

Abstract
Tuberculosis (TB) is still a major public health issue in developing countries, and its chemotherapy is compromised by poor drug compliance and severe side effects. This study aimed to synthesize and characterize new multimodal PEGylated liposomes encapsulated with clinically commonly used anti-TB drugs with linkage to small interfering RNA (siRNA) against transforming growth factor-β1 (TGF-β1). The novel NP-siRNA liposomes could target THP-1-derived human macrophages that were the host cells of mycobacterium infection. The biological effects of the NP-siRNA liposomes were evaluated on cell cycle distribution, apoptosis, autophagy, and the gene silencing efficiency of TGF-β1 siRNA in human macrophages. We also explored the proteomic responses to the newly synthesized NP-siRNA liposomes using the stable isotope labeling with amino acids in cell culture approach. The results showed that the multifunctional PEGylated liposomes were successfully synthesized and chemically characterized with a mean size of 265.1 nm. The novel NP-siRNA liposomes functionalized with the anti-TB drugs and TGF-β1 siRNA were endocytosed efficiently by human macrophages as visualized by transmission electron microscopy and scanning electron microscopy. Furthermore, the liposomes showed a low cytotoxicity toward human macrophages. There was no significant effect on cell cycle distribution and apoptosis in THP-1-derived macrophages after drug exposure at concentrations ranging from 2.5 to 62.5 μg/mL. Notably, there was a 6.4-fold increase in the autophagy of human macrophages when treated with the NP-siRNA liposomes at 62.5 μg/mL. In addition, the TGF-β1 and nuclear factor-κB expression levels were downregulated by the NP-siRNA liposomes in THP-1-derived macrophages. The Ingenuity Pathway Analysis data showed that there were over 40 signaling pathways involved in the proteomic responses to NP-siRNA liposome exposure in human macrophages, with 160 proteins mapped. The top five canonical signaling pathways were eukaryotic initiation factor 2 signaling, actin cytoskeleton signaling, remodeling of epithelial adherens junctions, epithelial adherens junction signaling, and Rho GDP-dissociation inhibitor signaling pathways. Collectively, the novel synthetic targeting liposomes represent a promising delivery system for anti-TB drugs to human macrophages with good selectivity and minimal cytotoxicity.
Keywords: tuberculosis, cytokine, liposome, apoptosis, autophagy, cell cycle, proteomics, SILAC, NF-κB, interleukin
Introduction
Tuberculosis (TB), caused by Mycobacterium tuberculosis, remains a major global health problem and ranks as the second leading cause of death from an infectious disease worldwide after human immunodeficiency virus (HIV), despite the discovery of effective and affordable chemotherapy more than 50 years ago.1–6 According to the data of World Health Organization (WHO),5 an estimated 9 million people developed TB, and 1.5 million (including HIV-positive patients who died of TB) died from the disease worldwide, in 2013. The deaths included 1.1 million among HIV-negative and 360,000 among HIV-positive, people. Approximately 78% of the overall total TB deaths, and 73% of TB deaths among HIV-negative people occurred in the African and Southeast Asian Regions in 2013.5 Of the estimated 9 million new TB cases worldwide in 2013, about 57% of TB cases occurred in men; there were an estimated 3.3 million cases and 510,000 TB deaths among women, as well as an estimated 550,000 cases and 80,000 deaths among children.5 The African and Southeast Asian Regions accounted for 69% of the cases among women. Additionally, half of the HIV-positive people who died from TB in 2013 were women.5 Most of the estimated number of cases in 2013 occurred in Asia (56%) and the African Region (29%); smaller proportions of cases occurred in the Eastern Mediterranean Region (8%), the European Region (4%), and the Region of the Americas (3%).5 India and the People’s Republic of China alone accounted for 24% and 11% of the total cases in 2013, respectively. In the US, there were 9,582 new TB cases in 2013, with an incidence of 3.0 cases/100,000 population.7 About 65% of these TB cases were in people born outside the US, and more than half came from five countries including Mexico, the Philippines, India, Vietnam, and the People’s Republic of China. Notably, TB was the leading killer of people with HIV infection, accounting for about one-fifth of HIV deaths. In particular, an estimated 480,000 people developed multidrug resistant (MDR)-TB globally in 2013, including 3.5% of new and 20.5% of previously treated TB cases (5% of total cases).5 There were ~210,000 deaths from MDR-TB (range, 130,000–290,000). On average, an estimated 9% of people with MDR-TB have extensive MDR-TB (XDR-TB). Globally, the TB mortality rate fell by an estimated 45% between 1990 and 2013, and the TB prevalence rate fell by 41% during the same period.5
TB is most commonly transmitted from a person with infectious pulmonary TB to others by droplet nuclei, which are aerosolized by coughing, sneezing, or speaking.8 M. tuberculosis normally enters into the pulmonary alveolus via aerosol delivery of 2–5 μm particles, containing the bacterium. About one-third of the world’s population (~2 billion) is estimated to have been exposed to TB bacteria and potentially infected.5 TB typically affects the lungs, but it also can affect any other organ of the body including lymph nodes, bones, kidneys, brain, spine, liver, skin, and intestine.9,10 WHO adopted the DOTS (Directly Observed Therapy, Short Course) strategy as the standard approach to address the global TB epidemic in 1993. The key component of the DOTS strategy recommended by WHO is the standard chemotherapy regimen for drug-susceptible TB, which requires continual oral administration of isoniazid (INH), rifampicin (RIF), pyrazinamide (PZA), and ethambutol (EMB) for 6 months. In the intensive phase, the treatment consists of 2 months of RIF, INH, PZA, and EMB, followed by 4 months of RIF and INH during the continuation phase.11 In the continuation phase, EMB is added in countries with high levels of INH resistance in new TB patients, and in those where INH susceptibility testing in new patients is not conducted. The dosing frequency can be daily or 3 times/week. Rifabutin (RBT) and rifapentine (RPT) may also be considered first-line drugs under certain circumstances.12,13 RBT is used as a substitute for RIF in the treatment of all forms of TB caused by organisms that are known or presumed to be susceptible to this agent. RBT is generally reserved for patients for whom drug–drug interactions preclude the use of RIF. Streptomycin (SM) was formerly considered to be a first-line drug and is now used as a second-line anti-TB drug in the US due to increasing prevalence of resistance to SM. Other second-line anti-TB drugs approved by the US Food and Drug Administration (FDA) include cycloserine, capreomycin, ρ-aminosalicylic acid, and ethionamide. In the US, the FDA has approved fixed-dose combinations of 150 mg INH and 300 mg RIF (Rifamate®, Sanofi-Aventis Pharmaceuticals, Bridgewater, NJ, USA) and of 50 mg INH, 120 mg RIF, and 300 mg PZA (Rifater®, Sanofi-Aventis Pharmaceuticals). In view of the seriousness of TB infection, the People’s Republic of China established the China National Tuberculosis Prevention and Control Scheme in 1990 and has been implementing DOTS since 1991, which constitutes the cornerstone of the current strategy for TB control and covers the country’s entire population.14 From 1990 to 2010, the prevalence of smear-positive TB decreased from 170 to 59 cases/100,000 population in the People’s Republic of China.15 Although the treatment success rate is often more than 85% for newly diagnosed TB using the standard regimen, MDR-TB still develops in many patients due to resistance to INH and RIF.16–19 XDR-TB is a form of MDR-TB that responds to even fewer available medicines, including the most effective second-line anti-TB drugs.2,20,21 MDR-TB is treated with a combination of second-line drugs including fluoroquinolones, aminoglycosides, ethionamide, cycloserine, ρ-aminosalicyclic acid, linezolid, and clofazimine for up to 2 years. Two new anti-TB drugs belonging to two novel classes of antibiotics have been recently approved: bedaquiline, by the FDA,22–24 and delamanid, by Japan and the European Union.25–28 These two drugs offer hope of better outcomes in severe cases of MDR-TB, but have not yet been tested to shorten treatment of drug-susceptible TB or as preventive treatment. Only 48% of the MDR-TB patients in the 2011 cohort of detected cases were successfully treated, and the treatment success rate for XDR-TB patients in the 2011 cohort was only 22% since second-line treatment options are limited and recommended medicines are not always available.20 Existing and newly approved anti-TB drugs are generally tolerated well by most patients, but they may cause side effects including liver injury, skin reactions, and gastrointestinal and neurological disorders in a proportion of patients that can be alleviated by symptomatic therapy.29–31 Thus, there is a further need to develop highly effective and safe anti-TB drugs that can improve the clinical outcome and overcome multidrug resistance in the chemotherapy of TB.
To date, there has been increasing interest in the design and use of nanotechnology-based new delivery platforms with the potential for improving the efficacy and reducing the side effects associated with the treatment of newly diagnosed and MTD-TB.32–38 Many types of nanocarriers have been tested against TB, including vesicular drug delivery systems (liposomes, niosomes, and solid lipid nanoparticles), particulate drug delivery systems (nanoparticles, microparticles, and dendrimers), supramolecular drug delivery systems (polymeric micelles), specialized drug delivery systems (nanosuspensions, nanoemulsions, microemulsions, and dry powders), complex conjugate drug delivery systems (immunostimulating complex and cyclodextrin inclusion complexes), and other carrier-based drug delivery systems (eg, nanotubes, nanobeads, nanofibers, quantum dots, etc).32,36,37 These nanometric delivery systems are expected to offer advantages over conventional systems by producing optimum effectiveness to the target site, enhanced therapeutic efficacy, uniform distribution of the drug throughout the target site, increased bioavailability and sustainability of anti-TB drugs, fewer side effects, and increased patient compliance. Nanoparticles as drug carriers enable higher stability and carrier capacity along with immense improvement of drug bioavailability, which further leads to reduction in dosage frequency. Liposomes are safe and effective drug carriers and can be functionalized with a great variety of ligands for targeting. Importantly, the bilayer of liposomes can be utilized to combine both hydrophobic and hydrophilic drugs through self-assembling; liposomes are biodegradable with low cytotoxicity and immunogenicity, and enter into mononuclear phagocytic macrophages preferentially.39–41
The host response to M. tuberculosis infection is complex and multifaceted, involving many components of the immune system, mainly the result of interplay between macrophages, T-cells, B cells, and neutrophils.42–49 The primary pathologic feature of M. tuberculosis infection is the formation of a granuloma.50 Macrophages are the main target of anti-TB drugs since they are the host of M. tuberculosis, and M. tuberculosis is internalized through phagocytosis by the resident alveolar macrophages.42–44,51,52 M. tuberculosis infection is an inflammatory progress through the host immune system to release cytokines and chemokines that stimulate and recruit a wide range of cells involved in immunity and inflammation.53,54 The actions of chemokines could be homeostatic, where they guide cells during immune surveillance for pathogens by interacting with antigen presenting cells residing in these tissues. Studies in mice and humans showed that interferon-γ (IFN-γ), tumor necrosis factor-α (TNF-α), and interleukin-12 (IL-12) are key cytokines involved in the control of M. tuberculosis infections.42–44,55 Transforming growth factor-β1 (TGF-β1) primarily produced by mononuclear phagocytes is known as the immunoregulatory cytokines, inhibiting T-cell responses and deactivating the macrophages in patients with TB.51,56 Overexpression of TGF-β1 results in tissue damage and fibrosis in TB patients. In view of the key role of immune response in TB development, targeting the inflammatory response in TB may represent an effective approach to control TB.57 For example, silencing of TGF-β1 by small interfering RNA (siRNA) in macrophages results in a significant decrease in titers of the intracellular M. tuberculosis.58 siRNA can efficiently and specifically block the expression of homologous genes in vitro and in vivo when it promotes homologous mRNA degradation and induces cells to exhibit specific gene deletion phenotype. A recent study has shown that IL-1 confers host resistance through the induction of eicosanoids that limit excessive type I IFN production and foster bacterial containment.59 In TB-infected mice and patients, reduced IL-1 responses and/or excessive type I IFN induction are linked to an eicosanoid imbalance associated with disease exacerbation, while host-directed immunotherapy with clinically approved drugs that augment prostaglandin E2 levels prevents acute mortality of M. tuberculosis-infected mice.59 These findings indicate that IL-1 and type I IFNs represent two major counter-regulatory classes of inflammatory cytokines that control the outcome of M. tuberculosis infection and are functionally linked via eicosanoids.
Our previous studies have shown that an ultrashort-course chemotherapy regimen produced good efficacy and was well tolerated in spinal TB patients, with a mean duration of 5.5 months.60–62 In the present study, we propose that a combination of INH, RIF, PZA, SM, and TGF-β1 siRNA with the utilization of nanofabrication using liposomes can improve the anti-TB therapeutic effect and reduce the side effect in the treatment of TB. We aimed to synthesize multifunctional polyethylene glycosylated (PEGylated) liposomes that encapsulated four anti-TB drugs including SM, INH, RIF, and PZA linked to TGF-β1 siRNA to deliver the multifunctional liposomes to human macrophages that were differentiated from the human leukemic (THP-1) cells. To explore the molecular targets, a quantitative proteomic study using stable isotope labeling with amino acids in cell culture (SILAC) was also conducted and the data initially validated.
Materials and methods
Chemicals and reagents
INH, RIF, PZA, SM, phorbol 12-myristate 13-acetate (PMA), lipopolysaccharide (LPS), fetal bovine serum (FBS), ribonuclease A (RNase A), propidium iodide (PI), dimethyl sulfoxide (DMSO), and the primary antibody against human β-actin were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). Cholesterol, 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP), high-performance liquid chromatography-grade acetone, butanol, acetonitrile, thin-layer chromatography plates (1,000 and 200 mm), nuclear and cytoplasmic extraction reagent (NE-PER) kit, and Western blotting substrate were purchased from Thermo Fisher Scientific Inc. (Waltham, MA, USA). 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(polyethylene glycol)-2000] (DSPE-PEG 2000) was purchased from Avanti Polar Lipids Inc. (Alabaster, AL, USA). The primers for the detection of TGF-β1, IL-1, IL-6, IL-8, TNF-α, and IFN-γ mRNAs, TGF-β1 siRNA, negative control siRNA, and primary antibody against human β-actin were obtained from Santa Cruz Biotechnology Inc. (Dallas, TX, USA). Roswell Park Memorial Institute (RPMI)-1640 medium were obtained from Corning Cellgro Inc. (Herndon, VA, USA). Dulbecco’s phosphate buffered saline (PBS), thiazolyl blue tetrazolium bromide (MTT), and the annexin V: phycoerythrin (PE) apoptosis kit was purchased from BD Biosciences Inc. (San Jose, CA, USA). The Cyto-ID® autophagy detection kit was obtained from Enzo Life Sciences Inc. (Farmingdale, NY, USA). Spectra/Por® Dialysis Membranes with molecular weight (MW) of 1,000 Da was purchased from Spectrum Laboratories Inc. (Rancho Dominguez, CA, USA). Polyvinylidene difluoride (PVDF) membrane was purchased from EMD Millipore Inc. (Bedford, MA, USA). Primary antibodies against human phosphorylated phosphoinositide 3-kinase (p-PI3K), phosphorylated p38 mitogenactivated protein kinase (p-p38 MAPK), p38 MAPK, mammalian target of rapamycin (mTOR), p-mTOR, phosphorylated extracellular signal-regulated kinase (ERK), p-ERK, phosphatase and tensin homologue (PTEN), beclin 1, microtubule-associated protein 1A/1B-light chain 3 (LC3), nuclear factor-κB (NF-κB), and nuclear factor (erythroid-derived 2)-like 2 (Nrf2) were purchased from Cell Signaling Technology Inc. (Beverly, MA, USA). The kits for detecting human TGF-β1, IL-6, IL-8, TNF-α, and IFN-γ were purchased from eBioscience Inc. (San Diego, CA, USA).
Cell culture
THP-1 cells were purchased from American Type Culture Collection (Manassas, VA, USA). The cells were maintained at 2×105 cells/mL, cultured in RPMI 1640 medium supplemented with 10% FBS, and incubated at 37°C in a 5% CO2 humidified incubator. Medium renewal was carried out 2–3 times/week, and cells were subcultured when they achieved 80%–90% confluence. The THP-1 cells were differentiated into macrophages by incubation with 100 ng/mL PMA for 48 hours in RPMI-1640 medium with 10% FBS, as described previously.63 After 48-hour treatment, suspended THP-1 cells were differentiated into adherent macrophages, the cells were washed with PBS three times, and suspended in fresh RPMI-1640 medium with 10% FBS for 3 hours.
Chemical synthesis of PEGylated liposomes loaded with four anti-TB drugs and TGF-β1 siRNA
The conjugation of SM to DSPE-PEG 2000
The nanoparticles were formulated using DSPE-PEG 2000 and SM at a 1:10 molar ratio, which were dissolved in 2 mL DMSO, followed by adding 500 μL pyridine. The mixture was sonicated at 40°C for 2 hours, and then stirred at room temperature (RT) for 24 hours. The product was dialyzed in DMSO and then double deionized water (dd-H2O) to filter off the small molecule substances with MW less than 1,000 Da. Subsequently, the resultant product DSPE-PEG2000-SM was lipolyzed at −50°C.
PEGylated liposome formation and INH, RIF, and PZA encapsulation
DOTAP, cholesterol, DSPE-PEG2000-SM, INH, RIF, and PZA were formulated in a 20:10:1:21:5.3:5.9 molar ratio and dissolved in chloroform (800 μL) and methanol (200 μL) (4:1 v/v) after the self-assembly of the liposomal nanoparticles. The organic solvent was evaporated under pressure gradually for 30 minutes at 30°C using a rotary evaporator. The resultant products were flushed with a stream of nitrogen gas to remove the residue of organic solvent. The products were stored in nitrogen gas at 4°C overnight.
Cationic PEGylated liposomes with INH, RIF, and PZA encapsulated TGF-β1 siRNA
Cationic PEGylated liposome with INH, RIF, and PZA were codissolved in 500 μL DMSO and 500 μL RNase-free dd-H2O at RT. TGF-β1 siRNA was then added into the lipid film, and hydrated in a sonicated water bath for 1 minute, followed by filtration through a 0.20 μm Anotop 10 filter (Whatman, GE Healthcare Life Sciences, Little Chalfont, UK), which was incubated at RT for 30 minutes to allow it to be stabilized. The amount of TGF-β1 siRNA was formulated into cationic PEGylated liposomes at a 1:20 molar ratio for static neutralization with a final concentration of TGF-β1 siRNA at 80 nM. The final liposomal complex (NP-siRNA) was maintained in a sterile environment for subsequent gene silencing experiments.
Compound characterization by ultraviolet spectroscopy, fluorescent spectroscopy, and Fourier transform infrared spectroscopy
The instrument used for ultraviolet (UV) and fluorescent (FL) analysis was a BioTek microplate reader (Bio-Rad, Winooski, VT, USA). The software used for collecting the UV and FL data was the Gen5™. An Agilent Cary 630 FTIR (Fourier transform infrared) spectrometer (Agilent Technologies Inc., Santa Clara, CA, USA) was used for the IR spectra of the diamond module (solid) or transmission module.
Scanning electron microscopy and transmission electron microscopy
The morphology of the novel anti-TB liposomes (NP-siRNA) were observed by scanning electron microscopy (SEM) (Hitachi High-Technologies Corp, Tokyo, Japan). Samples were prepared on stubs and coated with gold and palladium prior to examination. The morphology and size of the NP-siRNA liposomes were also evaluated by transmission electron microscopy (TEM) (JEM 100CX; JEOL Ltd, Tokyo, Japan). An aqueous solution of liposomes was directly trickled onto a 300-mesh copper grid and dried in a vacuum oven at 37°C for 4 hours, and the microimages were observed by TEM with an accelerating voltage of 80 kV. Meanwhile, the endocytosis of NP-siRNA liposomes by human macrophages was observed using TEM within 5 minutes at 4°C and 37°C as compared with the blank control without drug exposure.
Size distribution and zeta potential measurement of the novel NP-siRNA liposomes
The mean size and zeta potential of the liposomes were determined by dynamic light scattering using a zetasizer 1000 HSA (Malvern Instruments, Malvern, UK). As a measure of particle size distribution of the dispersion, the system reported the polydispersity index (PDI) ranging from 0.0 for an entirely monodisperse sample up to 1.0 for a polydisperse liposomal mixture.
Liquid chromatography mass spectrometry
The samples were cleaned and the concentration of anti-TB drugs analyzed by 6400 Series Triple Quadrupole LC/MS (Agilent Technologies Inc.) conducted on a Zorbax® Eclipse C18 column (Agilent Technologies Inc.) with a pore size of 3.5 μm (4.6×100 mm). The analysis was conducted using a mobile phase consisting of acetonitrile and 0.1% formic acid in water, which was gradually increased from 20% to 80% over the course of the run at a flow rate of 1 mL/minute. The column was maintained at RT with an injection volume of 20 μL.
Drug release assay
The in vitro release profiles and kinetics of the novel NP-siRNA liposomes were determined by a dialysis method at various time points (0, 4, 8, 12, 24, 48, and 72 hours), as described previously by us.64 Briefly, 3 mL of the aqueous liposomes or corresponding parent drug was mixed with 1 mL PBS, and the mixture was dialyzed against water to filter off the small molecule substance with MW less than 1,000 Da. The resultant products were then dialyzed against 10 mL PBS containing 10% FBS at 37°C with gentle shaking for 3 days. A 20 μL aliquot of the sample was withdrawn from the incubation medium and replaced with an equal volume of buffer at designated time points and stored frozen for further analysis. The released drugs were quantified by validated liquid chromatography mass spectrometry (LC–MS) methods. Calibration curves were prepared using different concentrations of individual free anti-TB drugs including INH, RIF, PZA, and SM.
Cell viability assay
The MTT assays were performed to examine the effect of the novel NP-siRNA liposomes on cell viability of human macrophages. THP-1 cells were seeded into 96-well plates at a density of 6,000 cells/well, which were differentiated into macrophages after treatment with PMA at 100 ng/mL for 48 hours and maintained in RPMI medium. The macrophages were treated with a single anti-TB drug, a combination of four anti-TB drugs, or the multimodal NP-siRNA liposomes at concentrations of 2.5, 12.5, and 62.5 μg/mL, and then incubated for 24 hours at 37°C. Following the drug treatment, 10 μL of MTT stock solution (5 mg/mL) was added to each well and incubated for 4 hours. Then, the medium was carefully removed, and 100 μL DMSO was added into each well. The plate was placed on a rocker to mix the solution thoroughly for 30 seconds, and incubated at 37°C for 10 minutes. The absorbance was measured using a Synergy™ H4 Hybrid microplate reader (BioTek Inc, Winooski, VT, USA) at wavelengths of 560 and 670 nm. The 50% inhibitory potency (IC50) values were calculated from dose–response curves obtained in multireplicated experiments by GraphPad Prism 5 (GraphPad Software Inc., La Jolla, CA, USA).
Cell cycle distribution analysis
The effect of the novel NP-siRNA liposomes on the cell cycle of macrophages was determined using PI as the DNA stain using flow cytometry, as described previously.65 Briefly, macrophages were treated with the novel NP-siRNA liposomes at concentrations of 2.5, 12.5, and 62.5 μg/mL for 24 hours at 37°C. Cells were trypsinized, washed by PBS, and suspended in 1 mL PBS containing 1 mg/mL RNase A and 50 μg/mL PI, fixed by 70% ethanol at −20°C overnight. A total number of 1×105 cells were subject to cell cycle analysis using a flow cytometer (Becton Dickinson Immunocytometry Systems, San Jose, CA, USA). The flow cytometer collected 10,000 events.
Quantification of cellular apoptosis using flow cytometry
Annexin V is a 35–36 kDa Ca++-dependent phospholipid-binding protein that has a high affinity for negatively charged phospholipid phosphatidylserine, and binds to cells that are actively undergoing apoptosis with exposed phospholipid phosphatidylserine. Apoptosis of human macrophages derived from THP-1 cells was quantitated using the BD Pharmingen™ annexin V:PE apoptosis detection kit (BD Biosciences Inc.) after cells were treated with the NP-siRNA liposomes at different concentrations of 2.5, 12.5, and 62.5 μg/mL for 24 hours, as described previously.63,66,67 Briefly, macrophages were trypsinized and washed with cold PBS. Following that, the cells were pelleted and resuspended in 1× binding buffer (BD Biosciences Inc.) with 5 μL of annexin V:PE and 5 μL of 7-aminoactinomycin D (7-AAD, a vital nucleic acid dye) at 1×105 cells/mL in a total volume of 100 μL. The cells were gently mixed and incubated in the dark for 15 minutes at RT. The sample was then mixed with 150 μL 1× binding buffer and transferred into a clean test tube, and the number of apoptotic cells was quantified using the flow cytometer within 1 hour. Cells that stain positive for PE and annexin V, and negative for 7-AAD are undergoing apoptosis; cells that stain positive for PE, annexin V, and 7-AAD are either in the end stage of apoptosis, are undergoing necrosis, or are already dead; and cells that stain negative for PE annexin V and 7-AAD are alive and not undergoing measurable apoptosis.
Determination of autophagy of human macrophages by flow cytometry
The autophagy level in macrophages was examined using flow cytometry as described previously.67,68 THP-1-derived macrophages were treated with the novel NP-siRNA liposomes at different concentrations of 2.5, 12.5, and 62.5 μg/mL for 24 hours. After 24-hour incubation, the cells were collected and resuspended in a 250 μL phenol red-free culture medium containing 5% FBS. Following the resuspension, the cells were pelleted and 250 μL diluted Cyto-ID® (Enzo Life Sciences Inc.) Green stain solution was added to each sample and mixed well. Macrophages were incubated for 30 minutes at 37°C in the dark. Then, the cells were collected by centrifugation, washed with 1× assay buffer, and resuspended in 250 μL fresh 1× assay buffer. Cells were analyzed using the green (FL1) channel of the flow cytometer that collected 10,000 events.
Confocal fluorescence microscopy
Confocal fluorescence microscopy was performed to further examine the cellular autophagy using a Cyto-ID® (Enzo Life Sciences Inc.) autophagy detection kit according to the manufacturer’s instruction. The kit was used to measure cellular autophagic vacuoles and autophagic flux using a novel dye that selectively labels autophagic vacuoles. The assay provides a rapid and quantitative approach to monitoring autophagy in live cells without the need for cell transfection, and allows the measurement and differentiation between autophagic flux and autophagolysosome accumulation. Briefly, THP-1-derived macrophages were seeded into an 8-well chamber slide at 30% confluence for 24 hours. Then, the cells were treated with the NP-siRNA liposomes at 2.5, 12.5, and 62.5 μg/mL for 24 hours. The confocal images were recorded with a Leica TCS SP2 laser scanning confocal microscope (Leica Microsystems, Wetzlar, Germany) at wavelengths of 405/488 nm.
Enzyme-linked immunosorbent assay
Macrophages differentiated from THP-1 cells were treated with fresh medium alone (control), 0.1% DMSO, NP (the liposomes without TGF-β1 siRNA), NP-siRNA-control (the liposomes with control TGF-β1 siRNA), NP-siRNA liposomes with TGF-β1 siRNA linked, and the liposomes at a concentration of 12.5 μg/mL for 24 hours. A volume of 500 μL medium was collected at 24 hours and another 500 μL fresh medium was supplemented. Cytokines including TGF-β1, IL-6, IL-8, TNF-α, and IFN-γ in medium were quantified using ELISA (enzyme-linked immunosorbent assay) kits according to manufacturer’s protocol. LPS at a concentration of 100 ng/mL was added to the cell culture medium as a positive control.
Quantitative proteomic study using SILAC approach
Proteomic experiments were performed using SILAC protein quantitation kits from Thermo Fisher Scientific Inc., as described previously by us.69,70 Briefly, THP-1 cells were cultured in the medium with (heavy) or without (light) stable isotope labeled amino acids (13C6 L-lysine and 13C6 15N4 L-arginine). THP-1 cells were passaged for five times by changing the medium or splitting the cells. THP-1 cells were differentiated into macrophages by treatment with 100 ng/mL PMA for 48 hours. Then, macrophages were treated with the novel NP-siRNA liposomes at a concentration of 12.5 μg/mL for 24 hours, which were maintained in heavy medium. Following that, the cells were harvested and lysated with hot lysis buffer (100 mM Tris base, 4% sodium dodecyl sulfate [SDS], and 100 mM dithiothreitol). The protein was denatured at 95°C for 5 minutes and sonicated at 20% amplitude for 3 seconds with 6 pulses. The samples were centrifuged at 15,000× g for 20 minutes, and the supernatant was collected in clean tubes. The protein concentration was determined using the ionic detergent compatibility reagent from Thermo Fisher Scientific Inc. Subsequently, equal amounts of heavy and light protein samples were combined to reach a total volume of 30–60 μL containing 300–600 μg protein. The combined protein sample was digested using FASP™ protein digestion kit from Protein Discovery Inc. (Knoxville, TN, USA). After the protein was digested, the resultant sample was acidified to pH 3.0 and desalted using a C18 solid-phase extraction column. The protein IDs were identified using Scaffold 4.3.2 from Proteome Software Inc. (Portland, OR, USA), and the pathway was analyzed using Ingenuity Pathway Analysis (IPA) from QIAGEN (Redwood City, CA, USA).
Western blotting analysis
The levels of various cellular proteins were determined using Western blotting assays as described previously.65 Macrophages were washed with PBS after 24-hour treatment with 0.1% DMSO, NP, NP-siRNA-control, and NP-siRNA, lysed with the radioimmunoprecipitation assay buffer (50 mM 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid [HEPES] at pH 7.5, 150 mM NaCl, 10% glycerol, 1.5 mM MgCl2, 1% Triton-X 100, 1 mM ethylenediaminetetraacetic acid [EDTA] at pH 8.0, 10 mM sodium pyrophosphate, and 10 mM sodium fluoride) containing the protease inhibitor and phosphatase inhibitor cocktails. Nuclear–cytoplasmic fractionation was conducted using the NE-PER kit according to the manufacturer’s protocol. Protein concentrations were determined using the bicinchoninic acid assay kit from Thermal Fisher Scientific Inc. An equal amount of protein sample was electrophoresed on 7%–12% SDS polyacrylamide gel electrophoresis (SDS–PAGE) mini-gel after thermal denaturation for 5 minutes at 95°C. Proteins were transferred onto immobilon PVDF membrane at 400 mA for 2 hours at 4°C. Membranes were probed with indicated primary antibody overnight at 4°C and then blotted with secondary antibody. Visualization was performed using the Bio-Rad ChemiDoc™ XRS system (Hercules, CA, USA) with an electrochemiluminescent substrate. The blots were analyzed using Image Lab 3.0 (Bio-Rad Inc.). Protein level was normalized to the matching densitometric value of the internal control β-actin.
Quantitative real-time polymerase chain reaction (RT-PCR) assay
The THP1-derived macrophages were treated with different drugs for 6 hours, and total RNA was extracted by TRIzol® reagent and single-strand cDNA synthesized by a high-capacity cDNA reverse transcription kit (Invitrogen Inc., Carlsbad, CA, USA). The quality of the RNA was examined by determining the 260/280 absorbance ratio using a GeneQuant™ Pro spectrophotometer (Amersham Pharmacia Biotech Inc., Arlington Heights, IL, USA). The reaction was performed in triplicate in a total volume of 10 μL QuantiTest™ SYBR® Green PCR Kit (Qiagen Inc., Valencia, CA, USA), 2 μL cDNA, 2 μL each of the primers, and 6 μL RNase-free water on a 7500 Fast Real-Time PCR System (Applied Biosystems) according to the manufacturer’s instructions. The PCR reaction condition was 95°C for 3 minutes, followed by 40 cycles at 95°C for 15 seconds, and 60°C for 60 seconds. Gene expression levels were analyzed using the comparative Ct method (2−ΔΔCt) with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as the normalizer.
Statistical analysis
All experiments were performed in triplicate, with the results presented from representative experiments. Data are presented as the mean ± standard deviation (SD). Multiple comparisons were analyzed by one- or two-way analysis of variance (ANOVA). Differences between two groups were analyzed using unpaired Student’s t-test. Statistical difference was considered significant at P<0.05. Statistical probability is indicated as follows: *P<0.05; **P<0.01; and ***P<0.001.
Results
Synthesis of the novel NP-siRNA liposomes
SM (Figure 1A) was conjugated to the amino terminals of the PEG segment in DOTAP to obtain the polymerized SM. The SM-PEG was then dialyzed against DMSO and dd-H2O alternatively to remove the free drugs with MW cutoff at 3,000 Da. The physicochemical properties of the synthesized particles were characterized by the UV, FL, and FTIR spectroscopy. UV and FL spectra showed characteristic absorption peaks for the SM conjugation (Figure 1A–C). With regard to the UV spectra, the extreme absorption at 320 nm for SM, it preserved the same extreme at 320 nm after the conjugation (Figure 1A). In the FL spectrum, SM showed a maximum peak at Em of 450 nm when excited at 390 nm, while the conjugated SM displayed a maximum peak at Em of 450 nm as well, which was absent for the un-functionalized polymer (Figure 1B).
Figure 1.

Representative spectra of SM, PEG, SM-PEG, DMSO, and dd-H2O.
Notes: Representative UV and chemical structure of SM (A), FL (B), and FTIR (C) spectra for SM, PEG, SM-PEG, DMSO, and dd-H2O. FTIR shows SM-PEG formed by the −OH of SM connected with -NH2 of PEG. Arrow 1 shows the spectral peaks of PEG, and arrow 2 points to the spectral peaks of SM.
Abbreviations: dd-H2O, distilled deionized water; DMSO, dimethyl sulfoxide; FL, fluorescence; FTIR, Fourier transform infrared spectroscopy; PEG, polyethylene glycol; SM, streptomycin; UV, ultraviolet.
In FTIR spectra of SM-PEG, the successful conjugation was indicated by the −OH groups of the sugar structures of SM (Figure 1C). In addition, the absorption peak at 3,385 cm−1 of SM-PEG showed characteristic PEG residue for the O–H bending vibration, which confirmed a successful conjugation of SM with PEG. Additionally, the peak at 1,538 cm−1 of SM-PEG represented the stretching vibration of C=N of SM.
Finally, the liposomes were formulated through a self-assembly process with DOTAP, cholesterol, chloroform, methanol, and encapsulated anti-TB drugs including INH, RIF, and PZA, and SM-PEG to form cationic PEGylated liposomes. The TGF-β1 siRNA was linked to the cationic PEGylated liposomes via the electrostatic interaction.
Particle size and morphology of the novel liposomal nanoparticles
The morphology and size distribution of the novel NP-siRNA liposomes were examined using SEM (Figure 2A) and TEM (Figure 2B). The morphology of the NP-siRNA liposomes was spherical with double layers encapsulated, compared with the randomly orientated PEG particles (Figure 2C).
Figure 2.

The morphology of newly synthesized NP-siRNA liposomes under SEM and TEM.
Notes: (A) SEM images showing the round shapes of newly synthesized NP-siRNA liposomes. (B) TEM images showing the spherical shapes of the newly synthesized NP-siRNA liposomes. (C) TEM images showing the irregular shapes of PEG. (D) TEM images showing the macrophages without exposure to the NP-siRNA liposomes. (E) TEM images showing the endocytosis of NP-siRNA liposomes by macrophages at 37°C. (F) TEM images showing the endocytosed NP-siRNA liposomes in macrophages at 37°C. (G) TEM images showing lack of endocytosis of NP-siRNA liposomes by macrophages at 4°C. Arrows point to NP-siRNA liposomal nanoparticles.
Abbreviations: NP, nanoparticle; PEG, polyethylene glycol; SEM, scanning electron microscopy; siRNA, small interfering RNA; TEM, transmission electron microscopy.
The internalization of the NP-siRNA liposomes to the macrophages had been examined using TEM. The macrophages were observed without drug exposure under TEM (Figure 2D). The novel NP-siRNA liposomes at 12.5 μg/mL was endocytosed by macrophages at 37°C after drug exposure for 5 minutes (Figure 2E and F), whereas this process could not be duplicated at 4°C (Figure 2G).
Finally, the mean size and zeta potential were measured using a Malvern Zeta Nanosizer ZS 90 (Malvern Instruments). The zeta potential of TGF-β1 siRNA was −12.71 kV, which represented the stability of the liposomal formulation. The mean size of the nanoparticles was 265.1 nm with a PDI of 0.290 and zeta potential of 31.8 kV (Figure 3).
Figure 3.
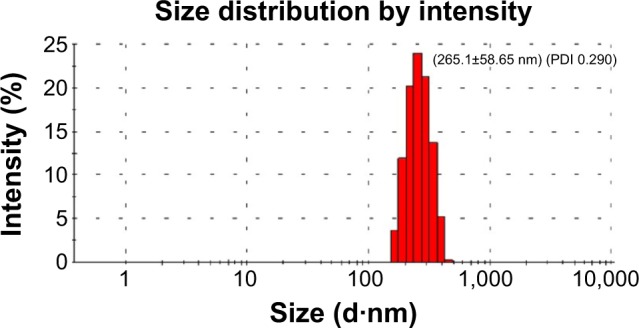
The size distribution of the novel NP-siRNA liposomes.
Notes: The size and the zeta potential of the novel NP-siRNA liposomes were measured using a Malvern Zeta Nanosizer ZS 90. The peak size of the novel NP-siRNA liposomes was 265.1 nm with a PDI of 0.290 and zeta potential was 31.8 mV.
Abbreviations: NP, nanoparticle; PDI, polydispersity index; siRNA, small interfering RNA.
The release profile in vitro of the liposomal nanoparticles
The release profiles of the anti-TB drugs from the NP-siRNA liposomes are shown in Figure 4. The amounts of the anti-TB drugs released from the NP-siRNA liposomes were quantified using validated LC–MS methods. Figure 4A–D show the drug release profiles for the liposomal nanoparticles and free INH, RIF, PZA, and SM over 72 hours. Overall, the encapsulated anti-TB drugs were released from the novel NP-siRNA liposomes in a sustained manner for up to 48 hours (Figure 4A–D). However, the free anti-TB drugs were released in a burst between 8 and 12 hours, and most of the drugs were significantly released after 24 hours (Figure 4A–D).
Figure 4.

Drug release profiles of NP-siRNA liposomes containing the anti-TB drugs and free antidrugs determined by a dialysis method.
Notes: (A) The drug release profile of liposomes and free INH over 72 hours. (B) The drug release profile of liposomes and free RIF over 72 hours. (C) The drug release profile of liposomes and free PZA over 72 hours. (D) The drug release profile of liposomes and free SM over 72 hours. Drug concentrations are measured by validated LC–MS methods. Data are the mean ± SD of three independent experiments. Data were analyzed by two-way ANOVA. Statistical probability is indicated as follows: **P<0.01; ***P<0.001 by two-way ANOVA.
Abbreviations: ANOVA, analysis of variance; INH, isoniazid; LC–MS, liquid chromatography mass spectrometry; NP, nanoparticle; PZA, pyrazinamide; RIF, rifampicin; SD, standard deviation; siRNA, small interfering RNA; SM, streptomycin; TB, tuberculosis.
As shown in Figure 4A, the free INH exhibited a faster release than that of the novel NP-siRNA liposomes with an 8.2- and 3.7-fold change at the time points of 4 and 8 hours, respectively (P<0.001). In contrast, the novel NP-siRNA liposomes showed a sustainable releasing prolife for INH, and the novel NP-siRNA liposomes released 1.5, 2.5, and 4.8 times higher concentrations of INH than that of the free INH at time points of 24, 48, and 72 hours, respectively (P<0.001; Figure 4A). Similarly, free RIF showed a faster release profile than that of the novel NP-siRNA liposomes with a 1.5- and 1.6-fold change at time points of 8 and 12 hours, respectively (P<0.001; Figure 4B). In contrast, the novel NP-siRNA liposomes exhibited 2.4, 4.0, and 5.3 times higher concentrations of RIF than that of the free RIF at the time points of 24, 48, and 72 hours, respectively (P<0.001; Figure 4B). As shown in Figure 4C, the concentration of PZA was 1.5 times higher in the free PZA than that of the novel NP-siRNA liposomes at the time point of 8 hours (P<0.001), whereas the concentrations of PZA were 3.3, 8.5, and 13.7 times higher in the novel NP-siRNA liposomes than that of the free PZA at the time points of 24, 48, and 72 hours, respectively (P<0.001; Figure 4C).
In addition, SM showed a similar release profile as INH, RIF, and PZA in free SM and the novel NP-siRNA liposomes (Figure 4C). The concentrations of SM were 5.6 and 2.6 times higher for free SM than that of the novel NP-siRNA liposomes at the time points of 8 and 12 hours, respectively (P<0.001), whereas the concentrations of SM were 1.4, 5.7, and 15.0 times higher in the novel NP-siRNA liposomes than that of the free SM at the time points of 24, 48, and 72 hours, respectively (P<0.001; Figure 4D).
Cytotoxicity of the novel NP-siRNA liposomal nanoparticles in THP-1-derived macrophages
The cytotoxicity of the NP-siRNA liposomal nanoparticles on human macrophages differentiated from THP-1 cells was examined using the MTT assay. The IC50 value in 24 hours of the novel NP-siRNA liposomes was 269.5 μg/mL (Figure 5A). When the four anti-TB drugs were used in combination, the IC50 value was 241.8 μg/mL, which was lower than individual free anti-TB drugs (Figure 5B). In addition, the IC50 values for INH, RIF, PZA, and SM were 92.55, 132.5, 144.0, and 79.96 μM, respectively (Figure 5C–F). Collectively, the newly synthesized anti-TB liposomes showed a lower cytotoxicity in comparison with the anti-TB drugs in combination.
Figure 5.

Cytotoxicity of the novel NP-siRNA liposomes and free anti-TB drugs toward THP-1-derived macrophages determined by the MTT assay.
Notes: Cells were cultured and treated with NP-siRNA liposomes or the anti-TB drugs for 24 hours. The IC50 values were determined and compared. (A) Survival of THP-1-derived macrophages treated with 2.5–1,000 μg/mL NP-siRNA liposomes for 24 hours. (B) Survival of THP-1-derived macrophages treated with 2.5–200 μg/mL combined anti-TB drugs including INH, RIF, PZA, and SM for 24 hours. (C) Survival of THP-1-derived macrophages treated with 0.1–100 μM INH for 24 hours. (D) Survival of THP-1-derived macrophages treated with 1–100 μM RIF for 24 hours. (E) Survival of THP-1-derived macrophages treated with 1–100 μM PZA for 24 hours. (F) Survival of THP-1-derived macrophages treated with 0.1–100 μM SM for 24 hours. Data are the mean ± SD of three independent experiments.
Abbreviations: INH, isoniazid; MTT, thiazolyl blue tetrazolium bromide; NP, nanoparticle; PZA, pyrazinamide; RIF, rifampicin; SD, standard deviation; siRNA, small interfering RNA; SM, streptomycin; TB, tuberculosis.
Effects of the NP-siRNA liposomal nanoparticles on cell cycle distribution, apoptosis, and autophagy in THP-1-derived macrophages
The effects of the novel NP-siRNA liposomal nanoparticles on cell cycle distribution in THP-1-derived macrophages were examined using flow cytometry. The results showed there was no significant difference in the percentage of cell populations in G1, S, and G2/M phases when the macrophages were treated with the novel NP-siRNA liposomes at concentrations of 2.5, 12.5, and 62.5 μg/mL (Figure 6A and B). In order to measure the apoptosis-inducing effect of the novel NP-siRNA liposomal nanoparticles in THP-1-derived macrophages, the number of apoptotic macrophages was quantified using flow cytometry. As shown in Figure 6C and D, no significant apoptosis-inducing effect of NP-siRNA liposomes was observed in human macrophages.
Figure 6.


Effects of the novel NP-siRNA liposomes on cell cycle distribution, apoptosis, and autophagy in THP-1-derived macrophages.
Notes: Cells were treated with the novel NP-siRNA liposomes at 2.5, 12.5, and 62.5 μg/mL for 24 hours, and the effects on cell cycle distribution, apoptosis, and autophagy in macrophages were examined using flow cytometry and confocal microscopy. (A) Representative DNA fluorescence histograms show the effect of treatment with NP-siRNA liposomes on cell cycle distribution. (B) Bar graphs show the percentage of macrophages in G1, S, and G2/M phases. (C) Representative flow cytometric dot plots show the distribution of macrophages undergoing early or late apoptosis when treated with the NP-siRNA liposomes at 2.5, 12.5, and 62.5 μg/mL for 24 hours. (D) Bar graphs show the percentage of macrophages undergoing early or late apoptosis when treated with the NP-siRNA liposomes at 2.5, 12.5, and 62.5 μg/mL for 24 hours. (E) Representative flow cytometric dot plots show the autophagic cells when treated with the NP-siRNA liposomes at 2.5, 12.5, and 62.5 μg/mL for 24 hours. (F) Bar graphs show the percentage of autophagic macrophages when the cells were treated with the NP-siRNA liposomes at 2.5, 12.5, and 62.5 μg/mL for 24 hours. (G) The confocal microscopic images show the Cyto-ID® stained autophagic cells with green fluorescence when the cells were treated with the NP-siRNA liposomes at 2.5, 12.5, and 62.5 μg/mL for 24 hours. (H) Bar graphs show the fluorescence level, reflecting the level of autophagic macrophages when the cells were treated with the NP-siRNA liposomes at 2.5, 12.5, and 62.5 μg/mL for 24 hours. Data are present as the mean ± SD of three independent experiments. ***P<0.001 by one-way ANOVA followed by Tukey’s post hoc test.
Abbreviations: ANOVA, analysis of variance; NP, nanoparticle; SD, standard deviation; siRNA, small interfering RNA.
We also examined the effect of the novel NP-siRNA liposomes on autophagy in THP-1-derived macrophages using flow cytometry (Figure 6E and F) and confocal fluorescence microscopy (Figure 6G and H). Notably, treatment of macrophages with 62.5 μg/mL NP-siRNA liposomes for 24 hours remarkably increased the percentage of autophagic cells 7.5-fold (P<0.001; Figure 6F), which was consistent with confocal microscopic results when compared with the control cells (2.5-fold; P<0.001; Figure 6H).
Next, we investigated the underlying mechanisms for the autophagy-inducing effect of the novel NP-siRNA liposomes in macrophages. First, we examined the phosphorylation levels of PI3K at Tyr199, and p38 MAPK at Thr180/Tyr182 that are upstream signaling molecules of Akt/mTOR pathway and play important roles in the regulation of cell proliferation and death (Figure 7A). In this study, exposure of macrophages to 2.5, 12.5, and 62.5 μg/mL NP-siRNA liposomes for 24 hours did not significantly affect the phosphorylation level of p-PI3K at Tyr 199 (Figure 7B). However, treatment of macrophages with 62.5 μg/mL NP-siRNA liposomes for 24 hours reduced the phosphorylation level p38 MAPK at Thr180/Tyr182. p38 MAPK has bifurcated roles in the regulation of cell fate: it can either mediate cell survival or cell death, depending not only on the type of stimulus but also in a cell type specific manner.71,72 In comparison with the control cells, the phosphorylation level of p38 MAPK and the ratio of p-p38 MAPK/p38 MAPK were decreased 15.8% when cells were treated with NP-siRNA liposomes at 62.5 μg/mL (P<0.05; Figure 7A and B). mTOR plays a key role in cell growth, autophagic cell death, and homeostasis. Exposure of cells to 62.5 μg/mL NP-siRNA liposomes resulted in a 38.3% decrease in the ratio of p-mTOR/mTOR at Ser2448 (Figure 7A).
Figure 7.

Effects of the novel NP-siRNA liposomes on the expression levels of pro- and antiautophagic proteins in THP-1-derived macrophages determined by Western blotting assay.
Notes: (A) Representive blots of p-PI3K, p-mTOR, mTOR, p-p38 MAPK, p38 MAPK, PTEN, beclin 1, and LC3 in macrophages treated with the NP-siRNA liposomes at 2.5, 12.5, and 62.5 μg/mL for 24 hours. (B) Bar graphs show the effect of NP-siRNA liposomes on the levels of p-PI3K, p-mTOR, mTOR, p-p38 MAPK, p38 MAPK, PTEN, beclin 1, and LC3 in macrophages. β-actin was used as the internal control. Data are the mean ± SD of three independent experiments. *P<0.05 by one-way ANOVA followed by Tukey’s post hoc test.
Abbreviations: ANOVA, analysis of variance; LC3, microtube-associated protein 1A/1B light chain 3; MAPK, mitogen-activated protein kinase; mTOR, mammalian target of rapamycin; NP, nanoparticle; PI3K, phosphoinositide 3-kinase; PTEN, phosphatase and tensin homologue; SD, standard deviation; siRNA, small interfering RNA.
PTEN, a dual-specificity phosphatase and tumor suppressor gene, inhibits Akt/mTOR and MAPK signaling pathways, leading to cell death and growth regulation.73 Autophagy is tightly regulated by beclin 1 (a mammalian homologue of yeast Atg6) that forms a complex with vacuolar sorting protein 34, and serves as a platform for recruitment of other Atgs that are critical for autophagosome formation.74 LC3, a mammalian homologue of yeast Atg8, is associated with the autophagosome membranes after processing and has been used as a specific marker monitor autophagy.75,76 In comparison with the control cells, the expression levels of PTEN, beclin 1, and LC3 increased 1.3-, 1.5-, and 1.6-fold when treated with 62.5 μg/mL NP-siRNA liposomes, respectively (P<0.05; Figure 7A and B), whereas there was no significant alteration in the expression levels of PTEN, beclin 1, and LC3 in macrophages with the treatment of 2.5 and 12.5 μg/mL novel NP-siRNA liposomes for 24 hours (Figure 7A and B).
Effects of the novel NP-siRNA liposomal nanoparticles on the expression levels of TGF-β1, IL-6, IL-8, TNF-α, and IFN-γ in THP-1-derived macrophages
TGF-β1, IL-6, IL-8, TNF-α, and IFN-γ can be secreted by macrophages. We examined whether the novel NP-siRNA liposomes were able to decrease the expression levels of TGF-β1, IL-6, IL-8, TNF-α, and IFN-γ. The ELISA results showed the expression level of TGF-β1 in supernatants obtained from macrophages culture medium was decreased in macrophages treated with the novel NP-siRNA liposomes at 12.5 μg/mL for 24 hours, whereas the level of TFG-β1 was not significantly altered in control, DMSO, NP, NP–siRNA-control groups (Figure 8). LPS treatment increased the level of TGF-β1. Moreover, the expression levels of IL-6, IL-8, TNF-α, and IFN-γ (Figure 8) in NP-siRNA treated cells were elevated compared with blank control.
Figure 8.

Effects of the novel NP-siRNA liposomes on the expression levels of TGF-β1, IL-6, IL-8, TNF-α, and IFN-γ in THP-1-derived macrophages determined by ELISA.
Notes: Macrophages were cultured and treated with 12.5 μg/mL NP-siRNA liposomes, DMSO, NP-siRNA-control or medium for 24 hours. The levels of cytokines were determined using ELISA. LPS was included as the positive control. Data are the mean ± SD of three independent experiments. *P<0.05; **P<0.01; and ***P<0.001 by one-way ANOVA followed by Tukey’s post hoc test.
Abbreviations: ANOVA, analysis of variance; DMSO, dimethyl sulfoxide; IFN, interferon; IL, interleukin; ELISA, enzyme-linked immunosorbent assay; LPS, liposaccharide; NP, nanoparticle; SD, standard deviation; siRNA, small interfering RNA; TGF-β1, transforming growth factor-β1; TNF-α, tumor necrosis factor-α.
Effects of the novel NP-siRNA liposomes on the mRNA levels of TGF-β1, IL-1, IL-6, IL-8, TNF-α, and IFN-γ in THP-1-derived macrophages
Furthermore, the mRNA expression levels of key cytokines were assessed in THP-1-derived macrophages treated with NP-siRNA liposomes by RT-PCR. The expression level of TGF-β1 mRNA was markedly decreased in the macrophages after treatment with NP-siRNA compared with the controls (Figure 9). In contrast, TNF-α and IFN-γ mRNA expression levels were increased in macrophages (Figure 9), whereas there was no significant difference in the expression levels of IL-1, IL-6, and IL-8 mRNA (Figure 9).
Figure 9.

Effects of the novel NP-siRNA liposomes on the mRNA expression levels of TGF-β1, IL-6, IL-8, TNF-α, and IFN-γ in THP-1-derived macrophages determined by RT-PCR assay.
Notes: THP-1-derived macrophages were cultured and treated with 12.5 μg/mL NP-siRNA liposomes, DMSO, NP-siRNA-control or medium for 6 hours. The mRNA levels of cytokines were determined using RT-PCR assay. Data are the mean ± SD of three independent experiments. *P<0.05 and **P<0.01 by one-way ANOVA followed by Tukey’s post hoc test.
Abbreviations: ANOVA, analysis of variance; DMSO, dimethyl sulfoxide; IFN, interferon; IL, interleukin; NP, nanoparticle; RT-PCR, real-time polymerase chain reaction; SD, standard deviation; siRNA, small interfering RNA; TGF-β1, transforming growth factor-β1; TNF-α, tumor necrosis factor-α.
Proteomic responses to NP-siRNA liposome treatment in THP-1-derived macrophages
In order to reveal the proteomic responses to NP-siRNA liposomes in THP-1-derived macrophages, a SILAC-based quantitative proteomic study was conducted to determine the molecular targets of NP-siRNA liposomes. Our study showed that there were numerous molecules and signaling pathways that responded to NP-siRNA liposome treatment (Figure 10A and B, Tables 1–3).
Figure 10.

The proteomic profiling of human macrophages treated with the novel NP-siRNA liposomes at 12.5 μg/mL for 24 hours determined using SILAC.
Notes: The proteomic responses of macrophages to NP-siRNA were examined using SILAC, and the pathways were analzyed by IPA. This figure shows the top pathways from the proteomic study with the threshold of 1.2 of −log(P-value) (A and B).
Abbreviations: IPA, ingenuity pathway analysis; NP, nanoparticle; SILAC, stable isotope labeling with amino acids in cell culture; siRNA, small interfering RNA.
Table 1.
The top five canonical signaling pathways regulated by the novel NP-siRNA liposomes in THP-1-derived in macrophages
| Pathway | P-value | Ratio |
|---|---|---|
| EIF2 signaling | 2.93×10−9 | 9/201 (0.045) |
| Actin cytoskeleton signaling | 1.90×10−7 | 8/242 (0.033) |
| Remodeling of epithelial adherens junctions | 1.61×10−6 | 5/70 (0.071) |
| Epithelial adherens junction signaling | 4.19×10−6 | 6/154 (0.039) |
| RhoGDI signaling | 1.15×10−5 | 6/202 (0.030) |
Note: Data in the parenthesis shows the ratio of regulated proteins over the total proteins in the targeted signaling pathway.
Abbreviations: EIF2, eukaryotic initiation factor 2; RhoGDI, Rho GDP-dissociation inhibitor.
Table 2.
The top eleven upstream proteins regulated by the novel NP-siRNA liposomes in THP-1-derived macrophages
| Upstream regulator | P-value of overlap | Predicted activation state |
|---|---|---|
| APP | 5.87×10−17 | |
| CD3 | 4.90×10−3 | Inhibited |
| CD437 | 1.50×10−5 | Inhibited |
| HSF1 | 3.89×10−6 | Inhibited |
| MAPT | 1.90×10−18 | |
| MYC | 2.30×10−12 | Activated |
| MYCN | 1.16×10−12 | Activated |
| PSEN1 | 1.40×10−15 | |
| Sirolimus | 3.77×10−11 | Inhibited |
| TGF-β1 | 4.82×10−5 | |
| TNF-α | 1.27×10−4 |
Abbreviations: APP, β-amyloid (A4) precursor protein; CD3, T-cell surface glyco-protein; CD437, a cell-permeable, selective agonist of retinoic acid receptor; HSF1, heat shock transcription factor 1; MAPT, microtubule-associated protein tau; MYC, v-myc avian myelocytomatosis viral oncogene homologue; MYCN, v-myc avian myelocytomatosis viral oncogene neuroblastoma derived homologue; PSEN1, presenilin 1; TGF-β1, transforming growth factor-β1; TNF-α, tumor necrosis factor-α.
Table 3.
The upregulated and downregulated proteins involved in TGF-β1-mediated signaling pathway in THP-1-derived macrophages treated with the novel NP-siRNA liposomes at 12.5 μg/mL for 24 hours
| ID | Gene in dataset | Prediction (based on expression direction) | Fold changea | Findingsb |
|---|---|---|---|---|
| P68104 | EEF1A1 | Affected | 1.676 | Regulated (1) |
| Q13509 | TUBB3 | Activated | 1.638 | Upregulated (3) |
| B7Z6Z4 | MYL6 | Activated | 1.465 | Upregulated (1) |
| Q01469 | FABP5 | Activated | 1.402 | Upregulated (1) |
| P06753 | TPM3 | Activated | 1.392 | Upregulated (9) |
| P28482 | MAPK1 | Activated | 1.310 | Upregulated (4) |
| P00558 | PGK1 | Inhibited | 1.262 | Downregulated (1) |
| P16070 | CD44 | Inhibited | 1.261 | Downregulated (3) |
| E9PK25 | CFL1 | Activated | 1.155 | Upregulated (1) |
| P06396-2 | GSN | Inhibited | 1.142 | Downregulated (2) |
| Q01518 | CAP1 | Activated | 1.072 | Upregulated (2) |
| P50552 | VASP | Activated | 1.031 | Upregulated (2) |
| P26038 | MSN | Activated | 1.012 | Upregulated (1) |
| P07900-2 | HSP90AA1 | Activated | 1.005 | Upregulated (1) |
Notes:
The value indicates the ratio of heavy over light signal.
The number in the brackets indicates the number of proteins involved.
Abbreviations: CAP1, CAP, adenylate cyclase-associated protein 1 (yeast); CD44, CD44 molecule (Indian blood group); CFL1, cofilin 1 (nonmuscle); EEF1A1, eukaryotic translation elongation factor 1 α1; FABP5, fatty acid binding protein 5 (psoriasis-associated); GSN, gelsolin; HSP90AA1, heat shock protein 90 kDa α (cytosolic), class A member 1; MAPK1, mitogen-activated protein kinase 1; MSN, moesin; MYL6, myosin, light chain 6, alkali, smooth muscle and nonmuscle; PGK1, phosphoglycerate kinase 1; TPM3, tropomyosin 3; TUBB3, tubulin, β3 class III; VASP, vasodilator-stimulated phosphoprotein.
The IPA data showed that there were over 40 signaling pathways involved in the proteomic responses to NP-siRNA liposome exposure in human macrophages, with 160 proteins mapped (Figure 10A and B). The top five canonical signaling pathways were eukaryotic initiation factor 2 (EIF2) signaling (Figure 11), actin cytoskeleton signaling, remodeling of epithelial adherens junctions, epithelial adherens junction signaling, and Rho GDP-dissociation inhibitor (RhoGDI) signaling pathways (Table 1). Of note, treatment of macrophages with NP-siRNA liposomes regulated programmed cell death pathways including apoptosis and PI3K/Akt/mTOR signaling pathways (Figure 10A and B). We further studied the EIF2 signaling pathway and found that the expression level of ERK1/2, the key protein involved in the EIF2 signaling pathway, was increased (Figure 11). Futhermore, the IPA data presented that ERK1/2 played a pivotal role in the ERK/MAPK signaling pathway. Treatment of macrophages with the novel NP-siRNA liposome increased the expression level of ERK1/2, which in turn enhanced a number of gene transcriptions in the nucleus (Figure 12). Of note, the IPA results showed that NF-κB, PI3K, and heat shock protein 90 (HSP90) were involved in the ERK1/2 signaling pathway in response to the novel NP-siRNA liposome treatment, which may contribute to the immune response, control of viral gene expression, antiapoptosis, antiviral response, and antimicrobial response (Figure 13) and cell growth (Figure 14).
Figure 11.

EIF2 signaling pathway was regulated when THP-1-derived macrophages were treated with the novel NP-siRNA liposomes at 12.5 μg/mL for 24 hours.
Notes: The proteomic response was determined by SILAC, and the pathways were analyzed using IPA. The figure shows that ERK1/2 was upregulated in the EIF2 signaling pathway. Red indicates an upregulation; green indicates a downregulation. The intensity of green and red colors indicates the degree of down- or upregulation. Solid arrow indicates direct interaction, and dashed arrow indicates indirect interaction.
Abbreviations: EIF2, eukaryotic initiation factor 2; ER, endoplasmic reticulum; ERK, extracellular signal-regulated kinase; IPA, ingenuity pathway analysis; NP, nanoparticle; SILAC, stable isotope labeling with amino acids in cell culture; siRNA, small interfering RNA.
Figure 12.

The ERK/MAPK signaling pathway was regulated when THP-1-derived macrophages were treated with the novel NP-siRNA liposomes at 12.5 μg/mL for 24 hours.
Notes: The proteomic response was determined by SILAC, and pathways were analyzed using IPA. Red indicates an upregulation; green indicates a downregulation. The intensity of green and red colors indicates the degree of down- or upregulation. Solid arrow indicates direct interaction, and dashed arrow indicates indirect interaction.
Abbreviations: ERK, extracellular signal-regulated kinase; IPA, ingenuity pathway analysis; MAPK, mitogen-activated protein kinase; NP, nanoparticle; SILAC, stable isotope labeling with amino acids in cell culture; siRNA, small interfering RNA.
Figure 13.

The NF-κB signaling pathway was regulated via ERK1/2 when THP-1-derived macrophages were treated with the novel NP-siRNA liposomes at 12.5 μg/mL for 24 hours.
Notes: The proteomic response was determined by SILAC, and pathways were analyzed using IPA. Red indicates an upregulation; green indicates a downregulation. The intensity of green and red colors indicates the degree of down- or upregulation. Solid arrow indicates direct interaction.
Abbreviations: ERK, extracellular signal-regulated kinase; IPA, ingenuity pathway analysis; NF-κB, nuclear factor-κB; NP, nanoparticle; SILAC, stable isotope labeling with amino acids in cell culture; siRNA, small interfering RNA.
Figure 14.

The PI3K/AKT and HSP90 interacted with ERK1/2 when THP-1-derived macrophages were treated with the novel NP-siRNA liposomes at 12.5 μg/mL for 24 hours.
Notes: The proteomic response was determined by SILAC, and pathways were analyzed using IPA. Red indicates an upregulation; green indicates a downregulation. The intensity of green and red colors indicates the degree of down- or upregulation. Solid arrow indicates direct interaction, and dashed arrow indicates indirect interaction.
Abbreviations: AKT, protein kinase B; ERK, extracellular signal-regulated kinase; HSP90, heat shock protein 90; IPA, ingenuity pathway analysis; NF-κB, nuclear factor-κB; NP, nanoparticle; PI3K, phosphoinositide 3-kinase; SILAC, stable isotope labeling with amino acids in cell culture; siRNA, small interfering RNA.
ERK1/2 has a crucial role in the regulation of cellular processes, including cell cycle progression, cell proliferation and differentiation, cytokine generation, gene transcription, cell migration and invasion, senescence, cell death, GAP junction formation, actin and microtubule networks, and neurite extension.77 Aberrations in ERK1/2 signaling have been implicated in many pathologies including many cancers, diabetes, and infectious and cardiovascular diseases.78 Thus, we analyzed the ERK signaling pathways, and the upstream regulators are given in Table 2, in which TGF-β1 was involved. Then, we further detected the upregulated and downregulated genes involved in the TGF-β1-mediated signaling pathways (Table 3).
The subsequent validation experiments showed that the phosphorylation level of ERK1/2 was increased 1.2- and 1.3-fold in human macrophages with the treatment of NP-siRNA-control and NP-siRNA liposomes at 12.5 μg/mL for 24 hours, respectively (P<0.05 or 0.01; Figure 15A and B). However, there was no significant alteration in the expression level of ERK1/2 in control cells (Figure 15A and B). There was a 49.2% decrease in the expression level of NF-κB in the macrophages when treated with the novel NP-siRNA liposomes at 12.5 μg/mL for 24 hours, whereas there was no marked change in the expression level of NF-κB in macrophages with the treatment of NP and NP-siRNA-control (P<0.01; Figure 15A and B). Of note, in comparison with the NP treatment group, NP-siRNA liposomes decreased 55.9% in the expression level of NF-κB (P<0.001; Figure 15A and B). In contrast, the level of Nrf2 was remarkably increased 1.4- and 1.6-fold in human macrophages with the treatment of NP-siRNA-control and NP-siRNA liposomes at 12.5 μg/mL for 24 hours, respectively (P<0.05; Figure 15A and B). Taken together, NF-κB plays a critical role in the siRNA-mediated therapy targeting the TGF-β1 inflammatory signaling pathway in the treatment of TB.
Figure 15.

Effect of the novel NP-siRNA liposomes on the expression levels of p-ERK, ERK, NF-κB, and Nrf2 in THP-1-derived macrophages determined using Western blotting analysis.
Notes: (A) Representative blots for p-ERK, ERK, NF-κB, and Nrf2 in macrophages when treated with DMSO, NP, NP-siRNA-control, and NP-siRNA at 12.5 μg/mL for 24 hours. (B) Bar graphs show the relative expression levels of the proteins in macrophages. (C) Representative blot of NF-κB when macrophages were treated with the novel NP-siRNA liposomes at 2.5, 12.5, and 62.5 μg/mL for 24 hours. (D) Bar graphs show the relative expression level of NF-κB in macrophages. The β-actin was used as the internal control. Data are the mean ± SD of three in dependent experiments. *P<0.05; **P<0.01; and ***P<0.001 by one-way ANOVA followed by Tukey’s post hoc test.
Abbreviations: ANOVA, analysis of variance; DMSO, dimethyl sulfoxide; ERK, extracellular signal-regulated kinase; NF-κB, nuclear factor-κB; NP, nanoparticle; Nrf2, nuclear factor (erythroid-derived 2)-like 2; SD, standard deviation; siRNA, small interfering RNA.
Effects of the NP-siRNA liposomes on NF-κB production in THP-1-derived macrophages
The interplay between TGF-β1 and NF-κB is involved in the inflammatory response to M. tuberculosis infection; TGF-β1 is involved in the TB growth by activating the NF-κB, which in turn reactivates TGF-β1 and other cytokine pathways in macrophages.42–44,51,58 Thus, we examined the effect of NP-siRNA liposomes on the expression of NF-κB in THP-1 -derived macrophages. As shown in Figure 15C and D, there was a remarkable decrease in the expression level of NF-κB when treated with NP-siRNA liposomes in THP1-derived macrophages. There was a reduction of 18.0%, 15.2%, and 44.0% in the expression levels of NF-κB in macrophages treated with NP-siRNA liposomes at 2.5, 12.5, and 62.5 μg/mL, respectively (P<0.05 or 0.001; Figure 15D). Moreover, in comparison with cells treated with DMSO, NP, or NP-siRNA-control, the NP-siRNA liposomes exhibited a potent inhibitory effect on the expression of NF-κB, and the 62.5 μg/Ml NP-siRNA liposomes exerted a maximum suppressing effect on NF-κB expression in macrophages (Figure 15A and B). Collectively, the results show that NP-siRNA liposomes suppress the expression of NF-κB, probably contributing to the attenuated inflammatory response to M. tuberculosis infection.
Discussion
The majority of infected individuals develop asymptomatic latent TB, while ~5%–10% of latently infected individuals will progress to active pulmonary TB.79,80 Due to the poor compliance, drug resistance, and potential side effects of the current chemotherapy for TB treatment, TB remains the second leading cause of mortality worldwide due to a single infectious agent, resulting in a substantial burden on individuals and the healthcare system. Therefore, it requires new approaches and therapies to achieve a better therapeutic effect and minimal side effects for the treatment of TB. With the development of new therapeutic drugs and/or novel drug delivery systems for TB treatment, it is expected to improve therapeutic effect and reduce side effects in clinical practice. In this study, we have synthesized and characterized the multifunctional PEGylated liposomes that encapsulated four anti-TB drugs including SM, INH, RIF, and PZA and linked to TGF-β1 siRNA. The newly synthesized NP-siRNA liposomes exhibit favorable physicochemical and biological properties in THP-1 differentiated macrophages. Our results show that human macrophages can engulf the novel NP-siRNA liposomes. Of importance, our SILAC-based quantitative proteomic study has revealed specific proteomic responses to the treatment of the novel NP-siRNA liposomes in human macrophages, and the initial validation experiments verify the involvement of the ERK1/2/NF-κB signaling pathways in response to the exposure of human macrophages to the novel NP-siRNA liposomes.
As stated above, we have synthesized novel liposomal nanoparticles to encapsulate the three first-line anti-TB drugs, including INH, RIF, and PZA, and one second-line anti-TB drug, SM. These drugs are commonly used in clinical practice.5,81 SM was conjugated to PEG, and then encapsulated INH, RIF, and PZA and was formulated to be an orally administered complex. The N-terminal covalent bonding between SM and PEG facilitates the stabilization of the complex and has a substantial influence on the pharmacological activities of the complex. In comparison with the free drugs, the conjugated complex exhibits a low cytotoxic effect on human macrophages. Therefore, it will be clinically helpful to minimize the side effects. Furthermore, the newly synthesized liposomes can improve patient compliance and overcome the limitations of current anti-TB therapy.
Given the physicochemical properties of the novel NP-siRNA liposomes, the results have demonstrated that the liposomes can be used to encapsulate the anti-TB drugs. The novel NP-siRNA liposomes are facile to be prepared, stabilized, and controlled in a reasonable size. These physicochemical properties facilitate its internalization through the endocytosis by human macrophages. Moreover, the physicochemical properties determine the pharmacological and toxicological features of the novel NP-siRNA liposomes, such as absorption, distribution, metabolism, excretion, and toxicity. The favorable physicochemical properties of the newly synthesized NP-siRNA liposomes ensure their pharmacological effect in the treatment of TB. Indeed, this study showed that the novel NP-siRNA liposomes exhibit a slow drug release profile within the initial 12 hours compared with the free drugs. Notably, the novel NP-siRNA liposomes showed a sustained drug release profile between 12 and 72 hours, which has great clinical importance in terms of enhancing the therapeutic effect and minimizing the side effects in the treatment of TB. Our biochemical assays demonstrated that the novel NP-siRNA liposomes were safe and nontoxic to the host cells. The IC50 of the novel NP-siRNA liposomes was 269.5 μg/mL, which is lower than that of the combined four anti-TB drugs together. The novel NP-siRNA liposomes did not exert a significant impact on cell cycle distribution, apoptosis, and autophagy of human macrophages derived from THP-1 cells. There was an autophagy-inducing effect of NP-siRNA liposomes on human macrophages only at a high concentration of 62.5 μg/mL, but such high concentrations are not achieved in clinical practice. Collectively, the novel NP-siRNA liposomes exhibit favorable physicochemical and biochemical properties that make it a promising therapy for TB treatment.
Combination chemotherapy is a basic principle to cure TB patients. Multiple drug chemotherapy is effective in curing TB, but with challenges and limitations. The major hurdles compromising the therapeutic outcome are poor compliance, drug resistance, and potential side effects. Current anti-TB drugs mainly interfere with the nucleotide metabolism and/or the building of new bacterial walls; the treatment period of current anti-TB therapy is long (even the most potent medications have to be taken at least 6 months);82 and the anti-TB drugs development is slow. Therefore, increasing evidence shows that future studies need to focus on new therapeutic strategies to overcome the shortcomings of the current TB therapies, including polymeric drug delivery systems, vector construction, and genetic engineering methods. Anti-TB drugs formulated with nanoparticles are widely studied, such as polymer micelles, liposomes, nanospheres, nanotubes, nanobeads, etc, and there are some achievements in the treatment of TB in clinical settings.83 However, these nanomaterials combine only one or two kinds of commonly used anti-TB drugs, which limits their clinical application in the treatment of newly diagnosed TB and MDR-TB. In this study, the data showed that the newly synthesized NP-siRNA liposomes encapsulated four anti-TB drugs and exhibited good physicochemical and biochemical properties toward human macrophages differentiated from THP-1 cells, which ensures its favorable pharmacological and toxicological profiles in the treatment of TB.
Although the ultrashort chemotherapy regimen to treat spinal TB has been applied in clinic,5,84 it has been noted that some patients experienced difficulty in adherence to the treatment in view of the long period involved. The poor patient compliance for anti-TB drugs often results in TB relapse, persisting disease, and the development of multi-drug resistance. Therefore, novel drug delivery systems are incorporated to improve the existing therapy in terms of better efficacy, reduced drug toxicity, administration frequency, and shortened period of treatment.85 Encapsulating anti-TBs within nanoparticles can greatly enhance the bioavailability of the drugs, reduce the frequency of drug administration, and thereby achieve good compliance. As shown in this study, the novel NP-siRNA liposomes exert a sustainable drug release profile over 72 hours, indicating that the novel NP-siRNA liposomes may have great potential in reducing the frequency of drug administration and the drug treatment period.
While the therapeutic effect of anti-TB drugs can be improved with nanotechnology, it is important and indispensable to exploit host immune functions for the recognition of and protection against TB, especially in immunocompromised hosts and HIV-infected patients.82,86 In 10% of those infected patients, TB will erupt into full-blown disease in response to various stresses if the immune system is compromised.87 It has been well recognized that anti-TB progress happens mainly in macrophages that reflect the equilibrium between the host and pathogen. This leads to the attenuation of several cellular processes that include fusion of phagosomes with lysosomes, antigen presentation, apoptosis, and the bactericidal responses initiated by the macrophages. The process is mediated mainly by T-cells with the involvement of inflammatory responses, including inflammatory cytokines generation, such as TGF-β1; TNF-α; IFN-γ; and IL-1, 6, 8, and 10.56,58,59
After entry into the lungs, M. tuberculosis interacts with airway epithelial cells, alveolar Type II pneumocytes, alveolar macrophage, dendritic cells, and neutrophils.48,53,54,88 Once in contact with M. tuberculosis, both alveolar Type II pneumocytes and airway epithelial cells may contribute to early host immune defense by generating various types of cytokines, chemokines, and other molecules including TNF-α, IL-1α, IL-1β, IFN-γ, and chemoattractant molecules that may directly kill M. tuberculosis or enhance anti-TB activities of infected macrophages.53 In response to M. tuberculosis infection, chemokine (C-X-C motif) ligand 8 (CXCL8) production by pulmonary epithelium and lung fibroblasts rapidly promotes the recruitment of neutrophils. In addition, infected dendritic cells migrating to local lymph nodes initiate an adaptive immune response, required for the arrest of growth of the bacterium and its entry into a latent phase. Other important players of the host response directed against M. tuberculosis infection are T lymphocytes and B cells.48,49,88 Notably, the primary pathologic feature of M. tuberculosis infection is the formation of granulomas.88 The formation of an early granuloma is mainly regulated by TNF-α, and IFN-γ released from the infected cells to contain bacterial spread.54 At this early stage, the structure is composed of a central core of infected macrophages surrounded by other macrophages differentiated in giant cells together with infected neutrophils.88 Accessory not-infected macrophages surround this structure powered by blood vessels, which bring on the site of infection several cell types equipped to contain bacterial spread. At a later stage, the granuloma is composed primarily of CD4+ and CD8+ T-cells, but other T lymphocytes, including γδ T-cells, natural killers, and CD1-restricted αβ T-cells, are also present.49,53 TGF-β1 plays a central role in the formation of TB granulomas, and TGF-β1 overexpression can cause tissue damage and fibrosis in TB patients.89,90 Excessive production of tuberculous granulomas compromises the therapeutic effect of anti-TB drugs. Consequently, M. tuberculosis can evade the host immune system and remain in the dormant stage for a long time, and TB can be reactivated to be a virulent form under the immunocompromised condition.91 Macrophage–pathogen interactions play a central role in TB pathogenesis. Owing to the key role of TGF-β1 in the pathogenesis of TB, it will be feasible to control TB infection by suppressing the expression of TGF-β1 with a combination of anti-TB drugs.
siRNA can efficiently and specifically silence the expression of homologous genes at the cellular level, promoting homologous mRNA degradation and inducing specific gene deletion phenotype.92,93 Cationic liposomes are the most studied nonviral delivery system used for nucleic acid delivery owing to the simplicity of the electrostatic interaction between cationic liposomes and negatively charged nucleic acids to form the vector complex. It has been used successfully for the intracellular delivery of siRNA. Therefore, siRNA with a negative charge is combined with positively charged liposomes to form stable nanoparticles. The employment of the TGF-β1 siRNA-mediated gene silencing approach can reduce the expression of TGF-β1, thereby decreasing its translational products. The reduction of TGF-β1 leads to a decrease in the tuberculous granuloma formation.94 After M. tuberculosis infection, proinflammatory cytokines, including TNF-α, IL-1, IL-6, and IL-18, and anti-inflammatory (IL-10) cytokines are secreted rapidly at high and sustained levels preferentially by M. tuberculosis-infected macrophages in the infected lung, while dendritic cells produce low or undetectable levels of these cytokines.53 Our data showed that the novel NP-siRNA liposomes were endocytosed by human macrophages and that the secretion of TGF-β1 was significantly reduced, whereas the ELISA results showed that the novel NP-siRNA liposomes significantly increased the generation of IL-6, IL-8, TNF-α, and IFN-γ in THP-1-derived macrophages. Moreover, the gene expression level of TNF-α and IFN-γ was increased, but there was no significant alteration in the gene expression level of IL-1, 6, and 8. Notably, it has been recently reported that IL-1 exerts a beneficial effect on the control of TB via the positive regulation of the eicosanoid network.59 In this study, the diverse regulatory effects of the novel NP-siRNA liposomes on the generation of cytokines may enhance the therapeutic effect in the treatment of TB. However, the carriers of the novel NP-siRNA complex may contribute to the increase in the expression of the proinflammatory cytokines, although the encapsulated anti-TB drugs may play a role. Thus, these regulatory effects of the novel NP-siRNA liposomes on the inflammatory response in human macrophages need to be further elucidated. Collectively, the novel NP-siRNA liposomes exhibit a potent modulating effect on the cytokine production, which may have substantial contributions to the treatment of TB in clinical practice.
Furthermore, we explored the molecular targets and the underlying mechanisms of the therapeutic effect of the novel NP-siRNA liposomes in human macrophages using the SILAC-based quantitative proteomic approach. The SILAC-based quantitative proteomic approach can reveal a global and comprehensive molecular interactome including the therapeutic and toxicological targets, and discriminate the differential responses to drug exposure.69,70,95,96 In this study, the proteomic results showed that inflammatory response associated signaling pathways were profoundly regulated by the novel NP-siRNA liposome treatment in human macrophages. In particular, ERK1/2 and NF-κB were closely orchestrated in response to the exposure of human macrophages to the novel NP-siRNA liposomes. Subsequently, our initial validation results demonstrated that the novel NP-siRNA enhanced the phosphorylation of ERK1/2 while suppressing the expression of NF-κB in THP-1-differentiated macrophages, indicating that the newly synthesized NP-siRNA liposomes regulated ERK1/2 and NF-κB signaling pathways, contributing to the diverse regulatory effects on the expression of TNF-α, IFN-γ, IL-1, 6, and 8. Indeed, extensive studies show that ERK1/2/NF-κB play a critical role in a variety of cellular processes, such as cytokine generation,77 and that aberrations in ERK1/2/NF-κB signals have been implicated in many pathologies, including infectious diseases.78 Thus, the regulatory effects of the novel NP-siRNA liposomes on ERK1/2/NF-κB signaling pathways make a substantial contribution to the cytokines production in macrophages with the dependence of TGF-β1. However, these effects need to be further examined. Taken together, given the critical role of TGF-β1 in the formation of tuberculous granuloma, combining TGF-β1 siRNA is more conducive to the enhancement of the therapeutic effect of anti-TB drugs in macrophages by targeting ERK1/2/NF-κB signaling pathways.
In summary, a facile approach has been developed to fabricate a liposomal nanoparticle with favorable physicochemical properties for the codelivery of multiple anti-TB drugs as well as the anti-TGF-β1 siRNA simultaneously. The TEM measurements monitoring the drug uptake process at both 37°C and 4°C reveal that the liposomal nanoparticles are engulfed through endocytosis by THP-1-derived macrophages. The liposomal nanoparticles possess a sustained drug release profile over 72 hours, which may help to improve compliance in conventional anti-TB therapy. Notably, the biochemical assays and SILAC-based quantitative proteomics show that the NP-siRNA liposomes display minimal cytotoxicity and exhibit maximum transfection efficiency against the target gene in human macrophages. Furthermore, the novel NP-siRNA liposomes exhibit diverse modulating effects on cytokine generation with the involvement of ERK1/2 and NF-κB signaling pathways in human macrophages. Collectively, the novel synthetic targeting liposomes represent a promising delivery system for anti-TB drugs that may improve the therapeutic effect and reduce the side effects in clinical practice.
Acknowledgments
The authors appreciate the financial support from the Startup Fund of the College of Pharmacy, University of South Florida, Tampa, Florida, USA. This work has been supported in part by the Florida Center of Excellence for Drug Discovery and Innovation at the University of South Florida, Tampa, FL, USA.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Murray CJ, Ortblad KF, Guinovart C, et al. Global, regional, and national incidence and mortality for HIV, tuberculosis, and malaria during 1990–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. 2014;384(9947):1005–1070. doi: 10.1016/S0140-6736(14)60844-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dheda K, Gumbo T, Gandhi NR, et al. Global control of tuberculosis: from extensively drug-resistant to untreatable tuberculosis. Lancet Respir Med. 2014;2(4):321–338. doi: 10.1016/S2213-2600(14)70031-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tanne JH. Some progress and some missed targets in the TB epidemic. BMJ. 2014;348:g2626. doi: 10.1136/bmj.g2626. [DOI] [PubMed] [Google Scholar]
- 4.Glaziou P, Sismanidis C, Floyd K, Raviglione M. Global epidemiology of tuberculosis. Cold Spring Harb Perspect Med. 2014;5(2):a017798. doi: 10.1101/cshperspect.a017798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.World Health Organization . Global Tuberculosis Report 2014. Geneva, Switzerland: World Health Organization; 2015. (Report No 9241564652). [Google Scholar]
- 6.Russell DG, Barry CE, 3rd, Flynn JL. Tuberculosis: what we don’t know can, and does, hurt us. Science. 2010;328(5980):852–856. doi: 10.1126/science.1184784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alami NN, Yuen CM, Miramontes R, et al. Trends in tuberculosis − United States, 2013. MMWR Morb Mortal Wkly Rep. 2014;63(11):229–233. [PMC free article] [PubMed] [Google Scholar]
- 8.Turner RD, Bothamley GH. Cough and the Transmission of Tuberculosis. J Infect Dis. 2015;211(9):1367–1372. doi: 10.1093/infdis/jiu625. [DOI] [PubMed] [Google Scholar]
- 9.Kulchavenya E. Extrapulmonary tuberculosis: are statistical reports accurate? Ther Adv Infect Dis. 2014;2(2):61–70. doi: 10.1177/2049936114528173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chaudhary P. Hepatobiliary tuberculosis. Ann Gastroenterol. 2014;27(3):207–211. [PMC free article] [PubMed] [Google Scholar]
- 11.World Health Organization . Treatment of Tuberculosis: Guidelines. 4th ed. Geneva, Switzerland: World Health Organization; 2010. [Google Scholar]
- 12.Chan JG, Bai X, Traini D. An update on the use of rifapentine for tuberculosis therapy. Expert Opin Drug Deliv. 2014;11(3):421–431. doi: 10.1517/17425247.2014.877886. [DOI] [PubMed] [Google Scholar]
- 13.Field SK, Fisher D, Jarand JM, Cowie RL. New treatment options for multidrug-resistant tuberculosis. Ther Adv Respir Dis. 2012;6(5):255–268. doi: 10.1177/1753465812452193. [DOI] [PubMed] [Google Scholar]
- 14.China Tuberculosis Control Collaboration The effect of tuberculosis control in China. Lancet. 2004;364(9432):417–422. doi: 10.1016/S0140-6736(04)16764-0. [DOI] [PubMed] [Google Scholar]
- 15.Wang L, Zhang H, Ruan Y, et al. Tuberculosis prevalence in China, 1990–2010; a longitudinal analysis of national survey data. Lancet. 2014;383(9934):2057–2064. doi: 10.1016/S0140-6736(13)62639-2. [DOI] [PubMed] [Google Scholar]
- 16.Millard J, Ugarte-Gil C, Moore DA. Multidrug resistant tuberculosis. BMJ. 2015;350:h882. doi: 10.1136/bmj.h882. [DOI] [PubMed] [Google Scholar]
- 17.Gunther G. Multidrug-resistant and extensively drug-resistant tuberculosis: a review of current concepts and future challenges. Clin Med. 2014;14(3):279–285. doi: 10.7861/clinmedicine.14-3-279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jenkins HE, Tolman AW, Yuen CM, et al. Incidence of multidrug-resistant tuberculosis disease in children: systematic review and global estimates. Lancet. 2014;383(9928):1572–1579. doi: 10.1016/S0140-6736(14)60195-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goldstein BP. Resistance to rifampicin: a review. J Antibiot (Tokyo) 2014;67(9):625–630. doi: 10.1038/ja.2014.107. [DOI] [PubMed] [Google Scholar]
- 20.World Health Organization . Guidelines for the Programmatic Management of Drug-resistant Tuberculosis, 2011 Update. Geneva, Switzerland: World Health Organization; 2011. [PubMed] [Google Scholar]
- 21.Matteelli A, Roggi A, Carvalho AC. Extensively drug-resistant tuberculosis: epidemiology and management. Clin Epidemiol. 2014;6:111–118. doi: 10.2147/CLEP.S35839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kakkar AK, Dahiya N. Bedaquiline for the treatment of resistant tuberculosis: promises and pitfalls. Tuberculosis (Edinb) 2014;94(4):357–362. doi: 10.1016/j.tube.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 23.Cox E, Laessig K. FDA approval of bedaquiline − the benefit-risk balance for drug-resistant tuberculosis. N Engl J Med. 2014;371(8):689–691. doi: 10.1056/NEJMp1314385. [DOI] [PubMed] [Google Scholar]
- 24.Wong EB, Cohen KA, Bishai WR. Rising to the challenge: new therapies for tuberculosis. Trends Microbiol. 2013;21(9):493–501. doi: 10.1016/j.tim.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ryan NJ, Lo JH. Delamanid: first global approval. Drugs. 2014;74(9):1041–1045. doi: 10.1007/s40265-014-0241-5. [DOI] [PubMed] [Google Scholar]
- 26.Kwon YS, Jeong BH, Koh WJ. Delamanid when other anti-tuberculosis-treatment regimens failed due to resistance or tolerability. Expert Opin Pharmacother. 2015;16(2):253–261. doi: 10.1517/14656566.2015.973853. [DOI] [PubMed] [Google Scholar]
- 27.Blair HA, Scott LJ. Delamanid: a review of its use in patients with multidrug-resistant tuberculosis. Drugs. 2015;75(1):91–100. doi: 10.1007/s40265-014-0331-4. [DOI] [PubMed] [Google Scholar]
- 28.Szumowski JD, Lynch JB. Profile of delamanid for the treatment of multidrug-resistant tuberculosis. Drug Des Devel Ther. 2015;9:677–682. doi: 10.2147/DDDT.S60923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yee D, Valiquette C, Pelletier M, Parisien I, Rocher I, Menzies D. Incidence of serious side effects from first-line antituberculosis drugs among patients treated for active tuberculosis. Am J Respir Crit Care Med. 2003;167(11):1472–1477. doi: 10.1164/rccm.200206-626OC. [DOI] [PubMed] [Google Scholar]
- 30.Forget EJ, Menzies D. Adverse reactions to first-line antituberculosis drugs. Expert Opin Drug Saf. 2006;5(2):231–249. doi: 10.1517/14740338.5.2.231. [DOI] [PubMed] [Google Scholar]
- 31.Hosford JD, von Fricken ME, Lauzardo M, et al. Hepatotoxicity from antituberculous therapy in the elderly: a systematic review. Tuberculosis (Edinb) 2014;95(2):112–122. doi: 10.1016/j.tube.2014.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Choudhary S, Kusum Devi V. Potential of nanotechnology as a delivery platform against tuberculosis: current research review. J Control Release. 2015;202C:65–75. doi: 10.1016/j.jconrel.2015.01.035. [DOI] [PubMed] [Google Scholar]
- 33.Griffiths G, Nystrom B, Sable SB, Khuller GK. Nanobead-based interventions for the treatment and prevention of tuberculosis. Nat Rev Microbiol. 2010;8(11):827–834. doi: 10.1038/nrmicro2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Banyal S, Malik P, Tuli HS, Mukherjee TK. Advances in nanotechnology for diagnosis and treatment of tuberculosis. Curr Opin Pulm Med. 2013;19(3):289–297. doi: 10.1097/MCP.0b013e32835eff08. [DOI] [PubMed] [Google Scholar]
- 35.Dube A, Lemmer Y, Hayeshi R, et al. State of the art and future directions in nanomedicine for tuberculosis. Expert Opin Drug Deliv. 2013;10(12):1725–1734. doi: 10.1517/17425247.2014.846905. [DOI] [PubMed] [Google Scholar]
- 36.Kaur M, Garg T, Rath G, Goyal AK. Current nanotechnological strategies for effective delivery of bioactive drug molecules in the treatment of tuberculosis. Crit Rev Ther Drug Carrier Syst. 2014;31(1):49–88. doi: 10.1615/critrevtherdrugcarriersyst.2014008285. [DOI] [PubMed] [Google Scholar]
- 37.Mehanna MM, Mohyeldin SM, Elgindy NA. Respirable nanocarriers as a promising strategy for antitubercular drug delivery. J Control Release. 2014;187:183–197. doi: 10.1016/j.jconrel.2014.05.038. [DOI] [PubMed] [Google Scholar]
- 38.Kaur IP, Singh H. Nanostructured drug delivery for better management of tuberculosis. J Control Release. 2014;184:36–50. doi: 10.1016/j.jconrel.2014.04.009. [DOI] [PubMed] [Google Scholar]
- 39.Vyas SP, Khatri K. Liposome-based drug delivery to alveolar macrophages. Expert Opin Drug Deliv. 2007;4(2):95–99. doi: 10.1517/17425247.4.2.95. [DOI] [PubMed] [Google Scholar]
- 40.Pinheiro M, Lucio M, Lima JL, Reis S. Liposomes as drug delivery systems for the treatment of TB. Nanomedicine (Lond) 2011;6(8):1413–1428. doi: 10.2217/nnm.11.122. [DOI] [PubMed] [Google Scholar]
- 41.Pham DD, Fattal E, Tsapis N. Pulmonary drug delivery systems for tuberculosis treatment. Int J Pharm. 2015;478(2):517–529. doi: 10.1016/j.ijpharm.2014.12.009. [DOI] [PubMed] [Google Scholar]
- 42.Berrington WR, Hawn TR. Mycobacterium tuberculosis, macrophages, and the innate immune response: does common variation matter? Immunol Rev. 2007;219:167–186. doi: 10.1111/j.1600-065X.2007.00545.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zuniga J, Torres-Garcia D, Santos-Mendoza T, Rodriguez-Reyna TS, Granados J, Yunis EJ. Cellular and humoral mechanisms involved in the control of tuberculosis. Clin Dev Immunol. 2012;2012:193923. doi: 10.1155/2012/193923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Torrado E, Cooper AM. Cytokines in the balance of protection and pathology during mycobacterial infections. Adv Exp Med Biol. 2013;783:121–140. doi: 10.1007/978-1-4614-6111-1_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Matucci A, Maggi E, Vultaggio A. Cellular and humoral immune responses during tuberculosis infection: useful knowledge in the era of biological agents. J Rheumatol Suppl. 2014;91:17–23. doi: 10.3899/jrheum.140098. [DOI] [PubMed] [Google Scholar]
- 46.de Martino M, Galli L, Chiappini E. Reflections on the immunology of tuberculosis: will we ever unravel the skein? BMC Infect Dis. 2014;14(Suppl 1):S1. doi: 10.1186/1471-2334-14-S1-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Parandhaman DK, Narayanan S. Cell death paradigms in the pathogenesis of Mycobacterium tuberculosis infection. Front Cell Infect Microbiol. 2014;4:31. doi: 10.3389/fcimb.2014.00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chan J, Mehta S, Bharrhan S, et al. The role of B cells and humoral immunity in Mycobacterium tuberculosis infection. Semin Immunol. 2014;26(6):588–600. doi: 10.1016/j.smim.2014.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Urdahl KB. Understanding and overcoming the barriers to T cell-mediated immunity against tuberculosis. Semin Immunol. 2014;26(6):578–587. doi: 10.1016/j.smim.2014.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cooper AM. Cell-mediated immune responses in tuberculosis. Annu Rev Immunol. 2009;27:393–422. doi: 10.1146/annurev.immunol.021908.132703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rajaram MV, Ni B, Dodd CE, Schlesinger LS. Macrophage immunoregulatory pathways in tuberculosis. Semin Immunol. 2014;26(6):471–485. doi: 10.1016/j.smim.2014.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bruns H, Stenger S. New insights into the interaction of Mycobacterium tuberculosis and human macrophages. Future Microbiol. 2014;9(3):327–341. doi: 10.2217/fmb.13.164. [DOI] [PubMed] [Google Scholar]
- 53.Etna MP, Giacomini E, Severa M, Coccia EM. Pro- and anti-inflammatory cytokines in tuberculosis: a two-edged sword in TB pathogenesis. Semin Immunol. 2014;26(6):543–551. doi: 10.1016/j.smim.2014.09.011. [DOI] [PubMed] [Google Scholar]
- 54.Monin L, Khader SA. Chemokines in tuberculosis: the good, the bad and the ugly. Semin Immunol. 2014;26(6):552–558. doi: 10.1016/j.smim.2014.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dorhoi A, Kaufmann SH. Tumor necrosis factor-α in mycobacterial infection. Semin Immunol. 2014;26(3):203–209. doi: 10.1016/j.smim.2014.04.003. [DOI] [PubMed] [Google Scholar]
- 56.Toossi Z, Ellner JJ. The role of TGFβ in the pathogenesis of human tuberculosis. Clin Immunol Immunopathol. 1998;87(2):107–114. doi: 10.1006/clin.1998.4528. [DOI] [PubMed] [Google Scholar]
- 57.Friedland JS. Targeting the inflammatory response in tuberculosis. N Engl J Med. 2014;371(14):1354–1356. doi: 10.1056/NEJMcibr1408663. [DOI] [PubMed] [Google Scholar]
- 58.Wu M, Aung H, Hirsch CS, Toossi Z. Inhibition of Mycobacterium tuberculosis-induced signalling by transforming growth factor-β in human mononuclear phagocytes. Scand J Immunol. 2012;75(3):301–304. doi: 10.1111/j.1365-3083.2011.02668.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mayer-Barber KD, Andrade BB, Oland SD, et al. Host-directed therapy of tuberculosis based on interleukin-1 and type I interferon crosstalk. Nature. 2014;511(7507):99–103. doi: 10.1038/nature13489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jin W, Wang Q, Wang Z, Geng G. Complete debridement for treatment of thoracolumbar spinal tuberculosis: a clinical curative effect observation. Spine J. 2014;14(6):964–970. doi: 10.1016/j.spinee.2013.07.466. [DOI] [PubMed] [Google Scholar]
- 61.Wang Z, Shi J, Geng G, Qiu H. Ultra-short-course chemotherapy for spinal tuberculosis: five years of observation. Eur Spine J. 2013;22(2):274–281. doi: 10.1007/s00586-012-2536-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang Z, Ge Z, Jin W, et al. Treatment of spinal tuberculosis with ultrashort-course chemotherapy in conjunction with partial excision of pathologic vertebrae. Spine J. 2007;7(6):671–681. doi: 10.1016/j.spinee.2006.07.016. [DOI] [PubMed] [Google Scholar]
- 63.Qin Y, Zhou ZW, Pan ST, et al. Graphene quantum dots induce apoptosis, autophagy, and inflammatory response via p38 mitogen-activated protein kinase and nuclear factor-kappaB mediated signaling pathways in activated THP-1 macrophages. Toxicology. 2015;327:62–76. doi: 10.1016/j.tox.2014.10.011. [DOI] [PubMed] [Google Scholar]
- 64.Yin JJ, Sharma S, Shumyak SP, et al. Synthesis and biological evaluation of novel folic acid receptor-targeted, β-cyclodextrin-based drug complexes for cancer treatment. PLoS One. 2013;8(5):e62289. doi: 10.1371/journal.pone.0062289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li YC, He SM, He ZX, et al. Plumbagin induces apoptotic and autophagic cell death through inhibition of the PI3K/Akt/mTOR pathway in human non-small cell lung cancer cells. Cancer Lett. 2014;344(2):239–259. doi: 10.1016/j.canlet.2013.11.001. [DOI] [PubMed] [Google Scholar]
- 66.Ding YH, Zhou ZW, Ha CF, et al. Alisertib, an Aurora kinase A inhibitor, induces apoptosis and autophagy but inhibits epithelial to mesenchymal transition in human epithelial ovarian cancer cells. Drug Des Devel Ther. 2015;9:425–464. doi: 10.2147/DDDT.S74062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yuan CX, Zhou ZW, Yang YX, et al. Inhibition of mitotic Aurora kinase A by alisertib induces apoptosis and autophagy of human gastric cancer AGS and NCI-N78 cells. Drug Des Devel Ther. 2015;9:487–508. doi: 10.2147/DDDT.S74127. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 68.Zhou ZW, Li XX, He ZX, et al. Induction of apoptosis and autophagy via sirtuin1- and PI3K/Akt/mTOR-mediated pathways by plumbagin in human prostate cancer cells. Drug Des Devel Ther. 2015;9:1511–1554. doi: 10.2147/DDDT.S75976. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 69.Qiu JX, Zhou ZW, He ZX, et al. Plumbagin elicits differential proteomic responses mainly involving cell cycle, apoptosis, autophagy, and epithelial-to-mesenchymal transition pathways in human prostate cancer PC-3 and DU145 cells. Drug Des Devel Ther. 2015;9:349–417. doi: 10.2147/DDDT.S71677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pan ST, Zhou ZW, He ZX, et al. Proteomic response to 5,6-dimethylxanthenone 4-acetic acid (DMXAA, vadimezan) in human non-small cell lung cancer A549 cells determined by the stable-isotope labeling by amino acids in cell culture (SILAC) approach. Drug Des Devel Ther. 2015;9:937–968. doi: 10.2147/DDDT.S76021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Koul HK, Pal M, Koul S. Role of p38 MAP kinase signal transduction in solid tumors. Genes Cancer. 2013;4(9–10):342–359. doi: 10.1177/1947601913507951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wagner EF, Nebreda AR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer. 2009;9(8):537–549. doi: 10.1038/nrc2694. [DOI] [PubMed] [Google Scholar]
- 73.Baker SJ. PTEN enters the nuclear age. Cell. 2007;128(1):25–28. doi: 10.1016/j.cell.2006.12.023. [DOI] [PubMed] [Google Scholar]
- 74.Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011;18(4):571–580. doi: 10.1038/cdd.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Green DR, Levine B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell. 2014;157(1):65–75. doi: 10.1016/j.cell.2014.02.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hurley JH, Schulman BA. Atomistic autophagy: the structures of cellular self-digestion. Cell. 2014;157(2):300–311. doi: 10.1016/j.cell.2014.01.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Deschenes-Simard X, Kottakis F, Meloche S, Ferbeyre G. ERKs in cancer: friends or foes? Cancer Res. 2014;74(2):412–419. doi: 10.1158/0008-5472.CAN-13-2381. [DOI] [PubMed] [Google Scholar]
- 78.Ramos JW. The regulation of extracellular signal-regulated kinase (ERK) in mammalian cells. Int J Biochem Cell Biol. 2008;40(12):2707–2719. doi: 10.1016/j.biocel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 79.Garcia-Monco JC. Tuberculosis. Handb Clin Neurol. 2014;121:1485–1499. doi: 10.1016/B978-0-7020-4088-7.00100-0. [DOI] [PubMed] [Google Scholar]
- 80.Horsburgh CR., Jr Tuberculosis. Eur Respir Rev. 2014;23(131):36–39. doi: 10.1183/09059180.00008213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Alliance T. Streptomycin. Tuberculosis (Edinb) 2008;88(2):162–163. doi: 10.1016/S1472-9792(08)70027-1. [DOI] [PubMed] [Google Scholar]
- 82.Chaisson RE, Churchyard GJ. Recurrent tuberculosis: relapse, reinfection, and HIV. J Infect Dis. 2010;201(5):653–655. doi: 10.1086/650531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Salem II, Flasher DL, Duzgunes N. Liposome-encapsulated antibiotics. Methods Enzymol. 2005;391:261–291. doi: 10.1016/S0076-6879(05)91015-X. [DOI] [PubMed] [Google Scholar]
- 84.Sarkar S, Suresh MR. An overview of tuberculosis chemotherapy – a literature review. J Pharm Pharm Sci. 2011;14(2):148–161. doi: 10.18433/j33591. [DOI] [PubMed] [Google Scholar]
- 85.Wilczewska AZ, Niemirowicz K, Markiewicz KH, Car H. Nanoparticles as drug delivery systems. Pharmacol Rep. 2012;64(5):1020–1037. doi: 10.1016/s1734-1140(12)70901-5. [DOI] [PubMed] [Google Scholar]
- 86.Korbel DS, Schneider BE, Schaible UE. Innate immunity in tuberculosis: myths and truth. Microbes Infect. 2008;10(9):995–1004. doi: 10.1016/j.micinf.2008.07.039. [DOI] [PubMed] [Google Scholar]
- 87.Helmuth L. Microbiology – a weak link in TB bacterium is found. Science. 2000;289(5482):1123–1125. doi: 10.1126/science.289.5482.1123a. [DOI] [PubMed] [Google Scholar]
- 88.Orme IM, Basaraba RJ. The formation of the granuloma in tuberculosis infection. Semin Immunol. 2014;26(6):601–609. doi: 10.1016/j.smim.2014.09.009. [DOI] [PubMed] [Google Scholar]
- 89.Ellner JJ. Immunoregulation in TB: observations and implications. Clin Transl Sci. 2010;3(1):23–28. doi: 10.1111/j.1752-8062.2010.00180.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Li H, Liang CZ, Tao YQ, Shen CC, Chen QX. Elevated local TGF-β1 level predisposes a closed bone fracture to tuberculosis infection. Med Hypotheses. 2012;79(3):400–402. doi: 10.1016/j.mehy.2012.06.005. [DOI] [PubMed] [Google Scholar]
- 91.Cambier CJ, Falkow S, Ramakrishnan L. Host evasion and exploitation schemes of Mycobacterium tuberculosis. Cell. 2014;159(7):1497–1509. doi: 10.1016/j.cell.2014.11.024. [DOI] [PubMed] [Google Scholar]
- 92.Carthew RW, Sontheimer EJ. Origins and mechanisms of miRNAs and siRNAs. Cell. 2009;136(4):642–655. doi: 10.1016/j.cell.2009.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lima WF, Prakash TP, Murray HM, et al. Single-stranded siRNAs activate RNAi in animals. Cell. 2012;150(5):883–894. doi: 10.1016/j.cell.2012.08.014. [DOI] [PubMed] [Google Scholar]
- 94.Hwang M, Kim H-J, Noh H-J, et al. TGF-β1 siRNA suppresses the tubulointerstitial fibrosis in the kidney of ureteral obstruction. Exp Mol Pathol. 2006;81(1):48–54. doi: 10.1016/j.yexmp.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 95.Mann M. Functional and quantitative proteomics using SILAC. Nat Rev Mol Cell Biol. 2006;7(12):952–958. doi: 10.1038/nrm2067. [DOI] [PubMed] [Google Scholar]
- 96.Ong SE, Mann M. A practical recipe for stable isotope labeling by amino acids in cell culture (SILAC) Nat Protoc. 2006;1(6):2650–2660. doi: 10.1038/nprot.2006.427. [DOI] [PubMed] [Google Scholar]