

Abstract
Block II of Plasmodium vivax merozoite surface protein 3α (PvMSP3α) is conserved and has been proposed as a potential candidate for a malaria vaccine. The present study aimed to compare sequence diversity in PvMSP3a block II at a local microgeographic scale in a village as well as from larger geographic regions (countries and worldwide). Blood samples were collected from asymptomatic carriers of P. vivax in a village at the western border of Thailand and PvMSP3α was amplified and sequenced. For population genetic analysis, 237 PvMSP3α block II sequences from eleven P. vivax endemic countries were analyzed. PvMSP3α sequences from 20 village-level samples revealed two length variant types with one type containing a large deletion in block I. In contrast, block II was relatively conserved; especially, some non-synonymous mutations were extensively shared among 11 parasite populations. However, the majority of the low-frequency synonymous variations were population specific. The conserved pattern of nucleotide diversity in block II sequences was probably due to functional/structural constraints, which were further supported by the tests of neutrality. Notably, a small region in block II that encodes a predicted B cell epitope was highly polymorphic and showed signs of balancing selection, signifying that this region might be influenced by the immune selection and may serve as a starting point for designing multi-antigen/stage epitope based vaccines against this parasite.
Introduction
Vaccine is a long-term hope to combat malaria—a major infectious disease responsible for more than half a million deaths annually around the world. The alarming signals of artemisinin resistant parasites seemingly to follow the same path initially laid down by chloroquine-resistant parasites across international borders in Southeast Asia further urge the development of vaccines against malaria [1, 2]. Vaccine research has been largely focused on Plasmodium falciparum—a species responsible for the majority of malaria-related deaths. However, vaccine research for P. vivax has trailed far behind [3, 4]. Yet, P. vivax is the most widespread human malaria parasite and it causes 50–70 million infections annually [5]. This co-called ‘benign tertian’ malaria parasite has been increasingly recognized as the cause of significant morbidity and mortality. The changing malaria epidemiology worldwide with increasing proportions of P. vivax malaria further highlights the difficulty for controlling this parasite and emphasizes the need to develop integrated control strategies including vaccine for this parasite [6].
Several antigens have been proposed as potential vaccine candidates for P. falciparum [7], and their orthologs in P. vivax (PvAMA-1, PvMSP-1, PvDBP, PvCSP, PvMSP3α, etc.) have also been characterized. Antigenic diversity in these genes [8–14] has significantly hindered the progress in vaccine research [15–17], since multiple antigenic alleles could evade vaccine-induced, allele-specific immunity. In contrast, antigens with low variability or the conserved functional regions of polymorphic antigens are attractive vaccine targets [17], as these regions are assumed to be under functional constraints and possibly have slower evolutionary mechanisms. This approach has been used for MSP3-LSP and GMZ2 vaccines which include conserved C-terminal region of the P. falciparum merozoite surface protein 3 (PfMSP3) gene [18, 19]. Furthermore, various genomics and proteomics approaches are being exploited to identify such conserved regions to overcome the challenges imposed by genetic variations [20].
MSP3 in P. vivax is a family of 11 members with a complex evolutionary history [21, 22]. Two of the loci, MSP3α and 3β, have been widely used as population genetic markers for typing P. vivax isolates based on polymorphisms depicted by PCR/RFLP analysis [10, 23–25]. Previous studies analyzing PvMSP3α gene sequences have observed differential pattern of diversity across different domains of the gene [10, 26, 27]. PvMSP3α is composed of an N-terminal signal sequence, a central alanine rich region and an acidic C-terminus. The alanine rich repeat region of PvMSP3α encodes block I (residues 104–396) and block II (434–687). Block II has been shown to be relatively conserved with non-random variations clustered in two structural motifs; motif I from amino acid position 533 to 538 and motif II from 580 to 587 [26, 27]. Interestingly, the variations within each motif are tightly linked that have generated dimorphic alleles for each motif (motif I: MSELEK/LSKLEE and motif II: TAANVVKD/KEATAAKL). All of these alleles have been found equally prevalent in natural P. vivax populations [10, 24, 26, 27]. Based on this peculiar pattern of variation, block II has generated considerable interest as a potential vaccine candidate. Block II is also known to elicit a pronounced antibody response against clinical malaria infections reported from Papua New Guinea [28] and Brazil Amazon [29]. In fact one of the studies suggested that block II specific antibodies compared to other regions of the gene are more responsive to high density natural infections [28]. All these features point to block II as a potential vaccine candidate or target for sero-epidemiology studies in P. vivax.
A conserved pattern of variation in an antigenic sequence has been widely associated with purifying (negative) selection that can be the result of either structural constraints or strong immune directional selection [30, 31]. On the contrary, high diversity has been usually associated with balancing selection by the immune system [32, 33] but alternatively could be the result of relaxation. Since antigenic diversity is generally influenced by local endemic settings, comparative analysis of the gene in diverse population backgrounds is more informative. Genetic diversity in block II of PvMSP3α has been previously characterized only in a few laboratory adapted strains [26], and limited clinical samples from Thailand [10, 34] and Venezuela [24, 27]. Thus, this study aimed to define the patterns of variation in PvMSP3α block II in samples from a small village (local diversity) compared with the larger geographic structures. By studying the extent and distribution of polymorphisms in PvMSP3α block II among P. vivax samples from 11 countries, we hope to understand the evolutionary mechanism underlying the variation patterns. Genetic diversity in block II preferentially revealed large number of rare alleles, and high frequency variants were restricted to specific genetic regions. The prominent allelic forms of block II were extensively shared by diverse P. vivax populations.
Material and Methods
Study area and sampling
The present study was conducted in a small village Suan Oi in Tha Song Yang district of Tak province located at western border of Thailand (Fig 1A), which is known to contribute highest number of malaria cases in the country [35]. Malaria in this region is hypo-endemic and seasonal [35, 36]. P. falciparum and P. vivax infections are more prevalent and notably, we have reported a large number of asymptomatic infections (only detected by expert microscopists and by PCR assay) in that area recently [35, 36]. Asymptomatic infections usually remain undetected by passive case detection by hospitals and clinics [35], thus present study conducted mass blood surveys in Suan Oi village between June 2011 and June 2013 to determine the magnitude of P. vivax infections at a micro-geographic scale.
Fig 1. Location of sampling sites with PvMSP3α block II allelic forms.

(A) The location of Suan Oi village at the western border of Thailand. (B) A map showing the distribution of block II allelic forms across eleven countries. The black portion of the pie chart indicates shared alleles (with one or more populations), while the white portion shows population specific alleles. Population specific alleles that were observed only once (singletons) are shown as pattern-filled portion in the pie chart. (C) Haplotype network constructed from block II alleles generated using only non-synonymous variations that were seen in more than two isolates. Both the maps of the world and Thailand are taken from Wikimedia Commons.
Finger-pricked blood samples were obtained from all the residents of Suan Oi during mass blood surveys. The presence of malaria parasites in some of the blood samples from participants were confirmed by microscopic examination of Giemsa-stained blood films and by PCR [36]. Genomic DNA was extracted from dried blood spots on Whatman filter paper using a QiaAmp DNA Mini Kit (Qiagen, Germany). Plasmodium species identification was carried out by species-specific rDNA based primers following method described in Snounou et al., 1993. Twenty four samples showing single P. vivax infections were included in sequencing analysis.
Ethics statement
Written informed consent was obtained from the participants or guardians. This study was approved by the Institutional Review Boards of Pennsylvania State University and Thai Ministry of Public Health.
Sequencing of PvMSP3α gene
PvMSP3α in P. vivax samples was amplified using primers described previously [10]. Amplified fragments were visualized on 1.5% agarose gel for approximate size estimation. PCR amplified fragments were further purified using the High Pure PCR cleanup microkit (Roche) and sequenced in both directions using BigDye Terminator v3.1. DNA sequences obtained were assembled using Lasergene software (DNASTAR) with manual editing, and aligned with the Sal I reference gene sequence (PVX_097720) using ClustalW. The sequences corresponding to block II region of PvMSP3α present in all samples were extracted for analysis.
Data collection
PvMSP3α block II sequences generated in the present study were compared and analyzed together with the sequences retrieved from GenBank (http://www.ncbi.nlm.nih.gov/genbank/) and Plasmodb (http://plasmodb.org/plasmo/) database. Totally, 237 sequences were derived from 11 parasite populations, which included 52 samples from Thailand (including 20 samples from the present study [10, 34]; 25 from Myanmar [9], 6 from India, 6 from China, 25 from South Korea [37], 17 from Sri Lanka [38], 12 from Brazil, 22 from Colombia, 23 from Peru, 26 from Venezuela [27] and 15 from Mexico (S1 Table). Excluding indels and multiple alleles, the 695 bp region encoding block II (nucleotide 2078 to 2773) from all 237 samples were used for analysis.
Population genetic analysis
Within population, polymorphism was quantified by total number of segregating sites and haplotypes. Genetic diversity was measured by average pairwise nucleotide diversity (θπ) and haplotype diversity (Hd) [39]. Local diversity measures estimated for each population were compared with overall worldwide diversity. Genetic differentiation between populations was estimated using Wright’s FST−a measure of fixation index [40] and the statistical significance of the FST values was tested through 1000 random permutations. All the above analyses were performed using DnaSP v5 software [41].
Phylogeographic clustering of the isolates was evaluated by Maximum Likelihood (ML) tree in MEGA6 [42] using Tamura and Nei’s model of nucleotide substitution. Support for individual nodes was obtained by performing 500 bootstrap replicates. In order to visualize the distribution of immunologically relevant polymorphisms across populations, haplotypes were constructed from non-synonymous SNPs that were observed in more than two isolates (excluding singletons and doubletons), as singletons and low frequency alleles are not generally considered informative for vaccine design [43]. Haplotype network was drawn by NETWORK (fluxus-engineering.com) using the median joining algorithm [44].
To examine departure from neutrality, we estimated the numbers of synonymous substitutions per synonymous site (dS) and of nonsynonymous substitutions per nonsynonymous site (dN) using the Nei and Gojobori method [45] as implemented in MEGA6. Significance of the difference between dN and dS was estimated with a Z-test of selection. A dN significantly higher than dS is consistent with positive selection, while dS higher than dN is expected under purifying selection. We also used Tajima’s D [46] and Fu and Li’s F* [47] frequency-based tests of neutrality implemented in DnaSP v5 to examine departure from neutrality. Tajima’s D test compares average pairwise nucleotide diversity (θπ) with the standardized number of polymorphic sites per site (θS), whereas Fu and Li’s F* tests excess or lack of singletons by comparing number of singletons and the average number of nucleotide differences between two sequences. Significantly positive values of these tests suggest a recent population bottleneck or balancing selection, whereas negative values indicate population growth or directional selection.
We used an array of methods to detect recombination signals viz; RDP, MaxChi, GENECONV, GARD and minimum number of recombination events (Rm) according to the four-gamete test by Hudson & Kaplan [48]. We used the RDP3 package [49] for RDP [50], MaxChi [51], and GENECONV [52]. These methods are designed to detect recombination breakpoints. RDP and MaxChi are used in sliding window analysis, whereas GENECONV scans for long regions of identity between sequences. We also used another tree based method of recombination detection, Genetic Algorithm for Recombination Detection (GARD) [53] implemented in Datamonkey (datamonkey.org) [54], which identifies recombination breakpoints by searching for significant change in the nodes of the tree constructed from all possible partitions. Recombination rate (ρ) and mutation rate (θ) were calculated using LDhat package [55].
Results
Polymorphisms in PvMSP3α gene in the western Thai village
The PvMSP3α gene displays enormous genetic diversity in P. vivax populations and consequently has been used as a molecular marker for differentiating field parasite strains [24]. To investigate the genetic diversity of PvMSP3α gene on a microgeographic scale, we collected P. vivax samples from asymptomatic carriers in a small village Suan Oi (~500 residents) in western Thailand (Fig 1A) during mass blood surveys conducted in this area. The PvMSP3α gene was successfully amplified in 22 of 24 P. vivax samples. Two PCR length variant types A (1.9 Kb) and C (1.1 Kb) were observed, whereas type B (1.5 Kb) was not observed in the tested samples [34]. Two of the samples produced two bands, suggesting of mixed strain infections.
In order to determine the details of sequence diversity of PvMSP3α, PCR products of 20 samples were sequenced and aligned with the Sal I reference gene. PCR fragment type C contained a ~750 bp deletion in block I, whereas block II was relatively conserved in all 20 samples. PvMSP3α block II in the 20 samples had 28 single nucleotide polymorphisms (SNPs), 24 of which were parsimony informative sites (SNPs observed in more than one sequence). Among these SNPs, 15 were non-synonymous mutations which changed 15 amino acids (12 as parsimony informative). Nucleotide diversity of block II was 0.013 and haplotype diversity was 0.800 with 10 haplotypes/allelic forms of block II.
Genetic diversity of PvMSP3α in Thailand
We compared the genetic diversity of the PvMSP3α block II sequences from the Suan Oi isolates with 32 publically available PvMSP3α sequences from Thailand. A total of 85 mutations were observed in 32 sequences, of which 59 were singletons. These sequences differed from the Suan Oi samples in the excess number of singletons. While singletons could be potential consequences of sequencing errors or population expansions [56], low frequency alleles are not generally considered very informative in vaccine design [43]. Twelve parsimony informative amino acid changes were observed in the 32 sequences, of which 10 were shared with the Suan Oi samples, indicating that these high-frequency variants are commonly present in samples from diverse regions of Thailand and these parasites persisted during the last ten years. When all the 52 samples from Thailand were analyzed together (hereafter named as Thai samples), a total of 87 SNPs were observed with 50 amino acid changes, of which only 14 were parsimony informative. As expected from previous studies, nine of the 14 amino acid changes were clustered in two structural motifs (motif I: 533 to 538 and motif II: 580 to 587). The SNPs in each motif are tightly linked and formed two major alleles for each motif (motif I: MSELEK/LSKLEE and motif II: TAANVVKD/KEATAAKL) [34].
Worldwide genetic diversity in PvMSP3α block II
We further investigated the worldwide extent of genetic diversity in a total of 237 sequences of PvMSP3α block II from 11 parasite populations. The sequences included 52 obtained from Thailand and 185 of worldwide isolates retrieved from the GenBank and Plasmodb database (S1 Table). The parasite populations of the 237 samples included 139 sequences from Asian countries (China, India, Sri Lanka, Myanmar, South Korea, and Thailand) and 98 from America (Brazil, Colombia, Mexico, Peru, and Venezuela) (Fig 1B). These sequences contained 158 SNPs ranging from 24–86 in each country, which resulted in 76 amino acid changes (Table 1). Asian samples showed a relatively higher number of singletons compared to American populations (Table 1). However, the high-frequency non-synonymous mutations were extensively shared by all populations from Asia and America. Twenty nine of the total amino acid changes were parsimony informative, of which only 13 had a frequency of more than 5% and 10 of them were present in two aforementioned structural motifs (Fig 2). A similar pattern of amino acid variations was observed in each population. Population-specific polymorphisms were mostly singletons and synonymous in nature.
Table 1. Single nucleotide polymorphisms and summary statistics of PvMSP3α block II in different geographical regions.
| Population | No. of isolates | SNPs | Singletons | Amino acid changes (Parsimony informative) | Haplotypes | θπ | Haplotype diversity | Tajima’s D | Fu & Li’s F |
|---|---|---|---|---|---|---|---|---|---|
| Sua Oi | 20 | 28 | 4 | 12 | 10 | 0.013 | 0.800 | 0.7558 | 0.797 |
| Thailand# | 52 | 87 | 60 | 14 | 31 | 0.017 | 0.947 | -1.3265 | -3.835** |
| India | 6 | 39 | 25 | 7 | 5 | 0.023 | 0.933 | -0.4033 | -0.4219 |
| China | 6 | 27 | 6 | 9 | 5 | 0.019 | 0.933 | 0.7700 | 1.0298 |
| Myanmar | 25 | 60 | 33 | 13 | 21 | 0.021 | 0.977 | -0.2238 | -1.5189 |
| Sri Lanka | 17 | 36 | 9 | 15 | 11 | 0.017 | 0.926 | 0.4825 | 0.4395 |
| South Korea | 33 | 36 | 14 | 10 | 13 | 0.016 | 0.816 | 1.1073 | -0.3151 |
| Brazil | 12 | 29 | 8 | 9 | 10 | 0.015 | 0.955 | 0.4974 | 0.4652 |
| Mexico | 15 | 38 | 5 | 17 | 6 | 0.020 | 0.819 | 0.9332 | 1.1185 |
| Peru | 23 | 29 | 1 | 13 | 9 | 0.016 | 0.897 | 1.7677* | 1.8257** |
| Colombia | 22 | 35 | 3 | 15 | 15 | 0.016 | 0.961 | 0.6206 | 1.1887 |
| Venezuela | 26 | 24 | 0 | 10 | 7 | 0.015 | 0.818 | 2.4208** | 2.2213** |
| World-wide | 237 | 158 | 108 | 29 | 100 | 0.019 | 0.972 | -1.5136* | -6.5208** |
Fig 2. Polymorphism and its pattern in PvMSP3α and block II.

Left panel shows a schematic representation of different domains of PvMSP3α and two genotypes of the locus observed after PCR amplification of the Suan Oi isolates. Right panel shows distribution of amino acid substitutions (excluding singletons in all 237 sequences) in each country. Amino acid positions are numbered corresponding to Salvador I reference strain and the changes in two structural motifs are boxed. Mutations that are observed only once in particular country are highlighted in red and country-specific mutations are circled.
Nucleotide diversity was 0.019 in worldwide samples, ranging from 0.015 to 0.023 in 11 parasite populations (Table 1). θπ was relatively high in India (0.023) and lowest in Brazil and Venezuela (0.015). A sliding window plot of θπ revealed a peak (0.069) at nucleotide positions 2477–2577 (positions corresponding to the Sal I sequence) (Fig 3A), and a similar trend was observed in all the populations. Again, this region encodes structural motif II, where 6 of 13 high-frequency amino acid variants (minor allele frequency >5%) were identified.
Fig 3. Sliding window plot analysis of nucleotide diversity and tests of selection on PvMSP3α block II in worldwide sequences.
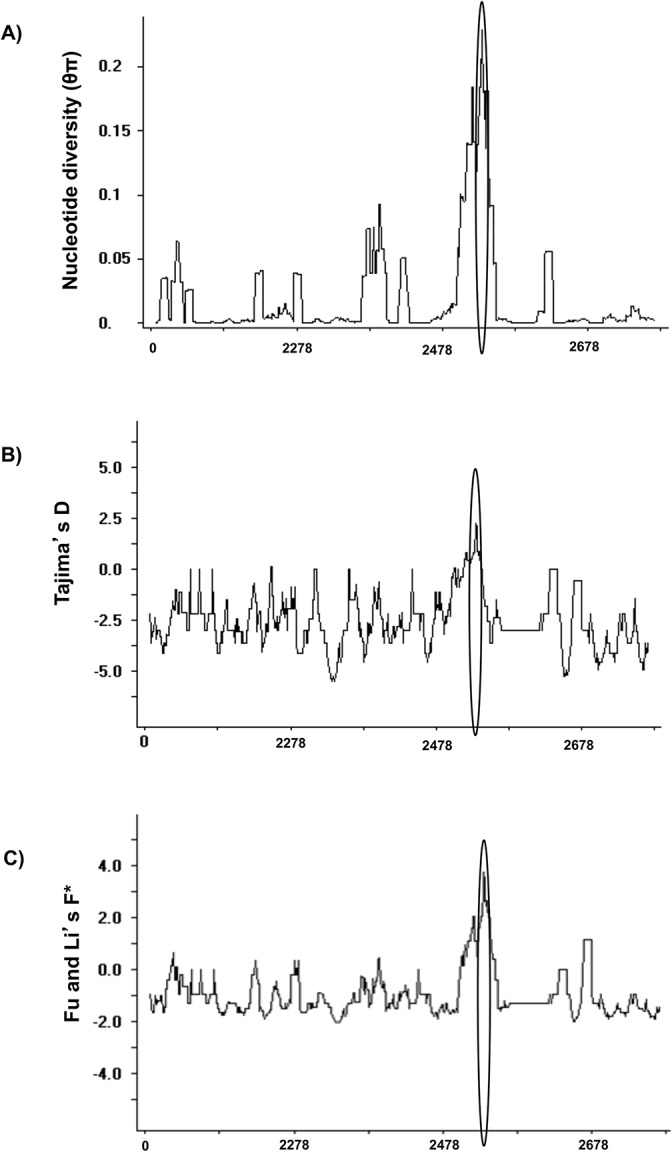
(A) Average pairwise nucleotide diversity (θπ). (B) Tajima’s D values. (C) Fu & Li’s F* test values. A window size of 11 and step size of 1 bp were used. The region with the highest peak of significant values is circled.
The 237 sequences had a total of 100 allelic forms of block II (from 5 in India and China to 31 in Thai samples) with an overall allelic diversity of 0.9724 (0.816–0.977 country-wise). Fifteen block II alleles were shared between populations and 85 were population specific. Both shared and population-specific alleles were observed in each population. Interestingly, all the population-specific alleles were singletons in five out of the 11 populations (Fig 1B). Population specific alleles are not the preferred choice for formulating vaccines aiming to control pathogens worldwide. Haplotype network constructed from the block II allelic forms observed in more than two isolates revealed 15 alleles, of which 6 were shared among four or more populations (Fig 1C). Highly frequent block II allelic forms were shared by diverse population samples from Asia and America (Fig 1C).
Population differentiation
Genetic differentiation between worldwide populations estimated using FST showed a modest genetic structure (0.083), which means population genetic differentiation accounting for only 8% of the total variations in the gene. The highest degree of population differentiation was observed between Thail and Mexico (FST = 0.2559, P<0.001) and the lowest between Mexico and China (FST = -0.0025, P>0.05; Table 2), suggesting that geographic distance is not significantly responsible for genetic differentiation. Moreover, FST values did not correlate with the geographic distance between the populations (Spearman correlation coefficient = -0.0336, P = 0.8).
Table 2. Pairwise FST estimates for 11 Plasmodium vivax endemic countries using PvMSP3α block II sequences.
| Populations | Brazil | China | Colombia | India | Mexico | Peru | Sri-Lanka | Myanmar | South Korea | Venezuela |
|---|---|---|---|---|---|---|---|---|---|---|
| China | 0.0166 | |||||||||
| Colombia | -0.0126 | 0.1035 | ||||||||
| India | 0.1046 | -0.1018 | 0.1323 | |||||||
| Mexico | 0.1734 | -0.0025 | 0.2271 | -0.0041 | ||||||
| Peru | -0.0051 | 0.0233 | -0.0083 | 0.0329 | 0.1542 | |||||
| Sri-Lanka | -0.0314 | 0.0610 | -0.0032 | 0.1180 | 0.1975 | 0.0308 | ||||
| Myanmar | 0.0363 | -0.0951 | 0.1114 | 0.0005 | 0.0948 | 0.0567 | 0.0717 | |||
| South Korea | 0.1653 | 0.0588 | 0.1344 | 0.0363 | 0.1902 | 0.0801 | 0.1472 | 0.1140 | ||
| Venezuela | 0.0942 | -0.0699 | 0.2087 | 0.0435 | 0.1083 | 0.1402 | 0.1606 | 0.0002 | 0.2297 | |
| Thailand | -0.0200 | 0.0847 | 0.0398 | 0.1864 | 0.2559 | 0.0630 | 0.0135 | 0.0835 | 0.2090 | 0.1500 |
Bold values are statistically significant with P <0.05.
Absence of population structuring was further supported by ML analysis of the 237 PvMSP3α block II sequences. Phylogenetic relations between isolates revealed three robust clusters (with bootstrap value more than 75%) based on sequence variations. Group I included 21 sequences from only four countries (Thailand, Myanmar, Brazil and Venezuela), whereas Group II and III included 112 and 104 sequences from all the 11 populations, respectively (Fig 4). High-frequency variations in the regions of two structural motifs mainly determined the pattern of clustering, though other variations defined few sequences that formed sub-clusters within the three major groups (Fig 4). Group I sequences contained the Sal I allele (wild type) in both motifs I and II (LSKLEE and TAANVVKD). In comparison, group II showed mutated allele only in motif I (MSELEK and TAANVVKD), whereas group III contained both alleles of motif I (LSKLEE/MSELEK) and only mutated allele in motif II (KEATAAKL).
Fig 4. Maximum likelihood phylogeny of PvMSP3α block II DNA sequences.

An unrooted phylogeny of the 237 PvMSP3α block II sequences was inferred with maximum likelihood using Tamura and Nei’s model of nucleotide substitution implemented in MEGA6. Bootstrapping was performed with 500 replicates and tree was condensed using 75% bootstrap as a threshold. The label of each sequence is color coded corresponding to the country of origin. Three major clusters were identified as group I, II and III and several sub-clusters were observed within each group. Consensus protein sequences generated for each cluster/sub-cluster using 29 parsimony informative amino acid changes have been shown. Salvador I sequence has been shown as a reference. Amino acids highlighted in red are indicating wild alleles of structural motif I and II while amino acids in green are the mutated alleles. Dashes '-' are representing amino acids that are similar in all the clusters.
Selection and recombination
The rate of synonymous substitutions was found significantly higher than the rate of non-synonymous substitutions in worldwide sequences as well as in each population (data not shown), which indicates purifying/negative selection on block II. This observation was further supported by the significant negative values of Tajima’s D and Fu and Li’s F* tests observed in worldwide sequences. The Tajima’s D and Fu and Li’s F* values were positive in eight populations, but the deviation was significant only in Peru and Venezuela populations (Table 2). Interestingly, window plot analysis of Tajima’s D and Fu and Li’s F* in each population observed significantly positive values in a small region (from 582–591) that covers structural motif II (Fig 3B and 3C). This was further supported by performing the comparative analysis of structural motif II region (24bp) and rest of the block II (672bp) from all 237 sequences. Block II sequences (672 bp) showed highly significant negative values of Tajima's D (-1.9064, P<0.05) and Fu & Li's F (-7.1944, P<0.02), while motif II sequences (24 bp) showed significant positive values of Tajima's D (2.7062, P<0.05) and Fu & Li's F (1.3084, P>0.05). These observations suggest the influence of purifying selection on the entire block II possibly due to structural constraint forced by alanine heptad repeats, whereas a small region containing motif II might have experienced balancing selection. Interestingly, previous in vitro studies have localized a B-cell epitope in motif II (IDEB database; http://www.iedb.org/). Moreover, in silico B Cell Epitope Prediction server [57] also predicted both alleles of motif II as B cell epitopes with >75% specificity.
Intragenic recombination has been repeatedly reported as a prominent evolutionary force in maintaining genetic diversity in PvMSP3α. Since recombination rates estimated by different methods vary with the number of sequences, rate of recombination, and the number of recombination sites, we analyzed all 237 sequences together using five different tests for detecting recombination events. Among phylogenetic approaches of recombination detection, RDP, GENECONV and GARD tests failed to identify any breakpoints, while MaxChi identified one recombination breakpoint. In contrast to these phylogenetic approaches, 13 recombination events were identified by population genetics based estimator using DnaSP. We assumed that singletons might have influenced the DnaSP results, but singleton-free data (replaced singletons with major alleles) produced similar results. Recombination is also evident by eye, since any combinations of dimorphic alleles of the two structural motifs were observed among 237 sequences, implying that recombination in block II has taken place. This was further supported by the recombination event observed by DnaSP in the region between the two motifs (data not shown). Moreover, the estimated recombination rate (ρ = 0.06) was higher than mutation rate (θ = 0.0375) leading to a ρ/θ ratio of 1.6. Recombination to mutation ratio exceeding 1 signifies that the recombination is more prevalent in the dataset than mutation.
Discussion
Analyzing the diversity of gene encoding antigens and the mechanisms involved in the maintenance of such variation is a necessary step for prioritizing vaccine candidates and monitoring their efficacy [33]. The importance of this can be illustrated by studies in P. falciparum that identified considerable diversity in the haplotypes used for designing MSP3-LSP and MSP119 vaccines [58–61]. Significant genetic diversity was assumed to be one of the plausible reasons for the failures of these vaccines in clinical trials. The issue is particularly important in P. vivax since its antigens are still understudied and it has been observed that many genes encoding vaccine candidates in P. vivax show different patterns than P. falciparum [22, 61–64]. The block II of PvMSP3α, being relatively conserved with restricted variations has been proposed as a good vaccine candidate, but defining immunological relevance of the region is required. We analyzed PvMSP3α block II in 11 P. vivax endemic countries worldwide to highlight the extent and distribution of polymorphisms and the potential mechanisms generating these variation patterns. Block II was found less diverse compared to other vaccine candidate genes and many mutations were singletons. The pattern of variation was extensively shared by diverse P. vivax populations suggesting functional/structural constraint on block II, however, each population maintained different allelic forms of block II.
Though a large number of SNPs were observed in PvMSP3α block II of worldwide P. vivax populations, 66% (104/158) of them were singletons. Singletons and low-frequency alleles are generally excluded from diversity analysis of vaccine candidate genes [43] to avoid sequencing and/or PCR artifacts, especially for data retrieved from public databases when sequence accuracy cannot be confirmed. It is worth noting that singletons are expected in a population under expansion, a pattern that has been found in other studies [56, 65]. Alternatively, negative selection on functional genes also increases singletons [66]. The nucleotide diversity of worldwide samples was 0.019 with slight variations among 11 diverse populations (ranging from 0.015 to 0.023). The nucleotide diversity of PvMSP3α block II in each population was found lower than that of the full-length PvMSP3α gene [24, 27] as well as many other merozoite surface proteins analyzed in P. vivax populations, e.g., PvMSP1 (θπ = 0.027) [67], PvMSP7C, PvMSP7H and PvMSP7I (θπ = 0.057, 0.0357 & 0.043) [68], PvMSP3β (θπ = 0.0367) [23] and PvMSP5 (θπ = 0.0375) [8]. However, the level of diversity in block II was high as compared to PvMSP8 and PvMSP10 (θπ = 0.0033 & 0.0022) [69]. This reflects that PvMSP3α block II is relatively conserved among many merozoite surface proteins in P. vivax populations. Additionally, nucleotide diversity in block II showed a non-random pattern, as peaks of nucleotide diversity were restricted to certain regions of the block II. A similar trend was observed when samples from different geographical regions were analyzed, suggesting of functional/structural constraint, since block II is rich in alanine heptad repeats that are predicted to form coiled coil structures possibly needed for the functioning of PvMSP3α gene [26].
Clustering analysis of block II sequences revealed a general lack of geographic structure. However, three robust clusters, each comprising of mixture of sequences from diverse populations were also observed. Moreover, highly frequent non-synonymous SNP-based haplotypes were shared by multiple populations irrespective of their geographical locations, which suggest either extensive gene flow between populations, or independent convergence of variations due to their functional/structural importance. The phylogenetic grouping was found to be based on the type of sequence variations especially influenced by the presence/absence of dimorphic alleles of two structural motifs, suggesting of selective pressure on these motifs.
Antigenic genes are generally expected to be under diversifying/balancing selection, genes under such selective pressure could show lower levels of genetic differentiation between populations than the one expected by genetic drift alone. Accordingly, the overall FST value in worldwide samples was 0.09 with the highest estimate was observed between Thailand and Mexico as 0.2559, P<0.05. Moreover, an FST value of 0.012 between Asian (n = 139 sequences) and American (n = 98) samples analyzed in this study is remarkably low as compared to the FST estimates previously observed between Asian and American P. vivax populations using mitochondrial DNA (FST = 0.15–0.50) [56] and silent SNPs (FST = 0.228) [70]. This might be due to shared non-synonymous variations between populations which tend to maintain low FST values [71], a pattern consistent with balancing selection, but these results need to be interpreted with caution given our small sample sizes.
Though recombination seems to play an important role in generating new genetic variants, polymorphisms in block II are largely clustered in particular regions. Moreover, the pattern of amino acid changes is shared by diverse populations, possibly due to purifying selection that might be acting on alanine heptad repeats for structural conservation. However, a small region in block II encoding motif II showed evidences of balancing selection. Particularly, two alleles of this motif seem to maintain intermediate frequencies in the study populations. This motif might be involved in host pathogen interaction since one of the epitope identified by in vitro studies is localized in this region. Moreover, both alleles were predicted as B cell epitopes with >75% specificity. Blocking this region might prevent merozoite invasion of reticulocytes, and inclusion of both alleles in a vaccine design might be able to induce immune responses recognizing both alleles. However, immunological studies are required to assess the immunogenicity and protection-inducing capability of both alleles in natural P. vivax infections in diverse population backgrounds.
Conserved pattern of amino-acid variations in block II of PvMSP3α compared to the full-length PvMSP3α as well as many other merozoite surface proteins provides a strong support for vaccine development based on block II. An important observation is the common and extensively shared pattern of polymorphisms among diverse populations, which increase the possibility of formulating vaccines effective against worldwide P. vivax populations. Though conserved regions in malaria antigens are generally not highly immunogenic and protective [72, 73], antibodies against block II have been significantly associated with protection against clinical P. vivax infections [28]. This study also identified a B cell epitope in a small region in block II, which was also predicted to be under immune selection, suggesting that it is probably involved in direct interactions with the host cells. Noticeably, only two prominent alleles were observed in this epitope worldwide and both of them showed equivalent specificity as a B cell epitope. Functional studies are needed to determine the immunogenicity and protection ability of these small polypeptides against P. vivax infections.
Supporting Information
(DOC)
Acknowledgments
This work was supported by NIH grants U19AI089672 to LC and GM080586 to AE.
Data Availability
Sequences have been submitted to GenBank under the accession numbers of KR902511–KR902530.
Funding Statement
This work was supported by the National Institutes of Health, US (http://www.nih.gov), grants U19AI089672 to LC and National Institutes of Health, US (http://www.nih.gov) GM080586 to AE.
References
- 1. Tun KM, Imwong M, Lwin KM, Win AA, Hlaing TM, Hlaing T, et al. Spread of artemisinin-resistant Plasmodium falciparum in Myanmar: a cross-sectional survey of the K13 molecular marker. Lancet Infect Dis. 2015; 15: 415–21. 10.1016/S1473-3099(15)70032-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ashley EA, Dhorda M, Fairhurst RM, Amaratunga C, Lim P, Suon S, et al. Spread of artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med. 2014; 371: 411–23. 10.1056/NEJMoa1314981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Herrera S, Corradin G, Arevalo-Herrera M. An update on the search for a Plasmodium vivax vaccine. Trends Parasitol. 2007; 23: 122–8. [DOI] [PubMed] [Google Scholar]
- 4. Beeson JG, Crabb BS. Towards a vaccine against Plasmodium vivax malaria. PLoS Med. 2007; 4: e350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gething PW, Elyazar IR, Moyes CL, Smith DL, Battle KE, Guerra CA, et al. A long neglected world malaria map: Plasmodium vivax endemicity in 2010. PLoS Negl Trop Dis. 2012; 6: e1814 10.1371/journal.pntd.0001814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cotter C, Sturrock HJ, Hsiang MS, Liu J, Phillips AA, Hwang J, et al. The changing epidemiology of malaria elimination: new strategies for new challenges. Lancet. 2013; 382: 900–11. 10.1016/S0140-6736(13)60310-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Carvalho LJ, Daniel-Ribeiro CT, Goto H. Malaria vaccine: candidate antigens, mechanisms, constraints and prospects. Scand J Immunol. 2002; 56: 327–43. [DOI] [PubMed] [Google Scholar]
- 8. Gomez A, Suarez CF, Martinez P, Saravia C, Patarroyo MA. High polymorphism in Plasmodium vivax merozoite surface protein-5 (MSP5). Parasitology. 2006; 133: 661–72. [DOI] [PubMed] [Google Scholar]
- 9. Moon SU, Lee HW, Kim JY, Na BK, Cho SH, Lin K, et al. High frequency of genetic diversity of Plasmodium vivax field isolates in Myanmar. Acta Trop. 2009; 109: 30–6. 10.1016/j.actatropica.2008.09.006 [DOI] [PubMed] [Google Scholar]
- 10. Mascorro CN, Zhao K, Khuntirat B, Sattabongkot J, Yan G, Escalante AA, et al. Molecular evolution and intragenic recombination of the merozoite surface protein MSP-3alpha from the malaria parasite Plasmodium vivax in Thailand. Parasitology. 2005; 131: 25–35. [DOI] [PubMed] [Google Scholar]
- 11. Kang JM, Ju HL, Cho PY, Moon SU, Ahn SK, Sohn WM, et al. Polymorphic patterns of the merozoite surface protein-3beta in Korean isolates of Plasmodium vivax. Malar J. 2014; 13: 104 10.1186/1475-2875-13-104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nobrega de Sousa T, Carvalho LH, Alves de Brito CF. Worldwide genetic variability of the Duffy binding protein: insights into Plasmodium vivax vaccine development. PLoS One. 2011; 6: e22944 10.1371/journal.pone.0022944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cerritos R, Gonzalez-Ceron L, Nettel JA, Wegier A. Genetic structure of Plasmodium vivax using the merozoite surface protein 1 icb5-6 fragment reveals new hybrid haplotypes in southern Mexico. Malar J. 2014; 13: 35 10.1186/1475-2875-13-35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chenet SM, Tapia LL, Escalante AA, Durand S, Lucas C, Bacon DJ. Genetic diversity and population structure of genes encoding vaccine candidate antigens of Plasmodium vivax. Malar J. 2012; 11: 68 10.1186/1475-2875-11-68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Crompton PD, Pierce SK, Miller LH. Advances and challenges in malaria vaccine development. J Clin Invest. 2010; 120: 4168–78. 10.1172/JCI44423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fluck C, Smith T, Beck HP, Irion A, Betuela I, Alpers MP, et al. Strain-specific humoral response to a polymorphic malaria vaccine. Infect Immun. 2004; 72: 6300–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Takala SL, Plowe CV. Genetic diversity and malaria vaccine design, testing and efficacy: preventing and overcoming 'vaccine resistant malaria'. Parasite Immunol. 2009; 31: 560–73. 10.1111/j.1365-3024.2009.01138.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Audran R, Cachat M, Lurati F, Soe S, Leroy O, Corradin G, et al. Phase I malaria vaccine trial with a long synthetic peptide derived from the merozoite surface protein 3 antigen. Infect Immun. 2005; 73: 8017–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Belard S, Issifou S, Hounkpatin AB, Schaumburg F, Ngoa UA, Esen M, et al. A randomized controlled phase Ib trial of the malaria vaccine candidate GMZ2 in African children. PLoS One. 2011; 6: e22525 10.1371/journal.pone.0022525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chaudhuri R, Ahmed S, Ansari FA, Singh HV, Ramachandran S. MalVac: database of malarial vaccine candidates. Malar J. 2008; 7: 184 10.1186/1475-2875-7-184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jiang J, Barnwell JW, Meyer EV, Galinski MR. Plasmodium vivax merozoite surface protein-3 (PvMSP3): expression of an 11 member multigene family in blood-stage parasites. PLoS One. 2013; 8: e63888 10.1371/journal.pone.0063888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rice BL, Acosta MM, Pacheco MA, Carlton JM, Barnwell JW, Escalante AA. The origin and diversification of the merozoite surface protein 3 (msp3) multi-gene family in Plasmodium vivax and related parasites. Mol Phylogenet Evol. 2014; 78C: 172–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Putaporntip C, Miao J, Kuamsab N, Sattabongkot J, Sirichaisinthop J, Jongwutiwes S, et al. The Plasmodium vivax merozoite surface protein 3beta sequence reveals contrasting parasite populations in southern and northwestern Thailand. PLoS Negl Trop Dis. 2014; 8: e3336 10.1371/journal.pntd.0003336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rice BL, Acosta MM, Pacheco MA, Escalante AA. Merozoite surface protein-3 alpha as a genetic marker for epidemiologic studies in Plasmodium vivax: a cautionary note. Malar J. 2013; 12: 288 10.1186/1475-2875-12-288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rungsihirunrat K, Chaijaroenkul W, Siripoon N, Seugorn A, Na-Bangchang K. Genotyping of polymorphic marker (MSP3alpha and MSP3beta) genes of Plasmodium vivax field isolates from malaria endemic of Thailand. Trop Med Int Health. 2011; 16: 794–801. 10.1111/j.1365-3156.2011.02771.x [DOI] [PubMed] [Google Scholar]
- 26. Rayner JC, Corredor V, Feldman D, Ingravallo P, Iderabdullah F, Galinski MR, et al. Extensive polymorphism in the plasmodium vivax merozoite surface coat protein MSP-3alpha is limited to specific domains. Parasitology. 2002; 125: 393–405. [DOI] [PubMed] [Google Scholar]
- 27. Ord R, Polley S, Tami A, Sutherland CJ. High sequence diversity and evidence of balancing selection in the Pvmsp3alpha gene of Plasmodium vivax in the Venezuelan Amazon. Mol Biochem Parasitol. 2005; 144: 86–93. [DOI] [PubMed] [Google Scholar]
- 28. Stanisic DI, Javati S, Kiniboro B, Lin E, Jiang J, Singh B, et al. Naturally acquired immune responses to P. vivax merozoite surface protein 3alpha and merozoite surface protein 9 are associated with reduced risk of P. vivax malaria in young Papua New Guinean children. PLoS Negl Trop Dis. 2013; 7: e2498 10.1371/journal.pntd.0002498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mourao LC, Morais CG, Bueno LL, Jimenez MC, Soares IS, Fontes CJ, et al. Naturally acquired antibodies to Plasmodium vivax blood-stage vaccine candidates (PvMSP-1(1)(9) and PvMSP-3alpha(3)(5)(9)(-)(7)(9)(8) and their relationship with hematological features in malaria patients from the Brazilian Amazon. Microbes Infect. 2012; 14: 730–9. 10.1016/j.micinf.2012.02.011 [DOI] [PubMed] [Google Scholar]
- 30. Parobek CM, Bailey JA, Hathaway NJ, Socheat D, Rogers WO, Juliano JJ. Differing patterns of selection and geospatial genetic diversity within two leading Plasmodium vivax candidate vaccine antigens. PLoS Negl Trop Dis. 2014; 8: e2796 10.1371/journal.pntd.0002796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Conway DJ, Polley SD. Measuring immune selection. Parasitology. 2002; 125 Suppl: S3–16. [DOI] [PubMed] [Google Scholar]
- 32. Conway DJ. Natural selection on polymorphic malaria antigens and the search for a vaccine. Parasitol Today. 1997; 13: 26–9. [DOI] [PubMed] [Google Scholar]
- 33. Escalante AA, Cornejo OE, Rojas A, Udhayakumar V, Lal AA. Assessing the effect of natural selection in malaria parasites. Trends Parasitol. 2004; 20: 388–95. [DOI] [PubMed] [Google Scholar]
- 34. Cui L, Mascorro CN, Fan Q, Rzomp KA, Khuntirat B, Zhou G, et al. Genetic diversity and multiple infections of Plasmodium vivax malaria in Western Thailand. Am J Trop Med Hyg. 2003; 68: 613–9. [DOI] [PubMed] [Google Scholar]
- 35. Parker DM, Matthews SA, Yan G, Zhou G, Lee MC, Sirichaisinthop J, et al. Microgeography and molecular epidemiology of malaria at the Thailand-Myanmar border in the malaria pre-elimination phase. Malar J. 2015; 14: 198 10.1186/s12936-015-0712-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li P, Zhao Z, Wang Y, Xing H, Parker DM, Yang Z, et al. Nested PCR detection of malaria directly using blood filter paper samples from epidemiological surveys. Malar J. 2014; 13: 175 10.1186/1475-2875-13-175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nam DH, Oh JS, Nam MH, Park HC, Lim CS, Lee WJ, et al. Emergence of new alleles of the MSP-3alpha gene in Plasmodium vivax isolates from Korea. Am J Trop Med Hyg. 2010; 82: 522–4. 10.4269/ajtmh.2010.08-0332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wickramarachchi T, Premaratne PH, Dias S, Handunnetti SM, Udagama-Randeniya PV. Genetic complexity of Plasmodium vivax infections in Sri Lanka, as reflected at the merozoite-surface-protein-3alpha locus. Ann Trop Med Parasitol. 2010; 104: 95–108. 10.1179/136485910X12607012374190 [DOI] [PubMed] [Google Scholar]
- 39. Nei M. (1987) Molecular Evolutionary Genetics.: Columbia University Press. [Google Scholar]
- 40. Wright S. The genetical structure of populations. Ann Eugen. 1949; 15: 323–54. [DOI] [PubMed] [Google Scholar]
- 41. Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009; 25: 1451–2. 10.1093/bioinformatics/btp187 [DOI] [PubMed] [Google Scholar]
- 42. Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol. 2013; 30: 2725–9. 10.1093/molbev/mst197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Barry AE, Arnott A. Strategies for designing and monitoring malaria vaccines targeting diverse antigens. Front Immunol. 2014; 5: 359 10.3389/fimmu.2014.00359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bandelt HJ, Forster P, Rohl A. Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol. 1999; 16: 37–48. [DOI] [PubMed] [Google Scholar]
- 45. Nei M, Gojobori T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol. 1986; 3: 418–26. [DOI] [PubMed] [Google Scholar]
- 46. Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989; 123: 585–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fu YX, Li WH. Statistical tests of neutrality of mutations. Genetics. 1993; 133: 693–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hudson RR, Kaplan NL. Statistical properties of the number of recombination events in the history of a sample of DNA sequences. Genetics. 1985; 111: 147–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Martin DP, Lemey P, Lott M, Moulton V, Posada D, Lefeuvre P. RDP3: a flexible and fast computer program for analyzing recombination. Bioinformatics. 2010; 26: 2462–3. 10.1093/bioinformatics/btq467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Martin D, Rybicki E. RDP: detection of recombination amongst aligned sequences. Bioinformatics. 2000; 16: 562–3. [DOI] [PubMed] [Google Scholar]
- 51. Smith JM. Analyzing the mosaic structure of genes. J Mol Evol. 1992; 34: 126–9. [DOI] [PubMed] [Google Scholar]
- 52. Padidam M, Sawyer S, Fauquet CM. Possible emergence of new geminiviruses by frequent recombination. Virology. 1999; 265: 218–25. [DOI] [PubMed] [Google Scholar]
- 53. Kosakovsky Pond SL, Posada D, Gravenor MB, Woelk CH, Frost SD. GARD: a genetic algorithm for recombination detection. Bioinformatics. 2006; 22: 3096–8. [DOI] [PubMed] [Google Scholar]
- 54. Pond SL, Frost SD. Datamonkey: rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics. 2005; 21: 2531–3. [DOI] [PubMed] [Google Scholar]
- 55. Auton A, McVean G. Recombination rate estimation in the presence of hotspots. Genome Res. 2007; 17: 1219–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Taylor JE, Pacheco MA, Bacon DJ, Beg MA, Machado RL, Fairhurst RM, et al. The evolutionary history of Plasmodium vivax as inferred from mitochondrial genomes: parasite genetic diversity in the Americas. Mol Biol Evol. 2013; 30: 2050–64. 10.1093/molbev/mst104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. El-Manzalawy Y, Dobbs D, Honavar V. Predicting linear B-cell epitopes using string kernels. J Mol Recognit. 2008; 21: 243–55. 10.1002/jmr.893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Takala SL, Coulibaly D, Thera MA, Dicko A, Smith DL, Guindo AB, et al. Dynamics of polymorphism in a malaria vaccine antigen at a vaccine-testing site in Mali. PLoS Med. 2007; 4: e93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Takala SL, Smith DL, Thera MA, Coulibaly D, Doumbo OK, Plowe CV. Short report: rare Plasmodium falciparum merozoite surface protein 1 19-kda (msp-1(19)) haplotypes identified in Mali using high-throughput genotyping methods. Am J Trop Med Hyg. 2007; 76: 855–9. [PMC free article] [PubMed] [Google Scholar]
- 60. Polley SD, Tetteh KK, Lloyd JM, Akpogheneta OJ, Greenwood BM, Bojang KA, et al. Plasmodium falciparum merozoite surface protein 3 is a target of allele-specific immunity and alleles are maintained by natural selection. J Infect Dis. 2007; 195: 279–87. [DOI] [PubMed] [Google Scholar]
- 61. Pacheco MA, Poe AC, Collins WE, Lal AA, Tanabe K, Kariuki SK, et al. A comparative study of the genetic diversity of the 42kDa fragment of the merozoite surface protein 1 in Plasmodium falciparum and P. vivax. Infect Genet Evol. 2007; 7: 180–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Tanabe K, Escalante A, Sakihama N, Honda M, Arisue N, Horii T, et al. Recent independent evolution of msp1 polymorphism in Plasmodium vivax and related simian malaria parasites. Mol Biochem Parasitol. 2007; 156: 74–9. [DOI] [PubMed] [Google Scholar]
- 63. Pacheco MA, Ryan EM, Poe AC, Basco L, Udhayakumar V, Collins WE, et al. Evidence for negative selection on the gene encoding rhoptry-associated protein 1 (RAP-1) in Plasmodium spp. Infect Genet Evol. 2010; 10: 655–61. 10.1016/j.meegid.2010.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Gunasekera AM, Wickramarachchi T, Neafsey DE, Ganguli I, Perera L, Premaratne PH, et al. Genetic diversity and selection at the Plasmodium vivax apical membrane antigen-1 (PvAMA-1) locus in a Sri Lankan population. Mol Biol Evol. 2007; 24: 939–47. [DOI] [PubMed] [Google Scholar]
- 65. Miao M, Yang Z, Patch H, Huang Y, Escalante AA, Cui L. Plasmodium vivax populations revisited: mitochondrial genomes of temperate strains in Asia suggest ancient population expansion. BMC Evol Biol. 2012; 12: 22 10.1186/1471-2148-12-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ezawa K, Innan H. Theoretical framework of population genetics with somatic mutations taken into account: application to copy number variations in humans. Heredity (Edinb). 2013; 111: 364–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zeyrek FY, Tachibana S, Yuksel F, Doni N, Palacpac N, Arisue N, et al. Limited polymorphism of the Plasmodium vivax merozoite surface protein 1 gene in isolates from Turkey. Am J Trop Med Hyg. 2010; 83: 1230–7. 10.4269/ajtmh.2010.10-0353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Garzon-Ospina D, Lopez C, Forero-Rodriguez J, Patarroyo MA. Genetic diversity and selection in three Plasmodium vivax merozoite surface protein 7 (Pvmsp-7) genes in a Colombian population. PLoS One. 2012; 7: e45962 10.1371/journal.pone.0045962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Pacheco MA, Elango AP, Rahman AA, Fisher D, Collins WE, Barnwell JW, et al. Evidence of purifying selection on merozoite surface protein 8 (MSP8) and 10 (MSP10) in Plasmodium spp. Infect Genet Evol. 2012; 12: 978–86. 10.1016/j.meegid.2012.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Orjuela-Sanchez P, Karunaweera ND, da Silva-Nunes M, da Silva NS, Scopel KK, Goncalves RM, et al. Single-nucleotide polymorphism, linkage disequilibrium and geographic structure in the malaria parasite Plasmodium vivax: prospects for genome-wide association studies. BMC Genet. 2010; 11: 65 10.1186/1471-2156-11-65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Polley SD, Chokejindachai W, Conway DJ. Allele frequency-based analyses robustly map sequence sites under balancing selection in a malaria vaccine candidate antigen. Genetics. 2003; 165: 555–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Patarroyo MA, Calderon D, Moreno-Perez DA. Vaccines against Plasmodium vivax: a research challenge. Expert Rev Vaccines. 2012; 11: 1249–60. 10.1586/erv.12.91 [DOI] [PubMed] [Google Scholar]
- 73. Giraldo MA, Arevalo-Pinzon G, Rojas-Caraballo J, Mongui A, Rodriguez R, Patarroyo MA. Vaccination with recombinant Plasmodium vivax MSP-10 formulated in different adjuvants induces strong immunogenicity but no protection. Vaccine. 2009; 28: 7–13. 10.1016/j.vaccine.2009.09.046 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOC)
Data Availability Statement
Sequences have been submitted to GenBank under the accession numbers of KR902511–KR902530.