

Abstract
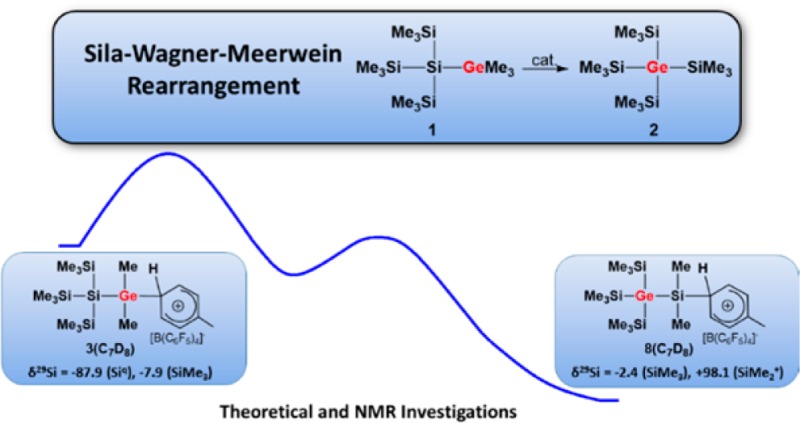
The rearrangement of tris(trimethylsilyl)silyltrimethylgermane 1 to give tetrakis(trimethylsilyl)germane 2 was investigated as a typical example for Lewis acid catalyzed Wagner–Meerwein-type rearrangements of polysilanes and polygermasilanes. Direct 29Si NMR spectroscopic evidence is provided for several cationic intermediates during the reaction. The identity of these species was verified by independent synthesis and NMR characterization, and their transformation was followed by NMR spectroscopy.
Introduction
Despite the most interesting properties and the widespread potential use of oligo- and polysilanes, synthetic access to this class of compounds is mainly limited to the Wurtz-type coupling reaction, in which the Si – Si chain is built up in a reductive process using silicon halides and alkali metals.1 Up to date alternative methods for the synthesis of oligosilanes such as dehydrocoupling of hydrosilanes2 or electrochemically mediated Si–Si bond forming processes3 do not reach the prominence of the Wurtz-type coupling. While this process works fairly well for the preparation of dialkylated linear polysilane polymers and small cyclosilanes, it does not provide satisfactory access to compounds of even slightly enhanced structural complexity. In particular, the generation of well-defined polycyclic molecules in acceptable yields has proven to be very challenging by this method.1 Regarding the potential for such materials being useful for technical applications, there is intrinsic interest in novel approaches that open the possibility to develop new synthetic tools for the preparation of open chain, cyclic, and polyhedral compounds with silicon containing backbones. Pioneering work by the group of Kumada and Ishikawa,4 further developed by the groups of West,5 Märkl,6a and Pannell,6b revealed that the Lewis acid catalyzed Wagner–Meerwein-type rearrangement of oligosilanes is a powerful method to create structures of higher complexity. The high yield synthesis of an all-silicon analogue of adamantane from a structural isomer, reported previously by two of us, is certainly one of the more spectacular examples for the potential of this synthetic methodology.7
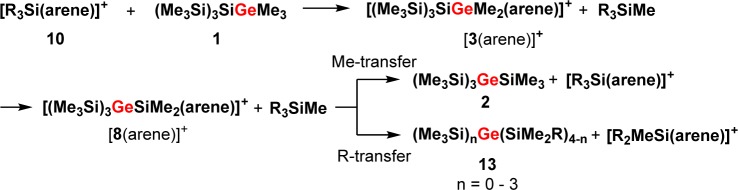
The description of this process as being a silicon variation of the Wagner–Meerwein rearrangement suggests the presence of cationic intermediates.4,5 However, despite the fact that the chemistry of silyl cations has experienced immense popularity in recent years,8 experimental evidence for polysilanyl-substituted silyl cations and their possible involvement in rearrangement or fragmentation reactions is indeed rather scarce. Lambert and co-workers reported on the formation of tris(trimethylsilyl)silylium, (Me3Si)3Si+, most probably in the form of its arene complexes; the obtained 29Si NMR spectra suggest, however, substantial decomposition of the material.9 Structural aspects and the question of aromaticity and homoaromaticity had been the focus in several publications of the Sekiguchi group concerning some cyclic polysilasilyl cations.10,11 Sekiguchi and co-workers also demonstrated the occurrence of a degenerated intramolecular 1,3-methyl shift occurring in a dialkyloligosilanylsilyl cation.12 Interestingly, in none of these reports were parallels drawn between the subject of study and the likely involvement of similar species in the sila-Wagner–Meerwein rearrangement. During our own experimental studies of the Wagner–Meerwein rearrangement in oligosilanes,13 we found that for germylsilanes a particular preference exists for the formation of products in which the germanium atoms occupy central positions in the oligosilane frameworks.14 A deeper insight was provided by a computational study of the fundamental reaction of germylsilane 1 to give germane 2 (reaction 1, Scheme 1). The results of density functional theory (DFT) calculations suggested an exothermic multistep rearrangement via several isomeric silyl- and germyl cations 3–8 (Scheme 2). According to this investigation, the isomerization occurs via consecutive 1,2-silyl and 1,2-methyl shifts with an overall barrier of less than 50 kJ mol–1.14
Scheme 1. Rearrangement of Germylsilane 1 to Silylgermane 2 Using Lewis Acids.
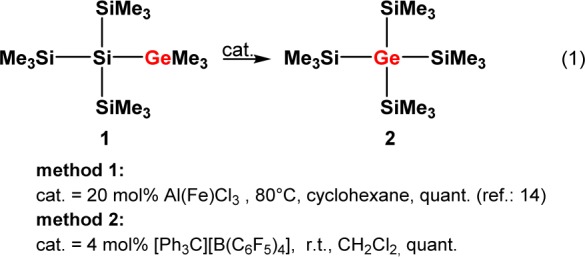
Scheme 2. Suggested Isomerization Cascade of Germylium Ion 3 to Silylium Ion 8(14).
An experimental investigation on the course of the principal reaction 1 would greatly benefit from the simplicity of the reactants, proposed intermediates, and products and from the straightforward interpretation of their NMR spectra. The close chemical relationship between silicon and germanium suggests that conclusions drawn from a mechanistic study of reaction 1 will be of significance also in the case of polysilanes. Here, we report on a 29Si NMR investigation of the exemplary reaction 1, which provides direct evidence for cationic intermediates in the sila-Wagner–Meerwein rearrangement and for the clean transformation of the incipient germyl cation 3 into the product-forming silyl cation 8 at low temperatures along a cascade of isomeric cations 4–7 (Scheme 2).
Results and Discussion
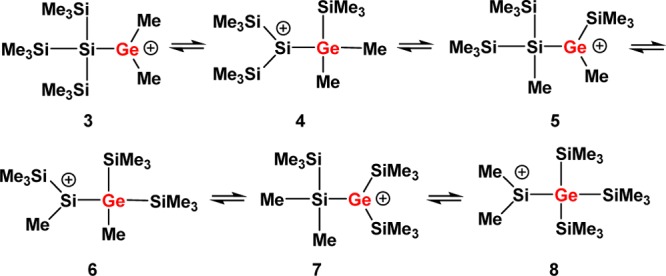
As a starting point for our experimental investigation, we tested additional cationic Lewis acids as stoichiometric reagents or catalysts for the rearrangement of model compound 1. The idea behind using a cationic Lewis acid is to introduce weakly coordinating anions such as the tetrakis(pentafluorophenyl)borate, [B(C6F5)4]−,15 which will be beneficial for the stabilization of the postulated cationic intermediates. Indeed, we found that already 4 mol % of trityl borate [Ph3C][B(C6F5)4], 9, induces the complete transformation of germylsilane 1 into the symmetric germane 2 in dichloromethane at room temperature (Scheme 1).16 For the generation of stoichiometric amounts of the postulated cationic intermediates in the rearrangement reaction shown in Scheme 1, we switched to the even more electrophilic trialkylsilyl arenium borates, [R3Si(arene)] [B(C6F5)4] (R = Me, Et, iPr; arene = C6H5CH3, C6H6, C6H5Cl), 10,8 and used arenes as solvents. In the past, it was shown8 that the combination of arene solvents and weakly coordinating anions is suitable for the detection of highly reactive silylium ions. In order to allow the NMR spectroscopic detection of the postulated cationic intermediates, we tested at first the stoichiometric reaction between germylsilane 1 and triethylsilyl benzenium borate, [Et3Si(C6D6))] [B(C6F5)4], 10a. The reaction in benzene at 6 °C was completed after 2 h and resulted in a biphasic reaction mixture that is typical for solutions of tetrakis(pentafluorophenyl)borates in aromatic hydrocarbons.829Si NMR spectra obtained from the ionic phase showed many signals, which indicated the formation of a complicated reaction mixture. The 29Si NMR analysis of the second, nonpolar layer showed the presence of at least nine different neutral compounds. These nine compounds were identified by GC/MS spectrometry and NMR spectroscopy as the expected germane 2, three different methylethylsilanes, and a series of silylated germanes 11 and 12 (Scheme 3 and Supporting Information for details). Noteworthy, germanes 11 and 12, which all have cation 8 as a common intermediate, are substituted with a different number of ethyl groups. When the same reaction was performed with tri-iso-propylsilyl toluenium borate, [iPr3Si(C7D8)][B(C6F5)4], 10b, only two side products were detected, namely, (Me3Si)3GeSiMe2iPr and iPr3SiMe. Finally, when trimethylsilyl toluenium borate [Me3Si(C7D8)] [B(C6F5)4], 10c, was applied as reaction partner, only the expected rearrangement product 2 and tetramethylsilane were identified in the nonpolar phase (see Supporting Information). Although with all three silylarenium borates 10 the clean production of cationic species at room temperature was not achieved, the results of these stoichiometric reactions indicate that the initial step of the rearrangement reaction is cation formation by cleavage of either a Ge–C- or Si–C bond. Judged from the calculated bond strengths, the cleavage of a Ge–C bond is preferred over that of a Si–C bond (De(Ge–C) = 305 kJ mol–1; (De(Si–C) = 347 kJ mol–1).14 This clearly supports our initial assumption that the first cationic intermediate formed should be germyl cation 3 which undergoes a rearrangement reaction to give silyl cation 8. Furthermore, the results suggest that the terminating step consists in back transfer of an alkyl group to the rearranged cation which yields besides germane 2 the alkylated products 13 (Scheme 4). The multiple substitution5 in the case of the ethyl substituted silyl arenium ion demonstrates the ease of alkyl group exchange under the applied conditions, which is only limited by the steric requirements of the exchanging groups.
Scheme 3. Stoichiometric Reaction between Triethylsilyl Benzenium Borate 10a and Silylgermane 1 in Benzene at T = 6°C.
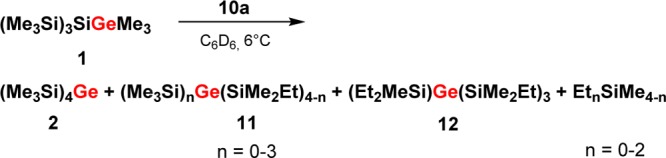
Scheme 4. Reaction of Silylgermane 1 with Different Triakylsilyl Arenium Borates 10 at r.t. (Borate Anion Not Shown).
In contrast to the reaction at room temperature, the reaction of silylgermane 1 with tri-iso-propylsilyl toluenium borate, 10b, in toluene-d8 at −20 °C yielded cleanly a single ionic compound as indicated by only two resonances that were detected in the 29Si NMR spectra of the ionic phase. The NMR data (δ29Si = −87.9 (Siq) and δ29Si = −7.9 (SiMe3), integral ratio in the 29Si{1H} inverse gated experiment, 1:3) is consistent with the structure of germyl toluenium ion 3(C7D8). This assignment was supported by the fact that the same cationic species was formed by the conventional Bartlett Condon Schneider hydride transfer17 reaction of hydridogermane 14 with trityl borate 9 (Scheme 5). In addition, the results of quantum mechanical calculations18,19 of 29Si NMR chemical shifts for an optimized molecular structure of 3(C7H8) predict values that are very close to the experiment (see Table 1).
Scheme 5. Formation of Silyl Toluenium Ion 8(C7D8) from Different Precursor Compounds.
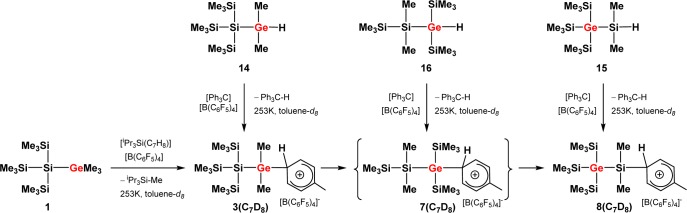
Table 1. Experimental and Calculated 29Si NMR Chemical Shifts of Solvent Complexes of Germylium Ions 3 and 7 and Silylium Ion 8a.
| compound | δ29Si (Siq) | δ29Si (SiMe3) | δ29Si (Si+Me2) | δ29Si (SiMe2) |
|---|---|---|---|---|
| [3(C7D8)] | –87.9 (−93) | –7.9 (−7) | ||
| 7(C7D8)] | (8; −13) | (−13) | ||
| 8(C7D8)] | –2.4 (0) | 98.1 (108) | ||
| [8(ClC6D5)]b | –1.5 (0) | 154.3 (157) |
Calculated values for the nondeuterated compounds at GIAO/M06-L/6-311G(2d,p)//M06-2X/6-311+G(d,p) are in parentheses.
Calculated for the chloronium ion structure [8–Cl-C6H5]. Chloronium ions are the dominating species when silyl cations are generated in chlorinated arenes; see ref (22).
Interestingly, already after 2 h at −20 °C two new 29Si NMR resonances were detected in both cases (δ29Si = −2.4 and +98.1, integral ratio in the 29Si{1H} inverse gated experiment, 3:1; see Figure 1 and Supporting Information).20 Over time, the intensity of the new signals increased significantly while the original 29Si NMR signals of germyl cation 3(C7D8) vanished (see Figure 1), thereby confirming the clean conversion of silylgermyl toluenium ion 3(C7D8) into a new cationic species in toluene at −20 °C. The new high field signal appears in the typical region for trimethylsilyl groups attached to germanium,21 but the measured chemical shift is not specific. More informative is the low field resonance at δ29Si = 98.1, which is characteristic for silyl arenium ions.8 The 29Si NMR chemical shifts of these cationic species are reported to be very sensitive to changes of the solvent due to the replacement of the arene molecule attached to the silicon atom. Indeed, the 29Si NMR chemical shift of the low field resonance varies significantly with the arene solvent from δ29Si = 98.1 in toluene-d8 to δ29Si = 154.3 in chlorobenzene-d5 (see Table 1).22 On the basis of these results, the two new 29Si NMR signals were tentatively assigned to silyl toluenium ion 8(C7D8). This assignment was supported by its independent synthesis by hydride transfer reaction from the appropriate silane 15 (see Scheme 5 and Figure 2) and by quantum mechanical calculations of the 29Si NMR chemical shifts of optimized molecular structures of arene complexes of 8 (see Table 1). According to the NMR results, solutions of the silyl toluenium borate [8(C7D8)][B(C6F5)4] in toluene at T = −20 °C are stable for at least 3 days. When the temperature of the NMR probe was gradually raised in steps of 10 °C, already at T = −10 °C new signals in the 29Si NMR spectrum appeared, and exposure of the NMR sample to temperatures of about +20 °C resulted in complete decomposition of cation 8(C7D8) (see Supporting Information, Figure S21).23 The thermal instability of arenium borate [8(C7D8)][B(C6F5)4] in solution prevents its isolation in substance and additional characterization. These NMR experiments demonstrated the clean conversion of silylgermyl toluenium ion 3(C7D8) to germylsilyl toluenium 8(C7D8) in toluene at −20 °C. According to our previous computational study, this sila-variant of the Wagner–Meerwein rearrangement is expected to proceed via several cationic intermediates (see Scheme 2).14 Additional DFT computations at the B3LYP/6-311+G(d,p) level of theory18,19 indicated that complexation of the formed silyl and germyl cations with the arene solvent has no significant influence on the calculated reaction coordinate. The calculated relative energies of the toluene complexes of cations 3–8 reveal only slight modifications of the previously predicted stability sequence for the free silylium and germylium ions (see Figure S22, Supporting Information). In particular, the silyltoluenium ion 8(C7H8) is predicted to be the most stable along the complete reaction sequence, and the overall reaction 3(C7H8) → 8(C7H8) is found to be exothermic by 44 kJ mol–1. Experimentally, however, no indications for the cationic intermediates 4–7 or for the related arenium ions were detected. In order to test our mechanistic proposal, we decided to generate one specific cation along the reaction cascade, shown in Scheme 2, and to monitor its rearrangement. Therefore, we subjected silylgermane 16 to the hydride transfer reaction in toluene in an attempt to generate the corresponding germyltoluenium 7(C7D8) (Scheme 5). This last step of the reaction sequence, 7(C7H8) → 8(C7H8), is predicted by the calculations to be exothermic by 15 kJ mol–1 (Figure S22, Supporting Information). 29Si{1H} NMR spectra recorded after 15 min at −20 °C showed no signals expected for cation 7(C7H8) (compare Figure S19c (Supporting Information) and calculated values in Table 1), but the characteristic resonances for silyltoluenium 8(C7D8) already dominated. In agreement with the computational results, this experiment shows that germyltoluenium ion 7(C7D8) undergoes a fast 1,2-shift of the trimethylsilyl group to form the more stable cation 8(C7D8).
Figure 1.

99 MHz 29Si{1H} NMR spectra (toluene-d8, 253 K) of the rearrangement of germyl toluenium ion 3(C7D8) (•) to silyl toluenium 8(C7D8) (*) (↓ iPr3SiMe). (a) Spectrum 2 h after the addition of [iPr3Si(C7D8)][B(C6F5)4]; (b) after 9 h; (c) after 25 h; (d) after 120 d at 210 K.
Figure 2.
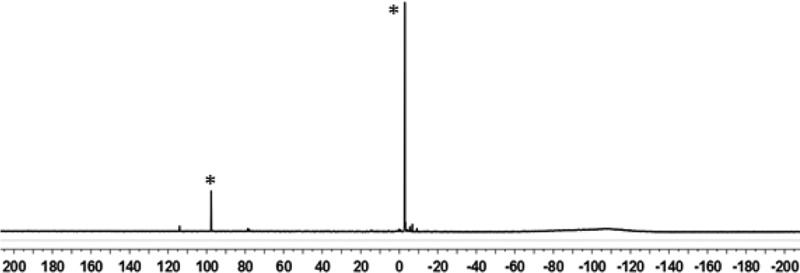
99 MHz 29Si{1H} NMR spectrum (toluene-d8, 253 K) of silyl toluenium ion 8(C7D8) at 253 K in toluene synthesized by hydride transfer from silane 15.
Obviously, the barrier for this rearrangement is rather low and in a similar range as the 18 kJ mol–1 that is predicted for the transformation of the free cations 7 → 8 (Figure S22, Supporting Information),14 and therefore, germyltoluenium ion 7(C7D8) cannot be detected even at temperatures as low as −20 °C.
Conclusions
We were able to synthesize polysilanyl-substituted silyl and germyl cations, such as silyl cation 8 and germyl cation 3 in the form of their arene complexes at low temperatures using the weakly coordinating [B(C6F5)4]− anion. The cation synthesis was done using either the stoichiometric reaction of germa-oligosilanes with a cationic Lewis acid such as tri-iso-propylsilyltoluenium, or, in a more regio-controlled way, by the standard hydride transfer reaction between hydrido-silanes and -germanes with the trityl cation. The cationic species were identified by low-temperature NMR spectroscopy supported by the results of quantum mechanical NMR chemical shift calculations. Applying these techniques, we were able to verify several silyl- and germyl cations and their solvent complexes as intermediates in the sila-Wagner–Meerwein rearrangement of silagermane 1 (eq 1, Scheme 1) and to provide strong evidence for their clean interconversion at low temperatures. We are convinced that this reaction is archetypical for many synthetic useful Lewis acid catalyzed skeletal rearrangements in polysilanes and polygermasilanes. Additional stable ion studies for sila-Wagner–Meerwein rearrangement of oligosilanes of higher complexity are currently under investigation in our laboratory to identify general rules and reaction patterns.
Experimental Section
All manipulations of air- and moisture-sensitive compounds were carried out under argon or nitrogen atmosphere using Schlenk techniques or a standard glovebox (Braun Unilab). Glassware was dried in an oven at 120 °C and evacuated prior to use. The solvents tetrahydrofuran (THF), dimethoxyethane (DME), n-pentane, benzene, and toluene were dried over sodium and distilled under nitrogen atmosphere. Chlorobenzene was dried over CaCl2 and stored over molecular sieves. Deuterated benzene and toluene were stored over molecular sieves after drying over sodium. Dichlorodimethylgermane, chlorodimethylsilane, triethylsilane, tri-iso-propylsilane and trimethylchlorosilane were obtained from commercial suppliers, and the silanes were dried over molecular sieves. Sodium methanolate was prepared by addition of sodium to an excess of abs. methanol. After all sodium was consumed, the solvent was removed in vacuo. Triphenylmethyl tetrakis(pentafluorophenyl) borate 9 ([Ph3C][B(C6F5)4]) was prepared according to a modified literature procedure.24 Tetrakis(trimethylsilyl)silane,25 tetrakis(trimethylsilyl)germane262, tris(trimethylsilyl)silylpotassium,27 tris(trimethylsilyl)germylpotassium,28 tris(trimethylsilyl)silyltrimethylgermane291, chloropentamethyldisilane,30 and trimethylsilane31 were synthesized according to reported procedures. GC-MS spectra were performed on a Thermo Focus DSQ. NMR spectra were recorded on Bruker Avance 500, Avance III 500 and Varian Inova 300 spectrometers. 1H NMR spectra were calibrated against the residual proton signal of the solvent as internal reference (benzene-d6/δ1H(C6D5H) = 7.20; toluene-d8/δ1H(CD2H) = 2.08; chloroform-d1/δ1H(CHCl3) = 7.24; chlorobenzene-d5/δ1H(C6D4HCl) = 7.14) and 13C NMR spectra by using the central line of the solvent signal (benzene-d6/δ13C(C6D6) = 128.0, toluene-d8/δ13C(C6D5CD3) = 20.4, chloroform-d1/δ13C(CDCl3) = 77.0, chlorobenzene-d5/δ13C(C6D5Cl) = 134.2). 29Si{1H} NMR spectra were calibrated against an external standard (29Si(Me2SiHCl) = 11.1 versus tetramethylsilane (TMS)). The 29Si{1H} NMR inverse gated spectra were recorded with a relaxation delay D1 = 10 s. On the basis of our experiences, at −20 °C this delay is long enough to allow a reliable integration of the peaks. The 29Si{1H} INEPT spectra were recorded with delays D3 = 8.4 ms and D4 = 31.3 ms. IR spectra were recorded on a Bruker Tensor 27 instrument. Analysis values for carbon show often too low values, which we attribute to the formation and incomplete combustion of silicon carbide, although vanadium pentoxide as combustion aid was used.
Dimethoxydimethylgermane (17)
This compound was prepared according to slightly modified literature procedures.32 NaOMe (2.84 g) (3.5 eq., 52.57 mmol) was suspended in 40 mL of pentane, and 1.74 mL (15.00 mmol) of dichlorodimethylgermane was slowly added with a syringe. The mixture was stirred overnight at room temperature. The excess of NaOMe and formed NaCl were separated from the solution by using a centrifuge (20 min, 2000 rpm), and then the product-containing pentane solution was decanted using a Teflon tube. The salts were washed with 10 mL of pentane and again centrifuged and decanted. The pentane solutions were combined, and the product was separated from the solvent by fractionated distillation (bp.: 118 °C at normal pressure (1.45 g, 58%). 1H NMR (500.13 MHz, 297.9 K, C6D6, δ): 0.30 (s, 6H, (CH3)2Ge), 3.54 (s, 6H, Ge(OCH3)2). 13C{1H} NMR (125.77 MHz, 298.1K, C6D6, δ): −2.9 ((CH3)2Ge), 51.6 (Ge(OCH3)2). Mass required for C4H12GeO2: 166.0. Mass found GC/MS: 164.9 (0.5) [M+-H], 150.8 (100) [M+-Me], 135.9 (72) [M+-OMe], 120.9 (88) [M+-OMe-Me], 104.9 (84) [M+-OMe-2Me].
Tris(trimethylsilyl)silyldimethylmethoxygermane (18)
A solution of 4.50 mmol tris(trimethylsilyl)silylpotassium27 in 40 mL of pentane and a solution of 0.75 g (4.50 mmol) of dimethoxydimethylgermane 17 in 10 mL of pentane were cooled to 0 °C. The silyl potassium compound was added dropwise to the germane solution. The ice bath was allowed to warm to room temperature overnight. The reaction mixture was then hydrolyzed with 1 M hydrochloric acid. The organic layer was separated and dried over sodium sulfate. The solvent was removed under reduced pressure, and the product was purified by Kugelrohr distillation (0.62 g, 36%). Because of the use of hydrochloric acid, about 14% of the corresponding germyl chloride was formed as a byproduct, which was detected in the GC chromatograms and NMR spectra. 1H NMR (499.87 MHz, 305.0 K, C6D6, δ ppm): 0.34 (s, 27H, (CH3)3Si), 0.60 (s, 6H, (CH3)2Ge), 3.54 (s, 3H, CH3OGe). 13C{1H} NMR (125.69 MHz, 305.0 K, C6D6, δ ppm): 2.6 ((CH3)3Si), 4.1 ((CH3)2Ge), 52.6 (CH3OGe). 29Si{1H} INEPT NMR (99.31 MHz, 305.0 K, C6D6, δ ppm): −124.6 (((CH3)3Si)3Si), −9.9 (((CH3)3Si)3Si). Mass required for C12H36GeOSi4: 382.1. Mass found GC/MS: 367.2 (1) [M+-Me], 351.2 (0.5) [M+-OMe], 278.0 (8) [M+-SiMe3-OMe], 205.1 (13) [M+-2SiMe3-OMe], 73.0 (100) [Me3Si+]. No satisfactory combustion analysis was available due to contamination with the chloride side-product.
Tris(trimethylsilyl)silyldimethylgermane (14)
A solution of 0.62 g (1.62 mmol) tris(trimethylsilyl)silyldimethylmethoxygermane 18 in 30 mL of THF and a suspension of 0.062 g (1.62 mmol) of LiAlH4 in 50 mL of THF were cooled to 0 °C with an ice bath. The solution of silagermane 18 was added to the LiAlH4 suspension and the reaction mixture was stirred for 20 min at 0 °C before it was allowed to warm to room temperature and stirred for another 20 min. The mixture was slowly added to ice cold 2 M sulfuric acid. The phases were separated, and the aqueous phase was extracted two times with 50 mL of diethyl ether. The combined organic phases were dried over sodium sulfate and filtered, and the solvent was removed under reduced pressure. The product was crystallized from ethanol as a waxy, colorless solid (0.38 g, 1.09 mmol, 67%). 1H NMR (499.87 MHz, 305.0 K, C6D6, δ ppm): 0.30 (s, 27H, (CH3)3Si), 0.50 (d, 3JH,H = 4.2 Hz, 6H, (CH3)2Ge), 4.04 (sept, 3JH,H = 4.2 Hz, 1H, GeH). 13C{1H} NMR (125.69 MHz, 305.0 K, C6D6, δ ppm): −2.3 ((CH3)2Ge), 2.5 ((CH3)3Si). 29Si{1H} NMR (99.31 MHz, 305.0 K, C6D6, δ ppm): −128.3 (((CH3)3Si)3Si), −9.4 (((CH3)3Si)3Si). Mass required for C11H34GeSi4: 352.1. Mass found GC/MS: 351.1 (0.1) [M+-H], 337.1 (0.6) [M+-Me-H], 278.0 (30) [M+-SiMe3-H], 189.1 (13) [M+-2SiMe3-Me-H], 174.0 (4) [M+-SiMe3-GeMe2H], 73.1 (100) [Me3Si+]. IR (ATR, neat): νGe–H 1982 cm–1. Anal. found/calcd. for C11H34GeSi4: C 37.63/37.60, H 10.67/9.75.
Tris(trimethylsilyl)germyldimethylsilane (15)34
Solutions of 2.99 mmol tris(trimethylsilyl)germylpotassium28 in 30 mL of DME and of 0.6 mL (excess, 5.52 mmol) of chlorodimethylsilane in 30 mL of DME were cooled to 0 °C with an ice bath. The germylpotassium compound was slowly added to the chlorosilane solution during 1 h. The ice bath was allowed to warm to room temperature overnight. The reaction mixture was then hydrolyzed with 1 M sulfuric acid. The organic layer was separated, and the aqueous phase was extracted with 10 mL of diethyl ether. The combined organic phases were dried over sodium sulfate, and the filtrate was concentrated to 5 mL under reduced pressure. The product was crystallized by adding 2 mL of acetonitrile as a colorless, waxy solid (0.847 g, 80.6%). 1H NMR (499.87 MHz, 305.0 K, CDCl3, δ ppm):34 0.22 (s, 27H, (CH3)3Si), 0.27 (d, 3JH,H = 4.2 Hz, 6H, (CH3)2Si), 4.12 (sept, 3JH,H = 4.2 Hz, 1H, SiH). 13C{1H} NMR (125.71 MHz, 305.0 K, CDCl3, δ ppm): −1.4 ((CH3)2Si), 3.1 ((CH3)3Si). 29Si INEPT NMR (99.31 MHz, 305.0 K, CDCl3, δ ppm): −29.8 (dsept, 1JSi,H = 180.5 Hz, 2JSi,H = 7.0 Hz, SiH), −4.7 ((CH3)3Si). Mass required for C11H34GeSi4: 352.1. Mass found GC/MS: m/z (%) = 351.1 (0.3) [M+-H], 337.1 (2.5) [M+-Me-H], 278.1 (64) [M+-SiMe3-H], 189.9 (22) [M+-2SiMe3-Me-H], 174.0 (2) [M+-SiMe3-GeMe2H], 73.0 (100) [Me3Si+]. IR (ATR, neat): νSi–H 2085 cm–1. Anal. found/calcd. for C11H34GeSi4: C 36.31/37.60, H 9.98/9.75.
Tris(trimethylsilyl)pentamethyldisilanylgermane (19)
A solution of 1.37 mmol tris(trimethylsilyl)germylpotassium·18-crown-628 in 3 mL of benzene was added dropwise to a solution of 0.25 g (1.51 mmol) of chloropentamethyldisilane30 in 3 mL of benzene. After 5 h, the solution mixture was quenched with 1 M sulfuric acid, and the phases were separated. The aqueous phase was extracted with pentane, and the combined organic phases were dried over sodium sulfate and filtered, and the solvent was removed under reduced pressure. The product was obtained as colorless crystals by crystallization from methanol/diethyl ether 1:2 (0.42 g, 73%). 1H NMR (299.94 MHz, 298.0 K, C6D6, δ ppm): 0.22 (s, 9H, Si(CH3)2Si(CH3)3), 0.36 (s, 27H, ((CH3)3Si)3Ge), 0.40 (s, 6H, Si(CH3)2Si(CH3)3). 13C{1H} NMR (75.43 MHz, 298.0 K, C6D6, δ ppm): −0.8 (Si(CH3)2Si(CH3)3), −0.4 (Si(CH3)2Si(CH3)3), 4.0 (((CH3)3Si)3Ge). 29Si{1H} INEPT NMR (59.59 MHz, 295.0 K, C6D6, δ ppm): −34.0 (Si(CH3)2Si(CH3)3), −15.5 (Si(CH3)2Si(CH3)3), −5.2 (((CH3)3Si)2Ge). Mass required for C14H42GeSi5: 424.1. Mass found GC/MS: m/z (%) = 424 (1) [M+]; 408 (1) [M+-Me-H]; 351 (3) [M+-SiMe3]; 278 (10) [M+-2SiMe3]; 259 (1) [GeSi3C7H17+]; 243 (1) [GeSi3C6H13+]; 219 (3) [GeSi2C6H17+]; 203 (11) [M+-3SiMe3-2H]; 187(8)[M+-3SiMe3-Me-4H]; 147 (7) [GeSiMe3+]; 131 (35) [SiMe3SiMe2+]; 73 (100) [SiMe3+]. Anal. found/calcd. for C14H42GeSi5 C 39.33/39.70, H 9.50/9.99.
Bis(trimethylsilyl)pentamethyldisilanylgermane (16)
A mixture of 0.21 g (0.49 mmol) of germapolysilane 19, 0.062 g (0.51 mmol) of KOtBu, and 0.134 g (0.51 mmol) of 18-crown-6 ether was dissolved in 2 mL of benzene. After the complete formation of germylpotassium compound 20 was confirmed by NMR spectroscopy, the solution was added to a stirred mixture of 10 mL of degassed diethyl ether and 20 mL of degassed 2 M sulfuric acid cooled with an ice bath. The phases were separated, the aqueous phase was extracted with degassed diethyl ether, and the combined organic phases dried over sodium sulfate. The solvents were removed under reduced pressure, and the product was obtained as a colorless oil (0.15 g, 91%). The compound is sensitive to oxygen and should be stored under argon at −20 °C.
Germylpotassium Compound 18-Crown-6 (20)
1H NMR (299.94 MHz, 298.0 K, C6D6, δ ppm): 0.39 (s, 9H (Si(CH3)2)Si(CH3)3), 0.59 (s, 18H ((CH3)3Si)2GeK), 0.64 (s, 6H, Si(CH3)2Si(CH3)3), 3.25 (s, 24H, CH2O). 13C{1H} NMR (75.43 MHz, 295.0 K, C6D6, δ ppm): 0.0 (Si(CH3)2Si(CH3)3), 3.5 (Si(CH3)2Si(CH3)3), 8.5 (((CH3)3Si)2GeK), 70.0 (CH2O). 29Si{1H} INEPT NMR (59.59 MHz, 295.0 K, C6D6, δ ppm): −33.0 (Si(CH3)2Si(CH3)3), −16.6 (Si(CH3)2Si(CH3)3), −3.4 (((CH3)3Si)2GeK). Mass required for C13H38GeSi4 after ethyl bromide derivatization: 380.1. Mass found after ethyl bromide derivatization GC/MS: m/z (%) = 380 (1)[M+]; 365 (1) [M+-Me]; 350 (3) [M+-Et-H]; 307 (1) [M+-SiMe3]; 292 (1) [M+-SiMe3-Me]; 277 (3) [M+-SiMe3-2Me]; 262 (1) [M+-SiMe3-3Me]; 234 (3) [M+-2SiMe3]; 219 (8) [M+-2SiMe3-Me]; 203 (17) [M+-2SiMe3-2Me-H]; 187 (6) [M+-2SiMe3-3Me-2H]; 159 (4) [M+-3SiMe3-H]; 145 (10) [M+-3SiMe3-Me]; 131 (33) [GeSiC2H5+]; 115 (9) [GeSiCH+]; 73 (100) [SiMe3+].
Bis(trimethylsilyl)pentamethyldisilanylgermane (16)
1H NMR (499.87 MHz, 305.1 K, C6D6, δ ppm):): 0.22 (s, 9H, Si(CH3)2Si(CH3)3), 0.35 (s, 18H, ((CH3)3Si)2Ge), 0.40 (s, 6H, Si(CH3)2Si(CH3)3), 2.25 (s, 1H, GeH). 13C{1H} NMR (125.71 MHz, 305.0 K, CDCl3, δ ppm): −1.7 (Si(CH3)2Si(CH3)2), −1.6 (Si(CH3)3Si(CH3)2), 3.1 (((CH3)3Si)2Ge). 29Si{1H} NMR (99.31 MHz, 305.0 K, C6D6, δ ppm): −34.3 (Si(CH3)2Si(CH3)3), −16.2 (Si(CH3)2Si(CH3)3), −5.7 (((CH3)3Si)2Ge). Mass required for C11H34GeSi4: 352.1. Mass found GC/MS: 335 (2) [M+-Me-2H]; 278 (27) [M+-SiMe3-H]; 263 (2) [M+-SiMe3-Me-H]; 203 (13) [M+-2SiMe3-2H]; 189 (10) [M+-2SiMe3-Me-2H]; 173 (2) [M+-2SiMe3-2Me-2H]; 131 (24) [SiMe3SiMe2+]; 115 (14) [Si2C4H11+]; 73 (100) [SiMe3+]. IR (ATR, neat) νGe–H 1951 cm–1. Anal. found/calcd. for C11H34GeSi4 C 38.85/37.60, H 9.41/9.75.
General Preparation of Trialkylsilyl Arenium Borates (10a–d)33
Triphenylmethyl tetrakis(pentafluorophenyl)borate was dissolved in 3 mL of the indicated solvent, and the silane was added. The formation of two phases could be observed, and the biphasic reaction mixture was vigorously stirred for 30 min. The upper, nonpolar phase was removed, and the lower, polar phase was washed with 2 mL of the used solvent, and again the nonpolar phase was removed. The polar phase was dried under reduced pressure for 30 min and then dissolved in the respective deuterated solvent.
Triethylsilyl Benzenium Borate (10a)
Triphenylmethyl tetrakis(pentafluorophenyl)borate (0.50 g (0.54 mmol)) was dissolved in 3 mL of benzene, and 0.14 mL (1.6 eq., 0.87 mmol) of triethylsilane was added.
Tri-iso-propylsilyl Toluenium Borate (10b)
Triphenylmethyl tetrakis(pentafluorophenyl)borate (0.46 g (0.50 mmol)) was dissolved in 3 mL of toluene, and 0.11 mL (1.1 eq., 0.55 mmol) of triiso-propylsilane was added.
Trimethylsilyl Toluenium Borate (10c)
Triphenylmethyl tetrakis(pentafluorophenyl)borate (0.46 g (0.50 mmol)) was dissolved in 3 mL of toluene, and 0.06 mL (1.1 eq., 0.55 mmol) of trimethylsilane was added.
General Procedure for the Rearrangement of Tris(trimethylsilyl)silyltrimethylgermane (1) with Trialkylsilyl Arenium Borates (10a–c)
A solution of 0.18 g (0.50 mmol) of silagermane 1 in 1 mL of the named deuterated solvent was added to a precooled solution of the named trialkylsilyl arenium borate 10a–c. The reaction mixture was stirred for 2 h at the specified temperature and then allowed to warm to room temperature. The polar phase and the nonpolar phase were each transferred to separate NMR tubes to be analyzed independently. In the following reactions, the NMR spectra of the polar phase showed too may signals to be analyzable, but the compounds in the nonpolar phase were identified by NMR and GC/MS spectroscopy (see Figures S8–11 in the Supporting Information for details).
Rearrangement of Tris(trimethylsilyl)silyldimethylgermyl Toluenium Borate (3(C7D8)[B(C6F5)4]) to Tris(trimethylsilyl)germyldimethylsilyl Toluenium Borate (8(C7D8)[B(C6F5)4]) Starting from Silagermane (1)
To a solution of 0.18 g (0.50 mmol) of tris(trimethylsilyl)silyltrimethylgermane 1 in 1 mL of toluene-d8 cooled to −20 °C, 1 equiv of tri-iso-propylsilyl toluenium borate 10b was slowly added via a Teflon tube. The mixture was stirred for 1 h at −20 °C. The brown polar phase and the light yellow nonpolar phase were each transferred to separate NMR tubes at −20 °C and stored at −60 °C overnight until the NMR spectra were recorded the next morning. The polar phase contained borates [3(C7D8)][B(C6F5)4] and [8(C7D8)][B(C6F5)4]). The nonpolar phase contained methyl-triiso-propylsilane and the rearrangement product 2. Polar phase1H NMR (499.87 MHz, 253.0 K, C7D8, δ ppm): −0.15 (((CH3)3Si)3GeSi(CH3)2+), 0.07 (((CH3)3Si)3GeSi(CH3)2+), 0.12 (((CH3)3Si)3SiGe(CH3)2+), 0.28 (((CH3)3Si)3SiGe(CH3)2+). 13C{1H} NMR (125.71 MHz, 253.0 K, C7D8, δ ppm): 1.6 (((CH3)3Si)3SiGe(CH3)2+), 2.5 (((CH3)3Si)3GeSi(CH3)2+), 5.5 (((CH3)3Si)3GeSi(CH3)2+), 12.1 (((CH3)3Si)3SiGe(CH3)2+). 29Si{1H} NMR (99.31 MHz, 253.0 K, C7D8, δ ppm): −87.9 (((CH3)3Si)3SiGe(CH3)2+), −7.9 (((CH3)3Si)3SiGe(CH3)2+), −2.4 (((CH3)3Si)3GeSi(CH3)2+), 98.1 (((CH3)3Si)3GeSi(CH3)2+) (see Figures S12a–c in the Supporting Information). Nonpolar phase1H NMR (499.87 MHz, 305.0 K, C7D8, δ ppm): −0.17 (s, ((CH3)2CH)3SiCH3), 0.26 (s, ((CH3)3Si)4Ge), 0.86 (sept, 3JH,H = 7.3 Hz, ((CH3)2CH)3SiCH3), 0.97 (d, 3JH,H = 7.3 Hz, ((CH3)2CH)3SiCH3). 13C{1H} NMR (127.71 MHz, 305.0 K, C7D8, δ ppm): −10.1 (((CH3)2CH)3SiCH3), 3.5 (((CH3)3Si)4Ge), 11.8 (((CH3)2CH)3SiCH3), 18.8 (((CH3)2CH)3SiCH3). 29Si{1H} NMR (99.31 MHz, 305.0 K, C7D8, δ ppm): −5.1 (((CH3)3Si)4Ge), 9.0 (((CH3)2CH)3SiCH3) (see Figures S13a–d in the Supporting Information).
Rearrangement of Tris(trimethylsilyl)silyldimethylgermyl toluenium Borate (3(C7D8)[B(C6F5)4]) to Tris(trimethylsilyl)germyldimethylsilyl Toluenium Borate (8(C7D8)[B(C6F5)4]) Starting from Hydrogen Substituted Silagermane (14)
tris(trimethylsilyl)silyldimethylgermane 14 (0.14 g (0.40 mmol)) and 0.37 g (0.40 mmol) of triphenylmethyl tetrakis(pentafluorophenyl)borate were each dissolved in 1 mL of toluene-d8 and cooled to −20 °C. Silagermane 14 was slowly added to the borate salt via a Teflon tube, and the mixture was stirred at −20 °C for 1.5 h. The brown polar phase and the light yellow nonpolar phase were each transferred to NMR tubes at −20 °C. The NMR spectra were recorded at −20 °C. The polar phase contained borates [3(C7D8)][B(C6F5)4] and [8(C7D8)][B(C6F5)4]). The nonpolar phase contained triphenylmethane and the rearrangement product 2. Polar phase1H NMR (499.87 MHz, 253.0 K, C7D8, δ ppm): −0.15 (((CH3)3Si)3GeSi(CH3)2+), 0.07 (((CH3)3Si)3SiGe(CH3)2+), 0.12 (((CH3)3Si)3GeSi(CH3)2+), 0.28 (((CH3)3Si)3SiGe(CH3)2+). 13C{1H} NMR (125.71 MHz, 253.0 K, C7D8, δ ppm): 1.6 (((CH3)3Si)3SiGe(CH3)2+), 2.4 (((CH3)3Si)3GeSi(CH3)2+), 5.5 (((CH3)3Si)3GeSi(CH3)2+), 12.1 (((CH3)3Si)3SiGe(CH3)2+). 29Si{1H} NMR (99.31 MHz, 253.0 K, C7D8, δ ppm): −87.9 (((CH3)3Si)3SiGe(CH3)2+), −7.9 (((CH3)3Si)3SiGe(CH3)2+), −2.4 ((CH3)3Si)3GeSi(CH3)2+)), 98.1 (((CH3)3Si)3GeSi(CH3)2+) (see Figures S14a–c and S15a–b in the Supporting Information). Nonpolar phase1H NMR (499.87 MHz, 305.0 K, C7D8, δ ppm): 0.30 (s, ((CH3)3Si)4Ge), 5.38 (s, Ph3CH), 6.98–7.09 (m, Ph3CH). 13C{1H} NMR (127.71 MHz, 305.0 K, C7D8, δ ppm): 3.5 ((CH3)3Si)4Ge), 57.1 (Ph3CH), 125.4 (Ph3CH), 128.3 (Ph3CH), 129.2 (Ph3CH), 144.3 (Ph3CH). 29Si{1H} NMR (99.31 MHz, 305.0 K, C7D8, δ ppm): −5.1 ((CH3)3Si)4Ge) (see Figures S16a–c in the Supporting Information).
Tris(trimethylsilyl)germyldimethylsilyl Toluenium Borate (8(C7H8)[B(C6F5)4]) from Silane 15
Tris(trimethylsilyl)germyldimethylsilane 15 (0.18 g (0.50 mmol)) and 0.46 g (0.50 mmol) of triphenylmethyl tetrakis(pentafluorophenyl)borate were both dissolved in 1 mL of toluene-d8 and cooled to −20 °C. Germylsilane 15 was slowly added to the borate salt via a Teflon tube, and the mixture was stirred at −20 °C for 1.5 h. The brown polar phase and the light yellow nonpolar phase were each transferred to separate NMR tubes at −20 °C and stored at −60 °C overnight until the NMR spectra were recorded the next morning. The polar phase contained borate [8(C7D8)][B(C6F5)4]. The nonpolar phase contained triphenylmethane and the rearrangement product 2. Polar phase1H NMR (499.87 MHz, 253.0 K, C7D8, δ ppm): −0.15 (((CH3)3Si)3GeSi(CH3)2+), 0.12 (((CH3)3Si)3GeSi(CH3)2+). 13C{1H} NMR (125.71 MHz, 253.0 K, C7D8, δ ppm): 2.5 (((CH3)3Si)3GeSi(CH3)2+), 5.5 (((CH3)3Si)3GeSi(CH3)2+). 29Si{1H} NMR (99.31 MHz, 253.0 K, C7D8, δ ppm): −2.4 (((CH3)3Si)3GeSi(CH3)2+), 98.1 (((CH3)3Si)3GeSi(CH3)2+) (see Figures S17a–c in the Supporting Information). Nonpolar phase1H NMR (499.87 MHz, 305.0 K, C7D8, δ ppm): 0.29 (s, ((CH3)3Si)4Ge), 5.38 (s, Ph3CH), 6.98–7.10 (m, Ph3CH). 13C{1H} NMR (125.69 MHz, 305.0 K, C7D8, δ ppm): 3.5 (((CH3)3Si)4Ge), 57.2 (Ph3CH), 125.4 (Ph3CH), 128.3 (Ph3CH), 129.2 (Ph3CH), 144.3 (Ph3CH). 29Si{1H} NMR (99.31 MHz, 305.0 K, C7D8, δ ppm): −5.1 (((CH3)3Si)4Ge) (see Figure S18a–c in the Supporting Information).
Tris(trimethylsilyl)germyldimethylsilyl-phenylchloronium Borate (8(C6D5Cl)[B(C6F5)4]) from Silane 15
Tris(trimethylsilyl)germyldimethylsilane 15 (0.09 g (0.25 mmol)) and 0.23 g (0.25 mmol) of triphenylmethyl tetrakis(pentafluorophenyl)borate were both dissolved in 0.5 mL of chlorobenzene-d5 and cooled to −20 °C. Germylsilane 15 was slowly added to the borate salt via a Teflon tube, and the mixture was stirred at −20 °C for 1.5 h. The brown solution was transferred into a NMR tube at −20 °C and stored at −60 °C overnight until the NMR spectra were recorded the next morning. The mixture contained borate [8(C6D5Cl)][B(C6F5)4], the rearrangement product 2, and triphenylmethane. 29Si{1H} NMR (99.31 MHz, 253.0 K, C6D6Cl, δ ppm): −5.1 (((CH3)3Si)4Ge), −1.4 (((CH3)3Si)3GeSi(CH3)2+), 154.3 (((CH3)3Si)3GeSi(CH3)2+).
Tris(trimethylsilyl)germyldimethylsilyl Toluenium Borate (8(C7D8)[B(C6F5)4]) from Germane 16
Bis(trimethylsilyl)pentamethyldisilanylgermane 16 (0.11 g (0.32 mmol)) and 0.29 g (0.32 mmol) of triphenylmethyl tetrakis(pentafluorophenyl)borate were both dissolved in 1 mL of toluene-d8 and cooled to −20 °C. Silylgermane 18 was slowly added to the borate salt via a Teflon tube, and the mixture was stirred at −20 °C for 5 min. The brown polar phase and the light yellow nonpolar phase were each transferred to separate NMR tubes at −20 °C and stored at −60 °C for 5 h until the NMR spectra were recorded. At −60 °C, the polar phase solidifies, and no further reaction is expected. The NMR spectra of the polar phase recorded at −20 °C contained nearly exclusively borate [8(C7D8)][B(C6F5)4]. The nonpolar phase contained triphenylmethane and the rearrangement product 2. Polar phase1H NMR (499.87 MHz, 253.0 K, C7D8, δ ppm): −0.16 (((CH3)3Si)3GeSi(CH3)2+), 0.12 (((CH3)3Si)3GeSi(CH3)2+). 13C{1H} NMR (125.71 MHz, 253.0 K, C7D8, δ ppm): 2.5 (((CH3)3Si)3GeSi(CH3)2+), 5.5 (((CH3)3Si)3GeSi(CH3)2+). 29Si{1H} NMR (99.31 MHz, 253.0 K, C7D8, δ ppm): −2.4 (((CH3)3Si)3GeSi(CH3)2+)), 98.2 (((CH3)3Si)3GeSi(CH3)2+)) (see Figures S19a–c in the Supporting Information). Nonpolar phase1H NMR (499.87 MHz, 305.0 K, C7D8, δ ppm): 0.30 (s, ((CH3)3Si)4Ge), 5.38 (s, Ph3CH), 6.98–7.10 (m, Ph3CH). 13C{1H} NMR (125.69 MHz, 305.0 K, C7D8, δ ppm): 3.5 ((CH3)3Si)4Ge), 57.1 (Ph3CH), 125.4 (Ph3CH), 128.2 (Ph3CH), 129.1 (Ph3CH), 144.3 (Ph3CH). 29Si{1H} NMR (99.31 MHz, 305.0 K, C7D8, δ ppm): −5.1 (((CH3)3Si)4Ge) (see Figure S20a–c in the Supporting Information).
Acknowledgments
This work was supported by the ERA-chemistry program (DFG-Mu1440/8-1, FWF-I00669) and by Carl von Ossietzky University Oldenburg. The simulations were performed at the HPC Cluster HERO (High End Computing Resource Oldenburg), located at the University of Oldenburg (Germany) and funded by the DFG through its Major Research Instrumentation program (INST 184/108-1 FUGG) and the Ministry of Science and Culture (MWK) of the Lower Saxony State.
Supporting Information Available
Experimental and theoretical characterization, including the preparation of all compounds of interests and computational details. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.organomet.5b00431.
The authors declare no competing financial interest.
Supplementary Material
References
- Recent review:Marschner C. Struct. Bonding 2014, 155, 163–288. [Google Scholar]
- Recent review:Corey J. Y. Adv. Organomet. Chem. 2004, 51, 1–52. [Google Scholar]
- Ishifune M.; Kashimura S.; Kogai Y.; Fukuhara Y.; Kato T.; Bu H.-B-; Yamashita N.; Murai Y.; Murase H.; Nishida R. J. Organomet. Chem. 2000, 611, 26–31 10.1016/S0022-328X(00)00327-2. [DOI] [Google Scholar]
- a Ishikawa M.; Kumada M. J. Chem. Soc., Chem. Commun. 1969, 567–568. [Google Scholar]; b Ishikawa M.; Kumada M. J. Chem. Soc., Chem. Commun. 1970, 157. [Google Scholar]; c Ishikawa M.; Iyoda J.; Ikeda H.; Kotake K.; Hashimoto T.; Kumada M. J. Am. Chem. Soc. 1981, 103, 4845–4850 10.1021/ja00406a029. [DOI] [Google Scholar]; d Ishikawa M.; Watanawe M.; Iyoda J.; Ikeda H.; Kumada M. Organometallics 1982, 1, 317–322 10.1021/om00062a015. [DOI] [Google Scholar]
- Blinka A.; West R. Organometallics 1986, 5, 128–133 10.1021/om00132a023. [DOI] [Google Scholar]
- a Märkl G.; Wagner R. Tetrahedron Lett. 1986, 27, 4015–4018 10.1016/S0040-4039(00)84898-4. [DOI] [Google Scholar]; b Sharma S.; Caballero N.; Li H.; Pannell K. H. Organometallics 1999, 18, 2855–2860 10.1021/om990169p. [DOI] [Google Scholar]
- Fischer J.; Baumgartner J.; Marschner C. Science 2005, 310, 825. 10.1126/science.1118981. [DOI] [PubMed] [Google Scholar]
- Recent reviews:; a Müller T. Struct. Bonding 2014, 155, 107–162 10.1007/430_2013_132. [DOI] [Google Scholar]; b Müller T. In Science of Synthesis: Knowledge Updates 2013/3; Oestreich M., Ed.; Thieme: Stuttgart, Germany, 2013; pp 1–42. [Google Scholar]; c Klare H. F. T.; Oestreich M. Dalton Trans. 2010, 39, 9176–9184 10.1039/c003097j. [DOI] [PubMed] [Google Scholar]; d Müller T. Adv. Organomet. Chem. 2005, 155–215 10.1016/S0065-3055(05)53005-3. [DOI] [Google Scholar]
- a Lambert J. B.; Zhang S. J. Chem. Soc., Chem. Commun. 1993, 383–384 10.1039/c39930000383. [DOI] [Google Scholar]; b Lambert J. B.; Zhang S. J.; Ciro S. M. Organometallics 1994, 13, 2430–2443 10.1021/om00018a041. [DOI] [Google Scholar]; c Ottosson C.-H.; Cremer D. Organometallics 1996, 15, 5495–5501 10.1021/om960179f. [DOI] [Google Scholar]; d Ottosson C.-H.; Szabó K. J.; Cremer D. Organometallics 1997, 16, 2377–2385 10.1021/om9701635. [DOI] [Google Scholar]
- a Sekiguchi A.; Matsuno T.; Ichinohe M. J. Am. Chem. Soc. 2000, 122, 11250–11251 10.1021/ja002344v. [DOI] [Google Scholar]; b Inoue S.; Ichinohe M.; Yamaguchi T.; Sekiguchi A. Organometallics 2008, 27, 6056–6058 10.1021/om800912v. [DOI] [Google Scholar]
- a Ichinohe M.; Igarashi M.; Sanuki K.; Sekiguchi A. J. Am. Chem. Soc. 2005, 127, 9978–9979 10.1021/ja053202+. [DOI] [PubMed] [Google Scholar]; b Igarashi M.; Ichinohe M.; Sekiguchi A. J. Am. Chem. Soc. 2007, 129, 12660–12661 10.1021/ja075740n. [DOI] [PubMed] [Google Scholar]
- Nakamoto M.; Fukawa T.; Sekiguchi A. Chem. Lett. 2004, 33, 38–39 10.1246/cl.2004.38. [DOI] [Google Scholar]
- Wagner H.; Wallner A.; Fischer J.; Flock M.; Baumgartner J.; Marschner C. Organometallics 2007, 26, 6704–6717 10.1021/om7007524. [DOI] [Google Scholar]
- Wagner H.; Baumgartner J.; Müller T.; Marschner C. J. Am. Chem. Soc. 2009, 131, 5022–5023 10.1021/ja809270m. [DOI] [PubMed] [Google Scholar]
- Reviews:; a Reed C. A. Acc. Chem. Res. 2010, 43, 121–128 10.1021/ar900159e. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Krossing I.; Raabe I. Angew. Chem., Int. Ed. 2004, 43, 2066–2090 10.1002/anie.200300620. [DOI] [PubMed] [Google Scholar]
- No silyl- or germylchlorides have been detected as by-products. Obviously, the rearrangement of the intermediate cations and alkylation of the final cation is faster than chloride abstraction from the solvent.
- a Bartlett P. D.; Condon F. E.; Schneider A. J. Am. Chem. Soc. 1944, 66, 1531–1539 10.1021/ja01237a037. [DOI] [Google Scholar]; b Corey J. Y. J. Am. Chem. Soc. 1975, 97, 3237–3238 10.1021/ja00844a063. [DOI] [Google Scholar]
- All computations were performed using the Gaussian 09 package: Frisch M. J.et al. Gaussian 09, revision B.01; Gaussian, Inc.: Wallingford, CT, 2009. [Google Scholar]
- See Supporting Information for further details.
- At temperatures lower than −20 °C, the ionic phase became solid, which excludes further NMR investigations.
- Krempner C. Polymers 2002, 4, 408–447 10.3390/polym4010408. [DOI] [Google Scholar]
- In chlorobenzene solution, usually the corresponding silylated chloronium ions predominate over the silylated arenium ion, see:; a Schäfer A.; Saak W.; Haase D.; Müller T. Angew. Chem., Int. Ed. 2012, 51, 2881–2984 10.1002/anie.201107958. [DOI] [PubMed] [Google Scholar]; b Hoffmann S. P.; Kato T.; Tham F. S.; Reed C. A. Chem. Commun. 2006, 767–769 10.1039/b511344j. [DOI] [PubMed] [Google Scholar]
- In addition, NMR detection of toluenium ion 8(C7D8) at temperatures around 0 °C is severely hampered by significant line broadening of the 29Si NMR resonances, most probably due to fast exchange of the toluene molecule in cation 8(C7D8) with the solvent (for example, the line width at half height, w1/2 of the 29Si NMR signal at δ29Si = 98.1 is w1/2 = 17 Hz at T = −20 °C and w1/2 = 40 Hz at T = +10 °C).
- a Massey A. G.; Park A. J. J. Organomet. Chem. 1964, 2, 245–250 10.1016/S0022-328X(00)80518-5. [DOI] [Google Scholar]; b Chien J. C. W.; Tsai W. M.; Rausch M. D. J. Am. Chem. Soc. 1991, 113, 8570–8571 10.1021/ja00022a081. [DOI] [Google Scholar]
- Gilman H.; Smith C. L. J. Organomet. Chem. 1967, 8, 245–253 10.1016/S0022-328X(00)91037-4. [DOI] [Google Scholar]
- Brook A. G.; Abdesaken F.; Söllradl H. J. Organomet. Chem. 1986, 299, 9–13 10.1016/0022-328X(86)84028-1. [DOI] [Google Scholar]
- Marschner C. Eur. J. Inorg. Chem. 1998, 1998, 221–226. [DOI] [Google Scholar]
- Fischer J.; Baumgartner J.; Marschner C. Organometallics 2005, 24, 1263–1268 10.1021/om0491894. [DOI] [Google Scholar]
- Mallela S. P.; Ghuman M. A.; Geanangel R. A. Inorg. Chim. Acta 1992, 202, 211–217 10.1016/S0020-1693(00)86836-X. [DOI] [Google Scholar]
- Gluyas J. B. G.; Burschka C.; Kraft P.; Tacke R. Organometallics 2010, 29, 5897–5903 10.1021/om100720k. [DOI] [Google Scholar]
- Lehmann M.; Schulz A.; Villinger A. Angew. Chem., Int. Ed. 2009, 48, 7444–7447 10.1002/anie.200902992. [DOI] [PubMed] [Google Scholar]
- a West R.; Hunt H. R.; Whipple R. O. J. Am. Chem. Soc. 1954, 76, 310–310 10.1021/ja01630a105. [DOI] [Google Scholar]; b Moedritzer K.; Van Wazer J. R. Inorg. Chem. 1965, 4, 1753–1760 10.1021/ic50034a018. [DOI] [Google Scholar]
- Schäfer A.; Schäfer A.; Müller T. Dalton Trans. 2010, 39, 9296–9303 10.1039/c0dt00313a. [DOI] [PubMed] [Google Scholar]
- Sharma S.; Pannell K. H. Organometallics 2000, 19, 1225. 10.1021/om990897c. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.