage-gated channels and TRP channels has functional significance. Very few sequences of the high identity of this S4–S5 linker region have previously been noted between TRP and voltage-gated channels. While this similarity in the S4–S5 linker is remarkable on its own, it also promotes our confidence in an S6 helix facing T635 despite a low overall sequence identity. Additionally, the high percentage identity at the end of helix S6 between rTRPV1 and mTRPC3 encourages our analysis here. In hypothesizing that T635 of the S4–S5 linker of mTRPC3 plays a significant functional role, we are not positing that the S1–S4 linker necessarily also has a strong functional role, as it was seen in the TRPV1 structures that its movement is minimal upon ligand binding and channel opening. In fact, we suggest that the S4–S5 linker plays a role in stabilizing the S6 helix, which the side chain of residue T635 seems to face, even upon placement in a bilayer.
Together, these studies have resulted in further insights into a likely mechanism of the Mwk mouse phenotype. However, further refinement of the homology models, including more extensive membrane-embedded simulations, could help in the more robust exploration of the conformational stability and dynamics of these models. It would also be of possible interest to include the recent TRPA1 channel structure as an additional template, and possibly to compare the output of membrane protein-specific tools such as MEDELLER40 and/or Rosetta Membrane41 with those of the used MODELLER. Additionally, a more exhaustive alanine scan of the region of the protein identified in this study and/or the use of non-natural amino acids in mutations might yield further insights into a definitive hydrogen bonding for the T635 residue of the TRPC3 channel. Ultimately, the future crystallization of additional TRP channels including TRPC3 should provide a better template for modeling the functional effect of the Mwk mutation and other ataxia-causing TRPC3 mutations.
Overall, it remains clear from the current studies that understanding the structural–functional significance of the S4–S5 linker of TRP channels has important implications to understanding the mechanistic basis of certain diseases. For the first time, with access to structural information through the recent high-resolution TRP channel structures, we are able to hypothesize more clearly about this mechanistic basis, at least in the case of the Mwk mouse model of cerebellar ataxia. Mechanistic insight like this might help in the development of targeted treatments to ion channels including TRPC3 for cerebellar ataxia in the future.
Acknowledgments
We thank Dr. Matthias Schmidt and Dr. Phillip J. Stansfeld for help with parallelizing homology model building and CG2AT scripts, respectively.
Glossary
Abbreviations
- TRP
transient receptor potential
- Mwk
Moonwalker
- Kv channel
voltage-gated potassium channel
- PKCγ
protein kinase C γ
- SEM
standard error of the mean
- ANOVA
analysis of variance
- PDB
Protein Data Bank.
Supporting Information Available
Tables with the frequency of any hydrogen bonds involving the hydroxyl of T635 as seen in all homology models, Ramachandran plots illustrating the reasonableness of homology models, experimental cell death results in Neuro-2a cells upon overexpression of mTRPC3 mutants S735L and S735V, and PDB files of six relevant homology models. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.5b00235.
Author Present Address
∥ S.M.H.: Computational Biology Program, Memorial Sloan Kettering Cancer Center, New York, New York 10065.
E.B.E.B. is the recipient of a Royal Society Research Fellowship. S.M.H. was supported by the NIH-Oxford Scholars Program during much of this work. Research in M.S.P.S.'s group is supported by The Wellcome Trust.
The authors declare no competing financial interest.
Supplementary Material
References
- Venkatachalam K.; Montell C. (2007) TRP channels. Annu. Rev. Biochem. 76, 387–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey I. S.; Delling M.; Clapham D. E. (2006) An introduction to TRP channels. Annu. Rev. Physiol. 68, 619–647. [DOI] [PubMed] [Google Scholar]
- Sekerková G.; Kim J.-A.; Nigro M. J.; Becker E. B. E.; Hartmann J.; Birnbaumer L.; Mugnaini E.; Martina M. (2013) Early onset of ataxia in moonwalker mice is accompanied by complete ablation of type II unipolar brush cells and Purkinje cell dysfunction. J. Neurosci. 33, 19689–19694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann J.; Dragicevic E.; Adelsberger H.; Henning H. A.; Sumser M.; Abramowitz J.; Blum R.; Dietrich A.; Freichel M.; Flockerzi V.; Birnbaumer L.; Konnerth A. (2008) TRPC3 channels are required for synaptic transmission and motor coordination. Neuron 59, 392–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker E. B. E.; Oliver P. L.; Glitsch M. D.; Banks G. T.; Achilli F.; Hardy A.; Nolan P. M.; Fisher E. M. C.; Davies K. E. (2009) A point mutation in TRPC3 causes abnormal Purkinje cell development and cerebellar ataxia in moonwalker mice. Proc. Natl. Acad. Sci. U.S.A. 106, 6706–6711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker E. B. E. (2014) The moonwalker mouse: New insights into TRPC3 function, cerebellar development, and ataxia. Cerebellum 13, 628–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogel B. L.; Hanson S. M.; Becker E. B. E. (2015) Do mutations in the murine ataxia gene TRPC3 cause cerebellar ataxia in humans?. Mov. Disord. 30, 284–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck A.; Speicher T.; Stoerger C.; Sell T.; Dettmer V.; Jusoh S. A.; Abdulmughni A.; Cavalié A.; Philipp S. E.; Zhu M. X.; Helms V.; Wissenbach U.; Flockerzi V. (2013) Conserved gating elements in TRPC4 and TRPC5 channels. J. Biol. Chem. 288, 19471–19483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H.; Delling M.; Li L.; Dong X.; Clapham D. E. (2007) Activating mutation in a mucolipin transient receptor potential channel leads to melanocyte loss in varitint-waddler mice. Proc. Natl. Acad. Sci. U.S.A. 104, 18321–18326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Z.; Zhou X.; Haynes W. J.; Loukin S. H.; Anishkin A.; Saimi Y.; Kung C. (2007) Yeast gain-of-function mutations reveal structure function relationships conserved among different subfamilies of transient receptor potential channels. Proc. Natl. Acad. Sci. U.S.A. 49, 19607–19612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Z.; Chen Q.; Lee M.; Cao X.; Zhang J.; Ma D.; Chen L.; Hu X.; Wang H.; Wang X.; Zhang P.; Liu X.; Guan L.; Tang Y.; Yang H.; Tu P.; Bu D.; Zhu X.; Wang K.; Li R.; Yang Y. (2012) Exome sequencing reveals mutations in TRPV3 as a cause of Olmsted syndrome. Am. J. Hum. Genet. 90, 558–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock M. J.; Prenen J.; Funari V. A.; Funari T. L.; Merriman B.; Nelson S. F.; Lachman R. S.; Wilcox W. R.; Reyno S.; Quadrelli R.; Vaglio A.; Owsianik G.; Janssens A.; Voets T.; Ikegawa S.; Nagai T.; Rimoin D. L.; Nilius B.; Cohn D. H. (2008) Gain-of-function mutations in TRPV4 cause autosomal dominant brachyolmia. Nat. Genet. 40, 999–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao M.; Cao E.; Julius D.; Cheng Y. (2013) Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature 504, 107–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao E.; Liao M.; Cheng Y.; Julius D. (2013) TRPV1 structures in distinct conformations reveal activation mechanisms. Nature 504, 113–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu C.; Jensen K.; Sokolova I.; Wang D.; Li M.; Deshpande P.; Davidson N.; Mody I.; Quick M. W.; Quake S. R.; Lester H. A. (2002) Number, density, and surface/cytoplasmic distribution of GABA transporters at presynaptic structures of knock-in mice carrying GABA transporter subtype 1–green fluorescent protein fusions. J. Neurosci. 22, 10251–10266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh A.; Larsson B.; von Heijne G.; Sonnhammer E. L. (2001) Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 305, 567–580. [DOI] [PubMed] [Google Scholar]
- Linding R. (2003) GlobPlot: Exploring protein sequences for globularity and disorder. Nucleic Acids Res. 31, 3701–3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones D. T. (1999) Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 292, 195–202. [DOI] [PubMed] [Google Scholar]
- Cole C.; Barber J. D.; Barton G. J. (2008) The Jpred 3 secondary structure prediction server. Nucleic Acids Res. 36, W197–W201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vannier B.; Zhu X.; Brown D.; Birnbaumer L. (1998) The Membrane Topology of Human Transient Receptor Potential 3 as Inferred from Glycosylation-scanning Mutagenesis and Epitope Immunocytochemistry. J. Biol. Chem. 273, 8675–8679. [DOI] [PubMed] [Google Scholar]
- Yang Z. R.; Thomson R.; McNeil P.; Esnouf R. M. (2005) RONN: The bio-basis function neural network technique applied to the detection of natively disordered regions in proteins. Bioinformatics 21, 3369–3376. [DOI] [PubMed] [Google Scholar]
- Linding R.; Jensen L. J.; Diella F.; Bork P.; Gibson T. J.; Russell R. B. (2003) Protein disorder prediction: Implications for structural proteomics. Structure 11, 1453–1459. [DOI] [PubMed] [Google Scholar]
- Katoh K.; Misawa K.; Kuma K.; Miyata T. (2002) MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiser A.; Sali A. (2003) Modeller: Generation and refinement of homology-based protein structure models. Methods Enzymol. 374, 461–491. [DOI] [PubMed] [Google Scholar]
- Baker E. N.; Hubbard R. E. (1984) Hydrogen Bonding in Globular Proteins. Prog. Biophys. Mol. Biol. 44, 97–179. [DOI] [PubMed] [Google Scholar]
- Mcgibbon R. T.; Beauchamp K. A.; Schwantes C. R.; Wang L.; Hern C. X.; Herrigan M. P.; Lane T. J.; Swails J. M.; Pande V. S. (2014) MDTraj: A modern, open library for the analysis of molecular dynamics trajectories. bioRxiv 10.1101/008896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski R. A.; MacArthur M. W.; Moss D. S.; Thornton J. M. (1993) PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26, 283–291. [Google Scholar]
- Stansfeld P. J.; Sansom M. S. P. (2011) From Coarse Grained to Atomistic: A Serial Multiscale Approach to Membrane Protein Simulations. J. Chem. Theory Comput. 7, 1157–1166. [DOI] [PubMed] [Google Scholar]
- Marrink S. J.; Risselada H. J.; Yefimov S.; Tieleman D. P.; de Vries A. H. (2007) The MARTINI force field: Coarse grained model for biomolecular simulations. J. Phys. Chem. B 111, 7812–7824. [DOI] [PubMed] [Google Scholar]
- Hess B.; Kutzner C.; van der Spoel D.; Lindahl E. (2008) GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 4, 435–447. [DOI] [PubMed] [Google Scholar]
- Hogan P. G.; Chen L.; Nardone J.; Rao A. (2003) Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 17, 2205–2232. [DOI] [PubMed] [Google Scholar]
- Dulneva A.; Lee S.; Oliver P. L.; Di Gleria K.; Kessler B. M.; Davies K. E.; Becker E. B. E. (2015) The mutant Moonwalker TRPC3 channel links calcium signaling to lipid metabolism in the developing cerebellum. Hum. Mol. Genet. 24, 4114–4125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatachalam K.; Zheng F.; Gill D. L. (2003) Regulation of canonical transient receptor potential (TRPC) channel function by diacylglycerol and protein kinase C. J. Biol. Chem. 278, 29031–29040. [DOI] [PubMed] [Google Scholar]
- Poteser M.; Schleifer H.; Lichtenegger M.; Schernthaner M.; Stockner T.; Kappe C. O.; Glasnov T. N.; Romanin C.; Groschner K. (2011) PKC-dependent coupling of calcium permeation through transient receptor potential canonical 3 (TRPC3) to calcineurin signaling in HL-1 myocytes. Proc. Natl. Acad. Sci. U.S.A. 108, 10556–10561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trebak M.; Hempel N.; Wedel B. J.; Smyth J. T.; Bird G. S. J.; Putney J. W. (2005) Negative regulation of TRPC3 channels by protein kinase C-mediated phosphorylation of serine 712. Mol. Pharmacol. 67, 558–563. [DOI] [PubMed] [Google Scholar]
- Adachi N.; Kobayashi T.; Takahashi H.; Kawasaki T.; Shirai Y.; Ueyama T.; Matsuda T.; Seki T.; Sakai N.; Saito N. (2008) Enzymological analysis of mutant protein kinase Cγ causing spinocerebellar ataxia type 14 and dysfunction in Ca2+ homeostasis. J. Biol. Chem. 283, 19854–19863. [DOI] [PubMed] [Google Scholar]
- Labro A. J.; Raes A. L.; Grottesi A.; Van Hoorick D.; Sansom M. S. P.; Snyders D. J. (2008) Kv channel gating requires a compatible S4-S5 linker and bottom part of S6, constrained by non-interacting residues. J. Gen. Physiol. 132, 667–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu F. H.; Catterall W. A. (2004) The VGL-chanome: A protein superfamily specialized for electrical signaling and ionic homeostasis. Sci. STKE 2004, re15. [DOI] [PubMed] [Google Scholar]
- Paulsen C. E.; Armache J.-P.; Gao Y.; Cheng Y.; Julius D. (2015) Structure of the TRPA1 ion channel suggests regulatory mechanisms. Nature 520, 511–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelm S.; Shi J.; Deane C. M. (2010) MEDELLER: Homology-based coordinate generation for membrane proteins. Bioinformatics 26, 2833–2840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarov-Yarovoy V.; Schonbrun J.; Baker D. (2006) Multipass membrane protein structure prediction using Rosetta. Proteins 62, 1010–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.