

Abstract
Genomic rearrangements are increasingly recognized as important contributors to human disease. Here we report on an 11½-year-old child with myopia, Duane retraction syndrome, bilateral mixed hearing loss, skeletal anomalies including multiple epiphyseal dysplasia, and global developmental delay, and a complex 6p25 genomic rearrangement. We have employed oligonucleotide-based comparative genomic hybridization arrays (aCGH) of different resolutions (44 and 244K) as well as a 1 M single nucleotide polymorphism (SNP) array to analyze this complex rearrangement. Our analyses reveal a complex rearrangement involving a ~2.21 Mb interstitial deletion, a ~240 kb terminal deletion, and a 70–80 kb region in between these two deletions that shows maintenance of genomic copy number. The interstitial deletion contains eight known genes, including three Forkhead box containing (FOX) transcription factors (FOXQ1, FOXF2, and FOXC1). The region maintaining genomic copy number partly overlaps the dual specificity protein phosphatase 22 (DUSP22) gene. Array analyses suggest a homozygous loss of genomic material at the 5′ end of DUSP22, which was corroborated using TaqMan® copy number analysis. It is possible that this homozygous genomic loss may render both copies of DUSP22 or its products non-functional. Our analysis suggests a rearrangement mechanism distinct from a previously reported replication-based error-prone mechanism without template switching for a specific 6p25 rearrangement with a 1.22 Mb interstitial deletion. Our study demonstrates the utility and limitations of using oligonucleotide-based aCGH and SNP array technologies of increasing resolutions in order to identify complex DNA rearrangements and gene disruptions.
Keywords: 6p25, FOXC1, epiphyseal dysplasia, rearrangement, aCGH, SNP array, TaqMan®
INTRODUCTION
Genomic disorders are a group of human genetic disorders caused by segmental DNA rearrangements resulting in gain, loss, or disruption of dosage-sensitive critical genes within a genomic region [Lupski, 1998]. In the last few years, array-based technologies have demonstrated that small chromosomal rearrangements are very common and contribute to human genetic and phenotypic variations as well as disease [Zhang et al., 2009]. In many cases, the mechanisms underlying these chromosomal rearrangements remain poorly characterized.
Genomic rearrangements of the short arm of chromosome 6 represent a group of disorders with gene dosage attributable phenotypes [Gould et al., 2004; Lin et al., 2005; Maclean et al., 2005; Kannu et al., 2006; DeScipio, 2007]. The phenotypes of patients with terminal deletion of the short arm of chromosome 6 (6p25-pter) include the Axenfeld–Rieger syndrome spectrum of ocular anomalies, progressive hearing loss, developmental delay, mental retardation, language impairment, as well as cardiac, renal, skeletal, and craniofacial anomalies [Gould et al., 2004; Lin et al., 2005; Maclean et al., 2005; Kannu et al., 2006; DeScipio, 2007]. Skeletal anomalies, including scoliosis and other vertebral anomalies, clubfeet, joint positional defects, and epiphyseal dysplasia of the femur and tibia, may also occur [Koshino et al., 1989; Garcia-Cruz et al., 1990; Chitty et al., 1991; Gould et al., 2004; Rodriguez-Rojas et al., 2004; Maclean et al., 2005; Caluseriu et al., 2006; Kannu et al., 2006].
High-resolution oligonucleotide-based comparative genomic hybridization array (aCGH) technologies offer improved sensitivity to detect genomic rearrangements, more precise delineation of breakpoints, and enhanced insights into the rearrangement mechanism. Such technologies have provided insight into several mechanisms that contribute to genomic disorders [Inoue et al., 2002; Toffolatti et al., 2002; Shaw and Lupski, 2004, 2005; Hastings et al., 2009]. In this report, we describe the phenotype of a child with a complex genomic rearrangement of 6p25. We performed array analyses at three resolutions in order to map the breakpoints of this complex rearrangement and employed TaqMan® copy number analysis to investigate genomic loss in specific rearrangement regions. Our study demonstrates the utility and limitations of using oligonucleotide-based aCGH and single nucleotide polymorphism (SNP) array technologies of increasing resolutions in order to identify complex DNA rearrangements and gene disruptions, and provide some insight into rearrangement mechanisms as well as phenotypes associated with 6p25 rearrangements.
CLINICAL REPORT
The proband is an 11½-year-old girl (Fig. 1) with a history of myopia, Duane retraction syndrome, bilateral mixed hearing loss, skeletal anomalies including multiple epiphyseal dysplasia, and global developmental delay. She was born at term with a birth weight of 3,200g (30th centile) to a 32-year-old mother via induced vaginal delivery. There were no known teratogenic exposures during the pregnancy. In infancy, the patient was noted to have a 25° right lateral torticollis; however, computed tomography (CT) scan of the head and cervical region at 6 months of age did not identify any skeletal or brain abnormalities. She was also noted to have ocular abnormalities, including prominent eyes, myopia, and a congenital left VI nerve palsy with esotropia. These findings were consistent with Duane syndrome type I involving the left eye and a mild strabismic amblyopia. The patient also demonstrated mixed conductive and sensorineural hearing loss. She had a long-standing history of recurrent middle ear infections, but had persistent significant hearing loss despite functioning tympanostomy tubes. She has used hearing aids since age 6 years. Family history was significant for myopia in the father, paternal grandmother, and paternal half aunt and half uncle. The patient also had a 14-year-old brother with history of myopia and learning disabilities. The family was non-consanguineous.
FIG. 1.
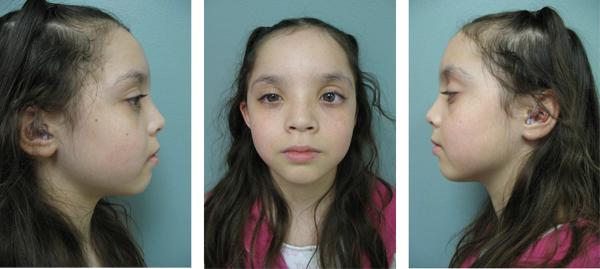
Pictures of our patient at 11½ years of age. Our patient exhibited mild frontal bossing, hypertelorism, prominent eyes, bilateral epicanthal folds, a broad nasal bridge, malar hypoplasia, and slightly low set and posteriorly rotated ears.
A series of diagnostic evaluations were performed at 6 years of age. Renal ultrasound and temporal bone CT did not demonstrate any significant abnormalities. Echocardiogram revealed only a patent ductus arteriosus. MRI of the hips demonstrated decreased femoral head epiphyseal height with bilateral fattening and fragmentation of the proximal femoral epiphysis without evidence of avascular necrosis. These findings were suggestive of multiple epiphyseal dysplasia. A skeletal survey showed anterior wedging of a midthoracic vertebra without evidence of vertebral flattening; the long bone epiphyses, particularly at the proximal femurs (Fig. 2), proximal humeri, and the knees and ankles were small, also consistent with multiple epiphyseal dysplasia. Standard karyotyping at the 550 band level of resolution as well as COL2A1 and COL11A2 sequencing (performed because of earlier concerns for spondyloepiphyseal dysplasia congenita and Stickler syndrome, respectively) were normal. The differential for other genes involved in multiple epiphyseal dysplasia also includes COMP, MATN3, COL9A1, COL9A2, COL9A3, and DTDST, but additional testing for these genes were not pursued because of the abnormal aCGH showing a complex 6p25 rearrangement.
FIG. 2.

Radiograph of patient at 10 years of age. AP view of pelvis showing bilateral flattening of the femoral epiphyses (right more severely affected than left). The right femoral head is also fragmented and irregular with increased sclerosis.
The patient's clinical course was notable for developmental delays from early infancy. She spoke her first word at 12 months and used simple phrases at 4½ years of age. Despite daily speech, occupational, and physical therapies at school, a school-based cognitive assessment at 11½ years of age noted that global cognitive skills were delayed approximately 2–3 years.
At 11½ years of age, her weight, height, and head circumference were at the 29th (35.7 kg), 39th (145.9 cm), and 45th (52.1 cm) centiles, respectively. She was noted to have several minor dysmorphic features, including mild frontal bossing, hypertelorism, prominent eyes, bilateral epicanthal folds, a broad nasal bridge, malar hypoplasia, and slightly low-set and posteriorly rotated ears (Fig. 1). There were no morphological abnormalities of the external ears. Her palate and dentition were normal. She had left lateral gaze palsy consistent with Duane retraction syndrome and esotropia. The patient also exhibited hyperextensibility at the elbows and had long tapered fingers. Her feet appeared long with long toes and bilateral pes planovalgus. She demonstrated good muscle tone, bulk, and symmetric strength in all four extremities.
MATERIALS AND METHODS
The proposita and her mother were enrolled in a research protocol after acquiring informed consent approved by the Institutional Review Board for Human Subject Research at the University of Michigan. Genomic DNA samples from the proband and her mother were analyzed after isolation from blood samples using a standard, semi-automated method (Biorobot M48 workstation, Qiagen, Inc., Valencia, CA).
Array CGH
Chromosomal microarray analyses were conducted in the Michigan Medical Genetics Laboratories (MMGL) at the University of Michigan using two oligonucleotide-based array platforms with whole genome coverage: a custom-designed EMArray Cyto6000 chip containing 43,103 oligonucleotide probes with an average inter-probe spacing of 75 kb implemented on the Agilent 44K platform [Baldwin et al., 2008], and an off-the-shelf, Agilent human genome G4411B 244K array platform containing 236,000 oligonucleotide probes with a spacing of ~9 kb (Agilent Technologies, Inc., Santa Clara, CA). The procedures for DNA digestion, labeling, and comparative genomic hybridization were as described in Agilent Oligonucleotide-Based Array CGH for Genomic DNA Analysis, Protocol version 4.0 June 2006 (Agilent Technologies, Inc., Santa Clara, CA) with some modifications [Baldwin et al., 2008]. The fluorescent signals on the array slides were detected and scanned into image files using the GenePix 4200A scanner and GenePix-Pro 6.1 software (Axon Instruments/Molecular Devices Corp., Union City, CA). The array images were imported and converted into data by Agilent Feature Extraction 9.5 software. Data were analyzed by Agilent's CGH Analytics 3.5 software to determine copy number differences or aberrations between the patient DNA and the sex mismatched DNA. Patient DNA was labeled with Cy3-dUTP and sex-mismatched pooled reference DNA was labeled with Cy5-dUTP. All the labeled DNA samples were cleaned up of reagents and unincorporated dyes by vacuum filtration. Purified fluorescently labeled patient DNA and reference DNA were mixed together and hybridized to the arrays [Baldwin et al., 2008]. Data were analyzed by interpreting the resulting Cy3/Cy5 ratio. Numbering of the Cyto6000 44K EMArray and the 244K Agilent array results were according to Genome Build UCSC hg 17 (Build 35, May 2004) and UCSC hg18 (Build 36.1, March 2006) assemblies, respectively. Chr6: 108,083–2,669,844 coordinates in hg17 remain unchanged in hg18 (100% of bases and 100% of span) as determined using the Convert function on the UCSC Genome Browser, simplifying data comparison between the 44 and 244K arrays.
SNP Array
SNP array analysis was performed using a HumanOmni1-Quad BeadChip array containing 1,140,419 SNP loci (Illumina, Inc., San Diego, CA) at the University of Michigan DNA Sequencing Core. Genomic DNA was quantified using the Quant-iT™ PicoGreen ® dsDNA Kit (Invitrogen Corporation, Carlsbad, CA). The HumanOmni1-Quad v1.0 DNA Analysis Kit (Illumina, Inc.) was used to assay 1,140,419 markers for SNP tag genotypes and/or copy numbervariants (CNVs).The HumanOmni-1 assay wasperformed according to the Infinium® HD Assay Super protocol provided by the manufacturer (Illumina, Inc.). SNPs were called using Illumina's GenomeStudio Genotyping Analysis Module (v1.6.3) and SNP cluster positions were defined by using the Illumina HumanOmni1-Quad_v1-0_B cluster file (Illumina, Inc.). GenomeStudio threshold configuration setting was modified to allow for aneuploidy information. The UCSC hg18 assembly (Build 36.1, March 2006) was used for coordinates during the SNP analysis in order to directly compare and contrast results with those from the 44 and 244K arrays.
FISH Analysis
Fluorescent in situ hybridization (FISH) analysis was performed at the Emory Genetics Laboratory's (EGL) Cytogenetics Laboratory by standard methods using a chromosome 6 subtelomeric probe mix (ToTelVysion, Mix 6; VYSIS, Inc., Downers Grove, IL).
TaqMan® Copy Number Assay
TaqMan® copy number assay (Applied Biosystems, Foster City, CA) was performed according to the manufacturer protocol (TaqMan® Copy Number Assays Protocol, P/N 4397425, Rev. C, 02/2009). Briefly, 20 ng of genomic DNA were transferred into a 96-well MicroAmp® Optical Reaction Plate (Applied Biosystems). TaqMan® Genotyping Master Mix (Product # 4371355, Applied Biosystems), TaqMan® Copy Number Assay (Hs03587293_cn, Hs03600180_cn, or Hs02910583_cn, hg19.v9; Applied Biosystems), and TaqMan® Copy Number RNaseP Reference Assay (Product # 4403326, Applied Biosystems) were mixed and pipetted into the 96-well plate and real time PCR was performed using the Step-OnePlus RT PCR System (Software v2.1, Applied Biosystems) at 95°C for 10 min followed by 40 cycles at 95°C for 5 sec and 60°C for 1 min. Four technical replicates were run for the patient and the control samples. The StepOnePlus RT PCR data files were imported into and analyzed by CopyCaller software v1.0 (Applied Biosystems) and the copy number for each sample was calculated [Livak and Schmittgen, 2001]. Pooled control genomic DNA (used at MMGL) was from 10 different normal individuals. A co-author (RKI) provided the control single normal individual DNA sample.
RESULTS
Array CGH Analyses
The 44K EMArray analysis identified a deletion (Chr6: 108,083–2,669,844; hg18) of the short arm of chromosome 6 characterized by a 2.56 Mb loss of genomic material (represented by 45 oligonucleotide probes) at 6p25 with minimum and maximum proximal deletion breakpoints at Chr6: 2,472,373–2,669,844; hg18 (represented by three probes including the boundary probes; Fig. 3A). This region of loss contains eight known genes, including the three tandemly arranged paralogs of the Forkhead box containing (FOX) transcription factors FOXQ1, FOXF2, and FOXC1 (Fig. 3A). There was a 197,471 bp region of uncertainty (Fig. 3A) at the proximal (centromeric) breakpoint region which completely overlaps a region with DNA polymerase α pause (GAG) and topoisomerase I consensus cleavage (CAT/GTC) sites within (GTG)n repeats (Motifs I, II, and III; TGGTGGTGGTGGC, GTGGTGGTGGTGGTGGTAGTCGTGGTGGTAGT and GTGGTGGTAGTGGTAGTGCTGCTGGTGGTGGTGGTGGTGATGGTGGTAGTG, respectively). This region was implicated in the recently proposed replication-based rearrangement mechanism [Chanda et al., 2008] that does not involve fork stalling and template switching (FoSTeS) [Lee et al., 2007]. A BLAT search (using the UCSC Genome Build hg18 March 2006) for Motifs II and III throughout the genome revealed such sequences with 100% identity distributed among several chromosomes including, as expected, chromosome 6 (Table I, in bold).
FIG. 3.

aCGH analyses of patient's DNA. A: 44K aCGH result showing the 6p25 terminal deletion (purple) with uncertainties in the most terminal regions shown (oligonucleotide probes reflecting possible two genomic copy number loss (blue circle) or maintenance of genomic copy number (red circle)). The 197 kb region of uncertainty involving the proximal (centromeric) breakpoint spans 22 probes for the 244K array, as compared to three probes for the 44K array. B: 244K aCGH result with insets b1 and b2 expanding the most terminal and proximal (centromeric) breakpoint 6p25 regions, respectively. Inset b1 shows the regions with one (1 CL), two (2 CL) and no (no CL) copy losses. The eight oligonucleotide probes which are almost completely overlapping DUSP22 are shown (circled in inset b1; orientation of DUSP22 noted). Inset b2 is an expansion showing the most proximal (centromeric) breakpoint boundaries resolved to ~9 kb and the positions of Motifs I, II, and III relative to the breakpoint.
Table I.
Locations of Motifs II and III with 100% Identity Throughout the Human Genome.
| Chr | Strand | Start | End | Span |
|---|---|---|---|---|
| Motif II | ||||
| 3 | - | 196980441 | 196980460 | 20 |
| 5 | + | 175951772 | 175952237 | 466 |
| 6 | + | 2639727 | 2639800 | 74 |
| 6 | - | 25035168 | 25035187 | 20 |
| 16 | - | 57483910 | 57483929 | 20 |
| 22 | - | 18530629 | 18530648 | 20 |
| 22 | - | 18531250 | 18531269 | 20 |
| Motif III | ||||
| 1 | + | 2076839 | 2076858 | 20 |
| 1 | + | 151482424 | 151482450 | 27 |
| 1 | + | 212450625 | 212450647 | 23 |
| 1 | - | 7551995 | 7552017 | 23 |
| 2 | + | 128814416 | 128814555 | 140 |
| 2 | - | 112821042 | 112821070 | 29 |
| 3 | + | 129646964 | 129647007 | 44 |
| 5 | - | 7425363 | 7425390 | 28 |
| 6 | + | 2639790 | 2639903 | 114 |
| 6 | + | 83437089 | 83437111 | 23 |
| 7 | - | 51398102 | 51398130 | 29 |
| 7 | - | 126686884 | 126686912 | 29 |
| 8 | + | 143598819 | 143598899 | 81 |
| 10 | + | 115018486 | 115018506 | 21 |
| 11 | + | 3184563 | 3184744 | 182 |
| 11 | + | 15758934 | 15758954 | 21 |
| 16 | + | 85055214 | 85055662 | 449 |
| 16 | - | 26461468 | 26461496 | 29 |
| 17 | + | 76288768 | 76288791 | 24 |
| 22 | - | 18529953 | 18529978 | 26 |
| 22 | - | 25138800 | 25138821 | 22 |
| X | - | 3452647 | 3452672 | 26 |
| X | - | 108403340 | 108403362 | 23 |
Motifs II and III are GTGGTGGTGGTGGTGGTAGTCGTGGTGGTAGT and GTGGTGGTAGTGGTAGTGCTGCTGGTGGTGGTGGTGGTGATGGTGGTAGTG, respectively. Chr, chromosome
The 244K oligonucleotide-based aCGH (Fig. 3B) revealed the rearrangement involved a ~2.21 Mb interstitial deletion with minimum and maximum boundaries of 2,205,945 bp (Chr6: 320,890–2,526,835; hg18) and 2,221,500 bp (Chr6: 314,479–2,535,979; hg18), respectively, as well as a ~240 kb terminal deletion with minimum and maximum proximal breakpoints at Chr6: 238,493–252,939; hg18 (Fig. 3B). This terminal deletion was not observed in over 600 chromosomal microarrays performed in our lab. The minimum and maximum deletion boundaries at the most proximal (centromeric) breakpoint on 6p was Chr6: 2,526,835– 2,535,979; hg18, resulting in an uncertainty of 9,144 bp of the breakpoint. There were 22 probes on the 244K aCGH covering the ~197 kb proximal breakpoint region compared with only three probes on the 44K aCGH (Fig. 3A). The 244K aCGH analysis also showed that the positions of Motifs I, II, and III were >100 kb removed from the most centromeric breakpoint (Fig. 3B). No Motif II or III sequences were found around the remaining breakpoints of the interstitial and terminal deletion regions using a BLAT search (UCSC Genome Build hg18 March 2006).
In the 44K EMArray analysis, two oligonucleotide probes close to the terminal end of 6p partly overlapped the dual specificity protein phosphatase 22 (DUSP22) gene and raised uncertainty about the copy number of a small terminal region of 6p including DUSP22 (Fig. 3A). Eight probes on the 244K aCGH almost completely overlapped DUSP22 at the distal terminal end of 6p and showed maintenance of copy number of ~70 kb genomic material in that region (compare Fig. 3A,B). Log2 ratios for the four oligonucleotide probes at the 5′ end of DUSP22 (probes immediately telomeric to the eight probes showing maintenance of copy number noted earlier; see Fig. 3B, inset b1) were −1.332, −1.983, −1.550, and −1.688 from most proximal to distal (start and stop boundaries of all four probes range between Chr6: 204,528–238,493; hg18). Because the theoretical log2 ratio for two copy loss is −1.585, the ratios observed (−1.332, −1.983, −1.550, and −1.688) were consistent with loss of two copies of genomic material at the 5′ end of DUSP22 (genomic sequence range Chr6: 237,101–296,355; hg18; see Fig. 3B, inset b1).
SNP Array Analysis
A SNP array analysis was performed in order to more carefully investigate the potential loss of two copies of genomic material at the 5′ end of DUSP22 as determined by the 244K array (see above section) and provide better resolution of the 6p25 rearrangement breakpoints. The call rate and logR deviation in our SNP array analysis were 99.7% and 0.188%, respectively. The analysis revealed that the complex rearrangement involved a ~2.21 Mb interstitial deletion with maximum boundaries at Chr6: 323,970 (hg18) and Chr6: 2,536,488 (hg18), consistent with that noted using the 244K array. The SNP analysis also showed maintenance of copy number of ~80 kb genomic material partially overlapping DUSP22 with minimum coordinates Chr6: 243,915 (hg18) and Chr6: 323,970 (hg18), as well as a terminal deletion beginning between Chr6: 241,218 (hg18) and Chr6: 243,915 (hg18), consistent with the 244K array analysis (compare Figs. 3B and 4A). The SNP array analysis allowed for better resolution of the most proximal (centromeric) breakpoint boundary with the minimum and maximum deletion boundaries at Chr6: 2,534,138 (hg18) and Chr6: 2,536,488 (hg18), respectively, resulting in an uncertainty of 2,350 bp of the breakpoint (first proximal boundary showing change in copy number value from 2 to 1, Fig. 4A).
FIG. 4.

SNP array and TaqMan® copy number analyses of patient's DNA. A: SNP array result showing the ~2.21 Mb region of interstitial deletion, a more telomeric region of ~80 kb with maintenance of genomic copy number, and the most terminal region of genomic loss. The red oval delineates probes in the 5′ region of DUSP22 used to further explore potential copy number losses in the region and compare with 244K aCGH result in Figure 3B. CL, copy loss. B: SNP array analysis using Nexus Copy Number version 5 software from BioDiversity, Inc. (El Seguando, CA). Three sets of 16 probes within the red ovals were statistically analyzed for difference in log2 R ratios (i.e., copy number value differences; see the Results Section). The region defined by the central oval is 5′ of DUSP22 and partially overlaps the 5′ end of the DUSP22 genomic boundary (green vertical line showing the 5′ DUSP22 boundary). Positions of primers A, B, and C used in the TaqMan® copy number assay (below) are indicated with coordinates Chr6: 155,627–155,651 (hg18), Chr6: 210,832–210,856 (hg18), and Chr6: 280,147–280,171 (hg18), respectively. C: TaqMan® copy number assay of patient's DNA versus control DNA from normal individuals. Coordinates for primers A, B, and C noted as above. SC and PC represent control (single) normal individual DNA sample (co-author RKI) and control (pooled) DNA sample from 10 normal individuals (used by MMGL), respectively. Pt; patient DNA.
Sixteen loci were identified on the SNP array between coordinates Chr6: 202,216 (hg18) and Chr6: 240,956 (hg18) 5′ of DUSP22 (i.e., telomeric to DUSP22); namely, rs1011328, rs2025158, rs6596874, rs12212311, rs6925262, rs815574, rs815576, cnvi0155642, rs1166328, rs1150764, rs815583, rs4959170, rs1849044, rs3800239, cnvi0151240, and rs9503190. These 16 loci were distinct from the 4 loci (probes) in the 244K array within the same coordinates (data not shown). Most were considered “intensity only probes” (similar to cnvi probes) and represented as “NC” (no calls) by GenomeStudio (Fig. 4A). Only three SNPs, rs12212311 (Chr6: 219,847; hg18), rs815576 (Chr6: 226,566; hg18), and rs9503190 (Chr6: 240,956; hg18) were informative with log2 ratios of −0.879, −0.473, and −0.939, respectively. The mean ± standard deviation (SD) of log2 ratios of the 16 probes at Chr6: 202,216–240,956 (hg18) 5′ of DUSP22 was −0.766 ± 0.176 (n = 16) and was significantly different than the log2 ratios of probes in regions immediately adjacent (i.e., telomeric and centromeric), −0.431 ± 0.242 (n = 16; Chr6: 181,731–201,281; hg18) and −0.182 ± 0.118 (n = 16; Chr6: 243,915–293, 102; hg18), respectively, using one-way ANOVA with a Fisher F-value = 39.842 and P < 0.001 between the groups (Fig. 4B). This suggested three regions of variable copy number values within Chr6: 181,731–293,102 (hg18).
TaqMan® Copy Number Analysis
TaqMan® copy number assay indicated a one copy genomic DNA loss at Chr6: 155,627–155,651 (hg18), two copy loss at Chr6: 210,832-210,856 (hg18), and no copy loss at Chr6: 280,147–280,171 (hg18) (Fig. 4C), corroborating the results from the 244K and SNP arrays (see above sections).
FISH Analysis
FISH analysis of the proband's DNA detected an abnormal hybridization pattern consistent with a terminal deletion of 6p (data not shown). FISH analysis on DNA from the mother showed normal hybridization patterns on both homologs of chromosome 6 (data not shown) suggesting that the 6p25.3 aberration seen in the patient is not of maternal origin. A paternal DNA sample was not available, so the origin of this aberration (whether paternal or de novo) remains uncertain.
Simple and Microsatellite Repeats
A UCSC Genome Browser analysis of the breakpoint boundary regions revealed several nearby simple and microsatellite repeat sequences: (AC)19.5 at Chr6: 254,680–254,718; (T)30 at Chr6: 309,796–309,825; (AGCAGCAGTTGCAGTAGCTGTGGCAGG AGGAGTAGCAGC)37.4 at Chr6: 316,050–317,456; (A)29 at Chr6: 320,676–320,704; (AAAG)22.8 at Chr6: 2,523,368–2,523,456; (AT TTGTCTGCTTA)2.2 at Chr6: 2,526,196–2,526,225; (TTTG)6.2 at Chr6: 2,528,605–2,528,629; (TATTCCATCATATACA)2.2 at Chr6: 2,529,080–2,529,114; (A)29 at Chr6: 2,539,174–2,539,202; and (CCCATCTTCATGTGACCTTTCCCTGTGTATTTCTATATGT TTC AGATCTCCCTTT)2 at Chr6: 2,540,932–2,541,045. All above coordinates are from hg18.
DISCUSSION
Identifying architectural features that result in instability of genomic regions may provide insight into genetic variation underlying a number of clinical disorders. Several mechanisms that depend on local genome architecture are thought to underlie genomic rearrangements involving 6p25 [Chanda et al., 2008]. Non-homologous end joining (NHEJ) has been proposed as the mechanism for six ancestral non-recurrent 6p25 rearrangements [Chanda et al., 2008]. These rearrangements contained simple repeat and microsatellite sequences, but lacked LCRs1 in regions immediately adjacent to the breakpoints. A replication-based rearrangement mechanism was proposed for a specific 6p25 rearrangement with a 1.22 Mb interstitial deletion. This mechanism involves error-prone repair steps using sites for DNA polymerase α pausing and topoisomerase I cleavage without template switching but mediated by Motifs I–III [Chanda et al., 2008]. This mechanism was distinct from FoSTeS—a replication-based rearrangement mechanism with template switching [Lee et al., 2007].
Here, we report on the results of 244K aCGH and 1M SNP array analyses that provide additional information about the breakpoint boundaries and some insight into the rearrangement mechanism. For example, a reduction in the uncertainty of the proximal (centromeric) interstitial deletion breakpoint was observed with each array of higher resolution; ~197 kb (44K array), ~9.1 kb (244K array), and ~2.4 kb (1 M SNP array). Furthermore, the array analyses not only demonstrated a 2.21 Mb interstitial deletion but also a terminal region with one, two, and no genomic material copy losses (see Fig. 3B, inset b1 and Fig. 4A–C). The terminal region, overlapping DUSP22 (Fig. 2B), has many reported CNVs (copy number gains and losses; Database of Genomic Variants, http://projects.tcag.ca/variation/), which could explain why this region maintains genomic copy number while other adjacent regions show variable copy number losses. In fact, one complex 6p25 rearrangement involving ring chromosome 6 reported by Chanda et al. [2008] (Proband 11 in Fig. 1 of Chanda et al. [2008]) also exhibited a short region of no genomic loss (about DUSP22 and IRF4; between ~0.2 and 0.3 Mb chromosomal coordinates) though characterization of the breakpoints was not available.
The genomic region between Chr6: 108,000–2,540,000 (hg18) is essentially devoid of LCRs (Database of Genomic Variants, http://projects.tcag.ca/variation/). These likely rules out non-allelic homologous recombination (NAHR) for the complex rearrangement in our patient. A number of simple repeats and microsatellite sequences were found within and adjacent to the (maximum) breakpoint boundaries (see the Results Section), but their involvement in this rearrangement is unclear. The distance of Motifs I–III from the most proximal breakpoint strongly argues against the replication-based rearrangement mechanism proposed by Chanda et al. [2008], in which the centromeric breakpoint was mapped to be within Motif II. Direct sequencing of the breakpoints are needed to draw definitive conclusions about mechanisms for this rearrangement.
Despite our findings, the replication-based rearrangement mechanism described by Chanda et al. [2008] may be involved in rearrangements at other loci. Motifs II and III are also found on other chromosomes (Table I). Interestingly, the locations of Motifs II and III on 22q11.2 (Chr22: 18,529,953–18,531,269; hg18, see Table I, underlined) are very close to the recently reported proximal (Chr22: 17,036,078–17,398,178; hg18) and distal (Chr22:19,830,112–20,122,222; hg18) breakpoints of the 22q11.2 syndrome (DiGeorge syndrome) as determined by a 244K oligonucleotide-based aCGH analysis [Bittel et al., 2009]. However, unlike the 6p25 rearrangement(s) reported here and elsewhere [Chanda et al., 2008], the region on either side of the 22q11.2 breakpoints contain LCRs [Bittel et al., 2009]. Accordingly, the mechanism for recurrent 22q11.2 rearrangements is NAHR [Shaikh et al., 2007; Emanuel, 2008].
We speculate that several different mechanisms may be involved in rearrangements within the terminal 6p25 region. While this region is devoid of LCRs, microhomologies, other short repetitive sequences, specific pause or cleavage sites, or other structures such as chromatin higher-order structures or genomic copy number changes in the vicinity of potential breakpoints, may become important constraining elements favoring one rearrangement mechanism over another in different circumstances.
A number of genes of potential importance in morphogenesis within our patient's rearrangement may be related to her phenotype. Among the dosage-sensitive developmental genes in this region, FOXC1 (OMIM #601090) is involved in a number of ocular anomalies [Kume et al., 1998; Lehmann et al., 2000; Nishimura et al., 2001; Lehmann et al., 2002; Gould et al., 2004; Maclean et al., 2005; Chanda et al., 2008]. FOXC1 mutations have been identified in individuals with a variety of anterior chamber defects such as the Rieger eye anomaly, Peters anomaly, posterior embryotoxon, and iridogoniodysgenesis [Nishimura et al., 2001; Lin et al., 2005]. Both increased and decreased gene dosage of FOXC1 are associated with anterior-chamber defects of the eye [Nishimura et al., 2001]. FOXC1, along with FOXC2, influences cochlear morphogenesis through direct activation of TBX1 expression [Yamagishi et al., 2003; Braunstein et al., 2009]. FOXC1 is also involved in cardiac, renal, and skeletal development [Nishimura et al., 2001]. Furthermore, murine models with homozygous and heterozygous FOXC1 mutations show anterior chamber and skeletal anomalies similar to humans [Kume et al., 1998; Smith et al., 2000].
DUSP22 appears to be involved in activating the c-Jun N-terminal kinase (JNK) signaling pathway as well as dephosphorylating and deactivating p38. JNK activity is specifically required for the late-stage differentiation of osteoblasts, required for normal bone formation and development [Matsuguchi et al., 2009]. The p38 mitogen-activated protein kinase pathway and specifically p38alpha play critical roles in skeletal muscle formation and differentiation [Perdiguero et al., 2007]. Our array analyses and TaqMan® copy number analysis suggest a homozygous loss of genomic material at the 5′ end of DUSP22 (Figs. 3B and 4A–C). It is possible that this homozygous genomic loss may render both copies of DUSP22 or its products non-functional but this remains to be proven. Whether the observed skeletal anomalies of our patient are also partially the result of DUSP22 insufficiency or loss of function, or interruption of expression of other genes in addition to haploinsufficiency of FOXC1, is unclear at this point and requires functional testing and/or additional careful characterization of DUSP22 and adjacent regions from other patients with similar phenotype. Kannu et al. [2006] report a patient with multiple skeletal anomalies of the lower limb who had a 2.2–2.4 Mb terminal 6p25 microdeletion analyzed by FISH that was remarkably similar to that found in our patient. However, the patient described in Kannu et al. [2006] declined to participate in further DNA testing.
Genomic rearrangements are increasingly recognized as an important contributor to human disease. The use of increasingly higher resolution oligonucleotide-based aCGHs to interrogate genomic rearrangements will help identify complex rearrangements, provide additional opportunities for determining the rearrangement mechanisms, and examine genotype–phenotype relationships, especially in instances where breakpoints disrupt known genes or as yet unstudied transcripts. However, these opportunities are limited by the uneven distribution in the number of informative probes mapped to various regions of a chromosome as noted in this report.
ACKNOWLEDGMENTS
We would like to thank Dr. Donna M. Martin and Dr. Thomas W. Glover for critically reading this manuscript and for their comments. We also thank Dr. Susan L. Dagenais and Dr. Robert H. Lyons from the University of Michigan DNA Sequencing Core and Dr. Thomas W. Glover for help with the SNP array analysis. We thank Tracy Simmons for her technical help with the TaqMan® Copy Number Assay. We also thank Dr. Salim Aftimos for looking into further testing of one of his patient's DNA—a patient with a similar 6p25 rearrangement as noted in this report. This study was supported in part by funds from MMGL, the Department of Pediatrics, and the Department of Human Genetics at the University of Michigan. Part of this work was presented at the 51st Annual Short Course on Medical and Experimental Mammalian Genetics, The Jackson Laboratory, Bar Harbor, ME. JKB was supported in part by NICHD K12-grant HD028820. VKM was supported by an NIH Mentored Scientist Award (K08-HD045609).
Footnotes
Present address of Vinod K. Misra is Division of Genetic and Metabolic Disorders, Children's Hospital of Michigan, 3901 Beaubien Blvd., Detroit, MI 48201.
How to Cite this Article:
Bedoyan JK, Lesperance MM, Ackley T, Iyer RK, Innis JW, Misra VK. 2011. A complex 6p25 rearrangement in a child with multiple epiphyseal dysplasia.
Am J Med Genet Part A 155:154–163.
LCRs are low copy repeats also called duplicons or segmental duplications and are defined as repetitive DNA segments 10-500 kb in size with ≥95 sequence identity.
REFERENCES
- Baldwin EL, Lee JY, Blake DM, Bunke BP, Alexander CR, Kogan AL, Ledbetter DH, Martin CL. Enhanced detection of clinically relevant genomic imbalances using a targeted plus whole genome oligonucleotide microarray. Genet Med. 2008;10:415–429. doi: 10.1097/GIM.0b013e318177015c. [DOI] [PubMed] [Google Scholar]
- Bittel DC, Yu S, Newkirk H, Kibiryeva N, Holt A III, Butler MG, Cooley LD. Refining the 22q11.2 deletion breakpoints in DiGeorge syndrome by aCGH. Cytogenet Genome Res. 2009;124:113–120. doi: 10.1159/000207515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braunstein EM, Monks DC, Aggarwal VS, Arnold JS, Morrow BE. Tbx1 and Brn4 regulate retinoic acid metabolic genes during cochlear morphogenesis. BMC Dev Biol. 2009;9:31. doi: 10.1186/1471-213X-9-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caluseriu O, Mirza G, Ragoussis J, Chow EW, MacCrimmon D, Bassett AS. Schizophrenia in an adult with 6p25 deletion syndrome. Am J Med Genet Part A. 2006;140A:1208–1213. doi: 10.1002/ajmg.a.31222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanda B, Asai-Coakwell M, Ye M, Mungall AJ, Barrow M, Dobyns WB, Behesti H, Sowden JC, Carter NP, Walter MA, Lehmann OJ. A novel mechanistic spectrum underlies glaucoma-associated chromosome 6p25 copy number variation. Hum Mol Genet. 2008;17:3446–3458. doi: 10.1093/hmg/ddn238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitty LS, McCrimmon R, Temple IK, Russell-Eggitt IM, Baraitser M. Dominantly inherited syndrome comprising partially absent eye muscles, hydrocephaly, skeletal abnormalities, and a distinctive facial phenotype. Am J Med Genet. 1991;40:417–420. doi: 10.1002/ajmg.1320400407. [DOI] [PubMed] [Google Scholar]
- DeScipio C. The 6p subtelomere deletion syndrome. Am J Med Genet Part C. 2007;145C:377–382. doi: 10.1002/ajmg.c.30156. [DOI] [PubMed] [Google Scholar]
- Emanuel BS. Molecular mechanisms and diagnosis of chromosome 22q11.2 rearrangements. Dev Disabil Res Rev. 2008;14:11–18. doi: 10.1002/ddrr.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Cruz D, Mendoza R, Villar V, Sanchez-Corona J, Garcia-Cruz MO, Rojas Q, Chavez-Anaya F, Nazara Z, Barrios MT, Cantu JM. A distinct dysmorphic syndrome with congenital glaucoma and probable autosomal recessive inheritance. Ophthalmic Paediatr Genet. 1990;11:35–40. doi: 10.3109/13816819009012947. [DOI] [PubMed] [Google Scholar]
- Gould DB, Jaafar MS, Addison MK, Munier F, Ritch R, MacDonald IM, Walter MA. Phenotypic and molecular assessment of seven patients with 6p25 deletion syndrome: Relevance to ocular dysgenesis and hearing impairment. BMC Med Genet. 2004;5:17. doi: 10.1186/1471-2350-5-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings PJ, Ira G, Lupski JR. A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet. 2009;5:e1000327. doi: 10.1371/journal.pgen.1000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue K, Osaka H, Thurston VC, Clarke JT, Yoneyama A, Rosenbarker L, Bird TD, Hodes ME, Shaffer LG, Lupski JR. Genomic rearrangements resulting in PLP1 deletion occur by nonhomologous end joining and cause different dysmyelinating phenotypes in males and females. Am J Hum Genet. 2002;71:838–853. doi: 10.1086/342728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannu P, Oei P, Slater HR, Khammy O, Aftimos S. Epiphyseal dysplasia and other skeletal anomalies in a patient with the 6p25 microdeletion syndrome. Am J Med Genet Part A. 2006;140A:1955–1959. doi: 10.1002/ajmg.a.31411. [DOI] [PubMed] [Google Scholar]
- Koshino T, Konno T, Ohzeki T. Bone and joint manifestations of Rieger's syndrome: A report of a family. J Pediatr Orthop. 1989;9:224–230. [PubMed] [Google Scholar]
- Kume T, Deng KY, Winfrey V, Gould DB, Walter MA, Hogan BL. The forkhead/winged helix gene Mf1 is disrupted in the pleiotropic mouse mutation congenital hydrocephalus. Cell. 1998;93:985–996. doi: 10.1016/s0092-8674(00)81204-0. [DOI] [PubMed] [Google Scholar]
- Lee JA, Carvalho CM, Lupski JR. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell. 2007;131:1235–1247. doi: 10.1016/j.cell.2007.11.037. [DOI] [PubMed] [Google Scholar]
- Lehmann OJ, Ebenezer ND, Ekong R, OcakaL, Mungall AJ, Fraser S, McGill JI, Hitchings RA, Khaw PT, Sowden JC, Povey S, Walter MA, Bhattacharya SS, Jordan T. Ocular developmental abnormalities and glaucoma associated with interstitial 6p25 duplications and deletions. Invest Ophthalmol Vis Sci. 2002;43:1843–1849. [PubMed] [Google Scholar]
- Lehmann OJ, Ebenezer ND, Jordan T, Fox M, Ocaka L, Payne A, Leroy BP, Clark BJ, Hitchings RA, Povey S, Khaw PT, Bhattacharya SS. Chromosomal duplication involving the forkhead transcription factor gene FOXC1 causes iris hypoplasia and glaucoma. Am J Hum Genet. 2000;67:1129–1135. doi: 10.1016/s0002-9297(07)62943-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin RJ, Cherry AM, Chen KC, Lyons M, Hoyme HE, Hudgins L. Terminal deletion of 6p results in a recognizable phenotype. Am J Med Genet Part A. 2005;136A:162–168. doi: 10.1002/ajmg.a.30784. [DOI] [PubMed] [Google Scholar]
- Livak K, Schmittgen T. Analysis of relative gene expression data using real-time quantitation PCR and the 2−ΔΔCt method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Lupski JR. Genomic disorders: Structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet. 1998;14:417–422. doi: 10.1016/s0168-9525(98)01555-8. [DOI] [PubMed] [Google Scholar]
- Maclean K, Smith J, St. Heaps L, Chia N, Williams R, Peters GB, Onikul E, McCrossin T, Lehmann OJ, Ades LC. Axenfeld-Rieger malformation and distinctive facial features: Clues to a recognizable 6p25 micro-deletion syndrome. Am J Med Genet Part A. 2005;132A:381–385. doi: 10.1002/ajmg.a.30274. [DOI] [PubMed] [Google Scholar]
- Matsuguchi T, Chiba N, Bandow K, Kakimoto K, Masuda A, Ohnishi T. JNK activity is essential for Atf4 expression and late-stage osteoblast differentiation. J Bone Miner Res. 2009;24:398–410. doi: 10.1359/jbmr.081107. [DOI] [PubMed] [Google Scholar]
- Nishimura DY, Searby CC, Alward WL, Walton D, Craig JE, Mackey DA, Kawase K, Kanis AB, Patil SR, Stone EM, Sheffield VC. A spectrum of FOXC1 mutations suggests gene dosage as a mechanism for developmental defects of the anterior chamber of the eye. Am J Hum Genet. 2001;68:364–372. doi: 10.1086/318183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perdiguero E, Ruiz-Bonilla V, Gresh L, Hui L, Ballestar E, Sousa-Victor P, Baeza-Raja B, Jardi M, Bosch-Comas A, Esteller M, Caelles C, Serrano AL, Wagner EF, Munoz-Canoves P. Genetic analysis of p38 MAP kinases in myogenesis: Fundamental role of p38alpha in abrogating myoblast proliferation. EMBO J. 2007;26:1245–1256. doi: 10.1038/sj.emboj.7601587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Rojas LX, Garcia-Cruz D, Mendoza-Topete R, Barba LB, Barrios MT, Patino-Garcia B, Lopez-Cardona MG, Nuno-Arana I, Garcia-Ortiz JE, Cantu JM. Familial iridogoniodysgenesis and skeletal anomalies: A probable new autosomal recessive disorder. Clin Genet. 2004;66:23–29. doi: 10.1111/j.0009-9163.2004.00271.x. [DOI] [PubMed] [Google Scholar]
- Shaikh TH, O'Connor RJ, Pierpont ME, McGrath J, Hacker AM, Nimmakayalu M, Geiger E, Emanuel BS, Saitta SC. Low copy repeats mediate distal chromosome 22q11.2 deletions: Sequence analysis predicts breakpoint mechanisms. Genome Res. 2007;17:482–491. doi: 10.1101/gr.5986507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw CJ, Lupski JR. Implications of human genome architecture for rearrangement-based disorders: The genomic basis of disease. Hum Mol Genet. 2004;13:R57–R64. doi: 10.1093/hmg/ddh073. [DOI] [PubMed] [Google Scholar]
- Shaw CJ, Lupski JR. Non-recurrent 17p11.2 deletions are generated by homologous and non-homologous mechanisms. Hum Genet. 2005;116:1–7. doi: 10.1007/s00439-004-1204-9. [DOI] [PubMed] [Google Scholar]
- Smith RS, Zabaleta A, Kume T, Savinova OV, Kidson SH, Martin JE, Nishimura DY, Alward WL, Hogan BL, John SW. Haploinsufficiency of the transcription factors FOXC1 and FOXC2 results in aberrant ocular development. Hum Mol Genet. 2000;9:1021–1032. doi: 10.1093/hmg/9.7.1021. [DOI] [PubMed] [Google Scholar]
- Toffolatti L, Cardazzo B, Nobile C, Danieli GA, Gualandi F, Muntoni F, Abbs S, Zanetti P, Angelini C, Ferlini A, Fanin M, Patarnello T. Investigating the mechanism of chromosomal deletion: Characterization of 39 deletion breakpoints in introns 47 and 48 of the human dystrophin gene. Genomics. 2002;80:523–530. [PubMed] [Google Scholar]
- Yamagishi H, Maeda J, Hu T, McAnally J, Conway SJ, Kume T, Meyers EN, Yamagishi C, Srivastava D. Tbx1 is regulated by tissue-specific forkhead proteins through a common Sonic hedgehog-responsive enhancer. Genes Dev. 2003;17:269–281. doi: 10.1101/gad.1048903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Gu W, Hurles ME, Lupski JR. Copy number variation in human health, disease, and evolution. Annu Rev Genomics Hum Genet. 2009;10:451–481. doi: 10.1146/annurev.genom.9.081307.164217. [DOI] [PMC free article] [PubMed] [Google Scholar]