

Significance
Our changed lifestyle, including decreased physical activity and increased food consumption, is leading to a pandemic of obesity and type 2 diabetes, the metabolic syndrome. Impaired release of hormones, such as insulin, from excitable cells usually is considered a symptom, not a cause, of this syndrome. Here, however, we show that a small genetic modification, replacing synaptosomal-associated protein of 25 kDa (SNAP-25), SNAP-25b with SNAP-25a in the machinery mediating stimulus-dependent release of hormones and neurotransmitters is sufficient to provoke hypothalamic dysfunction, obesity, and type 2 diabetes. When combined with a Western diet, this genetic condition triggers severe diabesity. Our work expands previous knowledge supporting the notion that many individuals have an increased susceptibility to developing metabolic disease because of a genetic predisposition.
Keywords: SNARE, insulin secretion, glucose metabolism, hypothalamus, regulated exocytosis
Abstract
Synaptosomal-associated protein of 25 kDa (SNAP-25) is a key molecule in the soluble N-ethylmaleimide–sensitive factor attachment protein (SNARE) complex mediating fast Ca2+-triggered release of hormones and neurotransmitters, and both splice variants, SNAP-25a and SNAP-25b, can participate in this process. Here we explore the hypothesis that minor alterations in the machinery mediating regulated membrane fusion can increase the susceptibility for metabolic disease and precede obesity and type 2 diabetes. Thus, we used a mouse mutant engineered to express normal levels of SNAP-25 but only SNAP-25a. These SNAP-25b–deficient mice were exposed to either a control or a high-fat/high-sucrose diet. Monitoring of food intake, body weight, hypothalamic function, and lipid and glucose homeostases showed that SNAP-25b–deficient mice fed with control diet developed hyperglycemia, liver steatosis, and adipocyte hypertrophy, conditions dramatically exacerbated when combined with the high-fat/high-sucrose diet. Thus, modified SNARE function regulating stimulus-dependent exocytosis can increase the vulnerability to and even provoke metabolic disease. When combined with a high-fat/high-sucrose diet, this vulnerability resulted in diabesity. Our SNAP-25b–deficient mouse may represent a diabesity model.
On-going lifestyle changes, including oversized meals with excessive amounts of sugar and fat, have led to a worldwide pandemic of obesity and type 2 diabetes (T2D) (1). These diseases and their comorbidities cause individual suffering and represent a heavy financial burden on society (2, 3). The term “diabesity” is used to define the coincidence of obesity with T2D under conditions of exaggerated intake of energy-dense diets (4–6). Moreover, genomewide association studies (GWAS) have identified polymorphisms associated with obesity and T2D, indicating that genetic factors predispose certain individuals to diabesity (7–11).
Signs of metabolic diseases include impaired regulated release of hormones, particularly insulin as in T2D (4, 12). Likewise, the secretion of inflammatory markers and other peripheral bioactive peptides is either increased (e.g., leptin, resistin, and adipsin) or decreased (e.g., ghrelin and adiponectin) (13–16). Synaptic and nonsynaptic transmission involving the release of neuropeptides and neurotransmitters, especially in hypothalamic areas controlling eating behavior and energy balance, are involved too (17–19).
In excitable cells, the release of messenger molecules is a consequence of regulated membrane fusion and involves soluble N-ethylmaleimide–sensitive factor attachment proteins (SNAREs) (20, 21), including synaptosomal-associated protein of 25 kDa (SNAP-25). SNAP-25 is expressed as two developmentally regulated and alternatively spliced isoforms, SNAP-25a and SNAP-25b, which differ in only 9 of 206 amino acids (22, 23). In the mouse brain, SNAP-25a precedes SNAP-25b expression during development, but by the second postnatal week SNAP-25b becomes the major splice variant, concomitantly with a dramatic increase in total SNAP-25 expression (23). In endocrine and neuroendocrine cells SNAP-25a appears to be the dominant isoform throughout life, although the expression patterns of the two splice variants are not dedicated exclusively to certain cell types (24, 25). Either SNAP-25a or SNAP-25b can participate in the core SNARE complex, which in addition comprises the two proteins syntaxin and vesicle-associated membrane protein (VAMP) (21, 26). Normal expression levels of total SNAP-25 appear to be critical for precise synaptic function (27, 28), and a reduction affects SNARE protein assembly and synaptic plasticity and even can cause neurodegeneration (28, 29). Even if both SNAP-25 splice variants mediate fast Ca2+-triggered release, SNAP-25b–containing SNARE complexes are believed to have a higher degree of stability and therefore increase the pool of primed vesicles, resulting in release dynamics different from those involving SNAP-25a (26, 30).
Polymorphisms in the human Snap25 gene have been associated with weight gain after antipsychotic treatment, with altered levels of serum triglycerides (31, 32), and also with the severity of the metabolic syndrome in T2D (33). Furthermore, polymorphisms in genes expressing proteins directly interacting with SNAP-25 have been implicated in childhood obesity, impaired glucose metabolism and obesity, age of onset of T2D, and insulin requirement in T2D (34–36). Moreover, polymorphisms in the human L-type channel (37) and the KATP channel (38) have been directly associated with T2D. These channels are indispensable for glucose-stimulated insulin secretion from pancreatic β cells and thus directly influence SNARE protein function and efficiency. Thus, emerging evidence suggests that direct or indirect alterations of SNARE protein function can increase the susceptibility and predisposition to serious metabolic illness.
To test this hypothesis experimentally, we used a genetically engineered mutant mouse expressing normal levels of SNAP-25 but with the nine amino acids specific for the SNAP-25b splice variant converted, by knockout/knockin replacement, to the SNAP-25a sequence (30). Using these mice, we studied the interplay between modified SNARE function and metabolic parameters during a 7-wk diet intervention involving a control standard food diet (CoD) and a high-fat/high-sucrose diet (Western diet, WeD). Our results demonstrate that SNAP-25b deficiency alone predisposes to metabolic disease and that the progressing pathology is accelerated dramatically by a high-fat/high-sucrose diet, leading to diabesity.
Results
SNAP-25–Deficient Mice Develop an Obese Phenotype.
To investigate whether SNAP-25b deficiency results in an increased risk for developing obesity and related metabolic conditions, SNAP-25b–deficient (MT) and WT mice were subjected to a 7-wk diet intervention starting in the fifth postnatal week (Fig. S1 and Table S1). The body weights of MT and WT mice in the different experimental groups were similar before the start of the diet intervention, although sex differences were observed (Table S2). The CoD-fed male SNAP-25b–deficient mice developed a significant increase of body weight compared with WT controls around week 5 of the diet intervention (P < 0.05) (Fig. 1A). In CoD-fed female SNAP-25b–deficient mice, differences in body weight increase, as compared with WT females (P < 0.05), were already statistically significant after 4 wk of diet intervention (Fig. 1B). The increased body weight in male and female CoD-fed MT mice, as compared with their WT controls, was sustained until the end of the study (P < 0.05) (Fig. 1 A and B; also see Fig. S3 D and E). Interestingly, the increase in body weight in both male and female CoD-fed MT mice followed a pattern similar to that observed in WeD-fed WT mice (Fig. 1 A and B). WeD-fed SNAP-25b–deficient MT mice exhibited a dramatic increase in body weight compared with all other experimental groups (P < 0.001) (Fig. 1 A and B), the difference being statistically significant in males after 3 wk on the WeD (P < 0.01) (Fig. 1A) and in females after only 1 wk on the WeD (P < 0.01) (Fig. 1B). All differences increased progressively in both sexes until the end of the study. After 7 wk of diet intervention, the length (nose to tail root) of the mice in the experimental groups did not differ from that of control mice (Fig. 1C); however, the body mass index, BMI, was increased significantly (P < 0.01 to P < 0.001) (Fig. 1D).
Fig. S1.

Summary of the experimental procedures followed in male and female mice of all genotypes from birth to the end of the diet-intervention study, matching the age of the animals with the corresponding human age. (This figure is related to Materials and Methods.)
Table S1.
Nutritional and energetic profiles of the CoD and WeD
| Diet profile | CoD | WeD |
| Nutritional profile | ||
| Proteins, % | 14.5 | 20.0 |
| Nonessential amino acids | 8.0 | 12.8 |
| Essential amino acids | 5.5 | 7.3 |
| Fat, % | 4.5 | 21.0 |
| Cholesterol, % | 0.018 | 0.21 |
| Linoleic acid, % | 1.0 | 5.39 |
| Linolenic acid, % | 0.19 | 1.48 |
| Arachidonic acid, % | 0.2 | 0.0 |
| Saturated fatty acids, % | 22.0 | 25.8 |
| Monounsaturated fatty acids, % | 23.0 | 12.6 |
| Polyunsaturated fatty acids, % | 53.8 | 2.86 |
| Fiber, % | 4.9 | 0.2 |
| Carbohydrates, % | 60.1 | 50.0 |
| Minerals, % | 2.65 | 0.04 |
| Vitamins, % | <5 | 0.01 |
| Water, % | <11 | <9 |
| Energy profile | ||
| Energy, kcal/g | 3.0 | 4.7 |
| Proteins, % of kcal | 17.7 | 17.0 |
| Fat, % of kcal | 10.5 | 40.0 |
| Carbohydrates, % of kcal | 71.7 | 43.0 |
Nutritional composition is expressed in the percentage (wt/wt) of the different nutrients. The contribution of each nutrient to the energy content of the diets is expressed both in kilocalories and percentage of total calories. Data were provided by the manufactures (CoD: R70, Lantmännen; WeD: D12079B, Research Diets Inc.). (This table is related to Materials and Methods.)
Table S2.
Body weight and blood parameters in animals before diet intervention at age 5 wk
| Males | Females | |||
| WT mice | MT mice | WT mice | MT mice | |
| Body weight, g | 19.77 ± 0.43 | 18.88 ± 0.42 | 16.25 ± 0.35 | 16.26 ± 0.51 |
| Blood glucose, mg/dL | 108.90 ± 3.64 | 109.80 ± 4.60 | 104.04 ± 1.92 | 104.91 ± 5.86 |
| Blood triglycerides, mg/dL | 111.61 ± 2.58 | 108.63 ± 2.74 | 110.71 ± 2.19 | 108.42 ± 3.61 |
| Blood cholesterol, mg/dL | 117.2 ± 4.92 | 116.21 ± 3.49 | 112.67 ± 2.95 | 114.242 ± 4.03 |
Body weight, preprandial blood glucose, triglycerides, and cholesterol levels were similar in male and female SNAP-25b–deficient mice and their corresponding WT controls before starting the diet-intervention study. Blood glucose levels were measured using a FreeStyle Lite Glucometer (Abbot Diabetes Care). Blood lipids were determined using a multiCare-in device (Biochemical Systems International Srl). The number of animals used in this experiment was as follows: body weight: males, WT n = 17; MT n = 16; females, WT n = 18; MT n = 14; nonfasting blood parameters: males, WT n = 10; MT n = 12; females, WT n = 11; MT n = 11. (This table is related to Figs. 1, 2, and 5.)
Fig. 1.

Overweight and impaired eating habits. (A and B) Monitoring of body weight. Compared with other groups, a significant increase in body weight is seen in male WeD-fed SNAP-25b–deficient MT mice after 3 wk of diet intervention (A) and in females after 1 wk of diet intervention (B). CoD-fed MT mice become significantly heavier than CoD-fed WT mice after 5 wk of the diet intervention in males (A) and after 4 wk in females (B), as indicated by red numbering on the y axes of A and B. Discontinuous red line in A and B marks the body weight of male (A) and female (B) WT-CoD mice at the time point of the diet study when all experimental groups became overweight. (C) Nose-tip to tail-root length was measured at the end of the 7-wk diet intervention and demonstrated no significant differences between experimental groups. (D) All experimental groups demonstrate a higher BMI than CoD-fed WT mice. (E) The average weight-matched daily food intake (grams of food consumed per gram of body weight) is significantly lower in male CoD-fed MT mice than in male CoD-fed WT mice, whereas female MT mice are hyperphagic. All animals on the CoD show a decrease in food ingested independent of genotype and sex. (F) Male CoD-fed MT mice exhibit lower calorie intake than CoD-fed WT mice but become hypercaloric when fed the WeD. Female CoD- or WeD-fed MT mice show a higher calorie intake than CoD-fed WT animals. (G) Average calorie efficiency is increased in all experimental groups compared with CoD-fed WT mice. *P < 0.05; **P < 0.01; ***P < 0.001 compared with CoD-fed WT mice. See also Figs. S2 and S3.
Fig. S3.

Eating habits in CoD-fed male MT mice. (A) Daily food and calorie intake was monitored during the last week of the diet-intervention study (6–7 wk of diet intervention;11–12 wk of age) in CoD-fed male SNAP-25b–deficient mice and the corresponding WT littermates. The absolute values of food and calorie intake were corrected by the body weight of the animals. (B) The measurement of these parameters reveals that WT mice under physiological conditions consumed 77.56 ± 1.28% of the daily amount of food and calories during the dark period, and only 22.44 ± 1.27% during the day. In contrast, male MT mice consumed approximately the same amount of food and calories during the light (53.64 ± 2.90%) and dark (46.36 ± 2.92%) hours, suggesting that the obese phenotype that developed in CoD-fed male MT mice resulted from impaired eating behavior. (C) Daily calorie efficiency was calculated using the formula: Δ body weight (in grams)/[daily calorie intake (in kilocalories)/body weight (in grams)]. (D and E) Body weight (D) and body weight gain (E) were determined in CoD-fed male SNAP-25b–deficient mice and their WT littermates in the same week and in the same light and dark periods as specified for the daily food and calorie intake. WT/MT-CoD7:00 PM vs. WT/MT-CoD7:00 AM: *P < 0.05; ***P < 0.001; MT-CoD7:00 PM vs. WT-CoD7:00 PM: ##P < 0.01; ###P < 0.001; MT-CoD7:00 AM vs. WT-CoD7:00 AM: §P < 0.05; §§P < 0.01; §§§P < 0.001. (This figure is related to Fig. 1E.)
To investigate possible differences in eating habits, food and calorie intake were measured and corrected by the body weight of each individual animal (Fig. 1E and Fig. S2 A and B). The average food and calorie intake of male CoD-fed MT mice was significantly lower than that of CoD-fed WT mice (P < 0.01 to P < 0.001) (Fig. 1 E and F and Figs. S2 A and C and S3 A and B). In contrast, CoD-fed female SNAP-25b–deficient MT mice demonstrated an increased food and calorie intake relative to WT controls (P < 0.05 to P < 0.01) (Fig. 1 E and F and Fig. S2 B and D). Moreover, the average calorie efficiency in CoD-fed MT mice was greater than in their corresponding WT controls (males, P < 0.05; females, P < 0.01) (Fig. 1G and Figs. S2 E and F and S3C), possibly as a result of increased adiposity (see below). The daily food consumption of WeD-fed WT mice was reduced compared with that of CoD-fed WT animals (P < 0.001) (Fig. 1E and Fig. S2 A and B). Thus, both male and female WeD-fed WT mice were isocaloric (Fig. 1F and Fig. S2 C and D). In any case, the WeD induced significantly increased calorie efficiency (P < 0.05 to P < 0.0010 (Fig. 1G and Fig. S2 E and F) in WT mice of both sexes. The food intake of WeD-fed SNAP-25b–deficient MT mice was reduced significantly compared with their control CoD-fed WT counterparts (P < 0.01 to P < 0.001) (Fig. 1E and Fig. S2 A and B). Furthermore, both male and female WeD-fed MT mice were hypercaloric (P < 0.05 to P < 0.001) (Fig. 1 E and F and Fig. S2 B and D) and presented an increased calorie efficiency compared with corresponding WT controls.
Fig. S2.

Cumulative food and calorie intake. Cumulative food intake (A and B), calorie intake (C and D), and calorie efficiency (E and F) were determined in male and female mice from all experimental groups during the whole time course of the diet intervention. Absolute values from each parameter were corrected by the body weight of the animals. *P < 0.05; **P < 0.01; ***P < 0.001 compared with CoD-fed WT mice. (This figure is related to Fig. 1 E–G.)
Thus, SNAP-25b deficiency, without the WeD, was sufficient to induce an obese phenotype. In WeD-fed WT animals the increased body weight apparently was related directly to diet composition, whereas SNAP-25b deficiency generated an impaired energy balance with elevated caloric efficiency, resulting in higher body weight. Additionally, CoD-fed male MT mice demonstrated a changed eating behavior which most likely contributed to obesity (SI Results).
SNAP-25b Deficiency Impairs Insulin Secretion and Glucose Homeostasis.
To characterize insulin secretion and glucose homeostasis, we performed standard glucose tolerance tests (GTT) by i.p. injection of glucose (Fig. 2 A and B). During the time course of the experiment both blood glucose and serum insulin levels were measured (Fig. 2 C and D). In addition, we monitored blood glucose in nonfasting mice during the entire diet intervention (Fig. 2 E and F).
Fig. 2.

Impairment of glucose homeostasis and insulin secretion. (A and B) GTT analyses show decreased blood glucose levels in both male (A) and female (B) CoD-fed MT mice after 15 min. Both male and female WeD-fed WT and MT mice exhibit hyperglycemia at the time points analyzed. (C and D) Serum insulin levels evaluated at the same time points as blood glucose are increased significantly only in WeD-fed male MT (C) and female WT (D) mice. (E and F) SNAP-25b deficiency elicits hyperglycemia that is more pronounced and debuts earlier in males. (G) Total pancreas insulin content is increased significantly in all experimental groups compared with CoD-fed WT mice. (H) The HOMAIR is statistically significant only in WeD-fed male MT mice but is significantly higher in all female groups as compared with CoD-fed WT female mice. *P < 0.05; **P < 0.01; ***P < 0.001 compared with CoD-fed WT controls. See also Fig. S4.
There were significant differences in fasting basal glucose levels between SNAP-25b–deficient MT and WT mice (Fig. 2 A and B and Fig. S4A). In females, all groups demonstrated a significant increase in basal blood glucose compared with CoD-fed WT mice (P < 0.05 to P < 0.001) (Fig. 2B and Fig. S4A), but the effect was significant only in WeD-fed MT mice (P < 0.001) (Fig. 2B and Fig. S4A). Both male and female CoD-fed MT mice exhibited significantly lower blood glucose levels 15 min after the glucose challenge compared with CoD-fed WT mice (P < 0.001) (Fig. 2 A and B). When fed the WeD, both WT and MT mice had severely increased blood glucose levels at every time point measured, in a sex-independent manner (P < 0.05 to P < 0.001) (Fig. 2 A and B). The differences in blood glucose concentrations during the GTT also were obvious when the increase in total blood glucose was analyzed as area under the curve (AUC) (Fig. S4B).
Fig. S4.

Glucose homeostasis. (A) Basal blood glucose levels measured after 12-h overnight starvation revealed that all experimental groups presented mild hyperglycemia, independent of sex, except for the WeD-fed male WT mice. Although WeD-fed male WT animals presented a tendency of increased basal glucose levels compared with CoD-fed WT mice, no statistical significance was found. (B) AUC for the increase from basal total blood glucose levels during the entire GTT. (C) Basal serum insulin levels measured after 12-h overnight starvation showed a tendency of increased insulin levels in all experimental groups compared with CoD-fed WT controls. Male and female WeD-fed MT mice showed the most dramatic effects in the basal serum insulin levels. (D) AUC for the increase from basal total serum insulin levels during the entire GTT. Values for basal blood glucose levels (A) and basal serum insulin levels (C) were taken from the 0 time point of the GTT (see Fig. 2 A and B) and the 0 time point of the pattern of insulin secretion (see Fig. 2 C and D), respectively. *P < 0.05; **P < 0.01; ***P < 0.001 compared with CoD-fed WT controls. (This figure is related to Fig. 2.)
The influence of SNAP-25b deficiency and/or diet on basal serum insulin levels was evaluated in animals starved for 12 h after 7 wk of diet intervention (Fig. 2 C and D and Fig. S4C). Here SNAP-25b deficiency combined with the WeD resulted in a dramatic increase in basal serum insulin levels in males (males, P < 0.01; females, P = 0.08) (Fig. 2 C and D and Fig. S4C), but significant changes were not seen in the other groups (Fig. 2 C and D and Fig. S4C). Furthermore, the pattern of insulin secretion appeared to be affected both by genetics and diet (Fig. 2 C and D), with a significant genotype–diet interaction that was increased dramatically, especially in males (P < 0.001) (Fig. 2C). This interaction also was obvious when the AUC for serum insulin levels was analyzed during the entire GTT (P < 0.05) (Fig. S4D).
Preprandial blood glucose levels were normal in all experimental groups before the diet intervention (nonsignificant, NS) (Table S2), but MT mice developed hyperglycemia compared with CoD-fed WT mice (P < 0.01 to P < 0.001) (Fig. 2 E and F). Both primary (genetic) and secondary (dietary) preprandial hyperglycemia became significant after 3 wk of diet intervention in males (P < 0.001) (Fig. 2E) and at 6 wk in females (P < 0.01) (Fig. 2F). This increase continued until the end of the study (P < 0.01 to P < 0.001) (Fig. 2 E and F).
Total pancreas insulin content was determined in all experimental groups (Fig. 2G). SNAP-25b deficiency in itself increased the insulin content, resulting in insulin levels similar to those observed in WT mice at the end of the 7-wk-long diet intervention (P < 0.001) (Fig. 2G). It is noteworthy that the WeD in combination with SNAP-25b deficiency did not result in significantly higher insulin levels than seen with the WeD or SNAP-25b deficiency alone (Fig. 2G). Calculation of the insulin resistance index (the homeostatic model assessment of insulin resistance, HOMAIR) revealed a tendency (NS) to increase in male CoD-fed MT and WeD-fed WT mice, reaching significance in WeD-fed MT male mice (P < 0.05) and in all female groups as compared with CoD-fed WT female mice (P < 0.05) (Fig. 2H).
In summary, although CoD-fed SNAP-25b–deficient mice initially appeared to handle a glucose challenge during a standard GTT better than WT controls, the basal and preprandial blood glucose levels showed that SNAP-25b deficiency alone results in a state similar to T2D diabetes. In males, SNAP-25b deficiency plus the WeD resulted in a severe inability to maintain normal blood glucose levels despite a 16-fold increase in insulin secretion during the GTT, suggesting insulin resistance (SI Results).
Impaired Hypothalamic Feeding Signals in SNAP-25b–Deficient Mice.
The discrepancy between preprandial blood glucose levels and those found in SNAP-25b–deficient MT mice after 12-h starvation indicated uncharacteristic eating habits, possibly resulting from disturbed feeding signals from the brain and in particular from the hypothalamus. Hypothalamic dysfunction associated with obesity and T2D is currently an important area of investigation (18). A diagnostic marker for metabolic impairment is the activation/phosphorylation of a master metabolic regulator, AMP-activated protein kinase (AMPK). We evaluated this switch in the hypothalamus by monitoring AMPK-α1/2 (39, 40). Immunoblotting demonstrated that AMPK activation was compromised in a sex-independent manner in all experimental groups as compared with CoD-fed WT mice (P < 0.001) (Fig. 3 A and B). It is well known that both genetic- and diet-induced obesity account for increasing leptin levels and elicit leptin resistance in a tissue-specific manner (41). Immunoblotting experiments were performed on the leptin receptor, ObR, in hypothalamic lysates (Fig. 3 C and D). As shown in Fig. 3 C and D, the long isoform of hypothalamic ObR, ObRb, was severely down-regulated in WeD-fed animals compared with CoD-fed WT mice, independent of sex and genotype (P < 0.05 to P < 0.01). The WeD-fed SNAP-25b–deficient MT mice at 7 wk of diet intervention displayed the most dramatic ObRb deficiency (P < 0.01) (Fig. 3 C and D). However, ObRb expression remained unchanged in CoD-fed MT mice, independent of the sex (nonsignificant, NS) (Fig. 3 C and D). To characterize further the hypothalamic intracellular signaling pathways regulated by leptin and insulin, we investigated hypothalamic STAT3 and ERK1/2 activation by phosphorylation of these proteins in animals after 7 wk of diet intervention. As shown in Fig. 3 E and F, STAT3 phosphorylation was reduced significantly in all experimental groups compared with CoD-fed WT mice, except for male CoD-fed MT mice (P < 0.05 to P < 0.001) (Fig. 3 E and F). Moreover, there was an increased protein content of hypothalamic phospho-ERK1/2 in all experimental groups, independent of sex (P < 0.05 to P < 0.01) (Fig. 3 G and H). Phosphorylation of ERK1/2 has been described as a response to low-grade inflammation in the hypothalamus of individuals with obesity and T2D (42).
Fig. 3.

Hypothalamic function. Representative semiquantitative Western blots and densitometry analyses of hypothalamic homogenates. (A and B) AMPK-α1/2 phosphorylation is significantly compromised in all experimental groups compared with CoD-fed WT mice, independent of sex. (C and D) Both WeD-fed male and female mice develop ObRb deficiency compared with CoD-fed WT mice. (E and F) Hypothalamic pSTAT3 is decreased in all experimental groups compared with CoD-fed WT mice, except for CoD-fed MT mice, in which only a tendency to decreased pSTAT3 levels was found. (G and H) ERK1/2 phosphorylation was significantly increased in all experimental groups compared with CoD-fed WT animals. β-Actin served as a loading control. (*)P = 0.05–0.09; *P < 0.05; **P < 0.01; ***P < 0.001 compared with CoD-fed WT mice.
Taken together, our results show that SNAP-25b deficiency alone was sufficient to inactivate the regulatory subunit of AMPK partially but did not significantly affect the levels of ObRb expression in the hypothalamus. However, in combination with the WeD, the absence of SNAP-25b expression resulted in both a significant decrease of AMPK phosphorylation and reduced expression of ObRb. These changes were accompanied by defective hypothalamic leptin signaling caused by STAT3 dephosphorylation. The increased phosphorylation of ERK1/2 suggested the presence of a low-grade inflammatory process within the hypothalamus.
Enhanced Accumulation of White Adipose Tissues.
Abdominal obesity, also known as “central obesity,” is the accumulation of excessive abdominal fat around the stomach and abdomen. Visceral (intraabdominal) white adipose tissue (WAT) is located inside the peritoneal cavity and torso and includes mesenteric adipose tissue (MsAT), peri-gonadal AT (PgAT), and retroperitoneal AT (RpAT), as opposed to the subcutaneous adipose tissue (ScAT) located underneath the skin. SNAP-25b–deficient MT mice exhibited a significant increase in all visceral and nonvisceral fat depots (P < 0.05 to P < 0.001) (Fig. 4A and Fig. S5) in both sexes as compared with CoD-fed WT mice. The increase in WAT accumulation in MT mice was even greater than in WeD-fed WT mice (P < 0.05 to P < 0.001) (Fig. 4). Consequently, all types of WAT were increased in all WeD-fed SNAP-25b–deficient MT mice as compared with the other experimental groups (P < 0.05 to P < 0.001) (Fig. 4).
Fig. 4.

Body fat distribution. All experimental groups show increased amounts of ScAT (A), PgAT (B), RpAT (C), and MsAT (D) fat (corrected by body weight). *P < 0.05; **P < 0.01; ***P < 0.001 compared with CoD-fed WT controls. See also Fig. S5.
Fig. S5.
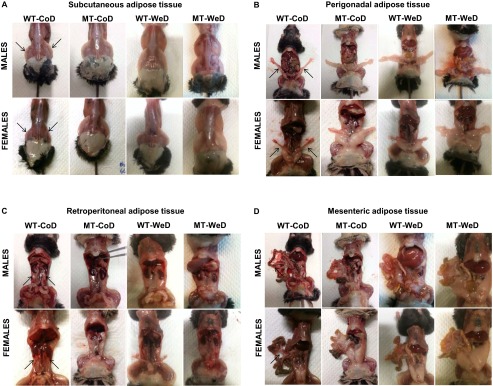
Representative pictures showing body fat distribution. There are evident differences in ScAT (A), PgAT (B), RpAT (C), and MsAT (D) WAT in all experimental groups compared with CoD-fed WT mice. (This figure is related to Fig. 4.)
In summary, SNAP-25b deficiency resulted in central obesity, a condition aggravated when mice were fed the WeD.
Dyslipidemias and Adipocyte Hypertrophy.
Hyperlipidemia, the most common form of dyslipidemia, involves abnormally elevated levels of triglycerides, cholesterol, and/or lipoproteins in the blood (43). These lipid parameters are prognostic/diagnostic markers for metabolic disturbances. Analyses of triglycerides (Fig. 5A) and cholesterol (Fig. 5B) after 12 h of starvation demonstrated a significant increase (P < 0.01 to P < 0.001) in all groups except in WeD-fed female WT mice (NS) (Fig. 5A). Moreover, preprandial monitoring of lipid homeostasis parameters revealed the time of primary hyperlipidemia debut (Fig. 5 C and D). Thus, at 5 wk of age, before the diet intervention, male and female MT mice demonstrated normal triglycerides and cholesterol levels compared with age-matched WT controls (Table S2). During the diet intervention signs of disturbances in lipid homeostasis developed in CoD-fed SNAP-25b–deficient MT mice but did so later in males than in females. Thus, males exhibited hypertriglyceridemia in week 5 of the diet intervention (P < 0.001) (Fig. 5C), vs. week 3 for females (P < 0.001) (Fig. 5D), and higher blood cholesterol levels were observed at week 3 for the males (P < 0.001) (Fig. 5E), vs. week 4 for females (P < 0.001) (Fig. 5F). The differences in triglycerides and cholesterol levels were maintained and increased progressively until the end of the study. Interestingly, the dyslipedemias seen after SNAP-25b deficiency alone followed the same pattern as observed in WT mice on WeD (NS) (Fig. 5 C–F). Finally, the primary dyslipidemias detected in the MT mice caused by the mutation, together with the secondary ones caused by WeD, became severe when WeD concurred with SNAP-25b deficiency (P < 0.001) (Fig. 5 C–F).
Fig. 5.

Lipid homeostasis. (A) Basal blood triglycerides levels after 7 wk of diet intervention are increased significantly in all experimental groups compared with CoD-fed WT animals, except for WeD-fed female WT mice. (B) All experimental groups demonstrate hypercholesterolemia compared with levels in CoD-fed WT mice after 7 wk of diet intervention. (C and D) Preprandial blood triglycerides levels are significantly increased in all groups as compared with CoD-fed WT mice, being most pronounced in the WeD-fed MT mice, and starting after 5 wk of diet intervention in males (C) and after 4 wk of diet intervention in females (D). (E and F) Preprandial blood cholesterol levels show development similar to that of triglycerides but are statistically significant after 3 wk of diet intervention in males (E) and after 4 wk of diet intervention in females (F). *P < 0.05; **P < 0.01; ***P < 0.001 compared with CoD-fed WT mice.
Adipocyte size and turnover are important factors underlying the onset of obesity and metabolic disorders (44). Here we observed that both genetics and diet contributed to adipose cell hypertrophy. Male and female CoD-fed MT mice exhibited enlarged adipose cell size in the ScAT (P < 0.001) (Fig. 6 A and B) and PgAT (P < 0.001) (Fig. 6 C and D) compared with WT mice receiving the same diet. In addition, WeD feeding increased the size of ScAT and PgAT adipocytes compared with CoD-fed WT animals (P < 0.001) (Fig. 6), independent of sex and genotype.
Fig. 6.

Adipocyte hypertrophy. Qualitative (A and C) and quantitative (B and D) analyses of adipocyte area are carried out on H&E-stained cryosections of ScAT (A) and PgAT (C). Both male and female CoD-fed MT mice and WeD-fed WT mice exhibit enlarged adipocytes compared with CoD-fed WT animals, with the strongest effect seen in WeD-fed MT mice (female ScAT and male and female PgAT). (Scale bars: 200 μm.) *P < 0.05; **P < 0.01; ***P < 0.001 compared with CoD-fed WT mice.
In summary, SNAP-25b deficiency elicited dyslipidemias that were severely aggravated in combination with the WeD. In addition, a pronounced increase in the size of adipocytes accompanied obesity.
Impaired Leptin Levels, Liver Lipotoxicity, and Hepatic Leptin Receptor Deficits.
Obesity and its associated metabolic disorders are characterized by changes in the release of humoral factors such as leptin and ghrelin (41). We analyzed basal serum leptin and ghrelin levels after diet intervention (Fig. 7 A and B). Female, but not male, CoD-fed MT mice demonstrated increased leptin levels compared with WT controls (P < 0.01) (Fig. 7A). WeD-fed MT mice of both sexes demonstrated a strong genetic–diet synergistic interaction with an approximately eightfold increase in basal leptin levels (P < 0.01) (Fig. 7A). Ghrelin was decreased in all experimental groups compared with CoD-fed WT mice (P < 0.01 to P < 0.05), except for male WeD-fed MT mice (P = 0.08) (Fig. 7B). Because increased leptin levels can elicit leptin resistance in a tissue-specific manner (41), we also studied ObRs in liver lysates with immunoblotting (Fig. 7C). SNAP-25b deficiency by itself was sufficient to down-regulate liver ObRb, independent of sex and diet (P < 0.05) (Fig. 7 C and D). Interestingly, ObRa, a short isoform of the ObR, was detectable only in MT mice (Fig. 7C). The role of the short isoforms of the ObR is not fully understood but possibly could compensate for a deficiency in ObRb expression.
Fig. 7.

Increased serum leptin levels and liver disease. (A) Basal serum leptin levels are markedly increased (<10-fold) in male and female WeD-fed MT mice compared with CoD-fed WT mice. Significant effects also are detected in CoD-fed MT females and in WeD-fed WT males. (B) Basal serum ghrelin levels are decreased significantly in all experimental groups compared with CoD-fed WT mice, except for WeD-fed male WT mice (P < 0.05). (C) The ObRa isoform is present only in MT mice liver homogenates. β-Actin served as a loading control. (D) There is a significant decrease in liver ObRb protein content in all experimental groups compared with CoD-fed WT mice. (E) ORO-stained liver cryosections show steatohepatitis in all experimental groups compared with CoD-fed WT mice (both sexes). (Scale bars: 50 μm.) (F) Spectrophotometry analyses confirm data obtained with ORO staining. (*)P = 0.05–0.09; *P < 0.05; **P < 0.01; ***P < 0.001 compared with CoD-fed WT mice.
To investigate further whether metabolic organs were affected, we performed qualitative (Fig. 7E) and quantitative (Fig. 7F) analyses of total liver triglycerides. SNAP-25b deficiency alone elicited hepatic steatosis that in WT mice was secondary to diet-induced diabesity (P < 0.001) (Fig. 7 E and F). When SNAP-25b deficiency coincided with WeD administration, the hepatic steatosis turned into severe fatty liver disease (P < 0.001) (Fig. 7 E and F).
In summary, SNAP-25b deficiency resulted in elevated serum leptin levels and impaired expression of ObRb in liver and also in an increase in liver triglycerides and hepatic steatosis. Serum ghrelin levels were decreased in all groups as compared with CoD-fed WT mice. All effects observed in SNAP-25b–deficient MT mice were exaggerated by the WeD (Table S3).
Table S3.
Summary table showing all events affecting body weight, energy balance, glucose metabolism, and lipid homeostasis in both male and female CoD-fed MT, WeD-fed WT, and WeD-fed MT mice
| Males | Females | |||||
| CoD-fed MT | WeD-fed WT | WeD-fed MT | CoD-fed MT | WeD-fed WT | WeD-fed MT | |
| Body weight | ↑ | ↑ | ↑↑↑ | ↑ | ↑↑ | ↑↑↑ |
| BMI | ↑ | ↑ | ↑↑↑ | ↑ | ↑↑ | ↑↑↑ |
| Glycemia | (↑) | — | ↑↑↑ | ↑ | ↑ | ↑↑↑ |
| Insulinemia | (↑) | (↑) | ↑↑ | (↑) | ↑ | (↑) |
| HOMAIR | — | (↑) | ↑↑↑ | ↑ | ↑ | ↑↑↑ |
| Total pancreas insulin content | ↑↑↑ | ↑↑↑ | ↑↑↑ | ↑↑↑ | ↑↑↑ | ↑↑↑ |
| Hepatic triglycerides content | ↑↑↑ | ↑↑↑ | ↑↑↑ | ↑↑↑ | ↑↑↑ | ↑↑↑ |
| Triglyceridemia | ↑↑↑ | ↑↑ | ↑↑↑ | ↑↑↑ | — | ↑↑↑ |
| Cholesterolemia | ↑↑↑ | ↑↑↑ | ↑↑↑ | ↑↑↑ | ↑↑↑ | ↑↑↑ |
| Leptinemia | — | ↑ | ↑↑↑ | ↑↑ | — | ↑↑ |
| Ghrelinemia | ↓ | ↓ | (↓) | ↓ | ↓ | ↓↓↓ |
SI Results
Feeding Behavior and Obesity in SNAP-25b–Deficient Mice.
Cumulative food intake (Fig. S2 A and B), calorie intake (Fig. S2 C and D), and calorie efficiency (Fig. S2 E and F) verify the data presented in Fig. 1 E and G. The fact that SNAP-25b–deficient CoD-fed male MT mice became overweight, even when they ate less than the corresponding WT mice fed the same diet, raises the possibility that the eating pattern is dependent on the light/dark period (63). As shown in Fig. S3 A and B, food and calorie intake in male CoD-fed WT animals was associated mainly with the dark period (active hours), and, consumption during the day amounted to only 22.68 ± 1.24% of the daily intake (Fig. S3B), both in terms of the amount of food eaten and the calories ingested. In contrast, the eating pattern of male SNAP-25b–deficient mice was distributed equally during dark and light hours (Fig. S3 A and B). These results suggest that the obese phenotype of the SNAP-25b–deficient male MT mice is associated with a derangement of the circadian feeding behavior. Interestingly, the calorie efficiency of the CoD was significantly greater in male MT mice than in WT mice, during both the light and the dark periods (P < 0.05) (Fig. S3C). A significantly lower increase in body weight gain was detected in CoD-fed WT mice during the light period compared with the dark period (P < 0.05) (Fig. S3E). Although the increase in body weight was significantly higher in male MT mice than in CoD-fed WT mice, independent of the light cycle (P < 0.001) (Fig. S3 D and E), no statistical differences were found in COD-fed SNAP-25b–deficient mice between 7:00 AM and 7:00 PM (Fig. S3 D and E). Taken together, these results suggest that the higher calorie efficiency detected in CoD-fed male SNAP-25b–deficient mice is caused by increased adiposity, probably the result of altered catabolic pathways.
Glucose Homeostasis.
SNAP-25b deficiency by itself induces higher basal blood glucose levels (expressed in mg*dl−1) in a sex-dependent manner (males, CoD-fed WT, 104.32 ± 9.54; CoD-fed MT, 123.59 ± 4.22, P = 0.06; females, CoD-fed WT, 89.70 ± 5.65; CoD-fed MT, 108.27 ± 4.49, P < 0.05) (Fig. S4A). However, CoD-fed male and female MT mice seem to have improved glucose handling during the GTT challenge as compared with CoD-fed WT mice (Fig. 2 A and B). We therefore calculated the AUC for the increase in blood glucose from basal blood glucose levels for the entire GTT in all experimental groups (Fig. S4B). CoD-fed female MT mice demonstrated a decreased rise of blood glucose at 15 min into the GTT (P < 0.05) (Fig. S4B), whereas in males only a nonsignificant tendency was observed (Fig. S4B). Interestingly, the values for the blood glucose AUC were significantly higher in the WeD-fed animals, independent of genotype and sex (P < 0.05 to P < 0.001) (Fig. S4B). The higher basal serum insulin levels detected (P < 0.05 to P < 0.01) (Fig. S4C) appeared to be insufficient to counteract the increase in blood glucose in WeD-fed SNAP-25b–deficient MT mice. Thus, the AUC for the increase in total serum insulin from basal levels revealed that CoD-fed male MT mice do not increase insulin secretion (Fig. S4D). In CoD-fed female MT animals, there was a tendency (NS) to increase both basal serum insulin levels (P = 0.08) (Fig. S4C) and the total amount of insulin secretion during the GTT (P = 0.06) (Fig. S4D). Similar results were obtained in male and female WeD-fed WT mice (Fig. S4 C and D), indicating a state close to T2D diabetes. Finally, WeD-fed MT mice showed a dramatic increase in basal serum insulin levels that reached statistical significance only in male mice (P < 0.01) (Fig. S4C). A significant increase in insulin secretion was observed in both sexes during the time course of the GTT (P < 0.05; Fig. S4 C and D). For WeD-fed male MT mice this increase was noticeable: Insulin secretion increased 16-fold during the GTT (P < 0.001) (Fig. 2C). These results demonstrate impaired glucose and insulin homeostasis in WeD-fed MT mice, suggesting insulin resistance.
Discussion
SNAP-25 isoforms fine-tune the kinetics of regulated membrane fusion. Here, we took advantage of a SNAP-25b–deficient mouse mutant (30) and explored whether small modifications in SNARE function in excitable cells predispose to obesity and metabolic disease. In CoD-fed MT mice multiple impairments were observed, such as increased body weight, a pronounced effect on calorie efficiency and WAT mass, adipocyte hypertrophy, steatohepatitis, and hypothalamic dysfunction. As expected, WT animals fed the WeD for 7 wk also developed impairments in the parameters analyzed. Moreover, when SNAP-25 deficiency was combined with the WeD a dramatic, synergistic effect was observed. The results we observed are all established features of the human metabolic syndrome (1, 45) and diet-induced diabesity (4–6). Indeed, it appeared that the genetic mutation in combination with the WeD triggered a vicious circle of increased lipid accumulation and impaired glucose homeostasis. Interestingly, although SNAP-25b deficiency and diet generally affected both sexes equally dramatically, there were certain distinct sex-based differences (SI Discussion).
Deficiencies of hormonal secretion, e.g., the impaired release of insulin from pancreatic β cells, usually are considered secondary signs and consequences of the progressing pathology in metabolic disease, such as in T2D (12). Here we asked instead if a minor alteration in membrane fusion dynamics can lead to obesity and metabolic disease, a hypothesis not previously tested experimentally, to our knowledge. SNAP-25, together with VAMP/synaptobrevin and syntaxin, form a core complex essential for mediating regulated membrane fusion. These proteins exist as several splice variants or isoforms, some of which also operate at intracellular membrane-trafficking steps. However, because many of them appear to be functionally interchangeable, the physiological differences, if any, are yet not fully understood (21).
SNAP-25 is essential for stimulus-dependent exocytosis, and disruption of the gene results in death at birth (46). However, SNAP-25 exists as two splice variants, SNAP-25a and SNAP-25b, both of which can support insulin release (24). SNAP-25b–deficient mice are viable and live until adulthood (30). SNAP-25b forms a SNARE complex with a higher degree of stability than SNAP-25a, thereby increasing the pool of primed vesicles (26). Indeed, when Sørensen et al. (26) separately overexpressed either SNAP-25a or SNAP-25b in embryonic adrenal chromaffin cells from SNAP-25–null mice, the burst of Ca2+-evoked catecholamine release differed. Furthermore, impairment of vesicle trafficking and reduced insulin secretion was observed in the blind-drunk mouse, a mutant with a dominant mutation in the SNAP-25b–specific exon, resulting in a further increased stability of the SNARE complex (47). Although SNAP-25a has a lower capacity to keep vesicles in a primed state, little is known how this molecular difference between the splice variants influences animal physiology.
We started the diet intervention at 5 wk of age, a developmentally critical window in the life of mice (i.e., puberty), and assessed different metabolic parameters before, during, and after the intervention. First we characterized glucose homeostasis and insulin secretion. Surprisingly, we detected an apparently improved ability to handle a glucose challenge in CoD-fed SNAP-25b–deficient MT mice. When injected with glucose, starved MT mice did not demonstrate the typical rise in blood glucose, usually peaking after 15 min. The lack of increase in blood glucose was accompanied by facilitated insulin release, suggesting that the presence of SNAP-25a alone does favor insulin exocytosis, perhaps at a lower stimulatory threshold (24, 47). In sharp contrast, in males, SNAP-25b deficiency in combination with the WeD resulted in a 16-fold increase in insulin secreted during GTT. Nonetheless, the preprandial measurements of blood glucose demonstrated a severe inability to maintain normal blood glucose levels, suggesting insulin resistance. However, a closer investigation of basal blood glucose levels during the diet intervention showed that CoD-fed MT mice had preprandial levels comparable with those in WeD-fed WT mice. This finding suggests that the increased nonfasting blood glucose levels in both CoD- and WeD-fed SNAP-25b–deficient mice likely were caused by impaired eating behavior and peripheral insulin resistance.
Altered eating habits usually involve dysfunction in the hypothalamic areas involved in the maintenance of metabolic homeostasis, and it is likely that SNAP-25b deficiency introduces changes in peptidergic/neurotransmitter signaling in those brain centers (18, 19). Therefore, we investigated the expression of the long isoform of the leptin receptor, ObRb, and the phosphorylation status of AMPK, a key switch in leptin signaling in the hypothalamus. To explore further hypothalamic intracellular cascades involving leptin and insulin signaling, we also investigated the phosphorylation status of STAT3 and ERK1/2. Our results indicate that the dramatic synergistic effects of combined SNAP-25b deficiency and the WeD likely are the result of cross-talk between periphery and the brain, in particular hypothalamic dysfunction. This notion is in line with previous studies reporting that the onset of diet-induced obesity and insulin resistance induces permanent effects in both the periphery and the brain (48–50). The impaired eating habits demonstrated by the SNAP-25b–deficient MT mice also included increased feeding during the light hours, suggesting that the circadian systems regulating sleep and/or feeding cycles did not follow a normal pattern. Indeed, dysregulation of these hypothalamic-dependent rhythms alone can induce an obesogenic development (51).
Surprisingly, SNAP-25b deficiency in itself significantly increased baseline serum cholesterol and triglyceride levels and liver triglycerides. Previously, elevated serum triglyceride levels and body weight gain have been described in patients with polymorphism in the human Snap25 gene, although, notably, these patients had received antipsychotic treatment for schizophrenia (31, 32). The hypothalamus controls hepatic lipid metabolism and storage via the autonomic nervous system (52, 53). It is believed that increased sympathetic activity augments the production of triglycerides and that increased parasympathetic activity enhances lipid accumulation in liver (53, 54). Thus, it is plausible that in SNAP-25b–deficient mice neuroexocytosis from autonomic nerves innervating the liver is less well controlled and thus contributes to the phenotype with increased serum triglycerides and liver steatosis (51, 54).
We propose that our SNAP-25b–deficient mice might represent a model for studies of the mechanisms associated with obesity and metabolic conditions. At 5 wk of age SNAP-25b–deficient mice are similar to their WT littermates in body weight and blood triglycerides and cholesterol levels. However, a more detailed characterization of young mice is necessary to determine if differences exist that could explain the mechanism of obesity. Unlike most other rodent obesity models used in diabetes research (e.g., ob/ob and db/db mice and certain fa/fa rats) (55–57), our model does not have a mutation in the genes for leptin or its receptor, but leptin signaling is affected, nevertheless. Only a handful of mutations have been identified in human genes encoding leptin or the leptin receptor in patients with obesity and insulin resistance; however, the majority of patients lack such mutations but still demonstrate defects in leptin signaling (58).
The severe diabese phenotype found in WeD-fed SNAP-25b–deficient mice suggests that any polymorphism in genes directly or indirectly regulating Ca2+-dependent membrane fusion potentially could be associated with an increased risk of developing metabolic disease, especially in combination with increased calorie intake and insufficient physical activity. Our mutant mouse expresses only SNAP-25a, and thus those excitable cells that normally express SNAP-25b in the exocytosis process are affected (23, 25, 26, 59). In this respect, it is intriguing that several genes identified in GWAS or in functional studies as carrying a risk for susceptibility for T2D encode proteins that are important for insulin secretion from β cells, including potassium channels, voltage-gated Ca2+ channels, and G protein-coupled receptors (34–38). These genes also are expressed in other excitable cells, including neurons, and therefore it is possible to envisage a scenario similar to that of our SNAP-25b–deficient mice in which a small imbalance in stimulus-dependent exocytosis mechanisms in excitable cells deregulates the interplay of metabolic signals and is followed by obesity and T2D. A recent publication also links the severity of T2D to an SNP in the human Snap25 gene (33). Therefore it would be interesting to investigate mouse mutants targeting other genes identified in GWAS as susceptibly genes for T2D and obesity and directly or indirectly involved in the control of regulated membrane fusion. Would such mice develop diabesity when exposed to a diet intervention as the SNAP-25b–deficient mice does, as shown in this study? In contrast to conditional knockouts, our model is in agreement with most humans who have a genetic predisposition to metabolic disease; i.e., they have the same mutation in all cells in the body (7–11).
In conclusion, our data suggest that mutations or minor alterations in the expression of the proteins regulating membrane fusion can increase the vulnerability to develop obesity and precede T2D. We hypothesize that in humans, also, such mutations may participate in the initial phase of developing a metabolic disease and, in combination with a hypercaloric diet, may in fact be a factor initiating the development of diabesity.
SI Discussion
SNAP-25b deficiency and diet mostly affected both sexes equally, but there were certain differences (Table S3). For example, (i) the WeD affected body weight and BMI more in female than male WT mice, and triglycerides evolution demonstrated a quicker and higher increase in female than in male WT mice; (ii) in both CoD- and WeD-fed MT mice the increase in body weight occurred earlier in females than in males; (iii) the increased adiposity, mainly affecting the ScAT and the PgAT, was more accentuated in female than in male CoD-fed MT mice; (iv) leptin levels were high in female but not in male CoD-fed MT mice; and (v) all female experimental groups showed significantly reduced levels of ghrelin, but in male mice this change reached statistical significance only in CoD-fed MT and WeD-fed WT mice as compared with CoD-fed WT mice.
Another sex-dependent difference is the feeding pattern of CoD-fed SNAP-25b–deficient mice. In female CoD-fed MT mice, the obese phenotype could be explained by oversized meal consumption. However, obesity in CoD-fed MT mice is related to a changed circadian feeding behavior, i.e., a switch from eating mainly during the dark period at night to continuous snacking. Furthermore, WeD-fed female MT mice demonstrated hyperphagia, whereas the intake of WeD-fed MT males was the same as that of CoD-fed WT mice.
In summary, several sex dissimilarities were observed in our SNAP-25b–deficient mice that likely could be explained by sexual hormones, but the overall outcome of the diet intervention was comparable in males and females.
Materials and Methods
Animals and Diets.
The generation of SNAP-25b–deficient MT mice and their breeding, maintenance, and genotyping were performed as previously described (30). Weight-matched daily food and calorie intake, as well as calorie efficiency, were determined during the entire diet-intervention study (Fig. S1). Body fat distribution was determined in all genotypes after 7 wk of diet intervention. Animals were killed by decapitation, and ScAT, PgAT, RpAT, and MsAT WAT were dissected and weighed. Tissue weight was corrected by the body weight of each individual animal. All animal studies were done in accordance with the guidelines from local authorities and ethical committees, i.e., the Stockholm Northern Animal Experiments Ethics Board, and in accordance with Directive 2010/63/EU of the European Parliament and of the Council on the Protection of Animals Used for Scientific Purposes.
GTTs and Serum and Pancreas Insulin Levels.
Intraperitoneal GTTs were carried out after 12-h overnight starvation. Blood glucose levels were determined using a FreeStyle Glucometer (Abbot Diabetes Care). Serum and total pancreas insulin levels were analyzed using an ultrasensitive mouse insulin ELISA kit (Crystal Chem Inc.). A separate cohort of animals was used to evaluate hyperglycemia, defined as nonfasting blood glucose ≥250 mg/dL. Nonfasting blood glucose was determined in blood obtained from the tail vein (SI Materials and Methods).
Leptin and Ghrelin Measurements in Serum.
Basal leptin and ghrelin levels were determined after 12-h overnight starvation in serum samples of age-matched mice belonging to all experimental groups at the end of the diet-intervention study, using multiplex immunoassays specific for mouse serum samples (BioPlex Pro) (SI Materials and Methods).
Adipocyte Size Quantification.
Adipocyte size quantification was carried out as previously described (SI Materials and Methods) (60).
Triglycerides Content and Oil Red “O” Staining in Liver.
Hepatic triglycerides content were determined in liver samples (61) and lipid visualization by Oil Red “O” (ORO) staining (SI Materials and Methods) (62).
Monitoring Blood Triglycerides and Cholesterol Levels.
A separate cohort of animals was used to analyze basal blood levels of triglycerides and cholesterol. Nonfasting blood triglycerides and cholesterol were monitored in the same cohort used for hyperglycemia studies. Blood lipid levels were measured using a multiparameter diagnostic device for triglycerides and cholesterol (multiCare-in; Biochemical Systems International Srl) (SI Materials and Methods).
Western Blotting.
Western blotting was performed as described previously (SI Materials and Methods) (30).
Statistical Analysis.
Quantifications of the data are presented in bar and line graphs created with StatView 5.0 (SAS Institute Inc.). Data represent mean values ± SEM. We used two-way ANOVA and repeated measures ANOVA with a corrected Bonferroni multiple comparison test to calculate statistical significance in all our experiments (SI Materials and Methods).
SI Materials and Methods
We used four different cohorts of animals, each composed of three sets of mice. We consider one set of animals when all experimental groups and controls were present at the same time in each sex for each experimental procedure. The total sets of mice used in each experiment constitute a cohort of animals.
Animals and Diets.
The diet intervention started at the age of 5 wk (Fig. S1) (64), i.e., at adolescence, a critical window for the development of metabolic disorders (65). SNAP-25b–deficient MT mice and WT controls from each sex were divided randomly into two groups with a similar average body weight (Table S2) and were assigned to either (i) the CoD (10.5% of kilocalories from fat, 17.7% from proteins; and 71.7% from carbohydrates) or (ii) the high-fat/high-sucrose WeD (40.0% of kilocalories from fat; 17.0% from proteins; and 43.0% from carbohydrates) (Table S1). Free-feeding animals received these diets for 7 wk (Fig. S1). The mice used were maintained in an environmentally controlled room (22–24 °C) under a regular12 h/12 h light/dark cycle (light on 7:00 AM) with food and water ad libitum. Animal care and experimental procedures were carried out according to the guidelines from local authorities and ethical committees and in accordance with Directive 2010/63/EU of the European Parliament and the Council on the Protection of Animals Used for Scientific Purposes.
Body Weight and BMI.
Mice in cohort 1 were housed three per cage, and body weight was monitored for each genotype and sex twice a week during the whole time course of the diet intervention. At the end of the study, the length of the animals was measured from the tip of the nose to the root of the tail, and the BMI (expressed as grams per square millimeter) was calculated as the relationship between the body weight (in grams) of each animal and its squared length (in square millimeters). These experiments were replicated in three different sets of animals. The number of animals belonging to the first cohort was as follows: males, CoD-fed WT, n = 5; CoD-fed MT, n = 7; WeD-fed WT, n = 12; WeD-fed MT, n = 9; females, CoD-fed WT, n = 8; CoD-fed MT, n = 6; WeD-fed WT, n = 10; WeD-fed MT, n = 8.
Feeding Pattern.
Daily food intake was determined twice a week. Because the calorie intake differed between the CoD- and Wed-fed mice (Table S1), daily calorie consumption was calculated as the kilocalories ingested daily by each individual animal. Finally, calorie efficiency was determined to establish the relationship between body weight gain and calories consumed by each animal (Δ body weight/kcal). All parameters determined for characterization of the feeding pattern were corrected for the body weight of each individual animal in each experimental group. For these experiments, we used a separate cohort of animals (cohort 2), which were placed in individual cages. These experiments were replicated in three different sets of mice. The number of animals belonging to cohort 2 was as follows: males, CoD-fed WT, n = 5; CoD-fed MT, n = 4; WeD-fed WT, n = 5; WeD-fed MT, n = 7; females, CoD-fed WT, n = 8; CoD-fed MT, n = 6; WeD-fed WT, n = 5; WeD-fed MT, n = 5.
As shown in Results, male CoD-fed SNAP-25b–deficient MT mice became overweight (Fig. 1A), even though their average food intake was significantly lower than that of the corresponding WT controls (P < 0.001) (Fig. 1E). To investigate this finding, we carried out a separate experiment with five male WT mice and five male SNAP-25b–deficient mice fed with the CoD diet and daily monitored food intake, body weight, and body weight gain at 7:00 AM (after the active period) and at 7:00 PM (after the inactive period). Calorie intake and calorie efficiency were calculated as previously specified. To ensure that the increase in body weight detected in CoD-fed SNAP-25b–deficient male MT mice was stable (Fig. 1A), this experiment was conducted between wk 6 and 7 of CoD administration.
GTT and Serum Insulin Levels.
A GTT was performed in the animals in cohort 3 after 12-h overnight starvation. Basal blood glucose (0 time point) was measured in blood samples collected from the tail vein. Thereafter, mice received an i.p. glucose injection (2 g/kg body weight), and blood glucose was measured after 15, 30, 60, 90, and 120 min. Blood glucose levels were determined using a FreeStyle Glucometer (Abbot Diabetes Care). Serum insulin levels were analyzed using an ultrasensitive mouse insulin ELISA kit (Crystal Chem Inc.). Insulin levels were determined in serum from blood collected at the same time points as in the GTT. The HOMAIR was calculated using the formula: fasting insulin (mU/L) × fasting blood glucose (mmol/L)/22.5. The AUC was calculated using the basal levels of blood glucose and serum insulin as baselines, respectively, using GraphPad Prism 6 statistical software (GraphPad Software Inc.). These experiments were replicated in three different sets of animals. The number of animals belonging to the third cohort was as follows: males, CoD-fed WT, n = 6; CoD-fed MT, n = 7; WeD-fed WT, n = 11; WeD-fed MT, n = 9; females, CoD-fed WT, n = 8; CoD-fed MT, n = 5; WeD-fed WT, n = 11; WeD-fed MT, n = 9. The number of animals in which serum insulin levels were checked during the time course of the GTT was as follows: males, CoD-fed WT, n = 5; CoD-fed MT, n = 7; WeD-fed WT, n = 7; WeD-fed MT, n = 6; females, CoD-fed WT, n = 8; CoD-fed MT, n = 5; WeD-fed WT, n = 7; WeD-fed MT, n = 5.
Development of Hyperglycemia.
Cohort 4 was used to evaluate hyperglycemia, defined as nonfasting blood glucose ≥250 mg/dL (66). Nonfasting blood glucose was determined once a week during the 7-wk diet intervention in blood obtained from the tail vein at the start of the dark cycle (7:00 PM). The measurements were carried out using a FreeStyle Glucometer (Abbot Diabetes Care). These preprandial blood glucose values measured along the whole time course of the diet intervention provided valuable information on the starting time point of hyperglycemia. The number of animals belonging to cohort 4 was as follows: males, CoD-fed WT, n = 5; CoD-fed MT, n = 6; WeD-fed WT, n = 5; WeD-fed MT, n = 6; females, CoD-fed WT, n = 6; CoD-fed MT, n = 5; WeD-fed WT, n = 5; WeD-fed MT, n = 6.
Total Pancreas Insulin Content.
The whole pancreas was dissected out quickly and was washed once in PBS. Pancreas preparations were resuspended in 1.4 mL of acid ethanol containing 75% (vol/vol) ethanol in 0.2 M HCl. Thereafter, the pancreas preparations were homogenized by an automatic tissue homogenizer (Sigma-Aldrich) until no big pieces were observed. Subsequently, pancreas homogenates were vortexed and kept at −20 °C until use. The total pancreas insulin content of these extracts was measured using an ultrasensitive mouse insulin ELISA kit (Chrystal Chem Inc.). Before insulin determination by ELISA, pancreas homogenates were defrosted at 4 °C and sonicated (frequency: 30 Hz; 12 pulses) (Branson Sonifier Cell Disruptor B15; Branson Ultrasonic). Pancreas homogenates then were diluted at 1:5,000, 1:10,000, and 1:20,000 followed by the ELISA experiment. Protein levels in each pancreas homogenate were measured by the Bradford method, and the total pancreas insulin content (in nanograms per milliliter) obtained from ELISA immunoassay was normalized with the total protein concentration (in milligrams per milliliter) of each extract. Pancreas samples were obtained from animals belonging to the first cohort of mice. The number of animals used in this experiment was as follows: males, CoD-fed WT, n = 5; CoD-fed MT, n = 6; WeD-fed WT, n = 6; WeD-fed MT, n = 4; females, CoD-fed WT, n = 7; CoD-fed MT, n = 7; WeD-fed WT, n = 7; WeD-fed MT, n = 6.
Body Fat Distribution.
Subcutaneous (ScAT) and visceral (MsAT, RpAT, PgAT) WAT from animals in all experimental groups belonging to cohort 1 was dissected quickly. MsAT and RpAT WAT were removed and quantified as previously described (67). For isolation and quantification of SbAT, fat tissue located under the skin and around the waist and legs of the animal was dissected. PgAT is located on both sides of the abdominal cavity surrounding the internal genitalia. Because both SbAT and PgAT are paired, we weighed both pads independently. The number of mice per experimental group and controls was the same as previously specified in Body Weight and BMI above.
Blood Triglycerides and Cholesterol Levels.
Development of hyperlipidemia was monitored in the same animals and with the same procedures used for the nonfasting blood glucose. Preprandial blood triglycerides and cholesterol levels were determined using a multiparameter diagnostic device for triglycerides and cholesterol (multiCare-in, Biochemical Systems International Srl) from a drop of blood obtained from the tail vein. At the end of the diet-intervention study, this cohort of animals was subjected to 12-h overnight starvation, and then basal blood triglycerides and cholesterol levels were determined. The number of mice per experimental groups and controls was the same as previously specified in Development of Hyperglycemia above.
Adipocyte Size Quantification.
ScAT and PgAT tissues were dissected quickly, washed once immediately in PBS, and formalin-fixed by immersion for 2 wk. After fixation, both ScAT and PgAT were placed in 10% (vol/vol) sucrose-impregnated cardboard blocks, frozen in dry ice, and stored at −80 °C until use. Tissues were sectioned at 20-μm thickness using a cryostat (Microm HM500M/Cryostar NX70; Thermo Scientific) and thawed onto SuperFrost Plus microscope slides (VWR International). Slides were stained with H&E (Histolab) and mounted in VectaMount permanent mounting medium (Vector Laboratories, Inc.). The stained adipocytes were analyzed in an optical microscope (Leica), and images were obtained at 10× and 40× magnification objectives. The area of the adipocytes (in square micrometers) was analyzed using the ImageJ program (National Institutes of Health), and images were obtained with the 10× magnification objective. ScAT and PgAT were taken randomly from three individual animals from each experimental group in the first cohort of mice. These experiments were replicated three times.
Multiplex Experiments.
For multiplex experiments we used blood samples taken randomly from animals belonging to the different cohorts of mice, always after a 12-h period of overnight starvation. The number of animals used in this experiment was as follows: males, CoD-fed WT, n = 6; CoD-fed MT, n = 7; WeD-fed WT, n = 11; WeD-fed MT, n = 11; females, CoD-fed WT, n = 8; CoD-fed MT, n = 5; WeD-fed WT, n = 6; WeD-fed MT, n = 11.
Liver Triglycerides Content.
For the current protocol, 100–300 mg of the left lobe of each liver was dissected out and frozen immediately in liquid nitrogen. Samples from animals belonging to all experimental groups were stored at −80 °C until use. For triglyceride extraction, the tissues were defrosted at 4 °C, immersed quickly in 350 μL of ethanolic KOH [0.1 M potassium hydroxide (Sigma-Aldrich) in absolute ethanol (Merck)], and incubated overnight at 55 °C until no oil layer was visible. After homogenization, each sample was brought up to 1 mL with a 1:1 ethanol:water solution, vortexed, and centrifuged at 4 °C (7,000 × g) for 5 min. The supernatant was moved to another tube, and again the volume was brought up to 1.2 mL with a 50% (vol/vol) ethanol solution. Two hundred microliters of each sample were neutralized with 215 μL of 1 M MgCl2 solution, vortexed, and left on ice for 10 min. After incubation, samples were centrifuged under the conditions described above, and the supernatant was moved to a new tube. Free liver triglycerides were analyzed using Free Glycerol Reagent (Sigma-Aldrich) and Glycerol Standards (Sigma-Aldrich) to construct the standard curve. The glycerol concentration in each cuvette (triolein equivalents) was measured by spectrophotometry (SAFAS-MONACO spectrophotometer; Monaco) at a wavelength of 540 nm and was determined by extrapolation with the standard curve. Total triglyceride content expressed in milligrams per gram of liver was calculated. Liver samples were taken randomly from animals belonging to cohorts 1 and 2. The number of animals used in this experiment was as follows: males, CoD-fed WT, n = 7; CoD-fed MT, n = 7; WeD-fed WT, n = 7; WeD-fed MT, n = 8; females, CoD-fed WT, n = 7; CoD-fed MT, n = 7; WeD-fed WT, n = 7; WeD-fed MT, n = 7.
ORO Staining in Liver.
For lipid visualization, ORO staining was conducted on 14-μm-thick liver sections. Before sectioning, liver samples from the left lobe of each animal were dissected out, washed once in PBS, and formalin-fixed for 24 h. After fixation, tissues were placed on cardboard blocks impregnated with 10% (vol/vol) sucrose, frozen in dry ice, and stored at −80 °C until use. Sections were obtained in a cryostat (Microm HM500M/Cryostar NX70) and thawed onto SuperFrost Plus microscope slides (VWR International). After 1 h at room temperature slides were rinsed in 60% (vol/vol) isopropyl alcohol, stained in freshly prepared 0.1% ORO solution (Sigma-Aldrich) for 15 min, rinsed in 60% (vol/vol) isopropyl alcohol, washed in distilled water, and mounted with 0.25% DABCO mounting medium (Sigma-Aldrich). Liver sections from all experimental groups were analyzed, and lipid droplets were identified using a 5× objective (Leica) followed by amplification to 10× and 40× magnification. The ORO staining was conducted in three individual animals from each experimental group and controls in triplicate. Liver samples were taken randomly from animals belonging to cohorts 1 and 2.
Western Blotting.
Hypothalamus and liver samples from all experimental groups were homogenized in ice-cold buffer containing 0.42 mM NaCl, 20 mM Hepes (pH 7.9), 1 mM Na4P2O7, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, 20% (vol/vol) glycerol, 1 μg/mL aprotinin, 1 μg/mL leupeptin, 20 mM sodium fluoride, 1 mM trisodium orthovanadate, and 2 mM phenylmethylsulfonyl fluoride. Tubes containing homogenates were exposed to a thermal shock at −80 °C in liquid nitrogen and thawed to 37 °C three consecutive times, and centrifuged at 10,000 × g for 20 min. Then the supernatant was collected. Protein levels in the supernatant of both liver and hypothalamus homogenates were measured by the Bradford method, and volumes were adjusted in Laemmli buffer [50 mM Tris (pH 6.8), 10% (vol/vol) SDS, 10% (vol/vol) glycerol, 5% (vol/vol) mercaptoethanol, and 2 mg/mL bromophenol blue] to 2 μg/μL of protein concentration. Equivalent amounts of proteins (40 μg) were loaded using a Trans-Blot apparatus (Bio-Rad). Each sample was size-separated in 10% (vol/vol) SDS/PAGE and transferred to PVDF membranes (GE Healthcare). For immunoblotting, membranes were blocked with 5% (wt/vol) nonfat dried milk. Primary antibodies directed against total and phosphorylated (Thr172) forms of AMPK-α1/2 (1:1,000 dilution; Cell Signaling), total and phosphorylated (Tyr705) STAT3 (1:1,000 dilution; Santa Cruz Biotechnology), total and phosphorylated (Thr202/Tyr204) ERK1/2 (1:1,000 dilution; Cell Signaling), or ObR (1:1,000 dilution; Santa Cruz Biotechnology) were applied overnight. After incubation with IgG–peroxidase complexes, blots were incubated in commercial enhanced chemiluminescence reagents (ECL-Prime; GE Healthcare), and membranes were exposed to a luminescent image analyzer (Las-1000 Plus; Fuji). Obtained images were quantified using ImageJ software. Values for all proteins were normalized to β-actin (1:1,000; Sigma-Aldrich). Western blots for hypothalamus and liver lysates were carried out on four individual animals from each experimental group and controls; experiments were performed in triplicate. Hypothalamus and liver samples were taken randomly from cohort 1.
Statistical Analysis.
StatView 5.0 software was used for statistical analysis. Cell-line charts were analyzed by repeated-measures ANOVA. For time point by time point analysis, two-way ANOVA was used. Cell bar charts were analyzed by two-way ANOVA. Unpaired t-test analysis was used to compare one experimental group with its corresponding control group during physiological conditions. P < 0.05 was considered to be statistically significant. Statistical significance was verified with nonparametric statistics using the Kruskal–Wallis H-test, followed by the post hoc Mann–Whitney, Fisher’s protected least significant difference (PLSD), and Student–Newman–Keuls tests. Bonferroni correction was applied in all statistical analyses. When the cell counts were not equal, the harmonic mean was used to estimate the number. Data are presented as mean ± SEM. The variance between groups was tested using Bartlett’s test and was examined in all datasets. We did not use statistical methods to predetermine sample size. Sample sizes were based on pilot experiments and previous studies of similar nature. We did not use randomization, but all genotypes were represented in each experiment. Normal distribution was verified by using the Kolmogorov–Smirnov normality test.
Acknowledgments
We thank Charlotte Mattsson for help with the BioPlex analyses, Åse Mattsson for initial studies of glucose homeostasis, Mingdong Zhang for his help with the submission process, and Jessica Lundgren, Torun Wallgren, Sandra Mezei, and Sandra Olsson for help with the animals. This work was supported by grants from the Swedish Research Council, the Family Erling-Persson Foundation, Karolinska Institutet funds, the Magnus Bergvalls Foundation, the Gun and Bertil Stohnes Foundation, Längmanska Kulturfonden, Peter and Augusta Hedlund’s Foundation, the Novo Nordisk Foundation, the Fogelströms Foundation, and the Sven Mattssons Foundation.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1511951112/-/DCSupplemental.
References
- 1.Ng M, et al. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980-2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet. 2014;384(9945):766–781. doi: 10.1016/S0140-6736(14)60460-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang YC, McPherson K, Marsh T, Gortmaker SL, Brown M. Health and economic burden of the projected obesity trends in the USA and the UK. Lancet. 2011;378(9793):815–825. doi: 10.1016/S0140-6736(11)60814-3. [DOI] [PubMed] [Google Scholar]
- 3.Withrow D, Alter DA. The economic burden of obesity worldwide: A systematic review of the direct costs of obesity. Obesity reviews: An official journal of the International Association for the Study of Obesity. 2011;12(2):131–141. doi: 10.1111/j.1467-789X.2009.00712.x. [DOI] [PubMed] [Google Scholar]
- 4.Anonymous From the NIH: Successful diet and exercise therapy is conducted in Vermont for “diabesity”. JAMA. 1980;243(6):519–520. [PubMed] [Google Scholar]
- 5.Kral JG. Diabesity: Palliating, curing or preventing the dysmetabolic diathesis. Maturitas. 2014;77(3):243–248. doi: 10.1016/j.maturitas.2013.12.004. [DOI] [PubMed] [Google Scholar]
- 6.Schmidt MI, Duncan BB. Diabesity: An inflammatory metabolic condition. Clin Chem Lab Med. 2003;41(9):1120–1130. doi: 10.1515/CCLM.2003.174. [DOI] [PubMed] [Google Scholar]
- 7.Rosengren AH, et al. Reduced insulin exocytosis in human pancreatic β-cells with gene variants linked to type 2 diabetes. Diabetes. 2012;61(7):1726–1733. doi: 10.2337/db11-1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kebede MA, Attie AD. Insights into obesity and diabetes at the intersection of mouse and human genetics. Trends Endocrinol Metab. 2014;25(10):493–501. doi: 10.1016/j.tem.2014.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Langenberg C, et al. Gene-lifestyle interaction and type 2 diabetes: The EPIC interact case-cohort study. PLoS Med. 2014;11(5):e1001647. doi: 10.1371/journal.pmed.1001647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scott RA, et al. RISC Study Group EPIC-InterAct Consortium Common genetic variants highlight the role of insulin resistance and body fat distribution in type 2 diabetes, independent of obesity. Diabetes. 2014;63(12):4378–4387. doi: 10.2337/db14-0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Torres JM, Cox NJ, Philipson LH. Genome wide association studies for diabetes: Perspective on results and challenges. Pediatr Diabetes. 2013;14(2):90–96. doi: 10.1111/pedi.12015. [DOI] [PubMed] [Google Scholar]
- 12.Seino S, Shibasaki T, Minami K. Dynamics of insulin secretion and the clinical implications for obesity and diabetes. J Clin Invest. 2011;121(6):2118–2125. doi: 10.1172/JCI45680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leiter EH, Reifsnyder PC, Xiao Q, Mistry J. Adipokine and insulin profiles distinguish diabetogenic and non-diabetogenic obesities in mice. Obesity (Silver Spring) 2007;15(8):1961–1968. doi: 10.1038/oby.2007.234. [DOI] [PubMed] [Google Scholar]
- 14.Lo JC, et al. Adipsin is an adipokine that improves β cell function in diabetes. Cell. 2014;158(1):41–53. doi: 10.1016/j.cell.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Perret J, De Vriese C, Delporte C. Polymorphisms for ghrelin with consequences on satiety and metabolic alterations. Curr Opin Clin Nutr Metab Care. 2014;17(4):306–311. doi: 10.1097/MCO.0000000000000072. [DOI] [PubMed] [Google Scholar]
- 16.Müller TD, Tschöp MH. Ghrelin - a key pleiotropic hormone-regulating systemic energy metabolism. Endocr Dev. 2013;25:91–100. doi: 10.1159/000346590. [DOI] [PubMed] [Google Scholar]
- 17.Chee MJ, Colmers WF. Y eat? Nutrition. 2008;24(9):869–877. doi: 10.1016/j.nut.2008.06.007. [DOI] [PubMed] [Google Scholar]
- 18.Gao Q, Horvath TL. Neurobiology of feeding and energy expenditure. Annu Rev Neurosci. 2007;30:367–398. doi: 10.1146/annurev.neuro.30.051606.094324. [DOI] [PubMed] [Google Scholar]
- 19.Morton GJ, Meek TH, Schwartz MW. Neurobiology of food intake in health and disease. Nat Rev Neurosci. 2014;15(6):367–378. doi: 10.1038/nrn3745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Söllner T, Bennett MK, Whiteheart SW, Scheller RH, Rothman JE. A protein assembly-disassembly pathway in vitro that may correspond to sequential steps of synaptic vesicle docking, activation, and fusion. Cell. 1993;75(3):409–418. doi: 10.1016/0092-8674(93)90376-2. [DOI] [PubMed] [Google Scholar]
- 21.Südhof TC. The molecular machinery of neurotransmitter release (Nobel lecture) Angew Chem Int Ed Engl. 2014;53(47):12696–12717. doi: 10.1002/anie.201406359. [DOI] [PubMed] [Google Scholar]
- 22.Bark IC. Structure of the chicken gene for SNAP-25 reveals duplicated exon encoding distinct isoforms of the protein. J Mol Biol. 1993;233(1):67–76. doi: 10.1006/jmbi.1993.1485. [DOI] [PubMed] [Google Scholar]
- 23.Bark IC, Hahn KM, Ryabinin AE, Wilson MC. Differential expression of SNAP-25 protein isoforms during divergent vesicle fusion events of neural development. Proc Natl Acad Sci USA. 1995;92(5):1510–1514. doi: 10.1073/pnas.92.5.1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gonelle-Gispert C, et al. SNAP-25a and -25b isoforms are both expressed in insulin-secreting cells and can function in insulin secretion. Biochem J. 1999;339(Pt 1):159–165. [PMC free article] [PubMed] [Google Scholar]
- 25.Yamamori S, et al. Differential expression of SNAP-25 family proteins in the mouse brain. J Comp Neurol. 2011;519(5):916–932. doi: 10.1002/cne.22558. [DOI] [PubMed] [Google Scholar]
- 26.Sørensen JB, et al. Differential control of the releasable vesicle pools by SNAP-25 splice variants and SNAP-23. Cell. 2003;114(1):75–86. doi: 10.1016/s0092-8674(03)00477-x. [DOI] [PubMed] [Google Scholar]
- 27.Bark C, et al. Developmentally regulated switch in alternatively spliced SNAP-25 isoforms alters facilitation of synaptic transmission. J Neurosci. 2004;24(40):8796–8805. doi: 10.1523/JNEUROSCI.1940-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sharma M, et al. CSPα knockout causes neurodegeneration by impairing SNAP-25 function. EMBO J. 2012;31(4):829–841. doi: 10.1038/emboj.2011.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sharma M, Burré J, Südhof TC. CSPα promotes SNARE-complex assembly by chaperoning SNAP-25 during synaptic activity. Nat Cell Biol. 2011;13(1):30–39. doi: 10.1038/ncb2131. [DOI] [PubMed] [Google Scholar]
- 30.Johansson JU, et al. An ancient duplication of exon 5 in the Snap25 gene is required for complex neuronal development/function. PLoS Genet. 2008;4(11):e1000278. doi: 10.1371/journal.pgen.1000278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Müller DJ, et al. The SNAP-25 gene may be associated with clinical response and weight gain in antipsychotic treatment of schizophrenia. Neurosci Lett. 2005;379(2):81–89. doi: 10.1016/j.neulet.2004.12.037. [DOI] [PubMed] [Google Scholar]
- 32.Musil R, et al. SNAP-25 gene polymorphisms and weight gain in schizophrenic patients. J Psychiatr Res. 2008;42(12):963–970. doi: 10.1016/j.jpsychires.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 33.Chen YL, et al. Associations between genetic variants and the severity of metabolic syndrome in subjects with type 2 diabetes. Genet Mol Res. 2015;14(1):2518–2526. doi: 10.4238/2015.March.30.10. [DOI] [PubMed] [Google Scholar]
- 34.Goldlust IS, et al. Unique Rare Chromosome Disorder Support Group Mouse model implicates GNB3 duplication in a childhood obesity syndrome. Proc Natl Acad Sci USA. 2013;110(37):14990–14994. doi: 10.1073/pnas.1305999110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Romeo S, et al. Search for genetic variants of the SYNTAXIN 1A (STX1A) gene: The -352 A>T variant in the STX1A promoter associates with impaired glucose metabolism in an Italian obese population. Int J Obes. 2008;32(3):413–420. doi: 10.1038/sj.ijo.0803743. [DOI] [PubMed] [Google Scholar]
- 36.Tsunoda K, Sanke T, Nakagawa T, Furuta H, Nanjo K. Single nucleotide polymorphism (D68D, T to C) in the syntaxin 1A gene correlates to age at onset and insulin requirement in Type II diabetic patients. Diabetologia. 2001;44(11):2092–2097. doi: 10.1007/s001250100015. [DOI] [PubMed] [Google Scholar]
- 37.Reinbothe TM, et al. The human L-type calcium channel Cav1.3 regulates insulin release and polymorphisms in CACNA1D associate with type 2 diabetes. Diabetologia. 2013;56(2):340–349. doi: 10.1007/s00125-012-2758-z. [DOI] [PubMed] [Google Scholar]
- 38.Olson TM, Terzic A. Human K(ATP) channelopathies: Diseases of metabolic homeostasis. Pflugers Arch. 2010;460(2):295–306. doi: 10.1007/s00424-009-0771-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Long YC, Zierath JR. AMP-activated protein kinase signaling in metabolic regulation. J Clin Invest. 2006;116(7):1776–1783. doi: 10.1172/JCI29044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grahame Hardie D. AMP-activated protein kinase: A key regulator of energy balance with many roles in human disease. J Intern Med. 2014;276(6):543–559. doi: 10.1111/joim.12268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Friedman J. 20 years of leptin: Leptin at 20: An overview. J Endocrinol. 2014;223(1):T1–T8. doi: 10.1530/JOE-14-0405. [DOI] [PubMed] [Google Scholar]
- 42.Tanti JF, Jager J. Cellular mechanisms of insulin resistance: Role of stress-regulated serine kinases and insulin receptor substrates (IRS) serine phosphorylation. Curr Opin Pharmacol. 2009;9(6):753–762. doi: 10.1016/j.coph.2009.07.004. [DOI] [PubMed] [Google Scholar]
- 43.Chatterjee C, Sparks DL. Hepatic lipase, high density lipoproteins, and hypertriglyceridemia. Am J Pathol. 2011;178(4):1429–1433. doi: 10.1016/j.ajpath.2010.12.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Arner P, et al. Dynamics of human adipose lipid turnover in health and metabolic disease. Nature. 2011;478(7367):110–113. doi: 10.1038/nature10426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ford ES, Giles WH, Dietz WH. Prevalence of the metabolic syndrome among US adults: Findings from the third National Health and Nutrition Examination Survey. JAMA. 2002;287(3):356–359. doi: 10.1001/jama.287.3.356. [DOI] [PubMed] [Google Scholar]
- 46.Washbourne P, et al. Genetic ablation of the t-SNARE SNAP-25 distinguishes mechanisms of neuroexocytosis. Nat Neurosci. 2002;5(1):19–26. doi: 10.1038/nn783. [DOI] [PubMed] [Google Scholar]
- 47.Jeans AF, et al. A dominant mutation in Snap25 causes impaired vesicle trafficking, sensorimotor gating, and ataxia in the blind-drunk mouse. Proc Natl Acad Sci USA. 2007;104(7):2431–2436. doi: 10.1073/pnas.0610222104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bouret SG. Role of early hormonal and nutritional experiences in shaping feeding behavior and hypothalamic development. J Nutr. 2010;140(3):653–657. doi: 10.3945/jn.109.112433. [DOI] [PubMed] [Google Scholar]
- 49.Schwartz MW, et al. Cooperation between brain and islet in glucose homeostasis and diabetes. Nature. 2013;503(7474):59–66. doi: 10.1038/nature12709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Williams KW, Elmquist JK. From neuroanatomy to behavior: Central integration of peripheral signals regulating feeding behavior. Nat Neurosci. 2012;15(10):1350–1355. doi: 10.1038/nn.3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vieira E, Burris TP, Quesada I. Clock genes, pancreatic function, and diabetes. Trends Mol Med. 2014;20(12):685–693. doi: 10.1016/j.molmed.2014.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tiniakos DG, Lee JA, Burt AD. Innervation of the liver: Morphology and function. Liver. 1996;16(3):151–160. doi: 10.1111/j.1600-0676.1996.tb00721.x. [DOI] [PubMed] [Google Scholar]
- 53.Bruinstroop E, Fliers E, Kalsbeek A. Hypothalamic control of hepatic lipid metabolism via the autonomic nervous system. Best Pract Res Clin Endocrinol Metab. 2014;28(5):673–684. doi: 10.1016/j.beem.2014.05.001. [DOI] [PubMed] [Google Scholar]
- 54.Lam TK. Neuronal regulation of homeostasis by nutrient sensing. Nat Med. 2010;16(4):392–395. doi: 10.1038/nm0410-392. [DOI] [PubMed] [Google Scholar]
- 55.Frühbeck G. Intracellular signalling pathways activated by leptin. Biochem J. 2006;393(Pt 1):7–20. doi: 10.1042/BJ20051578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang Y, et al. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372(6505):425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 57.Wang B, Chandrasekera PC, Pippin JJ. Leptin- and leptin receptor-deficient rodent models: Relevance for human type 2 diabetes. Curr Diabetes Rev. 2014;10(2):131–145. doi: 10.2174/1573399810666140508121012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Farooqi IS, O’Rahilly S. 20 years of leptin: Human disorders of leptin action. J Endocrinol. 2014;223(1):T63–T70. doi: 10.1530/JOE-14-0480. [DOI] [PubMed] [Google Scholar]
- 59.Delgado-Martínez I, Nehring RB, Sørensen JB. Differential abilities of SNAP-25 homologs to support neuronal function. J Neurosci. 2007;27(35):9380–9391. doi: 10.1523/JNEUROSCI.5092-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9(7):671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Norris AW, et al. Muscle-specific PPARgamma-deficient mice develop increased adiposity and insulin resistance but respond to thiazolidinediones. J Clin Invest. 2003;112(4):608–618. doi: 10.1172/JCI17305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mehlem A, Hagberg CE, Muhl L, Eriksson U, Falkevall A. Imaging of neutral lipids by oil red O for analyzing the metabolic status in health and disease. Nat Protoc. 2013;8(6):1149–1154. doi: 10.1038/nprot.2013.055. [DOI] [PubMed] [Google Scholar]
- 63.Morales L, et al. Shift of circadian feeding pattern by high-fat diets is coincident with reward deficits in obese mice. PLoS One. 2012;7(5):e36139. doi: 10.1371/journal.pone.0036139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Valladolid-Acebes I, et al. Spatial memory impairment and changes in hippocampal morphology are triggered by high-fat diets in adolescent mice. Is there a role of leptin? Neurobiol Learn Mem. 2013;106:18–25. doi: 10.1016/j.nlm.2013.06.012. [DOI] [PubMed] [Google Scholar]
- 65.Shield J, Summerbell C. 2008. Obesity in childhood. Obesity, Science and Practice eds Williams G, Frühbeck G (Wiley-Blackwell, Chichester, UK), pp 509–542.
- 66.Leiter EH, et al. Comparison of two new mouse models of polygenic type 2 diabetes at the Jackson Laboratory, NONcNZO10Lt/J and TALLYHO/JngJ. J Diabetes Res. 2013;2013:165327. doi: 10.1155/2013/165327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gil-Ortega M, et al. Adaptative nitric oxide overproduction in perivascular adipose tissue during early diet-induced obesity. Endocrinology. 2010;151(7):3299–3306. doi: 10.1210/en.2009-1464. [DOI] [PubMed] [Google Scholar]