

Abstract
BACKGROUND
Children with 22q11.2 deletion syndrome (22q11.2DS) have a wide range of clinical features. TBX1 has been proposed as a candidate gene for some of the features in this condition. Polymorphisms in the non-deleted TBX1, which may affect the function of the sole TBX1 gene in individuals with the 22q11.2DS, may be a key to understanding the phenotypic variability among individuals with a shared deletion. Comprehensive single nucleotide polymorphism (SNP) discovery by resequencing candidate genes can identify genetic variants that influence a given phenotype. The purpose of this study was to further characterize the sequence variability in TBX1 by identifying all common SNPs in this gene.
METHODS
We resequenced TBX1 in 29 children with a documented 22q11.2 deletion and 95 non-deleted, healthy individuals. We estimated allele frequencies, performed tagSNP selection, and inferred haplotypes. We also compared SNP frequencies between 22q11.2DS and control samples.
RESULTS
We identified 355 biallelic markers among the 190 chromosomes resequenced in the control panel. The vast majority of the markers identified were SNPs (n=331), and the remainder indels (n=24). We did not identify SNPs or indels in the cis- regulatory element (FOX–binding site) upstream of TBX1. In children with 22q11.2DS we detected 187 biallelic markers, six of which were indels. Four of the seven coding SNPs identified in the controls were identified in children with 22q11.2DS.
CONCLUSIONS
This comprehensive SNP discovery data can be used to select SNPs to genotype for future association studies assessing the role of TBX1 and phenotypic variability in individuals with 22q11.2DS.
Keywords: 22q11.2 Deletion Syndrome, TBX1, Single nucleotide polymorphism discovery
INTRODUCTION
Common approaches to selecting single nucleotide polymorphisms (SNPs) for genotyping in genetic association studies of birth defects include reliance on information about SNPs’ putative functionality or to incorporation of data from publicly available SNP databases, such as dbSNP. For many genes, these approaches would miss a large proportion of genetic variation that could be related to the disease of interest (Kruglyak, 2008; Wiltshire et al., 2006). Thus, comprehensive SNP discovery by resequencing candidate genes can serve as an important first step for finding genetic variants that influence a given phenotype (Crawford et al., 2005).
To our knowledge, comprehensive SNP discovery has not been conducted in TBX1, a gene that appears likely to influence the developmental biology of chromosome 22q11.2 deletion syndrome (22q11.2DS), a genetic disorder with an estimated prevalence of 1 in 4000 to 6000 live births (Devriendt et al., 1998; Vittorini et al., 2001; Wilson et al., 1994) or higher (Botto et al., 2003; Shprintzen, 2005). Over 180 clinical features have been described in individuals with 22q11.2DS, and wide phenotypic variability is a hallmark of this condition (Robin and Shprintzen, 2005). The most commonly observed features are cleft palate and functional anomalies of the palate, such as velopharyngeal insufficiency (McDonald-McGinn et al., 1999; Ryan et al., 1997), cardiac anomalies such as interrupted aortic arch Tetrology of Fallot and ventricular septal defects (McDonald-McGinn et al., 1999; Ryan et al., 1997), a characteristic facies, abnormalities of the thymus gland, learning difficulties, and behavioral and psychiatric conditions. Several syndromes that were once thought to be distinct, including DiGeorge and Velo-cardio-facial syndrome, are now known to share a common genetic cause and are collectively referred to as 22q11.2DS (Robin and Shprintzen, 2005).
The typically deleted region on chromosome 22q11.2 harbors approximately 30 genes (Hoogendoorn et al., 2004), including TBX1. The TBX1 gene encodes a transcription factor from a gene family known as T-box genes, which contain a homologous T-box necessary for DNA binding and protein-protein interactions during early embryologic development (Chieffo et al., 1997; Jerome and Papaioannou, 2001; Yamagishi et al., 2003; Yamagishi and Srivastava, 2003). Mice homozygous for tbx1 mutations display anomalies encompassing almost all of the common features observed in 22q11.2DS, including cleft palate (Jerome and Papaioannou, 2001). Further evidence that TBX1 contributes to the phenotype in 22q11.2DS comes from the detection of four different TBX1 mutations in patients with characteristic manifestations of the syndrome but without the 22q11.2 deletion (Yagi et al., 2003; Zweier et al., 2007).
These studies demonstrate that alterations in the number of copies or sequence of TBX1 result in many of the features observed in 22q11.2 DS (Aggarwal and Morrow, 2008; Arnold et al., 2006a; Arnold et al., 2006b; Fagman et al., 2007; Lindsay et al., 2001; Merscher et al., 2001; Theveniau-Ruissy et al., 2008; Vitelli et al., 2002; Vitelli et al., 2003; Yagi et al., 2003; Zweier et al., 2007). In addition to rare mutations, relatively common polymorphisms in the remaining (non-deleted) copy of TBX1 may be one source of the phenotypic variability among children with 22q11.2DS (Rauch et al., 2004; Voelckel et al., 2004; Yamagishi et al., 2003). Furthermore, SNPs that do not directly influence phenotypic variability may be in linkage disequilibrium (LD), or correlated, with causal variants and can thereby serve as markers to detect gene-phenotype associations. Therefore, we performed genetic variation discovery in children with 22q11.2DS and in unaffected individuals to identify all common SNPs and insertion-deletion (indel) polymorphisms in TBX1 and the surrounding regulatory sequences. In addition to SNP discovery, we performed various analyses to facilitate tagSNP selection, including haplotype inference and estimation of tagSNP overlap between the two populations. We performed preliminary comparison of SNP frequencies between individuals with and without 22q11.2DS. This comprehensive SNP discovery data can be used to select SNPs to genotype for future association studies assessing the role of TBX1 and phenotypic variability in individuals with 22q11.2DS.
METHODS
Study Participants
SNP discovery was performed by resequencing TBX1 in two groups: 1) 29 children with a documented 22q11.2 deletion and 2) 95 apparently healthy individuals from Coriell DNA samples (Livingston et al., 2004). Based on population genetics and probability theory, we expected that these sample sizes would allow, on average, for a common SNP (minor allele frequency [MAF] ≥ 5%) detection rate of 99% (Kruglyak and Nickerson, 2001).
Participants with 22q11.2DS were identified from a tertiary care pediatric hospital (Seattle Children’s Hospital) through multiple subspecialty clinics to include children with a variety of phenotypic features. The 22q11.2 deletions were confirmed by a Clinical Laboratory Improvement Amendments-approved laboratory. The majority of the participants were self-described European Americans (EA; n=24), and the remaining five participants were of mixed race/ethnicity, which included various combinations of Asian, Pacific Islander, Native American, European American, and Hispanic heritage.
Population based reference samples were used from the Environmental Genome Project (EGP; egp.gs.washington.edu) and were made up of 95 individuals whose DNAs are available from Coriell Cell Repositories: 22 European Americans, 15 African Americans, 12 Yorubans, 24 Asians (representing Han Chinese and Japanese), and 22 Hispanics (Table 1). The 12 Yorubans are representative of West Africa, the ancestral home of most African Americans. Allele frequencies are highly concordant (r2>0.9) among allele frequencies between the Yoruban and African American samples, as well as between the Japanese and Han Chinese samples, respectively (Howie et al., 2006)). Therefore, samples from these subpopulations were combined to form “African” and “Asian” populations, respectively, to obtain an adequate sample size for allele and genotype frequency estimations and for reliable identification of tagSNPs (Howie et al., 2006)
Table 1.
Repository numbers for the control samples
| Population (number) | Coriell Cell Repository number |
|---|---|
| European American (n=24) | NA11995, NA12892, NA11882, NA11994, NA12815, NA12891, NA06985, NA11840, NA11881, NA11993, NA12751, NA12814, NA06993, NA07056, NA11832, NA11839, NA11992, NA12057, NA12156, NA12239, NA12750, NA12813 |
| African American (n=15) | NA17101 through NA17115 |
| Yoruban (n=12) | NA18502, NA19153, NA19223, NA19201, NA18504, NA18870, NA19137, NA19238, NA19144, NA19203, NA19200, NA18855 |
| Asian (n=24) | NA18526, NA18562, NA18545, NA18609, NA18566, NA18621, NA18577, NA18635, NA18524, NA18537, NA18572, NA18552, NA18942, NA18945, NA18964, NA18961, NA18967, NA18981, NA18994, NA18998, NA18940, NA18949, NA18953, NA18972 |
| Hispanic (n=22) | NA17438 through NA17446, NA17448 through NA17454, NA17456 through NA17461 |
Sample collection
Participants with 22q11.2DS provided either a buccal swab specimen (obtained by brushing Dacron cotton swabs along the inside of the oral cavity) or 10 mL of venous blood in an EDTA tube. DNA extractions on these buccal and blood specimens were performed using QIAamp DNA Blood Mini Kits (Qiagen, Inc., Valencia, CA).
Resequencing
We targeted 43,812 base pairs of TBX1 for resequencing in all participants. This included the longest genomic transcript for TBX1 (transcript B), including all exons and introns (Fig. 1). In addition, 15 kilobases upstream of the gene was also resequenced to capture sequence variation in the promoter and the FOX transcription factor binding site (Yamagishi et al., 2003). Mouse models demonstrate that Tbx1 transcription in pharyngeal endoderm and head mesenchyme is regulated by this upstream cis-element that binds Foxc1, Foxc2, and Foxa2 (Yamagishi et al., 2003). The TBX1 gene demonstrates several highly conserved regions between the human and mouse sequences (Gong et al., 2001). The ECR browser identified 40 regions of at least 90 base pairs that share at least 68% homology in the targeted region for this study (Chr22:18109240-18153016). Half these shared regions were located upstream of the coding sequence, which may harbor additional important regulatory elements (Ovcharenko et al., 2004).
Figure 1.
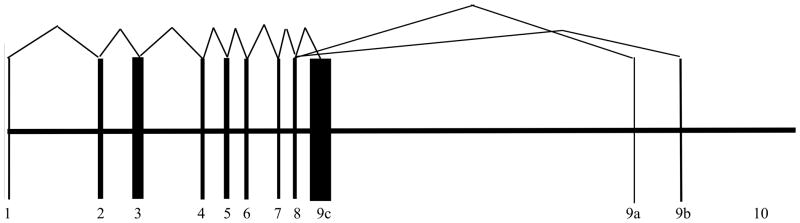
Illustration of the sequence for the three splice variants of TBX1 (A,B,C). The first 8 exons are shared by all transcripts.
We used over 200 primers (http://egp.gs.washington.edu/data/tbx1/TBX1X.primers.fasta) to resequence overlapping PCR amplicons with an average size of 500 base pairs instead of the standard size of 1 kb to ensure adequate coverage of this GC-rich gene. Direct sequencing with standard dye-terminator fluorescent signaling was performed on an ABI 3730 XL. Gene sequences were assembled as previously described (Carlson et al., 2004). Polymorphic sites were identified using PolyPhred v5.0 (Stephens et al., 2006). Each SNP was verified by visually inspecting the sequence data at each polymorphic site identified by PolyPhred v5.0. Indels were identified visually and genotyped manually. Primers were then re-designed to sequence past the polymorphism. All SNPs discovered in the EGP reference panel were deposited in dbSNP and GenBank (Accession No. DQ650705) and are available through the NIEHS SNPs website (egp.gs.washington.edu).
Statistical methods
All analyses were performed on sequence from chromosome 22 region:18109240-18153016 (genome build 18).
Estimation of allele frequencies, tagSNP selection, tagSNP overlap, and haplotype inference
For each variant detected, we estimated allele frequencies separately for each disease and ethnic subpopulation. We estimated pair-wise LD (r2) for the 22q11.2DS population and each ethnic reference population. By using the LDSelect software (Carlson et al., 2004) on the Genome Variation Server (GVS, http://gvs.gs.washington.edu/GVS), we clustered the SNPs by degree of LD, binning together SNPs in strong LD, defined as r2 >0.8, >0.9, or =1.0 (in three separate analyses). Only SNPs for which the sequence coverage was at least 70% in the subpopulation (70% of alleles sequenced in the population had sufficiently high quality to identify the nucleotide position) were included in the bins. From these bins we then selected tagSNPs such that every SNP detected through resequencing with MAF ≥ 5% was either tagged or was in strong LD with a tagged SNP. Only SNPs with minimum sequence coverage of 85% were candidates to be selected as tagSNPs.
SNPs, their allele frequencies, and LD patterns differ across populations. There are global estimates of tagSNP transferability from large portions of the genome; however, to our knowledge this has never been estimated for genes in individuals with 22Q11 DS, in whom the hemizygous state may alter types and frequencies of specific SNPs and the LD patterns among them. As a surrogate measure of tagSNP transferability, we selected SNPs from the EA reference population with a minor allele frequency of at least 5%, and we estimated the proportion of these SNPs that would also have been selected as tagSNPs in the individuals with 22q11.2DS.
We inferred haplotypes for SNPs with a MAF ≥ 5% for the reference population by using PHASEv2.0 (Stephens and Donnelly, 2003). PHASE implements a Bayesian algorithm and is coalescent-based; that is, it incorporates information about the inferred population history of the sample (Stephens and Donnelly, 2003). We evaluated the haplo-block structure using Haploview 4.1.
Exploration of possible functional significance of SNPs
We performed several different computational techniques to predict which SNPs are likely to be functional. Evolutionarily conserved regions were identified by using the ECR Browser at default settings (Ovcharenko et al., 2004). Two computational methods were used to predict the effects of nonsynonymous SNPs identified in TBX1. The Sorting Intolerant From Tolerant (SIFT) program predicts whether an amino acid substitution will affect protein function by aligning a queried amino acid change and associated protein sequence with related proteins (Ng and Henikoff, 2003). The program calculates the probability that an amino acid change at that position will be tolerated conditional on the most frequent amino acid being tolerated (Ng and Henikoff, 2003). Polymorphism Phenotyping (PolyPhen) uses an empirically derived set of rules, based on substitution site properties and position-specific independent counts, to predict whether a given amino acid substitution is damaging or benign based on phylogenetic relationships between this gene and the ortholog in other species (Ramensky et al., 2002). Tajima’s D was calculated as a measure of the influence of selection on sequence variants identified in our reference sample (Tajima, 1989).
Preliminary comparison of SNP frequencies between 22q11.2DS and controls
For SNPs with a MAF ≥ 5% in both the EA subpopulation (controls) and the EA patient subgroup with 22q11.2 DS, we compared allele frequencies by estimating odds ratios and exact p-values from a Fisher’s exact test (with the control population as the reference group). We did not correct p-values for the multiple tests performed, as this was an exploratory and low-powered analysis. We do not interpret the p-values in terms of a dichotomous significance test.
RESULTS
Sample Characteristics
The average age of the participants with 22q11.2DS was 8.5 years (Table 2). Approximately half the participants were male and the majority European American (Table 2). Sequence data confirmed that all cases had a single copy of the TBX1 gene. Information beyond sex and self-reported race/ethnicity is not available for the EGP reference samples.
Table 2.
Select characteristics of individuals with a positive FISH test for the 22q11.2 deletion
| Characteristics | Cases N=29 |
|---|---|
| Age in years, (Mean (SD)) | 8.52 (5.0) |
| Male (n (%)) | 14 (48) |
| Race/Ethnicity (n (%)) | |
| White/Non-Hispanic | 24 (83) |
| Other | 5 (17) |
| Palatal clefta (n (%)) | 10 (34) |
| Congenital Heart Diseaseb (n (%)) | 14 (48) |
Includes submucous cleft palate, overt secondary cleft, cleft lip and palate.
Includes double outlet right ventricle, pulmonic stenosis, ventricular septal defect, double aortic arch, tetralogy of fallot and others.
Variation discovery
For both groups combined—those with and without 22q11.2DS— we successfully resequenced >92% of the targeted reference sequence for variation discovery. Some portions of the TBX1 region were not successfully resequenced in the 22q11.2DS population, including exons 3 and 8 due to low complexity (i.e., repetitive) or GC-rich sequence (http://egp.gs.washington.edu/data/tbx1/tbx1.ColorFasta.html). Exon 3 is located within a region of high GC content (77%) and we had minimal success resequencing this region for all samples despite having used previously published primers (Rauch et al., 2004). Exon 8 was not adequately resequenced in the population with 22q11.2DS, as it is distally flanked by a repetitive element and also has high GC content (78%). We did not have sufficient DNA available for patient samples to repeat attempts to resequence this exon.
Sequence variation
We identified 355 biallelic markers (i.e., sequence variants for which two alleles were discovered in the population) among the 190 chromosomes resequenced in the reference panel (Table 3, Fig. 2, and a color-coded fasta representation of our findings is available at http://egp.gs.washington.edu/data/tbx1/tbx1.ColorFasta.html). The vast majority of the markers identified were SNPs (n=331), and the remainder indels (n=24). Consistent with previous reports, the size of the indels ranged from 1 to 16 base pairs (Bhangale et al., 2005). We also identified one tri-allelic marker in the African-descent subpopulation. We did not identify SNPs or indels in the cis- regulatory element (FOX–binding site) upstream of TBX1.
Table 3.
| Table 3A. Number of sequence variations detected in TBX1 through gene resequencing in a reference population | |||||
|---|---|---|---|---|---|
| Sample size | Copies of TBX1 | Variationsa | MAF ≥5% | TagSNPs | |
| Total population | 95 | 190 | 355 | 86 | 109b |
| European descent | 22 | 44 | 146 | 86 | 47 |
| African descent | 27 | 54 | 241 | 132 | 82 |
| Asian descent | 24 | 48 | 157 | 80 | 37 |
| Hispanic | 22 | 44 | 120 | 88 | 35 |
| Table 3B. Number of sequence variations detected in TBX1 through gene resequencing in children with 22q11.2DS | ||||
|---|---|---|---|---|
| Sample size | Copies of TBX1 | Variationsa | MAF≥5% | |
| Total populationc | 29 | 29 | 187 | - |
| European descent | 24 | 24 | 100 | 91 |
Sequence variations include single nucleotide polymorphisms and insertion/deletions
See Methods section for details of TagSNP selection. The MultiPop Algorithm was used to identify tagSNPs for the entire reference population
Includes five individuals with combinations of Asian, Pacific Islander, Native American, Hispanic and European descent
Figure 2.

Illustration of the location and frequencies of the SNPs identified in this study. Exons labeled in light grey represent untranslated regions. Image was modified from GeneSNPs (http://www.genome.utah.edu/genesnps/cgi-bin/main.cgi?gene_id=6899&set_id=36)
We identified a greater number of total genetic variants and common (≥ 5%) variants (241 and 132, respectively) in the African-descent population compared with the other populations resequenced in the reference sample (Table 3A; Crawford et al., 2005). Sixty-nine of the 355 biallelic variants (19%) identified in the entire population were shared among all subpopulations. This proportion was much higher for common variants (63% of the 86 total common variants). Pairwise comparisons across the ethnic groups revealed that the Europeans shared the greatest number of variants with the Hispanic (n=77) subpopulation. The Asian and African-American subpopulations shared the fewest number of variants (n=54; Table 4). Six SNPs were identified in the coding regions (for transcripts A, B, and C) in the EGP sample (Table 5), two of which were rare.
Table 4.
Number of common SNPs (MAF≥5%) shared between subpopulationsa
| European | African | Asian | Hispanic | |
|---|---|---|---|---|
| European | 86 | |||
| African | 65 | 132 | ||
| Asian | 66 | 54 | 79 | |
| Hispanic | 77 | 65 | 66 | 88 |
Limited to SNPs with a MAF ≥ 5% within each subpopulation in the comparison.
Table 5.
Sequence variations identified in the exons of transcripts A, B, and C of TBX1
| RS no., reference | Exon | SNP | Amino acid position | Residuea | Present Study (MAF)b | |
|---|---|---|---|---|---|---|
|
| ||||||
| 1 | N/A, Gong et al., 2001 | 2 | G to C | 5′ UTR (75 nt 5′ to ATG) | N/A | N/A |
|
| ||||||
| 2 | N/A, Gong et al., 2001; Torres-Juan et al., 2007 | 2 | C to T | 5′ UTR (39 nt 5′ to ATG) | N/A | N/A |
|
| ||||||
| 3 | rs737868, present study; Funke et al, 2007; Han et al., 2006a | 2 | C to G | 5′ UTR (mRNA position 45) | N/A | African (0.25) European American (0.43) Hispanic (0.41) Asian (0.25) 22Q European-American (0.13) |
|
| ||||||
| 4 | rs41298786, present study | 2 | A to G | 5′ UTR (mRNA position 127) | N/A | African (0.02) |
|
| ||||||
| 5 | rs28649236, Han et al., 2006a | 2 | A to G | 8 | Arginine to glycine | N/A |
|
| ||||||
| 6 | N/A, Rauch et al., 2004; Gong et al., 2001 | 3 | G to A | 99 | Arginine | N/A |
|
| ||||||
| 7 | N/A, Rauch et al., 2004 | 3 | G to A | 107 | Valine | N/A |
|
| ||||||
| 8 | rs41298814, present study; Funke et al., 2007; Gong et al., 2001 | 4 | T to C | 140 | Phenylalanine | African (0.06) European American (0.25) Hispanic (0.23) Asian (0.42) 22Q European-American (0.30) |
|
| ||||||
| 9 | N/A, Rauch et al., 2004 | 4 | C to T | 148 | Phenylalanine | N/A |
| rs28939675, Han et al., 2006b; Yagi et al., 2003 | 4 | T to A | 148 | Phenylalanine to Tyrosine | N/A | |
|
| ||||||
| 10 | rs41298816, present study | 4 | C to T | 154 | Alanine | Hispanic (0.02) |
|
| ||||||
| 11 | N/A, Gong et al., 2001 | 5 | C to T | 192 | Arginine | N/A |
|
| ||||||
| 12 | N/A, Zweier et al., 2007 | 194 | Histamine to glutamine | N/A | ||
|
| ||||||
| 13 | rs2301558, present study; Chieffo et al., 1997; Conti et al., 2003; Funke et al., 2007; Gong et al., 2001; HapMap; Rauch et al., 2004 | 5 | C to T | 222 | Leucine | African (0.62) European American (0.12) Hispanic (0.32) Asian (0.15) 22Q European-American (0.36) |
|
| ||||||
| 14 | N/A, Gong et al., 2001 | 7 | C to A | 296 | Arginine | N/A |
|
| ||||||
| 15 | rs41298838, present study; Yagi et al., 2003 | 8 | G to A | 310 | Glycine to serinec | Asian (0.07) |
|
| ||||||
| 16 | rs41298840, present study; Gong et al., 2001 | 8 | A to G | 311 | Alanine | African (0.03) European American (0.20) Hispanic (0.17) Asian (0.23) 22Q European-American (0.26) |
|
| ||||||
| 17 | rs13054377, Gong et al., 2001; Rauch et al., 2004 | 9C | A to G | 353 | Alanine | N/A |
|
| ||||||
| 18 | N/A., Rauch et al., 2004 | 9C | delGCCGG CGGC |
379–381 | Deletion of arganine-glycine- glycine | N/A |
|
| ||||||
| 19 | N/A, Conti et al., 2003 | 9C | C to A | 384 | Proline to glycine | N/A |
|
| ||||||
| 20 | N/A, Gong et al., 2001 | 9C | C to T | 396 | Proline to leucine | N/A |
|
| ||||||
| 21 | N/A, Rauch et al., 2004; Gong et al., 2001 | 9C | C to A | 397 | Asparagine to histidine | N/A |
|
| ||||||
| 22 | N/A, Yagi et al., 2003 | 9C | del C | 408 | Premature termination | N/A |
|
| ||||||
| 23 | N/A, Torres-Juan et al., 2007 | 9C | T to C | 411 | Leucine to proline | N/A |
|
| ||||||
| 24 | N/A, Gong et al., 2001 | 9C | delACTAT CTC |
Frameshift | N/A | |
|
| ||||||
| 25 | N/A, Gong et al., 2001 | del 15 bp | 445–449 | delGYHPH | ||
|
| ||||||
| 26 | N/A, Rauch et al., 2004 | 9C | T to C | 451 | Histidine | N/A |
|
| ||||||
| 27 | N/A, Gong et al., 2001 | insCAC | 457–458 | Insert histidine | ||
|
| ||||||
| 28 | N/A, Gong et al., 2001 | dup 30 bp | 466–476 | Duplicate Ala | ||
|
| ||||||
| 29 | N/A, Gong et al., 2001 | delCGCTG CCGC |
468–471 | Deletion of 3 Ala | ||
|
| ||||||
| 30 | rs4819522, present study; Cheiffo et al., 1997; Conti et al., 2003; Funke et al., 2007; Gong et al., 2001; Rauch et al., 2004; Thorisson et al., 2005 | 9A | C to T | 350 | Threonine to methionine | African (0.12) European American (0.07) Hispanic (0.25) Asian (0.15) 22Q European-American (0.20) |
|
| ||||||
| 31 | rs5993826 | 9A | G to A | 393 | Alanine to threonine | N/A |
|
| ||||||
| 32 | rs41298848, present study | 9B | C to T | 351 | Asparagine | African (0.02) |
Amino acid changes are monomorphic unless otherwise specified.
Minor Allele Frequency.
Predicted function PolyPhen/SIFT = tolerated.
Many of the SNPs that have been discovered in TBX1 can be found in the existing databases (dbSNP, HapMap) or the literature (Table 5); however, these resources are not yet comprehensive. Nine of the 33 exonic variations (28%) reported in the literature were also identified in this study (Table 5). Our study was designed to identify common SNPs (MAF ≥5%), and although allele frequencies of other variants are not always reported (see http://www.ncbi.nlm.nih.gov/SNP/) most publications indicate that the variants not detected in this study are rare (<1%). Conversely, we identified many more common variants in the TBX1 gene region that had not been previously reported. Thirty-nine of the 355 SNPs identified in the EGP sample were also found in HapMap. Comparison of linkage disequilibrium capture (r2 > 0.8) between HapMap SNPs and common SNPs (>5% allele frequency) from the resequencing data was low. Only 8, 24, and 32% of the SNPs identified in our SNP discovery were captured in HapMap African-descent, European-descent and Asian-descent samples, respectively. Only 27% of the SNPs from the European American 22q11.2DS samples were present in the HapMap European subpopulation.
Variation in individuals with 22q11.2DS
In children with 22q11.2DS we detected 187 biallelic markers, seven of which were indels.
We identified 100 variants in the European subpopulation, 91 of which were common (Table 3B). Four of the six coding SNPs (for transcripts A, B, and C) identified in the EGP reference sample were identified in children with 22q11.2DS (Table 5).
TBX1 linkage disequilibrium and tagSNPs
Forty-seven tagSNPs were identified for the EA subpopulation, which represents >40% reduction in the number of SNPs to be genotyped in a future genetic association study (Fig. 3). Similar reductions were realized for Hispanic and Asian subpopulations (Table 3A); however, the African-descent population has more tagSNPs and less reduction (~38%) from all common SNPs compared with the other subpopulations. TagSNP selection with more stringent r2 thresholds (i.e., 0.9 and 1.0) resulted in a similar number of tagSNPs in the EA subpopulation (54 and 56, respectively).
Figure 3.
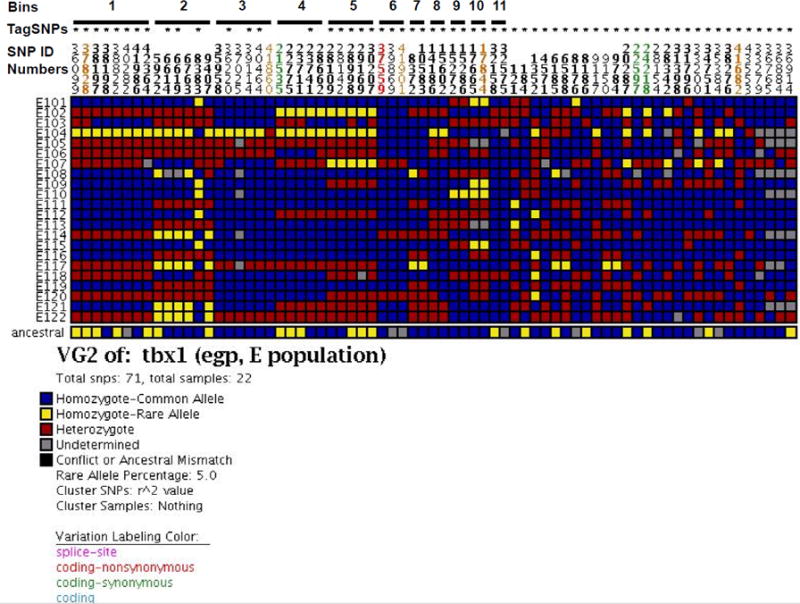
Visual representation of SNPs and tagSNPs identified in TBX1.
Overlap of SNPs from the reference population to 22q11.2DS
To evaluate how well the SNPs discovered in the reference population represented SNPs identified in the 22q11.2DS population, we compared the number and frequency of SNPs in the European American subpopulations among the cases and controls. Seventy eight (79%) of the common SNPs (MAF ≥ 5%) were shared between the two groups, 13 SNPs were unique to the 22q11.2DS population, and eight SNPs were unique to the reference population (Fig. 4). We observed a similar proportion of shared common SNPs at MAF ≥ 10% (81%); Fig. 4). The majority of the 21 SNPs that were unique to either the 22q11.2DS group or the EGP population were among the less common SNPs (MAF 5–10%). However, one common SNP (rs737869) in the EGP sample (MAF 36%) was not identified in the 22q11.2DS population.
Figure 4.

SNPs detected with minor allele frequencies ≥5 and 10% in individuals with 22q11.2 deletion syndrome and controls.
Haplotype Inference
One-hundred fifty-one haplotypes were inferred in the reference population, none of which had a frequency ≥5%. None of the participants with 22q11.2DS shared the same haplotype. Analysis of recombination breakpoints (haplo-blocks) revealed 24 distinct regions in this 43 kb segment confirming the extensive haplotype diversity.
Exploration of possible functional significance of SNPs
Of the seven coding SNPs detected in the reference population, only one resulted in a non-synonymous amino acid change (Table 5). Two computational methods, SIFT (Ng and Henikoff, 2003) and Polyphen (Ramensky et al., 2002), predicted that this non-synonymous SNP would be tolerated by the protein. Tajima’s D, a measure of selection, suggested genetic variation within TBX1 is consistent with neutrality, or lack of selection, in all subpopulations examined (AA −0.8, EA −0.6, Hispanic 0.5, Asian −0.1).
Preliminary comparison of SNP frequencies between 22q11.2DS and controls
Odds ratios comparing common SNP allele frequencies between the two groups ranged from 0.17 to 9.0, and exact p-values ranged from 0.01 to 1.0 (data not shown). There were several SNPs for which these comparisons yielded p-values <0.05 [rs5993820 (position 8383), rs2301558 (position 22597), rs4819843 (position35959), rs41297994 (position 36884), rs9798754 (position 36914), and rs13055776 (position 41503)] and a further five for which 0.05<p<0.1 [rs41295441 (position 8176), rs8138086 (position 9407,) rs737867 (position 17665), rs737868 (position 17844), and rs41299934 (position 28154)].
DISCUSSION
In this study, we compared the SNP content and structure of TBX1 between individuals who are hemizygous for the 22q11.2 deletion and a multi-ethnic reference population. Our findings did not reveal any common, distinctive polymorphisms, such as those associated with an amino acid substitution, or haplotypes that appear to be informative in individuals with 22q11.2DS. This comprehensive SNP discovery effort was designed to provide data needed to select SNPs to genotype for future studies assessing the role of TBX1 and phenotypic variability in individuals with the 22q11.2 deletion.
Our approach was likely to have identified most of the common variation in the reference population as well as individuals with the 22q11.2 deletion. The purpose of performing SNP discovery in both groups was twofold. First, there may be common SNPs in the study population of interest (individuals with 22q11.2DS) that are not common in the reference population. In particular, it is not known whether variants identified in a control population would be tolerated in hemizygous individuals.
Second, performing the SNP discovery in both a reference population and individuals with the 22q11.2 DS allowed us to perform some preliminary comparisons between the SNP discoveries in the two groups, and to investigate whether SNPs and minor allele frequencies (which are used to select tagSNPs) identified in a reference, non-deleted population were comparable to those in individuals with 22q11.2DS. Our data suggest that most of the common SNPs are not substantially different between individuals with and without the 22q11.2 deletion. Inconsistencies in regions of low sequence quality accounted for a small portion (2/21) of the SNPs that were unique to either the control or 22q11.2DS populations. However, given this limitation, the data obtained from our SNP discovery for TBX1 in the reference subpopulations and individuals with 22q11.2DS can be used to select a maximally informative set of tagSNPs to genotype for future genotype-phenotype association studies. Whether or not one can extrapolate these results regarding the overlap between tagSNPs identified in TBX1 in individuals with and without 22q11.2DS translate to other genes for which this population is hemizygous is not clear.
SNPs for genotyping in association studies have often been selected from existing databases, such as the International HapMap Project, which has characterized > 6 million validated and genotyped SNPs throughout the human genome with an approximate density of 1 SNP every 500 basepairs. (International HapMap Consortium, 2003). This SNP density may be inadequate for specific candidate genes or regions of interest in smaller association studies (Bhangale et al., 2005, 2008). Our analysis shows that only a small proportion of SNPs discovered in TBX1 (39 of 355) were present in the HapMap dataset. Since the HapMap dataset forms the basis for most whole genome platforms, it is unlikely that they would be able to capture the full extent of variation for association studies focused on TBX1. Furthermore, a comparison of linkage disequilibrium capture (r2 > 0.8) between HapMap SNPs against common SNPs (≥5% allele frequency) from the resequencing data in the reference samples and patients was low. The increased density of one SNP per 150 basepairs provided in this study will contribute to the design of a more powerful genetic association study that is less likely to miss associations between common variants in TBX1 and phenotypic diversity in children with 22q11.2DS.
Some investigators have selectively targeted coding SNPs for genotyping in large association studies, including studies of cardiac phenotypes, schizophrenia, and congenital heart defects. Most of these studies have yielded little evidence of associations (Rauch et al., 2004; Voelckel et al., 2004; Ma et al., 2007; Cabuk et al., 2007; Conti et al., 2003), although in one study there was an association between SNP rs28649236 in Exon 2 (Table 4) and conotruncal defects in individuals without 22q11.2DS (Han et al., 2006). These studies may have had falsely negative results due to the primary focus on polymorphisms in exons, which left much of the TBX1 genetic variation unaccounted for in the analyses.
Average nucleotide diversity (i.e.. frequency of genetic variation) also varies across ethnic/racial populations (Crawford et al., 2005). Results from our study were consistent with prior reports, in that individuals of African descent had more sequence variation in TBX1 than individuals of European descent (Crawford et al., 2005). Designing association studies based on data from only the European subpopulation could lead to loss of power for other populations (such as individuals of African-decent) in future association studies (Crawford et al., 2005).
Having selected tagSNPs based on the correlation among SNPs ensures that in future association studies all common SNPs detected through resequencing will either be tagged and genotyped directly or will be strongly correlated with a tagged, genotyped SNP. The strength of the association detected with any non-genotyped SNP will depend on the strength of correlation between the tagSNPs genotyped and a possible causal nongenotyped SNP. A second advantage to having selected tagSNPs on the basis of correlation is that the statistical power for detecting an association with a nongenotyped SNP is inversely related to the correlation between the tagSNPs and the unknown, putative causal SNP (Pritchard and Przeworski, 2001). Thus the sample size for future studies can be chosen such that they have sufficient statistical power to detected indirect associations with untagged SNPs as well as direct associations with tagSNPs.
This study is limited by the small sample size of the population with the 22q11.2 deletion. Although the sample was sufficiently large to capture most common variation in the European American subgroup, it is likely there are many remaining rare (<5%) variants that we did not detect, and some of these rare variants may be functional (Pritchard and Rosenberg, 1999). This may explain discrepancies between the number of exonic variations identified in our study and previous reports. In addition, common SNP discovery may also have been incomplete in regions with poor sequence quality (exons 3 and 8).
Finally, this study was designed to identify sequence variation in TBX1 by identifying common SNPs. It is possible that other forms of sequence variation (e.g., variable nucleotide repeats, rare single nucleotide polymorphisms, or copy number variations) that were not assessed in this study and not in LD with the common SNPs provide unique information and would be informative as markers in assessing the relationship between the phenotypic variability and TBX1 in individuals with the 22q11.2 deletion (Pritchard and Przeworski, 2001).
Conclusions
The results from this comprehensive SNP discovery effort and review of the literature provide an extensive catalogue of the sequence variation in TBX1. Comprehensive SNP discovery in this manner provides a solid basis for association studies because most common variation is identified and characterized so that a smaller subset of informative SNPs can be genotyped in a larger association study. These data will be useful in larger association studies to investigate the relationship between the genetic variability in TBX1 and phenotypic variability in individuals with the 22q11.2 deletion.
Acknowledgments
Sources of financial support CH was supported by the National Institute of Dental and Craniofacial Research grants T32 DE07132 and 5K23DE17741-2. We also received support from the American Academy of Pediatrics Section on Genetics and Birth Defects, as well as the General Clinical Research Center, Grant # M01- RR 00037.
We are grateful to the participants and their families, and the support of the Pediatric Clinical Research Center.
Footnotes
The authors of this work do not have any financial disclosures or commercial associations that might pose or create a conflict of interest with the information in this manuscript.
Contributor Information
Carrie L. Heike, Department of Pediatrics, Division of Craniofacial Medicine, University of Washington and Seattle Children’s Hospital; Seattle, WA.
Jacqueline R. Starr, Department of Pediatrics, Division of Craniofacial Medicine, and Department of Epidemiology, University of Washington and Seattle Children’s Hospital; Seattle, WA.
Mark J. Rieder, Department of Genome Sciences, University of Washington; Seattle, WA.
Michael L. Cunningham, Department of Pediatrics, Division of Craniofacial Medicine, University of Washington and Seattle Children’s Hospital; Seattle, WA.
Karen L. Edwards, Department of Epidemiology, Institute for Public Health Genetics, University of Washington; Seattle, WA.
Ian Stanaway, Department of Genome Sciences, University of Washington; Seattle, WA.
Dana C. Crawford, Molecular Physiology & Biophysics, Vanderbilt University; Nashville, Tennessee.
References
- Aggarwal VS, Morrow BE. Genetic modifiers of the physical malformations in velo-cardio-facial syndrome/DiGeorge syndrome. Dev Disabil Res Rev. 2008;14(1):19–25. doi: 10.1002/ddrr.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold JS, Braunstein EM, Ohyama T, Groves AK, Adams JC, Brown MC, Morrow BE. Tissue-specific roles of Tbx1 in the development of the outer, middle and inner ear, defective in 22q11DS patients. Human molecular genetics. 2006a;15(10):1629–1639. doi: 10.1093/hmg/ddl084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold JS, Werling U, Braunstein EM, Liao J, Nowotschin S, Edelmann W, Hebert JM, Morrow BE. Inactivation of Tbx1 in the pharyngeal endoderm results in 22q11DS malformations. Development. 2006b;133(5):977–987. doi: 10.1242/dev.02264. [DOI] [PubMed] [Google Scholar]
- Bhangale TR, Rieder MJ, Livingston RJ, Nickerson DA. Comprehensive identification and characterization of diallelic insertion-deletion polymorphisms in 330 human candidate genes. Human molecular genetics. 2005;14(1):59–69. doi: 10.1093/hmg/ddi006. [DOI] [PubMed] [Google Scholar]
- Bhangale TR, Rieder MJ, et al. Estimating coverage and power for genetic association studies using near-complete variation data. Nat Genet. 2008;40:841–843. doi: 10.1038/ng.180. [DOI] [PubMed] [Google Scholar]
- Botto LD, May K, Fernhoff PM, Correa A, Coleman K, Rasmussen SA, Merritt RK, O’Leary LA, Wong LY, Elixson EM, Mahle WT, Campbell RM. A population-based study of the 22q11.2 deletion: phenotype, incidence, and contribution to major birth defects in the population. Pediatrics. 2003;112(1 Pt 1):101–107. doi: 10.1542/peds.112.1.101. [DOI] [PubMed] [Google Scholar]
- Cabuk F, Karabulut HG, Tuncali T, Karademir S, Bozdayi M, Tukun A. TBX1 gene mutation screening in patients with non-syndromic Fallot tetralogy. Turk J Pediatr. 2007;49(1):61–68. [PubMed] [Google Scholar]
- Carlson CS, Eberle MA, Rieder MJ, Yi Q, Kruglyak L, Nickerson DA. Selecting a maximally informative set of single-nucleotide polymorphisms for association analyses using linkage disequilibrium. American journal of human genetics. 2004;74(1):106–120. doi: 10.1086/381000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chieffo C, Garvey N, Gong W, Roe B, Zhang G, Silver L, Emanuel BS, Budarf ML. Isolation and characterization of a gene from the DiGeorge chromosomal region homologous to the mouse Tbx1 gene. Genomics. 1997;43(3):267–277. doi: 10.1006/geno.1997.4829. [DOI] [PubMed] [Google Scholar]
- Conti E, Grifone N, Sarkozy A, Tandoi C, Marino B, Digilio MC, Mingarelli R, Pizzuti A, Dallapiccola B. DiGeorge subtypes of nonsyndromic conotruncal defects: evidence against a major role of TBX1 gene. Eur J Hum Genet. 2003;11(4):349–351. doi: 10.1038/sj.ejhg.5200956. [DOI] [PubMed] [Google Scholar]
- Crawford DC, Akey DT, Nickerson DA. The patterns of natural variation in human genes. Annu Rev Genomics Hum Genet. 2005;6:287–312. doi: 10.1146/annurev.genom.6.080604.162309. [DOI] [PubMed] [Google Scholar]
- Devriendt K, Fryns J, Mortier G, Van Thienen M, Keymolen K. The annual incidence of DiGeorge/velocardiofacial syndrome. Am J Med Genet. 1998;35:789–790. doi: 10.1136/jmg.35.9.789-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagman H, Liao J, Westerlund J, Andersson L, Morrow BE, Nilsson M. The 22q11 deletion syndrome candidate gene Tbx1 determines thyroid size and positioning. Human molecular genetics. 2007;16(3):276–285. doi: 10.1093/hmg/ddl455. [DOI] [PubMed] [Google Scholar]
- Gong W, Gottlieb S, Collins J, Blescia A, Dietz H, Goldmuntz E, McDonald-McGinn DM, Zackai EH, Emanuel BS, Driscoll DA, Budarf ML. Mutation analysis of TBX1 in non-deleted patients with features of DGS/VCFS or isolated cardiovascular defects. J Med Genet. 2001;38(12):E45. doi: 10.1136/jmg.38.12.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han XM, Lou Y, Zhu XY, Hu XF, Pang WY, Sun ZJ, Zhang H, Zhang DQ, Sun YX. Single nucleotide polymorphism and haplotype in TBX1 gene of patients with conotruncal defects: analysis of 130 cases. Zhonghua Yi Xue Za Zhi. 2006;86(22):1553–1557. [PubMed] [Google Scholar]
- Hoogendoorn B, Coleman SL, Guy CA, Smith SK, O’Donovan MC, Buckland PR. Functional analysis of polymorphisms in the promoter regions of genes on 22q11. Human mutation. 2004;24(1):35–42. doi: 10.1002/humu.20061. [DOI] [PubMed] [Google Scholar]
- Howie BN, Carlson CS, Rieder MJ, Nickerson DA. Efficient selection of tagging single-nucleotide polymorphisms in multiple populations. Hum Genet. 2006;120(1):58–68. doi: 10.1007/s00439-006-0182-5. [DOI] [PubMed] [Google Scholar]
- International HapMap Consortium. The International HapMap Project. Nature. 2003;426(6968):789–796. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- Jerome LA, Papaioannou VE. DiGeorge syndrome phenotype in mice mutant for the T-box gene, Tbx1. Nature genetics. 2001;27(3):286–291. doi: 10.1038/85845. [DOI] [PubMed] [Google Scholar]
- Kruglyak L. The road to genome-wide association studies. Nature reviews. 2008;9(4):314–318. doi: 10.1038/nrg2316. [DOI] [PubMed] [Google Scholar]
- Kruglyak L, Nickerson DA. Variation is the spice of life. Nature genetics. 2001;27(3):234–236. doi: 10.1038/85776. [DOI] [PubMed] [Google Scholar]
- Lindsay EA, Vitelli F, Su H, Morishima M, Huynh T, Pramparo T, Jurecic V, Ogunrinu G, Sutherland HF, Scambler PJ, Bradley A, Baldini A. Tbx1 haploinsufficieny in the DiGeorge syndrome region causes aortic arch defects in mice. Nature. 2001;410(6824):97–101. doi: 10.1038/35065105. [DOI] [PubMed] [Google Scholar]
- Livingston RJ, von Niederhausern A, Jegga AG, Crawford DC, Carlson CS, Rieder MJ, Gowrisankar S, Aronow BJ, Weiss RB, Nickerson DA. Pattern of sequence variation across 213 environmental response genes. Genome research. 2004;14(10A):1821–1831. doi: 10.1101/gr.2730004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma G, Shi Y, Tang W, He Z, Huang K, Li Z, He G, Feng G, Li H, He L. An association study between the genetic polymorphisms within TBX1 and schizophrenia in the Chinese population. Neurosci Lett. 2007;425(3):146–150. doi: 10.1016/j.neulet.2007.07.055. [DOI] [PubMed] [Google Scholar]
- McDonald-McGinn DM, Kirschner R, Goldmuntz E, Sullivan K, Eicher P, Gerdes M, Moss E, Solot C, Wang P, Jacobs I, Handler S, Knightly C, Heher K, Wilson M, Ming JE, Grace K, Driscoll D, Pasquariello P, Randall P, Larossa D, Emanuel BS, Zackai EH. The Philadelphia story: the 22q11.2 deletion: report on 250 patients. Genet Couns. 1999;10(1):11–24. [PubMed] [Google Scholar]
- Merscher S, Funke B, Epstein JA, Heyer J, Puech A, Lu MM, Xavier RJ, Demay MB, Russell RG, Factor S, Tokooya K, Jore BS, Lopez M, Pandita RK, Lia M, Carrion D, Xu H, Schorle H, Kobler JB, Scambler P, Wynshaw-Boris A, Skoultchi AI, Morrow BE, Kucherlapati R. TBX1 is responsible for cardiovascular defects in velo-cardio-facial/DiGeorge syndrome. Cell. 2001;104(4):619–629. doi: 10.1016/s0092-8674(01)00247-1. [DOI] [PubMed] [Google Scholar]
- Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31(13):3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovcharenko I, Nobrega MA, Loots GG, Stubbs L. ECR Browser: a tool for visualizing and accessing data from comparisons of multiple vertebrate genomes. Nucleic Acids Res. 2004;32(Web Server issue):W280–286. doi: 10.1093/nar/gkh355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK, Przeworski M. Linkage disequilibrium in humans: models and data. American journal of human genetics. 2001;69(1):1–14. doi: 10.1086/321275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK, Rosenberg NA. Use of unlinked genetic markers to detect population stratification in association studies. American journal of human genetics. 1999;65(1):220–228. doi: 10.1086/302449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramensky V, Bork P, Sunyaev S. Human non-synonymous SNPs: server and survey. Nucleic Acids Res. 2002;30(17):3894–3900. doi: 10.1093/nar/gkf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch A, Devriendt K, Koch A, Rauch R, Gewillig M, Kraus C, Weyand M, Singer H, Reis A, Hofbeck M. Assessment of association between variants and haplotypes of the remaining TBX1 gene and manifestations of congenital heart defects in 22q11.2 deletion patients. J Med Genet. 2004;41(4):e40. doi: 10.1136/jmg.2003.010975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robin NH, Shprintzen RJ. Defining the clinical spectrum of deletion 22q11.2. J Pediatr. 2005;147(1):90–96. doi: 10.1016/j.jpeds.2005.03.007. [DOI] [PubMed] [Google Scholar]
- Ryan AK, Goodship JA, Wilson DI, Philip N, Levy A, Seidel H, Schuffenhauer S, Oechsler H, Belohradsky B, Prieur M, Aurias A, Raymond FL, Clayton-Smith J, Hatchwell E, McKeown C, Beemer FA, Dallapiccola B, Novelli G, Hurst JA, Ignatius J, Green AJ, Winter RM, Brueton L, Brondum-Nielsen K, Scambler PJ, et al. Spectrum of clinical features associated with interstitial chromosome 22q11 deletions: a European collaborative study. J Med Genet. 1997;34(10):798–804. doi: 10.1136/jmg.34.10.798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shprintzen RJ. Velo-Cardio-Facial Syndrome. In: Cassidy SB, Allanson JE, editors. Management of Genetic Syndromes. 2. Wiley-Liss, Inc; 2005. pp. 615–631. [Google Scholar]
- Stephens M, Donnelly P. A comparison of bayesian methods for haplotype reconstruction from population genotype data. American journal of human genetics. 2003;73(5):1162–1169. doi: 10.1086/379378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens M, Sloan JS, Robertson PD, Scheet P, Nickerson DA. Automating sequence-based detection and genotyping of SNPs from diploid samples. Nature genetics. 2006;38(3):375–381. doi: 10.1038/ng1746. [DOI] [PubMed] [Google Scholar]
- Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123(3):585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theveniau-Ruissy M, Dandonneau M, Mesbah K, Ghez O, Mattei MG, Miquerol L, Kelly RG. The del22q11.2 candidate gene Tbx1 controls regional outflow tract identity and coronary artery patterning. Circ Res. 2008;103(2):142–148. doi: 10.1161/CIRCRESAHA.108.172189. [DOI] [PubMed] [Google Scholar]
- Vitelli F, Morishima M, Taddei I, Lindsay EA, Baldini A. Tbx1 mutation causes multiple cardiovascular defects and disrupts neural crest and cranial nerve migratory pathways. Human molecular genetics. 2002;11(8):915–922. doi: 10.1093/hmg/11.8.915. [DOI] [PubMed] [Google Scholar]
- Vitelli F, Viola A, Morishima M, Pramparo T, Baldini A, Lindsay E. TBX1 is required for inner ear morphogenesis. Human molecular genetics. 2003;12(16):2041–2048. doi: 10.1093/hmg/ddg216. [DOI] [PubMed] [Google Scholar]
- Vittorini S, Sacchelli M, Iascone MR, Collavoli A, Storti S, Giusti A, Andreani G, Botto N, Biagini A, Clerico A. Molecular characterization of chromosome 22 deletions by short tandem repeat polymorphism (STRP) in patients with conotruncal heart defects. Clin Chem Lab Med. 2001;39(12):1249–1258. doi: 10.1515/CCLM.2001.201. [DOI] [PubMed] [Google Scholar]
- Voelckel MA, Girardot L, Giusiano B, Levy N, Philip N. Allelic variations at the haploid TBX1 locus do not influence the cardiac phenotype in cases of 22q11 microdeletion. Ann Genet. 2004;47(3):235–240. doi: 10.1016/j.anngen.2004.04.002. [DOI] [PubMed] [Google Scholar]
- Wilson D, Cross I, Wren C, et al. Minimum prevalence of chromosome 22q11 deletions. Am J Med Genet. 1994;55:A169. [Google Scholar]
- Wiltshire S, de Bakker PI, Daly MJ. The value of gene-based selection of tag SNPs in genome-wide association studies. Eur J Hum Genet. 2006;14(11):1209–1214. doi: 10.1038/sj.ejhg.5201678. [DOI] [PubMed] [Google Scholar]
- Yagi H, Furutani Y, Hamada H, Sasaki T, Asakawa S, Minoshima S, Ichida F, Joo K, Kimura M, Imamura S, Kamatani N, Momma K, Takao A, Nakazawa M, Shimizu N, Matsuoka R. Role of TBX1 in human del22q11.2 syndrome. Lancet. 2003;362(9393):1366–1373. doi: 10.1016/s0140-6736(03)14632-6. [DOI] [PubMed] [Google Scholar]
- Yamagishi H, Maeda J, Hu T, McAnally J, Conway SJ, Kume T, Meyers EN, Yamagishi C, Srivastava D. Tbx1 is regulated by tissue-specific forkhead proteins through a common Sonic hedgehog-responsive enhancer. Genes Dev. 2003;17(2):269–281. doi: 10.1101/gad.1048903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagishi H, Srivastava D. Unraveling the genetic and developmental mysteries of 22q11 deletion syndrome. Trends Mol Med. 2003;9(9):383–389. doi: 10.1016/s1471-4914(03)00141-2. [DOI] [PubMed] [Google Scholar]
- Zweier C, Sticht H, Aydin-Yaylagul I, Campbell CE, Rauch A. Human TBX1 Missense Mutations Cause Gain of Function Resulting in the Same Phenotype as 22q11.2 Deletions. American journal of human genetics. 2007;80(3):510–517. doi: 10.1086/511993. [DOI] [PMC free article] [PubMed] [Google Scholar]