

Abstract
Cellular heparan sulfate (HS) has a dual role in scrapie pathogenesis; it is required for PrPSc (scrapie prion protein) formation and facilitates infection of cells, mediating cellular uptake of prions. We examined the involvement of heparanase, a mammalian endoglycosidase degrading HS, in scrapie infection. In cultured cells, heparanase treatment or over-expression resulted in a profound decrease in PrPSc. Moreover, disease onset and progression were dramatically delayed in scrapie infected transgenic mice over-expressing heparanase. Together, our results provide direct in vivo evidence for the involvement of intact HS in the pathogenesis of prion disease and the protective role of heparanase both in terms of susceptibility to infection and disease progression.
Keywords: Prion disease, heparanase, heparan sulfate, scrapie prion protein, transgenic mice, survival time
Introduction
Several amyloid diseases exist and their classification is based on the specific protein that makes up the amyloid fibril. Hitherto, more than 28 proteins have been identified to be able to form local or systemic amyloidosis in human [1,2]. Beside the amyloid-specific protein, other components such as serum amyloid P (SAP) and proteoglycans (PGs) are present in amyloid deposits, where both glycosaminoglycans (GAGs) and core proteins have been identified [3,4,5]. Heparan sulfate (HS) dominates as the most frequent encountered GAG in amyloid deposits [6,7,8,9,10], occurring as cell membrane associated syndecan and glypican [11], and perlecan and agrin that constitute major components of the extracellular matrix (ECM) and basement membrane [10]. The function for HS in amyloidogenesis is not entirely understood, but accumulated information points to an important role during initiation of amyloid formation [12]. We have generated heparanase over-expressing transgenic (Hpa-tg) mice [13] and demonstrated that in a mouse model of AA-amyloidosis, organ specific differences in human heparanase overexpression coincided with development of amyloids [14]. Thus, liver and kidney with high levels of heparanase showed little or no amyloid depositions whereas the spleen without heparanase expression displayed extensive deposits [14]. The hypothesis behind this finding is that the shorter fragments of HS produced in the liver of Hpa-tg mice due to extensive degradation by heparanase [13,14], failed to form complex with serum amyloid A, precluding its aggregation. Heparanase is the sole mammalian endoglycosidase that specifically cleaves HS chains [15,16], leading to reduced length of cell surface-bound and ECM associated HS. It is a major protagonist in pathophysiological settings such as cancer, inflammation, diabetic nephropathy, diabetes, atherosclerosis and other pathologies [15,16,17,18]. The protective role of heparanase in AA-amyloidosis [14] led us to investigate its effect on the onset and progression of prion disease. The transmissible spongiform encephalopathies that comprise infectious, familial and sporadic neurodegenerations such as Creutzfeldt-Jakob disease of humans, scrapie of sheep, and bovine spongiform encephalopathy are caused by prions [19,20]. These proteinaceous agents propagate by refolding the normal cell surface glycoprotein of the host, the cellular prion protein (PrPC), into an abnormal β-sheet-rich conformer PrP-scrapie (PrPSc) [19,21]. We have previously shown that cellular HS serves both as a cofactor in the propagation of PrPSc and as receptor for purified prion rods [22,23]. Notably, various sulfated glycans reduce the formation of PrPSc in infected cells and in some cases prolong the incubation time of experimental prion diseases [24,25,26]. In the present study, we investigated whether heparanase can affect PrPSc in vitro and disease progression in vivo. Our results indicate that heparanase treatment or over-expression resulted in a profound decrease in PrPSc. Moreover, disease onset and progression were dramatically delayed in scrapie infected transgenic mice over-expressing heparanase.
Materials and methods
Proteins
Recombinant 50+8 kDa active heparanase was kindly provided by Dr. Hua-Quan Miao (ImClone systems Inc., New York, NY) [27]. Latent 65 kDa heparanase was expressed in transfected CHO cells and purified as described [28].
Cells
Mouse neuroblastoma ScN2a-M are ScN2a cells that stably express the chimeric mouse/Syrian hamster MHM2 PrP that reacts with mAb 3F4 [22,23,29,30]. An uninfected version (N2a-M) was obtained by curing ScN2a-M cells with pentosan polysulfate (PPS, 5 μg/ml, 5 days) [31,32] and subsequently maintaining them without inhibitors for at least 1 month prior to use. ScGT1–1 cells are mouse hypothalamus cells infected with mouse RML prions [33]. Cells were maintained in low glucose Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FCS, as described [22,23,30].
Animals and scrapie infections
Heparanase transgenic (hpa-Tg) mice carrying human heparanase under the beta actin promoter have been described [13,34]. Mice were crossed for over 10 generations with C57BL/6J mice to produce pure genetic background [13,34]. All procedures were conducted using facilities and protocols approved by the Animal Care and Use Committee of the Hadassah-Hebrew University School of Medicine. Four- to six-week-old heparanase transgenic (hpa-Tg) mice, and age- and sex-matched C57BL mice (The Jackson Laboratory, Bar-Harbour, ME) were inoculated intraperitoneally (i.p) or intracerebrally (i.c.) with 100 μL or 50 μL, respectively, of diluted 1% (w/v) brain homogenate (in 4.5% BSA; w/v) derived from the Rocky Mountain Laboratory - Chandler (RML) strain of mice terminally sick with experimental scrapie [35,36]. All infections in the i.c. and i.p. groups were carried out on the same day using the same aliquot as inoculum. Animals were monitored at least twice a week for the development of clinical signs of the disease, which typically included a poor coat condition, a hunched posture, and development of a hind leg paralysis, weight loss and behavioral changes. Once clinical signs of the disease were noted, the mice were examined daily. The attack rate was 100% and the time when clinical signs first appeared is defined as ‘disease onset’. Mice were sacrificed by isoflurane at the terminal stage of the disease reached when the scrapie symptoms indicated that the animals would die within the next 72 h [36]. Brains and spleens were frozen for biochemical analysis. The survival times of i.c. and i.p. inoculated mice were converted to percent survived animals [36]. These values were then compared using the log-rank test (GraphPad Prizm). Brains and spleens were kept at −80°C until homogenized in PBS to 10% w/v, using an Ultra Turrax T8 IKA Labor Technic (Germany) homogenizer (10,000 rpm, 1 min, RT). The homogenates were lysed [8 μL of homogenate diluted to 200 μL with 2% N-octyl glucoside (NOG) in HEPES buffer, pH 7.4], insoluble material was removed by a 30s spin in a microcentrifuge, and 200 μL supernatants were brought to 1% Sarkosyl and incubated for 30 min on ice prior to proteolysis with proteinase K, SDS PAGE and Western blotting, as described under PrP analysis below (see Fig. S1) [36].
Antibodies
Rabbit antiserum RO73 recognizes both mouse PrP and MHM2-PrP (36). mAb 3F4 binds to residues Met108 and Met111 of chimeric MHM2-PrP, but does not recognize the endogenous wild-type mouse PrP of N2a cells [29]. Both antibodies were used at a dilution of 1:5000 for Western blot analysis [22,23]. Anti-heparanase pAb 733 which preferentially recognizes the active 50+8 kDa enzyme was used at a dilution of 1:1000 for Western blotting and 1:200 for immunostaining [37].
Histology and immunohistochemistry
Histological examination and immunostaining of formalin-fixed, paraffin-embedded 5 micron sections were performed essentially as described [37]. Images were acquired by Nikon ECLIPSE microscope and Digital Sight Camera (Nikon) with objectives 20x or 40x.
PrP isoforms and PrP analysis
The PrP isoforms were characterized and separated as described [22,23,38]. Cells were lysed in ice-cold lysis buffer (0.5% Triton X-100, 0.23% Na-deoxycholate, 150 mM NaCl, 10 mM Tris-HCl pH 7.5, 10 mM EDTA) and lysates were immediately centrifuged for 30s at 14000 rpm in a microcentrifuge. All biochemical analyses were performed on this post-nuclear supernatant also referred to as ‘cell lysate’. Protease-resistant PrPSc is defined as the PrP fraction resistant to standard proteolysis by proteinase K (Roche) (20 μg/ml, 37°C, 30 min for cultured cells, and 40 μg/ml, 37°C, 1 h for brain samples) [29]. The same proteolytic conditions were used to assay protease resistance of PrPC fractions [22,23]. Proteolysis was terminated by 2 mM phenylmethyl sulfonyl fluoride (PMSF). SDS-PAGE and Western immunoblotting of PrP isoforms were carried out as described [22,23,30]. Western blots were developed using an ECL system.
Results
Recombinant heparanase reduces the amount of PrPSc in ScN2a-M cells
We treated (3 days) chronically scrapie infected ScN2a-M mouse neuroblastoma cells with purified active 50+8 kDa human heparanase or with its latent, 65 kDa proenzyme. The amount of proteinase K-resistant (+PK) PrPSc was then examined (Fig. 1a). Treatment with active heparanase resulted in a profound decrease in the amount of protease-resistant PrPSc (Fig. 1a, 50, middle panel), while the latent 65 kDa enzyme had no effect, indicating that heparanase enzymatic activity is required. The same treatment of ScN2a-M cells that were first cured of scrapie by pentosan polysulfate (PPS) [22,23,39] had no effect on the amount PrPC (Fig. 1a, right panel). Next, we examined the effect of heparanase over-expression on scrapie. Stable transfection of persistently scrapie infected GT1-1 cells [33] with human heparanase resulted in a marked decrease in PrPSc compared to mock transfected cells (Fig. 1b, bottom).
Figure 1. The amount of PrPSc is reduced following treatment of scrapie-infected cells with recombinant heparanase, or over-expression of heparanase.
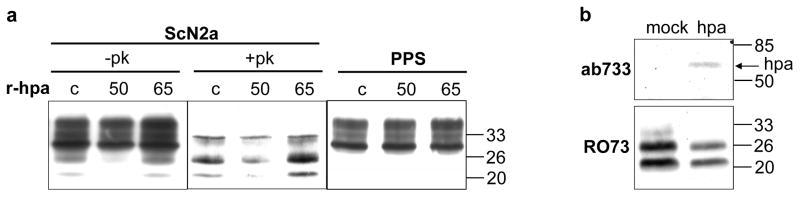
a) ScN2a-M cells (ScN2a) and pentosan polyphosphate treated N2a-M cells (PPS) growing in 6-well plates were left untreated (c) or treated (3 days, 0.1 μg/ml) with either active 50+8 kDa (50) or latent 65 kDa (65) recombinant heparanase (r-hpa). Subsequently, the cells were lysed, incubated (37 °C, 30 min) with (+PK) or without (−PK) 20 μg/ml proteinase K, and immunoblotted with mAb 3F4. b) Scrapie infected GT1-1 cells were stable transfected with human heparanase or mock-transfected. Cell lysates were subjected to SDS/PAGE and immunoblotting with anti-heparanase pAb 733 [37] (upper panel), or incubated (37°C, 30 min) with 20 μg/ml PK and immunoblotted with anti PrPSc pAb RO73 (lower panel). Notably, unlike the 3F4 mAb that recognizes the chimeric MHM2 PrPSc produced in ScN2a-M cells (Fig. 1a), the RO73 antibody detects the endogenous PrPSc produced in scrapie infected GT1-1 cells (Fig. 1b).
Heparanase over-expression delays prion disease
In order to examine the involvement of mammalian heparanase in scrapie infection in vivo, we applied transgenic mice that over-express human heparanase in most tissues [13], including the brain [40]. Since heparanase expression in the brain tissue of these hpa-Tg mice was lower relative to other tissues, we determined the levels of heparanase in different areas of the brain applying a sandwich ELISA [41]. The heparanase protein was markedly elevated in all areas with the highest levels detected in the hpa-Tg cerebellum (Fig. 2a). Immunostaining revealed that heparanase expression in the cerebellum of hpa-Tg mice was mainly localized in Purkinje cells and in the molecular layer which comprises the Purkinje cells’ dendritic tree (Fig. 2b), areas that preferentially express PrPC [42].
Figure 2. Heparanase protein is over-expressed in the brain of hpa-transgenic mice.

A) ELISA. Heparanase protein was assayed by sandwich ELISA [41] in extracts of different brain areas of hpa-Tg vs. control C57BL mice (control). Each bar graph is the mean + SD of triplicate determinations. B) Immunohistochemistry. Immunostaining with anti-heparanase pAb 733 antiserum showing intense heparanase expression primarily in Purkinje cells (PC) of the cerebellum of hpa-Tg (right panel; original magnification X20) vs. weak expression in control C57BL mice (left panel; original magnification X10). ML = molecular layer; GL = granular layer; WM = white matter; Purkinje cells = PC.
Both the hpa-Tg and their control wild type (wt) C57BL mice were inoculated intraperitoneally (i.p.) or intracerebrally (i.c.) with the Rocky Mountain Laboratory-Chandler (RML) strain of mouse adapted scrapie prions [35]. The mean survival time of i.p. inoculated male mice was 274 ± 14 days post infection (dpi) and 230 ± 18 dpi for hpa-Tg vs. control C57BL mice, respectively (Fig. 3A; P =0.0002) (Table 1). Similarly, the survival time of i.p. inoculated female mice was 249 ± 7 dpi vs. 225 ± 8 dpi for hpa-Tg and control mice, respectively (Fig. 3B; P <0.00001) (Table 1). Video presentation of i.p. infected hpa-TG and control C57BL mice on day 215 of the experiment is presented in figure S2.
Figure 3. Significantly prolonged survival times of scrapie-infected heparanase transgenic (hpa-Tg) mice vs. C57BL control mice.

A–C) Survival plots of RML inoculated mice. Survival times are presented as percentage of mice that did not die of scrapie at different time points post-inoculation. Significantly prolonged survival times of scrapie-infected hpa-Tg mice as compared to control C57BL mice are noted following intraperitoneal (A, males; B, females) and intracerebral (C, males) inoculation of RML prions. See video presentation of i.p. infected hpa-TG and control C57BL mice on day 215 of the experiment (Fig. S2).
Table 1.
Survival times of i.p. and i.c. RML inoculated hpa-tg vs. C57BL mice
| Mice | i.p. survival time (days ± SD) | i.c. survival time (days ± SD) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Male | Female | ||||||||
| Shortest | Longest | Mean | Shortest | Longest | Mean | Shortest | Longest | Mean | |
| hpa-tg | 255 | 297 | 274 ± 14 (n = 9) | 234 | 257 | 249 ± 7 (n = 10) | 191 | 246 | 211 ± 18 (n = 9) |
| C57BL | 205 | 267 | 230 ± 18 (n = 10) | 205 | 234 | 225 ± 9 (n = 10) | 172 | 185 | 178 ± 5 (n = 9) |
i.c., intracerebrally; i.p., intraperitoneally; SD, standard deviation; n, number of mice in each group.
In subsequent studies, mice were inoculated with RML intracerebrally. The hpa-Tg i.c. inoculated mice succumbed to the disease on average 211 ± 18 days post infection, whereas the control C57BL mice died already on day 178 ± 5 post infection (Table 1; Fig. 3C). Statistical analysis showed a significant difference (P <0.0001) in incubation time (duration from infection until disease onset) between hpa-Tg and control mice. The shortest and longest mean survival times for each group of mice are shown in Table 1. As expected, i.c. vs. i.p. inoculation gave rise to shorter incubation times. Mice inoculated i.c. or i.p. with PBS did not show any signs of prion disease for >400 days post injection. Notably, the highly significant difference in survival of hpa-Tg vs. control mice is equivalent to infection of the hpa-Tg mice with a 1000 fold higher infectious dose, as deduced from dose/scrapie survival time relations previously reported for wild-type C57BL mice [43].
While the disease onset (defined as the time when clinical signs first appeared) was significantly delayed in i.p RML inoculated hpa-Tg mice vs. control mice, no significant difference in disease onset was observed in i.c. inoculated mice. Instead, duration of the symptomatic phase was markedly prolonged in the i.c. inoculated hpa-Tg mice, significantly extending their survival compared to control C57BL mice (Table 1). The latter finding was confirmed by measuring weight loss in the i.c. inoculated mice. As expected, weight loss was observed prior to the appearance of any neurological symptoms. In order to obtain an objective comparison of incubation times between hpa-Tg and control mice, i.c. inoculated animals were monitored for their weight starting from inoculation until 70 dpi. The hpa-Tg mice started to loose weight at about the same time as the control C57BL mice, but at a slower rate (not shown). Clinical signs of the disease were the same in hpa-Tg and control mice, in both the i.c. and i.p. routes of infection. Thus, heparanase over-expression led to a delayed death compared with control mice, but did not modify the scrapie symptoms in the final stages of the disease.
Unlike the human enzyme, chicken heparanase is localized primarily at the cell surface and is readily secreted [44]. Moreover, cells transfected with a chimeric construct composed of the human enzyme and the chicken heparanase signal sequence exhibited cell surface localization and secretion of heparanase, similar to cells transfected with the full-length chicken enzyme [44]. We infected male and female mice transgenic for the chimeric construct (sphpa-Tg) by i.p. inoculation of experimental scrapie (RML). The mean survival time of the inoculated male mice was 238 ± 7 dpi and 215 ± 6 dpi for sphpa-Tg and control male mice (Fig. 4A; P = 0.0005) and 252 ± 26 dpi vs. 222 ± 14 dpi for sphpa-Tg and control C57BL female mice, respectively (Fig. 4B; P = 0.0087) (Table 2). The survival of scrapie infected sphpa-Tg mice was prolonged to a higher extent than that of hpa-Tg mice, further supporting the notion that extracellular heparanase is required to exert an anti-scrapie effect.
Figure 4. Survival plots of sphpa-Tg mice vs. C57BL control mice inoculated i.p. with RML prions.
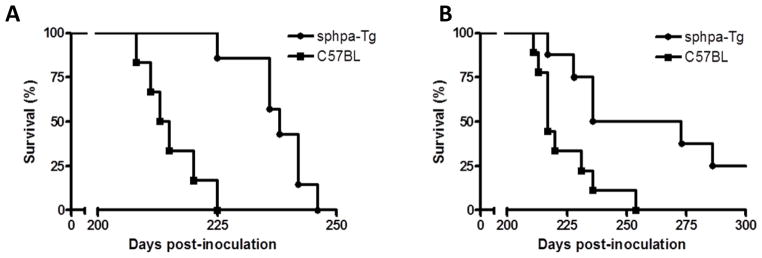
RML was inoculated (i.p.) into transgenic mice (A, males; B, females) over-expressing a secreted form of human heparanase (sphpa-Tg) vs. control C57BL mice. Mice inoculated i.c. or i.p. with PBS did not show any signs of prion disease >400 days post-inoculation.
Table 2.
Summary of survival times in i.p. sphpa-tg inoculated mice
| Mice | Survival time (dpi ± SD) | |||||||
|---|---|---|---|---|---|---|---|---|
| Male | Female | |||||||
| Shortest | Longest | Mean | Median | Shortest | Longest | Mean | Median | |
| sphpa-tg | 225 | 246 | 238 ± 7 (n = 7) | 238 | 211 | >370* | 252 ± 26 (n = 5) | 273 |
| C57BL | 208 | 225 | 215 ± 6 (n = 6) | 214 | 217 | 254 | 222 ± 14 (n = 8) | 217 |
SD, standard deviation; n, number of mice inoculated in each group.
2 of the female mice didn’t show any signs of disease until 370 days post infection (dpi).
Discussion
Our results provide the first in vivo evidence for the pivotal involvement of HS-degrading mammalian heparanase in scrapie disease. Recombinant heparanase reduced the amount of scrapie protein in infected mouse neuroblastoma cells, while its precursor PrPC was not affected. Importantly, only the processed active form of heparanase inhibited scrapie disease, while the latent enzymatically inactive precursor had no effect. Similarly, over-expression of heparanase by stable transfection of GT1-1 cells that are persistently infected with scrapie resulted in a marked decrease in PrPSc compared to mock-transfected cells.
Heparanase over-expression in hpa transgenic mice infected with experimental scrapie resulted in a dramatically prolonged survival as compared to control C57BL mice, irrespective of the inoculation route (i.p or i.c). Notably, heparanase over-expression had a more pronounced effect on the incubation time until disease onset in hpa-Tg mice that were infected by i.p. vs. i.c. inoculation. This suggests that heparanase over-expression in the peripheral tissues is capable of exerting potent anti-prion effect, taking into account that upon peripheral prion infection, the infectious agent accumulates in lymphoid organs, in particular follicular dendritic cells in spleen [45]. Neuroinvasion is thought to occur as a result of direct uptake of prions by the sympathetic nerve fibers which innervate the spleen or by an as yet unknown cell-mediated delivery system to the nerve termini of the sympathetic nerves [46]. Since HS, primarily syndecan 1, is known to mediate cellular uptake of heparin-binding proteins, including prions [23], it is conceivable that heparanase over-expression in peripheral organs would delay the entry of prions into the CNS and thereby delay disease onset, as in fact observed following i.p. infection. Once the symptoms appeared, disease progression was similar in hpa-Tg and control mice. In contrast, the i.c. route of infection prolonged the symptomatic phase in hpa-Tg vs. control mice. In both routes of infection, hpa-Tg mice eventually died of scrapie disease and revealed similar amounts of PK-resistant PrP in their brains and spleens, as determined by Western blot analysis (Figure S1). Similarly, the clinical signs of the disease were the same in hpa-Tg and control mice, further indicating that heparanase over-expression does not modify the scrapie symptoms at the final stage of the infection.
Collectively, our results provide a direct in vivo evidence for the role of HS and HS-degrading heparanase in prion disease both in terms of susceptibility to infection and disease progression. A protective effect of heparanase was previously demonstrated by showing that heparanase rich tissues of hpa-Tg mice are resistant to experimental amyloid protein A amyloidosis [14]. Notably, overexpression of heparanase also reduces amyloid load in animal model of Alzheimer’s disease (47) and formation of islet amyloid in vitro (48). It appears that the shorter fragments of HS produced due to extensive degradation by heparanase, fail to form complex with proteins that generate local or systemic amyloidosis, thereby precluding protein aggregation. Our results provide direct in vivo evidence for the involvement of intact heparan sulfate in the pathogenesis of prion disease and the protective role of mammalian heparanase in terms of disease onset and progression.
Supplementary Material
Intact heparan sulfate is involved in the pathogenesis of prion disease
Heparanase treatment/over-expression results in profound decrease in cellular PrPSc
Over-expression of mammalian heparanase delays prion disease onset and progression
Acknowledgments
This work was supported by grants from the Israel Science Foundation (601/14); National Cancer Institute, NIH (RO1-CA106456); the Israel Cancer Research Fund (ICRF); and the Horwitz Foundation (to A. Taraboulos). I. Vlodavsky is a Research Professor of the ICRF.
Abbreviations
- PrPC
cellular prion protein
- PrPSc
scrapie prion protein
- GAGs
glycosaminoglycans
- HS
heparan sulfate
- HSPGs
heparan sulfate proteoglycans
- PK
proteinase K
- hpa-Tg mice
heparanase over-expressing transgenic mice
- PPS
pentosan polysulfate
- RML
brain homogenate derived from scrapie infected Rocky Mountain Laboratory mice
- i.c
intracerebrally
- i.p
intraperitoneally
- ECM
extracellular matrix
- sphpa-Tg mice
mice overexpressing the secreted form of heparanase
Footnotes
AUTHOR CONTRIBUTIONS
O.K.B-Z, Y.T., S.T., I.N., and I.S. conducted the experiments. E.Z. and S.M. established the mouse models. I.V. wrote the manuscript. A.T. and I.V. coordinated the research and supervised the project.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Dumoulina M, Bader R. A short historical survey of developments in amyloid research. Protein Pept Lett. 2006;13:213–217. doi: 10.2174/092986606775338434. [DOI] [PubMed] [Google Scholar]
- 2.Gillmore JD, Hawkins PN. Pathophysiology and treatment of systemic amyloidosis. Nat Rev Nephrol. 2013;9:574–586. doi: 10.1038/nrneph.2013.171. [DOI] [PubMed] [Google Scholar]
- 3.Snow AD, Kisilevsky R, Willmer J, Prusiner SB, DeArmond SJ. Sulfated glycosaminoglycans in amyloid plaques of prion diseases. Acta Neuropathologica. 1989;77:337–342. doi: 10.1007/BF00687367. [DOI] [PubMed] [Google Scholar]
- 4.Snow AD, Willmer J, Kisilevsky R. Sulfated glycosaminoglycans: a common constituent of all amyloids? Lab Invest. 1987;56:120–123. [PubMed] [Google Scholar]
- 5.Stevens FJ, Kisilevsky R. Immunoglobulin light chains, glycosaminoglycans, and amyloid. Cellular Molecular Life Sci: CMLS. 2000;57:441–449. doi: 10.1007/PL00000706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Holmes BB, DeVos SL, Kfoury N, et al. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc Natl Acad Sci U S A. 2013;110:E3138–3147. doi: 10.1073/pnas.1301440110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McBride PA, Wilson MI, Eikelenboom P, Tunstall A, Bruce ME. Heparan sulfate proteoglycan is associated with amyloid plaques and neuroanatomically targeted PrP pathology throughout the incubation period of scrapie-infected mice. Exp Neurol. 1998;149:447–454. doi: 10.1006/exnr.1997.6740. [DOI] [PubMed] [Google Scholar]
- 8.O’Callaghan P, Sandwall E, Li JP, et al. Heparan sulfate accumulation with Abeta deposits in Alzheimer’s disease and Tg2576 mice is contributed by glial cells. Brain Pathol. 2008;18:548–561. doi: 10.1111/j.1750-3639.2008.00152.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Horssen J, Kleinnijenhuis J, Maass CN, et al. Accumulation of heparan sulfate proteoglycans in cerebellar senile plaques. Neurobiol Aging. 2002;23:537–545. doi: 10.1016/s0197-4580(02)00010-6. [DOI] [PubMed] [Google Scholar]
- 10.Zhang X, Li JP. Heparan sulfate proteoglycans in amyloidosis. Prog Mol Biol Trans Sci. 2010;93:309–334. doi: 10.1016/S1877-1173(10)93013-5. [DOI] [PubMed] [Google Scholar]
- 11.Taylor DR, Whitehouse IJ, Hooper NM. Glypican-1 mediates both prion protein lipid raft association and disease isoform formation. PLoS Pathog. 2009;5:e1000666. doi: 10.1371/journal.ppat.1000666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Linker A, Carney HC. Presence and role of glycosaminoglycans in amyloidosis. Lab Invest. 1987;57:297–305. [PubMed] [Google Scholar]
- 13.Zcharia E, Metzger S, Chajek-ShaulL T, Aingorn H, Elikn M, Friedmann Y, Weinstein T, Jin-Ping L, Lindahl U, Vlodavsky I. Transgenic expression of mammalian heparanase uncovers physiological functions of heparan sulfate in tissue morphogenesis, vascularization, and feeding behavior. FASEB J. 2004;18:252–263. doi: 10.1096/fj.03-0572com. [DOI] [PubMed] [Google Scholar]
- 14.Li JP, Galvis ML, Gong F, Zhang X, Zcharia E, Metzger S, Vlodavsky I, Kisilevsky R, Lindahl U. In vivo fragmentation of heparan sulfate by heparanase overexpression renders mice resistant to amyloid protein A amyloidosis. Proc Natl Acad Sci U S A. 2005;102:6473–6477. doi: 10.1073/pnas.0502287102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arvatz G, Shafat I, Levy-Adam F, Ilan N, Vlodavsky I. The heparanase system and tumor metastasis: is heparanase the seed and soil? Cancer Metastasis Rev. 2011;30:253–268. doi: 10.1007/s10555-011-9288-x. [DOI] [PubMed] [Google Scholar]
- 16.Ilan N, Elkin M, Vlodavsky I. Regulation, function and clinical significance of heparanase in cancer metastasis and angiogenesis. Int J Biochem Cell Biol. 2006;38:2018–2039. doi: 10.1016/j.biocel.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 17.Parish CR, Freeman C, Ziolkowski AF, He YQ, Sutcliffe EL, Zafar A, Rao S, Simeonovic CJ. Unexpected new roles for heparanase in Type 1 diabetes and immune gene regulation. Matrix Biol. 2013;32:228–233. doi: 10.1016/j.matbio.2013.02.007. [DOI] [PubMed] [Google Scholar]
- 18.Zhang X, Wang B, Li JP. Implications of heparan sulfate and heparanase in neuroinflammation. Matrix Biol. 2014;35:174–181. doi: 10.1016/j.matbio.2013.12.009. [DOI] [PubMed] [Google Scholar]
- 19.Fraser PE. Prions and prion-like proteins. J Biol Chem. 2014;289:19839–19840. doi: 10.1074/jbc.R114.583492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216:136–144. doi: 10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- 21.Prusiner SB. Prions. Proc Natl Acad Sci U S A. 1998;95:13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ben-Zaken O, Tzaban S, Tal Y, Horonchik L, Esko JD, Vlodavsky I, Taraboulos A. Cellular Heparan Sulfate Participates in the Metabolism of Prions. J Biol Chem. 2003;278:40041–40049. doi: 10.1074/jbc.M301152200. [DOI] [PubMed] [Google Scholar]
- 23.Horonchik L, Tzaban S, Ben-Zaken O, Yedidia Y, Rouvinski A, Papy-Garcia D, Barritault D, Vlodavsky I, Taraboulos A. Heparan sulfate is a cellular receptor for purified infectious prions. J Biol Chem. 2005;280:17062–17067. doi: 10.1074/jbc.M500122200. [DOI] [PubMed] [Google Scholar]
- 24.Gabizon R, Meiner Z, Halimi M, Ben-Sasson SA. Heparin-like molecules bind differentially to prion-proteins and change their intracellular metabolic fate. J Cell Physiol. 1993;157:319–325. doi: 10.1002/jcp.1041570215. [DOI] [PubMed] [Google Scholar]
- 25.Panegyres PK, Armari E. Therapies for human prion diseases. Am J Neurodegener Dis. 2013;2:176–186. [PMC free article] [PubMed] [Google Scholar]
- 26.Yudovin-Farber I, Azzam T, Metzer E, Taraboulos A, Domb AJ. Cationic polysaccharides as antiprion agents. J Med Chem. 2005;48:1414–1420. doi: 10.1021/jm049378o. [DOI] [PubMed] [Google Scholar]
- 27.Miao HQ, Navarro E, Patel SD, et al. Cloning, expression, and purification of mouse heparanase. Protein Expr Purif. 2002;26:425–431. doi: 10.1016/s1046-5928(02)00558-2. [DOI] [PubMed] [Google Scholar]
- 28.Zetser A, Bashenko Y, Miao HQ, Vlodavsky I, Ilan N. Heparanase affects adhesive and tumorigenic potential of human glioma cells. Cancer Res. 2003;63:7733–7741. [PubMed] [Google Scholar]
- 29.Kascsak RJ, Rubenstein R, Merz PA, Carp RI, Robakis NK, Wisniewski HM, Diringer H. Immunological comparison of scrapie-associated fibrils isolated from animals infected with four different scrapie strains. J Virol. 1986;59:676–683. doi: 10.1128/jvi.59.3.676-683.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Naslavsky N, Stein R, Yanai A, Friedlander G, Taraboulos A. Characterization of detergent-insoluble complexes containing the cellular prion protein and its scrapie isoform. T J Biol Chem. 1997;272:6324–6331. doi: 10.1074/jbc.272.10.6324. [DOI] [PubMed] [Google Scholar]
- 31.Honda H, Sasaki K, Minaki H, Masui K, Suzuki SO, Doh-Ura K, Iwaki T. Proteaseresistant PrP and PrP oligomers in the brain in human prion diseases after intraventricular pentosan polysulfate infusion. Neuropathology. 2012;32:124–132. doi: 10.1111/j.1440-1789.2011.01245.x. [DOI] [PubMed] [Google Scholar]
- 32.Tsuboi Y, Doh-Ura K, Yamada T. Continuous intraventricular infusion of pentosan polysulfate: clinical trial against prion diseases. Neuropathology. 2009;29:632–636. doi: 10.1111/j.1440-1789.2009.01058.x. [DOI] [PubMed] [Google Scholar]
- 33.Schatzl HM, Laszlo L, Holtzman DMJ, et al. A hypothalamic neuronal cell line persistently infected with scrapie prions exhibits apoptosis. J Virol. 1997;71:8821–8831. doi: 10.1128/jvi.71.11.8821-8831.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boyango I, Barash U, Naroditsky I, Li JP, Hammond E, Ilan N, Vlodavsky I. Heparanase cooperates with Ras to drive breast and skin tumorigenesis. Cancer Res. 2014;74:4504–4514. doi: 10.1158/0008-5472.CAN-13-2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chandler RL. Encephalopathy in mice produced by inoculation with scrapie brain material. Lancet. 1961;1:1378–1379. doi: 10.1016/s0140-6736(61)92008-6. [DOI] [PubMed] [Google Scholar]
- 36.Tal Y, Souan L, Cohen IR, Meiner Z, Taraboulos A, Mor F. Complete Freund’s adjuvant immunization prolongs survival in experimental prion disease in mice. J Neurosci Res. 2003;71:286–290. doi: 10.1002/jnr.10474. [DOI] [PubMed] [Google Scholar]
- 37.Zetser A, Levy-Adam F, Kaplan V, et al. Processing and activation of latent heparanase occurs in lysosomes. J Cell Sci. 2004;117:2249–2258. doi: 10.1242/jcs.01068. [DOI] [PubMed] [Google Scholar]
- 38.Meyer RK, McKinley MP, Bowman KA, Braunfeld MB, Barry RA, Prusiner SB. Separation and properties of cellular and scrapie prion proteins. Proc Natl Acad Sci U S A. 1986;83:2310–2314. doi: 10.1073/pnas.83.8.2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ehlers B, Diringer H. Dextran sulphate 500 delays and prevents mouse scrapie by impairment of agent replication in spleen. J Gen Virol. 1984;65(Pt 8):1325–1330. doi: 10.1099/0022-1317-65-8-1325. [DOI] [PubMed] [Google Scholar]
- 40.Zhang X, Wang B, O’Callaghan P, et al. Heparanase overexpression impairs inflammatory response and macrophage-mediated clearance of amyloid-beta in murine brain. Acta Neuropathol. 2012;124:465–478. doi: 10.1007/s00401-012-0997-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shafat I, Zcharia E, Nisman B, Nadir Y, Nakhoul F, Vlodavsky I, Ilan N. An ELISA method for the detection and quantification of human heparanase. Biochem Biophys Res Commun. 2006;341:958–963. doi: 10.1016/j.bbrc.2006.01.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rossi D, Cozzio A, Flechsig E, Klein MA, Rulicke T, Aguzzi A, Weissmann C. Onset of ataxia and Purkinje cell loss in PrP null mice inversely correlated with Dpl level in brain. Embo J. 2001;20:694–702. doi: 10.1093/emboj/20.4.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Klein MA, Kaeser PS, Schwarz P, et al. Complement facilitates early prion pathogenesis. Nature Med. 2001;7:488–492. doi: 10.1038/86567. [DOI] [PubMed] [Google Scholar]
- 44.Goldshmidt O, Zcharia E, Aingorn H, et al. Expression pattern and secretion of human and chicken heparanase are determined by their signal peptide sequence. J Biol Chem. 2001;276:29178–29187. doi: 10.1074/jbc.M102462200. [DOI] [PubMed] [Google Scholar]
- 45.Brown KL, Stewart K, Ritchie DL, et al. Scrapie replication in lymphoid tissues depends on prion protein-expressing follicular dendritic cells. Nature Med. 1999;5:1308–1312. doi: 10.1038/15264. [DOI] [PubMed] [Google Scholar]
- 46.Nuvolone M, Aguzzi A, Heikenwalder M. Cells and prions: a license to replicate. FEBS Lett. 2009;583:2674–2684. doi: 10.1016/j.febslet.2009.06.014. [DOI] [PubMed] [Google Scholar]
- 47.Jendresen CB, Cui H, Zhang X, Vlodavsky I, Nilsson LN, Li J-P. Overexpression of Heparanase lowers the amyloid burden in amyloid-β precursor protein transgenic mice. J Biol Chem. 2015;290:5053–64. doi: 10.1074/jbc.M114.600569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Oskarsson ME, Singh K, Wang J, Vlodavsky I, Li J-P, Westermark GT. Heparan sulfate proteoglycans are important for islet amyloid formation and islet amyloid polypeptideinduced apoptosis. J Biol Chem. 2015 Apr 28; doi: 10.1074/jbc.M114.631697. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.