

Abstract
A wide range of nanoparticles has been explored for the delivery of highly hydrophobic drugs, but very few publications provide comparative data of the performance of different nanoparticles. To address this need, this publication compares poly(lactic-co-glycolic acid) (PLGA) nanoparticles and nanospheres made from tyrosine-derived tri-block copolymers (termed TyroSpheres) for their respective performance as carriers for cyclosporine A (CSA). Using previously reported data on PLGA, we followed similar experimental protocols to evaluate the in vitro characteristics of TyroSpheres. Although there are some similarities between the two particle systems for the delivery of CSA, such as effective encapsulation and epidermal skin penetration, several differences were notable. First, the methods of preparation were different, i.e., self-assembly and emulsion-diffusion-evaporation process for TyroSpheres and PLGA, respectively. Second, TyroSpheres provided 7-day diffusion-controlled release, whereas PLGA nanoparticles provided >21-day erosion-controlled release. Third, the size of TyroSpheres was measured to be ~60–70 nm irrespective of drug loading, whereas the size of PLGA nanoparticles (~100–250 nm) was dependent on drug loading and the method of preparation. Overall, this publication provides a direct comparison between two different types of nanoparticles and illuminates the respective advantages and disadvantages, using CSA as a model for the release of highly hydrophobic drugs.
Nanoparticles have been developed as a promising approach to increase the bioavailability, solubility, circulation time, and resistance to metabolic degradation of lipophilic drugs and deliver these drugs into targeted organs or cells1,2,3. Among the many nanoparticle drug delivery systems (solid lipid and polymeric) reported in the literature4,5, nanoparticles made of poly(lactic-co-glycolic acid) (PLGA) have been studied extensively. PLGA is one of the most widely used polymeric biomaterials due to its (i) biodegradability and biocompatibility6, (ii) track record for Food and Drug Administration (FDA) and European Medicine Agency approval in drug delivery systems7, (iii) well described formulations and methods of production, (iv) ability to incorporate hydrophilic and hydrophobic molecules of various sizes8, (v) ability to protect drugs from degradation and provide sustained release9, and (vi) potential to provide better interaction with biological materials through the modification of its surface properties and targeting to specific organs or cells10,11. PLGA nanoparticles have been used as drug delivery vehicles for oral and parenteral administration12,13,14,15. In 2001, a detailed review of their utility in topical drug delivery applications was published by de Jalon and coworkers16. Despite these advantages, there are reports in the literature that PLGA nanoparticles have the potential to induce toxicity from dose dumping17, inconsistent release and drug-polymer interactions18.
Our laboratory has developed a nanoparticle system that is synthesized from a family of fully-degradable, ABA-type tri-block copolymers made of poly(ethylene glycol), oligomers of desaminotyrosyl-tyrosine esters and suberic acid (Fig. 1A)19,20. These tyrosine-derived copolymers are amphiphilic and spontaneously self-assemble to form supramolecular nanoparticles with a core-shell architecture in aqueous environments. Preparation via self-assembly is simple, time efficient and the particles exhibit a very low critical aggregation concentration (cac) of 2.6 × 10−7 g/mL19,20. The lipophilic drugs are incorporated into the hydrophobic core, which serves as a drug reservoir, and the hydrophilic shell enables stable dispersion in an aqueous environment21. In addition, the hydrophilic shell also offers prolonged circulation time, protection from protein adsorption and subsequent opsonization and clearance from the body22. The tyrosine-derived nanoparticles (referred to herein as TyroSpheres) have been shown to be nontoxic in vitro19 and in vivo23,24, bind and deliver lipophilic drugs21, substantially enhance the permeation of lipophilic agents into the epidermis25, and have been added to a gel formulation to maximize contact with the skin and prevent the “run-off” effect of non-viscous solutions or suspensions26.
Figure 1.

The chemical structures of (A) PEG-b-oligo(DTR-XA)-b-PEG tri-block copolymer, (B) Poly-L-lactic-co-glycolic acid (PLGA), and (C) Cyclosporine A (CSA).
Most often, one laboratory reports on multiple variations of the same basic nanoparticle system. For example, Sheihet et al. (2007) demonstrated that changes in the tyrosine-derived tri-block copolymer composition affect drug loading capabilities21. Knop et al. (2013) compared two amphiphilic star-shaped polymers, poly(ε-caprolactone)-block-poly(oligo(ethylene glycol)methacrylate)s and poly(ε-caprolactone)-block-poly(oligo(2-ethyl-2-oxazoline)methacrylate)s and showed that the molar mass of the macromonomer rather than the mere length was the crucial factor to form an efficiently stabilizing hydrophilic shell27. Zhao et al. (2013) compared the capability of 4-arm and 6-arm mPEG-PLA micelles to deliver a hydrophobic drug, indomethacin, and found that loading was higher in the 4-arm than the 6-arm copolymer28. According to our knowledge, there are only few literature reports available that directly compare two or more different nanoparticle formulations for the delivery of a specific drug. One study by Quan et al. (2014) showed a head-to-head comparison of four variations of a nanoparticle system for the delivery of dexamethasone in an adjuvant-induced arthritis rat model: liposomes, core-cross-linked micelles, slow releasing polymeric prodrugs, and fast releasing polymeric prodrugs29. In a second study, Abdel-Mottaleb et al. (2011) presented a direct comparison of two dissimilar nanoparticular systems (polymeric versus lipid-based) and showed that lipid and polymeric nanoparticles were more efficient for transdermal drug delivery and localized drug delivery to the dermis, respectively30. In a third study, Daman et al. (2014) demonstrated that self-assembled formulations promoted higher in vitro cellular uptake and cytotoxicity when compared to micellar formulations of the same polymer31.
In our work, we directly compared two different types of polymeric nanoparticles and their respective performance: TyroSpheres and PLGA nanoparticles. A recent report by Jain et al. on the topical delivery of CSA using PLGA nanoparticles provides a rich collection of characterization and performance data32,33. This made it possible for us to conduct a study, comparing PLGA nanoparticles (based on Jain’s published data) with the TyroSphere system. To facilitate this comparison, we adapted and followed as much as possible the procedures described by Jain et al.32. Specifically, we used the same drug (CSA) and the same target application (enhancement of skin penetration), and conducted similar in vitro evaluation studies (particle fabrication, particle size and distribution, drug release kinetics, stability, and skin distribution profile).
Results and Discussion
The polymer was synthesized using previously published protocols and polymer composition was confirmed using 1H-NMR21. The data obtained on CSA-loaded TyroSpheres was compared to two reports by Jain et al. on the evaluation of PLGA nanoparticles for CSA delivery32,33.
Characterization of nanoparticles
Nanoparticle preparation methods: Self-assembly vs. emulsion-diffusion-evaporation
The methods used to prepare TyroSpheres (self-assembly) and PLGA nanoparticles (emulsion-diffusion-evaporation) are substantially different. The preparation of CSA-TyroSpheres is a simple, single step method that depends on the self-assembly of a tri-block (ABA) tyrosine-derived copolymer in aqueous solution. In this method, the ABA-tri-block tyrosine-derived copolymer is added drop-wise to an aqueous solution containing CSA, and self-assembles into a nano-particle. No use of stabilizers or sonication/homogenization is required as opposed to other methods of nanoparticle preparation34.
The emulsion-diffusion-evaporation method used by Jain et al. results in the formation of nano to micron sized particles depending on the speed of shearing used in the process. Although this method results in the formation of spherical particles, which can be tuned in terms of their size and morphology, it requires the use of a stabilizer (such as poly(vinyl alcohol) or surfactants) for the prevention of aggregation or to increase the shelf life of the particles32.
Although both methods form spherical particles that can be tailored for size and morphology, the preparation method for TyroSpheres and the nature of the polymer offer several advantages. First, the TyroSpheres are prepared using a simpler, single-step method as compared to the PLGA nanoparticles, which requires a multi-step method and a stabilizer (e.g., poly(vinyl alcohol), surfactants, etc.). Second, the nature of the polymer composition (ABA tri-block) efficiently restricts the encapsulation of the lipophilic drug within the hydrophobic core of the TyroSpheres. This minimizes the amount of surface-bound drug, thus preventing a large bolus of drug from bursting out and leading to more controlled release. Third, the copolymer composition intentionally uses molecules of PEG to flank the oligomers of desaminotyrosyl-tyrosine esters and suberic acid. This chemical composition has been shown to decrease opsonization, which can result in increased drug retention time in the blood and reduced nonspecific distribution35,36.
Drug loading content and loading efficiency
Figure 2 shows the drug loading content and loading efficiency of CSA into TyroSpheres for theoretical loading content ranging from 10 to 60 wt.%.
Figure 2. Loading content and loading efficiency of CSA-TyroSpheres with theoretical loading concentrations ranging from 10–60 wt.%.
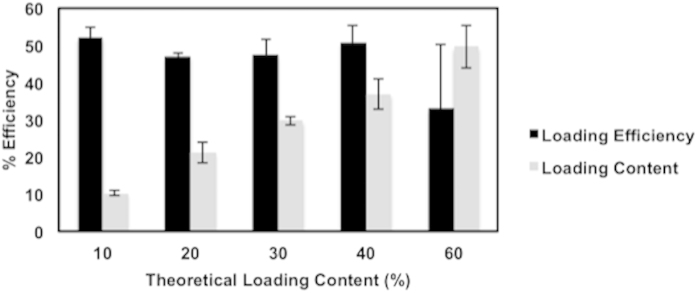
The loading efficiency was consistent at approximately 50%, irrespective of the theoretical loading content (10–60 wt.%). The 50% loss in drug concentration is attributed to the loss of both drug and polymer during the two washing and filtration steps. Despite this loss, the measured (actual) loading content equals the theoretical loading content (up to 40 wt.%), which suggests that the nanoparticles surviving the preparation process are loaded with the targeted concentration of CSA. In the case of 60 wt.% loading, the saturation limit was reached which caused the drug to precipitate, ultimately resulting in an average actual loading content of 35% with large variability (Table S1, see supplementary data). Based on these data, subsequent characterization was performed using TyroSpheres loaded with 30 wt.% CSA.
In contrast, the actual loading efficiencies of CSA into PLGA nanoparticles varied with theoretical loading concentrations. Jain et al. showed that loading efficiency increased to 86% only up to 10 wt.% target CSA loading and then decreased to 62% efficiency for 15 wt.% theoretical CSA loading. Although the range of theoretical loadings was quite limited (5, 10, and 15 wt.%), these data suggest that the actual loading of CSA into PLGA nanoparticles is substantially more limited than that of the TyroSpheres32.
Characterization of CSA within nanoparticles
The characteristic peak of CSA within the TyroSpheres was evaluated using XRD. Figure 3 shows the XRD patterns of the tri-block copolymer, empty TyroSpheres, 30 wt.% CSA-TyroSpheres, physical mixture of CSA and tri-block copolymer, and pure CSA. Characteristic peaks for the tri-block copolymer were observed in all samples containing polymer: empty TyroSpheres, polymer, physical mixture of CSA and polymer, and CSA-TyroSpheres (Fig. 3). These data confirm that CSA was successfully loaded into the cavity of the TyroSpheres, and was not present on the surface of the particles.
Figure 3. XRD patterns of the tri-block copolymer, empty TyroSpheres, CSA-TyroSpheres, a physical mixture of CSA and the tri-block copolymer, and pure CSA.
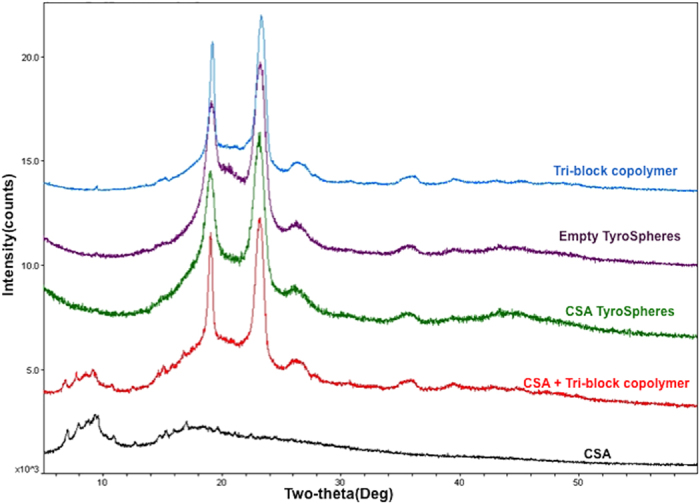
An explanation for the lack of a CSA peak in the CSA-TyroSpheres sample is that CSA is molecularly dispersed within the hydrophobic polymer domains and hence, does not scatter X-rays efficiently37. Similar data was observed by Jain et al. for the encapsulation of CSA by PLGA nanoparticles32.
Size, size distribution, and morphology
The hydrodynamic diameter of empty and CSA-TyroSpheres in aqueous solution was analyzed using DLS. Table S1 (see supplementary data) shows that the diameter ranges from 65–75 nm, irrespective of CSA loading up to 40 wt.%. It is important to note that the surface charge of the TyroSpheres is neutral, which confirms the presence of PEG on the outer core23.
The diameter of TyroSpheres loaded with 60 wt.% differs because the drug precipitated out showing that no more drug can be encapsulated at that given weight ratio. The size distribution remains narrow based on the PDI values, which were in the range of 0.2–0.3. The diameter of CSA-TyroSpheres-gel was ~2 times larger than CSA-TyroSpheres. This difference is likely due to the swelling of particles in the presence of HPMC gel resulting in an increased hydrodynamic diameter38. In a previous study, the morphology and diameter of lyophilized TyroSpheres and TyroSpheres-gel were studied using transmission electron microscopy (TEM) and it was shown that the average diameter was ~40 nm irrespective of CSA loading (0.5–5 wt.%) or the presence of HPMC gel26. An explanation for this is that TyroSpheres have a fixed cavity size regardless of the method of preparation and the type of stabilizer used, and as a result, there was no effect of drug loading on the particle size. In comparison, Jain et al. reported that the diameter of empty PLGA nanoparticles ranged from 100–250 nm, as measured by DLS, using various methods of preparation32. Also, the size of the particles was dependent on the kind of stabilizer used. In contrast, the average size of PLGA nanoparticles slightly increased by 9.5% (158 nm to 173 nm) when CSA loading was increased from 5 to 15 wt.%, using a standard method and stabilizer. This shows that the size of PLGA nanoparticles is highly dependent on the type of method and stabilizer used.
Nanoparticles less than 100 nm in size have been shown to enhance the penetration of the cargo into the cells and increase the efficiency of the delivery39. In this study, the size of the TyroSpheres was ~65–75 nm irrespective of CSA loading. This indicates that these particles have a fixed cavity size. Additionally, the size of the cavity may be changed by varying the length of the central hydrophobic block in the ABA-tri-block copolymer (Fig. 1A)40. On the other hand, the size of PLGA nanoparticles is highly dependent on the type of solvents and stabilizers used during the preparation process. While on one hand this may be an advantage for PLGA-based systems which makes them more tunable, on the other hand, one needs to optimize many parameters such as polymer concentration, type and concentration of stabilizer, speed and time for homogenization, concentration of drug, etc., to reach a targeted particle size32. Most data in the literature report a PLGA particle size above 100 nm, however some papers describe methods that can produce particles with diameters lower than 100 nm with or without drug loading41,42.
Solubility of CSA
Nanoparticles help in the dispersion of the drug into aqueous solutions by encapsulation and via hydrophobic-hydrophilic interactions39. For example, in the case of the self-assembly of the tri-block copolymer, the amphiphilic nature of the polymer allows for encapsulation of the hydrophobic drug inside the hydrophobic core. The hydrophilic outer surface of the polymer (comprising the PEG blocks) ensures interactions with polar solvents (such as water) and allows improved stabilization through hydrophilic interactions. Since the monomers are neutral, the interactions between PEG-water and the drug-hydrophobic core are not electrostatic. TyroSpheres are stable in water predominantly through hydrophobic-hydrophilic interactions. CSA is a hydrophobic drug and is known to have very low solubility (Log P = 4.12) in aqueous solutions. In this study, the solubility of free CSA in solution was compared to CSA encapsulated within TyroSpheres.
Table S2 shows that the solubility of CSA was ~1106 fold higher when encapsulated in TyroSpheres as compared to its free form in PBS. These data suggest that TyroSpheres are considerably more efficient at solubilizing CSA as compared to 1xPBS.
In vitro release of CSA from TyroSpheres
Previous reports on the release of hydrophobic moieties from TyroSpheres have shown ~30–40% release within 72 hours (for example, paclitaxel and nile red)20,23,25. The in vitro release of CSA from 30 wt.% CSA-TyroSpheres was studied for 7 days in 1xPBS at 37 °C under sink conditions. Figure 4A shows that the release of CSA from CSA-TyroSpheres was sustained for 7 days where 23 ± 1%, 52 ± 1% and 75 ± 10% was released at 1, 3 and 7 days, respectively (R2 = 0.76, rate constant, k = 0.52) (Table S3). In addition, the effect of CSA loading (10 vs. 30 wt.%) on the release from TyroSpheres was investigated. Table S3 shows that there was no significant difference (P > 0.05) in the release (R2 = 0.62, rate constant, k = 0.68), which indicates that CSA release is not affected by CSA loading in this range. Moreover, the cumulative release was not linear with time (Fig. 4A).
Figure 4.

(A) In vitro release of CSA for 7 days in PBS (pH 7.4, 37 °C) as a function of time from i) 30 wt.% CSA-TyroSpheres (open squares), and ii) 10 wt.% CSA-TyroSpheres (open triangles), (B) In vitro release of CSA for 7 days in PBS (pH 7.4, 37 °C) as a function of the square root of time (Higuchi model).
To better elucidate the mechanism of CSA release from the particle systems, data was fit to two rate equations:
Zero order release:
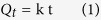 |
Higuchi model:
 |
where Q is the amount of drug released, t is time, and k is the rate constant. The release of CSA from TyroSpheres best fit the Higuchi square root model (Equation 2) with R2 > 0.9, irrespective of CSA loading. This indicates that the release of CSA from TyroSpheres is diffusion controlled (Fig. 4B). Jain et al. demonstrated that the PLGA nanoparticle system provided a longer (>21 vs. 7 days) and slower release of CSA and released only 8%, 15%, and 30% at 1, 3 and 7 days, respectively. In addition, the release profile was reported to have zero order kinetics (Equation 1), suggesting that the release of CSA from these PLGA nanoparticles was predominantly controlled by polymer erosion. However, others have reported that drug release from PLGA nanoparticles takes place by several mechanisms including surface and bulk erosion, disintegration, diffusion, and desorption, where the initial stages of drug release are predominantly controlled by diffusion of the drug, and the later phases by degradation of the polymer matrix43.
Overall, the data shows that TyroSpheres provide an initial burst (~23% within 24 h) followed by sustained release (~75% of the payload) of CSA for 7 days. On the other hand, Jain et al. showed that PLGA nanoparticles release CSA in a sustained manner for 21 days without an initial burst32. Both release profiles could be therapeutically beneficial depending on the targeted clinical indication. For example, a cutaneous wound healing application may benefit from an initial burst to provide immediate relief followed by prolonged release to promote gradual healing44. On the other hand, food coatings are used to protect flavors and aromas during processing and storage, but must provide rapid release when the product is consumed44.
Stability of TyroSpheres/CSA-TyroSpheres in solution
No significant difference in particle size was found when unloaded TyroSpheres were suspended in 1xPBS for 6 months at 4 °C: size = 65 ± 5 nm, PDI = 0.18 ± 0.05 at time 0, and size = 68 ± 7 nm, PDI = 0.17 ± 0.08 at 6 months (P > 0.05). Next, the stability of 30 wt.% CSA-TyroSpheres suspended in 1xPBS was assessed for 28 days at 4, 25, and 37 °C and compared to the stability of free CSA (not encapsulated in TyroSpheres). At all storage temperatures, the stability of encapsulated CSA was considerably longer than free CSA (Fig. 5). Specifically, TyroSpheres protected CSA from degradation at 4 °C and 25 °C up to four weeks and at 37 °C up to three weeks. Significant degradation was measured at 37 °C, where 50% of both encapsulated CSA and free CSA degraded at 4 weeks (P < 0.05). In contrast, PLGA nanoparticles are not stable in solution for periods longer than a few days45,46. Therefore, PLGA-based formulations often require stabilizers when used in solution and are transformed into dry for long-term stability45,46.
Figure 5.
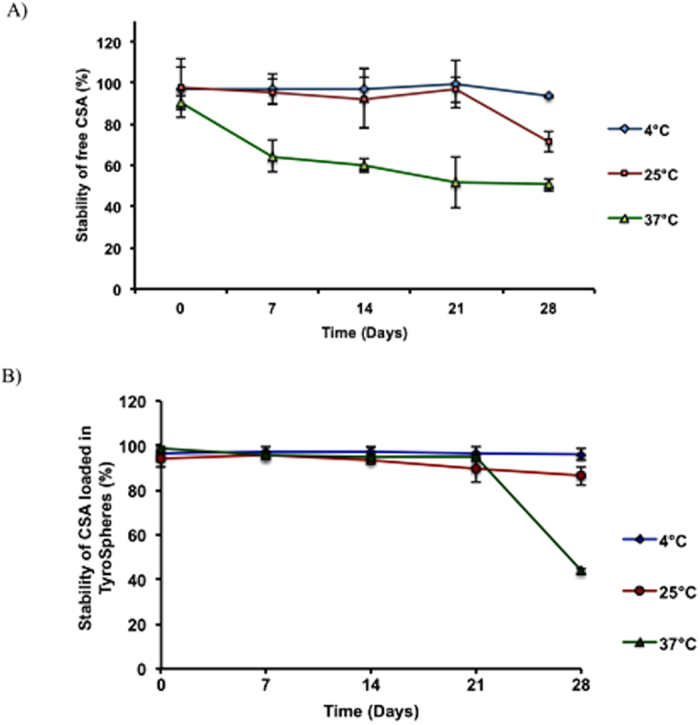
Stability of (A) free CSA and (B) 30 wt.% CSA-TyroSpheres in PBS for 28 days at 4°, 25° and 37 °C.
Freeze-thaw analysis of TyroSpheres
An analysis was done to determine the stability of empty TyroSpheres with and without 10% sucrose (used as a lyoprotectant) suspended in PBS after three freeze-thaw cycles (-80°C, 25°C). Particle size was measured before and after each freeze-thaw cycle and the ratio of the final particle size (Sf) to initial size (Si) was calculated. The Sf/Si ratio was calculated to be close to 1, which suggests minimal change in the particle size as a result of the freeze-thaw cycles (Table 1). Further, the presence of sucrose as a lyoprotectant did not notably change the Sf/Si ratio. Therefore, a stabilizer is not required to provide stability to the liquid formulations.
Table 1. The effect of freeze-thaw cycles on the particle size of TyroSpheres with and without the presence of sucrose as a lyoprotectant (Mean ± SD is reported, n = 3).
| Sample (n = 3) |
Before freezing |
After thawing |
Sf/Si | ||
|---|---|---|---|---|---|
| Particle size (nm) | PDI | Particle size (nm) | PDI | ||
| TyroSpheres alone | 74 ± 5 | 0.18 ± 0.01 | 84 ± 6 | 0.19 ± 0.01 | 1.1 |
| TyroSpheres + sucrose | 93 ± 3 | 0.18 ± 0.01 | 100 ± 4 | 0.21 ± 0.02 | 1.1 |
Preparation of dry formulations of TyroSpheres
Most nanoparticle formulations are suspensed in aqueous media for application in vivo. In addition, many polymeric nanoparticles, including TyroSpheres and PLGA nanoparticles, are susceptible to non-enzymatic hydrolytic degradation that is largely responsible for their instability in aqueous environments. Therefore, the development of a fully re-constitutable, dry formulation of nanoparticles is a suitbale method to increase product shelf-life. A freeze-drying technique was developed to convert aqueous suspensions of TyroSpheres into dry-formulated lyocakes. The optimized freeze-drying process was developed by B. Kilfoyle (PhD Thesis, 2011, New Jersey Center for Biomaterials, Rutgers University) and is summarized in section 2.12 of the materials and methods section of the thesis. This process requires the inclusion of a lyoprotectant (e.g., sugar) to provide stability during the drying process. In this way, the lyoprotectants interact with the nanoparticle surface via hydrogen bonding, displace water molecules during sublimation, prevent nanoparticle destruction, and maintain the nanoparticles’ properties upon rehydration. Various investigators have reported on the use of sugars as lyoprotectants during the freeze-drying process. Layre et al. determined that the incorporation of glucose, trehalose, maltose, and saccharose was insufficient for a reduction in agglomeration of polycaprolactone-PEG nanoparticles47. Saez et al. showed that sucrose, glucose, and trehalose maintained the integrity of PLGA nanoparticles without aggregation during freeze-thaw analysis but after lyophilization, the Sf/Si ratios were above the limit for non-significant changes (1.5)45.
In this work, we investigated the effects of sucrose concentration as a lyoprotectant for TyroSpheres during freeze-drying (Table S4). Overall, it was observed that TyroSphere particle size (Si) increased with increasing sucrose concentration. Therefore, 275 mM sucrose was determined to be the optimal concentration based on the initial particle size (<100 nm) and the Sf/Si ratio was close to 1 (1.1). Figure 6A shows a TEM micrograph of TyroSpheres suspended in water, where the TyroSpheres in solution are well dispersed and can be individually identified. The table in Fig. 6 contains the particle size measured using TEM micrographs (Image J software, National Institute of Health, USA) and DLS analysis.
Figure 6.

(A) A TEM micrograph of TyroSpheres suspended in water. SEM micrographs of lyocakes of TyroSpheres (B) without sucrose and (C) with 275 mM sucrose as a lyoprotectant. The table included in the figure shows the particle size measured using TEM and DLS. Scale bar = 100 nm for TEM and 10 μm for SEM micrographs.
Figure 6B,C show SEM micrographs of the TyroSpheres lyocake without and with 275 mM sucrose, respectively. The structure of the lyocake containing TyroSpheres without sucrose shows massive aggregation and a collapsed structure, which is supported by the very high Sf/Si ratio (88). On the other hand, the lyocakes containing TyroSpheres with 275 mM sucrose had a desirable cage-like structure and a Sf/Si ratio of 1.1. It is likely that the sucrose molecules interacted with the nanoparticle surface (via hydrogen bonding) and prevented their collapse45.
In a previous study by Saez et al., 20% glucose (w/v) or 20% sucrose (w/v) were found to be the most suitable lyoprotectants for stabilization of cyclosporine A-loaded PLGA nanoparticles45. On the other hand, Jain et al. used poly(vinyl alcohol) as a stabilizer within the liquid formulation and mannitol or dextrose (2.5 wt.%) as a lyoprotectant for making dry formulations which resulted in a sf/si ratio of ~1.132. Because various research groups have reported different findings with different sugars32, a more detailed study is required in this area to gain more insight into the use of lyoprotectants and their mechanism of action.
Penetration studies of CSA into cadaver skin via TyroSpheres-gel
Skin distribution studies were used to quantify the penetration of CSA into the dermal layer of cadaver skin (Fig. 7). After 6 h of treatment with 30 wt.% CSA-TyroSpheres-gel, ~22 μg of CSA penetrated through the epidermis and into the dermis. CSA-TyroSpheres-gel has an increased viscosity that allows for easy topical application of the formulation without run-off26. No CSA was detected in the receptor compartment, suggesting that CSA did not penetrate through the dermis and into the systemic compartment. These results are similar to a previous report by Black et al. where 32 μg of CSA was detected in the dermis with negligible systemic exposure when topical CSA was applied mixed with an oil-based vehicle48. It is important to note that Black et al. used a CSA dose of 25 mg/mL which is ~3 fold higher compared to the TyroSpheres-gel dose of ~8 mg/mL. Also Black et al. tested the oil-based formulation in a live mouse model, whereas TyroSpheres were applied to human cadaver skin. These data suggest that the CSA-TyroSpheres formulation holds promise as a topical drug delivery system for dermatological diseases where no systemic exposure is required. Our results are also comparable to Jain et al., who showed that ~16 μg of the total drug content was found in the epidermal and dermal portions of the skin after 24 h33. All the values were found to be statistically significant (*p < 0.05, **p < 0.005) except between the 1 h and 2 h time point (values indicated on Fig. 7). These results show that both the formulations may find applications in the clinical setup for use in therapies requiring topical immunosuppression.
Figure 7. Penetration of CSA eluted from 30 wt.% CSA-TyroSpheres gel through the epidermis and into the dermis of cadaver skin as a function of time.
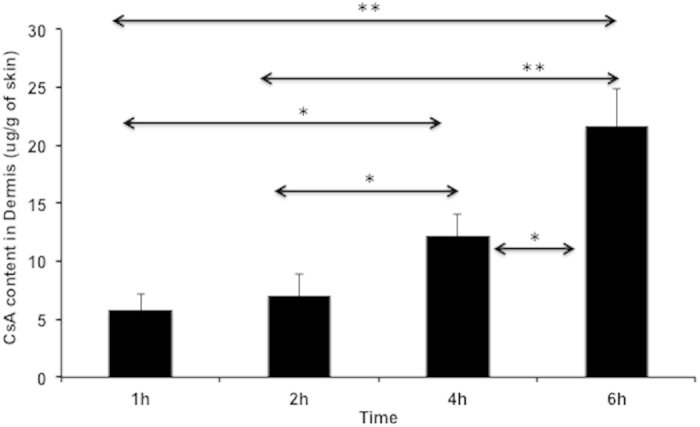
*p < 0.01, p < 0.001.
The performance of two particle systems – PLGA nanoparticles (formed via emulsion-diffusion-evaporation technique) and TyroSpheres (formed via self-assembly) was compared as topical delivery vehicles for cyclosporine A (CSA). CSA was used as a model drug because of its potential clinical use as a topical immunosuppressive agent in the treatment of various dermatological, immunological diseases49,50. Our data obtained for drug loading efficiency, release kinetics, effect of stabilizers and skin distribution profiles using TyroSpheres was directly compared to two recent reports by Jain et al.32,33, who reported on the use of PLGA nanoparticles as a topical drug delivery vehicle for CSA. It was observed that while TyroSpheres had the following properties (i) simple and single step self-assembly technique and no use of a stabilizer within the formulation, (ii) flexibility of loading varying amounts of drug into TyroSpheres, (iii) stability in both liquid and dry formulations, and (iv) less than 100 nm sized particles; PLGA was found flexible in terms of (i) choice of solvents and wide range of particle size and (ii) longer and sustained linear release profile. Also, both particle systems are able to deliver hydrophobic drugs to the upper layers of skin without any systemic exposure.
Materials and Methods
Materials
CSA was purchased from LC Laboratories (USA). Acetonitrile (ACN) [high performance liquid chromatography (HPLC) grade] and water (HPLC grade) were purchased from Fisher Scientific (Pittsburgh, PA). Dulbecco’s phosphate buffered saline (PBS, pH 7.4) was purchased from Aldrich Chemical Co. (Milwaukee, WI). N,N’-dimethylformamide (DMF) was obtained from Merck (EM Science, Darmstadt, Germany). Hydroxypropylmethylcellulose (HPMC) with 22% methoxyl and 8.1% hydroxypropyl content was purchased from Sigma (St. Louis, MO). All the experiments were carried out in accordance with the approved guidelines from New Jersey Center for Biomaterials.
Preparation of TyroSpheres loaded with CSA
ABA tri-block copolymer, PEG5K-b-oligo(desaminotyrosyl-tyrosine octyl ester suberate)-b-PEG5K (Fig. 1A), was synthesized, characterized, and prepared using previously established protocols21. Briefly, TyroSpheres loaded with and without CSA (referred to as CSA-TyroSpheres or empty, respectively) were prepared as follows. 20% (w/v) polymer (e.g., 60 mg/0.3 mL) and 10, 20, 30, 40 and 60 wt.% CSA (e.g., 20 mg/mL, equivalent to 10 wt.% initial loading) were dissolved in DMF, mixed in equal amounts (0.3 mL each) and added drop-wise to 1xPBS (14.4 mL) under constant stirring. The resulting suspension was passed through 0.22 μm PVDF filters (Millipore) and ultracentrifuged for 3 h at 18 °C and 65,000 rotations per minute (rpm) (290 000 × g ) (Beckman L8-70M ultracentrifuge, Beckman Coulter, Fullerton, CA). The supernatant was aspirated and the CSA-TyroSpheres were rinsed twice with 1 mL 1xPBS. Then, the CSA-TyroSpheres were re-suspended in 1 mL 1xPBS, ultracentrifuged under the same conditions, and re-suspended in 1 mL 1xPBS (pH 7.4) overnight at 25 ± 2 °C.
Characterization of CSA loaded TyroSpheres
Nanoparticle hydrodynamic diameter
The hydrodynamic diameters of the TyroSpheres with and without CSA were obtained by dynamic light scattering (DelsaNano S, Beckman Coulter Inc., NJ) as previously described25.
Determination of CSA concentration using HPLC
A 100 μL aliquot of CSA-TyroSpheres was lyophillzed and extracted into 2 mL of acetonitrile and filtered using 0.44 μm filters into the HPLC vials. The samples were analyzed on HPLC using the following parameters and a standard curve to determine the concentration of CSA in TyroSpheres. A Waters 2695 HPLC and Empower Pro software were used to quantify CSA in solution. Optimized chromatographic conditions included: column - Waters-C18, 2.1 mm × 50 mm, 5 μm particle size; column temperature −60 °C; injection volume −10 μL; detection wavelength −210 nm; mobile phase—A = HPLC water : B = acetonitrile (isocratic A:B (37.5:62.5)); flow rate −0.5 mL/min; run time −8 minutes; sample diluent - acetonitrile. The limit of detection was determined to be 1 μg. Concentrations of standards of CSA in acetonitrile ranged from 500–1.95 μg/mL.
Drug loading content and loading efficiency
Loading efficiency of CSA into TyroSpheres was determined by taking 0.1 mL aliquots of CSA-TyroSpheres (with various initial loadings), empty TyroSpheres and PBS and adding them to pre-weighed vials. The samples were frozen on dry ice and lyophilized (Labconco, MO, USA) for 24 h. The samples were re-weighed to determine the mass of salts, mass of empty TyroSpheres and mass of CSA-TyroSpheres. The concentration of CSA loaded in TyroSpheres was determined by adding 2 mL of acetonitrile to the lyophilized CSA-TyroSphere samples followed by vigorous vortexing for 1 h. The samples were then filtered using 0.45 μm filter (Millipore, USA) into HPLC vials. CSA was then quantified using HPLC and the standard curve. Drug loading content and loading efficiency were calculated using the following equations:
 |
 |
Solubility of CSA in PBS and TyroSpheres
Excess CSA was added to 1xPBS and stirred on a plate for 72 h at 37 °C in a water bath. The samples were filtered through 0.44 μm filters (PTFE, Puradisc Syringe Filters, Fisher Scientific) to remove non-solubilized drug, lyophilized, and re-dissolved in acetonitrile. The concentration of CSA dissolved in 1xPBS and TyroSpheres was determined as described above.
Release of CSA from TyroSpheres
The release of CSA from TyroSpheres was investigated using 0.5 mL dialysis cassettes with a 10,000 molecular weight cut off cellulose dialysis membrane (Slide-A-Lyzer, Fischer). The CSA-TyroSpheres were diluted with PBS to a CSA concentration of 400 μg/mL and 0.5 mL was added to the dialysis cassettes. The loaded cassettes were submerged in 200 mL of PBS (pH 7.4) and incubated in a shaking (60 rpm) water bath at 37 °C. At pre-determined time points for up to 7 days, the contents of the dialysis cassettes were collected, the compartment of the dialysis cassettes was rinsed three times with fresh PBS and added to the collected samples. At 24 h intervals, the receptor media was replaced with 200 mL fresh PBS. The concentration of CSA extracted from the donor compartment was determined as described above.
Preparation of 30 wt.% CSA-TyroSpheres-1% (w/v) HPMC gel formulation
Preparation of 1.5% (w/v) HPMC gel
150 mg of HPMC was suspended in 10 mL of 1xPBS and stirred at 400 rpm (slow enough to avoid the formation of bubbles) for 48 h at 25 ± 2 °C.
Preparation of 30 wt.% CSA-TyroSpheres-1% (w/v) HPMC gel
5 mL CSA-TyroSpheres were syringe filtered (0.22 μm, Millipore, USA) and mixed with 1.5% (w/v) HPMC gel (10 mL) to create a suspension of 30 wt.% CSA-TyroSpheres in 1% (w/v) HPMC gel (referred herein as CSA-TyroSpheres-gel). The suspension was stirred for 18 h at room temperature and stored at 4 °C until use.
Penetration studies of CSA-TyroSpheres-gel into cadaver skin
Human cadaver skin (skin bank number: LZ050911, split thickness, male) was cut into twelve 1 inch × 1 inch pieces. Each piece was soaked in PBS at 25 ± 2 °C for 45 min. The skin was then placed on a Franz cell with the epidermis facing the donor compartment. 5 mL 1xPBS was added to the receptor compartment with no air bubbles. 0.5 mL of CSA-TyroSpheres-gel was added to the donor compartment and placed on a 37 °C heating block for 1, 2, 4 and 6 h (n = 3). At each time point, the CSA-TyroSpheres-gel remaining on the skin was removed, the skin was washed several times with 1xPBS, and the dermis was manually separated from the epidermis using a surgical blade and a pair of scissors. Both the donor compartment and receptor compartment were collected and lyophilized for mass balance. The dermis and epidermis from each time point were soaked in 5 mL methanol for 48 h at 37 °C and homogenized (Polytron, Fisher Scientific, PA, USA) thereafter at 4 °C. The solution was then passed through a 0.45 μm filter and analyzed via liquid chromatography-mass spectrometry (LC-MS) at the Center for Integrative Proteomics Research at Rutgers University and a standard curve for CSA content.
Stability of CSA-TyroSpheres in solution
One mL samples of 30 wt.% CSA-TyroSpheres (30 μg/mL) in PBS (pH 7.4) were stored at three different temperatures (4, 25, and 37 °C) for 28 days. At 0 h, 7 d, 14 d, 21 d, and 28 d, the samples were collected, and freeze-dried. 2 mL of acetonitrile was then added to extract the CSA from the TyroSpheres. Samples were shaken on a vortex for 2 h and the concentration of CSA retained by the TyroSpheres as a function of time and storage temperature was determined as described above. Free CSA was dissolved in 100 μLof DMF and then diluted with 900 μL of 1xPBS to make a 30 μg/mL solution. The stability studies were conducted under identical conditions as the CSA-TyroSpheres.
Freeze-Thaw Analysis
TyroSpheres were prepared as above and mixed with an equal volume of cryo-protectant solution (550 mM sucrose in 1xPBS). Samples (1 mL) were analyzed using DLS to determine the particle size before freezing, placed in scintillation vials, and stored overnight at −80 °C. Samples were then thawed and allowed to equilibrate to 25 ± 2 °C before performing post-thawing particle size analysis. The procedure was repeated twice. The Sf/Si ratio was calculated by dividing the average post-thaw particle size (Sf) by the average pre-freeze particle size (Si).
Preparation of Dry Formulation
TyroSpheres were prepared as above and mixed with an equal volume of cryoprotectant solution (550 mM sucrose in 1xPBS). Samples were placed in scintillation vials and covered with KimWipes. A dry formulation was prepared using A MillRock Technology LD85 lyophilizer instrument with Opti-Dry Software with the following method. Step 1—. Freezing: Increase from room temperature to −45 °C as fast as possible, maintain −45 °C for 1 h, decrease temperature to −20 °C as fast as possible. Step 2—Annealing: Maintain at −20 °C for 6 h, decrease to −45 °C as fast as possible, and then remain at -45 °C for 0.5 h. Step 3 - Primary drying: Apply pressure 50 mTorr at −45 °C for 18 h, followed by an increase in temperature to −30 °C for 18 h. Step 4—Secondary drying: Maintain −30 °C for 18 h at 50 mTorr.
Following freeze-drying, the samples were removed from the lyophilizer, immediately sealed under nitrogen (to prevent moisture ingress), and placed under desiccation untill use. Lyophilized samples were analyzed for drug content as described above and samples reconstituted with deionized (DI) water were analyzed for particle size.
Characterization of Dry Formulation
Scanning Electron Microscopy (SEM)
Samples were dispersed lightly on tape mounted to aluminum stubs, sputter-coated with gold-palladium (Balzers SCD 004) and imaged with a Scanning Electron Microscope (AMRAY 1830 I). The microstructure of the cake was grossly analyzed for any appearance of collapse.
X-ray diffraction (XRD) Analysis
The XRD patterns of CSA, tri-block copolymer, empty TyroSpheres, CSA-TyroSpheres, and a physical mixture of CSA and tri-block copolymer were obtained using an X-ray diffractometer (Phillips XPert powder diffractometer with sample changer).
Statistical analysis
Statistical analysis, wherever needed, was carried out by Student’s t-test after ascertaining homogeneity of variance and normality of data. A value of p < 0.05 was considered statistically significant.
Additional Information
How to cite this article: Goyal, R. et al. Formulation Strategy for the Delivery of Cyclosporine A: Comparison of Two Polymeric Nanospheres. Sci. Rep. 5, 13065; doi: 10.1038/srep13065 (2015).
Supplementary Material
Acknowledgments
This work was supported by the Armed Forces Institute of Regenerative Medicine, (AFIRM) Department of Defense, under Award No. W81XWH-08-2-0034. The U.S. Army Medical Research Acquisition Activity, 820 Chandler Street, Fort Detrick MD 21702-5014 is the awarding and administering acquisition office. Opinions, interpretations, conclusions and recommendations are those of the author and are not necessarily endorsed by the Department of Defense.
Footnotes
Author Contributions All authors designed the experiments. R.G. carried out experiments and analyzed the results. All authors wrote and reviewed the manuscript.
References
- Kohane D. S. Microparticles and nanoparticles for drug delivery. Biotechnol. Bioeng. 96, 203–209 (2007). [DOI] [PubMed] [Google Scholar]
- Moinard-Checot D., Chevalier Y., Briancon S., Fessi H. & Guinebretiere S. Nanoparticles for drug delivery: review of the formulation and process difficulties illustrated by the emulsion-diffusion process. J. Nanosci. Nanotechno. 6, 2664–2681 (2006). [DOI] [PubMed] [Google Scholar]
- Sultana S., Khan M. R., Kumar M., Kumar S. & Ali M. Nanoparticles-mediated drug delivery approaches for cancer targeting: a review. J. Drug Target. 21, 107–125 (2013). [DOI] [PubMed] [Google Scholar]
- Tosi G. et al. Potential use of polymeric nanoparticles for drug delivery across the blood-brain barrier. Curr. Med. Chem. 20, 2212–2225 (2013). [DOI] [PubMed] [Google Scholar]
- Wissing S. A., Kayser O. & Muller R. H. Solid lipid nanoparticles for parenteral drug delivery. Adv. Drug Deliver. Rev. 56, 1257–1272 (2004). [DOI] [PubMed] [Google Scholar]
- Shive M. S. & Anderson J. M. Biodegradation and biocompatibility of PLA and PLGA microspheres. Adv. Drug Deliver. Rev. 28, 5–24 (1997). [DOI] [PubMed] [Google Scholar]
- Freiberg S. & Zhu X. X. Polymer microspheres for controlled drug release. Int. J. Pharm. 282, 1–18 (2004). [DOI] [PubMed] [Google Scholar]
- Dinarvand R., Sepehri N., Manoochehri S., Rouhani H. & Atyabi F. Polylactide-co-glycolide nanoparticles for controlled delivery of anticancer agents. Int. J. Nanomed. 6, 877–895 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vert M., Mauduit J. & Li S. Biodegradation of PLA/GA polymers: increasing complexity. Biomaterials 15, 1209–1213 (1994). [DOI] [PubMed] [Google Scholar]
- Betancourt T. et al. PEGylation strategies for active targeting of PLA/PLGA nanoparticles. J. Biomed. Mater. Res. A 91A, 263–276 (2009). [DOI] [PubMed] [Google Scholar]
- Wang M. & Thanou M. Targeting nanoparticles to cancer. Pharmacol. Res. 62, 90–99 (2010). [DOI] [PubMed] [Google Scholar]
- Bala I., Hariharan S. & Kumar M. N. PLGA nanoparticles in drug delivery: the state of the art. Crit. Rev. Ther. Drug 21, 387–422 (2004). [DOI] [PubMed] [Google Scholar]
- Sahana D. K., Mittal G., Bhardwaj V. & Kumar M. N. PLGA nanoparticles for oral delivery of hydrophobic drugs: influence of organic solvent on nanoparticle formation and release behavior in vitro and in vivo using estradiol as a model drug. J. Pharm. Sci. 97, 1530–1542 (2008). [DOI] [PubMed] [Google Scholar]
- Italia J. L., Bhatt D. K., Bhardwaj V., Tikoo K. & Kumar M. N. PLGA nanoparticles for oral delivery of cyclosporine: nephrotoxicity and pharmacokinetic studies in comparison to Sandimmune Neoral. J. Control. Release 119, 197–206 (2007). [DOI] [PubMed] [Google Scholar]
- Tang L. et al. Immunosuppressive Activity of Size-Controlled PEG-PLGA Nanoparticles Containing Encapsulated Cyclosporine A. J. Transplant. 2012, 896141 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Jalon E. G., Blanco-Prieto M. J., Ygartua P. & Santoyo S. PLGA microparticles: possible vehicles for topical drug delivery. Int. J. Pharm. 226, 181–184 (2001). [DOI] [PubMed] [Google Scholar]
- Jain A. et al. Peptide and Protein Delivery Using New Drug Delivery Systems. Crit. Rev. Ther. Drug 30, 293–329 (2013). [DOI] [PubMed] [Google Scholar]
- Kumari A., Yadav S. K. & Yadav S. C. Biodegradable polymeric nanoparticles based drug delivery systems. Colloid. Surface. B 75, 1–18 (2010). [DOI] [PubMed] [Google Scholar]
- Nardin C., Bolikal D. & Kohn J. Nontoxic block copolymer nanospheres: Design and characterization. Langmuir 20, 11721–11725 (2004). [DOI] [PubMed] [Google Scholar]
- Sheihet L., Dubin R. A., Devore D. & Kohn J. Hydrophobic drug delivery by self-assembling triblock copolymer-derived nanospheres. Biomacromolecules 6, 2726–2731 (2005). [DOI] [PubMed] [Google Scholar]
- Sheihet L., Piotrowska K., Dubin R. A., Kohn J. & Devore D. Effect of tyrosine-derived triblock copolymer compositions on nanosphere self-assembly and drug delivery. Biomacromolecules 8, 998–1003 (2007). [DOI] [PubMed] [Google Scholar]
- Haag R. Supramolecular drug-delivery systems based on polymeric core-shell architectures. Angew. Chem. Int. Edit. 43, 278–282 (2004). [DOI] [PubMed] [Google Scholar]
- Sheihet L. et al. Paclitaxel in tyrosine-derived nanospheres as a potential anti-cancer agent: In vivo evaluation of toxicity and efficacy in comparison with paclitaxel in Cremophor. Eur. J. Pharm. Sci. 45, 320–329 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel N. et al. Development of a Nanosphere Based Delivery of Sildenafil to Sites of Nerve Injury for Prevention of Post-Prostatectomy Erectile Dysfunction Using a Rat Model of Cavernous Nerve Injury. J. Urology 191, E481 (2014). [Google Scholar]
- Sheihet L. et al. Tyrosine-derived nanospheres for enhanced topical skin penetration. Int. J. Pharm. 350, 312–319 (2008). [DOI] [PubMed] [Google Scholar]
- Batheja P., Sheihet L., Kohn J., Singer A. J. & Michniak-Kohn B. Topical drug delivery by a polymeric nanosphere gel: Formulation optimization and in vitro and in vivo skin distribution studies. J. Control. Release 149, 159–167 (2011). [DOI] [PubMed] [Google Scholar]
- Knop K. et al. Star-shaped drug carriers for doxorubicin with POEGMA and POEtOxMA brush-like shells: a structural, physical, and biological comparison. Biomacromolecules 14, 2536–2548 (2013). [DOI] [PubMed] [Google Scholar]
- Zhao Y. P., Dan; Wang, Zheng. Comparison of four-arm and six-arm star-shaped mPEG-PLA block copolymer micelles for drug delivery. J. Control. Release 172, e84 (2013). [Google Scholar]
- Quan L. et al. Nanomedicines for inflammatory arthritis: head-to-head comparison of glucocorticoid-containing polymers, micelles, and liposomes. ACS nano 8, 458–466 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdel-Mottaleb M. M. A., Neumann D. & Lamprecht A. Lipid nanocapsules for dermal application: A comparative study of lipid-based versus polymer-based nanocarriers. Eur. J. Pharm. Biopharm. 79, 36–42 (2011). [DOI] [PubMed] [Google Scholar]
- Daman Z., Ostad S., Amini M. & Gilani K. Preparation, optimization and in vitro characterization of stearoyl-gemcitabine polymeric micelles: a comparison with its self-assembled nanoparticles. Int. J. Pharm. 468, 142–151 (2014). [DOI] [PubMed] [Google Scholar]
- Jain S., Mittal A., Jain A. K., Mahajan R. R. & Singh D. Cyclosporin A Loaded PLGA Nanoparticle: Preparation, Optimization, In-Vitro Characterization and Stability Studies. Curr. Nanosci. 6, 422–431 (2010). [Google Scholar]
- Jain S., Mittal A. & Jain A. K. Enhanced Topical Delivery of Cyclosporin-A Using PLGA Nanoparticles as Carrier. Curr. Nanosci. 7, 524–530 (2011). [Google Scholar]
- Pinto Reis C., Neufeld R. J., Ribeiro A. J. & Veiga F. Nanoencapsulation I. Methods for preparation of drug-loaded polymeric nanoparticles. Nanomed-Nanotechnol. 2, 8–21 (2006). [DOI] [PubMed] [Google Scholar]
- Greenwald R. B., Choe Y. H., McGuire J. & Conover C. D. Effective drug delivery by PEGylated drug conjugates. Adv. Drug Deliver. Rev. 55, 217–250 (2003). [DOI] [PubMed] [Google Scholar]
- Torchilin V. P. Structure and design of polymeric surfactant-based drug delivery systems. J. Control. Release 73, 137–172 (2001). [DOI] [PubMed] [Google Scholar]
- Seju U., Kumar A. & Sawant K. K. Development and evaluation of olanzapine-loaded PLGA nanoparticles for nose-to-brain delivery: in vitro and in vivo studies. Acta Biomater. 7, 4169–4176 (2011). [DOI] [PubMed] [Google Scholar]
- Lim J., Yeap S. P., Che H. X. & Low S. C. Characterization of magnetic nanoparticle by dynamic light scattering. Nanoscale Res. Lett. 8, 381–394 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uskokovic V. Entering the Era of Nanoscience: Time to Be So Small. J. Biomed. Nanotechnol. 9, 1441–1470 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohn J. et al. inventors; Rutgers University, assignee. Controlled releases of actives in skin. United States patent WO2008095144 A2. 2008 Aug 7.
- Danhier F. et al. PLGA-based nanoparticles: An overview of biomedical applications. J. Control. Release 161, 505–522 (2012). [DOI] [PubMed] [Google Scholar]
- Song K. C. et al. The effect of type of organic phase solvents on the particle size of poly(D,L-lactide-co-glycolide) nanoparticles. Colloid. Surface. A 276, 162–167 (2006). [Google Scholar]
- Hariharan S. et al. Design of estradiol loaded PLGA nanoparticulate formulations: A potential oral delivery system for hormone therapy. Pharm. Res. 23, 184–195 (2006). [DOI] [PubMed] [Google Scholar]
- Huang X. & Brazel C. S. On the importance and mechanisms of burst release in matrix-controlled drug delivery systems. J. Control. Release. 73, 121–136 (2001). [DOI] [PubMed] [Google Scholar]
- Saez A., Guzman M., Molpeceres J. & Aberturas M. R. Freeze-drying of polycaprolactone and poly(D,L-lactic-glycolic) nanoparticles induce minor particle size changes affecting the oral pharmacokinetics of loaded drugs. Eur. J. Pharm. Biopharm. 50, 379–387 (2000). [DOI] [PubMed] [Google Scholar]
- Fonte P. et al. Effect of cryoprotectants on the porosity and stability of insulin-loaded PLGA nanoparticles after freeze-drying. Biomatter. 2, 329–339 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layre A. M. et al. Freeze-drying of composite core-shell nanoparticles. Drug. Dev. Ind. Pharm. 32, 839–846 (2006). [DOI] [PubMed] [Google Scholar]
- Black K. S., Nguyen D. K., Proctor C. M., Patel M. P. & Hewitt C. W. Site-Specific Suppression of Cell-Mediated-Immunity by Cyclosporine. J. Invest. Dermatol. 94, 644–648 (1990). [DOI] [PubMed] [Google Scholar]
- Petruzzo P. et al. The International Registry on Hand and Composite Tissue Transplantation. Transplantation 90, 1590–1594 (2010). [DOI] [PubMed] [Google Scholar]
- Kaufman D. B. et al. Immunosuppression: practice and trends. Am. J. Transplant. 4, 38–53 (2004). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.